
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization-based downstream processing of ω-transaminase- and amine dehydrogenase-catalyzed reactions†
Feodor
Belov
a,
Andrea
Mildner
b,
Tanja
Knaus
 c,
Francesco G.
Mutti
c and
Jan
von Langermann
*d
c,
Francesco G.
Mutti
c and
Jan
von Langermann
*d
aBiocatalytic Synthesis Group, Institute of Chemistry, University of Rostock, Albert-Einstein-Str. 3A, 18059 Rostock, Germany
bInstitute for Chemical and Thermal Process Engineering, Center of Pharmaceutical Engineering, Technische Universität Braunschweig, D-38106 Braunschweig, Germany
cHIMS-Biocat Group, Van' t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, Netherlands
dBiocatalysis, Institute of Chemistry, Otto-von-Guericke-University Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: jan.langermann@ovgu.de
First published on 24th March 2023
Abstract
Biocatalytic synthesis is a powerful and frequently chosen method for the production of chiral amines. Unfortunately, these biocatalytic reactions often result in complex mixtures, bearing many components aside from the main product amine such as residual co-substrates, co-products, cofactors and buffer salts. This issue typically requires an additional effort during downstream processing towards the isolation of the desired chiral amine. For instance, transaminase- and amine dehydrogenase-catalyzed reactions, which often use high surpluses of amine or ammonia co-substrates, face complications in removing the residual amine donor or unreacted substrate and salts from the isolated amine products, thus complicating and increasing the costs of the process of product isolation and purification. This study explores the selective removal of chiral amines from model amine transaminase and amine dehydrogenase-catalyzed reactions via a salt-based specific crystallization step. The product amine is precipitated directly in one step from the reaction mixture as a product ammonium salt, which can easily be filtered from the reaction mixture, while the other reactants remain unchanged in solution for potential re-use.
Introduction
Enzyme catalysis is a frequently used approach in the synthesis of complex chiral compounds. The benefits of enzymatic synthesis approaches compared to conventional organic synthesis typically include high enantioselectivity towards the synthesis of relevant products. This often also includes mild and environmentally friendly reaction conditions, such as moderate temperatures, an aqueous (main) solvent system and typically non-toxic reagents and starting materials.1–3 Since the early 2000s biocatalysis found its way into many industrial applications and developed itself from an industrial niche of kinetic resolution of just a few substances, mainly catalyzed by hydrolases, to a global tool for pharmaceutical and chemical synthesis.1–7 With the global enzyme market reaching up to an estimated 11.47 billion $US in 2021, it is projected to reach 20.31 billion $US by 2030, while the market of enzyme derived products is even larger.8 However, even though the benefits of enzymatic synthesis often outweigh its limitations, there are some major challenges that the enzymatic processes face: biocatalyst stability, occasional dependency on expensive cofactors, unfavorable reaction equilibria and often rather complex downstream processing issues.1–3,6,9One significant class of chemical compounds increasingly produced through enzymatic reactions are chiral amines.1,10–15 Finding usage as building blocks especially in pharmaceutical synthesis, up to an estimated 40% of currently commercialized APIs contain an optically active amine group.16,17 Over the years, many enzymatic classes have been adopted for this purpose, among such transaminases (TAs), amine dehydrogenases (AmDHs), imine reductases (IREDs), P450 monooxygenases (P450s) and others.18–23
This work will focus on the reaction systems of TAs and AmDHs. TAs are known to occasionally face challenges, e.g. unfavorable equilibria and inhibitions, thus, the establishment of reaction systems for TAs can require additional effort.18,24–26 While the enzymes can be improved via enzyme engineering, process limitations still constrain their full potential.26,27 Examples of approaches to circumvent limitations of transaminase-catalyzed reaction systems involve raising the amine donor concentrations to very high excesses or shifting reaction equilibria through enzymatic cascades or more technical approaches.28–36
This statement is also true for AmDHs, which require an excess of ammonia as an amine donor to shift the equilibrium. In addition, these high concentrations are also needed due to the high KM (Michaelis–Menten constant) for ammonia. High KM values for ammonia are a distinguishing feature for most wild-type amino acid dehydrogenases and engineered AmDHs thereof, as well as wild-type AmDHs.37–43
Reagent excesses may in turn impede downstream processing since high concentrations of the substrates, such as amine donors, may complicate product recovery based on conventional means of product extraction and chromatography.44,45 In this context, crystallization of the desired product from the reaction broth may offer an alternative downstream processing route, as it focuses on a specific low solubility of a single target compound. Consequently, crystallization is often applied as a processing step for the production of chiral compounds. A major application example thereof is the diastereomeric salt resolution through selective crystallization,46–49 when a counterion of defined chirality is added to an enantiomer mixture, yielding diastereomeric salts with differing solubilities.
Noteworthy, a direct combination of asymmetric biocatalytic synthesis and crystallization is present within the concept of in situ product crystallization (ISPC), otherwise known as reactive crystallization, and has proven to be effective in transaminase-catalyzed reactions.27,50–54
However, ISPC may encounter challenges, such as enzyme inhibition by the applied counterion or mechanical challenges (e.g. stirring).53,55 Thus, as an alternative, post-reaction product crystallization can be used for downstream processing, which still retains the high selectivity of the product crystallization. It allows for the use of any compound to facilitate crystallization, hence a counter-ion for the product can be selected based on the factors deemed as necessary. This study aims to highlight the benefits of the post-reaction crystallization downstream processing approach. We focus on the development of a straight-forward crystallization strategy, which is applicable for product recovery from TA- and AmDH-derived reaction media. The study was performed around the well-established model reaction that converts acetophenone into (S)-1-phenylethylamine ((S)-1PEA), known in literature to be optimized for both enzyme classes. The amine products were removed as the ammonium salt of a carboxylic acid anion, while the amine donors, broadly applied in a significant surplus, as well as other reactants remained in solution. For that purpose, a screening of solubility margins between the amine donor and amine product salts was performed for a variety of possible carboxylic acid anions (see Fig. 1). With a selection of anions from the screening, the influence of several factors, such as temperature, amine donor concentrations and acid anion concentrations, on product isolation yields and purities was investigated. The results of this investigation were validated on real enzymatic reaction media.
