
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected evolution of Rhodotorula gracilisD-amino acid oxidase using single-cell hydrogel encapsulation and ultrahigh-throughput screening†
Christoph
Küng
 ab,
Rosario
Vanella
ab and
Michael A.
Nash
*ab
ab,
Rosario
Vanella
ab and
Michael A.
Nash
*ab
aInstitute of Physical Chemistry, Department of Chemistry, University of Basel, 4058 Basel, Switzerland. E-mail: michael.nash@unibas.ch
bDepartment of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland
First published on 20th April 2023
Abstract
Engineering catalytic and biophysical properties of enzymes is an essential step en route to advanced biomedical and industrial applications. Here, we developed a high-throughput screening and directed evolution strategy relying on single-cell hydrogel encapsulation to enhance the performance of D-Amino acid oxidase from Rhodotorula gracilis (RgDAAOx), a candidate enzyme for cancer therapy. We used a cascade reaction between RgDAAOx variants surface displayed on yeast and horseradish peroxidase (HRP) in the bulk media to trigger enzyme-mediated crosslinking of phenol-bearing fluorescent alginate macromonomers, resulting in hydrogel formation around single yeast cells. The fluorescent hydrogel capsules served as an artificial phenotype and basis for pooled library screening by fluorescence activated cell sorting (FACS). We screened a RgDAAOx variant library containing ∼106 clones while lowering the D-Ala substrate concentration over three sorting rounds in order to isolate variants with low Km. After three rounds of FACS sorting and regrowth, we isolated and fully characterized four variants displayed on the yeast surface. We identified variants with a more than 5-fold lower Km than the parent sequence, with an apparent increase in substrate binding affinity. The mutations we identified were scattered across the RgDAAOx structure, demonstrating the difficulty in rationally predicting allosteric sites and highlighting the advantages of scalable library screening technologies for evolving catalytic enzymes.
Introduction
D-Amino acid oxidase (DAAOx, EC.1.4.3.3.) from Rhodotorula gracilis is an 80 kDa homodimeric enzyme that utilizes flavin adenine dinucleotide (FAD) as a redox cofactor (Fig. 1A). The active dimer builds a head-to-tail complex and promiscuously oxidizes many D-amino acid substrates1 into their corresponding α-imino acids using molecular oxygen as an electron acceptor, producing one equivalent of hydrogen peroxide (H2O2) in the process. The imino acid product then spontaneously rearranges giving the respective α-keto acid and ammonia (Fig. 1B).2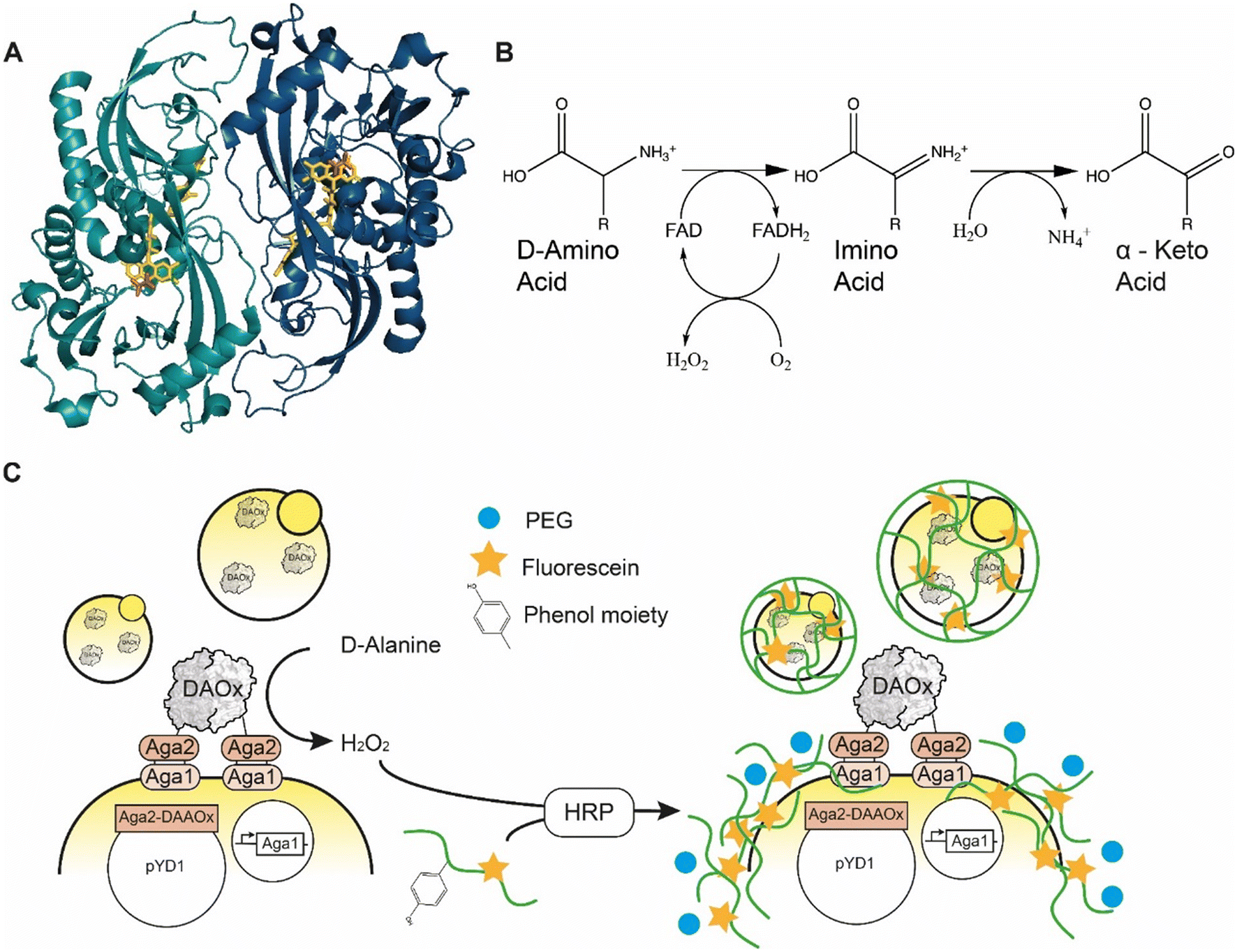 | ||
| Fig. 1 A) Quaternary structure of Rhodotorula gracilisD-amino acid oxidase dimer with D-alanine in active site (enzyme monomers: light and dark blue, FAD: yellow, D-ALA, orange) PDB: 1c0p. B) Oxidation reaction catalyzed by D-amino acid oxidase. C) Scheme of the yeast cell encapsulation method. The addition of PEG (blue circle) increases the viscosity and enables encapsulation of enzymes with lower turnover number. | ||
DAAOx is under investigation in a number of biomedical contexts. For example, human DAAOx is involved in regulating brain levels of D-serine, a neuromodulator and agonist of the NMDA receptor implicated in schizophrenia.3,4 DAAOx has been further used in biosensors for measuring levels of D-amino acids in food products,5 and in the context of engineered biological therapeutics variants of DAAOx are considered candidates for cancer therapy.6–9 In oxidative anticancer therapy, the generation of reactive oxygen species (i.e. H2O2) upon conversion of prodrug substrates can generate therapeutic cytotoxicity based on pre-sensitization of cancer cells to oxidative stress.10Rhodotorula gracilisD-Amino acid oxidase (RgDAAOx) has been suggested as one candidate enzyme for such an approach due to its high catalytic activity and stable interaction with its FAD cofactor (KD = 2 × 10−8 M).3 However, wild type (WT) RgDAAOx is limited by a relatively low affinity for O2 (Km = 1.9 mM)7 and weak binding to D-amino acid substrates, requiring high substrate doses to achieve measurable cytotoxicity. Rosini et al. used multiwell plate screening to engineer DAAOx towards higher oxygen binding affinity and screened their library under depleted D-alanine conditions, isolating a variant with a lower KM,O2.7 Indeed, the dependence of, and the low affinity towards molecular oxygen are factors limiting the industrial applications of not only D-amino acid oxidase but numerous O2-dependent enzymes.11 Other research was also conducted to engineer substrate specificity of RgDAAOx,12–14 however, large library screening over sequential rounds to improve substrate binding affinity and/or enhance KM to the D-amino acid substrate was not yet reported.
