
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContrasting thermoresponsiveness of stereoisomers of a dense 1,2,3-triazole polymer carrying amide side chains†
Koji
Okuno
a,
Junji
Miura
a,
Shota
Yamasaki
a,
Masaki
Nakahata
a,
Yuri
Kamon
b and
Akihito
Hashidzume
 *a
*a
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: hashidzume@chem.sci.osaka-u.ac.jp
bAdministrative Department, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 6th March 2023
Abstract
Since thermoresponsive polymers are an important class of smart polymer materials, it is an important subject of investigation to develop thermoresponsive polymers with a new polymer backbone for expanding their potential. Recently, we synthesized poly(N-ethyl-N-methyl-4-azido-5-hexynamide) (poly(ME)) by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) polymerization as a new lower-critical-solution-temperature (LCST) type thermoresponsive polymer. In this study, we synthesized two types of stereoregular poly(ME) (i.e., isotactic and syndiotactic) by CuAAC polymerization of ME dimers of the R- and R-isomers, and the R- and S-isomers, respectively, and investigated their thermoresponsive behavior. Binary mixtures of isotactic poly(ME) (iso-poly(ME)) with dimethyl sulfoxide (DMSO) and with N,N-dimethylformamide (DMF) underwent thermoresponsive gel-to-sol transition, in which the mixture turned from gel to sol at a certain temperature as the temperature was increased. On the other hand, aqueous solutions of syndiotactic poly(ME) (syndio-poly(ME)) underwent the LCST-type phase transition, in which the solution was transparent at lower temperatures whereas it became turbid at a certain temperature as the temperature was increased.
Introduction
Polymers, of which physical properties can be controlled in response to external stimuli, have attracted increasing interest from researchers as smart functional materials in recent decades.1–5 Since the physical properties of polymers depend on the conformation or aggregated state of polymer chains, the control of polymer properties requires marked changes in the conformation or aggregated state of polymer chains that respond to external stimuli. Various stimuli, e.g., temperature, electromagnetic wave, pH, and substances, have been utilized for the control of the physical properties of stimuli-responsive polymers so far.1–6 Of these stimuli, temperature is the most fundamental and has thus been investigated in the most detail. Typical examples of thermoresponsive polymers reported include (i) lower-critical-solution-temperature (LCST) or upper-critical-solution-temperature (UCST) type phase transition, in which the solubility of polymer chains in a solvent changes markedly at a critical temperature,7–21 (ii) thermoresponsive gel-to-sol transition, in which crosslinking density alters remarkably at a certain temperature,22–26 and (iii) thermoresponsive shape memory behavior, which is based on polymer networks containing thermally non-responsive covalent crosslinks and thermoresponsive noncovalent crosslinks.27–33 It is an important subject of investigation to develop thermoresponsive polymers with new polymer backbone for expanding their potential because the thermoresponsiveness of polymers should be strongly dependent on their polymer structure.Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) is one of the most important reactions in click chemistry because CuAAC produces selectively and efficiently 1,4-disubstituted 1,2,3-triazole from azide and alkyne moieties in the presence of a copper(I) compound with a wide tolerance for various functional groups.34–39 Thus, CuAAC has been widely utilized for synthesis of new polymers possessing 1,2,3-triazole moieties in their backbone.40–53 We have been also working on the synthesis of dense 1,2,3-triazole polymers by CuAAC polymerization of 3-azido-1-propyne derivatives possessing azide and alkyne moieties linked via a carbon atom.54–59 Recently, we prepared a series of dense 1,2,3-triazole polymers carrying amide side chains, aiming at synthesis of new thermoresponsive polymers.60 The characterization data have demonstrated that poly(N-ethyl-N-methyl-4-azido-5-hexynamide) (poly(ME)) is a new LCST-type thermoresponsive polymer exhibiting a large hysteresis. It should be noted that the fraction of polymer chains that underwent phase separation was only ca. 15% of the polymer sample. Since the monomer, N-ethyl-N-methyl-4-azido-5-hexynamide (ME), possesses a chiral carbon on the 4-position and its racemic mixture was used in our previous study, the poly(ME) obtained was an atactic polymer (a-poly(ME)) that possesses a statistical sequence of the R- and S-isomers. Thus, the poly(ME) chains that underwent phase separation should be characterized to elucidate their specific structural features, e.g., molecular weight and stereoregularity.
Among LCST-type thermoresponsive polymers, poly(N-isopropylacrylamide) (PNIPAM) has been studied in the most detail by a number of research groups.61,62 These researches have elucidated that the phase transition temperature for PNIPAM aqueous solutions depends on some structural parameters, e.g., stereoregularity, molecular architecture, and the terminal group. The phase separation temperature for PNIPAM aqueous solutions increases with increasing the fraction of r-diad.63–65 Since PNIPAM is usually prepared by conventional or controlled radical polymerization of a vinyl monomer, NIPAM, it is still difficult to obtain PNIPAM samples of ca. 100% m- and r-diad fractions.66–73
In contrast to PNIPAM, the R- and S-isomers of the monomer (ME) precursor of an enantiomer excess (ee) ≥96% can be synthesized through asymmetric reduction (Fig. S1, ESI†).74,75 Using the R- and S-isomers of ME precursor, it is possible to synthesize optically-pure stereoregular poly(ME) by CuAAC polymerization of the stereoregular dimers. In this study, we thus synthesized two types of poly(ME) (i.e., isotactic and syndiotactic) by CuAAC polymerization of ME dimers of the R- and R-isomers, and the R- and S-isomers (the RR and RS dimers), respectively, and investigated the LCST-type phase transition behavior of their aqueous solutions. It is noteworthy that a gel mixture was obtained in CuAAC polymerization of the RR dimer using N,N-dimethylformamide (DMF) as a solvent. Thus, the thermoresponsive gelation behavior of isotactic polymer (iso-poly(ME)) was also investigated in this study.