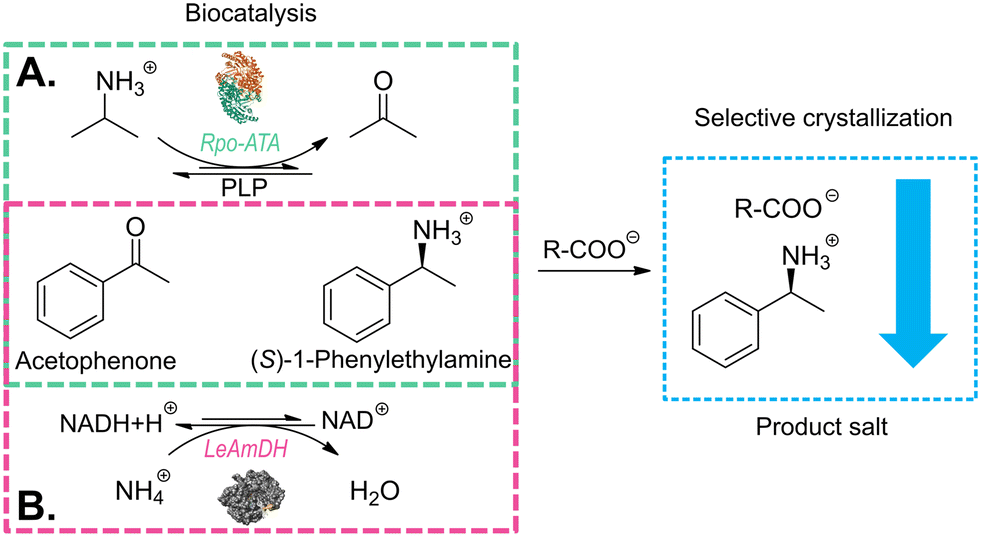 | ||
| Fig. 1 Reaction scheme of enzymatic approaches for the production of (S)-1-phenylethylamine used in this paper: A. scheme for the reaction catalyzed by the wild-type transaminase from Ruegeria pomeroyi with isopropylamine (IPA) as the amine donor (green box); B. scheme for the reaction catalyzed by the amine dehydrogenase (LE-AmDH-v1) engineered from the L-lysine-(ε-deaminating) dehydrogenase from Geobacillus stearothermophilus with ammonium ions as the amine donor (pink box). | ||
Results and discussion
Determination of the crystallizing agent
The determination of possible crystallizing agents is a crucial step for the subsequent crystallization. Taking into account that in the model systems there will always be at least two amine counterions available for crystallization, namely (S)-1PEA as the product and IPA/ammonium as the amine donor, one main consideration had to be made in the selection of the corresponding carboxylic acid anions as crystallizing agents. The solubility of the resulting (S)-1PEA salt had to be held as low as possible, while the solubility of the amine donor salt had to be held as high as possible. This principle of the crystallizing agent selection would allow for relatively high yields of product recovery from the reaction broth (minimal product salt solubility) whilst ensuring high purities of the crystallized product due to exclusion of isopropylamine (TA) or ammonia (AmDH) (via maximum amine donor salt solubility).Ten potential carboxylate anions (see Fig. 2) were selected based on a previous screening by Hülsewede et al. (ESI† Hülsewede et al.; pp. 14–16)53 and ranked according to solubility differences of the resulting salt pairs. The results of the solubility measurements are shown in Table 1.
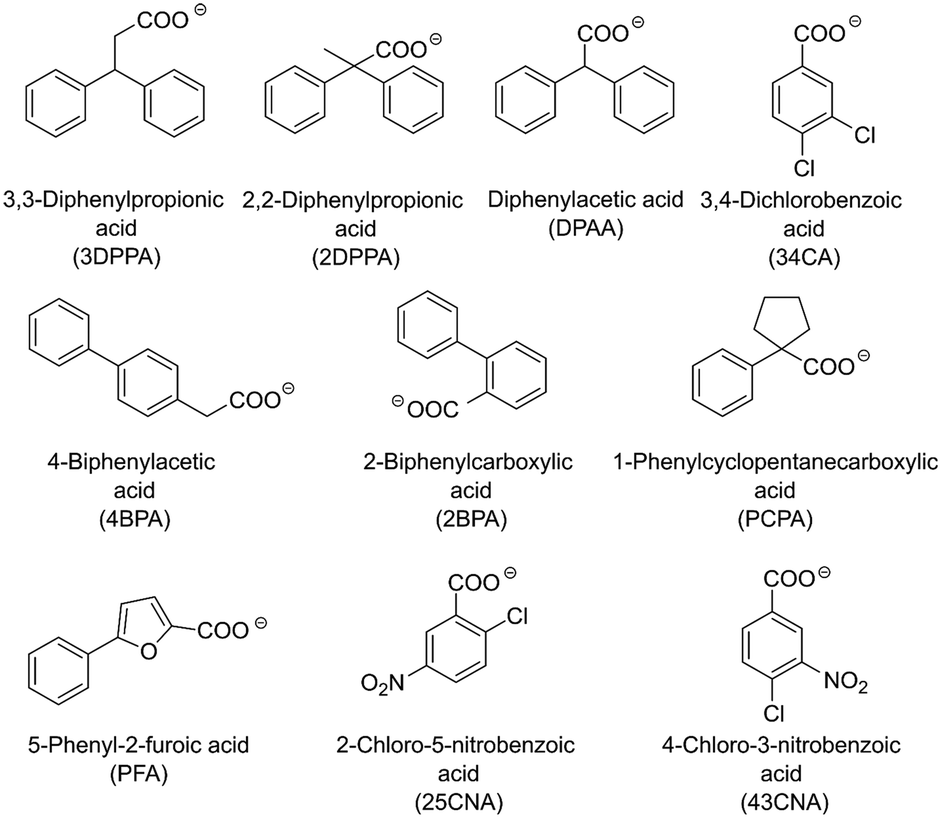 | ||
| Fig. 2 Carboxylate anions utilized in the solubility screening. | ||
|
|
|||||
|---|---|---|---|---|---|
| Acid anion | IPA-salt/(S)-1PEA salt quotient | IPA-salt solubility (mM) | (S)-1PEA salt solubility (mM) | Ammonium salt solubility (mM) | Ammonium salt/(S)-1PEA salt quotient |
| 3DPPA | 9.6 | 51.7 ± 0.3 | 5.4 ± 0.1 | 216.5 ± 87.5 | 40.1 |
| 2DPPA | 19.7 | 78.6 ± 0.2 | 4.0 ± 0.1 | 219.4 ± 23.2 | 54.9 |
| DPAA | 15.6 | 168.2 ± 1.3 | 10.8 ± 0.1 | 223.5 ± 47.8 | 20.7 |
| 4BPA | 6.3 | 83.6 ± 16.6 | 13.3 ± 0.5 | 236.6 ± 56.0 | 17.8 |
| 34CA | 8.5 | 203.1 ± 1.0 | 24.0 ± 0.7 | 165.3 ± 49.0 | 6.9 |
| PCPA | 4.8 | 89.8 ± 0.5 | 18.9 ± 0.1 | 192.8 ± 37.3 | 10.2 |
| 43CNA | 53.0 | 1066.1 ± 20.0 | 20.1 ± 0.4 | 2195.1 ± 121.7 | 109.2 |
| 25CNA | 24.5 | 1529.2 ± 384.6 | 62.5 ± 0.5 | 1714.8 ± 135.5 | 27.4 |
| 2BPA | 10.4 | 649.8 ± 24.3 | 62.5 ± 1.0 | 1092.8 ± 74.2 | 17.5 |
| PFA | 4.8 | 328.8 ± 35.4 | 69.1 ± 0.5 | 470.1 ± 250.5 | 6.8 |
The anions were sorted according to their respective quotient (see Table 1) of the amine donor (IPA and ammonium) salt solubility to the product salt solubility ((S)-1PEA). In addition, acid anions resulting in high product salt solubilities (e.g. 25CNA) >25 mM and low amine donor salt solubilities (e.g. 3DPPA) <150 mM were excluded to maximize product crystallization yields and amine donor impurities in the product salt. Moreover, the majority of the carboxylic acids screened in this work do not possess any noteworthy toxicities. Only 4BPA is known to be toxic to humans and was thus excluded from furthers experiments.
On this basis, three carboxylic acid anions matched those criteria and thus were selected for the subsequent crystallization conditions screening: DPAA, 34CA and 43CNA. It has to be mentioned, that the desired salt qualities for in situ product crystallization differ from its use as a downstream processing strategy. Please note that this screening was performed as a mean for qualitative evaluation of pure substance amine salts to narrow down a potential list of candidates for effective product crystallization. Thus, any effects of the solubility product and the influence of side components on the final solubility in the reaction media are not considered.
