
Supramolecular assembly of dendronized spiropyrans in aqueous solutions into nanospheres with photo- and thermo-responsive chiralities†
Shanbin
Qi
,
Xueting
Lu
,
Wenli
Mei
,
Guanglei
Gu
 ,
Wen
Li
* and
Afang
Zhang
*
,
Wen
Li
* and
Afang
Zhang
*
International Joint Laboratory of Biomimetic and Smart Polymers, School of Materials Science and Engineering, Shanghai University, Nanchen Street 333, Shanghai 200444, China. E-mail: wli@shu.edu.cn; azhang@shu.edu.cn
First published on 18th October 2023
Abstract
Tailoring the amphiphilicity of a molecule through external stimuli can alter the balance between self-association and repulsion, resulting in different propensities for its assembly. Here we report on the supramolecular assembly of a series of dendronized spiropyrans (DSPs) in water. These DSPs carry 3-fold dendritic oligoethylene glycols (OEGs) with either methoxyl or ethoxyl terminals for different hydrophilicities, and contain an Ala–Gly dipeptide to provide the chirality. These dendronized amphiphiles form supramolecular nanospheres in aqueous solutions with remarkable induced chirality to a level of 1.0 × 106 deg cm2 dmol−1. They can be tuned reversibly through photoisomerization of the spiropyran moieties from the hydrophobic SP form into the hydrophilic MC form, and can even become chirally silent through thermally mediated collapse of the dendritic OEGs. Photoisomerization of the spiropyran moieties in these DSPs is accompanied by simultaneous changes of UV absorption, fluorescence emission, supramolecular chirality and aqueous solution colors. These supramolecular nanospheres exhibit characteristic thermoresponsive behavior due to thermal collapse of the dendritic OEGs with their cloud point temperatures (Tcps) being dependent on the overall hydrophilicity of the molecules and also the aggregate morphologies resulting from how dendritic OEGs are wrapped around the aggregates. Both photo-irradiation-mediated isomerization of the spiropyran moieties and thermally mediated dehydration and collapse of the dendritic OEGs influence the amphiphilicity of these DSPs and their solvation by water, leading to varied driving forces for their assembly. NMR, circular dichroism (CD) and fluorescence spectroscopy techniques, as well as DLS and AFM techniques are combined to follow the supramolecular assembly and illustrate the aggregation mechanism. All experimental results demonstrate that the reversible chirality of the aggregates originates from the balance between dendritic OEGs and spiropyran moieties against water solvation.
1. Introduction
Amphiphilic dendritic macromolecules have been proven to show appealing features in supramolecular assembly.1–3 The major structural characteristics of dendritic macromolecules include their monodispersity, nonlinear topology and multivalency, which make a big difference compared to conventional polydisperse polymers in assemblies and their assembled morphology control.3 Non-linear topological structures from dendritic macromolecules afford the molecular segments different packing parameters, resulting in abundant ordered phases in bulk2,4–6 or in solutions.7,8 Multivalency from dendritic macromolecules is important in manipulating the assembly, especially when associations play pivotal roles in controlling morphologies of the assemblies.9 The supramolecular chiral assembly of amphiphilic dendritic macromolecules revealed that, due to the multivalent dendritic structures, amphiphilic dendritic macromolecules can form non-covalent hexameric macrocycles with variable diameters to fit guest molecules, which spontaneously stacked in one-dimension to form chiral tubules10 or were able to encapsulate a double-stranded DNA molecule through electrostatic interactions for helical co-assembly.11Stimuli-responsive supramolecular assemblies have attracted considerable attention recently in fabricating intelligent supramolecular materials.12–14 Among them, the photo-responsive supramolecular assembly has received particular attention,15–17 and photo-chromophores, such as azobenzene18,19 and spiropyran,20,21 have been extensively investigated. Spiropyran derivatives are interesting for their photo-responsive assembly, mainly because they can isomerize through UV irradiation from the spiropyran (SP) form (non-charged and more hydrophobic, non-fluorescent, colorless) to the merocyanine (MC) form (charged and more hydrophilic, fluorescent, colored pink). This isomerization is reversible, and the MC state can be isomerized back into the SP state upon heating or photo-irradiation with visible light.22–24 The isomerization kinetics of spiropyrans is dependent on steric hindrance generated through molecular interactions, and is also related to the physical microenvironment due to the dielectric constants of solvents.25–27 Therefore, photoisomerization of spiropyran derivatives has been utilized through switchable supramolecular assembly for various purposes, such as fabrication of photoresponsive organogels with switchable fluorescence,28 formation of vesicles in water showing light-fueled reversible expansion,29 fabrication of light-responsive micelles for drug delivery,30 and mimicking the dynamic nature of the native ECM to promote cell mechanotransduction.31
The combination of stimuli-responsive behaviors in supramolecular chiral assembly provides a convenient way to tune the supramolecular assembly and simultaneously modulate the supramolecular chirality.32–35 Photoresponsive chiral molecules have been found to be able to switch the helical orientation in cholesteric liquid crystals upon light stimulation.36,37 Temperature is a clean tool and can be easily manipulated, which has often been used as a stimulus in supramolecular chiral assembly of polymers, aimed at fabricating smart chiral materials.38,39 That's why thermoresponsive features are also attractive for the supramolecular assembly of small molecules.40,41 In addition, supramolecular assembly in aqueous solutions is a very important regulation of biological properties and functions,42,43 which makes thermoresponsive chiral assembly even more attractive for bioapplications.
We recently found that through dendronization with 3-fold dendritic oligoethylene glycols (OEGs), polymers exhibited unprecedent thermoresponsive behavior with fast phase transitions.44–46 Furthermore, crowded dendritic OEGs around the polymer backbones provided characteristic shielding effects to the interiors from protonation47 or host–guest interaction,48 and even biodegradation by enzymes.49,50 In addition, this shielding effect can be significantly enhanced in dendronized microgels51 or hydrogels52 through intermolecularly cooperative interactions. Through combination with 3-fold dendritic OEGs, amphiphilic dendronized tetraphenylethylenes form fluorescent thermoresponsive spheres in water with switchable chirality.53 In this work, a series of amphiphilic dendronized spiropyrans (DSPs) were synthesized, which were constructed from dendritic OEGs, dipeptides and spiropyran (Chart 1a). The dendritic OEGs afford hydrophilicity and the intrinsic thermo-responsiveness, the dipeptides from glycine and alanine contribute to the chirality, while the spiropyran moiety contributes to hydrophobicity and photo-responsiveness. These DPSs exhibit moderate water solubility, making it possible to investigate their supramolecular chiral assembly in aqueous solutions. Thanks to the dendritic OEGs and photo-responsive spiropyran moieties, the supramolecular assemblies in water from DSPs exhibit dually responsive behavior to both photo-irradiation and temperature, and their chiralities can be reversibly tuned (Chart 1b).