Results and discussion
Synthesis of stereoregular poly(ME) samples
In this study, two types of stereoregular poly(ME) samples were synthesized, i.e., iso-poly(ME) and syndio-poly(ME), which are homopolymer of the R-isomer and alternating copolymer of the R- and S-isomers of ME, respectively. Both the polymers were prepared by CuAAC polymerization of the corresponding dimers, i.e., the RR and RS dimers, respectively. The corresponding dimers were synthesized according to Scheme 1. The starting materials in this study, i.e., the t-butyldimethylsilyl(TBDMS)-protected stereoregular dimers of t-butyl 4-azido-5-hexynoate (tBuAH) (1RR and 1RS), were prepared according to our previous report.75 The TBDMS-protected tBuAH dimer, 1RR (or 1RS), was hydrolyzed with trifluoroacetic acid (TFA) to remove the t-butyl groups. The obtained 2RR (or 2RS) was coupled with N-methylethylamine in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), 1-hydroxybenzotriazole (HOBt), and diisopropylethylamine (DIPEA) to yield 3RR (or 3RS). The TBDMS protecting group of 3RR (or 3RS) was removed using tetrabutylammonium fluoride (TBAF) to form the monomer used in this study, i.e., the RR (or RS) dimer. All the steps proceeded in reasonable yields (ca. 60–90%). Both the RR and RS dimers were fully characterized by 1H and 13C NMR and electrospray ionization-mass spectrometry (ESI-MS) (see Experimental, ESI†). As an example, the 1H NMR spectra of RR and RS dimers are shown in Fig. 1a and b. These spectra indicate the signals due to the triazole and two methine protons at ca. 7.8, 5.7, and 4.8 ppm, respectively. The signals at ca. 2.6 ppm are assignable to the ethynyl proton. There are signals due to the methylene protons in the region of 2.2–2.5 ppm. These spectra also contain the signals of N-methyl protons at ca. 2.9 ppm and the signals due to the N-ethyl protons at ca. 3.3 and 1.1 ppm. It should be noted here that the signals due to N-methyl protons were observed as four singlets, indicating that the amide moieties take both cis and trans conformations in the dimers.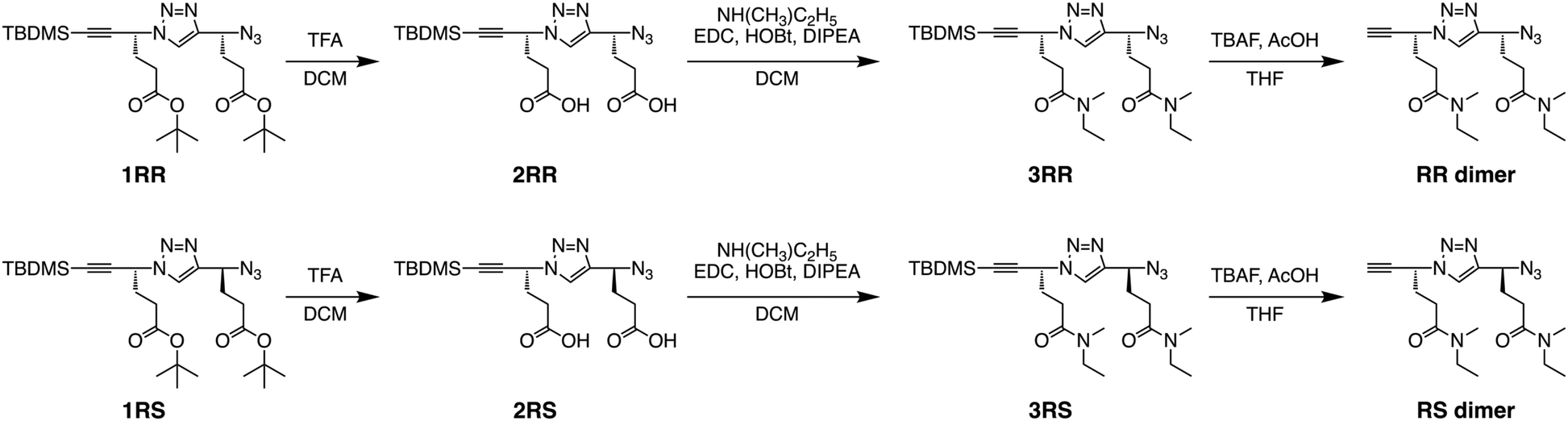 | ||
| Scheme 1 Synthesis of the RR and RS dimers of ME. | ||
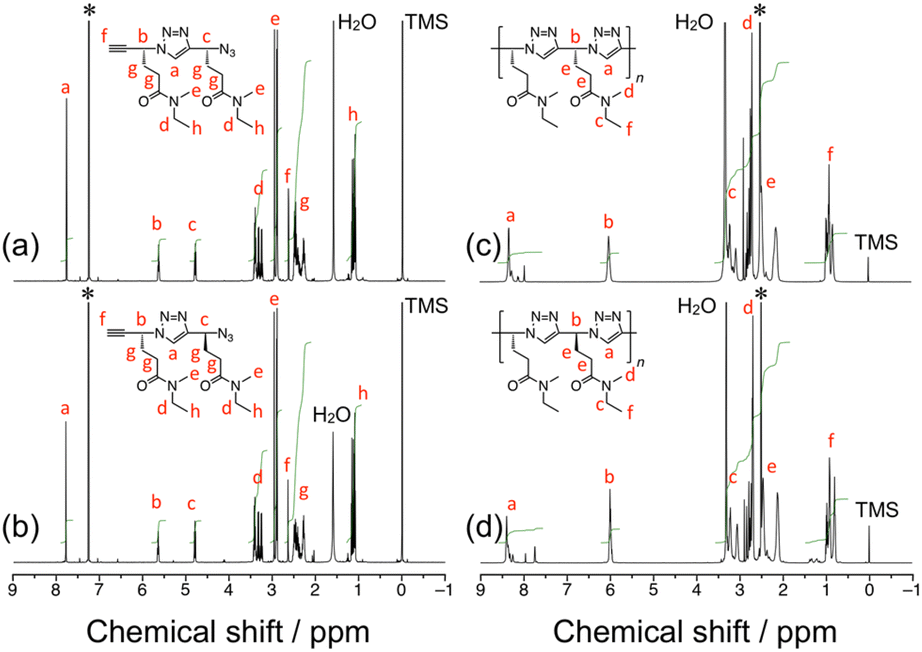 | ||
| Fig. 1 1H NMR spectra for the RR dimer (a) and RS dimer (CDCl3) (b), and for iso-poly(ME)-2 (c) and syndio-poly(ME)-3 (DMSO-d6) (d). | ||
CuAAC polymerizations of the RR and RS dimers were carried out in DMF at 60 °C for 48 h using CuBr as a copper(I) catalyst (Scheme 2 and Table S1, ESI†). It is noteworthy that CuAAC polymerization of the RR dimer yielded a gel mixture (Fig. S2, ESI†). Thus, the obtained iso-poly(ME) was fractionated into water-insoluble and water-soluble parts (iso-poly(ME)-1 and iso-poly(ME)-2, respectively). The total yield of two fractions of iso-poly(ME) was 67%. The samples of syndio-poly(ME) were obtained after reprecipitation using DMF and diethyl ether as good and poor solvents, as in the case of CuAAC polymerization of racemic ME monomer.60 The syndio-poly(ME) samples were obtained in modest yields (52–77%). It is known that the yield of CuAAC depends on the reaction conditions, i.e., substrates, solvent, copper(I) catalyst, temperature, and additive.37–39 In this study, however, the conditions of CuAAC polymerization were not fully optimized because only limited amounts of the RR and RS dimers were obtained. It is difficult to remove completely copper ions from dense 1,2,3-triazole polymers even by washing with an aqueous solution of ethylenediaminetetraacetic acid (EDTA),54,58,60 In this study, the amount of residual copper ions were roughly estimated for the polymer samples by elemental analysis (see Experimental, ESI†).