Fine tuning desired characteristics of enzymes may be achieved by rational design, using in silico models as basis for mutagenesis studies15 or higher throughput methodologies like directed evolution.16 Latter is a valuable approach that allows researchers to explore protein fitness landscapes and shape the characteristics of enzymes through artificial screening/selection. Although screening enzyme variant libraries in multiwell plates generates detailed characterization data, it is not realistically scalable to large libraries (>106 members) even with substantial automation, robotics and liquid handling infrastructure. Ultrahigh-throughput screening methods based on a variety of in vitro microscale (i.e. single-cell) compartmentalization17 strategies or alternatively growth based selections18,19 enable large library screening. However, these approaches may suffer from limitations such as requiring tailoring for a specific enzyme chemistry, necessitating microfluidic device design, fabrication, and utilization, or otherwise being limited in chemical scope. Approaches to achieve efficient and scalable reaction compartmentalization and enable genotype–phenotype linkage for the purposes of enzyme directed evolution are therefore still highly sought after.20
Here we report an approach to address this genotype–phenotype linkage and scalability challenge for ultrahigh-throughput RgDAAOx library screening. Our approach is based on peroxidase-mediated polymerization of hydrogel capsules around single cells where we used the ability of a particular variant sequence to trigger the hydrogel polymerization reaction under stringent conditions as a screening filter to identify active and stable variants. We previously described the concept of a similar single-cell hydrogel encapsulation platform for screening a library of glucose oxidase variants, which like RgDAAOx generate hydrogen peroxide21,22 during the catalytic cycle. In our system, enzyme variants displayed on yeast generate H2O2, which is used by horseradish peroxidase (HRP) to generate phenol radicals borne by crosslinkable fluorescent alginate macromonomer chains. The HRP in the reaction mixture oxidizes the phenolic residues of the modified alginate upon reaction with H2O2, forming highly reactive and short-lived phenoxyl radicals. For enzyme variants liberating sufficient quantities of H2O2, alginate molecules can be crosslinked and form a hydrogel around the cell expressing the enzyme (Fig. 1C).23 The fluorophores conjugated to the alginate allow the verification of encapsulation by single cell sorting. In a fluorescence activated cell sorting (FACS) experiment, successfully encapsulated cells can be differentiated from cells without a hydrogel shell, thus achieving a coupling of the phenotype to the underlying genetic sequence. This platform provides a high-throughput screening (HTS) system and enables large library screening. We used this platform to engineer DAAOx over several rounds towards higher D-alanine affinity and characterized the Km constants of the resulting variants.
Results and discussion
To use the hydrogel encapsulation system with RgDAAOx, we first cloned the gene encoding Rhodotorula gracilisD-amino acid oxidase into a pYD1 yeast display expression vector in-frame with the Aga2p a-agglutinin protein followed by a C-terminal hexa-histidine tag. The last three residues which form a peroxisomal targeting tag of the RgDAAOx sequence were excluded from the fusion construct.24 After sequence confirmation by Sanger sequencing, we transformed the plasmid into chemically competent Saccharomyces Cerevisiae (EBY100), which contained a genomic copy of Aga1p under control of a galactose promoter.25 This strain is amenable for yeast surface display, can mediate protein glycosylation and is capable of displaying relatively large proteins.26 We induced enzyme expression by transferring the cells to growth medium supplemented with galactose and used immunostaining with anti-His antibodies plus secondary antibodies conjugated to Alexa Fluor 594 to verify translocation and correct anchoring of the enzymes on the cell wall. By means of flow cytometry, we detected successful labeling of more than 70% of induced cells, thus verifying the presence of the C-terminal hexa-histidine at the outer cell wall. We confirmed the activity of displayed enzymes in triplicates in a plate based Amplex Red™ assay, which demonstrated correct assembly of active homodimers (Fig. S1†).During initial attempts to adapt the system from glucose oxidase to D-amino acid oxidase, we tested the same reaction conditions for encapsulation as previously reported.21 However, for cells displaying RgDAAOx, the protocol did not lead to any cell encapsulation, as observed by flow cytometry. A potential reason for this was that RgDAAOx has a significantly lower turnover rate than glucose oxidase. This resulted in a lower rate of H2O2 production and lower rate of alginate–phenol oxidation. The alginate molecules that reacted then likely diffused away before crosslinking was achieved, limiting encapsulation of yeast cells. Further contributing to this, it was reported that His-tag immobilized RgDAAOx had a lower apparent affinity towards D-alanine and a slower turnover rate than the free enzyme (23.3 and 52.4 s−1, respectively).27 For these reasons, we concluded that the yeast surface displayed enzyme was too slow for the encapsulation under standard conditions. To address this, we sought to slow the diffusion of all soluble components in the reaction mixture through the addition of viscosity enhancing compounds. Specifically, we added polyethylene glycol (PEG) to the reaction mixture and found that this additive successfully recovered single-cell labeling (Fig. 2).