Crystallization condition screening for transaminase-catalyzed reactions
After the determination of the respective salt pair solubilities and the selection of three possible carboxylic acid anions, a reaction model solution was created to allow for the investigation of the main factors for crystallization processes, mainly towards crystallized (S)-1PEA yields and obtained purities. As model conditions, the following parameters were chosen: the pH was kept at 7.5 with 50 mM phosphate buffer, (S)-1PEA concentration was kept at 50 mM to represent a somewhat high product concentration for the chosen reaction systems, starting from ≥100 mM substrate. With this constellation, the range of IPA, carboxylic anion concentrations, as well as the temperature were varied and tested as possible influencing parameters. The determined trends for those parameters are shown in Fig. 3. One must keep in mind that the reported results represent the initial precipitation of material without any additional washing steps that will lead to higher purities (see below for transaminase- and amine dehydrogenase-catalyzed reactions). As shown in Fig. 3, all three tested carboxylic anions chosen for this analysis basically follow the same trends, when the respective concentration is varied at a fixed amount of 500 mM isopropylamine (T = 22 °C). With increasing concentrations of the carboxylic anions in solution, product amine crystallization yield increases (1A through 3A). As expected, this leads to an increase of crystallization of the undesired isopropylamine salt, hence causing a decrease in the crystallized product purity. The effect is significant while using DPAA (1A), but only minor changes are found with 34CA and 43CNA (2A and 3A). The best combination was found to be 43CNA with almost 90% yield and 95% purity (without additional washing steps). The parallel effect of a variation of applied isopropylamine shows a similar trend (1B through 3B). An addition of considerable amounts of isopropylamine into solution clearly leads to a decrease in yield and purity for all three anions, although, for 34CA and 43CNA the crystallized purity could still be kept relatively high at ≥90% (2B and 3B). Those decreases can easily be explained with the increasing ratio of donor isopropylamine (IPA) to product amine ((S)-1PEA) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (500 mM IPA) and 20:1 (1000 mM IPA). An unexpected behavior was found with the shown effect of temperature on the investigated crystallization (1C through 3C). DPAA shows a remarkable increase of purity at higher temperatures, while the yield seems to not be affected at all (both at ca. 90%, 1C). This can partially be explained through the increase in solubility of the obtained IPA-DPAA salt, seeming to be heavily affected by temperature. The disproportion in the solubilities of the salt pairs causes the increase in crystallization purity, since the solubility increase affects the IPA-salts more significantly than the (S)-1PEA-salts. The other carboxylic ions remain relatively stable with a slight decrease of the observed yield over the same temperature range (2C and 3C). However, with these other two carboxylic ions a considerably higher purity was obtained with up to >99%, while still retaining a 70% yield (3C).
:1 (500 mM IPA) and 20:1 (1000 mM IPA). An unexpected behavior was found with the shown effect of temperature on the investigated crystallization (1C through 3C). DPAA shows a remarkable increase of purity at higher temperatures, while the yield seems to not be affected at all (both at ca. 90%, 1C). This can partially be explained through the increase in solubility of the obtained IPA-DPAA salt, seeming to be heavily affected by temperature. The disproportion in the solubilities of the salt pairs causes the increase in crystallization purity, since the solubility increase affects the IPA-salts more significantly than the (S)-1PEA-salts. The other carboxylic ions remain relatively stable with a slight decrease of the observed yield over the same temperature range (2C and 3C). However, with these other two carboxylic ions a considerably higher purity was obtained with up to >99%, while still retaining a 70% yield (3C).
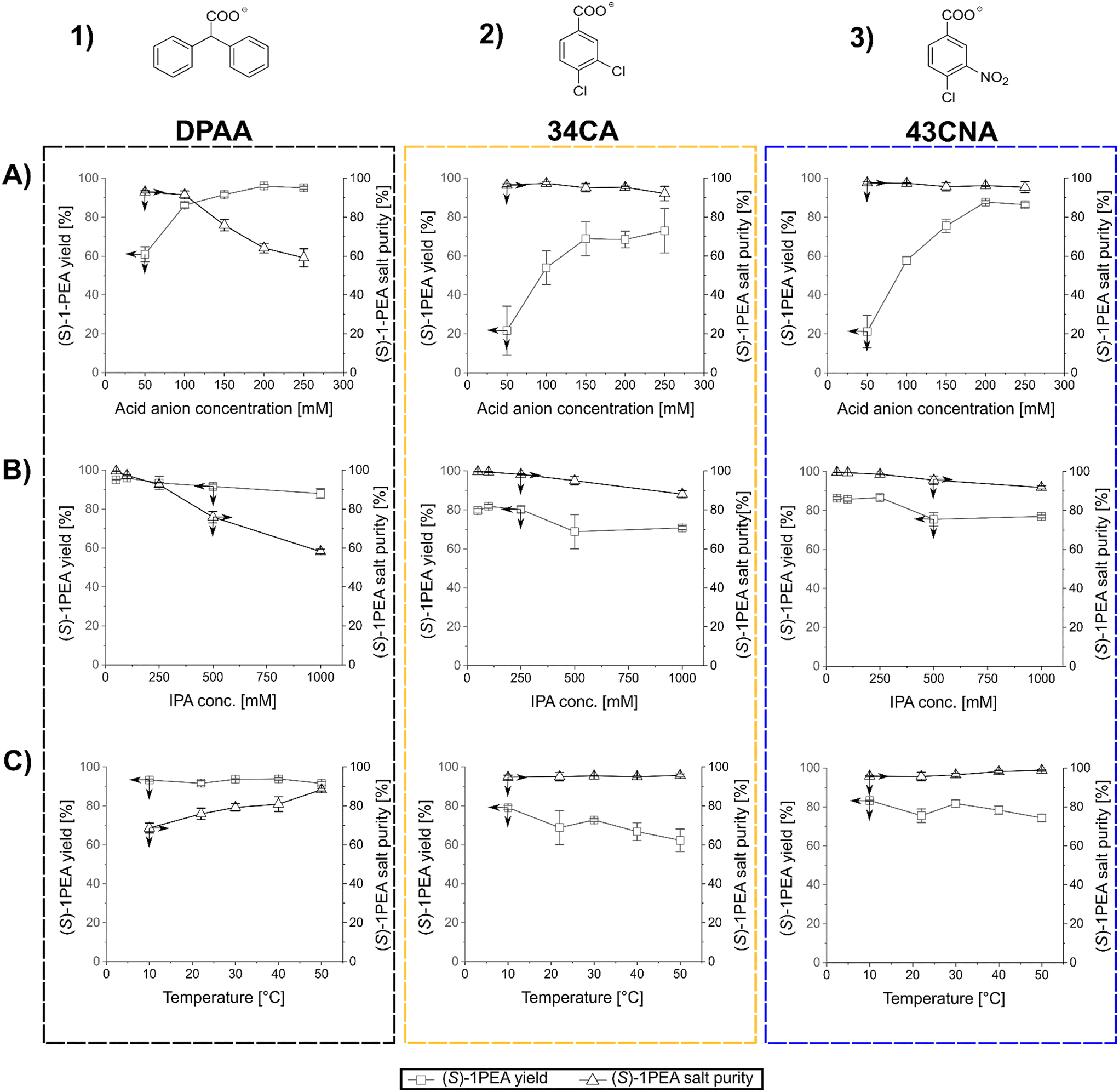 | ||
| Fig. 3 Results of the parameter screening for the IPA–(S)-1PEA salt pair (transaminase reaction system). The columns show all three screened parameters for a particular carboxylic anion (marked above), while the rows show a certain parameter: A) varied acid anion concentration with 50 mM (S)-1PEA, 500 mM IPA, pH 7.5, T = 22 °C; B) varied IPA concentration with 50 mM (S)-1PEA, 150 mM carboxylic anion, pH 7.5, T = 22 °C; C) varied crystallization temperature with 50 mM (S)-1PEA, 500 mM IPA, 150 mM carboxylic anion, pH 7.5. The shown arrows indicate the corresponding axes. | ||
In summary, 43CNA has proven to be the most suited for crystallization of product amines from transaminase reactions under chosen conditions, having its product crystallization behavior least affected by the parameter variation in terms of crystallized product purities, while also retaining high product yields. One must note that those observations are relevant for reaction setups with roughly 50 mM product amine, whereas the yields and purity will increase at higher and decrease at lower product concentrations. Hence, higher product concentrations are preferred.
