
Recent advances in self-healing polyurethane based on dynamic covalent bonds combined with other self-healing methods
Ze-Wei Ana,
Rui Xuea,
Kang Yea,
Hui Zhao *abde,
Yang Liua,
Peng Lib,
Zhen-Ming Chenb,
Chong-Xing Huangb and
Guo-Hua Huc
*abde,
Yang Liua,
Peng Lib,
Zhen-Ming Chenb,
Chong-Xing Huangb and
Guo-Hua Huc
aSchool of Light Industry and Food Engineering, Guangxi University, Nanning 530004, China
bGuangxi Key Laboratory of Calcium Carbonate Resources Comprehensive Utilization, College of Materials and Chemical Engineering, Hezhou University, Hezhou 542899, China
cLaboratory of Reactions and Process Engineering, CNRS-University of Lorraine, Nancy 54001, France
dNational Local Joint Laboratory for Advanced Textile Processing and Clean Production, Wuhan Textile University, Wuhan 430200, China
eLaboratory of Biocatalysis and Enzyme Engineering, School of Life Sciences, Hubei University, Wuhan 430062, China
First published on 9th February 2023
Abstract
To meet more application requirements, improving mechanical properties and self-healing efficiency has become the focus of current research on self-healing PU. The competitive relationship between self-healing ability and mechanical properties cannot be avoided by a single self-healing method. To address this problem, a growing number of studies have combined dynamic covalent bonding with other self-healing methods to construct the PU structure. This review summarizes recent studies on PU materials that combine typical dynamic covalent bonds with other self-healing methods. It mainly includes four parts: hydrogen bonding, metal coordination bonding, nanofillers combined with dynamic covalent bonding and multiple dynamic covalent bond bonding. The advantages and disadvantages of different self-healing methods and their significant role in improving self-healing ability and mechanical properties in PU networks are analyzed. At the same time, the possible challenges and research directions of self-healing PU materials in the future are discussed.
1. Introduction
Polyurethane (PU) is one of the most promising polymer materials. However, its service life is inevitably reduced due to damage during use, resulting in waste that is difficult to recycle and causes many environmental problems.7,14 At present, the introduction of a self-healing function to improve the durability of PU materials has become an effective way to solve the pollution problem.In a general sense, the self-healing methods in PU are divided into extrinsic and intrinsic self-healing. The extrinsic self-healing method refers to the addition of microcapsules/microvessels containing healing agents into the polymer.16–18 The intrinsic self-healing method refers to the introduction of dynamic reversible structures, including dynamic covalent bonds (such as disulfide bonds, Diels–Alder (DA) bonds, imine bonds, and boronic ester bonds) and non-dynamic covalent interactions (such as hydrogen bonds, metal coordination interactions, and host–guest interactions).21 Polymers containing dynamic covalent bonds are usually stable under environmental conditions. When dynamic covalent bonds are destroyed, they can heal after four reaction steps, including reversible cycloaddition reactions, exchange reactions, stable free-radical-mediated reshuffling reactions, and heterocyclic compound/carbohydrate facilitated bond reformations, stimulated by light, temperature, or pH.22 Covalent bonds in polymers that undergo cycloaddition reactions usually have higher bond energies. Although it can help improve the mechanical properties of materials, the reversible reaction of covalent bonds needs more energy for excitation.23 The exchange reaction of polymers takes advantage of the dynamic reversibility of covalent bonds. Due to the low reaction temperature of some exchange reactions, they show good self-healing characteristics under environmental conditions. However, such polymers usually do not have sufficient mechanical stability.24
With the wide application range of PU materials, the self-healing efficiency or mechanical properties of single dynamic covalent network polymers can no longer meet the current requirements.13 Therefore, researchers have designed different double-network or multi-network polymers (such as supramolecular networks, double dynamic networks, double dynamic polymer interpenetrating networks, and semi-interpenetrating polymer networks) to combine dynamic covalent bonds with other self-healing methods to synthesize polymers that simultaneously have a high self-healing efficiency and excellent mechanical strength.3,9,20,25 Hydrogen bonds, metal coordination bonds, nanofillers, and other dynamic covalent bonds are often introduced. Although the hydrogen bond energy is low, it has directionality and affinity. It can be used as a reinforcing filler in the low glass transition temperature (Tg) continuous matrix to improve the mechanical strength of the polymer.26 The coordination number and the conformation of the coordination center of metal-ion coordination bonds can be adjusted under stimulation, and its non-covalent binding force is variable.27 It can provide reversibility for constructing self-healing metal supramolecular networks similar to hydrogen bonds and can also use light reactivity to provide controllable self-healing ability. In addition, many non-covalent bonds can be used as sacrificial bonds to improve self-healing efficiency and as a supplement to covalent bonds. In addition, many non-covalent bonds can be used as sacrificial bonds to improve self-healing efficiency and as a supplement to covalent bonds. Many nanofillers can also be added to the polymers to promote the interfacial interaction between fracture surfaces and contribute to self-healing.28
To date, there have been no reviews on the recent progress in PU materials that combine covalent bonds with other methods. In order to fill this research gap, this paper systematically reviews the research on this kind of self-healing PU from the aspects of synthesis methods, self-healing efficiency, mechanical properties, and applications. The advantages and disadvantages of the different methods are also compared in detail, and future development trends and the possible challenges of such self-healing polymers are also pointed out.
2. Typical dynamic covalent bonds in PU
Some typical dynamic covalent bonds in self-healing PU include disulfide bonds, DA bonds, imine bonds and boronic ester bonds. The basic structure of a disulfide bond is R–S–S–R. Depending on the reaction environment, the reactions can be divided into those of thiol-disulfide polymers undergoing redox reactions and of disulfide-containing aromatic, aliphatic, or other polysulfide polymers undergoing disulfide exchange. The thiol-disulfide reaction occurs when the thiol is easily oxidized in a conventional environment, making it difficult for the reaction to proceed.29 Therefore, exchange reactions between polymers containing aromatic disulfide bonds or aliphatic disulfide bonds occurring through a free-radical-mediated mechanism are commonly seen when designing self-healing polymers.30 During the self-healing process, the disulfide bonds in the polymer chain are rearranged, as shown in Fig. 1a. In contrast to aliphatic disulfide bonds, which can be rearranged by external stimuli such as heating or ultraviolet light, aromatic disulfide bonds can be rearranged at room temperature (RT). Disulfide bonds are usually introduced into hard segments as chain expanders in PU, and they can affect the balance between the self-healing efficiency and mechanical properties.31–33 The combination of disulfide bonds and other self-healing methods to improve the self-healing efficiency and mechanical properties of PU has been widely researched.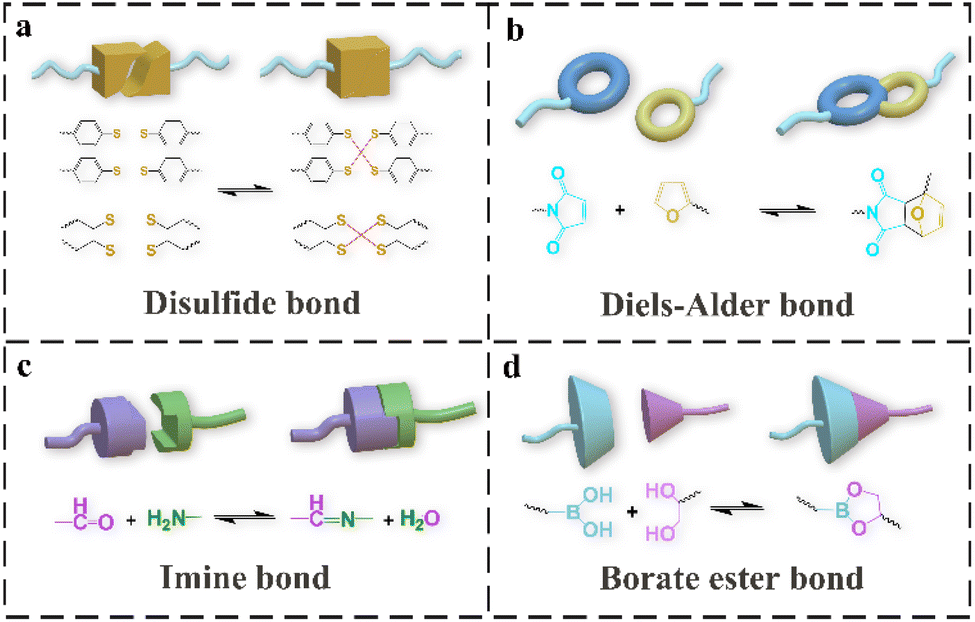 | ||
| Fig. 1 The schematic diagram of reversible reaction of disulfide bonds (a), DA bonds (b), imine bonds, (c) and boronic ester bonds (d). | ||
The DA reaction is a dynamic and reversible reaction based on a temperature reaction, in which compounds with reactive double or triple bonds (dienophile) undergo cycloaddition reactions with conjugated diene groups (pro-dienyl groups) at about 60 °C. When the temperature is increased to 100–150 °C, the retro-DA (rDA) reaction occurs to rebuild the covalent bonds to heal the cracks.34 In general, the DA bond is formed by the reaction of a linear PU with a furan side group and a maleimide group. Self-healing based on a DA reaction requires a higher temperature, which may damage the integrity of the material's performance or limit the material's applications. Improving the self-healing efficiency while possessing a high modulus and toughness and mild healing conditions is the current research hotspot of self-healing PU based on the DA reaction and other methods.35
The imine bond, also known as a Schiff base, generally refers to the R2C = NR′ (R′ ≠ H) structure, which is obtained by the reaction of aldehydes (or ketones) with primary amines.36 The synthesis mechanism is that the nitrogen atom with lone pair electrons in the amine compound acts as a nucleophilic reagent to attack the carbon atom of the carbonyl group and dehydrate to form the corresponding imine bond. This reaction is usually catalyzed by an acid but hydrolyzes at a higher acidity. The dynamic exchange of imine bonds also occurs in hard segment of the PU. The mechanism includes the hydrolysis reaction of imine bonds, the metathesis reaction between imine bonds, and an exchange reaction between imine bonds and amino groups.37 The dynamic activation energy of the imine bond is low, and the exchange reaction is easily triggered, which can be triggered under the stimulation of temperature and pH and can be water-assisted.38–40 Combining physically reversible non-covalent interactions with dynamic imine bonds to form multiple cross-linked networks to overcome the shortcomings of simple imine bonds that are unstable under acidic conditions and are sensitive to pH has attracted the attention of many researchers.
Boronic ester bonds are usually located in the PU side chain to form a dynamic reversible network. They can be rapidly and dynamically exchanged without any catalyst. Hydrogels containing boronic esters can self-heal under neutral and acidic conditions.41–43 Their stability generally depends on the pH of the environment. Typically, they have weak mechanical properties such as low stretch, brittleness, poor viscoelasticity, and poor formability.28 These problems can be improved by combining them with other self-healing methods to obtain stable materials.