Higher percentages of added PEG led to larger fractions of cells in the first (upper right) FACS quadrant (Fig. 2) that were both positive for expression and encapsulation. At 5% (w/v) PEG, we found 6% of gated single cells were positive for expression and encapsulation, while doubling the PEG concentration to 10% (w/v) increased this population to ∼19%. Finally, a concentration of 20% (w/v) PEG led to encapsulation of over 63% of the gated cells. Difficulties in accurate pipetting of high viscosity PEG mixtures imposed a practical limit on this approach to ∼20% (w/v) PEG. The encapsulation protocol with PEG as a viscosity enhancer maintained good specificity for DAAOx-expressing cells, as can be seen in Fig. 2 showing the negative population corresponding to the non-expressing cells remaining in the third (lower left) quadrant. Negative reaction controls either lacking HRP in the medium or containing yeasts displaying an empty cassette with only Aga2-His resulted in no encapsulation. Encapsulation reactions were repeated in triplicates and results are shown in a barplot in Fig. S2.†
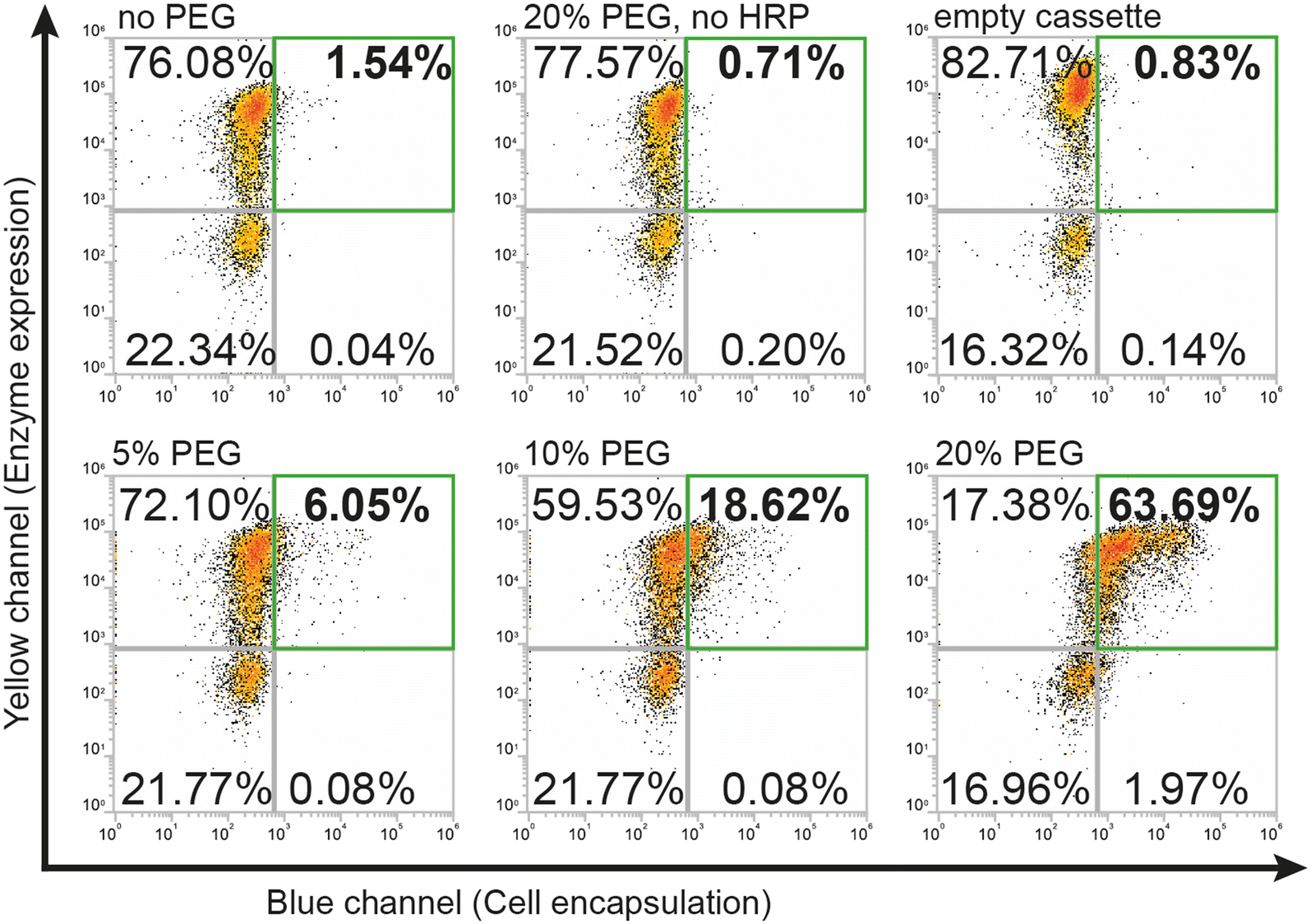 | ||
| Fig. 2 Flow cytometry density plots demonstrating the effect of increasing percentages (w/v) of PEG on encapsulation efficiency. Negative controls illustrate no shifts when PEG, HRP or the DAAOx enzyme are absent. | ||
To make use of this system for engineering RgDAAOx towards enhanced characteristics, we next constructed a variant library. Gene diversification was achieved by generating two libraries, one by error prone PCR and one by site saturation mutagenesis using NNK primers, both targeting nucleotides coding for positions 2 to 365 of the enzyme. We chose conditions for error prone PCR such that a mutational load of 3 mutations/kb was attained, which we considered a good complement to the saturation library mainly focusing on single substitution mutants (Fig. S3†). The mutagenized genes were transformed together with a linearized pYD1 backbone into yeast and assembled in vivo by gap repair. After growth of the yeast cells, the libraries were merged according to their complexity to avoid overrepresentation of one part of the library. We estimated the size of the combined library to be ∼1 × 106 through plating dilutions of the transformed yeast and counting colony forming units.