Crystallization condition screening for amine dehydrogenase-catalyzed reactions
The screening of crystallization conditions for amine dehydrogenase-catalyzed reactions was handled in a similar manner as shown above. The most relevant change is the replacement of isopropylamine with significantly higher amounts of ammonia, which is required for amine dehydrogenase-catalyzed reaction. Model reactions were conceptualized in a similar manner with the same parameters as shown above (pH 7.5, c ((S)-1PEA) = 50 mM, 50 mM phosphate buffer). Those parameters were chosen for comparability with the transaminase conditions, since usually other buffers and pH values are set for AmDH reactions. Since this study focuses on the downstream processing of the resulting reaction product, such relatively mild conditions could be easily adjusted for product crystallization after an AmDH reaction would reach its equilibrium. Temperature, carboxylic anion and ammonia concentrations were screened as variable parameters influencing crystallization. The determined trends for those parameters are shown in Fig. 4. | ||
| Fig. 4 Results of the parameter screening for the ammonium – (S)-1PEA salt pair. The columns show all three screened parameters for a particular carboxylic anion (marked above), while the rows show a certain parameter: A) varied acid anion concentration with 50 mM (S)-1PEA, 1000 mM NH4+, pH 7.5, T = 22 °C; B) varied ammonium concentration with 50 mM (S)-1PEA, 150 mM carboxylic anion, pH 7.5, T = 22 °C; C) varied crystallization temperature with 50 mM (S)-1PEA, 1000 mM NH4+, 150 mM carboxylic anion, pH 7.5, T = 22 °C. The shown arrows indicate the corresponding axes. | ||
The results with the chosen three carboxylic anions follow roughly the same trend in comparison to the IPA–(S)-1PEA salt pair discussed above, but in general higher yields and purities were obtained. The difference between isopropylamine and ammonia versus (S)-1PEA in precipitation is mainly caused by the higher solubilities of ammonium salts in comparison to isopropylammonium salts (also see Table 1).
An increased concentration of acid anion (at T = 22 °C) within the crystallization solution again produces a significant yield increase, as expected, with a decrease of purity (1A–3A). The use of DPAA shows the highest yields with only a slight decrease of purity at higher concentrations (1A). For 34CA and 43CNA the yields are lower, however it is similarly coupled to a moderate purity decrease (2A and 3A) with a noticeable, but currently unclear dip at 150 mM 34CA. Regardless, high yields and purities of ≥90% were eventually obtained with all three chosen counterions.
As for the variation of ammonium concentration (1B through 3B), DPAA showed a remarkable efficiency at even the highest ammonia concentrations with consistently high purities and yields (1B). These results allow to withstand enormously high ammonia concentrations and keep a high product salt purity with mounting ammonium concentrations. Lower, but still relatively good yields and purities were observed with the use of 43CNA, consistently at above 80 and 90% respectively (3B). The purities and yields with the use of 34CA experienced a relatively high decrease with an increase of ammonium in solution (2B). Here, only this anion follows the trends outlined for mounting donor amine concentration, as shown for isopropylamine in Fig. 3 with having both yield and purity decreased significantly. The point, that DPAA and 43CNA product salts could keep relatively high yields and product salt purities even at ammonium concentrations of 2 M can as well be associated with the solubility differences as shown in Table 1.
Finally, temperatures affect the results again along the trends as outlined for the first salt pairs for transaminases (1C through 3C). An increase of temperatures provoked only insignificant crystallization yield decreases for DPAA and 43CNA, while their product salt purities remained constant on a very high level. DPAA was found again to be the most versatile counterion for high yields and purities at ≥95% (1C). As for 34CA, here the crystallization yield dropped significantly with an increase in crystallization temperature, what would follow the same explanation, as for the IPA salt pairs (2C). This is explained through the highest (S)-1PEA-salt and lowest ammonium salt solubility among the three salt pairs (also see Table 1), meaning, that at the most extreme temperature of 50 °C the disproportion between the solubilities within the 34CA salt pair was not sufficient to keep the high purity of the crystallized product salt in check (2C).
In summary, considering the generated data, DPAA was proven to be the best suited for the amine dehydrogenase reaction system among the three tested anions, being able to generate the highest product salt purities throughout all tested condition extremes, while also generating the highest crystallization yields.
Preparative application of amine crystallization
After the determination of suitable crystallization conditions for all three selected carboxylic anions, two exemplary reactions were performed to show the efficacy of the proposed crystallization systems for downstream processing of biocatalyzed reactions.Transaminase-catalyzed reaction example
The enzyme chosen for the practical application example for biocatalytic transamination was the transaminase from Ruegeria pomeroyi (PDB-code: 3HMU; herein abbreviated as Rpo-TA), as E. coli whole cells. It has consistently shown to efficiently convert acetophenone into (S)-1-phenylethylamine with high stereoselectivity in the presence of isopropylamine as amine donor.53,55 The reaction (300 mM acetophenone, 1500 mM IPA, pH 7.5) was performed under a constant vacuum to enable a slight reaction equilibrium shift towards the product side by removing acetone as the by-product. The reaction was monitored via gas chromatography. After 96 h, the reaction mixture was cleared of the residual cells by centrifugation and a product concentration of approximately 72 mM was determined.The following crystallization as described in the experimental section initially produced an overall product crystallization yield of 89% with a product purity of 80%. The product salt was then washed with water, which increased the salt purity to 96%, corresponding to a yield of 84% after the washing step. To provide an explanation for the efficiency of the selective crystallization step we decided to determine the underlying ternary phase diagram. The phase diagram for the IPA/(S)-1PEA-43CNA salt pair is provided in Fig. 5. The phase diagram shows an extreme asymmetry between the solubilities of the IPA salt and the (S)-1PEA salt. Noteworthy, the position of the eutectic is found to be extremely to the left hand side, creating an enormous asymmetry in the phase behavior. Through such a disproportion coupled with very low (S)-1PEA salt saturation concentration (Fig. 5), a very broad zone for possible pure (S)-1PEA salt crystallization is created, with the eutectic point signifying the borderline crystallization proportions within the salt pair. Such a broad selection for the crystallization possibilities of pure (S)-1PEA salt crystallization again shows a broad applicability of the tested system on different reaction setups for efficient and specific amine product crystallization from the reaction broths.