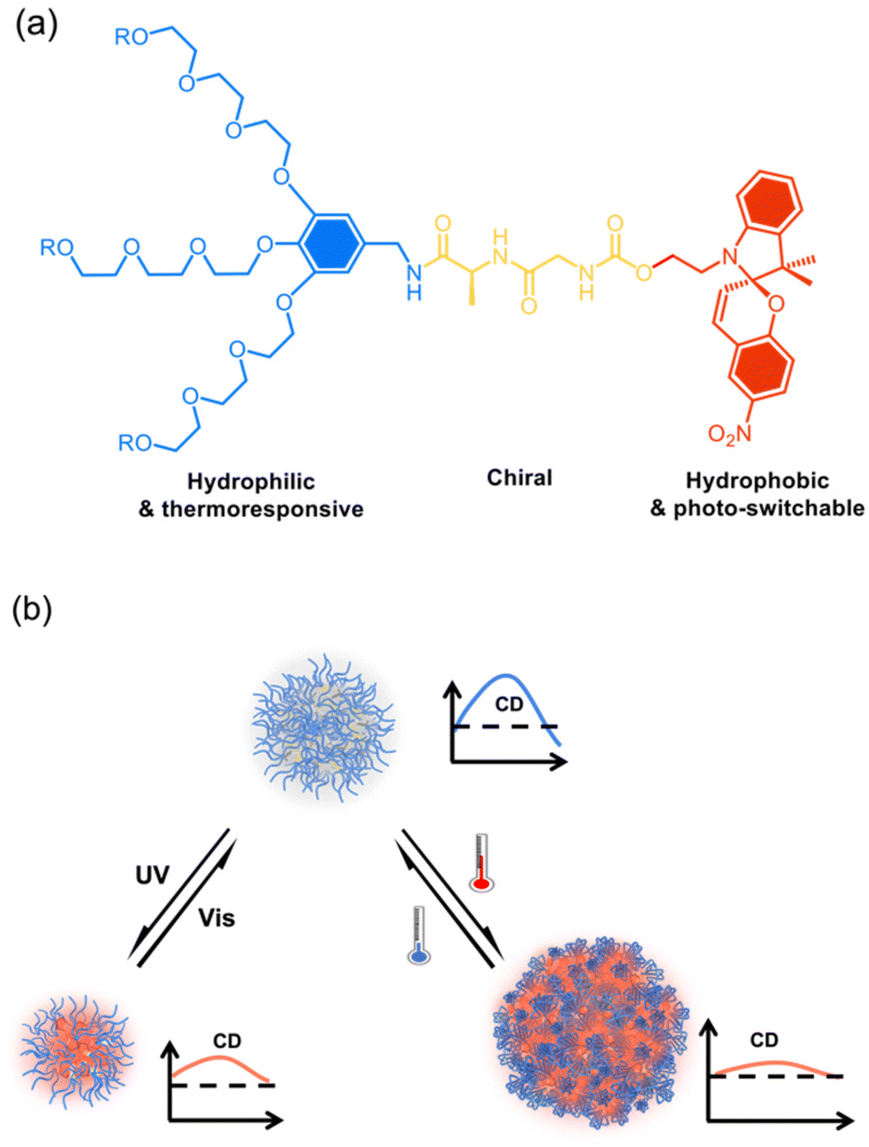 | ||
| Chart 1 Illustration of supramolecular assembly of the dendronized spiropyrans (DSPs) (a) in aqueous solutions into chiral spheres with chiralities dually tunable by irradiation and temperature (b). | ||
2. Experimental section
2.1 Materials
Active ester 1 was synthesized according to the literature method13 through esterification of 2-(3′,3′-dimethyl-6-nitrospiro[chromene-2,2′-indolin]-1′-yl)ethanol with 4-nitrophenyl chloroformate. Dendritic compounds MeG1, EtG1 and MeG1-OH were synthesized according to previous reports.44 Dipeptides Boc-GA-OH, Boc-AG-OH and H-GA-OMe were synthesized by typical solution peptide synthesis methods according to previous reports.54 Dichloromethane was distilled from CaH2 for drying. Tetrahydrofuran (THF) was distilled successively from Na and LiAlH4 for drying. Other reagents were purchased from Alfa Aesar, Sigma-Aldrich or TCI, and used without further purification. All syntheses were performed under a nitrogen atmosphere. Macherey-Nagel precoated TLC plates (silica gel 60 G/UV 254, 0.25 mm) were used for thin layer chromatography (TLC) analysis. Silica gel 60 M (200–300) was used as the stationary phase for column chromatography.2.2 Instrumentation and measurements
NMR spectra were recorded on a Bruker AV500 spectrometer (1H: 500 MHz; 13C: 125 MHz). Chemical shifts were given in ppm and referenced to residual solvent signals. Two-dimensional NOESY spectra were recorded in D2O with a mixing time of 400 ms. High resolution MALDI-TOF-MS analyses were performed on IonSpec Ultra instruments. AFM experiments were carried out on a Bruker NanoScope VIII Multi-Mode operated in peak force mode with an “E” scanner (scan range 15 μm × 15 μm) at room temperature in air. Samples were prepared by dropping on mica or through directly coating the solution onto mica using a pipette. Circular dichroism (CD) measurements were performed on a JASCO J-815 spectropolarimeter with a thermally controlled 1 mm quartz cell (1 accumulation, continuous scanning mode, scanning speed: 500 nm min−1, data pitch: 0.5 nm, response: 1 s, band width: 4.0 nm). Fluorescence emission spectra were recorded using a Horiba Jobin Yvon Fluorolog-3 fluorescence spectropolarimeter with excitation at 535 nm. Fluorescence measurements were carried out in a 10 mm path length cuvette with a solution concentration of 0.15 mg mL−1. UV/Vis measurements were recorded on a JASCO V-750 spectrophotometer equipped with a thermostatically regulated bath. Dynamic light scattering measurements were performed on a Zetasizer Nano-ZS analyzer (Malvern Instruments) with an integrated 4 mV He–Ne laser (λ = 633 nm), which used backscattering detection (scattering angle θ = 90°).2.3 Synthesis
3. Results and discussion
3.1 Synthesis and characterization of DSPs
Four DSPs, i.e., SP-GA-MeG1, SP-GA-EtG1, SP-AG-MeG1, and MeG1-GA-SP, were designed. They carry in their peripheral dendrons either ethoxyl or methoxyl units to contribute different hydrophilicities, while aiming at different phase transition temperatures. The dipeptides from glycine (G) and alanine (A) were selected due to their preference for strong hydrogen bond formation,54 aimed at transferring enhanced chirality to the spiropyran units. Both dendritic OEG and spiropyran units were connected to the dipeptides through different terminals, aimed at examining the structural effect on the supramolecular assembly and induced chirality. Their synthesis was based on conventional amidation or esterification, as outlined in Scheme 1. Amidation of Boc-GA-OH with amino-cored dendritic OEG afforded the corresponding dendronized dipeptides 2a and 2c, whose Boc groups were deprotected by TFA in quantitative yields. Through amidation with active ester 1, the deprotected dendronized dipeptides were transferred into the corresponding dendronized spiropyrans SP-GA-MeG1 and SP-GA-EtG1 in excellent yields. For the DSP with an inverted peptide sequence, SP-AG-MeG1 was prepared following a similar procedure affording an overall yield of around 50%. In order to examine the effects of the hydrogen bond on the assembly and supramolecular chirality, MeG1-GA-SP, which contains only two amide linkages, was synthesized while the former three DSPs contain three amide linkages. All new compounds were characterized by 1H and 13C NMR spectroscopy (Fig. S1–S8†) and mass spectrometry (Fig. S9–S16†), and their spectra are shown in the ESI.† | ||
| Scheme 1 Synthetic procedure for dendronized spiropyrans. Reagents and conditions: (a) DCM, TEA, 0–25 °C, overnight (53%); (b) DCM, HOBt, DiPEA, EDC·HCl, 0–25 °C, overnight (79%); (c) DCM, 1, TEA, 0–25 °C, overnight (56%); (d) DCM, TFA, 0–25 °C, overnight (100%); (e) 4b, EDC·HCl, DMAP (55%); and (f) MeOH, H2O, LiOH·H2O, 0–25 °C, overnight (90%). Abbreviations: DCM = dichloromethane, DiPEA = diisopropylethylamine, DMAP = 4-dimethylaminopyridine, EDC·HCl = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, HOBt = 1-hydroxy-1H-benzotriazole, TEA = triethylamine, and TFA = trifluoroacetic acid. Abbreviation for amino acids: G for glycine and A for alanine. | ||
3.2 Supramolecular assembly of the DSPs in aqueous solutions