3. Combining dynamic covalent bonding and hydrogen bonding
The hydrogen bond energy is much smaller than that of a dynamic covalent bond. According to the number of hydrogen bonds formed by the proton donor and the proton acceptor of the two structural units, it is divided into single and double hydrogen bonds with a weak interaction and multiple hydrogen bonds with a strong interaction obtained by superposition (Fig. 2). Hydrogen bonds are easy to design and have dynamic reversibility under external stimulation. The strong interaction force generated by the superposition of multiple hydrogen bonds can be used as a physical crosslinking point. Therefore, it is widely used in supramolecular polymers. Hydrogen bonds in PU are usually formed by carbamate, 2-ureido-4-pyrmidone (UPy), and amide groups.61,62 The common weak hydrogen bonds in PU originate from their characteristic groups. The strong hydrogen bonds come from the self-complementary quadruple hydrogen bond interactions between the UPy dimers introduced into the main chain.63,64 In addition to the self-healing ability, the introduction of strong hydrogen bonds will affect the tensile properties, and weak hydrogen bonds generally play a role in energy dissipation. The mechanical properties and self-healing abilities of PU materials combined with dynamic covalent bonds and hydrogen bonds are summarized in Table 1.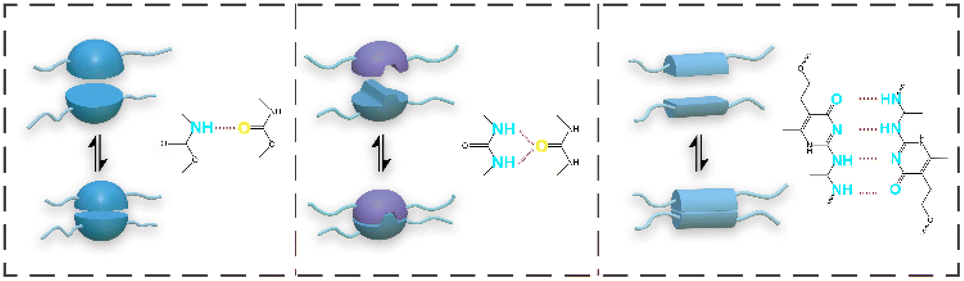 | ||
| Fig. 2 The three typical hydrogen bonds in PU. | ||
| Dynamic covalent bond | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (MPa) | Toughness (MJ m−3) | Healing conditions | Healing efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Aromatic disulfide bonds | 75.8 | 852 | 22 | 171.4 | 85 °C, 1 h | 71.50% recovery of tensile strength | 44 |
| 31 | 638.2 | — | — | 25 °C, 48 h | 101.9% recovery of tensile strength | 5 | |
| 4.8 | 489 | — | — | 75 °C, 2 h | 90% recovery of tensile strength | 45 | |
| 70% recovery of elongation at break | |||||||
| 3.37 | 518 | — | — | 60 °C, 12 h | 91% recovery of tensile strength | 46 | |
| — | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
— | — | 20 °C, 10 min | 86% recovery of tensile strength | 12 | |
| −40 °C, 12 h | 93% recovery of tensile strength | ||||||
| pH = 0 or 14 solution, 24 h | 88% or 84% recovery of tensile strength | ||||||
| 30% NaCl solution, −10 °C, 24 h | 89% recovery of tensile strength | ||||||
| Aliphatic disulfide bonds | >250 | 240 | 25.3 | — | UV 365 nm, 40 °C, 45 min | 100% recovery of tensile strength | 47 |
| 61.7 | 1068 | 1.76 | — | RT, 3 h | 96% recovery of tensile strength | 48 | |
| 27.83 | 2615.17 ± 3 | — | 164.36 | RT, 48 h | 92.76% recovery of tensile strength | 11 | |
| 25 | 1463 ± 32 | — | 101.0 ± 0.7 | 100 °C, 2 h | 92.4% recovery of tensile strength | 49 | |
| 13 | 160 | — | — | 200 mW cm−2, 1 h | 90% recovery of shear strength | 50 | |
| 9.67 ± 0.89 | — | 5.04 ± 0.05 | — | 100 mW cm−2, 24 h | 95.4 ± 0.9% recovery of Young's modulus | 31 | |
| 8.6 ± 0.6 | 224.2 ± 2.8 | 188.5 ± 11.2 | 16.3 ± 0.2 | 100 °C, 30 min | 96.5% recovery of tensile strength | 51 | |
| — | 812 | — | — | 130 °C, 1 h | 100% recovery of tensile strength | 52 | |
| DA bonds | 51.9 | 930 | 6.1 ± 0.3 | 166.7 | 130 °C, 1 h and 65 °C, 24 h | 91.2% recovery of tensile strength | 53 |
| 18.5 | 136 | 151 | — | 120 °C, 10 min and 60 °C, 24 h | 94% recovery of tensile strength | 6 | |
| 6 | 180 | 340 ± 30 | — | 90 °C, 1 h | 100% recovery of tensile strength | 54 | |
| Imine bonds | 41.2 | 823 | — | 127.2 | Water-enabled, RT, 72 h | 92.2% recovery of Young's modulus | 55 |
| 60 °C, 2 h | 93% recovery of Young's modulus | ||||||
| 10.2 | 51 | 79 | — | 80 °C, 6 h | 92% recovery of tensile strength | 56 | |
| 2.29 | 249 | — | — | 24 h | 100% recovery of tensile strength and elongation at break | 57 | |
| 1 | 1500 | — | — | RT, 1 h | 98.40% recovery of elongation at break | 58 | |
| 0.4 | 1670 | — | — | RT, 24 h | 95% recovery of tensile strength | 59 | |
| Water, 24 h | 89% recovery of tensile strength | ||||||
| Artificial sweat, 24 h | 78% recovery of tensile strength | ||||||
| Boronic ester bonds | 2.5 | 86.6 | — | 1.8 | 130 °C, 3 h | 120% recovery of tensile strength | 60 |
Disulfide and hydrogen bonds
The combination of dynamic disulfide and hydrogen bonds is a common self-healing method. Disulfide and hydrogen bonds, which function in concert as hard segments in the structure of PU, can have characteristics resembling those of a conjoined covalent adaptive network.65 Hydrogen bonds can act as physical crosslinking points along with the disulfide bonds to provide self-healing ability to the materials and improve the thermal stability, which allows the materials to be reprocessed and healed in the solid state.45 As shown in Fig. 3a, Yuanbo Cai et al. used 2,4-diamino-6-hydroxypyrimidine (DAHP) to synthesize PU elastomers to achieve a covalent-bonded crosslinking structure, in which there are sufficient hydrogen bonds at the crosslinking point.5 The tensile strength is up to 31.0 MPa and the self-healing efficiency is up to 93.6% after 48 h at RT. The hydrogen bond can cause a strong adhesion force to produce a better self-healing ability in the elastomer at RT, and the increase in DAHP content leads to the formation of a loose-phase separation structure and more disordered hydrogen bond transformation, which accelerates the formation of hydrogen bonds and provides space for the exchange of disulfide bonds.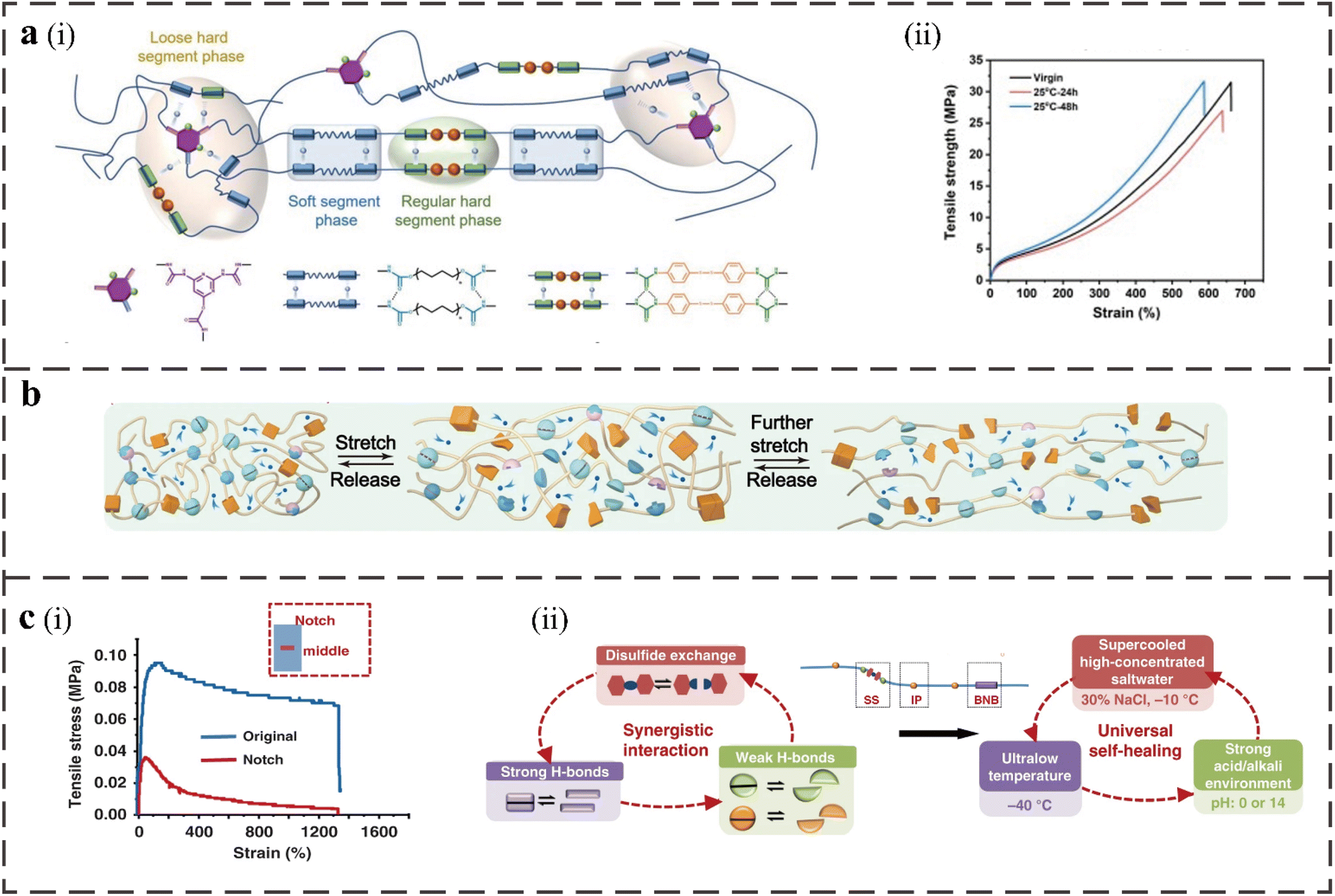 | ||
| Fig. 3 (a), (i) Schematic illustration of the structure of PIDA elastomers and the distribution of hydrogen bonds with regular and loose phase regions. (ii) Stress–strain curves of virgin and re-spliced PIDA-7.5 with different healing times at 25 °C.5 (b) The proposed mechanism for the superior toughness of DSICE upon stretching.11 (c) (i) The stress–strain curve of the notched film and the original film. (ii) The synergistic interaction of multiple dynamic bonds contributes to the universal self-healing capability of the elastomer.12 | ||
The reversible disulfide bond in the hard segment of PU leads to the self-healing ability, enhances the molecular chain's flexibility, and improves the elongation at the break of the material, but will relatively weaken the tensile strength.46 The tensile strength and toughness of the structure can be increased with the addition of UPy units, but the elongation at the break can be decreased. As shown in Fig. 3b, these synergistic interactions of several dynamic connections can produce the self-healing PU with strong mechanical properties when combined with disulfide bonds. Jing Chen et al. introduced aliphatic bis(2-hydroxyethyl) disulfide (HEDS) and UPy into PU; as a result, the self-healing efficiency can reach 99%, the elongation at break is 2615.17%, the tensile strength is 27.83 MPa and the toughness is164.36 MJ m−3.11
The presence of disulfide and multilevel hydrogen bonds in the structure provides a solid crosslinking and energy dissipation mechanism that allows the polymer network to produce multiple bond breaking, exchange and reorganization modes. The disordered weak hydrogen bonds can provide fluidity for the polymer chain and make it break to dissipate energy under tension. The ordered strong hydrogen bond limits the relaxation of the polymer chain and acts as a stable physical crosslinking point to resist deformation, thereby improving the material's toughness.44 There are intrachain and interchain hydrogen bond interactions between strong and weak hydrogen bonds. Intrachain hydrogen bond interactions lead to the folding of molecular chains and allow the extension of large chains; the interchain hydrogen bond interaction leads to a 3D crosslinked network and repeated bonding/breaking during the sliding process among the chains. Lei Zhang et al. designed a self-healing supramolecular elastomer, and its unnotched and notched films showed ultra-high elongation at break (Fig. 3c(i)). The polymer PDMS-SS-IP-BNB network contains disulfide bonds, strong hydrogen bonds (quadruple hydrogen bonds), and weak hydrogen bonds.12 They are derived from the cross-linking of 4,4′-dithiodianiline, 4,4′-bis(hydroxymethyl)-2,2′-bipyridine (BNB), and diisocyanate (IP) with other bonds. The dismutation of aromatic disulfides can be used as a sacrificial bond for material breakage, thereby improving the stability of the material. Dynamic strong hydrogen bonds, weak hydrogen bonds, and disulfide bonds play an important role in the self-healing process (Fig. 3c(ii)) and induce the main chain hydrophobicity and low Tg. This elastomer has excellent self-healing ability under a variety of conditions (as illustrated in Table 1).