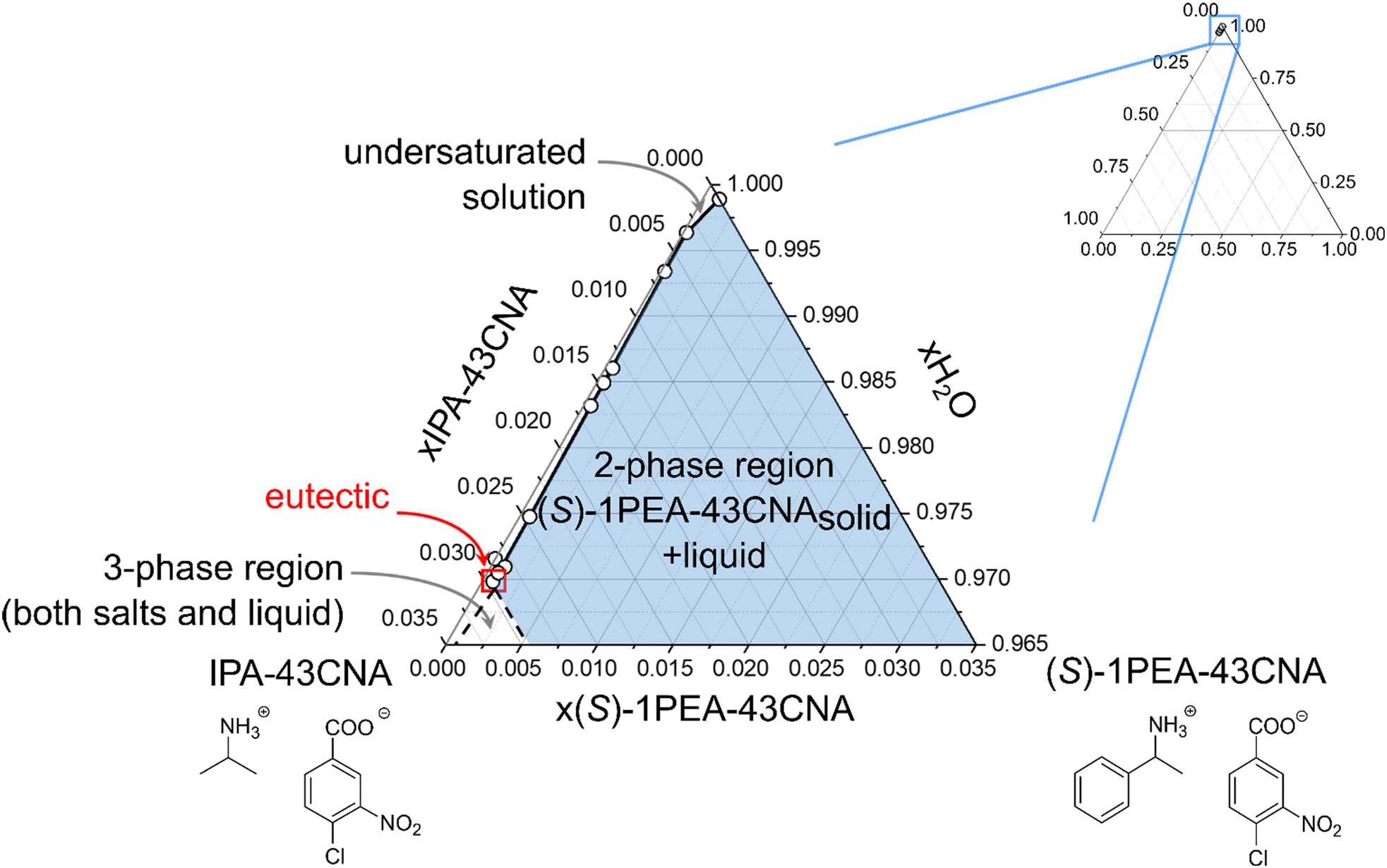 | ||
| Fig. 5 Ternary phase diagram of the IPA/(S)-1PEA-43CNA salt pair in 10 mM phosphate buffer, pH 7.5. | ||
Amine dehydrogenase-catalyzed reactions
A similar proof of concept for the selective product crystallization was obtained for the amine dehydrogenase-catalyzed reaction. An engineered amine dehydrogenase (LE-AmDH-v1) originated from the L-lysine-(ε-deaminating) dehydrogenase from Geobacillus stearothermophilus, as reported by Tseliou et al., was selected and used as a cell-free extract obtained from E. coli.37,56,57 LE-AmDH-v1 was previously shown to convert acetophenone to (S)-1-phenylethylamine with high stereoselectivity and activity. The catalyst was used as cell-free extract to achieve uniformity with the cofactor regenerating formate dehydrogenase (FDH) from Candida boidinii, used in the reaction system as a pure protein suspension. The reaction (100 mM acetophenone, 2 M ammonium, pH 8.5) was performed according to the experimental section and monitored via gas chromatography. After 72 h, the reaction mixture was cleared of the residual protein by centrifugation and the product concentration was determined at 55 mM. In the following crystallization step, an almost total yield could be obtained (99.9%) with an already very high purity of 94%, which approximately correlates with the screening study (see Fig. 4B). The crystallized product was again subjected to a washing step, yielding 97% purity with a remaining insignificant drop of yield to 98%.Similar to the 43CNA salt pair (see Fig. 5), a ternary phase diagram was prepared for the DPAA salt pair to prove the broad applicability of this crystallization system as a potential downstream processing strategy. The phase diagram for the ammonium/(S)-1PEA-DPAA salt pair is shown in Fig. 6. The disproportion and asymmetry of the phase diagram in Fig. 6 is even greater, than for the 43CNA salt pair in Fig. 5. The eutectic is barely measurable at x > 0.99 and thus the zone for selective (S)-1PEA salt crystallization is expanded, incl. an even higher potential concentration of ammonia in the reaction mixture.
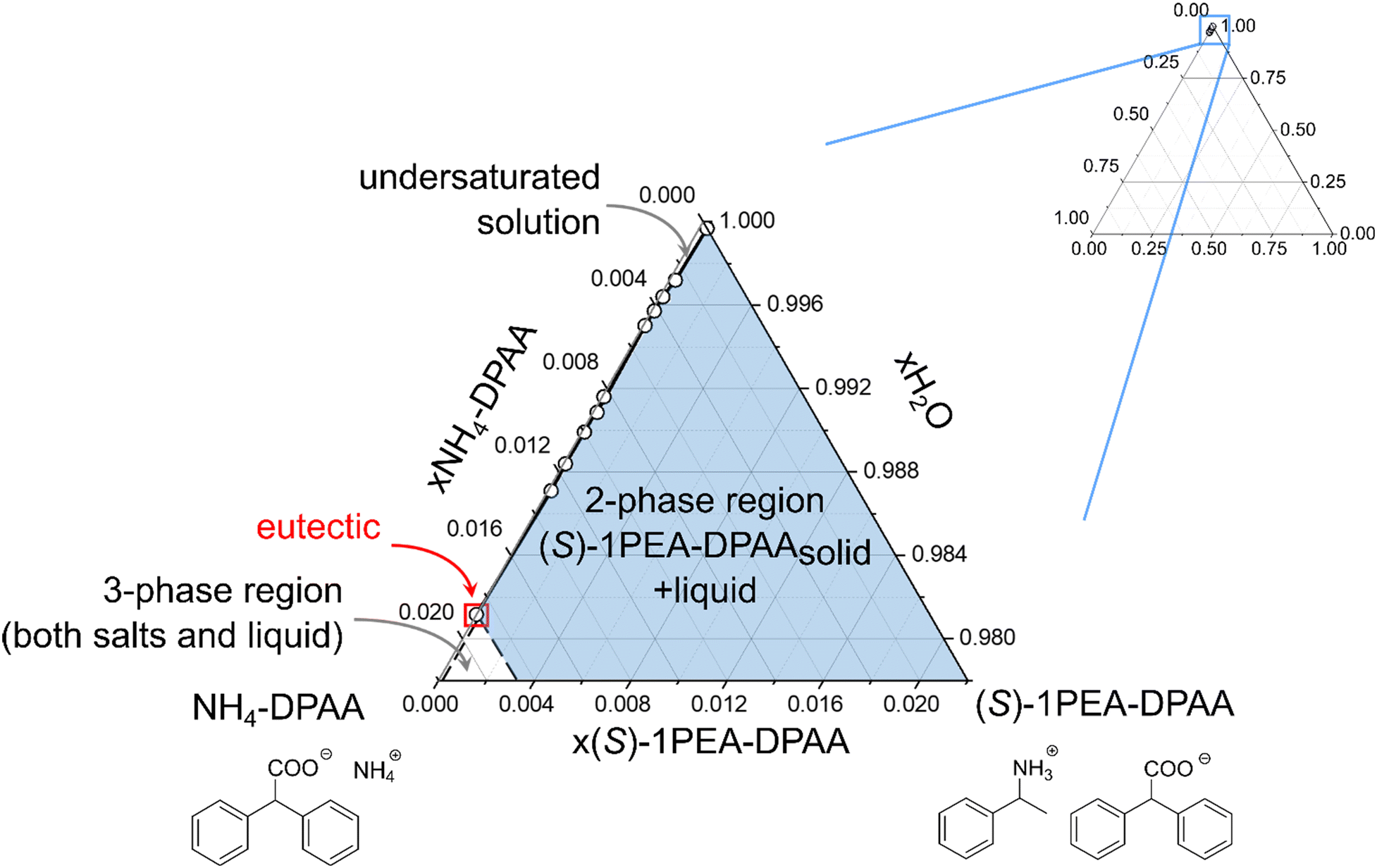 | ||
| Fig. 6 Ternary phase diagram of the ammonium/(S)-1PEA-DPAA salt pair in 10 mM phosphate buffer, pH 7.5. | ||
Experimental section
Salt preparation
5 mmol of the corresponding carboxylic acid were dissolved in 20 ml methyl tert-butyl ether (MTBE) at room temperature. Mild heating up to 60 °C was applied, if necessary, to speed up the dissolution process. After dissolution of the carboxylic acid, 5 mmol of the corresponding amine were slowly added to the solution under constant stirring/shaking to avoid partial oversaturation. While isopropylamine and (S)-1-phenylethylamine were added as a pure liquid, ammonia was utilized/applied as a 25% (v/v) aqueous solution. The resulting mixture/suspension was thoroughly stirred/shaken and left for one hour at room temperature. Afterwards, the mixture/suspension was filtered to isolate the desired amine salt. The isolated salts were dried at room temperature overnight and verified via1H- and 13C-NMR.Solubility measurements
An appropriate amount of the corresponding amine salt was dissolved in 2 ml of high-purity water. Salt was added, until solution saturation was achieved. The saturated solution was shaken at 30 °C and 1000 rpm for 72 hours to achieve the dissolution equilibrium. The pH value was kept at 7. In the following step, the solutions were centrifuged for 5 min at 14000 rpm and the resulting aqueous phase was filtered through 0.25 μm syringe filters to remove traces of crystalline salt. The aqueous phase was filtered into previously weighed vials. After that, the vials were weighted again to determine the exact amount of water in the sample. The water was evaporated at 40 °C under a constant argon stream in a Thermo Scientific Pierce ReactiTherm I & ReactiVap I heating and evaporation unit. The evaporated samples were weighed in the vials and the solubility of the amine salt was calculated. The experiments were performed in triplicates for each salt.
Precipitation from model solutions