 | ||
| Scheme 2 CuAAC polymerization of the RR and RS dimers of ME. | ||
Two samples of iso-poly(ME) and three samples of syndio-poly(ME) were utilized in this study. Table S1 in ESI† summarizes the Mw and Mw/Mn values estimated for the poly(ME) samples by size exclusion chromatography (SEC) calibrated with standard samples of poly(ethylene glycol) (PEG) and poly(ethylene oxide) (PEO). The Mw values for iso-poly(ME)-1, iso-poly(ME)-2, syndio-poly(ME)-1, syndio-poly(ME)-2, and syndio-poly(ME)-3 were 1.8 × 104, 1.8 × 103, 9.1 × 103, 6.6 × 103, and 2.5 × 103, respectively (see also Fig. S3, ESI†). Fig. 1c and d display 1H NMR spectra for iso-poly(ME)-2 and syndio-poly(ME)-3. The spectra contain the signals assignable to triazole and methine protons in the backbone at ca. 8.4 and 6.0 ppm, respectively. The signals in the region of 2.1–2.5 ppm are ascribable to the methylene protons. The spectra also indicate the signals of N-methyl protons at ca. 2.8 ppm and the signals due to the N-ethyl protons at ca. 3.3 and 0.9 ppm. These data are indicative of successful synthesis of the iso-poly(ME) and syndio-poly(ME) samples. As can be seen in Fig. S4 in ESI,† the circular dichroism (CD) spectrum for iso-poly(ME) shows a significant positive signal whereas that for syndio-poly(ME) does not contain any significant signals. The solubility of the obtained polymer samples in various solvents was tested (Table 1). This table also contains the data for poly(ME) of Mw = 7.1 × 103 obtained by CuAAC polymerization of racemic ME (a-poly(ME)) for comparison.60iso-Poly(ME)-2, syndio-poly(ME)-1, syndio-poly(ME)-2, and syndio-poly(ME)-3 exhibit almost the same solubilities as those of a-poly(ME); These four samples were soluble in many solvents, e.g., water, methanol, ethanol, dimethyl sulfoxide (DMSO), DMF, acetonitrile, chloroform, and DCM, but insoluble in ethyl acetate, toluene, diethyl ether, and hexane. On the other hand, iso-poly(ME)-1 was soluble only in DMSO and DMF at higher temperatures presumably because of the higher Mw.
| Solvent | a-Poly(ME)b | iso-Poly(ME)-1c | iso-Poly(ME)-2 | syndio-Poly(ME)-1 | syndio-Poly(ME)-2 | syndio-Poly(ME)-3 |
|---|---|---|---|---|---|---|
| a “++”, “+”, and “–” denote soluble at ≥10 g L−1, ≥1 g L−1, and insoluble, respectively. b Data from Ref. 60. c At a temperature close to the boiling point. d At ca. 0 °C. | ||||||
| Waterd | ++ | − | ++ | ++ | ++ | ++ |
| Methanol | ++ | − | ++ | ++ | ++ | ++ |
| Ethanol | ++ | − | ++ | ++ | ++ | ++ |
| 2-Propanol | − | − | + | − | − | − |
| DMSO | ++ | ++ | ++ | ++ | ++ | ++ |
| DMF | ++ | ++ | ++ | ++ | ++ | ++ |
| Acetonitrile | ++ | − | ++ | ++ | ++ | ++ |
| Acetone | − | − | ++ | ++ | ++ | ++ |
| Ethyl acetate | − | − | − | − | − | − |
| THF | − | − | + | − | − | − |
| Chloroform | ++ | − | ++ | ++ | ++ | ++ |
| DCM | ++ | − | ++ | ++ | ++ | ++ |
| Toluene | − | − | − | − | − | − |
| Diethyl ether | − | − | − | − | − | − |
| Hexane | − | − | − | − | − | − |
Thermoresponsive gel-to-sol transition
Since the CuAAC polymerization of the RR dimer yielded a gel mixture as described above, the thermoresponsive gelation behavior of iso-poly(ME)-1 was investigated using DMSO and DMF as solvent. The sample solutions of iso-poly(ME)-1 were prepared using DMSO and DMF, respectively, at higher temperatures. These binary mixtures turned gel at lower temperatures, as can be seen in Fig. 2a. These observations indicate that extended network structures were formed through noncovalent crosslinking between iso-poly(ME) chains at low temperatures. Since gelation was not observed for any binary mixtures of a-poly(ME) or syndio-poly(ME) with DMSO or DMF, it is likely that this gelation is based on longer sequences of the R-isomer units. As can be seen in Fig. S5 in ESI,† the powder X-ray diffraction pattern (PXRD) for iso-poly(ME)-1 exhibits resolved peaks, indicating that iso-poly(ME)-1 is crystalline. It is thus likely that the crosslinks in the binary gel mixtures are based on the partial crystallization of longer iso-poly(ME)-1 chains. Fig. 2b displays the gel-to-sol transition temperature (Tsol) determined by the sample inversion method for binary mixtures of varying polymer concentrations (C). In the case of the iso-poly(ME)-1/DMSO binary mixture, Tsol increased from 52 to 130 °C with increasing C from 1.2 to 4.9 wt%. In the case of the iso-poly(ME)-1/DMF binary mixture, on the other hand, Tsol increased only slightly from 103 to 114 °C with increasing C from 1.1 to 7.3 wt%. These observations indicate that the temperature dependencies of solubility of iso-poly(ME)-1 in the solvents should be different because Tsol should be lower for a better solvent. Both DMSO and DMF are aprotic polar solvents that dissolve well the dense 1,2,3-triazole polymers we prepared so far. These solvents may also exhibit a higher affinity for the N-ethyl-N-methylamide side chains of poly(ME) because of the larger dielectric constants (47.24 and 38.25 for DMSO and DMF, respectively, at 20 °C).76 At present, we do not have any reasonable explanations for the difference in gelation behavior of iso-poly(ME)-1 in DMSO and DMF, i.e., the C dependency of Tsol.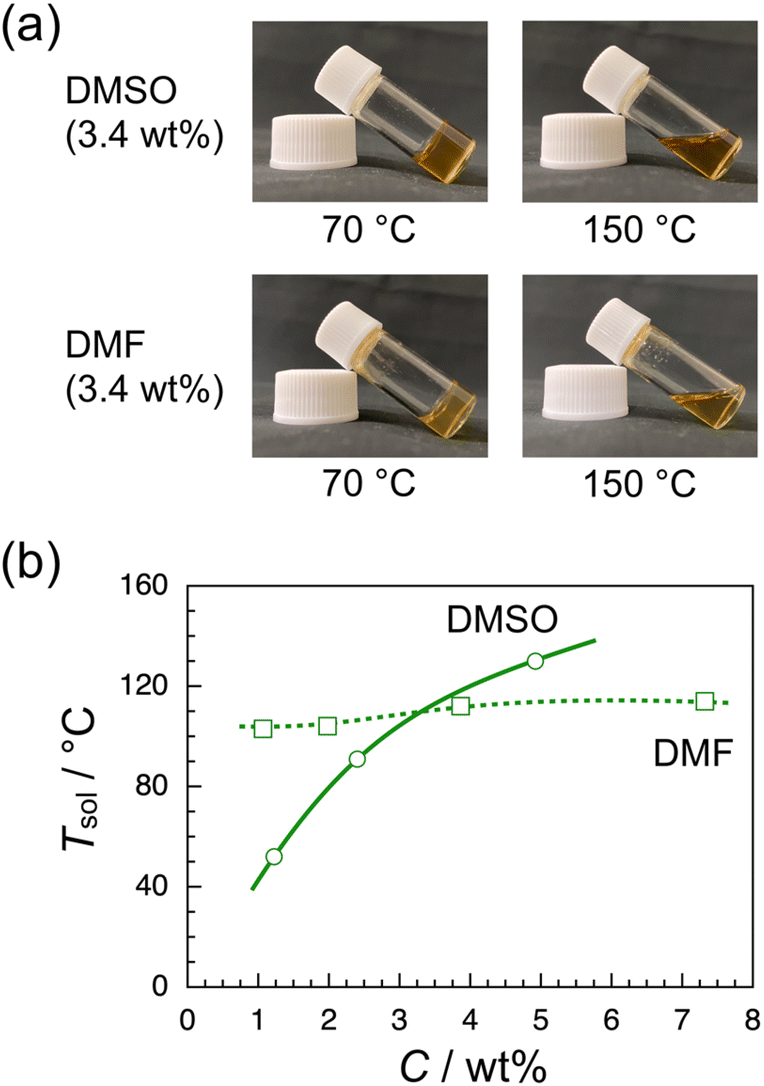 | ||