The supramolecular assembly of these DSPs in aqueous solutions was first examined with 1H NMR spectroscopy. As shown in Fig. 1a for SP-GA-MeG1, protons from the DSP were well resolved in THF-d8, indicating that there was no obvious aggregation in the good solvent of THF. However, the presence of poor solvent water led to worse resolutions from the protons. For example, when the water fraction in THF (fw) was higher than 80 vol%, signals for protons corresponding to the dendritic OEGs in the range of 3.2–3.8 ppm started to become broader. Even signals of protons from the peptide and spiropyran moieties disappeared when fw increased to 90%, indicating that these moieties should have been tightly wrapped under these conditions due to the obvious aggregation.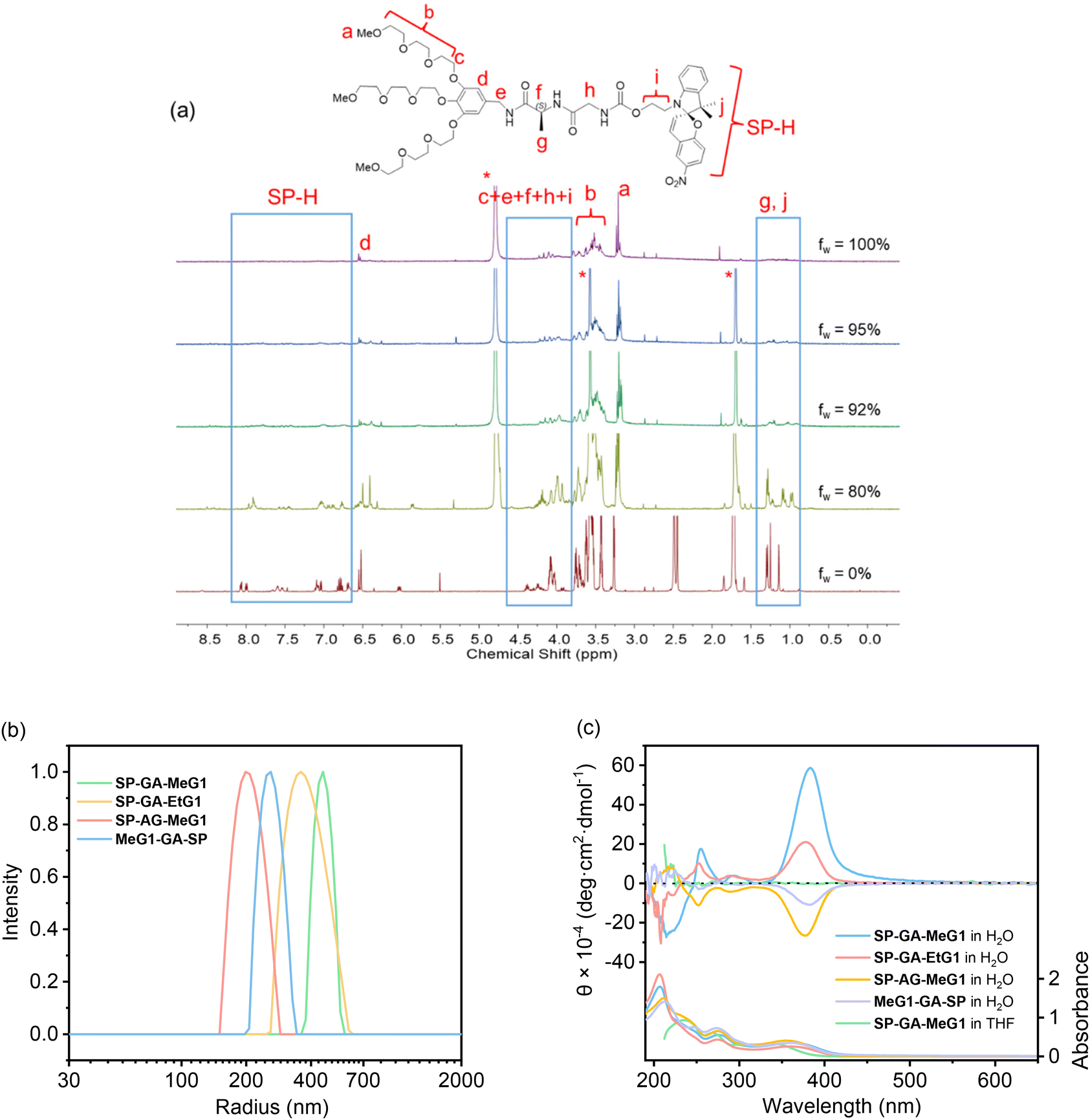 | ||
| Fig. 1 Assembly of DSPs in various solvents at 10 °C. (a) 1H NMR spectra of SP-GA-MeG1 in a mixture of D2O/THF-d8 at 10 °C with different volume fractions of water, fw. C = 1.0 mg mL−1. (b) DLS results of DSPs in aqueous solutions. C = 0.15 mg mL−1. (c) CD spectra of DSPs in different solvents. C = 0.3 mg mL−1. | ||
The assembly of these DSPs in aqueous solutions was further investigated with dynamic light scattering (DLS). In order to avoid the possible isomerization of the spiropyran moieties in water to form the merocyanine (MC), the aqueous solutions of the DSPs were first irradiated with visible light prior to the measurements. As shown in Fig. 1b, hydrodynamic radii (Rhs) for the four DSPs, SP-GA-MeG1, SP-GA-EtG1, SP-AG-MeG1, and MeG1-GA-SP, were found to be 481, 391, 213, and 276 nm, respectively, indicating the intensive aggregation of these DSPs in water. The Rh values for different DSPs are obviously different, suggesting that the molecular structure plays an important role in their assembly. Factors that influence the tendency for these DSPs to aggregate include the overall hydrophilicity and amphiphilicity of the DSPs. More hydrophilicity leads the DSPs to form aggregates with slightly larger sizes when compared to the more hydrophobic ones. Enhanced amphiphilicity for the DSPs with methoxyl terminals causes the pronounced aggregation to form aggregates with much larger sizes.
The chirality of the supramolecular assemblies was examined with circular dichroism (CD) spectroscopy. As shown in Fig. 1c, there was no induced Cotton effect for SP-GA-MeG1 in THF in the range of 350–450 nm, which corresponds to the spiropyran chromophore, suggesting that no ordered aggregates were formed in the good solvent THF. In contrast, intensive Cotton effects were observed for all four DSPs in the poor solvent water. As shown in Fig. 1c, the intensity of the positive Cotton effect at 380 nm for SP-GA-MeG1 reached 5.8 × 105 deg cm2 dmol−1, which is much larger than that for SP-GA-EtG1 (2.1 × 105 deg cm2 dmol−1). This large difference of the induced chirality between SP-GA-MeG1 and SP-GA-EtG1 suggests that the large polarity difference between dendritic OEGs with hydrophilic methoxyl terminals and the hydrophobic spiropyran moiety for SP-GA-MeG1, when compared to that between dendritic OEGs with fewer hydrophilic ethoxyl terminals and the hydrophobic spiropyran moiety for SP-GA-EtG1, should be helpful in enhancing the aggregation of DSPs in water, leading to tight packing of the molecules and enhanced chirality. Furthermore, the supramolecular chirality is dependent on the amphiphilic structures and how the dipeptide segment was connected. For example, when inverting the amino acid sequence in the dipeptide from GA to AG as in SP-AG-MeG1, the supramolecular chirality was inverted as shown in Fig. 1c. At the same time, the intensity of the Cotton effect also reduced to −2.6 × 105 deg cm2 dmol−1, suggesting that the alternating position of the chiral center in the peptide had significant influence on the supramolecular chiralities of DSPs. In the case of MeG1-GA-SP, which contains only two amide linkages, the weakest supramolecular chirality was obtained (−1.0 × 105 deg cm2 dmol−1), indicating that the H-bond of the amides plays a crucial role in the assembly of the DSPs and the chirality transfer. The concentration dependence of Cotton effects of the aggregates in aqueous solutions was checked, and the CD spectra of SP-GA-MeG1 are shown in Fig. S17.† When the concentration increased from 0.2 mg mL−1 to 0.7 mg mL−1, the induced chirality also increased greatly from 4.0 × 105 to 11.2 × 105 deg cm2 dmol−1, suggesting that the increase of DSP concentration should have enhanced the assembly for the spiropyran moieties with more tightly bound aggregations.
Atomic force microscopy (AFM) was used to check the morphologies of the assemblies from DSPs in water. Before measurements, all samples (the same as for DLS measurements) were spin-coated on mica substrates and kept overnight in the dark. Typical AFM images are shown in Fig. 2. Nanospheres with sizes of 430, 755, 510 and 325 nm were observed for SP-GA-MeG1, SP-GA-EtG1, SP-AG-MeG1, and MeG1-GA-SP, respectively. Due to the softness of the aggregates, they were strongly absorbed on the polar mica substrates with much larger diameters than their heights. It is necessary to point out that the aggregate sizes from AFM measurements are much smaller than those from DLS measurements, since partial dehydration can't be avoided during the sample preparation for AFM measurements.