In order to isolate mutant sequences with enhanced substrate affinity, we subjected the variant library to subsequent rounds of FACS-based selection while lowering the substrate concentration. We used FACS to sort cells that maintained the ability to auto-encapsulate in the fluorescent hydrogel while lowering the substrate concentration in the reaction mix in every round, expecting to enrich variants with higher affinity towards D-alanine from the library. We maintained a reaction incubation time of 10 minutes in every round, therefore a higher substrate affinity was necessary to generate sufficient crosslinking of fluorescent alginate to encapsulate the cell. Before the first sorting round, we encapsulated the cells in a reaction mixture with a PEG concentration of 20% (w/v) and a starting substrate concentration of 1.5 mM D-alanine. These starting conditions allowed a significant fraction of cells to be selected in the first round, and represented a substrate concentration ∼two times the Km of the wild type enzyme (Km,WT = 0.8 mM).13
On the FACS, we gated the yeast cells for singlets, excluding aggregates and background. We designed the sorting gate to separate cells displaying enzyme variants that were sufficiently active for encapsulation, however we excluded very strongly expressed variants (Fig. S4†). With this strategy we avoided selecting highly expressed enzymes with mediocre substrate affinity. We processed a total of 5 × 106 cells in a single sorting session, over sampling the library 5-fold. The sorted cells were able to be regrown and break free from the hydrogel shells, repopulating the growth media. Next, they were re-induced for DAAOx expression by transferring them to a growth medium supplemented with galactose in preparation for the next sorting round. The library was sorted two more times at substrate concentrations of 0.5 and 0.1 mM, and 1 × 106 and 225![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were sorted in each round, respectively. In the last sorting round, single cells were sorted into separate wells of 96-well plates.
000 cells were sorted in each round, respectively. In the last sorting round, single cells were sorted into separate wells of 96-well plates.
In order to characterize the selected variants, 95 monogenic samples plus wild type were grown and induced in a 96 well plate-based experiment. We detected initial velocities of the enzyme variants in an Amplex Red™ assay and compared them to wild type. The best 10 variants were retested from monogenic 5 mL expression cultures. Subsequently, we selected and analyzed in detail the 4 variants with the lowest apparent Km, termed V1 to V4. For more accurate measurements, we re-expressed the mutants and determined Michaelis Menten constants for yeast surface displayed enzymes in triplicates along with the wild type enzyme (Fig. 3A).
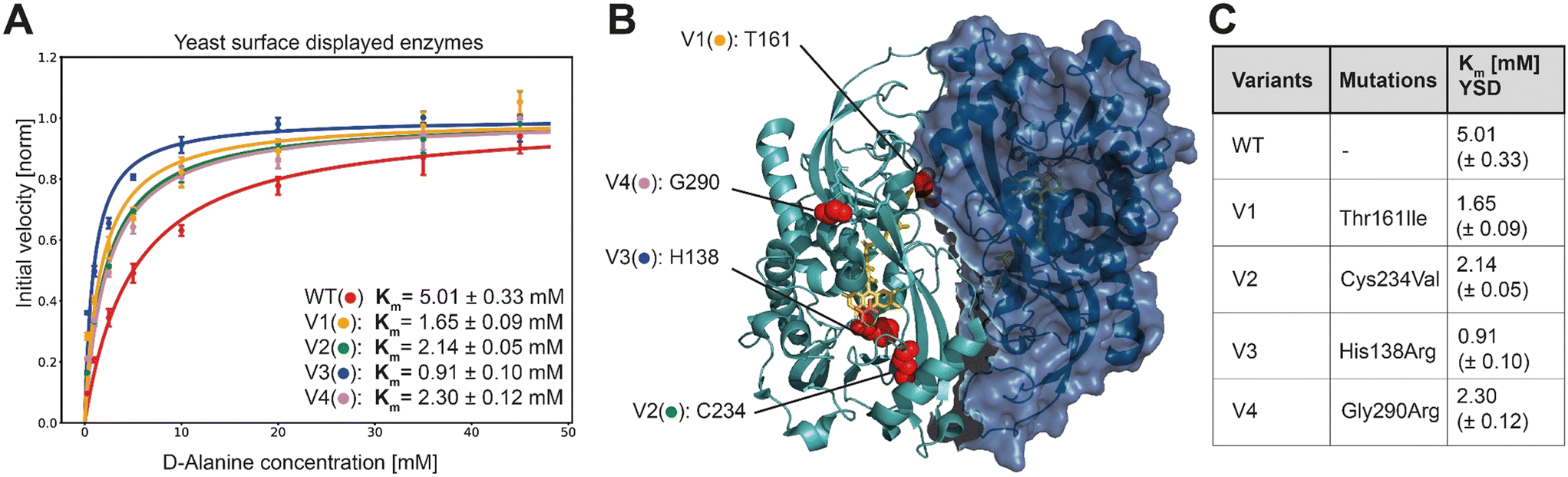 | ||
| Fig. 3 A) Extracted initial velocities plotted over substrate concentration. Michaelis–Menten equation is fitted on data for determination of Km constant. B) 3D structure of active dimer of RgDAAOx with one monomer showing the surface structure. Sites mutated in variants are highlighted as red spheres. FAD is colored in yellow and D-alanine substrate is shown in orange (PDB: 1c0p). C) Mutations and Michaelis–Menten constants for yeast surface displayed wild type and DAAOx-variants. | ||
Since the Michaelis–Menten parameter Km does not depend on enzyme concentration, we could determine the Km parameter by fitting initial velocity of enzymes at different substrate concentrations directly employing enzymes displayed on yeast cells at unknown absolute concentrations. Based on flow cytometry analysis, we knew the display level of the enzyme varied between the variants (Fig. S5†). Initial reaction velocities for each variant were normalized to their respective Vmax values. This allowed direct fitting of the Km parameter. We note that although Km does not reflect substrate binding (affinity) directly, it is closely related. Under an assumption that kcat is the rate limiting step in the reaction, Kd can be approximated by Km. Here, we use the term “affinity” to describe this apparent affinity, however we note it is different from the enzyme-substrate Kd. Since the library was selected under conditions of decreasing substrate concentration, we expect KM to be the dominant parameter influenced by the selection rounds. Complete characterization of soluble enzyme would be necessary to correctly determine kcat and Kd in addition to KM as done here for these variants.
K M-fitting of the four best performing variants to emerge from the sorting rounds showed an approximate two-fold increase in affinity and for mutant V3 a more than five fold lower Km was detected. Those findings reflect a successful screening strategy for variants with increased affinity towards D-alanine. Another consequence of the gating strategy is that we select not for stable enzymes, reflected in the lower than wildtype median fluorescence intensity (MFI) (Fig. S5†). However, the selected variants were, despite their low expression level, still able to trigger cell encapsulation, pointing to increased substrate affinity. This is also reflected in Fig. S5† where the differences in Vmax (not normalized) are apparent from the fitted curves, at low concentration however the initial velocities are very similar. While at the highest concentration used for Michaelis–Menten analysis (45 mM) the wild type was 4.9 times faster than the slowest variant (V3) it is only 1.4 times faster at the lowest assayed concentration (0.25 mM). In another example for V2, the higher affinity is even more evident, where the initial velocity is higher at the lowest concentration than for the wildtype although Vmax is 1.6 times lower. These results directly present the outcome of the sorting strategy, considering we sorted the last selection round at 0.1 mM D-alanine.