DA and hydrogen bonds
Scientists have synthesized macromolecular titin with dense hydrogen bonds and disulfide bonds formed by thiol groups between cysteines. The mixed physical crosslinking and covalent crosslinking network in the structure have synergistic enhancement effects that can make it elastic and tough.66 Inspired by the titin structure, Haopu Xie et al. introduced a double dynamic network to prepare PU, which included the physical crosslinking of the quadruple hydrogen bonds formed by the 5-(2-hydroxyethyl)-6-methyl-2-aminouracil dimers and the covalent crosslinking of the DA bond.3 By comparing the typical stress–strain curves and the healing efficiency of the obtained materials (Fig. 4a), it was found that when there is only a pure DA network, the covalent crosslinking bond and the polymer skeleton will break directly after the extension of the soft segment during the stretching process due to the absence of an effective energy-consuming sacrificial bond, resulting in lower fracture strain and poor toughness. The quadruple hydrogen bond sacrifices energy consumption for the dual-dynamic network when deformation occurs during the stretching process before the covalent bond breaks. A weak temporary quadruple hydrogen bond can be created by dimerizing the broken UPy motif with additional broken UPy motifs in the neighboring chain. The polymer network is more stable when DA covalent crosslinking is present, and the quadruple hydrogen bond can be more successfully rebuilt to boost the material's toughness. The produced material has excellent mechanical properties, including an ultra-high strength of 51.9 MPa, super toughness of 166.7 MJ m−3 and large elongation at break of 930%. The initial mechanical properties and integrity can also be restored by the hot pressing process.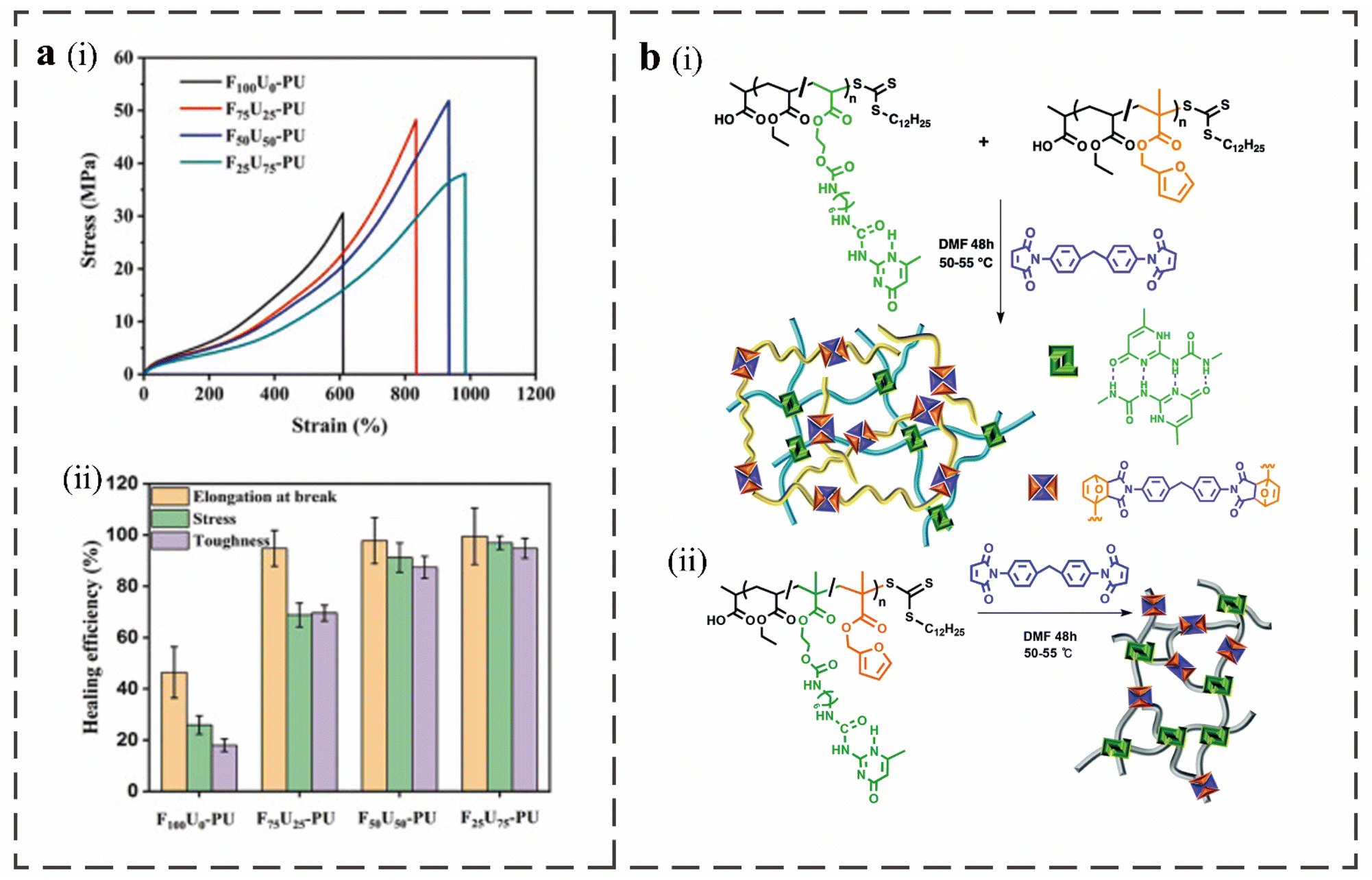 | ||
| Fig. 4 (a), (i) Typical stress–strain curves of FaUb-PU. (ii) Healing efficiency of breaking strain, stress, and toughness of FaUb-PU.3 (b) Crosslinkers and polymers used in the preparation of RAFT based IPN (i) and SN (ii) materials. Slashes indicate random incorporation of monomers.9 | ||
This produces a recyclable PU with improved toughness, elongation at break, and self-healing efficiency when the double dynamic polymer single network (SN) introduces the fast exchange of hydrogen bonds and thermally responsive DA bonds that can rely on the synergistic effect of the dual dynamic network. However, introducing multiple self-healing methods into interpenetrated networks (IPN) is also a way to improve the performance of self-healing materials. An IPN polymer is a combination of two or more topologically interlocked polymer chains that are physically or chemically crosslinked to produce two or more polymer networks that are independent of each other but remain together through network entanglement.67 Polymers with such networks usually have better mechanical properties. Using a reversible addition–fragmentation chain-transfer polymerization method, Borui Zhang et al. synthesized a novel self-healing dual-powered IPN material.9 The dynamic hydrogen bond of UPy is found in one chain of the material network, while the covalent bonds of the DA adduct and the furan-maleimide dynamic covalent crosslinking agent are found in the other chain. They designed IPN and SN materials and compared their properties, as shown in Fig. 4b. Through the self-healing performance test, it was found that the self-healing efficiency of IPN material at 90 °C for one hour was as high as 100%, which was significantly higher than that of the SN material. This is due to the increased chain mobility of the IPN material, which causes the self-healing molecular chain to exchange more rapidly. In addition, the overall non-bonding covalent interaction is stronger than that of the SN material because the IPN architecture provides the hydrogen bond network more degrees of freedom. Therefore, the tensile strength and peak stress of the IPN materials are better than those of the SN materials, as shown by the molecular dynamics simulation and tensile test of the two materials with the same molar density of the cross-linked material.68
From the perspective of green sustainable chemistry, Fengyu Zhao studied a series of PU and polyureas synthesized from CO2.69–71 Recently, a linear PU-urea DA adduct (PUUa-DA) was synthesized using CO2 and bio-based furfuryl amine as copolymers without isocyanate.72 The oligourea segment in the polymer structure provides a strong and ordered hydrogen bond interaction, while the carbamate group provides a weak and disordered hydrogen bond interaction, and an appropriate amount of cross-linked DA group makes it exhibit excellent mechanical and self-healing properties. PUUa-DA is semi-crystalline, with high thermal stability and excellent mechanical properties (tensile stress is 18.5 MPa, the modulus is 151 MPa, and elongation at break is 136%), and self-healing efficiency of 94%.
Imine and hydrogen bonds
Exchange reactions more easily form imine bonds under milder conditions (lower temperature and shorter time) than other dynamic covalent bonds.38 Moreover, polymers containing imine bonds have excellent mechanical properties and recyclable processing capabilities.73,74 However, due to the strong covalent crosslinking in the polymer structure, they also need heat for self-healing. The introduction of hydrogen bonds and imine bonds into PU to produce synergistic interactions can develop flexible or rigid polymers with double dynamic crosslinking networks that heal at lower temperatures or in a shorter time.Zhipeng Yang et al. synthesized a self-healing PDMS elastomer by a one-pot two-step approach of excess aminopropyl-terminated polydimethylsiloxane with isophorone diisocyanate (IPDI) and terephthalaldehyde. There are strong hydrogen bonds and reversible dynamic imine bonds formed by ureido groups in the elastomer structure.59 Hydrogen bonds play the role of physical crosslinking points, providing excellent stretchability and self-healing ability for elastomers. The imine bonds can further accelerate the self-healing of the elastomer at RT, and the healing efficiency reaches 95% after healing at RT for 24 h. The conjugated structure formed by the benzene ring and the imine bond helps to maintain the stability of the Schiff base in water and weak acid solutions. Therefore, the healing efficiency of the elastomer also reached 89% and 78% in water and artificial sweat, respectively. Peixuan Wu et al. synthesized a CO2-based poly(urea-imine) thermosetting material that combines intermolecular hydrogen bonds and reversible dynamic imine bonds. As shown in Fig. 5a, they confirmed the existence of intermolecular hydrogen bonds and their reversibility with the increase and decrease of temperature by temperature-dependent FTIR, and small-molecule model reactions confirmed the kinetic properties of imine bonds with temperature.56 Thermosetting materials with tri(2-aminoethyl)amine and CO2-sourced oligourea adjusted to an appropriate ratio can self-heal at 80 °C with an efficiency of more than 92%.