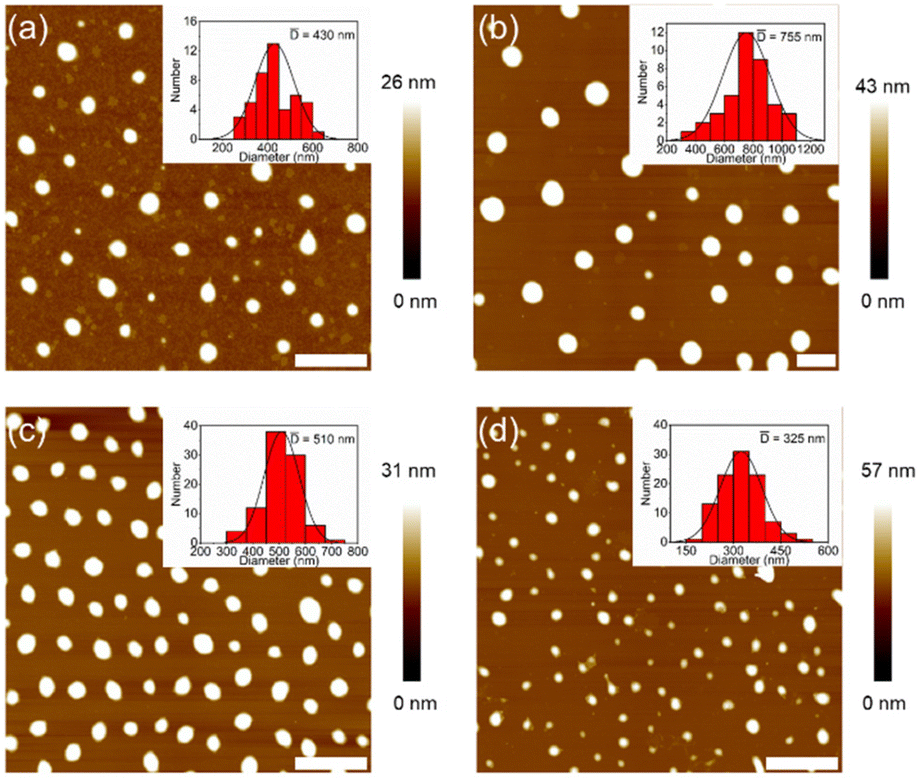 | ||
| Fig. 2 Morphologies of the assemblies from DSPs. AFM images on mica from aqueous solutions of SP-GA-MeG1 (a), SP-GA-EtG1 (b), SP-AG-MeG1 (c) and MeG1-GA-SP (d). C = 0.15 mg mL−1, scale bar = 2 μm. | ||
3.3 Isomerization of the DSPs in aqueous solutions through photo-irradiation
It has been well documented that the isomerization of spiropyrans is dependent on many factors, ranging from intrinsic features including molecular polarity or external conditions including temperature, photo-irradiation, and even solvent polarity. Therefore, the possible isomerization of these DSPs was checked first in their aqueous solutions in the dark. As shown in Fig. 3a, the absorbance at 516 nm increased gradually with time for all four DSPs in water. This indicates that DSPs can automatically isomerize into the MC state, suggesting that the strong polar environment created by water provides the driving force for these molecules to transfer from the hydrophobic SP state to the charged and more hydrophilic MC state. Alternatively, automatic isomerization of these DSPs from the SP to MC state in water indicates that the dendritic OEGs cannot provide sufficient shielding effects to the spiropyran interiors even in their aggregated states. This is quite surprising, since we found that densely packed dendritic OEGs can protect and provide shielding to their interiors,44–46 and this shielding effect can be significantly enhanced in the aggregated state.53 It is necessary to note that the isomerization of these DSPs in water in the dark is reversible. Through photo-irradiation with visible light, they transferred back to the initial hydrophobic SP state.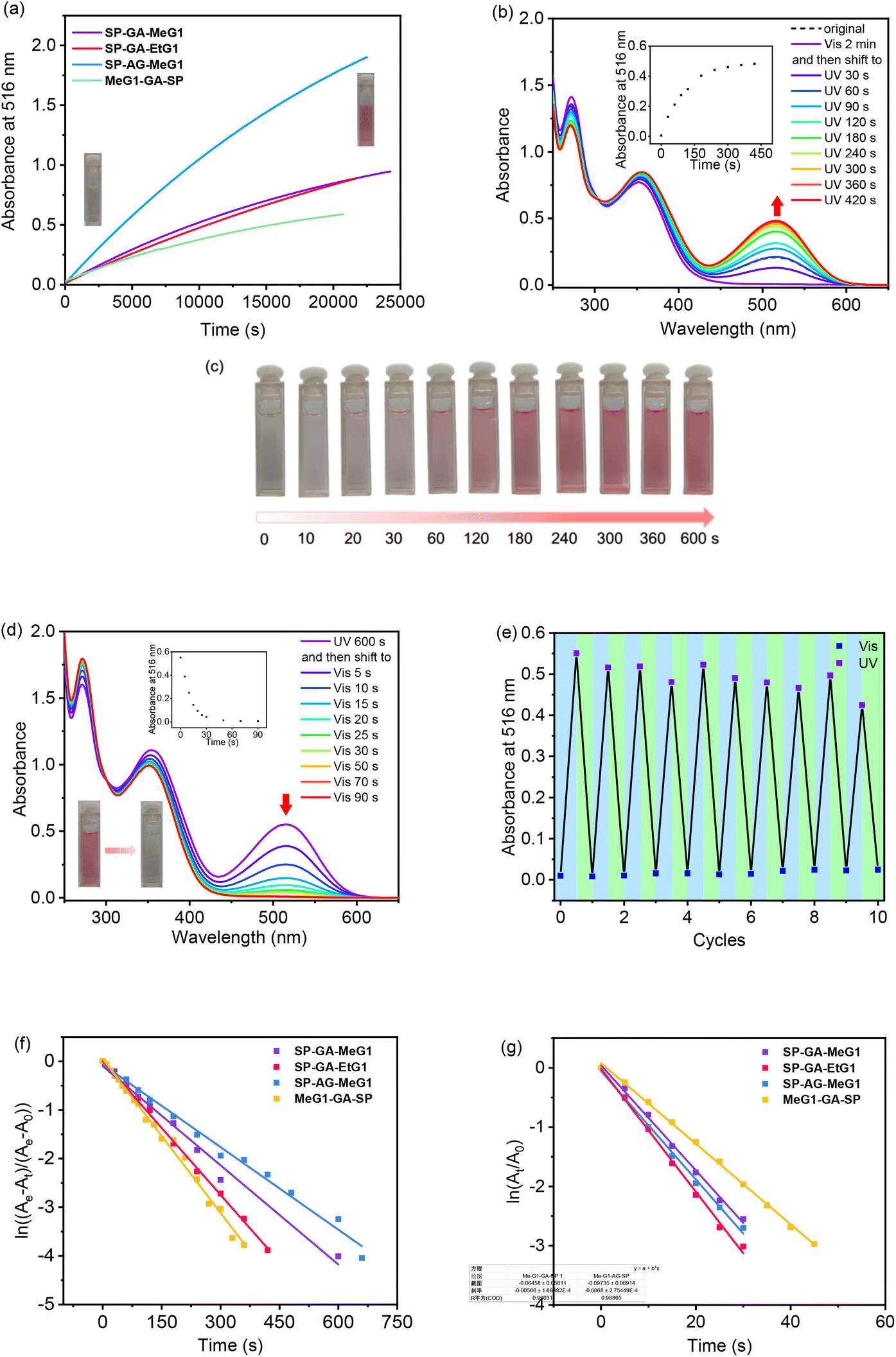 | ||
| Fig. 3 Isomerization of DSPs. Absorbance of DSPs at 516 nm with time in the dark (T = 22 °C) (a), UV/vis spectra of SP-GA-MeG1 in water after irradiation with UV light (b) and photographs of the corresponding solutions (c), UV/vis spectra of SP-GA-MeG1 in water after re-irradiation with visible light (d), absorbance at 516 nm after alternate irradiation with UV and visible light (e), and kinetics plots of isomerization for the DSPs through irradiation with UV light (f) and re-irradiation with visible light (g). Insets in (b) and (d): plots of absorbance at around 516 nm against irradiation time. λUV = 254 nm; λVis > 450 nm; T = 10 °C. C = 0.15 mg mL−1. | ||
The isomerization of these DSPs was further examined through photo-irradiation over different timescales with UV light (λ = 254 nm). SP-GA-MeG1 was taken as an example. As shown in Fig. 3b, the absorbance at 516 nm increased with UV irradiation time, indicating a continuous transformation from the SP state to the MC state. Simultaneously, the solution color gradually turned deep pink, as shown in Fig. 3c. This isomerization is reversible, since exposure to visible light (λ > 450 nm) can quickly induce the isomerization from the MC state back to the SP state (Fig. 3d). Reversibility of the transformation between SP and MC states was checked through alternate irradiation with UV and visible light. As shown in Fig. 3e, SP-GA-MeG1 exhibited excellent stability and can be reversibly isomerized between SP and MC states for more than ten cycles. Similar photoisomerization between SP and MC states through irradiation with UV and visible light was observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as shown in Fig. S18.† Therefore, the isomerization kinetics of the DSPs was investigated, and the results are plotted in Fig. 3f and g for SP → MC and MC → SP, respectively. The results are summarized in Table 1. For isomerization from the hydrophobic SP state to the hydrophilic MC state, both aggregates from SP-GA-MeG1 and SP-AG-MeG1 showed much slower kinetics than that from SP-GA-EtG1. This suggests that DSPs carrying dendritic OEGs with hydrophilic methoxyl terminals exhibit slower isomerization than their counterparts with hydrophobic ethoxyl terminals, mainly due to their tight aggregation. For MeG1-GA-SP with fewer amide bonds, its isomerization from SP to MC is also quite fast, suggesting that the weak hydrogen bonding in DSPs is helpful for the isomerization. This again suggests that loose packing of the molecules within the aggregates is favorable for their isomerization from the SP to MC state. However, SP-GA-MeG1, SP-GA-EtG1 and SP-AG-MeG1 showed similar isomerization kinetics from the MC to SP state when irradiated with visible light. Only MeG1-GA-SP exhibited much slower kinetics, suggesting that the loose packing of dendritic OEGs in the aggregates can't provide sufficient hydrophobic microenvironments to drive isomerization from the hydrophilic to hydrophobic state. The above observation indicates that dendronization with dendritic OEGs can provide a convenient platform to mediate the isomerization kinetics of SP moieties in the water phase through micro-confinement within the aggregation, similar to a literature report that the photoisomerization of hydrophobic azobenzene can be reversibly modulated through encapsulation in a water-soluble coordination cage.55