| Fig. 2 Photographs for binary mixtures of iso-poly(ME)-1/DMSO and iso-poly(ME)-1/DMF at 70 and 150 °C (a) and Tsol as a function of C for the iso-poly(ME)-1/DMSO (circle) and iso-poly(ME)-1/DMF binary mixtures (square) (b). | ||
Lower-critical-solution-temperature-type phase transition
Since our previous study has demonstrated that a-poly(ME) is an LCST-type thrmoresponsive polymer,60 the phase transition behavior of aqueous solutions of the water-soluble samples, i.e., iso-poly(ME)-2, syndio-poly(ME)-1, and syndio-poly(ME)-3, was investigated. Fig. 3a demonstrates photographs for 1.0 wt% aqueous solutions of iso-poly(ME)-2 (upper) and syndio-poly(ME)-1 (lower). The bluish green color of the iso-poly(ME)-2 aqueous solution was due to the residual copper ions. The aqueous solution of iso-poly(ME)-2 exhibited almost the same appearance at 0 and 100 °C. On the other hand, the aqueous solution of syndio-poly(ME)-1 was rather transparent at 0 °C whereas it was strongly turbid at 25 °C. These observations indicate that the aqueous solution of syndio-poly(ME)-1 underwent the LCST-type phase separation whereas the aqueous solution of iso-poly(ME)-2 did not. As can be seen in Fig. 3b, the transmittance data were monitored for the 1.0 wt% aqueous solutions of iso-poly(ME)-2 and syndio-poly(ME)-1 at 510 nm, at which the transmittance was close to the maximum at lower temperatures, with heating and cooling at 1.0 and 0.30 °C min−1, respectively. The transmittance of the aqueous solution of iso-poly(ME)-2 was almost constant at ca. 90% in the temperature range of 10–80 °C. On the other hand, as the temperature was increased from 10 to 40 °C at 0.30 °C min−1, the transmittance of the aqueous solution of syndio-poly(ME)-1 decreased abruptly from ca. 70% to 0% in a narrow temperature region of 22–23 °C. In the cooling process at 0.30 °C min−1, the transmittance for the syndio-poly(ME)-1 solution started to increase gradually at ca. 25 °C and recovered to the initial value at ca. 15 °C. (The transmittance was unstable in the region of 20–16 °C in the cooling process.) It should be noted here that the hysteresis observed for the aqueous solution of syndio-poly(ME)-1 in this study is different from the conventional one; The transmittance recovers in the cooling process in a temperature region lower than that of the transmittance decrease in the heating process.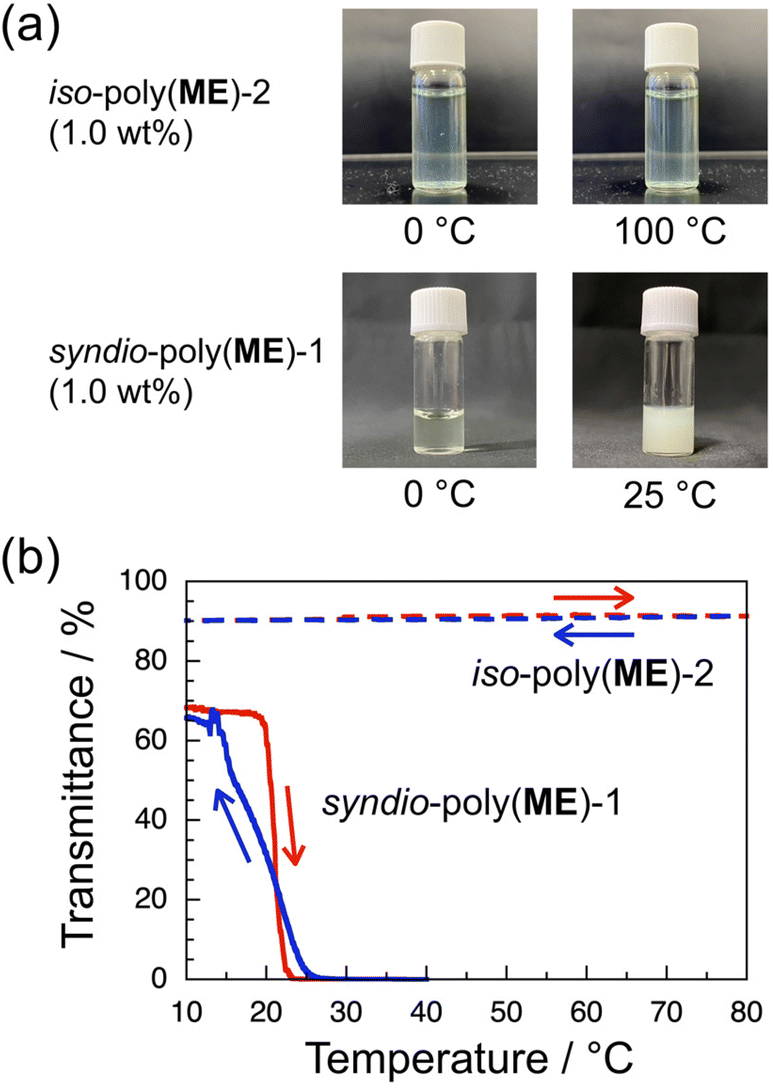 | ||
| Fig. 3 Photographs for 1.0 wt% aqueous solutions of iso-poly(ME)-2 and syndio-poly(ME)-1 at lower and higher temperatures (a) and transmittance data for 1.0 wt% aqueous solutions of iso-poly(ME)-2 (broken lines) and syndio-poly(ME)-1 (solid lines) monitored at 510 nm with heating (red) and cooling (blue) at 1.0 and 0.30 °C min−1, respectively (b). | ||
It is noteworthy that the transmittance data for the syndio-poly(ME)-1 solution were strongly dependent on the heating and cooling rates. The broken lines in Fig. 4a show the transmittance data obtained for a 1.0 wt% aqueous solution of syndio-poly(ME)-1 with heating and cooling at 3.0, 2.0, 1.0, 0.50, and 0.30 °C min−1. At heating and cooling rates of 3.0, 2.0, 1.0, and 0.50 °C min−1, the transmittance decreased abruptly at a certain temperature between ca. 22 and 23 °C in the heating process, whereas the transmittance commenced to increase at a certain temperature between ca. 21 and 20 °C in the cooling process, indicative of normal hysteresis. In the cooling process at 0.30 °C min−1, on the other hand, the transmittance started to recover at ca. 25 °C, which was higher than the temperature range of decrease in transmittance in the heating process. The clouding and clearing point temperatures (Tcloud and Tclear, respectively) were roughly estimated as the temperature at which the transmittance took half of the initial value of heating process, and plotted in Fig. 4b against the heating or cooling rate. This figure indicates that as the heating or cooling rate was decreased from 3.0 to 0.30 °C min−1, Tcloud decreased gradually from ca. 24 to 21 °C whereas Tclear increased gradually from ca. 13 to 15 °C in a range of 3.0–0.50 °C min−1, and then increased markedly to 20 °C in a narrow range of 0.50–0.30 °C min−1.