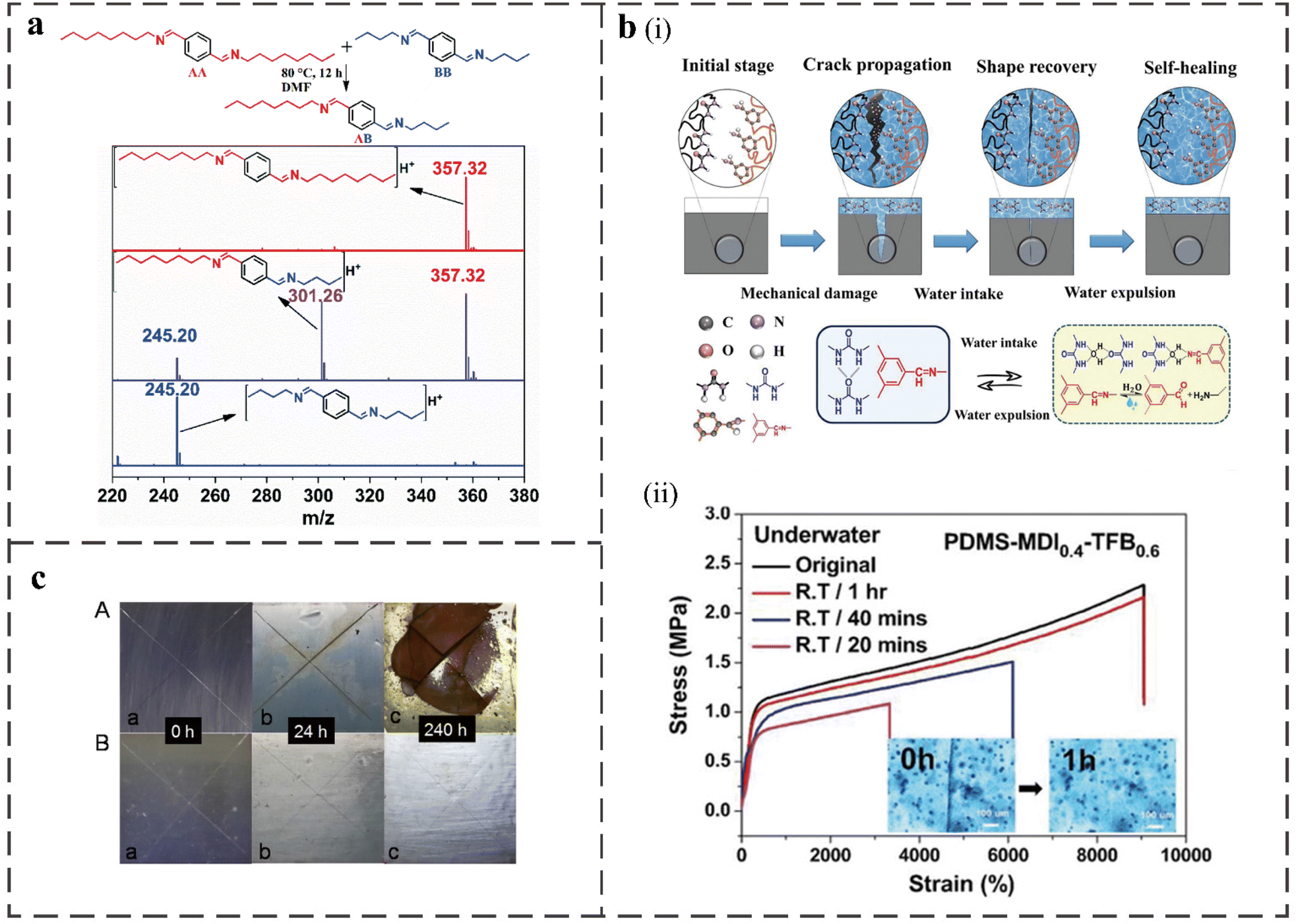 | ||
| Fig. 5 (a) Mass spectrum of the product of the model imine-based reaction of AA and BB to AB at 80 °C in DMF.56 (b) (i) Schematic diagrams of water-assisted self-healing mechanism of PDMS-MDIx-TFB1−x elastomer film and illustration of reversible imine bonds and dynamic hydrogen bonds dissociation–association with water molecules upon underwater healing process. (ii) Stress–strain curves and the inset displays optical microscope image of the PDMS-MDI0.4-TFB0.6 film underwater healing within different time at RT.75 (c) Corrosion images of carbon steel sheets coated with PDMS-U (A) and PDMS-UI-2 (B) immersed in 3.5% NaCl solution for 0, 24 and 240 h.58 | ||
When the UPy dimer was added to the PU that contained imine bonds, the energy was released during stretching by reversible hydrogen bond cleavage, but the relatively strong imine bonds (covalent bonds) maintained the network's integrity. It can further improve the material's mechanical properties by combining UPy units and imine-based elastomers. Xingyi Dai et al. prepared a self-healing triboelectric nanogenerator based on an imine bond and UPy unit by using the heat converted by human infrared radiation. The healing efficiency can reach 100% after 24 h at RT.57 In order to achieve the integration of the fracture surface, the active amino and aldehyde groups produced at the fracture interface can reform imine bonds and accompany imine exchange. In addition, the self-healing process can be accomplished by reforming UPy dimers through dissociated hydrogen-bonding.
The synthesis of water-induced self-healing polymers by combining imine bonds with hydrogen bonds is a promising self-healing process that can improve its instability and sensitivity. Cyuan-Lun He et al. proposed a new concept of a water-assisted RT self-healing mechanism based on synergistically dynamic covalent Schiff-base imine bonds with hydrogen bonds. The underwater healing process is shown in Fig. 5b(i).75 They used soft triformaldehyde benzene (TFB) units to conduct weak, covalent, and reversible Schiff base reactions involving imine metathesis so that strain energy can effectively diffuse along cracks. The hard methylene diphenyl diisocyanate (MDI) unit creates a crosslinked network through urea-based strong intermolecular hydrogen bonds to maintain the elastomer's robustness and elastic stability. The polymeric network is stabilized by polydimethyl siloxane (PDMS) to balance hydrophobicity and hydrophilicity, thereby preventing external degradation. The resulting water-assisted polymer has a healing efficiency after damage of 95% in 1 h under water (Fig. 5b(ii)). After underwater healing, the tensile properties reached 9050%, and the toughness recovered to 144.2 MJ m−3, which was several times higher than the material without water-assisted healing. The transparent polyurea material synthesized by Zhen Shi et al. can also heal by water at RT, and the tensile strength reaches 41.2 MPa.55 At the same time, it has a large fracture strain of 823.0% and a super toughness of 127.2 MJ m−3. They also explored the effects of hydrogen bonds with different densities and strengths (provided by different isocyanates), and imine bonds in the material on mechanical properties. It was concluded that the proper increase in the density of hydrogen bonds in the material significantly improves the tensile strength of PU. However, too many internal hydrogen bonds will lead to too dense crosslinking, resulting in a decrease in tensile properties. The higher the proportion of quadruple hydrogen bonds in the material, the greater the rigid domain of the material, the higher the tensile strength and the lower the elongation at break. When the ratio of quadruple hydrogen bonds to double hydrogen bonds is 1:1, the material has the maximum toughness. The introduction of dynamic imine covalent bonds greatly improved the mechanical strength, Young's modulus, and toughness of the material without necessarily sacrificing the material's tensile properties. In addition, an elastomer containing an imine bond and urea group was synthesized by Guangmeng Chen et al.58 The urea group that is easy to form hydrogen bonds is responsible for enhancing the mechanical strength of the polymer, and the imine group is responsible for self-healing performance at RT. The self-healing efficiency is 100% at 25 °C and it has excellent protection against the corrosion of carbon steel in 3.5% NaCl (Fig. 5c), which can be used as a protective coating in the marine environment.
Boronic ester and hydrogen bonds
Hydroxyl-terminated polybutadiene (HTPB) can replace polyether or polyester to react with isocyanate to synthesize PU with excellent properties such as low surface energy, low-temperature flexibility, high electrical insulation performance, high viscoelasticity, and high toughness and hydrolysis stability.76 However, HTPB-based PU has poor stability in air due to the presence of double bonds, and suffers from the common problem of thermosetting plastics being difficult to recycle. In order to solve this problem, researchers have developed HTPB-based PU vitrimers with disulfide bonds.77 Subsequently, Yanning Zeng's research group synthesized a rigid vitrimer monomer derived from rosin and synthesized 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (BDB).78 It was found that recyclable HTPB-based PU could be synthesized by introducing the crosslinking agent BDB and dynamic boronic ester to form a network because HTPB can undergo thiol–ene “click” reaction and addition reaction between HTPB and BDB and isocyanates (HDI).60 The double bond in HTPB reacts with the –SH group in BDB to form a highly stable network. The material can self-heal at 130 °C 3 h due to the dioxoborane metathesis reaction of the network's boronic ester bond and the network topology reconstruction caused by the hydrogen bond force.4. Dynamic covalent bonding and metal coordination bonding
The metal coordination bond is a special non-covalent bond formed between metal ions and organic ligands with a bond energy of 50–200 kJ mol−1, which can be used as an attractive crosslinking agent in supramolecular chemistry. Metal coordination bonds have the following advantages in developing supramolecular self-healing polymers: the formation of metal coordination bonds is spontaneous, and the bond energy is highly adjustable. Combining thermodynamic stability and kinetic variability to adjust the type of metal ions or ligands can afford different bonding strengths and functionality.79 The reversible reaction process is shown in Fig. 6a. The weaker bond is conducive to energy dissipation during stretching and self-healing during damage, while the intense metal ions remain near the ligand through stronger interactions, accelerating the re-formation of the bond.80 The common metal ions and ligands in PU include Fe3+, Zn2+, Cu2+ and catechol, carboxylic acid, pyridine, and imidazole. The coordination bond can enhance the intermolecular force and act as a sacrificial bond because the material's crosslinking density and energy dissipation during the stretching process increase.81,82 When subjected to an external force, the coordination bond will dissociate and recombine faster to dissipate energy, increasing the mechanical strength and tensile strain.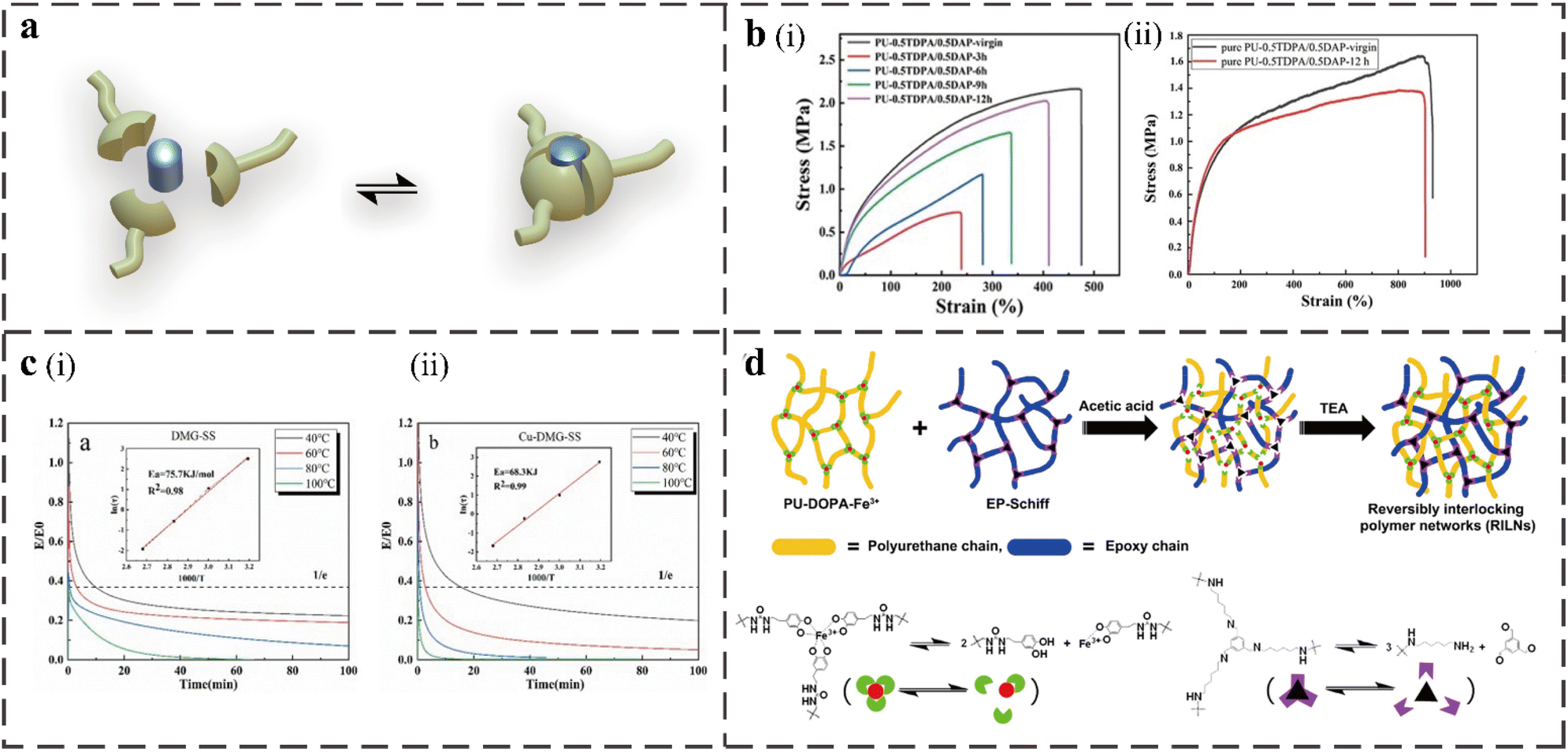 | ||