In Fig. 3B the mutations are mapped on the 3D structure (PDB: 1c0p) and highlighted in cyan.
As determined by Sanger sequence analysis, the improved affinity was achieved by single mutations listed in Fig. 3C. To the best of our knowledge no prior work reported any of these mutations leading to increased substrate affinity. The single point mutations do not lie together in a specific domain, but are scattered across the whole enzyme, implying that they would have been hard to detect by rational design (Fig. 3B). Variant V1 contains the single mutation of a threonine at site 161 to an isoleucine. This change from a polar to a neutral amino acid is far away from the active site, but interestingly is among the residues interacting at the dimer interface. It remains elusive however, how the mechanism for increased substrate affinity is achieved. Being a potential target for O-glycosylation we can also speculate that exchanging this residue causes a change in the glycosylation pattern of the enzyme with indirect influence in substrate affinity. Variant V4 introduces an arginine, exchanging the glycine at site 290. It is worth noting that together with R288, R289 and R293, the mutated R290 introduces another positive charge in the already strongly positive charged loop resulting in a –RR![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) GPR– motif. Moreover, the loop running in parallel, consists of the motif –DDQAAE– providing negative countercharges. This mutation more than halves the Km in our experiments with yeast displayed enzymes.
GPR– motif. Moreover, the loop running in parallel, consists of the motif –DDQAAE– providing negative countercharges. This mutation more than halves the Km in our experiments with yeast displayed enzymes.
Another mutation found among our top clones was the change of cysteine to glycine at position 234 (variant V2). While it is nowhere reported in literature or experimentally confirmed, prediction servers for disulfide bonds suggest the cysteines at site 234 and 259 form a disulfide bond. Furthermore, the online tool “PredDisulfideServer” suggests that a disulfide bond is formed between those cysteines with a probability of 99.5%.28 The described mutation would hence eliminate this disulfide bond. Moreover, worth noting is that a mutation at the same site (C234R) was reported previously in combination with a D40G mutation.12 This double mutant was selected from an error prone PCR library and showed increased modified substrate specificity with higher than wild type activity for D-Asp and D-Arg. However, it could not be further studied due to low expression yield, rendering purification impossible. The authors speculated that the D40G mutation caused this low expression level, since also a second variant containing D40G could not be expressed. Hence, the direct impact of the cysteine substitution mutation is unknown. It may be that the disruption of the disulfide bond is accompanied by a drop in enzyme stability, leading to a more accessible active site or more mobile catalytic site, leading to broader substrate promiscuity and in our case higher affinity towards D-alanine.
We can only conclude that these mutations generate changes in the structure with knock-on effects on the stability and substrate binding capability of the yeast displayed enzyme. Alteration of glycosylation of the enzyme in the host yeast strain can also not be ruled out as a possible mechanism by which the identified mutations lower KM.
We were interested in studying how the soluble expressed variants would perform in comparison with their yeast surface displayed counterparts. To achieve this, we expressed the enzyme variants and WT sequences lacking the Aga2 fusion domain using codon optimized DNA in E. coli and purified them as soluble proteins. Subsequently, we assayed the Michaelis Menten constants of the variants along with the WT enzyme. For all variants, we observed Km values that deviated somewhat from the results obtained for the same variants displayed as Aga2 fusion proteins anchored to the yeast surface.
While in the yeast displayed format we obtained a Km value of 5.01± 0.33 mM for WT, this value changed to 2.58 ± 0.09 mM when measured in the soluble form. Variant V3 with the lowest Km assayed in yeast display format (Km = 0.91 ± 0.10 mM) was found to have Km = 3.29 ± 0.06 mM when produced as a soluble enzyme in E. coli. For the other variants, we also detected deviations of the Km constants dependent on the expression format (i.e. soluble vs. displayed) (Fig. S6B†). There are several possible explanations as to why the Km values for the soluble variants differed from those measured for yeast displayed variants. For example, the expression environment in the host can influence post-translational modifications such as glycosylation and disulfide bond formation which can further affect the stability of the enzyme. Furthermore, the tethered, surface displayed enzymes experience a different microenvironment than soluble enzymes, which could influence enzyme-substrate interactions.29 Ultimately, the yeast displayed enzymes could be subject to interactions with other molecules on the yeast surface. Lowered Km constants of variants could also be the consequence of alterations in these surface interactions induced through mutations, and were therefore not detected in soluble versions.
Conclusion
In this work we adapted and optimized a one-pot hydrogel encapsulation platform to engineer RgDAAOx. We used the system for pooled large scale screening of a yeast surface displayed mutant library containing ∼106 clones and designed a FACS-based selection scheme to isolate variants with improved KM. This hydrogel polymerization technique, together with strategic FACS gating, lead to the isolation of single point mutants with increased apparent affinity towards a D-alanine substrate. The addition of PEG as a viscosity enhancer was shown to be effective at enabling this hydrogel-based method for compatibility with slower enzymes, likely by hindering diffusion of the substrates partaking in the encapsulation reaction. We validated the screening strategy and after selecting four final yeast displayed mutant variants were able to show they all had significantly lower Km, with the fold Km change ranging from 2.18 (variant V4) to 5.05 (variant V3). One limitation of our study was that since the absolute concentration of yeast display protein was not precisely known, we were not able to determine kcat or the catalytic efficiency for the isolated variant sequences. Soluble enzymes produced in E. coli that lacked the Aga2 fusion domain were found to have Km values different from those detected by yeast display. Taken together, the enzyme-mediated polymerization platform proved to be adaptable for slower fusion enzymes and provides high-scalability for enzyme engineering campaigns.Materials & methods
Materials