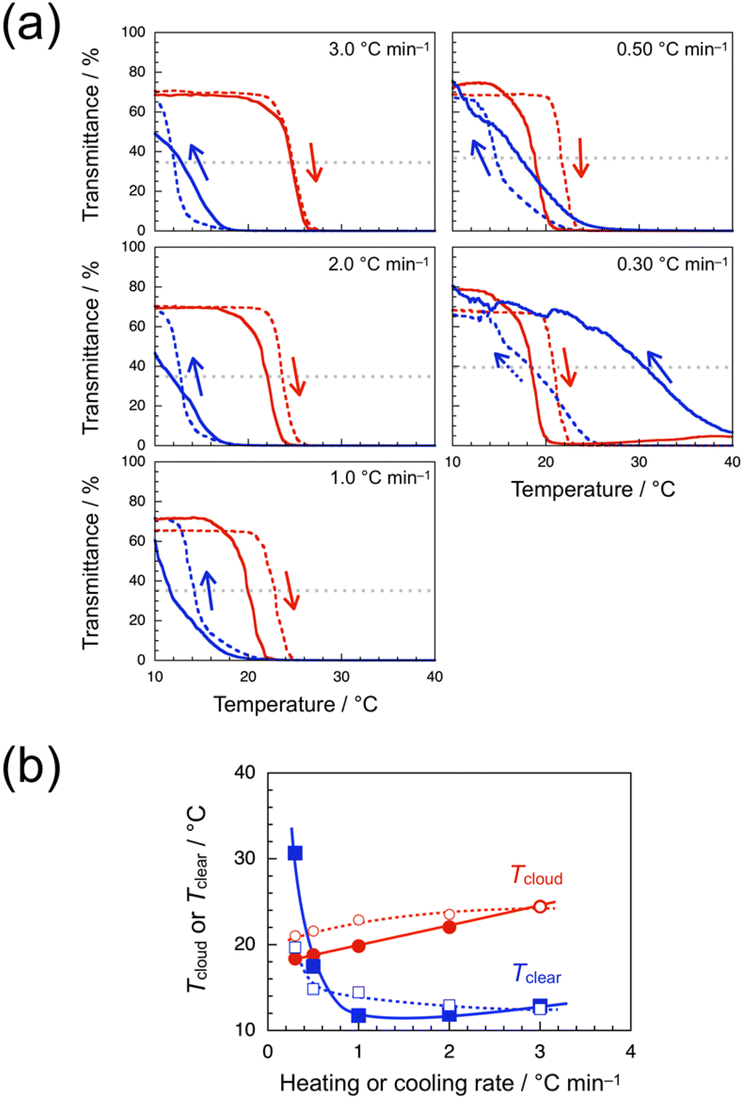 | ||
| Fig. 4 Transmittance data for a 1.0 wt% aqueous solution of syndio-poly(ME)-1 before (broken lines) and after treatment with 3-mercaptopropyl silica gel (solid lines) monitored at 510 nm with heating (red) and cooling (blue) at 3.0, 2.0, 1.0, 0.50, and 0.30 °C min−1 (a), and Tcloud (red circle) and Tclear (blue square) as a function of the heating and cooling rate determined for the 1.0 wt% aqueous solution of syndio-poly(ME)-1 before (unfilled symbols) and after treatment with 3-mercaptopropyl silica gel (filled symbols) in the heating and cooling process, respectively (b). | ||
Since syndio-poly(ME)-1 sample contained a small amount of residual copper ions (see Experimental, ESI†), the copper ions might cause the unique hysteresis observed in the slow heating and cooling processes.77 Thus, the sample was treated with 3-mercaptopyropyl silica gel. It was not possible to remove all the residual copper ions, but a syndio-poly(ME)-1 sample with ca. 25% copper ions removed was recovered. The transmittance data were recorded for a 1.0 wt% aqueous solution of the syndio-poly(ME)-1 sample treated (solid lines in Fig. 4a). When the temperature was increased and decreased at 3.0 °C min−1, the transmittance data were practically identical to those of the untreated sample. In the heating process at 2.0 or 1.0 °C min−1, the transmittance decreased in a lower temperature range than that for the untreated sample. In the cooling process at 2.0 or 1.0 °C min−1, the transmittance started to increase at ca. 20 °C, which was almost the same as that for the untreated sample, but the transmittance recovered more slowly and did not reach the initial value at 10 °C. The unique hysteresis was observed at heating and cooling rate of 0.50 °C min−1; The transmittance decreased in a wider temperature range of ca. 15–20 °C in the heating process, whereas the transmittance started to increase at ca. 26 °C in the cooling process and then recovered gradually to the initial value at 10 °C. It is noteworthy that the effect of copper ions was more pronounced in the slower heating and cooling processes at 0.30 °C min−1. In the heating process, the transmittance decreased from ca. 80% to 0% in the temperature range of 15–21 °C, and then increased slightly to 4% in a higher temperature range of 25–40 °C. In the subsequent cooling process, the transmittance gradually recovered from ca. 7% as the temperature was decreased from 40 °C and returned the initial value at 10 °C. These observations are indicative of inverse hysteresis. Tcloud and Tclear were also roughly estimated and plotted in Fig. 4b against the heating or cooling rate. As the heating or cooling rate was decreased from 3.0 to 0.10 °C min−1, Tcloud decreased gradually from ca. 24 to 18 °C whereas Tclear decreased slightly from ca. 13 to 12 °C in a range of 3.0–1.0 °C min−1, and then increased markedly to ca. 31 °C in a range of 1.0–0.30 °C min−1.
The transmittance data were also obtained for a 1.0 wt% aqueous solution of syndio-poly(ME)-3 of Mw lower than that of syndio-poly(ME)-1. Fig. 5a shows the transmittance data obtained for a 1.0 wt% aqueous solution of syndio-poly(ME)-3 with heating and cooling at 3.0, 2.0, 1.0, 0.40, and 0.10 °C min−1. In the faster heating and cooling processes at 3.0 and 2.0 °C min−1, the transmittance data are indicative of normal hysteresis. In the slower heating and cooling processes at 1.0, 0.40, and 0.10 °C min−1, on the other hand, the transmittance data indicate inverse hysteresis. These observations indicate that shorter chains of syndio-poly(ME) are rearranged more easily in polymer aggregates formed at higher temperatures. Tcloud and Tclear were also plotted in Fig. 5b against the heating or cooling rate. This figure indicates that as the heating or cooling rate was decreased from 3.0 to 0.10 °C min−1, Tcloud decreased gradually from 28 to 23 °C whereas Tclear increased gradually from 20 to 38 °C.