| Fig. 6 (a) The process of reversible reaction of metal ion coordination bonds. (b) (i) Comparison of the mechanical properties between virgin and full-cut PU-0.5TDPA/0.5DAP samples at different self-healing times. (ii) Typical strain–stress curves of pure PU-0.5TDPA/0.5DAP.4 (c) Normalized stress relaxation curves of Cu-DMG-SS (ii) and DMG-SS (i) at various temperatures and Linear fitting of the relaxation activation energy.10 (d) Formation mechanism of the RILNs from PU-DOPA-Fe3+ and EP-Schiff.16 | ||
Disulfide and metal coordination bonds
PU materials with a combination of disulfide and metal coordination bonds experience disulfide exchange and the interactions of metal ions and ligands in the process of self-healing. By increasing the proportion of metal coordination bonds, the material's cross-linking can be improved, which usually benefits the material's mechanical properties. The mechanical properties and self-healing ability of PU materials combined with disulfide bonds and metal coordination bonds are shown in Table 2.| Dynamic covalent bond | Tensile strength (MPa) | Elongation at break (%) | Healing conditions | Healing efficiency (%) | Ref. |
|---|---|---|---|---|---|
| Disulfide bonds | 19.5 ± 1.1 | 660 ± 30 | RT, 48 h | 83 ± 2% recovery of tensile strength | 10 |
| 9.40 ± 0.10 | 1663 ± 11 | 80 °C, 24 h | 96.6 ± 1.5% recovery of elongation at break | 83 | |
| 2.16 | 475.03 | 70 °C, 12 h | 93.48% recovery of tensile strength | 4 | |
| DA bonds | 9.5 | 725 | 65 °C, 3 h | >95% recovery of tensile strength | 85 |
| Imine bonds | 2.5 | 370.9 | pH = 4–9, 24 h | 75.9–96.3% recovery of tensile strength | 19 |
| Boronic ester bonds | >2.0 | — | pH = 9, water, 25 °C, 24 h | 92.0% recovery of tensile strength | 87 |
| pH = 7, water, 25 °C, 24 h | 86.4% recovery of tensile strength |
Xing Zhou prepared a self-healing PU elastomer using 3,3′-dithiodipropionic acid (TDPA) and iron ion-2,6-diaminopyridine (DAP) as dynamic chain extenders and ionic ligands, respectively.4 When the molar ratio of TDPA to DAP was 0.5:0.5, it could heal at 70 °C for 12 h, and the self-healing efficiency reached 93.48%. By testing the mechanical properties of the elastomers with different molar ratios of TDPA and DAP before and after self-healing, they proved that the presence of metal coordination bonds provided crosslinking for the polymer and improved the hardness of the material. By comparing the typical stress–strain curves of pure Pu-0.5TDPA/0.5DAP without FeCl3 and Pu-0.5TDPA/0.5DAP with FeCl3 before and after healing (Fig. 6b), it was found that the high content of the metal ligand coordination bond will lead to the increase in the crosslinking degree of the polymer, which improves the hardness of the material and reduces the overall self-healing efficiency. However, it is still more efficient than the material with only disulfide exchange. Then, a self-healing conductive elastomer with uniform conductivity was obtained by doping a certain amount of conductive graphene based on the PU-TDPA/DAP-Fe elastomer to construct a flexible sensor with sensitive and repeatable corresponding tensile and compressive strain.
Yu-han Li et al. developed a method to simultaneously improve the toughness and self-healing ability by adjusting the hard segment aggregation state and coordination bond.83 They incorporated aliphatic diamine-based disulfide bonds and diamine-based pyridine derivatives into the poly(urethane-urea) backbone to free some disulfide bonds from the hard phase, which facilitated the exchange of disulfide bonds. The coordination bond with Zn2+ as the coordination center not only provides self-healing ability, but also acts as a dynamic cross-linking joint to constrain mutual chain dislocation. This can improve the fracture elongation and tensile stress while having excellent self-healing ability.
In addition to forming coordination bonds to improve the self-healing efficiency, some metal ions may also play a catalytic role in the structure to accelerate covalent bond exchange. Minhui Wang et al. prepared PU elastomers with various reversible interactions, such as disulfide bonds, oxime carbamate bonds, hydrogen bonds, and coordination, that use IPDI as the hard segment, polytetramethylene ether glycol (PTMEG) as the soft segment, tri-HDI as the crosslinking agent, HEDS and dimethylglyoxime (DMG) as the chain extenders, and the solution of CuCl2, instead of the typical dibutyltin dilaurate, as a catalyst.10 Importantly, Cu2+ can be used not only as a coordination center but also as a catalyst to accelerate disulfide exchange and oxime carbamate exchange reactions. The dynamics of the Cu-containing PU system is increased because, as shown in Fig. 6c the final relaxation activation energy of the system is actually reduced. Due to the multiple interaction design and the effect of Cu2+, the elastomer has an excellent mechanical strength of 19.5 MPa and self-healing efficiency of 83% at RT.
DA and metal coordination bonds
Self-healing systems based on non-covalent interactions usually exhibit extraordinary healing and stimulus-responsive properties because they are usually more susceptible to external environments.79 Self-healing PU materials derived from metal ligand interactions have been widely studied, most of them being the combination of metal ions with other non-covalent interactions such as hydrogen bonds.84 There are few studies on the self-healing of the DA reaction and metal coordination bonds.Only Yuming Yang's group synthesized dual crosslinked network PU (PU-ND) with a metal coordination and DA bond, and the mechanical properties and self-healing ability are shown in Table 2.85 The metal ion coordination bond is formed by a pyridine-containing PU (PUN) reaction with Fe3+ containing triethylamine. The DA bond is provided by the furan pendant PU and a crosslinking agent with a maleimide end group. In order to better compare the difference between PU-DN and SN PU, they also prepared SN samples (PU-N) without DA bonds under the same conditions. Through the cyclic tensile measurement, it was found that PU-ND has a higher elastic deformation degree and it can return better to its original length, because, under the action of external load, the coordination bond breaks as a sacrificial bond to dissipate energy, and the DA bond maintains the shape of the sample. Although they found that the self-healing efficiency of PU-ND samples was lower than that of PU-N at RT, the mechanical properties of the material before and after healing at medium temperature and DA reaction temperature can be greatly enhanced by changing the concentration of coordination and DA bonds.
Imine and metal coordination bonds
The addition of metal coordination bonds also adds water or pH stimulation response ability, so the polymer can be self-healed in a special environment (underwater or in a specific pH environment). In addition, imines and metal coordination bonds have been shown to be highly dynamic and usually provide good self-healing properties in materials.86Mingqiu Zhang's group prepared homogeneous reversible interlocking polymer networks (RILNs).19 This RILN material was obtained by the topological recombination of two crosslinked polymers containing reversible catechol–Fe3+ coordination bonds and imine bonds (Fig. 6d). Table 2 shows that the material has superior underwater self-healing efficiency in a wide pH range. At higher pH, the catechol–Fe3+ coordination bond is responsible for self-healing, and the imine bond maintains the stability of the material. In neutral water, the imine bond mainly accounts for self-healing, and the hydrogen bonds and entanglement between the two networks prevent the collapse of the material. At lower pH, intermolecular hydrogen bonds and entanglements contribute to self-healing.