Restriction enzymes, DNA ligase kit and calf intestinal phosphatase (CIP) were purchased from New England Biolabs (Beverly, MA). GelRed™ dye nucleic acid gel stain was purchased from Biotium. Gene Morph II random mutagenesis kit (epPCR) was obtained from Agilent Technologies (Santa Clara, CA). The primary and secondary antibodies were purchased from Thermo Fisher Scientific (Waltham, MA). Custom oligonucleotides were obtained from Microsynth (Balgach, CH). The DNA miniprep, gel purification and PCR purification kits were purchased from Thermo Fisher Scientific. Chemicals, if not indicated otherwise, are from Sigma Aldrich (St. Louis, MO). Sodium alginate was purchased from Duchefa Biochemie (Haarlem, Netherlands).DAAOx mutant library construction and expression
A variant library of RgDAAOx (gene acquired codon optimized from Twist Bioscience, USA) was constructed by error prone PCR using the GeneMorph II random mutagenesis kit amplifying the gene using primers HR RgDAAOx for and HR RgDAAOx rev (Table 1). These primers contain overlaps with the expression vector. As an expression vector, we chose p111Y, a yeast plasmid based on pYD1 adapted in our lab containing a resistance gene for kanamycin instead of ampicilin.30 Further, p111Y contains a linker domain (GTPTPTPTPTGEF) connecting the cloning site with the yeast cell wall protein Aga2. This linker sequence was demonstrated to strongly withstand proteolysis which is why it was chosen for yeast display application.31 The p111Y vector backbone was digested with restriction enzymes BamHI-HF and XhoI for linearization. Then, insert and vector were combined at the overlapping sequences using a Gibson Assembly kit. After sequence confirmation, the error prone PCR library and the linearized backbone were transformed together into Saccharomyces cerevisiae EBY100 according to a lithium acetate transformation protocol.32 We selected positive clones subsequently on synthetic defined (SD) agar plates lacking tryptophan (−Trp) supplemented with 2% (wt/vol) glucose.| Name | Sequence 5′-3′ |
|---|---|
| HR_RgDAAOx_for | CCCACACCTACTCCGACACCTACTGGCGAATTTGGATCCATG |
| HR_RgDAAOx_rev | GGGTTAGGGATAGGCTTACCTTCGAAGGGCCCTCTAGACTCGAG |
In order to increase the chance of achieving a comprehensive library featuring all single mutants, a second library was prepared in a one pot mutagenesis reaction following a published protocol.33 We cloned the expression cassette for RgDAAOx in a smaller pUC19 plasmid and engineered two additional BbvCI restriction sites in the plasmid. Else following the protocol, we added an extra step, incubating the reaction mixture with 10 units of QuickCIP phosphatase (New England Biolabs) at 37 °C for 20 minutes after first digestion with Nt.BbvCI and exonuclease. For the PCR reaction introducing the mutations, we used NNK primers (Integrated DNA technologies) targeting codons 2 to 365 of the RgDAAOx wild type sequence (Primer design was conducted using the create_primers.py python script34). Then, by PCR amplification and HiFi DNA Assembly (New England Biolabs) the expression cassette was cloned in the same p111Y yeast expression vector as described above and the plasmid product was transformed in Saccharomyces cerevisiae EBY100.
The two libraries were cultivated for 24 h at 30 °C and shaking at 180 rpm in −TRP liquid medium containing 2% (wt/vol) glucose and were merged respecting their complexity. From combined libraries, aliquots of 108 cells were prepared and resuspended in 25% (vol/vol) glycerol and stored at −80 °C.
For the library expression, glycerol stocks were used to inoculate −TRP liquid medium containing 2% (wt/vol) glucose for 24 h at 30 °C and shaking at 180 rpm. Enzyme surface display was then induced by transferring the library to −TRP liquid medium containing 1.8% (wt/vol) galactose and 0.2% (wt/vol) glucose at an initial OD600 of 0.4. Cultures were grown for 48 h at 20 °C, shaking at 180 rpm. Expression medium was buffered by adding 100 mM of pH 7 sodium phosphate buffer.
Alginate modification with phenols and fluorophors
Fluorescent alginate with phenol moieties in the side chain was synthesized using aqueous phase carbodiimide activation chemistry, previously described.22,35 Sodium alginate (avg. MW 70000 Da) was dissolved at a final concentration of 10 mg mL−1 in 50 mM 2-(N-morpholino) ethanesulfonate (MES) buffer (pH 6). Subsequently, tyramine hydrochloride, N-hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were added to the sodium alginate solution at 7.0, 1.2, 3.9 mg mL−1, respectively. 6-Aminofluorescein was dissolved in DMS at a concentration of 50 mg mL−1 and added to the mixture at a final concentration of 0.25 mg mL−1. To protect the fluorophore from light, a beaker with the mixture was wrapped in tinfoil and the mixture was stirred overnight (16 h). The alginate solution was then added drop wise into cold 80% ethanol to precipitate alginate. Following stirring for 30 min on ice, the alginate was cleaned by filtration in fresh 80% ethanol solution. The precipitated alginate was solubilized in H2O resulting in a 1% concentrated solution and prepared for dialysis. Membranes with a 3.4 kDa cut-off were used to dialyse the alginate against water (5 L) for 16 h. Then, water was exchanged and dialysis continued for 2 h. After that, the water was again renewed. After another 2 h, the sample was collected in Falcon tubes and stored at −80 °C. The next day the samples were transferred in Falcon tubes with perforated tinfoil covers and dried by lyophilization for 5 days until all ice sublimated.
Optimized cell encapsulation in fluorescent hydrogel
In a 96-well round bottom plate, a total number of 4 × 106 yeast cells were added in each well. Subsequently, they were washed with 200 μL PBS containing 0.1% BSA and then resuspended and incubated at room temperature for 30 min with 1/500 dilution of the primary Anti His-tag antibody (stock concentration of 1.2 mg mL−1).Then, cells were washed with 200 μL of ice-cold PBS + 0.1% BSA and resuspended in ice cold buffer containing 1/500 dilution of the goat anti-mouse IgG secondary antibody (stock concentration 2 mg mL−1) conjugated with Alexa Fluor™ 594. After 20 min incubation on ice, the cells were pelleted, washed with cold buffer, and resuspended. As a negative control, the same yeast cells were treated only with the secondary antibody conjugated with Alexa Fluor™ 594. 200000 stained cells were suspended in a master mix consisting of 1.5–0.5 mM D-alanine, 4.5 μM horseradish peroxidase (HRP) and 0.125% (w/v) of modified alginate in 50 mM sodium phosphate buffer (pH 7.4), briefly vortexed and incubated for 10 min. Then, the reaction mixture was diluted fivefold with 50 mM sodium phosphate buffer (pH 7.4).
In these experiments D-alanine was used since it was reported to be the best RgDAAOx substrate with the highest turnover number found.13 Further it was shown to be a safe prodrug and provides concentration dependent tumor cell cytotoxicity in previous studies.7
While this encapsulation protocol has already been proved to work with glucose oxidase before, we observed no encapsulation under the same conditions as previously reported. Increasing the viscosity upon addition of filtered PEG 3350 to 20% (w/v) to the reaction mixture, improved conditions for slower enzymes could be obtained.