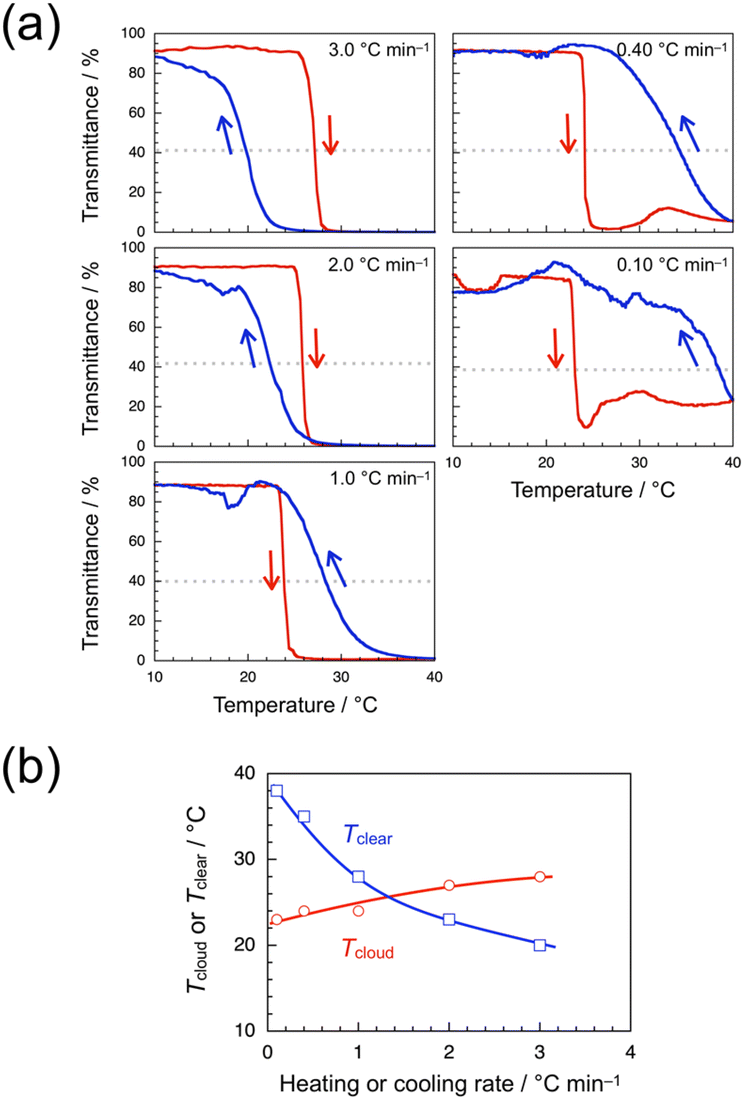 | ||
| Fig. 5 Transmittance data for a 1.0 wt% aqueous solution of syndio-poly(ME)-3 monitored at 510 nm with heating (red) and cooling (blue) at 3.0, 2.0, 1.0, 0.40, and 0.10 °C min−1 (a), and Tcloud (red circle) and Tclear (blue square) as a function of the heating and cooling rate determined for the 1.0 wt% aqueous solution of syndio-poly(ME)-3 solution in the heating and cooling process, respectively (b). | ||
The LCST-type phase transition behavior for aqueous solutions of syndio-poly(ME)-3 was also investigated by 1H NMR and differential scanning calorimetry (DSC). Fig. 6 displays 1H NMR spectra recorded for a 1.0 wt% solution of syndio-poly(ME)-3 in D2O at different temperatures, i.e., 20, 40, 60, and 80 °C. Here the temperature was increased to the measurement temperature in a few minutes and then the sample solution was equilibrated. As can be seen in Fig. 6c and d, new signals ascribable to the triazole and methine protons were observed in lower magnetic fields at higher temperatures, i.e., 60 and 80 °C. These signals are likely ascribed to more hydrated polymer chains in aggregates. The fraction of more hydrated polymer chain was roughly estimated to be ca. 28% at 80 °C from the area intensities of signals due to the triazole protons. Fig. S6 in ESI† shows the DSC data obtained for a 1.0 wt% aqueous solution of syndio-poly(ME)-3 at heating and cooling rates of 1.0 °C min−1. The DSC data did not exhibit any significant signals ascribable to the LCST-type phase transition of aqueous solution of syndio-poly(ME)-3 in the heating or cooling process, similar to the case of a-poly(ME).60 These observations indicate that only a small fraction of polymer chains underwent the phase transition or the phase transition of aqueous syndio-poly(ME) solution caused only small calorimetric changes.
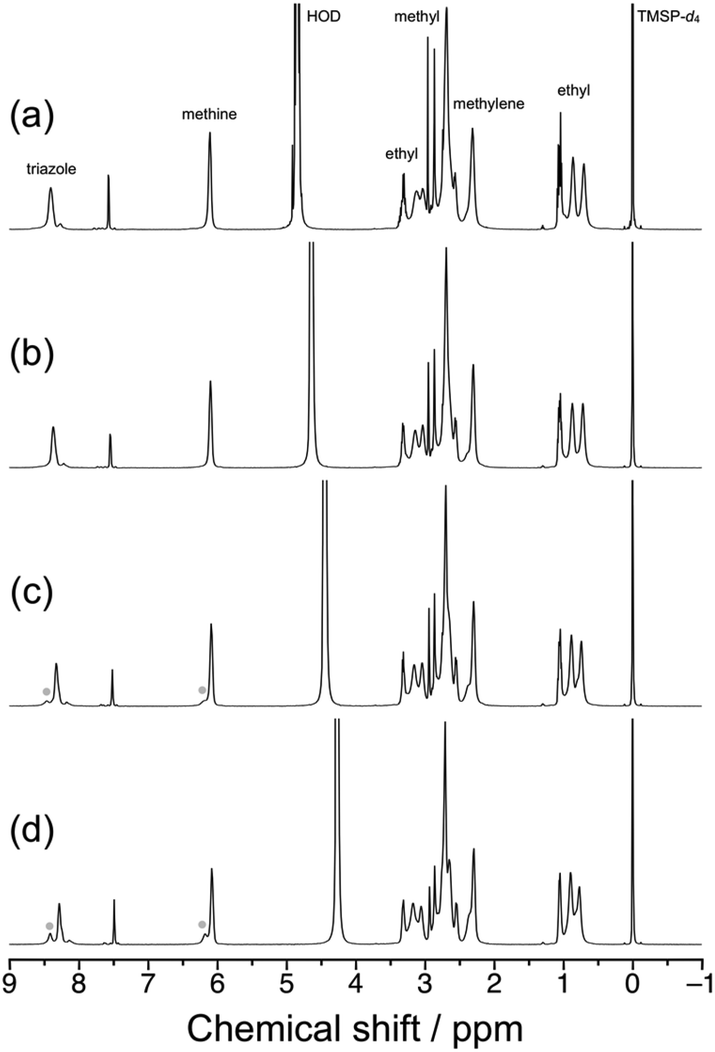 | ||
| Fig. 6 1H NMR spectra for a 1.0 wt% solution of syndio-poly(ME)-3 in D2O measured at 20 (a), 40 (b), 60 (c), and 80 °C (d). | ||
In LCST-type phase transition of aqueous solutions of thermoresponsive polymers, hysteresis is often observed; Once the phase separation occurs at higher temperatures, the transmittance recovers in the cooling process in the temperature region lower than the region in which the transmittance decreases in the heating process. The normal hysteresis is likely caused by slow rehydration in the cooling process presumably because of the restricted mobility of polymer chains in aggregates. To the best of our knowledge, there have been only a few examples of inverse hysteresis in the LCST-type phase transition.77,78 Yeshchenko et al.,77 reported that aqueous solutions containing a dextran-graft-PNIPAM copolymer and gold nanoparticles (AuNPs) underwent an LCST-type phase transition exhibiting inverse hysteresis. The authors concluded that AuNPs counteracted the formation of PNIPAM aggregates. In this study, aqueous solutions of syndio-poly(ME) samples underwent the LCST-type phase separation. The phase separation behavior depended on the heating and cooling rates, the amount of residual copper ions, and the molecular weight. It is noteworthy that inverse hysteresis was observed in the slow heating and cooling processes for a syndio-poly(ME) sample containing a smaller amount of residual copper ions or possessing a lower molecular weight. In the heating process, where the clear inverse hysteresis was observed, the transmittance increased slightly in a higher temperature region after phase separation occurred. It is likely that syndio-poly(ME) chains are rearranged in polymer aggregates during the slight increase in transmittance, resulting in the formation of polymer aggregates which are more readily rehydrated. Since the conformational change might occur rather slowly, the inverse hysteresis was observed only in slower processes. The residual copper ions inhibit the conformational change presumably because of coordination to the 1,2,3-triazole moieties and amide side chains in polymer chains.38,79syndio-Poly(ME) chains of lower molecular weights undergo more easily the conformational change. It should be noted here that the inverse hysteresis was not observed for any aqueous solutions of a-poly(ME) samples. It is thus likely that the syndiotactic sequence, i.e., the alternating sequence of the R- and S-isomers of ME, which is longer than a critical length, is necessary for the inverse hysteresis. The LCST-type phase separation behavior of aqueous solutions of uniform oligomers of syndio-poly(ME) will be further investigated to elucidate the molecular mechanism of inverse hysteresis in near future.
Conclusions