Boronic ester and metal coordination bonds
Coordination bonds are typically added to the network when designing PU containing boronic ester bonds that can be healed underwater. Because it possesses cationic coordination bonds due to electron attraction, the boronic ester bond at lower pH is not easy to hydrolyze. In order to reduce the pH sensitivity of the boronic ester bond, MingQiu Zhang's group synthesized a lipophilic hyperbranched PU.87 The structure of PU contains a small amount of quaternary ammonium salt and rich catechol–B3+ bonds. The quaternary ammonium cation plays a key role in attracting the electrons of the catechol–B3+ coordination bond, thereby stabilizing the boronic ester bond at a lower pH. This material has room temperature underwater (pH = 7–9) self-healing ability and recyclability. However, the mechanical strength of the synthesized polymers is insufficient for high performance structural applications. In the subsequent research, researchers can try to improve the crosslinking degree or introduce rigid segments to solve this problem.5. Dynamic covalent bonds and nanofillers
Combining dynamic covalent bonds and nanocomposites in PU structures is a promising research method. Commonly introduced fillers in the self-healing PU include graphene and CNT. Graphene has a large π–π conjugation network that can significantly improve the mechanical properties of the resulting nanocomposites at lower loadings.88 Carbon nanotubes generate a large amount of heat under near-infrared (NIR) irradiation.89 Materials have the ability to heal under infrared irradiation because the polymer matrix reacts reversibly upon thermal stimulation. Table 3 summarizes the research of PU materials combined with these fillers by dynamic covalent bonds.| Dynamic covalent bond | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (MPa) | Healing conditions | Healing efficiency (%) | Ref. |
|---|---|---|---|---|---|---|
| Disulfide bonds | 64.6 | 1306.8 | 56.1 | RT, 24 h | 88.6% | 2 |
| 4.22 | 451 | — | NIR, 24 h | 90% recovery of tensile strength | 91 | |
| DA bonds | 36 | 1100 | 127 | NIR, 1 min | 96% recovery of tensile strength | 95 |
| 14.79 | 603 | — | NIR, 20 min | 92.16% recovery of tensile strength | 6 | |
| 140 °C, 30 min | 90.06% recovery of tensile strength | |||||
| 8.01 ± 0.7 | 1449 ± 16 | 21.95 | 150 °C, 4 h and 65 °C, 24 h | 71.12% recovery of tensile strength | 93 | |
| 7.96 | 438 | — | 130 °C, 30 min and RT, 24 h | 75.8% recovery of tensile strength | 94 | |
| >0.25 | >275 | — | 120 °C, 10 min and RT, 30 min | 95% recovery of tensile strength | 96 |
Disulfide bonds and nanofillers
The incorporation of modified nanofillers into polyurethane improves self-healing efficiency by introducing hydrogen bonding or metal coordination. For example, adipic dihydrazide modified graphene oxide (AD/GO) with multiple hydrogen bonds can introduce high-density hydrogen bonds at the interface between PU matrix and nanosheets.2 Inspired by the structure of natural spider silk and nacreous layer, Xiaobo Zhu et al. incorporated AD/GO into a urea-containing PU. The network structure of the material is shown in Fig. 7a. The PU matrix without nanosheets has an excellent ultimate tensile strength (64.6 MPa) and Young's modulus (56.1 MPa) due to the disulfide bonds and abundant hydrogen bonds in the structure. The self-healing efficiency of the sample after fracture is as high as 89.8% at RT. The composites with a small amount of AD/GO showed a record ultimate tensile strength (78.3 MPa) and extremely high Young's modulus (81.8 MPa) due to the abundant interfacial hydrogen bonds between AD/GO and the PU matrix, while the self-healing ability was almost unchanged (88.6%). Zhang et al. synthesized a 3D binary conductive network silver nanowire@thiolated graphene foam/functionalized PU (AgNWs@TGF/FPU) strain sensor with RT self-healing ability.90 The strong intermolecular hydrogen bonds of the urea group in the structure and the dynamic exchange reaction of the aromatic disulfide afforded the material an excellent self-healing ability. In addition, the Ag–S coordination interaction from the disulfide bond and between AgNW also contributes to the self-healing ability.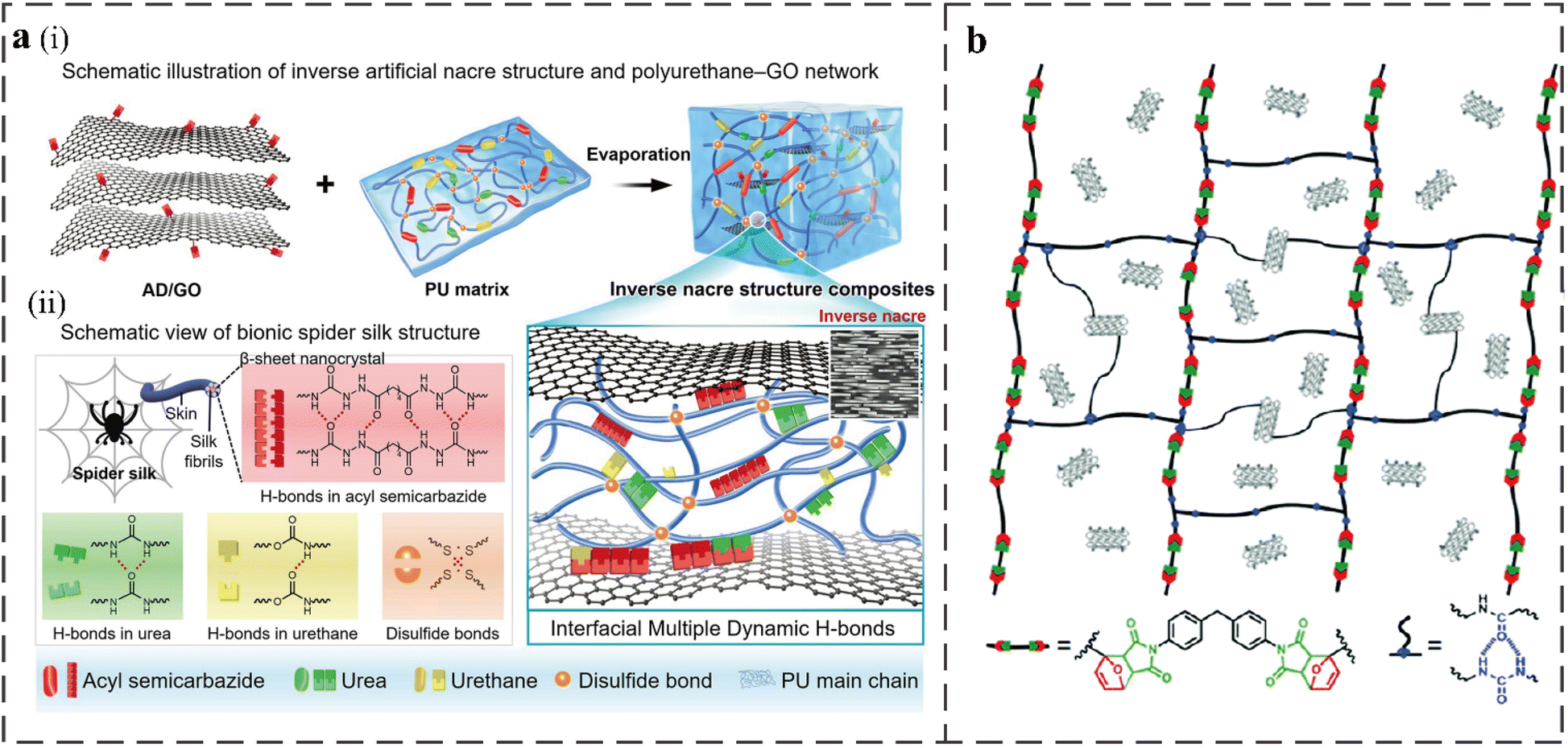 | ||
| Fig. 7 (a) (i) Schematic of the PU–graphene oxide network with an inverse artificial nacre structure. (ii) Dynamic bond interaction in the biomimetic composite.2 (b) The structure of cross-linked networks of RCFA.6 | ||
Multi-walled carbon nanotubes can provide local self-healing ability for polymers by NIR irradiation. In addition, it can also be used as a chemical crosslinking agent or physical crosslinking agent for polymer networks. Heqing Fu et al. designed a nanocomposite composed of urea-grafted multi-walled carbon nanotubes (urea-g-MWCNTs) and PU sulfide. The urea-g-MWCNTs were dispersed in the PU sulfide and formed urethane groups with ethanol-terminated PU reversible hydrogen bonds.91 In addition to the disulfide bonds and hydrogen bonds in PU, urea-g-MWCNTs also provide self-healing ability. This material can effectively restore more than 90% of the original strength, and also has excellent water resistance (1.01 MPa stable strength can be maintained after soaking in water for 6 h).
DA bonds and nanofillers
The PU of the DA network often combines with the modified graphene to produce 3D networked self-healing nanocomposites with significantly enhanced mechanical properties mechanical properties. Furfurylamine and BMI are usually used to connect PU to graphene. Reversible DA bonds are usually formed between PU and graphene in the synthesized materials, and these bonds are chemically cross-linked with graphene in the material. Both have good dispersion and Young's modulus and strain at break of these materials are enhanced.92–94Rong Sun et al. synthesized a new self-healing composite composed of functionalized graphene nanosheets and PU.95 They reduced water–amine-functionalized graphene oxide to obtain water–amine-functionalized graphene nanosheets, namely, hydroxylated graphene nanosheets (FGN). The Young's modulus of the nanocomposite increased to 127 MPa, the tensile strength reached 36 MPa, and the elongation at break could still exceed 1100%. The introduction of FGNS not only improves the mechanical properties, but also affords the film photothermal conversion properties for ultrafast and precise healing by NIR. After 1 minute of NIR irradiation, the healing efficiency of the material was higher than 96%. PU containing only DA bonds cannot self-heal under this infrared laser irradiation.
Heqing Fu's group prepared a reversible conductive flexible adhesive (RCFA).6 This RCFA was synthesized by combining furan-terminated four-arm PU (F4PU) resin, urea-g-MWCNTs and silver nanoparticle-coated multi-walled carbon nanotubes (Ag@MWCNTs). It has good mechanical strength; ultimate strength and fracture strain reach 14.79 MPa and 603%, respectively, and has a high conductivity under mechanical deformation. Additionally, it has a self-healing efficiency of more than 90% in 20 min of NIR irradiation. Its network structure is shown in Fig. 7b. The high specific surface area of MWCNTs enabled hydrogen bonding interactions among them and urea-g-MWCNTs and F4PU resin, and improved the mechanical properties of RCFA. They also introduced ZnO@MWCNTs nanoparticles into a PDMS elastomer containing DA bonds to synthesize a novel multifunctional nanocomposite.96 The material has excellent self-healing efficiency and microwave absorption performance. After repair, the mechanical strength can be restored to more than 90% (120 °C, 10 min), and even after three healing cycles, an excellent healing efficiency of 90% can be achieved.
6. Combination of two or more dynamic covalent bonds
The imine bond formed by amine and aldehyde under weak acid conditions produces a large amount of benzene in the polymer structure, resulting in a rigid skeleton.99 This rigid structure can be used to adjust the mechanical properties of the polymers related to moderate soft segments, so imine bonds are often combined with other dynamic covalent bonds to construct self-healing materials with excellent performance (Table 4). Considering that the disulfide bond has the advantages of simple operation, easy availability of raw materials, and reactivity to various stimuli, and its self-healing ability can be triggered under mild conditions, Dai-Soo Lee et al. prepared a PU network.1 They synthesized bio-based Schiff bases with double reversible covalent bonds (dynamic disulfide bonds and dynamic imine bonds) through the renewable biomaterials cystine and vanillin, and used them as chain extenders in PU networks. The structure of the PU is shown in the Fig. 8a. When the imine and disulfide molecules are chemically connected as closely as in the Schiff base, the synergistic effect of the dynamic methoxy compound reduces the activation energy of stress relaxation. In addition, scratch healing experiments also proved that the presence of double dynamic covalent bonds in Schiff bases is more effective for the rearrangement of PU networks than systems with only one type of dynamic covalent bond or a simple mixture of two dynamic covalent bonds. Additionally, due to the presence of disulfide bonds, the developed materials can also heal themselves under ultraviolet radiation. Xiaoyue Wang et al. synthesized a novel chain extensibility agent containing diimide bonds and disulfide bonds and incorporated them into thermosetting PU.97 The mechanical properties of PU containing double dynamic bonds are better than those of PU containing only disulfide bonds or imine bonds. Additionally, because of the low Tg of double dynamic bond PU, these PUs have a good low temperature flexibility. The maximum tensile strength and elongation at break were 33.70 ± 4.25 MPa and 1137.72 ± 81.29%, respectively, while also showing a high toughness of 197.35 ± 35.33 MJ m−3. Although the polymer structure contains disulfide bonds and imine bonds, the high crosslinking strength limits the movement of the molecular chain. Additionally, due to the slow speed of dynamic covalent bond exchange, the healing efficiency is low. Therefore, the breakage and recombination of dynamic covalent bonds and hydrogen bonds are promoted by heating. The healing efficiency of the final material reached about 100% after 24 h at 110 °C.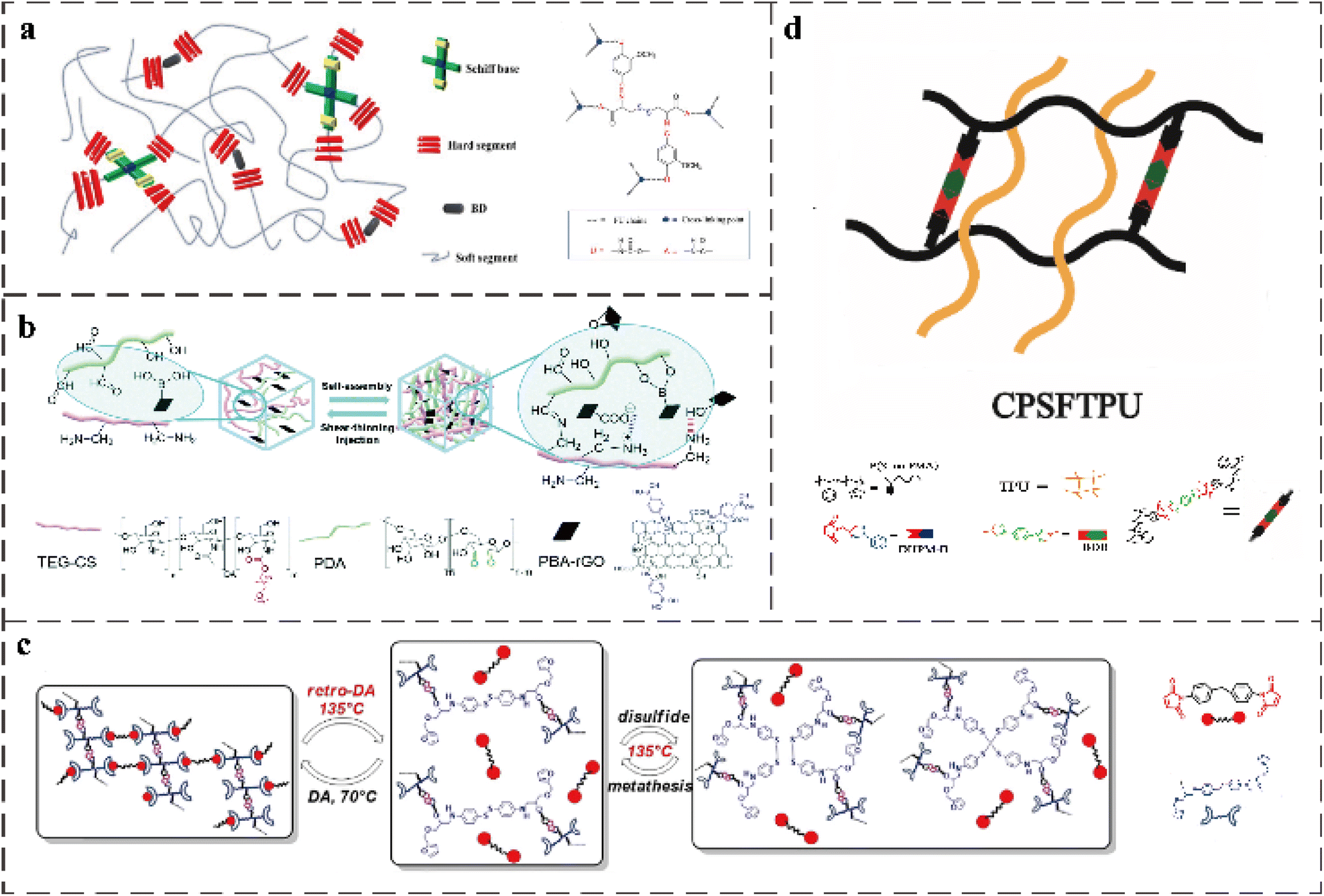 | ||