Fluorescent activated cell sorting
For the sorting experiments, the two libraries were merged according to their sizes, in order to avoid over-representing variants of one library. As the combined library was estimated to consist of roughly 1 × 106 variants and needs to be overrepresented to ensure complete screening of all of the variants, 24 × 106 cells were collected after expression and stained with the appropriate antibodies. After confirmation of correct labeling and presence of encapsulated cells, the reaction mixture was loaded on a BD FACSAria™ III cell sorter, equipped with a 488 nm and a 561 nm laser. In total, the library was sorted three times, with concentrations of substrate decreasing from 1.5 mM, to 0.5 mM and to 0.1 mM in every round. For the first two rounds, sorted cells were regrown in −TRP glucose (2%) medium supplemented with ampicillin at 30 °C. Then, enzymes were expressed again and cells were prepared for the next sorting step using fresh encapsulation reactions. In the last sorting round, 96-well plates were prepared with −TRP glucose (2%) media and inoculated with single cells. Plates were covered using sterile, gas-permeable breathseal seals (Greiner Bio-One, Germany) and placed at 30 °C with shaking (900 rpm).Kinetic experiments of surface displayed enzymes
The monoclonal cultures from the 96 well plates were expressed transferring them at an OD600 of 0.4 to 1 mL of induction media containing 0.2% (wt/vol) glucose and 1.8% (wt/vol) galactose, supplemented with 100 mM sodium phosphate buffer (pH 7). Plates were incubated at 20 C° for 24 h with shaking (900 rpm). In a plate based Amplex Red™ assay, the plate with variants was tested for activity on D-alanine substrate. Eight plates with eight different D-alanine concentrations ranging from 0.5–45 mM were prepared in 50 μL of reaction solution containing 0.1 mM Amplex Red™, 4.5 μM HRP in 50 mM sodium phosphate buffer pH 7.4. Solutions with cells were diluted 100 fold and 50 μL (20000 cells) of those dilutions added to plates resulting in a total volume of 100 μL. Subsequently, plates were loaded onto the fluorescence plate reader. Fluorescence was followed with excitation and emission wavelengths of 530 nm and 590 nm, respectively. Data obtained was normalized by subtracting the zero point fluorescence measured in the first cycle and converted to H2O2 turnover number by dividing through the slope of the calibration curve.
Enzyme expression in Escherichia coli
Variants of D-amino acid oxidase were expressed in Escherichia coli. Genes coding for the mutant variants were produced by site directed mutagenesis in Pet28 bacterial expression vector. The pET28 plasmids were transformed in BL21 (DE3) cells and plated on respective selective LB plates. From one colony a liquid LB Kanamycin culture has been started and grown overnight at 37 °C with shaking (200 rpm). 0.5 mL of this overnight culture was used to inoculate 50 mL LB Kan and let grow to an OD600 of 0.5. To induce expression, IPTG has been added to a final concentration of 0.5 mM and the culture was grown overnight for 18 h at 25 °C with shaking (200 rpm). The following day the cells were spun down at 4000 rpm for 15 min and the supernatant was discarded. The pellet was placed on ice for 15 min and then resuspended in 10 mL of lysis buffer (50 mM TRIS, pH 8.0, 50 mM NaCl, 0.1% (v/v) Triton X-100, 5 mM MgCl2). Cells were lysed by sonication for 10 min (Sonifier cell disruptor Branson Digital Sonifier, USA) and then spun down at 18000 g for 20 min. The supernatant was loaded on a gravitational flow column packed with HisPur Ni-NTA resin. Columns were washed with 5 column volumes of each, 5 mM and 10 mM imidazole PBS buffer and eluted with 5 column volumes of 500 mM Imidazole PBS buffer. Elution fractions were concentrated in vivaspin concentration columns (20 kDa cutoff) and salt was removed by dilution of the concentrate with PBS by a total factor of 800 (three consecutive 20 fold dilutions).
Author contributions
CK: conceptualization, investigation, formal analysis, writing – original draft. RV: conceptualization, supervision, investigation, writing – review & editing. MAN: project administration, supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors have no conflicts of interest to disclose.Acknowledgements
This work was supported by the University of Basel, ETH Zurich and the Swiss National Science Foundation (200021_191962).References
- G. Molla, L. Motteran, L. Piubelli, M. S. Pilone and L. Pollegioni, Regulation of D-amino acid oxidase expression in the yeast Rhodotorula gracilis, Yeast, 2003, 20(12), 1061–1069 CrossRef CAS PubMed
.
- L. Pollegioni, A. Falbo and M. S. Pilone, Specificity and kinetics of Rhodotorula gracillisd-amino acid oxidase, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120(1), 11–16 CrossRef CAS
- L. Pollegioni, L. Piubelli, S. Sacchi, M. S. Pilone and G. Molla, Physiological functions of D-amino acid oxidases: from yeast to humans, Cell. Mol. Life Sci., 2007, 64(11), 1373–1394 CrossRef CAS PubMed
- L. Verrall, P. Burnet, J. Betts and P. Harrison, The neurobiology of D-amino acid oxidase(DAO) and its involvement in schizophrenia, Mol. Psychiatry, 2010, 15(2), 122–137 CrossRef CAS
- M. Wcisło, D. Compagnone and M. Trojanowicz, Enantioselective screen-printed amperometric biosensor for the determination of d-amino acids, Bioelectrochemistry, 2007, 71(1), 91–98 CrossRef PubMed
- S. V. Khoronenkova and V. I. Tishkov, D-Amino acid oxidase: Physiological role and applications, Biochemistry, 2008, 73(13), 1511–1518 CAS
- E. Rosini, L. Pollegioni, S. Ghisla, R. Orru and G. Molla, Optimization of d-amino acid oxidase for low substrate concentrations – towards a cancer enzyme therapy, FEBS J., 2009, 276(17), 4921–4932 CrossRef CAS
- E. Rosini, N. A. Volpi, B. Ziffels, A. Grimaldi, S. Sacchi and D. Neri,
et al., An antibody-based enzymatic therapy for cancer treatment: The selective localization of D-amino acid oxidase to EDA fibronectin, Nanomedicine, 2021, 36, 102424 CrossRef CAS PubMed
- R. Vanella, C. Küng, A. A. Schoepfer, V. Doffini, J. Ren and M. A. Nash, Understanding Activity-Stability Tradeoffs in Biocatalysts by Enzyme Proximity Sequencing, bioRxiv, 2023, preprint, p. 2023.02.24.529916, Available from: https://www.biorxiv.org/content/10.1101/2023.02.24.529916v3 Search PubMed.
- L. D. Stegman, H. Zheng, E. R. Neal, O. Ben-Yoseph, L. Pollegioni and M. S. Pilone,
et al., Induction of cytotoxic oxidative stress by D-alanine in brain tumor cells expressing Rhodotorula gracilis D-amino acid oxidase: a cancer gene therapy strategy, Hum. Gene Ther., 1998, 9(2), 185–193 CrossRef CAS PubMed
- A. Al-Shameri, L. Schmermund and V. Sieber, Engineering approaches for O2-dependent Enzymes, Curr. Opin. Green Sustainable Chem., 2022, 100733 Search PubMed
- S. Sacchi, E. Rosini, G. Molla, M. S. Pilone and L. Pollegioni, Modulating d-amino acid oxidase substrate specificity: production of an enzyme for analytical determination of all d-amino acids by directed evolution, Protein Eng., Des. Sel., 2004, 17(6), 517–525 CrossRef CAS
- S. Sacchi, S. Lorenzi, G. Molla, M. S. Pilone, C. Rossetti and L. Pollegioni, Engineering the Substrate Specificity ofd-Amino-acid Oxidase*, J. Biol. Chem., 2002, 277(30), 27510–27516 CrossRef CAS
- A. Boselli, L. Piubelli, G. Molla, M. S. Pilone, L. Pollegioni and S. Sacchi, Investigating the role of active site residues of Rhodotorula gracilis d-amino acid oxidase on its substrate specificity, Biochimie, 2007, 89(3), 360–368 CrossRef CAS
- J. Cassidy, L. Bruen, E. Rosini, G. Molla, L. Pollegioni and F. Paradisi, Engineering substrate promiscuity in halophilic alcohol dehydrogenase(HvADH2) by in silico design, PLoS One, 2017, 12(11), e0187482 CrossRef PubMed
- P. A. Romero and F. H. Arnold, Exploring protein fitness landscapes by directed evolution, Nat. Rev. Mol. Cell Biol., 2009, 10(12), 866–876 CrossRef CAS
- N. Doi, S. Kumadaki, Y. Oishi, N. Matsumura and H. Yanagawa, In vitro selection of restriction endonucleases by in vitro compartmentalization, Nucleic Acids Res., 2004, 32(12), e95 CrossRef
- Z. J. Mays, K. Mohan, V. D. Trivedi, T. C. Chappell and N. U. Nair, Directed evolution of Anabaena variabilis phenylalanine ammonia-lyase(PAL) identifies mutants with enhanced activities, Chem. Commun., 2020, 56(39), 5255–5258 RSC
- S. Wu, C. Xiang, Y. Zhou, M. S. H. Khan, W. Liu and C. G. Feiler,
et al., A growth selection system for the directed evolution of amine-forming or converting enzymes, Nat. Commun., 2022, 13(1), 7458 CrossRef CAS PubMed
- R. Vanella, G. Kovacevic, V. Doffini, J. F. Santaella and M. A. Nash, High-throughput screening, next generation sequencing and machine learning: advanced methods in enzyme engineering, Chem. Commun., 2022, 58(15), 2455–2467 RSC
- R. Vanella, D. T. Ta and M. A. Nash, Enzyme-mediated hydrogel encapsulation of single cells for high-throughput screening and directed evolution of oxidoreductases, Biotechnol. Bioeng., 2019, 116(8), 1878–1886 CrossRef CAS PubMed
- R. Vanella, A. Bazin, D. T. Ta and M. A. Nash, Genetically encoded stimuli-responsive cytoprotective hydrogel capsules for single cells provide novel genotype–phenotype linkage, Chem. Mater., 2019, 31(6), 1899–1907 CrossRef
- Y. Liu, S. Sakai, S. Kawa and M. Taya, Identification of Hydrogen Peroxide-Secreting Cells by Cytocompatible Coating with a Hydrogel Membrane, Anal. Chem., 2014, 86(23), 11592–11598 CrossRef CAS PubMed
- L. Pollegioni, K. Diederichs, G. Molla, S. Umhau, W. Welte and S. Ghisla,
et al. Yeast d-Amino Acid Oxidase: Structural Basis of its Catalytic Properties, J. Mol. Biol., 2002, 324(3), 535–546 CrossRef CAS
- E. T. Boder and K. D. Wittrup, Yeast surface display for screening combinatorial polypeptide libraries, Nat. Biotechnol., 1997, 15(6), 553–557 CrossRef CAS
- D. Gonzalez-Perez, E. Garcia-Ruiz and M. Alcalde, Saccharomyces cerevisiae in directed evolution, Bioeng. Bugs, 2012, 3(3), 172–177 Search PubMed
- I. Kuan, R. Liao, H. Hsieh, K. Chen and C. Yu, Properties of Rhodotorula gracilis d-Amino Acid Oxidase Immobilized on Magnetic Beads through His-Tag, J. Biosci. Bioeng., 2008, 105(2), 110–115 CrossRef CAS PubMed
- X. Gao, X. Dong, X. Li, Z. Liu and H. Liu, Prediction of disulfide bond engineering sites using a machine learning method, Sci. Rep., 2020, 10(1), 10330 CrossRef CAS PubMed
- J. R. Klesmith, J. P. Bacik, E. E. Wrenbeck, R. Michalczyk and T. A. Whitehead, Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(9), 2265–2270 CrossRef CAS PubMed
- M. C. Kieke, B. K. Cho, E. T. Boder, D. M. Kranz and K. D. Wittrup, Isolation of anti-T cell receptor scFv mutants by yeast surface display, Protein Eng., 1997, 10(11), 1303–1310 CrossRef CAS PubMed
- M. Gustavsson, J. Lehtiö, S. Denman, T. T. Teeri, K. Hult and M. Martinelle, Stable linker peptides for a cellulose-binding domain-lipase fusion protein expressed in Pichia pastoris, Protein Eng., 2001, 14(9), 711–715 CrossRef CAS PubMed
- R. D. Gietz and R. A. Woods, Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method, Methods Enzymol., 2002, 350, 87–96 CAS
- E. E. Wrenbeck, J. R. Klesmith, J. A. Stapleton, A. Adeniran, K. E. J. Tyo and T. A. Whitehead, Plasmid-based one-pot saturation mutagenesis, Nat. Methods, 2016, 13(11), 928–930 CrossRef CAS
- J. D. Bloom, An Experimentally Determined Evolutionary Model Dramatically Improves Phylogenetic Fit, Mol. Biol. Evol., 2014, 31(8), 1956–1978 CrossRef CAS PubMed
- S. Sakai and K. Kawakami, Snthesis and characterization of both ionically and enzymatically cross-linkable alginate, Acta Biomater., 2007, 3(4), 495–501 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00002h |
| This journal is © The Royal Society of Chemistry 2023 |