In this study, two types of stereoregular poly(ME) samples, i.e., iso-poly(ME) and syndio-poly(ME), were synthesized by CuAAC polymerization of the RR and RS dimers, respectively. Since the CuAAC polymerization of the RR dimer yielded a gel mixture, the obtained iso-poly(ME) was fractionated into water-insoluble and water-soluble parts, i.e., iso-poly(ME)-1 (Mw = 1.8 × 104) and iso-poly(ME)-2 (Mw = 1.8 × 103), respectively. On the other hand, the three samples of syndio-poly(ME) (syndio-poly(ME)-1 (Mw = 9.1 × 103), syndio-poly(ME)-2 (Mw = 6.6 × 103), and syndio-poly(ME)-3 (Mw = 2.5 × 103)) were obtained after reprecipitation using DMF and diethyl ether as good and poor solvents. The poly(ME) samples used in this study contained a small amount of residual copper ions, because the dense 1,2,3-triazole backbones usually adsorb strongly copper ions. The thermoresponsive gel-to-sol transition behavior was investigated for iso-poly(ME)-1 using DMSO and DMF as solvent. The iso-poly(ME)-1/DMSO and iso-poly(ME)-1/DMF binary mixtures were gel at lower temperatures whereas the mixtures turned sol at a certain temperature as the temperature was increased, indicating that extended network structures were formed through noncovalent crosslinking between iso-poly(ME) chains at low temperatures, e.g., partial crystallization. The temperature-dependent transmittance data for aqueous solutions of iso-poly(ME)-2, syndio-poly(ME)-1, and syndio-poly(ME)-3 demonstrated that the syndio-poly(ME) samples were LCST-type thermoresponsive polymers whereas iso-poly(ME)-2 was not. The LCST-type phase separation behavior of aqueous solutions of the syndio-poly(ME) samples depended on the heating and cooling rates, the amount of residual copper ions, and the molecular weight. The noteworthy is that inverse hysteresis was observed in slow heating and cooling processes for a syndio-poly(ME) sample containing a smaller amount of residual copper ions or possessing a lower molecular weight. In the heating process of inverse hysteresis, the transmittance increased slightly in a higher temperature region after phase separation occurred. It is likely that syndio-poly(ME) chains are rearranged in polymer aggregates during the slight increase in transmittance, resulting in the formation of polymer aggregates which are more readily rehydrated.Author contributions
A. H. supervised the project; A. H., Y. K. and K. O. designed the project; K. O., J. M., S. Y. and M. N. performed the experiments; A. H., M. N. and K. O. analyzed data; A. H., Y. K., M. N. and K. O. discussed the results; A. H. and K. O. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Professor Hiroyasu Yamaguchi and Assistant Professor Yuichiro Kobayashi for allowing us to use the UV absorption spectrophotometer, the CD spectropolarimeter, and the chiral HPLC instrument. The authors acknowledge Professor Sadahito Aoshima and Associate Professor Arihiro Kanazawa, Department of Macromolecular Science, Graduate School of Science, Osaka University, for their kind support on the transmittance measurements and their valuable suggestions. The authors also thank Dr Yasuto Todokoro, Analytical Instrument Facility, Graduate School of Science, Osaka University, for his kind support on the DSC measurements.References
- Handbook of Stimuli-Responsive Materials, ed. M. W. Urban, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS.
- S. Wang, Q. Liu, L. Li and M. W. Urban, Macromol. Rapid Commun., 2021, 42, 2100054 CrossRef CAS PubMed.
- A. Hashidzume and A. Harada, in Supramolecular Polymer Chemistry, ed. A. Harada, Wiley-VCH, Weinheim, Germany, 2012, pp. 231–267 Search PubMed.
- A. Laukkanen, L. Valtola, F. M. Winnik and H. Tenhu, Macromolecules, 2004, 37, 2268–2274 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Adv. Polym. Sci., 2008, 210, 169–208 CrossRef CAS.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- Y. Kohno, S. Saita, Y. Men, J. Yuan and H. Ohno, Polym. Chem., 2015, 6, 2163–2178 RSC.
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 1700–1707 CrossRef CAS.
- D. N. Schulz, D. G. Peiffer, P. K. Agarwal, J. Larabee, J. J. Kaladas, L. Soni, B. Handwerker and R. T. Garner, Polymer, 1986, 27, 1734–1742 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromolecules, 2012, 45, 3910–3918 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS.
- J. Seuring and S. Agarwal, ACS Macro Lett., 2013, 2, 597–600 CrossRef CAS.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- J. Niskanen and H. Tenhu, Polym. Chem., 2017, 8, 220–232 RSC.
- Z. Xu and W. Liu, Chem. Commun., 2018, 54, 10540–10553 RSC.
- C. M. Papadakis, P. Müller-Buschbaum and A. Laschewsky, Langmuir, 2019, 35, 9660–9676 CrossRef CAS PubMed.