| Samples | SP-GA-MeG1 | SP-GA-EtG1 | SP-AG-MeG1 | MeG1-GA-SP |
|---|---|---|---|---|
| k SP→MC × 10−3 s−1 | −6.8 ± 0.276 | −9.18 ± 0.120 | −5.66 ± 0.169 | −10.54 ± 0.208 |
| R 2 SP→MC | 0.98865 | 0.99864 | 0.99031 | 0.9934 |
| k MC→SP × 10−2 s−1 | −8.861 ± 0.225 | −10.372 ± 0.269 | −9.126 ± 0.251 | −6.789 ± 0.0878 |
| R 2 MC→SP | 0.98865 | 0.99864 | 0.99031 | 0.9934 |
The isomerization of these DSPs was followed by fluorescence spectroscopy. As shown in Fig. 4a for SP-GA-MeG1, the fluorescence intensity at 630 nm increased significantly when irradiated with UV light, indicating enhanced isomerization from the SP state to MC state through continuous UV irradiation. This red fluorescence emission was reduced when irradiated with visible light (Fig. 4b). Upon alternate irradiation with UV and visible light, the fluorescence emission can be reversibly switched, indicating the high reversibility of the transformation between the two isomers within the aggregates. Similar fluorescence emission behavior according to photoisomerization was observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as shown in Fig. S19.†
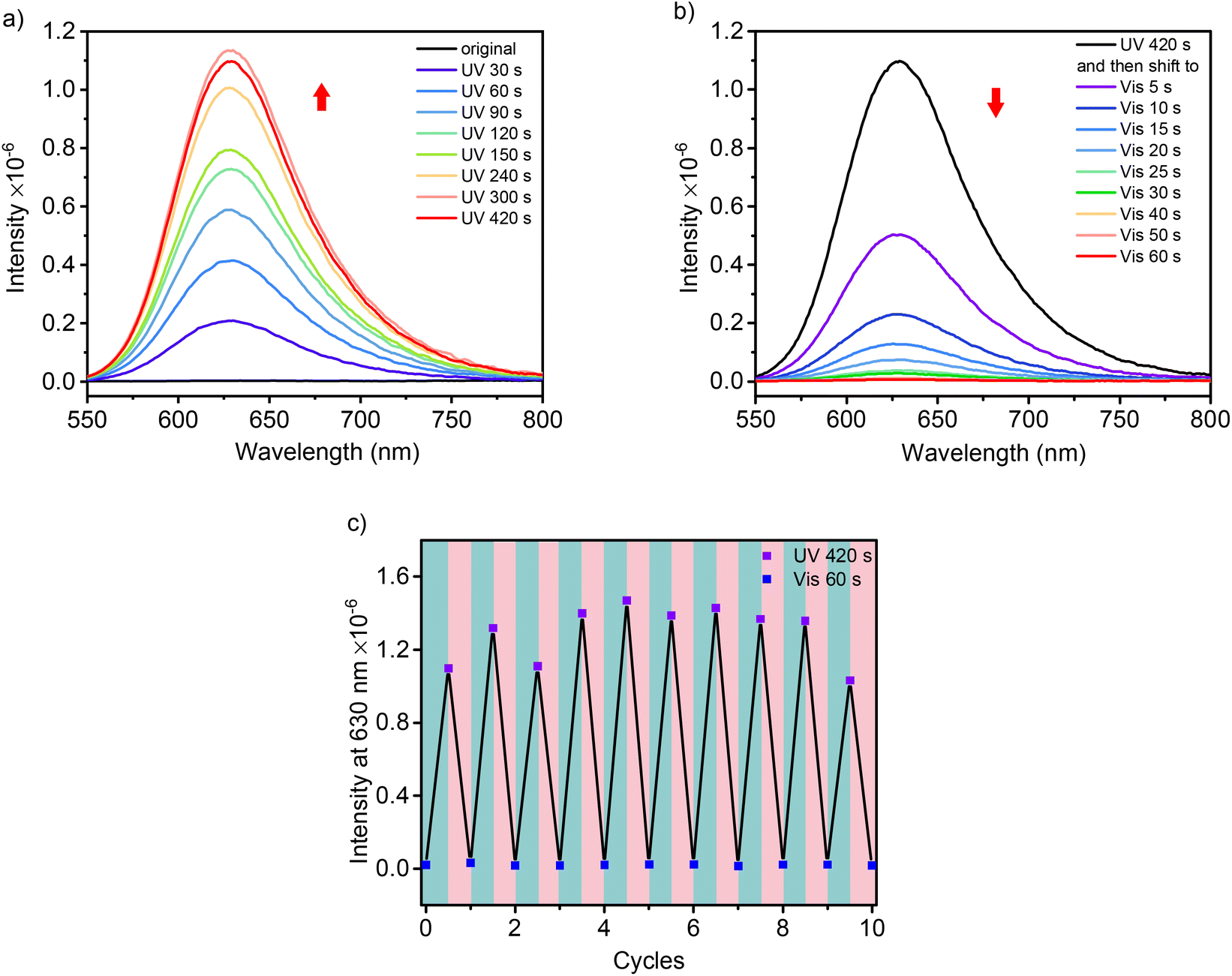 | ||
| Fig. 4 Fluorescence spectra of SP-GA-MeG1 aqueous solutions after irradiation with UV (a) and visible light (b), and its fluorescence intensity at 630 nm after alternate irradiation with UV and visible light (c). λex = 535 nm, λUV = 254 nm, λVis > 450 nm, T = 10 °C, C = 0.15 mg mL−1. | ||
Effects of photo-irradiation-mediated isomerization on the assembly of these DSPs were followed by DLS and AFM measurements. SP-GA-MeG1 was taken as an example. As shown in Fig. 5a, through UV irradiation, the aggregate size decreased to 192 nm, which can be increased to 481 nm via irradiation with visible light. Accordingly, the particle size from AFM measurements was found to be 285 nm after UV irradiation and this increased to 430 nm via irradiation with visible light (Fig. 5b and c). The above result indicates that isomerization from the hydrophobic SP state to the hydrophilic MC state leads to less propensity for these DSPs to aggregate, which can be recovered through isomerization back to the hydrophobic SP state via irradiation with visible light. Similar aggregation and deaggregation behaviors according to photoisomerization were observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as shown in Fig. S20 and S21.†
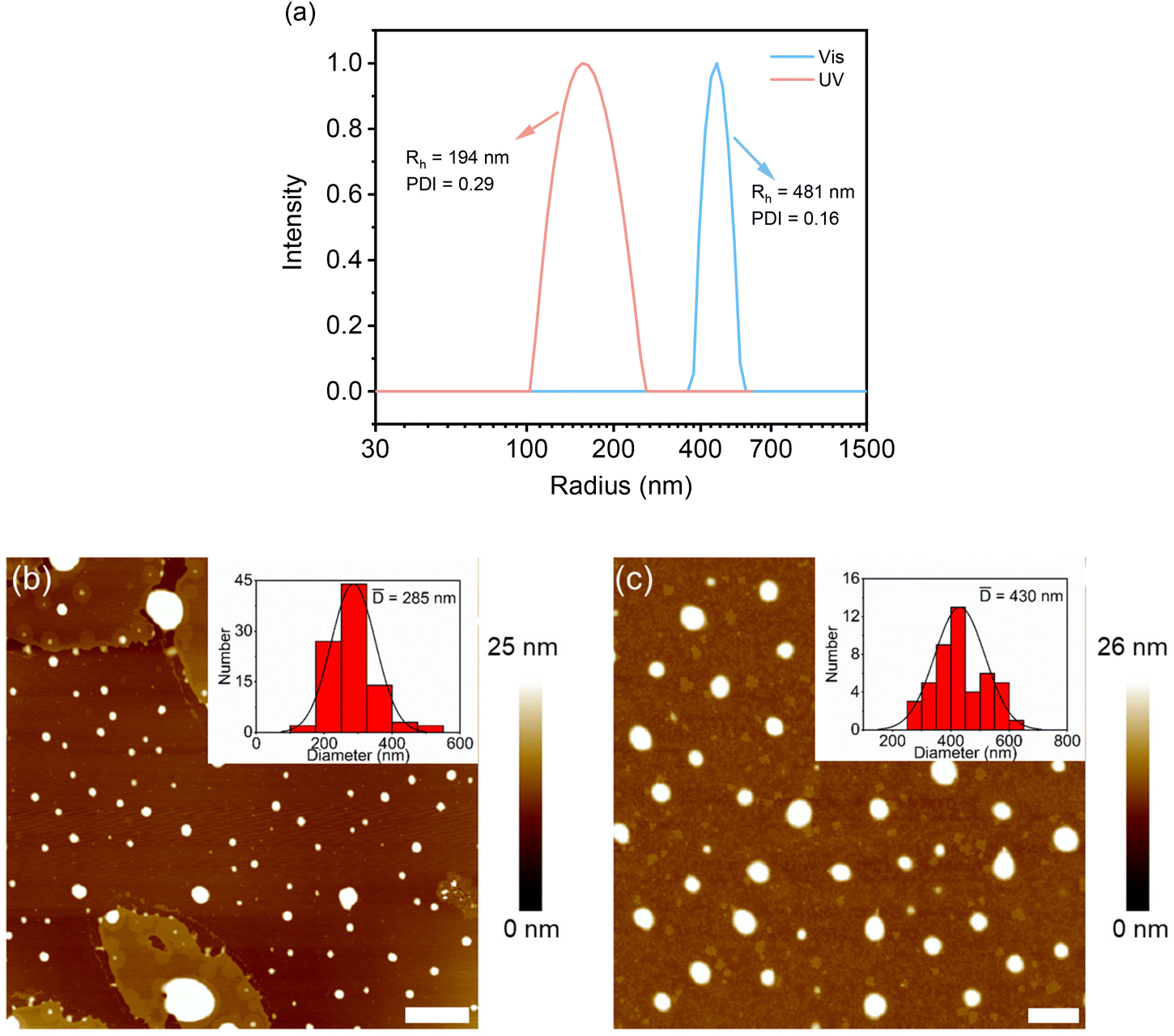 | ||
| Fig. 5 DLS plots of SP-GA-MeG1 aqueous solution after irradiation with UV light for 420 s and visible light for 120 s (a), and AFM images of SP-GA-MeG1 after irradiation with UV (b) and visible light (c). λUV = 254 nm; λVis > 450 nm; T = 10 °C, C = 0.15 mg mL−1, scale bar = 2 μm. | ||
3.4 Chirality transitions mediated by isomerization of the DSPs
CD spectra were recorded to follow the photoisomerization of these DSPs in aqueous solutions. SP-GA-MeG1 was taken as an example. As shown in Fig. 6a, through irradiation with UV light, the Cotton effect at 380 nm corresponding to the spiropyran moiety was reduced in its intensity greatly from its initial SP state (5.8 × 105 deg cm2 dmol−1) to the MC state (1.4 × 105 deg cm2 dmol−1) when irradiated for 70 seconds. Further elongation of the UV irradiation time only led to a slight decrease of the Cotton effect. The above result indicates that isomerization from the hydrophobic SP state to the more hydrophilic MC state should have reduced the amphiphilicity of the DSPs, resulting in less propensity for efficient aggregation. The reduced Cotton effect can be recovered through irradiation with visible light. As shown in Fig. 6b, through irradiation with visible light, the Cotton effect at 380 nm was increased in its intensity accordingly. After irradiation for about 30 seconds, the Cotton effect reached a plateau. The chirality recovery was also checked through alternate photo-irradiation with UV and visible light. As shown in Fig. 6c, the DSP aggregates well exhibited reversibility of its supramolecular chirality through alternate photo-irradiation. Similar chiroptical property transitions according to photoisomerization were observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as shown in Fig. S22.† | ||
| Fig. 6 CD and UV/vis spectra of SP-GA-MeG1 in aqueous solutions after irradiation with UV (a) and visible light (b) for different times, and its CD spectra after different irradiation times (c). For (a), before irradiation with UV light, the solution was first irradiated with visible light to guarantee that the content of the SP state in DSP was as high as possible. For (b), the solution was first irradiated with UV light to guarantee that the content of the MC state in DSP was as high as possible. For (c), the original solution was UV irradiated for 220 seconds before irradiating with visible light for 90 seconds. Insets in (a) and (b): plots of molar ellipticity at 380 nm (θ) against irradiation time. λUV = 254 nm; λVis > 450 nm; T = 10 °C, C = 0.3 mg mL−1. | ||
3.5 Thermoresponsiveness of the assemblies from DSPs
Recently, we found that low molar masses of dendritic OEGs exhibited characteristic thermoresponsive behavior,56 and dendritic OEGs within aggregates can even transfer from the interior to the periphery of the aggregates, leading to different phase transition temperatures.53 We therefore expect that aggregates from these DSPs should also inherit thermoresponsive behavior from their dendritic OEGs. Through heating, the aqueous solutions turned from colorless to red, indicating that the increase of solution temperature is helpful for the isomerization of the spiropyran moieties into the more hydrophilic MC state. SP-GA-MeG1 was taken as an example, and its typical turbidity curves are shown in Fig. 7a. From the heating curve, the cloud point temperature (Tcp) was found to be 43 °C.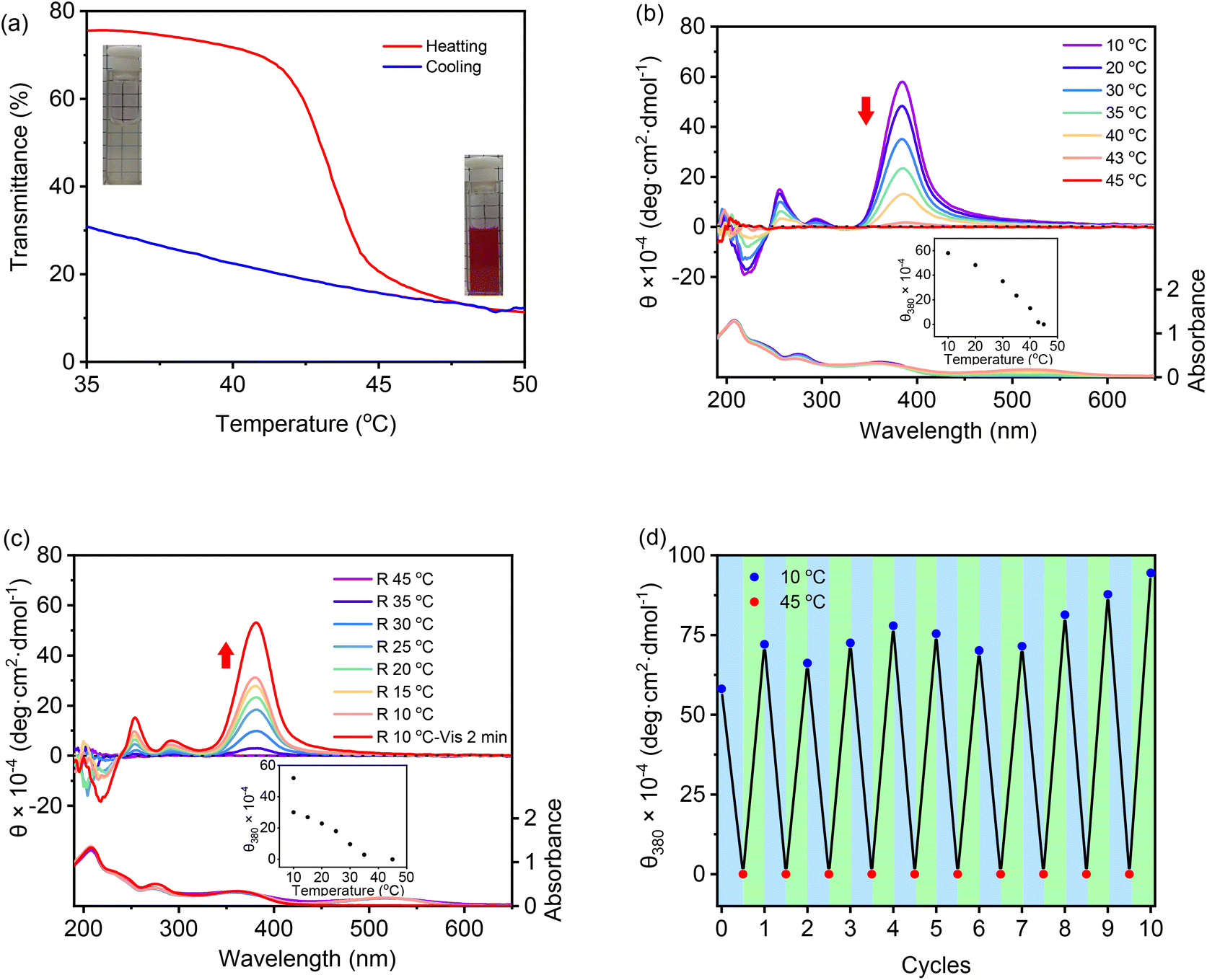 | ||
| Fig. 7 Plots of transmittance versus temperature for the aqueous solution (C = 1.0 mg mL−1) of SP-GA-MeG1 at a heating rate of 0.5 °C min−1 (a). CD and UV/vis spectra of aqueous solutions (0.3 mg mL−1) of SP-GA-MeG1 with the temperature increased from 10 to 45 °C (b) and decreased from 45 to 10 °C (c), and the first Cotton effect at 380 nm (θ380) after several cycles of heating and cooling through irradiation with visible light (d). Heating or cooling rate = 2.0 °C min−1; λVis > 450 nm. The inset in (a): photographs of the aqueous solutions below and above the Tcp. Insets in (b) and (c): plots of molar ellipticity (θ380) against temperature. | ||
A significant hysteresis between heating and cooling processes was observed. Actually, the cooling curve suggests that the aggregated molecules were tightly wrapped with each other and hard to release. In order to be dissolved from the thermally aggregated state, a much lower temperature and a longer time were needed. This situation is slightly different for different DSPs. As shown in Fig. S23 in the ESI,†Tcps for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP were found to be 28, 38, and 26 °C, respectively. These results indicate that the phase transition of the supramolecular assembly is mainly determined by the peripherals of the dendritic OEGs, and ethoxyl terminals contributed to significant hydrophobicity. More interestingly, the Tcps of both SP-AG-MeG1 and MeG1-GA-SP are much smaller than that for SP-GA-MeG1, although they all carry hydrophilic methoxyl-terminated dendritic OEGs. This suggests that the apparent hydrophobicity of the aggregates from DSPs is not solely dependent on the hydrophilicity of the molecules, but should be also related to their morphology, especially to how dendritic OEGs wrapped the aggregates. For SP-GA-MeG1, whose aggregates exhibited the strongest induced Cotton effects, spiropyran moieties must have been densely packed; therefore, more hydrophilic dendritic OEGs covered the aggregates to shield the hydrophobic spiropyran interiors, leading to a higher Tcp. For MeG1-GA-SP with the weakest propensity to form ordered assemblies, the hysteresis between heating and cooling was much smaller, since no strong driving force is needed to dissolute the aggregates. Through optical microscopy, the thermally induced aggregation of these DSPs in aqueous solutions was followed, and the results are shown in Fig. S24;† large aggregates appeared when the solution temperature increased above their Tcps.
Chiroptical properties of the aggregates from DSPs during the thermally induced phase transition process were examined with CD spectroscopy. SP-GA-MeG1 was taken as an example, and its CD spectra are shown in Fig. 7b. With the increase of solution temperature, the intensity of the Cotton effect at 380 nm decreased accordingly, and the DSP aggregates became chirally silent at around 45 °C. The increase of solution temperature at the Tcp is accompanied by the following changes: (1) a reduced H-bond, which reduced molecular interactions for tight packing, (2) enhanced isomerization from the SP state to the MC state, which reduced the amphiphilicity of the DSP, and (3) more hydrophobic-collapsed dendritic OEG domains, which may enhance their interaction with the spiropyran moieties. All these changes lead to weakened aggregation for the DSPs and diminish the induced chirality. This weakened chirality can be completely recovered when cooling the solution back to below their Tcps and can be helped by photo-irradiation with visible light, as shown in Fig. 7c. Through alternate heating and cooling, with the help of photo-irradiation by visible light, the supramolecular chirality from the assembly of DSPs is totally reversible. As shown in Fig. 7d, the Cotton effect at 380 nm corresponding to spiropyran moieties retains similar intensities after 10 heating and cooling cycles, demonstrating the excellent reversibility of the chiroptical properties for the assemblies from DSPs in water. Similar chiroptical property transitions according to thermally induced phase transitions were observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as shown in Fig. S25.†
NMR spectroscopy was utilized to follow the molecular level thermal aggregation of the DSPs. SP-GA-MeG1 was taken as an example, and its 1NMR spectra at different temperatures are shown in Fig. 8a. At room temperature, proton signals from spiropyran moieties were immersed inside the baseline, indicating that spiropyran moieties were tightly wrapped inside the aggregates. With the increase of solution temperature to 45 °C, resolution of the proton signals from dendritic OEGs became broader, indicative of their dehydration. However, with further increases of solution temperature, the proton signal intensities of spiropyran moieties increased, indicative of their improved mobility due to the increase of temperature. Simultaneously, the protons of dendritic OEGs split into two groups: the resolved (d and a) from the swollen moieties and the broad (d′ and a′) from the dehydrated ones. This finding is similar to our previous report that the swollen dendritic OEGs contribute to the solubility of the aggregates in water, while the dehydrated ones combined with the hydrophobic spiropyran moieties more intensively.53 We propose that these two states of dendritic OEGs should have exchanges with each other due to the dynamic feature observed during the thermoresponsive process, as demonstrated by others.57 Alternatively, dendritic OEGs in the two states should change their proportions according to the dehydration degree.46,53 The 1H NMR spectra of SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP illustrated a similar tendency for thermal dehydration, as shown in Fig. S26.† NOESY spectra further support the previous proposal. As shown in Fig. 8b and c, cross-coupling between protons from the spiropyran moieties and the dendritic OEGs was very weak at room temperature, but became significant at elevated temperatures. This again supports the theory that the hydrophobic dehydrated dendritic OEG domains tend to interact more strongly with the hydrophobic spiropyran moieties. Similar tendencies were observed for SP-GA-EtG1, SP-AG-MeG1 and MeG1-GA-SP, as illustrated in Fig. S27.†
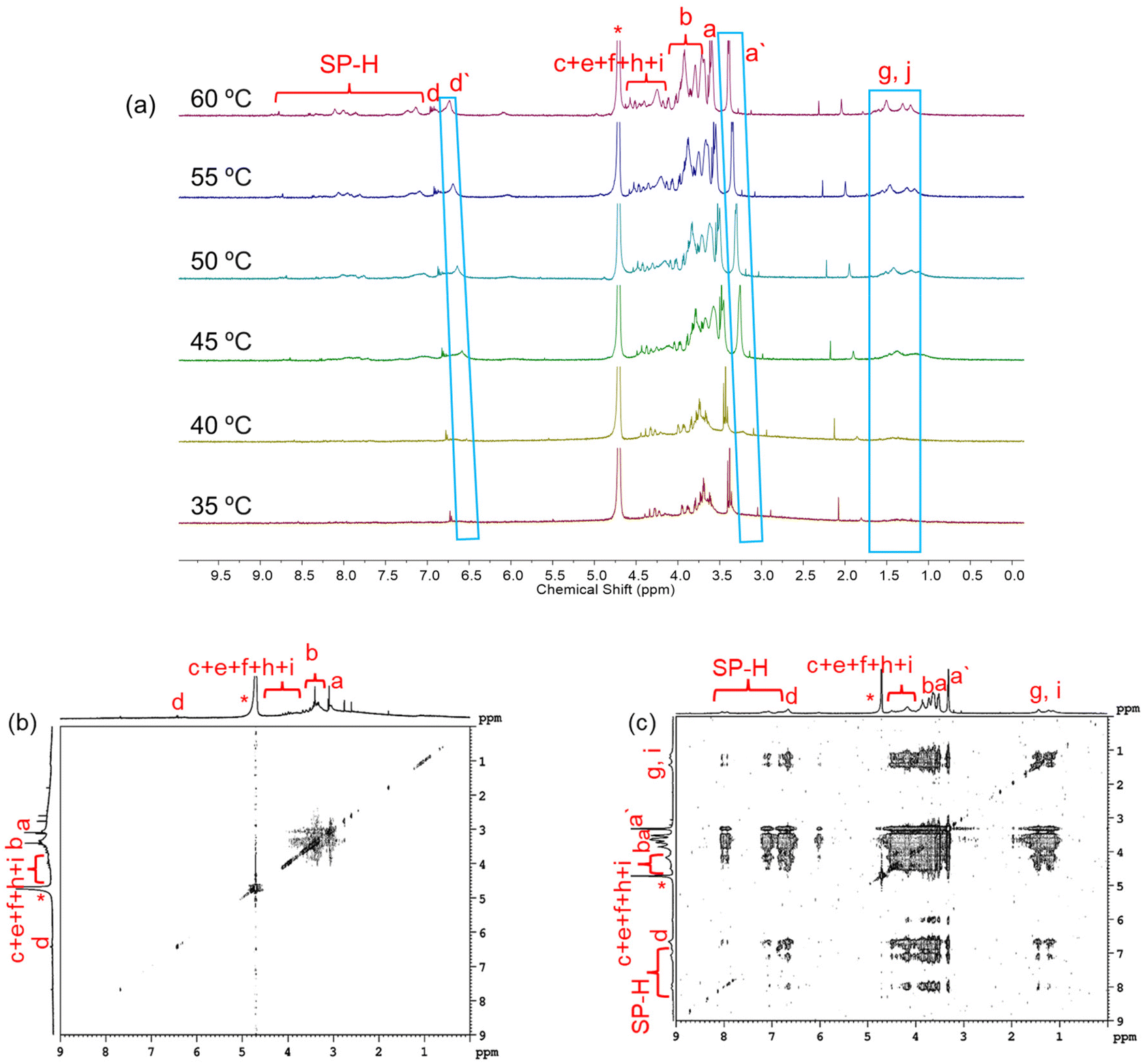 | ||
| Fig. 8 1H NMR spectra of SP-GA-MeG1 in D2O at varied temperatures (C = 2.5 mg mL−1) (a), and its NOESY spectra in D2O at 10 °C (below the Tcp) (b) and 50 °C (above the Tcp) (c) at a concentration of 5 mg mL−1. | ||
4. Conclusions
Creating intelligent supramolecular chiral assemblies is important not only for providing insights into the assembly mechanism, but also for providing new chiral materials for appealing applications. In this report, we designed and synthesized a series of DSPs that contain 3-fold dendritic OEGs through an Ala–Gly dipeptide, and investigated their supramolecular chiral assembly in water. Interestingly, supramolecular nanospheres with sizes in the range of a few hundreds of nanometers were formed, which exhibited remarkable supramolecular chirality to a level of 1.0 × 106 deg cm2 dmol−1. Thanks to the embedded spiropyran moieties, these supramolecular nanospheres showed photo-responsive chirality through isomerization between the non-conjugated SP state and the conjugated MC state, accompanied simultaneously by solution color, absorption and emission changes. In particular, the solution color changes make it possible to follow the isomerization process and chirality variations through apparent solution changes. Thermally mediated dehydration and collapse of the dendritic OEGs endowed the supramolecular nanospheres with unprecedent thermoresponsiveness, and their phase transition temperatures were found to be determined by the hydrophilicity of the DSPs and also the way dendritic OEGs shielded the interiors of the aggregates. The latter should be very important especially when utilizing the supramolecular assemblies for chiral recognition or chiral separation, since the shielding effects from densely packed dendritic OEGs may act as a molecular envelope to confine the process. Both photo-irradiation and thermoresponsiveness provided dual controls for the reversible tailoring of supramolecular chirality. Supramolecular chiral assembly in water may pave a way for wider bioapplication prospects of these smart chiral entities.Author contributions
W. L. and A. Z. designed and directed the project. S. Q., X. L., and W. M. conducted all the experiments. X. L., and W. M. and W. L. edited the schematic illustrations. W. L. and A. Z. cowrote the paper. All authors discussed the results, commented on the manuscript, and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21971160 and 22271183). The Program for Professor of Special Appointment (Eastern Scholar TP2019039) at Shanghai Institutions of Higher Learning is acknowledged.References
- D. K. Smith, Chem. Commun., 2006, 42, 34–44 RSC.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- J. Yan, W. Li and A. Zhang, Chem. Commun., 2014, 50, 12221–12233 RSC.
- C. Li, A. D. Schlüter, A. Zhang and R. Mezzenga, Adv. Mater., 2008, 20, 4530–4534 CrossRef CAS.
- V. Percec, S. Wang, N. Huang, B. E. Partridge, X. Wang, D. Sahoo, D. J. Hoffman, J. Malineni, M. Peterca, R. L. Jezorek, N. Zhang, H. Daud, P. D. Sung, E. R. McClure and S. L. Song, J. Am. Chem. Soc., 2021, 143, 17724–17743 CrossRef CAS PubMed.
- S. Fu, N. Niu, S. Song, D. Yan, J. Ge, J. Li, Z. Peng, L. Li, Y. Xiong, L. Wang, D. Wang and B. Z. Tang, Macromolecules, 2022, 55, 4742–4751 CrossRef CAS.
- Y. Wang, Z. Huang, Y. Kim, Y. He and M. Lee, J. Am. Chem. Soc., 2014, 136, 16152–16155 CrossRef CAS PubMed.
- B. Shen, Y. Zhu, Y. Kim, X. Zhou, H. Sun, Z. Lu and M. Lee, Nat. Commun., 2019, 10, 1080 CrossRef PubMed.
- D.-W. Lee, T. Kim, I.-S. Park, Z. Huang and M. Lee, J. Am. Chem. Soc., 2012, 134, 14722–14725 CrossRef CAS PubMed.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed.
- Y. Kim, H. Li, Y. He, X. Chen, X. Ma and M. Lee, Nat. Nanotechnol., 2017, 12, 551–556 CrossRef CAS PubMed.
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed.
- K.-S. Moon, H.-J. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 6807–6810 CrossRef CAS PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed.
- T. Saito, T. Kajitani and S. Yagai, J. Am. Chem. Soc., 2023, 145, 443–454 CrossRef CAS PubMed.
- J. S. Valera, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Glmez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 61, 202114290 CrossRef PubMed.
- G. Moncelsi and P. Ballester, ChemPhotoChem, 2019, 3, 304–317 CrossRef CAS.
- D. E. Williams, C. R. Martin, E. A. Dolgopolova, A. Swifton, D. C. Godfrey, O. A. Ejegbavwo, P. J. Pellechia, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2018, 140, 7611–7622 CrossRef CAS PubMed.
- J. B. Flannery Jr., J. Am. Chem. Soc., 1968, 90, 5660–5671 CrossRef.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1753 CrossRef CAS PubMed.
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS PubMed.
- K.-y. Tomizaki and H. Mihara, J. Mater. Chem., 2005, 15, 2732–2740 RSC.
- P. K. Kundu, A. Lerner, K. Kučanda, G. Leitus and R. Klajn, J. Am. Chem. Soc., 2014, 136, 11276–11279 CrossRef CAS PubMed.
- S. Wan, Y. Zheng, J. Shen, W. Yang and M. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 19515–19519 CrossRef CAS PubMed.
- Q. Chen, Y. Feng, D. Zhang, G. Zhang, Q. Fan, S. Sun and D. Zhu, Adv. Funct. Mater., 2010, 20, 36–42 CrossRef CAS.
- S. Kwangmettatam and T. Kudernac, Chem. Commun., 2018, 54, 5311–5314 RSC.
- S. Son, E. Shin and B.-S. Kim, Biomacromolecules, 2014, 15, 628–634 CrossRef CAS PubMed.
- L. Yu, Y. Hou, W. Xie, J. L. Cuellar-Camacho, Q. Wei and R. Haag, Adv. Mater., 2020, 32, 2006986 CrossRef PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- K. Nakamura, R. Kubota, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2023, 14, 1696 CrossRef CAS PubMed.
- X. Lang, Y. Huang, L. He, Y. Wang, U. Thumu, Z. Chu, W. T. S. Huck and H. Zhao, Nat. Commun., 2023, 14, 3084 CrossRef CAS PubMed.
- F. Yang, B. Yue and L. Zhu, Chem. – Eur. J., 2023, 29, 202203794 CrossRef PubMed.
- Y. Wang and Q. Li, Adv. Mater., 2012, 24, 1926–1945 CrossRef CAS PubMed.
- Y. Kim and N. Tamaoki, ChemPhotoChem, 2019, 3, 284–303 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Nat. Mater., 2016, 15, 318–325 CrossRef CAS PubMed; Y. Zhao, X. Zhang, W. Li and A. Zhang, Eur. Polym. J., 2019, 118, 275–279 CrossRef.
- S. Wu, Q. Zhang, Y. Deng, X. Li, Z. Luo, B. Zheng and S. Dong, J. Am. Chem. Soc., 2020, 142, 448–455 CrossRef CAS PubMed.
- L. Borsdorf, L. Herkert, N. Bäumer, L. Rubert, B. Soberats, P. A. Korevaar, C. Bourque, C. Gatsogiannis and G. Fernández, J. Am. Chem. Soc., 2023, 145, 8882–8895 CrossRef CAS PubMed.
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K.-C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed.
- X.-M. Chen, X.-F. Hou, H. K. Bisoy, W.-J. Feng, Q. Cao, S. Huang, H. Yang, D. Chen and Q. Li, Nat. Commun., 2021, 12, 4993 CrossRef CAS PubMed.
- W. Li, A. Zhang and A. D. Schlüter, Chem. Commun., 2008, 44, 5523–5525 RSC.
- G. Xu, J. Zhang, R. Jia, W. Li and A. Zhang, Macromolecules, 2022, 55, 630–642 CrossRef CAS.
- M. J. N. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 5683–5687 CrossRef CAS PubMed.
- L. Liu, W. Li, K. Liu, J. Yan, G. Hu and A. Zhang, Macromolecules, 2011, 44, 8614–8621 CrossRef CAS.
- J. Yan, K. Liu, W. Li, H. Shi and A. Zhang, Macromolecules, 2016, 49, 510–517 CrossRef CAS.
- D. Wu, J. Wu, P. Tao, Y. Yao, J. Wang, D. Liu, F. Chen, B. Xu, W. Li and A. Zhang, Polym. Chem., 2020, 11, 4105–4114 RSC.
- Y. Yao, J. Yang, W. Li and A. Zhang, Polym. Chem., 2022, 13, 5404–5411 RSC.
- J. Zhang, Y. Yao, Y. Zhang, D. Wu, W. Li, A. K. Whittaker and A. Zhang, Macromolecules, 2023, 56, 3931–3944 CrossRef CAS.
- Y. Yao, S. Cao, Q. Yang, A. Zhang and W. Li, ACS Appl. Bio Mater., 2022, 5, 5377–5385 CrossRef CAS PubMed.
- Y. Liu, Y. Cao, X. Zhang, Y. Lin, W. Li, B. Demir, D. J. Searles, A. K. Whittaker and A. Zhang, ACS Nano, 2021, 15, 20067–20078 CrossRef CAS PubMed.
- Y. Liu, Y. Lin, Y. Cao, A. Zhi, J. Chen, W. Li, B. Demir, D. J. Searles, A. K. Whittaker and A. Zhang, Chem. Commun., 2021, 57, 12780–12783 RSC.
- D. Samanta, J. Gemen, Z. Chu, Y. Diskin-Posner, L. J. W. Shimon and R. Klajn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9379–9384 CrossRef CAS PubMed.
- G. Xu, J. Zhang, M. Qi, X. Zhang, W. Li and A. Zhang, Phys. Chem. Chem. Phys., 2022, 24, 11848–11855 RSC.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03729k |
| This journal is © The Royal Society of Chemistry 2023 |