To survey the crystallization parameters, the following experimental model was used. Two separate 10 ml aqueous solutions were prepared in 50 mM phosphate buffer, adjusted to a pH of 7.5 after preparation. One solution, referred to as the “amine solution”, contained 100 mM (S)-1PEA and variable amounts of IPA or ammonia. The other solution, referred to as the “acid solution”, contained the carboxylic acid used for precipitation in the form of a Na+-salt. Such sodium salts were prepared by dissolving 1.5 g of the pure carboxylic acid in 50 ml of MTBE and adding 2 ml of a saturated NaOH-solution. The resulting sodium salt was filtered and dried overnight at RT.To initiate (S)-1PEA precipitation, the acid solution was added to the amine solution in a dropwise manner under constant stirring at 700 rpm. Beforehand, GC-samples were drawn from the amine solutions as the initial amine concentration marker. The resulting precipitating mixture of 20 ml was left stirring for 30 minutes, evening out the crystallization equilibrium. Thus, the experimental model worked with an end concentration of (S)-1PEA of 50 mM, thus halving the amine concentration of the amine solution and the carboxylic anion concentration of the acid solution. The equilibrated precipitated mixtures were analyzed for yield and purity of the resulting (S)-1PEA salt.
For crystallization yield analysis, GC-samples were taken according to the protocol below and compared to the initial concentration samples from prior to the crystallization procedure. Crystallization yield was calculated as:
| cr. yield = 100% − (n(product in solution)/n(product initial)) × 100% |
In order to analyze the purity of the obtained (S)-1PEA salts, those were filtered from the precipitation mixtures and dried prior to being analyzed via1H-NMR (in DMSO-d6). The purity was derived from the ratio of amine salt specific peaks (IPA/ammonium-salt to (S)-1PEA) to each other (see ESI† for further information).
The measurements were performed on a Thermo Fisher Trace 1310 gas chromatograph equipped with a flame ionization detector (FID). A J&W column (0.25 mm × 30 m × 0.25 μm, HP-5 phase) by Agilent Technologies (series number 19091J-433), shortened for 1 mm on each end was used. Helium and synth air were used as the carrier. The following temperature program was used: starting temperature: 90 °C; 1. 90–100 °C: rate of 2 °C min−1; 2. 100–130 °C: rate of 20 °C min−1; 3. 130–180 °C: rate of 10 °C min−1; Split factor 35.0. The chromatograms were refined and analyzed via the Thermo Xcalibur Qual Browser software by Thermo Fisher. For amine quantification, the obtained amine peak areas were normalized with the internal standard, scaled for the internal standard value of 100000 sqU and then the amine concentration was calculated according to the calibration parameters (see ESI† for amine calibration curve).
Biocatalyst preparation
Biocatalysts were prepared through overexpression in E. coli BL21 cells.:1000) were added. The cultures were inoculated to an OD600 of approximately 0.05 from overnight cultures. The cultures were first incubated at 37 °C for 5–6 hours and afterwards at 30 °C for 18 hours under constant shaking at 150 rpm. After a 24 hour cultivation the cells were harvested via centrifugation at 4000 × g for 10 min at 4 °C. The pellets were resuspended in 5 ml of utilized buffer (10 mM phosphate buffer for lyophilization) and centrifuged again at 4000 × g for 10 min at 4 °C. After resuspension in 5 ml of the utilized buffer, the pellet suspensions were unified and lyophilized or frozen at −20 °C (only prior to lysis).
AIM – auto-induction medium preparation: the following components were mixed together: 950 ml ddH2O, 10 g tryptone, 5 g yeast extract, 2.68 g NH4Cl, 0.71 g Na2SO4, 5 g glycerol 85%, 0.5 g glucose, 2 g lactose and the resulting solution sterilized by autoclave. After the cooling to RT, the following solutions were added: 1 ml 2 M MgSO4 (0.22 μm filter sterilized), 40 ml 1 M K2HPO4 (autoclaved), 10 ml 1 M KH2PO4 (autoclaved) and the appropriate antibiotic(s) were added before usage.
000 × g for 30 min at 4 °C. The cleared lysate supernatant was collected and lyophilized.
Enzyme activity assays
For the measurement, 639 μl of the 2 M HCOO− NH4+/NH3 buffer (pH 8.5) were mixed with 86 μl of the 350 mM acetophenone solution and 250 μl of the enzyme solution. The mixture was prewarmed at 60 °C for 2 min. To start the measurement, 25 μl of the 10 mM NADH solution were added to the sample. NADH absorption decrease at 360 nm was measured for 1 min, its linear slope being used to calculate enzymatic activity according to the Lambert–Beer law (NADH extinction at 360 nm ε = 4250 M cm−1). A mixture of 914 μl of 2 M HCOO− NH4+/NH3 buffer with 86 μl of the acetophenone solution were used as the reference. All measurements were performed in a triplicate.
Reaction procedure with (S)-selective transaminase from Ruegeria pomeroyi (Rpo-TA)
To obtain a practical demonstration for the described product crystallization, enzymatic batch reactions with an (S)-selective transaminase from Ruegeria pomeroyi were made. For this purpose, 1283 μl of pure IPA were dissolved in 10 ml 50 mM sodium phosphate buffer to a final concentration of 1500 mM IPA with the addition of 6.18 mg PLP (final concentration 2.5 mM) at pH 7.5. The pH was adjusted to 7.5 with conc. HCl and NaOH. 16.75 U ml−1 (670 mg) of cultivated dry weight E. coli cells bearing the overexpressed Rpo-TA (previously measured enzyme activity 250 mU mg−1, see ENZYME ACTIVITY ASSAYS) were added to the reaction mixture. The pH was again adjusted to 7.5. To start the reaction, 350 μl acetophenone were added (equals to 300 mM). The reaction was incubated at 30 °C under constant stirring and a vacuum of 300 mbar for 96 h. For reaction monitoring, 100 μl samples were drawn every 24 h (see GC-method). The reactions were prepared in a triplicate to ensure sufficient volumes for product crystallization.After determining the reaction yields, cell material was removed from the reaction solutions via two centrifugation steps. The first centrifugation was performed at 4000 × g and 4 °C for 10 min, whereafter the cleared supernatant was transferred into new tubes and centrifuged at 10000 × g and 4 °C for 10 min. 10 ml of the cleared unified supernatant were then subjected to product crystallization. For this purpose, pure 43CNA−Na+ salt was added to the supernatant to a final concentration of 150 mM. After the sodium salt dissolution, the pH was briefly adjusted to 7.5. The solution was left shaking at room temperature (22 °C) over night. Afterwards it was filtered to separate the product salt. The salt was dried and analyzed via NMR to determine its purity. The filtrate was analyzed via GC to determine the total yield. To ensure sufficient purity, the filtered salt was washed three times with 2 ml of pure H2O. After each washing step, samples were drawn for purity (NMR) and product loss (filtrate) analysis.
Reaction procedure with engineered amine dehydrogenase from L-lysine-(ε-deaminating) dehydrogenase from Geobacillus stearothermophilus (LE-AmDH-v1)
To obtain a practical demonstration for product crystallization from amine dehydrogenase reactions, an enzymatic batch reaction with the LE-AmDH-v1 was initiated. For this purpose, 132.7 mg NAD+ (final concentration 10 mM) were dissolved in 20 ml of a 2 M NH4+HCOO− buffer at a pH of 8.5. 30 U ml−1 (2 g) of freeze-dried cell-free extract from E. coli bearing the overexpressed LE-AmDH-v1 (previously measured enzyme activity 300 mU mg−1, see ENZYME ACTIVITY ASSAY) were added to the reaction mixture. 213 μL (16 U) FDH solution by Megazyme were added from a 75 U ml−1 stock solution. The pH was adjusted to 8.5 with conc. HCl and NaOH. To start the reaction, 234 μl pure acetophenone were added (equals to 100 mM). The reaction was incubated at 30 °C under constant shaking for 72 h. For reaction monitoring, 100 μl samples were drawn every 24 h (see GC-method).After determining the reaction yields, protein was removed from the reaction solutions via centrifugation at 10000 × g and 4 °C for 10 min. 10 ml of the cleared supernatant were then subjected to product crystallization. For this purpose, pure DPAA−Na+ salt was added to the supernatant to a final concentration of 150 mM. After the sodium salt dissolution, the pH was briefly adjusted to 7.5. The solution was left shaking at room temperature (22 °C) overnight. Afterwards it was filtered to separate the product salt. The salt was dried and analyzed via NMR to determine its purity. The filtrate was analyzed via GC to determine the total yield. To ensure sufficient purity, the filtered salt was washed three times with 2 ml of pure H2O. After each washing step, samples were drawn for purity (NMR) and product loss (filtrate) analysis.
Generation of chosen phase diagrams for utilized salt pairs
For further examination of the crystallization mechanisms, phase diagrams of the NH4+/(S)-1PEA-DPAA and IPA/(S)-1PEA-43CNA were prepared. For this purpose, the two salts of each salt pair were mixed in 9 different proportions (with additionally each pure salt as a control) and dissolved in 0.01 M phosphate buffer (pH 7.5) until a saturated solution was formed. To compensate for the dissolved parts, the same salt proportion mixture was added to each sample individually. The salts were left to dissolve at 25 °C and 150 rpm for 6 days (or until no further pH changes occurred) to reach solution saturation. Additional salt mixture was added on the second day, the pH was readjusted every 2 days.The salt solutions were centrifuged at 10000 × g for 5 min and the supernatant was filtered through 0.2 μm filters to remove all residual salt crystals. Afterwards, solution samples of approx. 1 ml were evaporated in a Thermo Scientific Pierce ReactiTherm I & ReactiVap I heating and evaporation unit under a constant argon stream. As was the case with other solubility measurements, the sample vials were weighed three times: empty, when filled with the samples (to determine the exact water amount) and after evaporation. The final ratios of the salt mixtures were determined via NMR.
This data was summarized and the mole fractions of the used salts and water could be calculated. Those mole fractions were then mapped against each other as a ternary diagram with the Origin 2021 software, resulting into a ternary phase diagram of the salt pair.
Conclusion
The claimed systems for post-reaction crystallization downstream processing were successfully tested for both the transaminase- and amine dehydrogenase-catalyzed reaction setups. Through a broad parameter variation, the ideal salt pairs within the tested scope could be determined. A proof of concept was obtained within realistic enzymatic reactions performed incl. high amine donor concentrations (1.5–2 M) for maximal interference simulation with the downstream processing. Nevertheless, very high purities of the amine product coupled with high yields in product recovery could be achieved. The following investigation into the salt pairs crystallization behavior by means of the ternary phase diagrams revealed a very broad spectrum of possible reaction setups, within which the amine products could be specifically crystallized despite extreme amine donor concentrations.The product itself can easily be recovered from the obtained product salt as described by Neuburger et al.54 While demonstrating a high efficiency, this method also allows for almost full recycling of the applied carboxylic acid through simple acidification, including its non-precipitated excesses for subsequent crystallizations.
Our proposed method for amine downstream processing from enzymatic reactions proved to be fairly versatile while also being relatively facile to accomplish. The broader selection of the tested salt pairs and their acquired solubility data offers an even broader palette to suit individual needs for post-reaction amine crystallization from enzymatic reactions.
Author contributions
Feodor Belov: conceptualization, data curation, formal analysis, methodology, investigation, validation, visualization, writing – original draft; Andrea Mildner: conceptualization, methodology, investigation, writing – review & editing; Tanja Knaus: methodology, writing – review & editing; Francesco G. Mutti: resources, supervision, writing – review & editing; Jan von Langermann: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for F. B. and J. v. L. by Central SME Innovation Programme (ZIM, projects 16KN073233 and ZF4402103CR9) and the Heisenberg Programme of the Deutsche Forschungsgemeinschaft (project number 450014604) is gratefully acknowledged. The authors thank the research group of Prof. Mirko Basen (Institute of Biological Science, University of Rostock) especially with Dr. Maria Lehmann and Dr. Ralf-Jörg Fischer and the research group of Prof. Kragl (Institute of Chemistry, University of Rostock) especially with Sandra Diederich for their ongoing support and fruitful discussions.Notes and references
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60(1), 88 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50(14), 8003 RSC.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12(13), 2859 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118(2), 801 CrossRef CAS PubMed.