- M. R. Matanović, J. Kristl and P. A. Grabnar, Int. J. Pharm., 2014, 472, 262–275 CrossRef.
- A. P. Constantinou and T. K. Georgiou, Eur. Polym. J., 2016, 78, 366–375 CrossRef CAS.
- M. J. Taylor, P. Tomlins and T. S. Sahota, Gels, 2017, 3, 4 CrossRef PubMed.
- S. Chatterjee, P. C. Hui and C.-w. Kan, Polymers, 2018, 10, 480 CrossRef PubMed.
- K. Zhang, K. Xue and X. J. Loh, Gels, 2021, 7, 77 CrossRef CAS PubMed.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- A. Garle, S. Kong, U. Ojha and B. M. Budhlall, ACS Appl. Mater. Interfaces, 2012, 4, 645–657 CrossRef CAS.
- Q. Zhao, M. Behl and A. Lendlein, Soft Matter, 2013, 9, 1744–1755 RSC.
- Y. Liu, Y. Li, G. Yang, X. Zheng and S. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 4118–4126 CrossRef CAS PubMed.
- B. Q. Y. Chan, S. J. W. Heng, S. S. Liow, K. Zhang and X. J. Loh, Mater. Chem. Front., 2017, 1, 767–779 RSC.
- Y. Xia, Y. He, F. Zhang, Y. Liu and J. Leng, Adv. Mater., 2021, 33, 2000713 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 14397–14402 CrossRef CAS.
- Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, Wiley & Sons, Chichester, UK, 2009 Search PubMed.
- Click Triazoles, ed. J. Košmrlj, Springer, Heidelberg, Germany, 2012 Search PubMed.
- Chemistry of 1,2,3-Triazoles, ed. W. Dehaen and V. A. Bakulev, Springer, Cham, Switzerland, 2015 Search PubMed.
- B. Le Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef CAS.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052–1072 CrossRef CAS.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2010, 43, 8693–8702 CrossRef CAS.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC.
- Click Polymerization, A. Qin and B. Z. Tang, ed. Royal Society of Chemistry, London, UK, 2018 Search PubMed.
- J. A. F. Joosten, N. T. H. Tholen, F. Ait El Maate, A. J. Brouwer, G. W. van Esse, D. T. S. Rijkers, R. M. J. Liskamp and R. J. Pieters, Eur. J. Org. Chem., 2005, 3182–3185 CrossRef CAS.
- M. van Dijk, M. L. Nollet, P. Weijers, A. C. Dechesne, C. F. van Nostrum, W. E. Hennink, D. T. S. Rijkers and R. M. J. Liskamp, Biomacromolecules, 2008, 9, 2834–2843 CrossRef CAS PubMed.
- S. van der Wal, C. J. Capicciotti, S. Rontogianni, R. N. Ben and R. M. J. Liskamp, MedChemComm, 2014, 5, 1159–1165 RSC.
- S. Binauld, F. Boisson, T. Hamaide, J.-P. Pascault, E. Drockenmuller and E. Fleury, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5506–5517 CrossRef CAS.
- C. Besset, S. Binauld, M. Ibert, P. Fuertes, J.-P. Pascault, E. Fleury, J. Bernard and E. Drockenmuller, Macromolecules, 2010, 43, 17–19 CrossRef CAS.
- M. M. Obadia and E. Drockenmuller, Chem. Commun., 2016, 52, 2433–2450 RSC.
- O. D. Montagnat, G. Lessene and A. B. Hughes, J. Org. Chem., 2010, 75, 390–398 CrossRef CAS PubMed.
- O. D. Montagnat, G. Lessene and A. B. Hughes, Aust. J. Chem., 2010, 63, 1541–1549 CrossRef CAS.
- A. Hashidzume, T. Nakamura and T. Sato, Polymer, 2013, 54, 3448–3451 CrossRef CAS.
- Y. Yang, A. Mori and A. Hashidzume, Polymers, 2019, 11, 1086 CrossRef PubMed.
- Y. Yang and A. Hashidzume, Macromol. Chem. Phys., 2019, 220, 1900317 CrossRef CAS.
- Y. Yang, Y. Kamon, N. A. Lynd and A. Hashidzume, Macromolecules, 2020, 53, 10323–10329 CrossRef CAS.
- S. Yamasaki, Y. Kamon, L. Xu and A. Hashidzume, Polymers, 2021, 13, 1627 CrossRef CAS PubMed.
- L. Xu, Y. Kamon and A. Hashidzume, Polymers, 2021, 13, 1614 CrossRef CAS PubMed.
- K. Okuno, T. Arisawa, Y. Kamon, A. Hashidzume and F. M. Winnik, Langmuir, 2022, 38, 5156–5165 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- C. S. Biswas, K. Mitra, S. Singh and B. Ray, J. Chem. Sci., 2016, 128, 415–420 CrossRef CAS.
- T. Tada, T. Hirano, K. Ute, Y. Katsumoto, T.-A. Asoh, T. Shoji, N. Kitamura and Y. Tsuboi, J. Phys. Chem. B, 2016, 120, 7724–7730 CrossRef CAS PubMed.
- K. Ushiro, T. Shoji, M. Matsumoto, T.-A. Asoh, H. Horibe, Y. Katsumoto and Y. Tsuboi, J. Phys. Chem. B, 2020, 124, 8454–8463 CrossRef CAS PubMed.
- B. Ray, Y. Isobe, K. Morioka, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2003, 36, 543–545 CrossRef CAS.
- B. Ray, Y. Isobe, K. Matsumoto, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2004, 37, 1702–1710 CrossRef CAS.
- T. Hirano, S. Ishii, H. Kitajima, M. Seno and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 50–62 CrossRef CAS.
- T. Hirano, H. Ishizu, M. Seno and T. Sato, Polymer, 2005, 46, 10607–10610 CrossRef CAS.
- T. Hirano, H. Kitajima, S. Ishii, M. Seno and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3899–3908 CrossRef CAS.
- T. Hirano, H. Kitajima, M. Seno and T. Sato, Polymer, 2006, 47, 539–546 CrossRef CAS.
- T. Hirano, H. Ishizu and T. Sato, Polymer, 2008, 49, 438–445 CrossRef CAS.
- T. Hirano, T. Kamikubo, Y. Fujioka and T. Sato, Eur. Polym. J., 2008, 44, 1053–1059 CrossRef CAS.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
- Y. Kamon, J. Miura, K. Okuno, M. Nakahata and A. Hashidzume, Macromolecules, 2023, 56, 292–304 CrossRef CAS.
- Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill, New York, 1999, pp. 5105–5128 Search PubMed.
- O. A. Yeshchenko, A. P. Naumenko, N. V. Kutsevol, D. O. Maskova, I. I. Harahuts, V. A. Chumachenko and A. I. Marinin, J. Phys. Chem. C, 2018, 122, 8003–8010 CrossRef CAS.
- P. Werner, M. Münzberg, R. Hass and O. Reich, Anal. Bioanal. Chem., 2017, 409, 807–819 CrossRef CAS.
- O. Clement, B. M. Rapko and B. P. Hay, Coord. Chem. Rev., 1998, 170, 203–243 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01528e |
| This journal is © The Royal Society of Chemistry 2023 |