| Fig. 8 (a) Schematic of synthesized PU crosslinked with a Schiff base (SPU).1 (b) Schematic illustration of ncDN hydrogels constructed with PDA, TEG-CS, and PBA-rGO through the formation of noncovalent interactions (i.e. electrostatic interactions (blue dashed line) and hydrogen bonds (red dashed line)) and dynamic covalent interactions (i.e. imine bonds and boronic ester bonds).8 (c) Dual self-healing mechanism (DA/rDA reaction and disulfide metathesis reaction).14 (d) Schematic of synthesized semi IPNs of CPSFTPU.17 | ||
| Dynamic covalent bond 1 | Dynamic covalent bond 2 | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (MPa) | Healing conditions | Healing efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Imine bonds | Disulfide bonds | 9.4 ± 1.8 | 34.6 ± 1.4 | 525 ± 5.2 | UV365 nm, 120 min | Scratch completely healed | 1 |
| 28.16 ± 1.95 | 822.49 ± 51.07 | 39.90 ± 0.33 | 110 °C, 24 h | 100% recovery of tensile strength | 97 | ||
| Boronic ester bonds | 48.86 ± 5.10kPa | — | 88.92 ± 0.67 | RT, 2 h | — | 8 | |
| DA bonds | Disulfide bonds | 39.5 ± 0.5 | 455 ± 10 | 131 ± 8 | 135 °C, 10 min and 65 °C, 24 h | 97.5 ± 1.5% recovery of tensile strength | 15 |
| 4.63 | 1092 | 1.2 | 60 °C | >97% recovery of tensile strength | 98 | ||
| Boronic ester bonds | 28.4 ± 1.37 | 661.7 ± 8.37 | 30.2 ± 3.38 | 120 °C, 20 min | The welding film can lift a weight of 0.5 kg without breaking at the welded part | 20 |
Tough hydrogel materials can be produced by combining nanocomposites with the twin-network concept considering that adding modified nanocomposites to the twin-network hydrogel effectively prevents crack diffusion and stress transfer between soft matrix and hard nanofiller.100,101 Tsan-Yu Tsai et al. developed a multifunctional double network (ncDN) hydrogel (Fig. 8b).8 By using a variety of interfacial cross-linking chemistry and surface-functionalized nanomaterials (phenylboronic-acid-modified reduced graphene oxide (PBA-rGO)), they solved the problem of lack of diversity and limited functionality of the network structure of nanocomposite ncDN hydrogels. Various interfacial cross-linking chemistry refers to electrostatic interactions, non-covalent interactions of hydrogen bonds, and dynamic covalent interactions of imine bonds and boronic bonds. The imine bond and boronic ester bond provide reversible crosslinking in the ncDN hydrogel network. The combined electrostatic interaction and hydrogen bonding for hydrogels provide shear-thinning and self-healing properties.
The self-healing PU based on DA bonds and disulfide bonds has different degrees of improvement in its mechanical properties and self-healing efficiency. For the first time, Ho-Hsiu Chou et al. integrated dynamically reversible DA bonds and disulfide bonds into a polymer. They used cystamine (CYS), 1,6-diaminohexane (HEX), and 4,40-diaminodiphenylmethane (DPM) as connectors to form PU-CYS, PUHEX, and PU-DPM, respectively.98 By comparison, it was found that PU-Cys containing disulfide bonds and DA bonds in the structure not only has a strain value of up to 1000% and transparency greater than 97%, but also has a fast and dual-mode healing ability. It can heal quickly within 5 min at 60 °C, and can also heal under the heating of a hair dryer or at 100 °C. Therefore, it can be repaired by different covalent dynamic reversible bonds according to the degree of damage. Nikhil K. Singha et al. obtained a novel chain extender (APDS-FGE diol) by reacting 4-aminodisulfide (APDS) with furfuryl glycidyl ether (FGE). This chain extender can be used to prepare functional PU with furan and disulfide moieties.15 Then, the PU is further crosslinked by the DA reaction using bismaleimide to obtain the PU with a double healing mechanism (Fig. 8c).
Similar to the IPN polymer synthesis method mentioned above, the semi-IPN polymer can be formed by adding a secondary polymer to the crosslinked SN. The secondary polymer composed of this structure can be separated from the primary crosslinking network without destroying any chemical bond.67 Yanning Zeng et al. constructed a new type of covalent adaptable network (CAN) with multiple dynamic linkers (DA bonds and boronic esters).20 Then, they introduced thermoplastic PU into CAN to provide a semi-IPN to enhance CAN performance. The structure of this semi-interpenetrating network is shown in Fig. 8d. Due to the ester exchange reaction between DA chemistry and boronic ester bonds in the CAN matrix, network rearrangement and bond rearrangement occur when the material is damaged, and covalent bonds can be re-established at the interface of the broken surface. The semi-IPN can achieve self-healing at high temperatures by exchange-induced network rearrangement without the addition of a self-healing agent (as shown in Table 4).
7. Conclusions
This paper summarized the research progress of self-healing PU materials using dynamic covalent bonds with other methods, including disulfide bonds, DA bonds, boronic ester bonds and imine bonds combined with hydrogen bonds, coordination bonds, and nanofillers. The self-healing principles of these dynamic covalent bonds were introduced. The methods of introducing various healing methods into PU structures and the properties and applications of synthetic materials were introduced. The problems that can be solved by various methods were briefly analyzed.Scientists have synthesized many PU/polyureas with excellent dynamic covalent bonds combined with various self-healing methods. Materials combining dynamic covalent bonds with hydrogen bonds have been widely studied. There are few studies on combining dynamic covalent bonds with coordination bonds or nanomaterials. The metal ion coordination bond can not only adjust the material's mechanical strength, but also stimulate the self-healing polymer and offer unique physical and chemical properties to the polymer. By focusing on this aspect, researchers can introduce more metal ion coordination bonds into self-healing polymer networks containing dynamic covalent bonds to develop more functional self-healing materials with excellent mechanical properties and self-healing efficiency. Adding nanocomposites to self-healing polymer networks can improve the mechanical properties of materials, and nanocomposites are diverse. Graphene, silver nanoparticles, chitosan, its derivatives, or various modified cellulose/carbon nanotubes can be examined in the future. As more physical embedding in the matrix or chemical bonding methods are combined with dynamic covalent bond networks, more important discoveries will be made.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the financial support provided by the National Natural Science Foundation of China (22268009), Guangxi Natural Science Foundation Program (2020GXNSFBA159023), Open Project Funding of the State Key Laboratory of Biocatalysis and Enzyme Engineering (SKLBEE2022029) and Opening Project of Guangxi Key Laboratory of Calcium Carbonate Resources Comprehensive Utilization (HZXYKFKT202204). The authors also would like to thank Xinfang Cui from the Shiyanjia lab (www.Shiyanjia.com).References
- S.-H. Lee, S.-R. Shin and D.-S. Lee, Mater. Des., 2019, 172, 107774 CrossRef CAS.
- X. Zhu, W. Zhang, G. Lu, H. Zhao and L. Wang, ACS Nano, 2022, 16, 16724–16735 CrossRef CAS PubMed.
- H. Xie, X. Liu, D. Sheng, H. Wu, Y. Zhou, X. Tian, Y. Sun, B. Shi and Y. Yang, Polymer, 2021, 230, 124096 CrossRef CAS.
- X. Zhou, H. Wang, S. Li and M. Liu, Eur. Polym. J., 2021, 159, 110769 CrossRef CAS.
- Y. Cai, L. Yan, Y. Wang, Y. Ge, M. Liang, Y. Chen, H. Zou and S. Zhou, Chem. Eng. J., 2022, 436, 135156 CrossRef CAS.
- Q. Yan, M. Zhou and H. Fu, J. Mater. Chem. C, 2020, 8, 7772–7785 RSC.
- S. Lambert and M. Wagner, Chem. Soc. Rev., 2017, 46, 6855–6871 RSC.
- T.-Y. Tsai, K.-H. Shen, C.-W. Chang, L. Jovanska, R. Wang and Y.-C. Yeh, Biomater. Sci., 2021, 9, 985–999 RSC.
- B. R. Zhang, J. Ke, J. R. Vakil, S. C. Cummings, Z. A. Digby, J. L. Sparks, Z. J. Ye, M. B. Zanjani and D. Konkolewicz, Polym. Chem., 2019, 10, 6290–6304 RSC.
- M. Wang, J. Zhou, X. Jiang, Y. Sheng, M. Xu and X. Lu, Eur. Polym. J., 2021, 146, 110257 CrossRef CAS.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CrossRef CAS PubMed.
- H. Guo, Y. Han, W. Zhao, J. Yang and L. Zhang, Nat. Commun., 2020, 11, 2037 CrossRef CAS PubMed.
- H. Zhao, W. C. Gao, Q. Li, M. R. Khan, G. H. Hu, Y. Liu, W. Wu, C. X. Huang and R. K. Y. Li, Adv. Colloid Interface Sci., 2022, 303, 102644 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 1700782 CrossRef PubMed.
- P. K. Behera, S. K. Raut, P. Mondal, S. Sarkar and N. K. Singha, ACS Appl. Polym. Mater., 2021, 3, 847–856 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2002, 415, 817–817 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS PubMed.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- W. L. Peng, Z. P. Zhang, M. Z. Rong and M. Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 27614–27624 CrossRef CAS PubMed.