- B. Hauer, ACS Catal., 2020, 10(15), 8418 CrossRef CAS.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2(12), 409 CrossRef.
- https://www.grandviewresearch.com/industry-analysis/enzymes-industry, access 20.10.2022 15:15.
- D. J. Timson, Fermentation, 2019, 5(2), 39 CrossRef CAS.
- E. E. Ferrandi and D. Monti, World J. Microbiol. Biotechnol., 2017, 34(1), 13 CrossRef PubMed.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1(1), 42 CrossRef.
- E. Cigan, B. Eggbauer, J. H. Schrittwieser and W. Kroutil, RSC Adv., 2021, 11(45), 28223 RSC.
- H. Gröger, Appl. Microbiol. Biotechnol., 2019, 103(1), 83 CrossRef PubMed.
- H. Kohls, F. Steffen-Munsberg and M. Höhne, Curr. Opin. Chem. Biol., 2014, 19, 180 CrossRef CAS PubMed.
- F. G. Mutti and T. Knaus, Enzymes Applied to the Synthesis of Amines, in Biocatalysis for Practitioners, ed. G. de Gonzalo and I. Lavandera, Wiley, 2021, pp. 143–180 Search PubMed.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57(5), 284 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43(7), 788 CrossRef CAS PubMed.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8(7), 254 CrossRef.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8(12), 10985 CrossRef CAS.
- A. Gomm and E. O'Reilly, Curr. Opin. Chem. Biol., 2018, 43, 106 CrossRef CAS PubMed.
- S. C. Cosgrove, J. I. Ramsden, J. Mangas-Sanchez and N. J. Turner, Biocatalytic Synthesis of Chiral Amines Using Oxidoreductases, in Methodologies in Amine Synthesis, ed. A. Ricci and L. Bernardi, Wiley, 2021, pp. 243–283 Search PubMed.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19 CrossRef CAS PubMed.
- L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2021, 363(2), 328 CrossRef CAS.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28(6), 324 CrossRef CAS PubMed.
- M. Voges, R. Abu, M. T. Gundersen, C. Held, J. M. Woodley and G. Sadowski, Org. Process Res. Dev., 2017, 21(7), 976 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19(2), 333 RSC.
- D. Hülsewede, L.-E. Meyer and J. von Langermann, Chemistry, 2019, 25(19), 4871 CrossRef PubMed.
- A. Gomm, W. Lewis, A. P. Green and E. O'Reilly, Chemistry, 2016, 22(36), 12692 CrossRef CAS PubMed.
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Green Chem., 2017, 19(2), 361 RSC.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53(40), 10714 CrossRef CAS PubMed.
- E. O'Reilly, C. Iglesias, D. Ghislieri, J. Hopwood, J. L. Galman, R. C. Lloyd and N. J. Turner, Angew. Chem., 2014, 126(9), 2479 CrossRef.
- D. Koszelewski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozzell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47(48), 9337 CrossRef CAS PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2016, 358(10), 1618 CrossRef.
- S. E. Payer, J. H. Schrittwieser and W. Kroutil, Eur. J. Org. Chem., 2017, 2017(17), 2553 CrossRef CAS.
- T. Börner, G. Rehn, C. Grey and P. Adlercreutz, Org. Process Res. Dev., 2015, 19(7), 793 CrossRef.
- M. Doeker, L. Grabowski, D. Rother and A. Jupke, Green Chem., 2022, 24(1), 295 RSC.
- V. Tseliou, T. Knaus, M. F. Masman, M. L. Corrado and F. G. Mutti, Nat. Commun., 2019, 10(1), 3717 CrossRef PubMed.
- L. Liu, D.-H. Wang, F.-F. Chen, Z.-J. Zhang, Q. Chen, J.-H. Xu, Z.-L. Wang and G.-W. Zheng, Catal. Sci. Technol., 2020, 10(8), 2353 RSC.
- A. Pushpanath, E. Siirola, A. Bornadel, D. Woodlock and U. Schell, ACS Catal., 2017, 7(5), 3204 CrossRef CAS.
- B. R. Bommarius, M. Schürmann and A. S. Bommarius, Chem. Commun., 2014, 50(95), 14953 RSC.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J.-L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2(4), 324 CrossRef CAS.
- M. J. Abrahamson, E. Vázquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51(16), 3969 CrossRef CAS PubMed.
- A. A. Caparco, E. Pelletier, J. L. Petit, A. Jouenne, B. R. Bommarius, V. Berardinis, A. Zaparucha, J. A. Champion, A. S. Bommarius and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2020, 362(12), 2427 CrossRef CAS.
- C. Matassa, D. Ormerod, U. T. Bornscheuer, M. Höhne and Y. Satyawali, Process Biochem., 2019, 80, 17 CrossRef CAS.
- I. Slabu, J. L. Galman, C. Iglesias, N. J. Weise, R. C. Lloyd and N. J. Turner, Catal. Today, 2018, 306, 96 CrossRef CAS.
- J. W. Westley, R. H. Evans and J. F. Blount, J. Am. Chem. Soc., 1977, 99(18), 6057 CrossRef CAS.
- M. H. T. Kwan, J. Breen, M. Bowden, L. Conway, B. Crossley, M. F. Jones, R. Munday, N. P. B. Pokar, T. Screen and A. J. Blacker, J. Org. Chem., 2021, 86(3), 2458 CrossRef CAS PubMed.
- F. C. Ferreira, N. F. Ghazali, U. Cocchini and A. G. Livingston, Tetrahedron: Asymmetry, 2006, 17(9), 1337 CrossRef CAS.
- F.-X. Gendron, J. Mahieux, M. Sanselme and G. Coquerel, Cryst. Growth Des., 2019, 19(8), 4793 CrossRef CAS.
- J. Urbanus, C. Roelands, D. Verdoes and J. H. ter Horst, Chem. Eng. Sci., 2012, 77, 18 CrossRef CAS.
- J. Ren, P. Yao, S. Yu, W. Dong, Q. Chen, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2016, 6(2), 564 CrossRef CAS.
- J. Bao, K. Koumatsu, K. Furumoto, M. Yoshimoto, K. Fukunaga and K. Nakao, Chem. Eng. Sci., 2001, 56(21–22), 6165 CrossRef CAS.
- D. Hülsewede, M. Tänzler, P. Süss, A. Mildner, U. Menyes and J. von Langermann, Eur. J. Org. Chem., 2018, 2018(18), 2130 CrossRef.
- J. Neuburger, F. Helmholz, S. Tiedemann, P. Lehmann, P. Süss, U. Menyes and J. von Langermann, Chem. Eng. Process., 2021, 168, 108578 CrossRef CAS.
- D. Hülsewede, J.-N. Dohm and J. von Langermann, Adv. Synth. Catal., 2019, 361(11), 2727 Search PubMed.
- V. Tseliou, T. Knaus, J. Vilím, M. F. Masman and F. G. Mutti, ChemCatChem, 2020, 12(8), 2184 CrossRef CAS PubMed.
- V. Tseliou, D. Schilder, M. F. Masman, T. Knaus and F. G. Mutti, Chemistry, 2021, 27(10), 3315 CrossRef CAS PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41(1), 207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00496h |
| This journal is © The Royal Society of Chemistry 2023 |