- Y. Zeng, W. Yang, S. Liu, X. Shi, A. Xi and F. Zhang, Polymers, 2021, 13, 1679 CrossRef CAS PubMed.
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- Y. Yao, M. Xiao and W. Liu, Macromol. Chem. Phys., 2021, 222, 2100002 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- Y. Deng, Q. Zhang, B. L. Feringa, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2020, 59, 5278–5283 CrossRef CAS PubMed.
- Y. He, D. Xie and X. Zhang, J. Mater. Sci., 2014, 49, 7339–7352 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- F. Seidi, Y. C. Jin, J. Q. Han, M. R. Saeb, A. Akbari, S. H. Hosseini, M. Shabanian and H. N. Xiao, Chem. Rec., 2020, 20, 1142–1162 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- B. X. Cheng, C. C. Lu, Q. Li, S. Q. Zhao, C. S. Bi, W. Wu, C. X. Huang and H. Zhao, J. Polym. Environ., 2022, 30, 5252–5262 CrossRef CAS.
- J. S. Chen, C. K. Ober and M. D. Poliks, Polymer, 2002, 43, 131–139 CrossRef CAS.
- S. K. Schoustra, T. Groeneveld and M. M. J. Smulders, Polym. Chem., 2021, 12, 1759–9954 RSC.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2021, 12, 293–302 RSC.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- S. W. Sinton, Macromolecules, 1987, 20, 2430–2441 CrossRef CAS.
- Y. Chen, D. Diaz-Dussan, D. Wu, W. Wang, Y.-Y. Peng, A. B. Asha, D. G. Hall, K. Ishihara and R. Narain, ACS Macro Lett., 2018, 7, 904–908 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- Y. Huang, H. Wu, W. Li, Z. Yuan, Q. Wu, R. Li and J. Wu, J. Mater. Chem. A, 2022, 10, 24290–24300 RSC.
- Z. Xiang, C. Chu, H. Xie, T. Xiang and S. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 1463–1473 CrossRef CAS PubMed.
- Y. Huang, Z. Shi, H. Wang, J. Wang and Z. Xue, Energy Storage Mater., 2022, 51, 1–10 CrossRef.
- C. Yan, F. Yang, M. Wu, Y. Yuan, F. Chen and Y. Chen, Macromolecules, 2019, 52, 9376–9382 CrossRef CAS.
- R.-P. Nie, H. Lin, Y. Li, H.-D. Huang, D.-X. Yan, K. Dai, J. Lei and Z.-M. Li, Chem. Eng. J., 2022, 443, 121912 Search PubMed.
- J. Hu, R. Mo, X. Jiang, X. Sheng and X. Zhang, Polymer, 2019, 183, 121912 CrossRef CAS.
- J. Ye, G. Lin, Z. Lin, H. Deng, J. Huang, H. Xiang, M. Z. Rong and M. Q. Zhang, Macromol. Mater. Eng., 2022, 307, 2100874 CrossRef CAS.
- Y. Zhang, L. Yuan, G. Liang and A. Gu, J. Mater. Chem. A, 2018, 6, 23425–23434 RSC.
- W. Kong, Y. Yang, Y. Wang, H. Cheng, P. Yan, L. Huang, J. Ning, F. Zeng, X. Cai and M. Wang, J. Mater. Chem. A, 2022, 10, 2012–2020 RSC.
- X. Haopu, L. Xiangdong, S. Dekun, W. Haohao, Z. Yan, T. Xinxin, S. Yinglu, S. Biru and Y. Yuming, Polymer, 2021, 230, 124096 CrossRef.
- B. Zhang, J. Ke, J. R. Vakil, S. C. Cummings, Z. A. Digby, J. L. Sparks, Z. Ye, M. B. Zanjani and D. Konkolewicz, Polym. Chem., 2019, 10, 6290–6304 RSC.
- Z. Shi, J. Kang and L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 23484–23493 CrossRef CAS PubMed.
- P. Wu, X. Wang, R. Shi, H. Cheng and F. Zhao, Green Chem., 2022, 24, 1561–1569 RSC.
- X. Dai, L.-B. Huang, Y. Du, J. Han, Q. Zheng, J. Kong and J. Hao, Adv. Funct. Mater., 2020, 30, 1910723 CrossRef CAS.
- G. Chen, Z. Sun, Y. Wang, J. Zheng, S. Wen, J. Zhang, L. Wang, J. Hou, C. Lin and Z. Yue, Prog. Org. Coat., 2020, 140, 105483 CrossRef CAS.
- Z. Yang, H. Li, L. Zhang, X. Lai and X. Zeng, J. Colloid Interface Sci., 2020, 570, 1–10 CrossRef CAS PubMed.
- J. Li, Z. Ning, W. Yang, B. Yang and Y. Zeng, ACS Omega, 2022, 7, 10156–10166 CrossRef CAS PubMed.
- D. W. R. Balkenende, C. A. Monnier, G. L. Fiore and C. Weder, Nat. Commun., 2016, 7, 10995 CrossRef PubMed.
- J. H. Xu, S. Ye, C. Di Ding, L. H. Tan and J. J. Fu, J. Mater. Chem. A, 2018, 6, 5887–5898 RSC.
- J. Xu, X. Wang, H. Ruan, X. Zhang, Y. Zhang, Z. Yang, Q. Wang and T. Wang, Polym. Chem., 2022, 13, 2420–2441 RSC.
- B. Qin, S. Zhang, P. Sun, B. Tang, Z. Yin, X. Cao, Q. Chen, J.-F. Xu and X. Zhang, Adv. Mater., 2020, 32, 2000096 CrossRef CAS PubMed.
- B.-X. Cheng, W.-C. Gao, X.-M. Ren, X.-Y. Ouyang, Y. Zhao, H. Zhao, W. Wu, C.-X. Huang, Y. Liu, X.-Y. Liu, H.-N. Li and R. K. Y. Li, Polym. Test., 2022, 107, 107489 CrossRef CAS.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- A. P. Dhand, J. H. Galarraga and J. A. Burdick, Trends Biotechnol., 2021, 39, 519–538 CrossRef CAS PubMed.
- M. B. Zanjani, B. Zhang, B. Ahammed, J. P. Chamberlin, P. Chakma, D. Konkolewicz and Z. Ye, Macromol. Theory Simul., 2019, 28, 1900008 CrossRef.
- S. Jiang, H.-Y. Cheng, R.-H. Shi, P.-X. Wu, W.-W. Lin, C. Zhang, M. Arai and F.-Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS PubMed.
- S. Jiang, R. Shi, H. Cheng, C. Zhang and F. Zhao, Green Energy Environ., 2017, 2, 370–376 CrossRef.
- P. Wu, H. Cheng, Y. Wang, R. Shi, Z. Wu, M. Arai and F. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 12677–12685 CrossRef CAS.
- P. X. Wu, H. Y. Cheng, X. C. Wang, R. H. Shi, C. Zhang, M. Arai and F. Y. Zhao, Green Chem., 2021, 23, 552–560 RSC.
- X. Lei, Y. Jin, H. Sun and W. Zhang, J. Mater. Chem. A, 2017, 5, 21140–21145 RSC.
- P. Taynton, C. Zhu, S. Loob, R. Shoemaker, J. Pritchard, Y. Jin and W. Zhang, Polym. Chem., 2016, 7, 7052–7056 RSC.
- C.-L. He, F.-C. Liang, L. Veeramuthu, C.-J. Cho, J.-S. Benas, Y.-R. Tzeng, Y.-L. Tseng, W.-C. Chen, A. Rwei and C.-C. Kuo, Adv. Sci., 2021, 8, 2102275 CrossRef CAS PubMed.
- F. M. B. Coutinho, M. C. Delpech, T. L. Alves and A. A. Ferreira, Polym. Degrad. Stab., 2003, 81, 19–27 CrossRef CAS.
- M. Khatib, O. Zohar, W. Saliba, S. Srebnik and H. Haick, Adv. Funct. Mater., 2020, 30, 1910196 CrossRef CAS.
- Y. Zeng, J. Li, S. Liu and B. Yang, Polymers, 2021, 13, 3386 CrossRef CAS PubMed.
- C.-H. Li and J.-L. Zuo, Adv. Mater., 2020, 32, 1903762 CAS.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. A. Bao, Nat. Chem., 2016, 8, 619–625 Search PubMed.
- W. Wang and A. Elbanna, Bone, 2014, 68, 20–31 CrossRef CAS PubMed.
- Z. Tang, J. Huang, B. Guo, L. Zhang and F. Liu, Macromolecules, 2016, 49, 1781–1789 CrossRef CAS.
- Y.-h. Li, W.-j. Guo, W.-j. Li, X. Liu, H. Zhu, J.-p. Zhang, X.-j. Liu, L.-h. Wei and A.-l. Sun, Chem. Eng. J., 2020, 393, 124583 CrossRef CAS.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Adv. Mater., 2020, 32, 2005759 CrossRef CAS PubMed.
- C. Lin, D. Sheng, X. Liu, S. Xu, F. Ji, L. Dong, Y. Zhou and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2228–2234 CrossRef CAS.
- Z. H. Williams, E. D. Burwell, A. E. Chiomento, K. J. Demsko, J. T. Pawlik, S. O. Harris, M. R. Yarolimek, M. B. Whitney, M. Hambourger and A. D. Schwab, Soft Matter, 2017, 13, 6542–6554 RSC.
- N. N. Xia, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 14122–14131 RSC.
- Q. Cheng, M. Wu, M. Li, L. Jiang and Z. Tang, Angew. Chem., 2013, 52, 3750–3755 CrossRef CAS PubMed.
- B. Han, Y.-L. Zhang, Q.-D. Chen and H.-B. Sun, Adv. Funct. Mater., 2018, 28, 201802235 Search PubMed.
- L. Zhang, H. Li, X. Lai, T. Gao and X. Zeng, ACS Appl. Mater. Interfaces, 2020, 12, 44360–44370 CrossRef CAS PubMed.
- Q. Yan, Q. Fu, J. Hu and H. Fu, J. Mater. Chem. C, 2020, 8, 607–618 RSC.
- G. Zhang, T. Patel, P. Nellepalli, S. Bhagat, H. Hase, A. M. Jazani, I. Salzmann, Z. Ye and J. K. Oh, Macromol. Rapid Commun., 2021, 42, 2000514 CrossRef CAS PubMed.
- J. Li, G. Zhang, L. Deng, S. Zhao, Y. Gao, K. Jiang, R. Sun and C. Wong, J. Mater. Chem. A, 2014, 2, 20642–20649 RSC.
- H. Tu, M. Zhou, Y. Gu and Y. Gu, Compos. Sci. Technol., 2022, 225, 109476 CrossRef.
- S. Wu, J. Li, G. Zhang, Y. Yao, G. Li, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 3040–3049 CrossRef CAS PubMed.
- M. Zhou, Q. Yan, Q. Fu and H. Fu, Carbon, 2020, 169, 235–247 CrossRef CAS.
- X. Wang, J. Xu, Y. Zhang, T. Wang, Q. Wang, Z. Yang and X. Zhang, Polym. Chem., 2022, 13, 3422–3432 RSC.
- C.-M. Yeh, C.-H. Lin, T.-Y. Han, Y.-T. Xiao, Y.-A. Chen and H.-H. Chou, J. Mater. Chem. A, 2021, 9, 6109–6116 RSC.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- J. R. McKee, J. Huokuna, L. Martikainen, M. Karesoja, A. Nykanen, E. Kontturi, H. Tenhu, J. Ruokolainen and O. Ikkala, Angew. Chem., Int. Ed., 2014, 53, 5049–5053 CrossRef CAS PubMed.
- C. Cui, C. Shao, L. Meng and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 39228–39237 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |