
Diverse CsPbI3 assembly structures: the role of surface acids†
Dandan
Yang
 *a,
Xuebin
Zhang
a,
Shijia
Liu
a,
Zhiheng
Xu
b,
Yang
Yang
a,
Xiaoming
Li
c,
Qiuyu
Ye
a,
Qin
Xu
*a and
Haibo
Zeng
*c
*a,
Xuebin
Zhang
a,
Shijia
Liu
a,
Zhiheng
Xu
b,
Yang
Yang
a,
Xiaoming
Li
c,
Qiuyu
Ye
a,
Qin
Xu
*a and
Haibo
Zeng
*c
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, China. E-mail: dandanyang@yzu.edu.cn
bDepartment of Nuclear Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing 211106, China
cMIIT Key Laboratory of Advanced Display Materials and Devices, Institute of Optoelectronics & Nanomaterials, College of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: zeng.haibo@njust.edu.cn
First published on 13th December 2022
Abstract
Surface ligand engineering, seed introduction and external driving forces play major roles in controlling the anisotropic growth of halide perovskites, which have been widely established in CsPbBr3 nanomaterials. However, colloidal CsPbI3 nanocrystals (NCs) have been less studied due to their low formation energy and low electronegativity. Here, by introducing different molar ratios of surface acids and amines to limit the monomer concentration of lead–iodine octahedra during nucleation, we report dumbbell-shaped CsPbI3 NCs obtained by the in situ self-assembly of nanospheres and nanorods with average sizes of 89 nm and 325 nm, respectively, which showed a high photoluminescence quantum yield of 89%. Structural and surface state analyses revealed that the strong binding of benzenesulfonic acid promoted the formation of a Pb(SO3−)x-rich surface of CsPbI3 assembly structures. Furthermore, the addition of benzenesulfonic acid increases the supersaturation threshold and the solubility of PbI2 in a high-temperature reaction system, and controls effectively the lead–iodine octahedron monomer concentration in the second nucleation stage. As a result, the as-synthesized CsPbI3-Sn NCs exhibited different assembly morphologies and high PLQYs, among which the role of sulfonate groups can be further verified by UV absorption and surface characteristics. The strategy provides a new frontier to rationally control the surface ligand-induced self-assembly structures of perovskites.
1. Introduction
Perovskite nanocrystals (NCs) can be self-assembled into more complex nanostructures that derive unique properties from their assembly structures (supercrystals, superlattices and so on), such as large Stokes shift and ultra-fast radiation decay, and corrective optical property,1 which make them promising candidates for various applications in light emitting diodes,2 lasers,3,4 and so on.5 However, despite successes achieved in synthesizing these assembly structures,6,7 the controlled self-assembly of perovskite NCs is also mainly limited to cubes8,9 or platelet shapes in their quantum confinement regimes.10 The construction of more complex assembly structures remains challenging, especially for iodine-containing perovskite NCs. In addition, the fast nucleation process11 and “highly dynamic surface” of these NCs are still the major obstacles to overcome when exploring the assembly mechanism.12–14 Hence, using the driving force to control the shape evolution of perovskite NCs remains a more fundamental understanding to be established, offering more possibilities for complex superstructure designs.For understanding more the self-assembly processes as related to the supporting chemistry and growth kinetics, different assembly reports on perovskite NCs were investigated. It was shown that the driving forces of inter-ligand or other types of interaction induce perovskite NCs to be assembled into ordered structures.1,15–17 For instance, among these “driving forces”, zwitterionic sulfobetaine,18 cations3 and octylphosphonic acid19 were all efficiently used to obtain basic cubic superstructures by inter-ligand interactions or by shortening the spacing between NCs. Unlike the aforementioned functional ligands, didodecyldimethylammonium sulfide,8 α-haloketones,20 and functional poly(ethylene glycols)21 as capping ligands stabilized NCs with low-dimensional shapes, and helped to regulate the assembly architectures of these NCs. Another study of note was the proposal of a seed-mediated method to synthesize CsPbX3 NCs with complex structures. An overgrowth of CsPbBr3 NCs could be obtained with the transformation process of Cs4PbBr6.22 In addition, intermediates consisting of CsPb2Br5 and Cs3In2Br9 as precursor reservoirs gradually released monomers, which could ensure both the slow growth rate and low defect content of CsPbBr3 nanorods.23 Recently, we proposed a two-step nucleation strategy to inhibit rapid nucleation and growth processes in which the complete coordination of oleic acid and 4-dodecylbenzenesulfonic acid (4-DBSA) promoted the evolution of CsPbBr3 from stacked to tongue-shaped nanoplatelets.24 Based on the above considerations, controlling the growth kinetics does help in constructing new shapes of perovskite NCs. However, the question remains of how to utilize surface ligand engineering to control the monomer concentration in the nucleation process. Optimizing the new shapes of CsPbI3 NCs and constructing more complex assembly structures may shed new light on meeting the needs of optical applications.
Here, we have utilized the second nucleation stage to control the monomer concentration when growing lead–iodine octahedra to realize the in situ self-assembly of dumbbell-shaped CsPbI3 NCs. The assembled structures synthesized deliver an absolute QY of 89% and good crystallinity. Meanwhile, it can be found that the strong binding between the benzenesulfonate ions and the surface lead cations can lead to the formation of a Pb(SO3−)x-rich surface, which is confirmed by surface chemical characterization studies. Furthermore, different lead–iodine octahedron monomer concentrations in the nucleation stages can be achieved by changing the molar ratio of 4-DBSA and OAm, and then CsPbI3-Sn NCs shaped into nanorods, four-leaf clovers and assembled nanospheres can be successfully prepared, and the role of benzenesulfonate ions further verified by UV absorption and Fourier transform infrared spectroscopy (FTIR). Also, dumbbell-shaped CsPbI3 NCs have a high PLQY after repeated dilutions, which is due to the Pb(SO3−)x-rich surface.
2. Results and discussion
CsPbI3 NCs were prepared using our previously reported method.25 Specifically, the lead iodide precursor was dissolved with the assistance of surface capping ligands and then injected into preheated cesium oleate to obtain a bright red CsPbI3 mixture (details are provided in the ESI†). Schematic representations of the process are shown in Fig. 1a and b.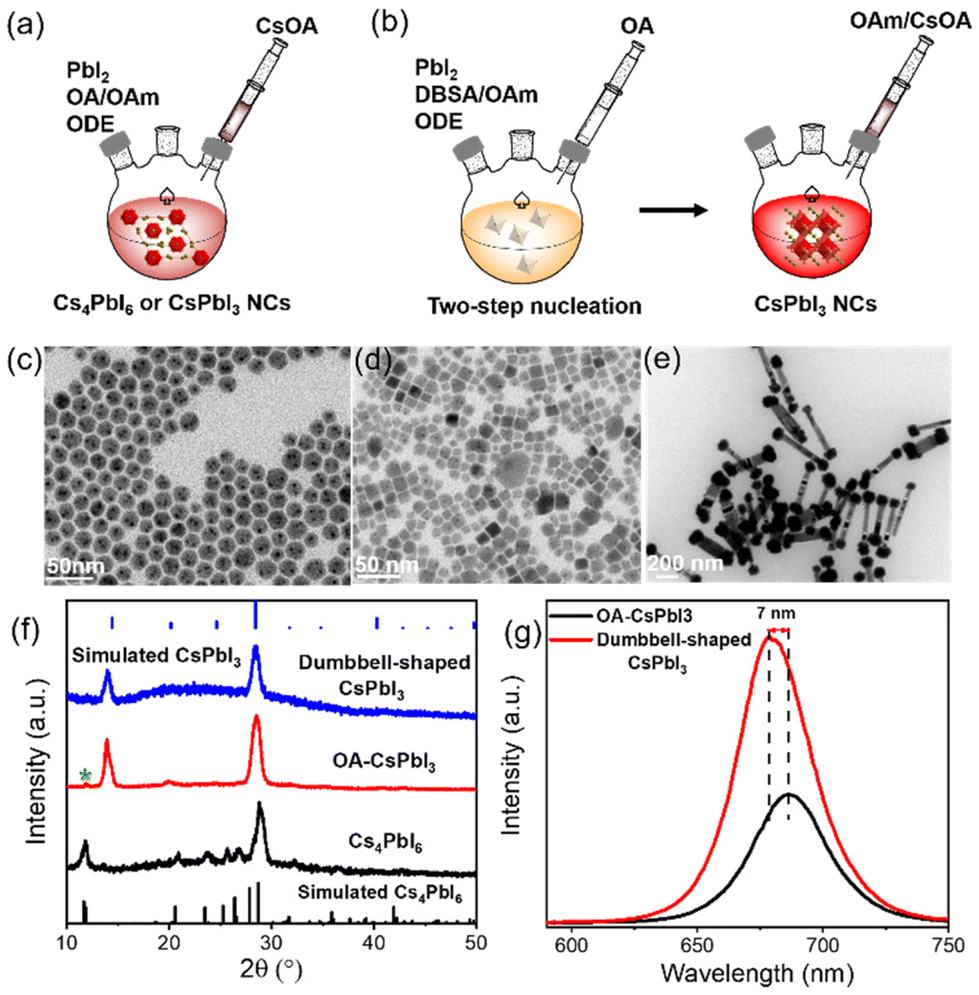 | ||
| Fig. 1 Construction process and optical properties of the complex self-assembled structures of CsPbI3 NCs at high temperature. Schematic representations of the synthesis of CsPbI3 NCs using (a) one-step nucleation and (b) two-step nucleation strategies. (c)–(e) TEM images and (f) XRD patterns of Cs4PbI6 NCs, OA-CsPbI3 and dumbbell-shaped CsPbI3 NCs, and (g) PL data of OA-CsPbI3 and dumbbell-shaped CsPbI3 NCs. | ||
Considering that surface ligands easily attain a highly dynamic bonding state at a high temperature, excess ligands are introduced into the precursors to promote the dissolution equilibrium of lead iodide, although more ligands will lead to a large consumption of lead iodide, resulting in the formation of a partial Cs4PbI6 phase.26,27 Cs4PbI6 NCs were prepared using more OA and OAm compared with the traditional synthesis approach for CsPbX3 NCs, as described in the ESI.† The transmission electron microscopy (TEM) image in Fig. 1c indicates the formation of nearly monodispersed hexagonal Cs4PbI6 NCs, and Fig. S1† shows that hexagonal Cs4PbI6 NCs with no photoluminescence exhibit an obvious characteristic absorption peak at 368 nm. Based on this synthesis, excess OA was added to the precursors to promote the phase transformation from Cs4PbX6 to CsPbX3 NCs.28,29 Then, cubic CsPbI3 NCs (OA-CsPbI3) with uneven shapes were prepared (Fig. 1d). The continuous addition of OA completely transforms Cs4PbI6 into pure CsPbI3 NCs (Fig. S2†). Fig. S3† shows that the stretching vibration peak of 1710 cm−1 belongs to the free OA. With an increase of OA content, the stretching vibration peak of 1710 cm−1 was enhanced, indicating that the addition of more OA results in it being adsorbed on the surfaces of NCs in the form of free groups, which tend to agglomerate and reduce the OY and stability of perovskite NCs in a long-term storage process, which is not conducive to an ordered self-assembly. Therefore, we used a two-step nucleation method to effectively separate the ligands with different coordination abilities, that is, the excess OA on the surface of OA-CsPbI3 NCs was replaced with an equal molar quantity of 4-DBSA ligands during the first step of precursor dissolution, and the remaining OA was added subsequently to maintain the dissolution equilibrium, as shown in Fig. 1b. As a result, dumbbell-shaped CsPbI3 NCs with a homogeneous morphology were obtained (Fig. 1e). The crystal structures of the as-synthesized NCs were further characterized by powder X-ray diffraction (XRD). Fig. 1f shows that OA-CsPbI3 NCs still retain the diffraction peak at 11.8° of the Cs4PbI6 phase (green star), while the only two diffraction peaks of dumbbell-shaped CsPbI3 NCs at 14.1° and 28.6° belong to the (100) and (200) facets of cubic phase CsPbI3 NCs, which coincided with the TEM images. It also shows that the introduction of benzenesulfonic acid is more beneficial in promoting the phase transition from Cs4PbI6 to CsPbI3 NCs, and inducing complex assembly structures. Interestingly, Fig. 1g and S1† show that the photoluminescence (PL) peak of dumbbell-shaped CsPbI3 modified by 4-DBSA located at 680 nm with a full width at half-maximum (FWHM) of 32 nm is blue-shifted 7 nm, and it has a good luminescence compared with OA-CsPbI3 NCs with a peak of 687 nm under conditions of the same concentration. This could be related to the strong quantum confinement effects due to the adsorption of –SO3− and –NH2 head group-containing ligands, as previously shown for perovskite NCs.21,30 In addition, they are diluted to a certain concentration, and the PLQYs measured with an integrating sphere were 59 ± 5% and 89 ± 5%, respectively.
Based on the above point of view, we further studied the crystal structures of dumbbell-shaped CsPbI3 NCs and their surface states, which were used to figure out the growth driving force of the structural assembly. Fig. 2a shows that monodisperse and uniformly arranged dumbbell-shaped NCs were prepared by a two-step nucleation method. Interestingly, it is found that the dumbbell-shaped NCs are formed by the self-assembly of nanospheres and nanorods, with average sizes of 89 nm and 325 nm, respectively (Fig. S4†). After the magnification of dumbbell-shaped NCs, the nanospheres were seen to be assembled from many nanoplates (Fig. 2b). In order to confirm the crystal phase of dumbbell-shaped NCs, high-resolution TEM (HRTEM) images of the as-prepared dumbbell-shaped NCs were studied (Fig. 2c). We observed a detailed crystallographic structure in which adjacent dumbbell-shaped NCs have the same lattice spacing of 0.63 nm, even at the interface between nanospheres and nanorods, which confirms the existence of lattice matching during the self-assembly process. The lattice spacing of 0.63 nm can be indexed to the (100) plane of the α-phase cubic CsPbI3 NCs, which further proves the formation of dumbbell-shaped CsPbI3 NCs (Fig. 1e and f).31 Based on the cubic CsPbI3 crystal structure, the driving force of the assembly structure of the as-prepared NCs was discussed. X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) spectroscopy measurements were performed to investigate the surface properties.
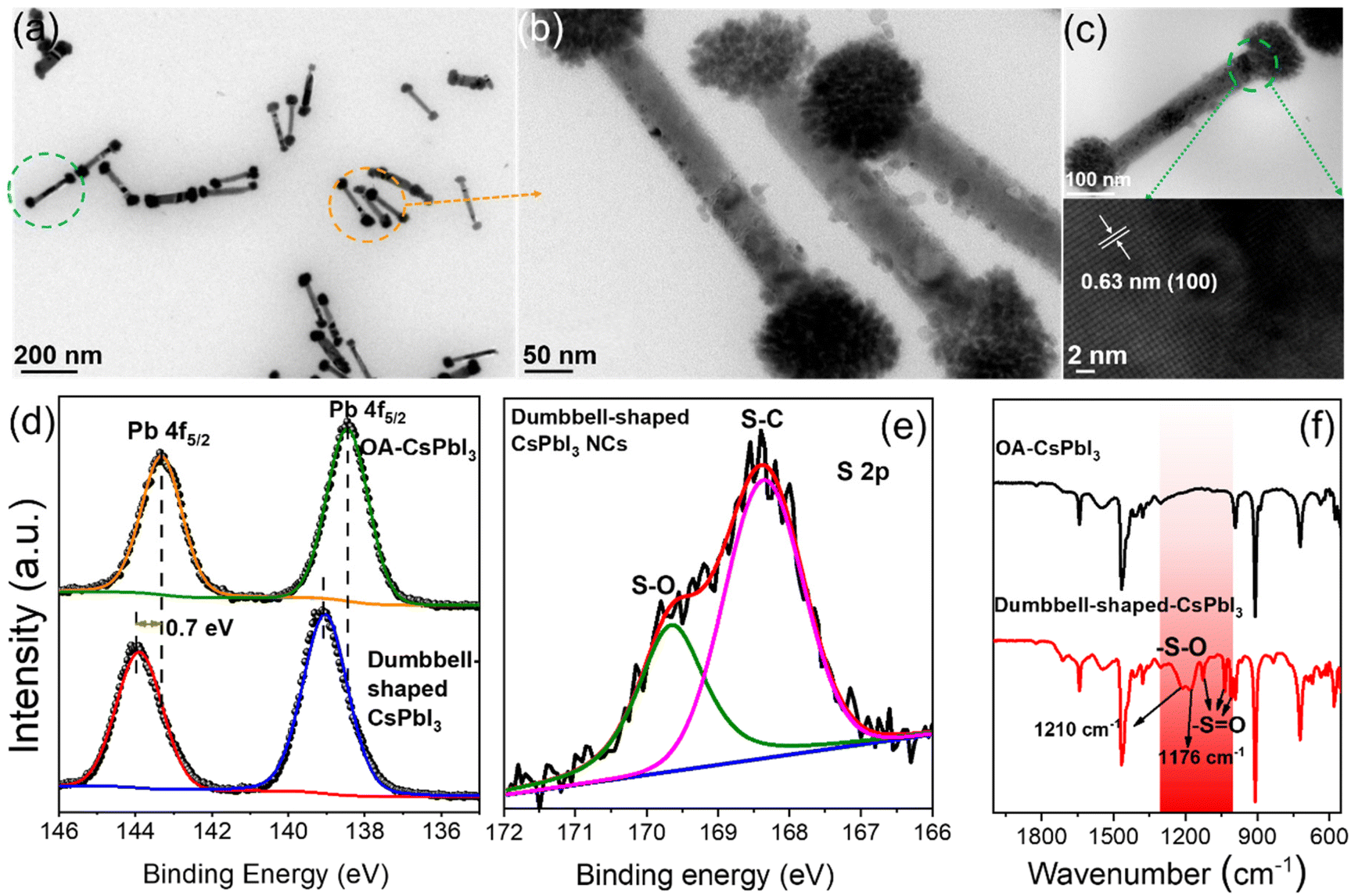 | ||
| Fig. 2 Crystal structure of dumbbell-shaped CsPbI3 and validation of the Pb(SO3−)x-rich surface. (a) TEM image, (b) enlarged TEM image and (c) HRTEM images of dumbbell-shaped CsPbI3 NCs. (d) Pb 4f and (e) S 2p XPS spectra, and (f) FTIR spectra of OA-CsPbI3 and dumbbell-shaped CsPbI3 NCs. | ||
Fig. 2d shows the binding energies of the surface Pb atoms in the two types of NCs. For OA-CsPbI3 NCs, the Pb 4f core-level signal can be fitted to Pb 4f5/2 and Pb 4f7/2 at 143.3 eV and 138.45 eV, respectively. A shift of 0.7 eV is observed for dumbbell-shaped CsPbI3 NCs. The shift can be ascribed to the fact that the binding of more benzenesulfonate ions to Pb atoms led to the formation of a Pb(SO3−)x-rich surface for dumbbell-shaped CsPbI3 NCs. Moreover, the analysis of the Pb/I ratio of the two samples can be used to support the XPS results, where the atomic ratio of Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I (1.4:3.75) is significantly lower than that of Pb:I (4:3.2) for the dumbbell-shaped CsPbI3 NCs. Preliminary results further confirm the existence of a Pb(SO3−)x-rich surface for dumbbell-shaped CsPbI3, which explains the high PLQY (89%). In comparison, OA-CsPbI3 NCs have a weaker binding environment, which is well consistent with the variation of the Pb binding energy. In addition, the existence of sulfonate groups and the binding motif of Pb–O were evident from the S 2p and O 1s XPS results (Fig. 2e and S5†) and the typical vibration modes of sulfonate groups.
:I (1.4:3.75) is significantly lower than that of Pb:I (4:3.2) for the dumbbell-shaped CsPbI3 NCs. Preliminary results further confirm the existence of a Pb(SO3−)x-rich surface for dumbbell-shaped CsPbI3, which explains the high PLQY (89%). In comparison, OA-CsPbI3 NCs have a weaker binding environment, which is well consistent with the variation of the Pb binding energy. In addition, the existence of sulfonate groups and the binding motif of Pb–O were evident from the S 2p and O 1s XPS results (Fig. 2e and S5†) and the typical vibration modes of sulfonate groups.
Fig. 2f shows the FTIR results of the two CsPbI3 NC samples. The wide stretching vibration peak at ∼1555 cm−1 can be assigned to the overlap of the carboxylate (R-COO−) asymmetric stretching vibration and symmetric NH3+ deformation.32 Obviously, partial oleylammonium ions were absorbed on the NC surface. In addition, the symmetric stretching vibration peak at 1411 cm−1 also belongs to R-COO−. The wavenumber difference between the two peaks was in the range of 100–150 cm−1, indicating a bridging or tilted coordination.33 Therefore, compared with the absorption peaks of Cs4PbI6 NCs at 1561 cm−1 and 1545 cm−1, the two CsPbI3 NC samples only show a wide infrared vibration peak at ∼1555 cm−1, and its intensity decreased significantly (Fig. S6†), which is due to the loss of oleylammonium carboxylate.34 Compared with OA-CsPbI3 NCs, the characteristic absorption peaks at 1125 cm−1, 1034 cm−1, and 1003 cm−1 of dumbbell-shaped CsPbI3 NCs are assigned to the stretching vibration of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, while the symmetrical absorption peaks at 1210 cm−1 and 1179 cm−1 belong to the stretching vibration of S–O and have an obvious red-shift compared with free 4-DBSA (Fig. S7†). It is indicative that the strong binding between the sulfonate group and the lead atom passivates the surface defects of the lead–iodine octahedron and promotes the formation of a Pb(SO3−)x-rich surface. Therefore, we speculate that the growth driving force of the self-assembly of complex structures can be provided by the monomer concentration of lead–iodine octahedra in the second nucleation stage with the assistance of the Pb(SO3−)x-rich surface.
O, while the symmetrical absorption peaks at 1210 cm−1 and 1179 cm−1 belong to the stretching vibration of S–O and have an obvious red-shift compared with free 4-DBSA (Fig. S7†). It is indicative that the strong binding between the sulfonate group and the lead atom passivates the surface defects of the lead–iodine octahedron and promotes the formation of a Pb(SO3−)x-rich surface. Therefore, we speculate that the growth driving force of the self-assembly of complex structures can be provided by the monomer concentration of lead–iodine octahedra in the second nucleation stage with the assistance of the Pb(SO3−)x-rich surface.
In order to investigate the self-assembly process of dumbbell-shaped CsPbI3 NCs, we used a two-step nucleation strategy to explore the growth process of CsPbI3 NCs by changing the molar ratio of 4-DBSA to OAm. As previously reported, the two-step nucleation strategy was proposed to prepare highly ordered CsPbBr3 nanoplatelets (NPLs).24 For the CsPbI3 system, the bond energy of Pb–I is weaker than that of Pb–Br, and the instability of its structure will make it more difficult to maintain the dissolution equilibrium of the PbI2 precursor at a high temperature and this eventually precipitates some lead iodide. In addition, the formation energy of the iodide perovskite is relatively low, which makes it easier to promote phase transformation.35 Therefore, by controlling the molar ratio of 4-DBSA to OAm in the precursor, an appropriate remaining monomer concentration during the nucleation stage can be provided to obtain more interesting morphologies and assembly structures for CsPbI3 NCs.36
As schematically shown in Scheme 1, we explore the role of surface-passivated ligands for the in situ self-assembly of CsPbI3 NCs by controlling the molar ratio of 4-DBSA and OAm. When the molar ratio of 4-DBSA and OAm is 2.6, the monomer concentration of large numbers of small nuclei (B1) remaining in the precursor mainly produces dendritic CsPbI3 assembly structures (CsPbI3-S1) with a size distribution of 18.2 nm (Fig. 3a and S8a†), and these are made up of very small nanowires. Surprisingly, the monomer concentration of large nuclei (A1) gradually increases with a decrease of its molar ratio during nucleation, but the monomer concentration of B1 is much greater than that of A1, which promotes the formation of nanosphere CsPbI3 assembly structures (CsPbI3-S2) with a size distribution of 36 nm (Fig. 3b and S8b†). These observations also indicate that many small nuclei in the second-step nucleation stage are helpful in the formation of the assembled structures. From the TEM data and particle size distributions, the as-synthesized assembly structures evolve from many small NPLs. In contrast, when the molar ratio of 4-DBSA and OAm is 0.79, the increase of the monomer concentration of B1 is exactly equal to the decrease of the A1 monomer concentration; four-leaf clover-like NCs (CsPbI3-S3) with a size distribution of 31 nm were prepared (Fig. 3c and S8c†), and these symmetrical assembly structures are composed of large and thick NPLs. Notably, when the molar ratio of 4-DBSA and OAm is less than 0.79, many nuclei can be formed in the precursor solution at 160 °C, which can continue to grow after heating (A2), thus reducing the monomer concentration of the remaining small nuclei in the system (B2). NPL-like structures (CsPbI3-S4) with a size distribution of 30 nm were prepared (Fig. 3d and S8d†). Now, more and more growing nuclei play a dominant role in the self-assembly system. Hence, the production of dumbbell-like CsPbI3 NCs (CsPbI3-S5) is mainly caused by the re-growth of many growing nuclei and small nuclei. Finally, with only a monomer concentration of A2 in the second-step nucleation, CsPbI3 nanorods (CsPbI3-S6) with an uneven size distribution of 61 nm and some small particles left at the head of the nanorods were prepared (Fig. S9†). Therefore, 4-DBSA increases the concentration of growing nuclei in the second-step nucleation stage, and promotes the formation of more Pb–I octahedra, with the size of nuclei determining the size of the assembly unit, forming assembled CsPbI3-S6 NCs of different sizes. In addition, when CsPbI3-Sn NCs were diluted to a certain concentration, the PLQY as measured by the integrating sphere increases gradually (Fig. S10†), which can be attributed to the formation of more Pb–I octahedra. XRD patterns of these assembled structures were recorded. Fig. 3e and S11a† show that the diffraction peaks of CsPbI3-Sn (n = 1, 2, …, 4) and CsPbI3-S6 at 2θ = 14.76° and 28.76° are consistent with the cubic CsPbI3 phase, indicating that they have good growth orientation at the (100) and (200) facets and the same crystal phase. Upon further characterization studies, the bright red CsPbI3 NCs show a sharp PL emission peak at 680 nm with an FWHM of ∼32 nm (Fig. 3f and S11b†). Although CsPbI3-Sn NCs have different assembly structures, the nature of the spectra in their cases remained identical.
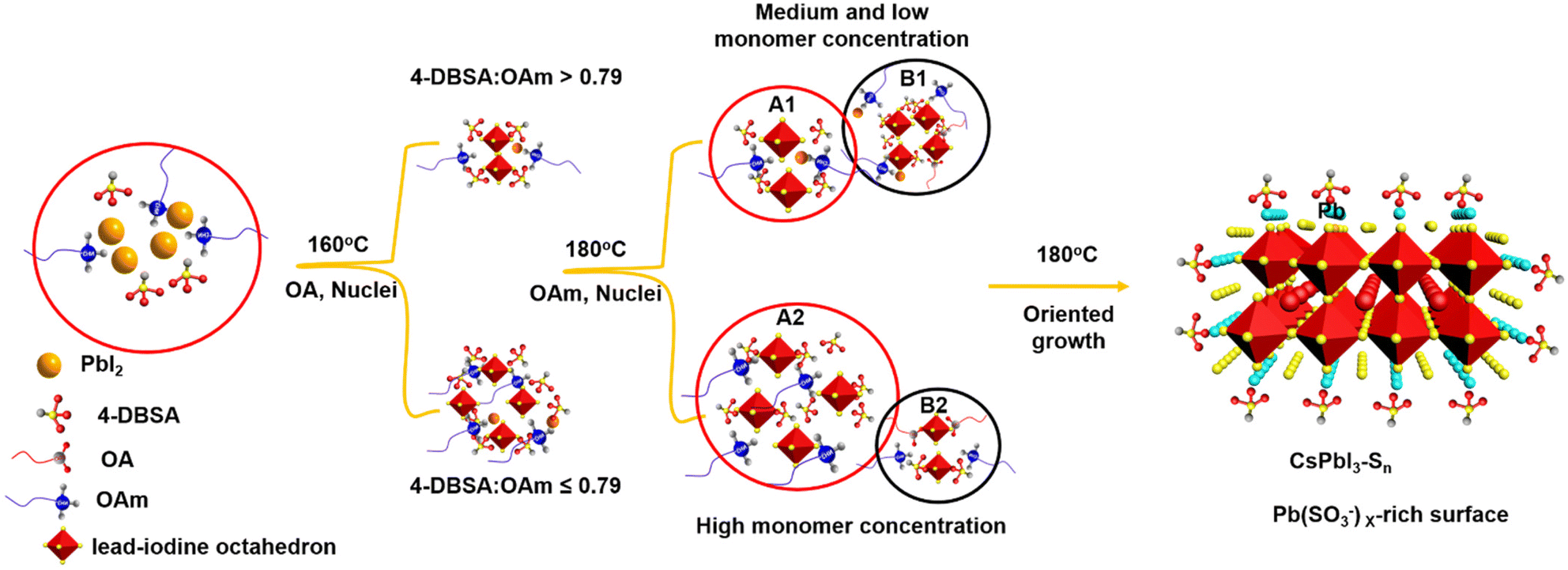 | ||
| Scheme 1 Nucleation and growth process diagrams of diverse assembly morphologies. When the 4-DBSA:OAm molar ratio is greater than 0.79, the first step involves a small amount of nucleation and then growth to form A1; the second step involves a larger amount of nucleation to form B1. When the 4-DBSA:OAm molar ratio is less than 0.79, the first step involves a larger amount of nucleation and then growth to form A2; the second step involves a smaller amount of nucleation to form B2. | ||
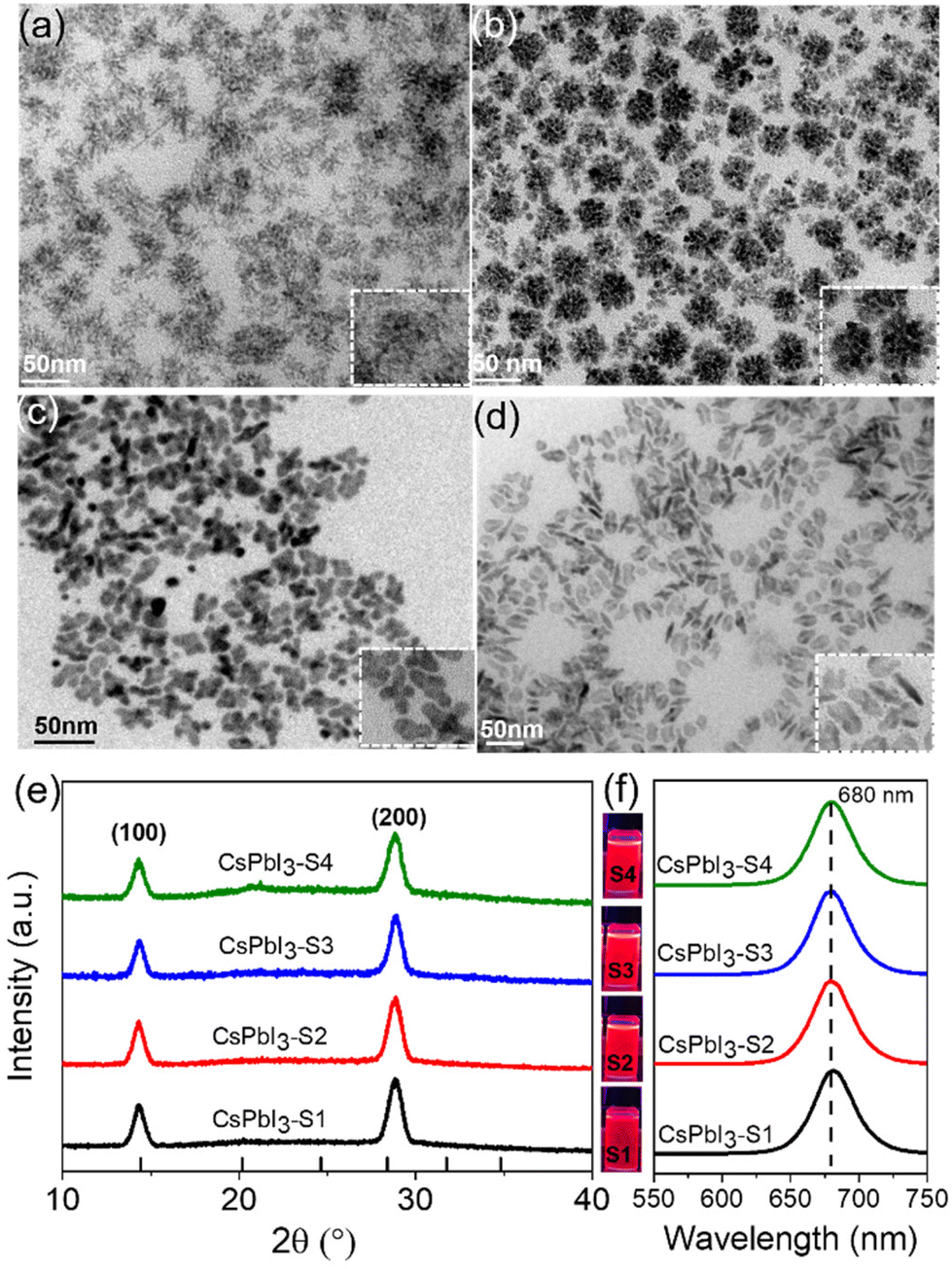 | ||
| Fig. 3 The crystal structure and optical performance of diverse assembly structures. (a)–(d) TEM images, (e) XRD patterns and (f) luminescence photographs (365 nm UV light) and corresponding PL spectra of CsPbI3-Sn NCs by adjusting the molar ratio of 4-DBSA and OAm. | ||
To further confirm the effect of 4-DBSA ligands on the assembly structure of CsPbI3 NCs, we also attempted to synthesize assembled CsPbI3 NCs by adding 4-DBSA ligands during the second nucleation stage, resulting in cubic CsPbI3 NCs with an uneven size distribution, as shown in Fig. S12a.† In addition, only adding 4-DBSA in the first nucleation leads to the formation of mixed morphologies, such as nanowires, nanorods, and nanodots (Fig. S12b†). Based on the above analysis, we found that it is difficult to form the CsPbI3 assembly structure without growing nuclei in the second nucleation stage. Therefore, the monomer concentration of Pb–I octahedra in the second nucleation stage determines the diverse assembly morphologies of CsPbI3 NCs.
This is clear evidence that 4-DBSA promotes the production of a lead–iodine octahedron and also strongly combines with surface lead ions to form a Pb(SO3−)x-rich surface, as shown in Fig. 4. As previously reported, the calculated absorption value (355 nm) of PbI637 was obtained. Fig. 4a shows that the obvious absorption peak at 362 nm is attributed to the PbI64− octahedron in the second nucleation stage. We also found that the concentration of the PbI64− octahedron increased with a decrease of the molar ratio of 4-DBSA and OAm, which may be due to more and more growing nuclei leading to the formation of more PbI64− octahedra. Meanwhile, the results coincide with the growth mechanism of the assembled CsPbI3-Sn as proposed in Scheme 1.
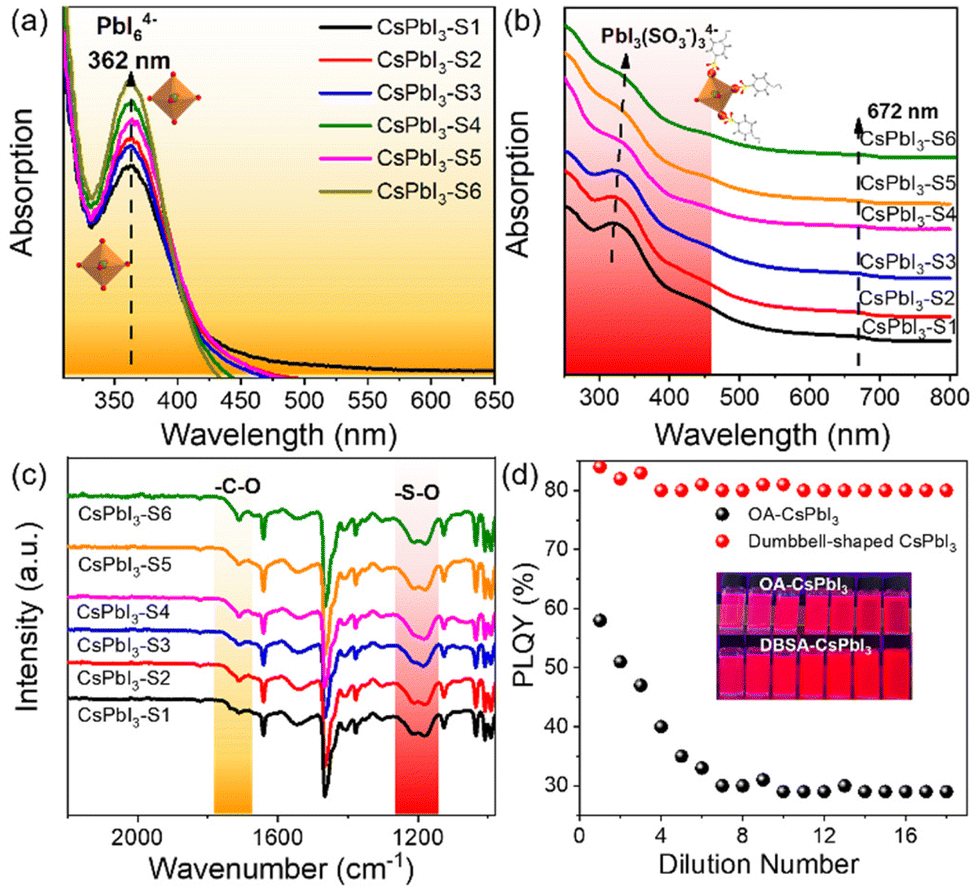 | ||
| Fig. 4 Verification of the role of benzenesulfonate ions. Absorption spectra of (a) the second nucleation precursor solutions containing PbI6 octahedra and (b) CsPbI3-Sn NC solutions containing PbI3(SO3)34−. (c) FTIR spectra of CsPbI3-Sn NCs by controlling the molar ratio of 4-DBSA and OAm; and (d) PLQY variation and luminescence photographs (365 nm UV light) of OA-CsPbI3 NCs and dumbbell-shaped CsPbI3 NCs after gradual dilutions. | ||
To illustrate this point of view, UV-vis absorption spectra and FTIR spectra of CsPbI3-Sn were recorded, as shown in Fig. 4b and c. Fig. 4b shows that CsPbI3-Sn has two obvious absorption peaks: one is the excitonic absorption peak at 672 nm, and the other peak is at 315–337 nm. The changes in UV-vis spectra of CsPbI3-Sn may be attributed to the formation of an intermediate PbIx(OA or 4-DBSA)6−x system.37 Specifically, the absorption peak of CsPbI3-Sn at 315 nm is red-shifted to 337 nm and gradually weakens as the ratio of 4-DBSA to OAm decreases. This may be due to the strong binding between more of the 4-DBSA and lead ions to form PbIx(SO3−)6−x for CsPbI3-Sn, which can be explained by the previously reported characteristics of sulfonates in a perfect perovskite lattice similar to those of natural bromine ions.22 In contrast, Fig. 4c shows that the characteristic adsorption peaks of CsPbI3-Sn in the range of 1000–1300 cm−1 and 1710 cm−1 vary with the molar ratio of 4-DBSA and OAm, which are attributed to the benzenesulfonate group and free carboxylic acid group, respectively. In addition, the asymmetric and symmetric stretching peaks at 1550 cm−1 and 1411 cm−1 are also attributed to the carboxylic acid group. The weak infrared vibration peaks of the carboxylic acid group remained unchanged with a decrease of the molar ratio of 4-DBSA and OAm, indicating that the binding types of the carboxylic acid group as a passivator agent remained unchanged on the perovskite surface. Therefore, the different characteristic adsorption peaks were further studied. With a decrease of the molar ratio of 4-DBSA and OAm, the intensity of the two infrared vibrational peaks is enhanced gradually. This implies that the benzenesulfonate ions replace some of the carboxylate ions and adhere to the surfaces of CsPbI3 NCs, which in turn leads to an increase in the number of free carboxylate ions on the surface. This also confirms that more benzenesulfonate ions combine with surfaces of CsPbI3 NCs, which coincides with a Pb(SO3−)x-rich surface. Fig. 4d shows that a simple dilution experiment provided an intuitive and controllable way to study the binding strength between surface ligands and CsPbI3 NCs. The dependence of the PLQY dilution concentration on halogen vacancy defects has been demonstrated before.38,39 Then, taking CsPbI3-S5 (dumbbell-shaped CsPbI3) as an example, in our system, we mainly analysed the PLQY changes of OA-CsPbI3 and dumbbell-shaped CsPbI3 NCs before and after dilution. We found that the PLQY decreases from 59 ± 5% to 29 ± 5% after excess OA dilution on the surfaces of OA-CsPbI3 NCs (Fig. S13†) and the crimson solution gradually changes to light rose red, while the PLQY and bright red luminescence of dumbbell-shaped CsPbI3 NCs remain basically unchanged. Therefore, the dilution process accelerates the desorption of weakly bound surface ligands, such as oleylammonium–halide ligand pairs, while 4-DBSA ligands remained bound to the surfaces of dumbbell-shaped CsPbI3 NCs.40,41 As a result, the dumbbell-shaped CsPbI3 NCs maintained a high PLQY of 83 ± 5% after gradual dilutions, which is consistent with the strong surface binding of dumbbell-shaped CsPbI3 NCs. In addition, the phase stabilities of OA-CsPbI3 and dumbbell-shaped CsPbI3 were also investigated under atmospheric pressure at room temperature of 25 °C with a humidity of 75%. Fig. S14† shows that the dumbbell-shaped CsPbI3 NCs can maintain its cubic phase and strong fluorescence under 365 nm UV light after one week, while the OA-CsPbI3 NCs transfer from the fluorescent cubic phase to the non-fluorescent orthorhombic phase in two days. Further work should focus on improving the phase stability prior to application in photoelectric devices.
3. Conclusion
In summary, we have provided deep insight into the second nucleation processes involving the in situ self-assembly of perovskite CsPbI3 NCs. A facile two-step nucleation method was employed to prepare high-quality dumbbell-shaped CsPbI3 NCs with a high PLQY and good crystallinity by controlling the monomer concentration of lead–iodine octahedra under high-temperature conditions. In differing from OA-CsPbI3 NCs, the strong binding between benzenesulfonate anions and lead atoms induces the formation of a Pb(SO3−)x-rich surface on dumbbell-shaped CsPbI3 NCs, which is further confirmed by XPS and FTIR data. Furthermore, the as-synthesized CsPbI3-Sn NCs with the same crystal phase and luminescence properties exhibit different assembly morphologies by controlling the molar ratio of 4-DBSA and OAm in the nucleation process. This indicates that the self-assembly of CsPbI3 NCs is related to the concentration of lead–iodine octahedra during nucleation, which is confirmed by adsorption data and FTIR spectra. The dumbbell-shaped CsPbI3 NCs have a high PLQY and solution stability after repeated dilutions due to their Pb(SO3−)x-rich surfaces. Based on this work, we believe that these diverse assembly structures can provide new ideas for assembly mechanisms and optoelectronic applications of perovskite NCs.4. Experimental section
4.1. Synthesis of OA-CsPbI3 NCs
PbI2 (0.54 mmol, 0.1242 g), ODE (7.5 mL), OAm (1.5 mL), and OA (2.36 mmol) were loaded into a 100 mL 3-neck flask and degassed for half an hour at 120 °C under an Ar flow. The temperature was increased to 160 °C under an Ar atmosphere. The preheated Cs-oleate solution (0.75 mL, 0.033 mmol) was swiftly injected into the transparent precursor solution. After 5 seconds, the reaction mixture was cooled down in an ice bath.4.2. Synthesis of dumbbell-shaped CsPbI3 NCs (CsPbI3-S5 NCs)
PbI2 (0.54 mmol, 0.1242 g), ODE (7.5 mL), and a 0.66 molar ratio of 4-DBSA to OAm (2.37 mol:3.6 mol) were loaded into a 100 mL 3-neck flask and degassed for half an hour at 120 °C under an Ar flow. The temperature was increased to 160 °C and OA (1.5 mL) was injected into the 3-neck flask, which was then heated up to 180 °C under an Ar atmosphere. With the temperature maintained at 180 °C, OAm (0.91 mol) was added into the reaction solution. The preheated Cs-oleate solution (0.75 mL, 0.033 mmol) was then injected into the transparent precursor solution swiftly. After 5 seconds, the reaction mixture was cooled down in an ice bath.
4.3. Purification of CsPbI3 NCs
Ethyl acetate was added into the crude solution at a volume ratio of 1:3 and the mixture was centrifuged for 1 min at 9000 rpm. The precipitate was dispersed in 1.5 mL of hexane to obtain a clear solution.
Author contributions
D. Yang conceived the idea for the study and designed the experiments. D. Yang, X. Zhang and S. Liu carried out the synthesis and characterization of NCs. The manuscript was mainly written by D. Yang, and revised by H. Zeng, Q. Xu, and Z. Xu. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (62204215 and 12275132) and Natural Science Research program of Jiangsu Higher Education Institutions of China (22KJB430012). The Yangzhou University Testing Center is also acknowledged for conducting the XPS and XRD characterization studies.References
- M. C. Brennan, S. Toso, I. M. Pavlovetc, M. Zhukovskyi, S. Marras, M. Kuno, L. Manna and D. Baranov, ACS Energy Lett., 2020, 5, 1465–1473 CrossRef CAS.
- D. Yang, Y. Zou, P. Li, Q. Liu, L. Wu, H. Hu, Y. Xu, B. Sun, Q. Zhang and S.-T. Lee, Nano Energy, 2018, 47, 235–242 CrossRef CAS.
- C. Zhou, J. M. Pina, T. Zhu, D. H. Parmar, H. Chang, J. Yu, F. Yuan, G. Bappi, Y. Hou, X. Zheng, J. Abed, H. Chen, J. Zhang, Y. Gao, B. Chen, Y. K. Wang, H. Chen, T. Zhang, S. Hoogland, M. I. Saidaminov, L. Sun, O. M. Bakr, H. Dong, L. Zhang and E. H. Sargent, Adv. Sci., 2021, 8, 2101125 CrossRef CAS PubMed.
- C. Zhou, Y. Zhong, H. Dong, W. Zheng, J. Tan, Q. Jie, A. Pan, L. Zhang and W. Xie, Nat. Commun., 2020, 11, 329 CrossRef CAS PubMed.
- J.-S. Benas, F.-C. Liang, W.-C. Chen, C.-W. Hung, J.-Y. Chen, Y. Zhou, S.-T. Han, R. Borsali and C.-C. Kuo, Chem. Eng. J., 2021, 431, 133701 CrossRef.
- G. Raino, M. A. Becker, M. I. Bodnarchuk, R. F. Mahrt, M. V. Kovalenko and T. Stoferle, Nature, 2018, 563, 671–675 CrossRef CAS PubMed.
- M. Cirignano, S. Fiorito, M. Barelli, V. Aglieri, M. De Franco, H. Bahmani Jalali, A. Toma and F. Di Stasio, Nanoscale, 2022, 14, 15525 RSC.
- J. Pan, X. Li, X. Gong, J. Yin, D. Zhou, L. Sinatra, R. Huang, J. Liu, J. Chen, I. Dursun, A. M. El-Zohry, M. I. Saidaminov, H.-T. Sun, O. F. Mohammed, C. Ye, E. H. Sargent and O. M. Bakr, Angew. Chem., Int. Ed., 2019, 58, 16077–16081 CrossRef CAS.
- H. Huang, M. W. Feil, S. Fuchs, T. Debnath, A. F. Richter, Y. Tong, L. Wu, Y. Wang, M. Döblinger and B. Nickel, Chem. Mater., 2020, 32, 8877–8884 CrossRef CAS.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS.
- V. M. Burlakov, Y. Hassan, M. Danaie, H. J. Snaith and A. Goriely, J. Phys. Chem. Lett., 2020, 11, 6535–6543 CrossRef CAS.
- S. Bera, S. Shyamal and N. Pradhan, J. Am. Chem. Soc., 2021, 143, 14895–14906 CrossRef CAS PubMed.
- D. Baranov, A. Fieramosca, R. X. Yang, L. Polimeno, G. Lerario, S. Toso, C. Giansante, M. Giorgi, L. Z. Tan, D. Sanvitto and L. Manna, ACS Nano, 2021, 15, 650–664 CrossRef CAS PubMed.
- R. Grisorio, F. Fasulo, A. B. Munoz-Garcia, M. Pavone, D. Conelli, E. Fanizza, M. Striccoli, I. Allegretta, R. Terzano, N. Margiotta, P. Vivo and G. P. Suranna, Nano Lett., 2022, 22, 4437–4444 CrossRef PubMed.
- A. R. Brill, M. K. Kuntumalla, G. de Ruiter and E. Koren, ACS Appl. Mater. Interfaces, 2020, 12, 33941–33949 CrossRef CAS.
- Y. Tang, L. Gomez, A. Lesage, E. Marino, T. E. Kodger, J. M. Meijer, P. Kolpakov, J. Meng, K. Zheng, T. Gregorkiewicz and P. Schall, Nano Lett., 2020, 20, 5997–6004 CrossRef CAS.
- H. Li, X. Liu, Q. Ying, C. Wang, W. Jia, X. Xing, L. Yin, Z. Lu, K. Zhang, Y. Pan, Z. Shi, L. Huang and D. Jia, Angew. Chem., Int. Ed., 2020, 59, 17207–17213 CrossRef CAS.
- F. Krieg, P. C. Sercel, M. Burian, H. Andrusiv, M. I. Bodnarchuk, T. Stoferle, R. F. Mahrt, D. Naumenko, H. Amenitsch, G. Raino and M. V. Kovalenko, ACS Cent. Sci., 2021, 7, 135–144 CrossRef CAS PubMed.
- A. A. M. Brown, T. J. N. Hooper, S. A. Veldhuis, X. Y. Chin, A. Bruno, P. Vashishtha, J. N. Tey, L. Jiang, B. Damodaran, S. H. Pu, S. G. Mhaisalkar and N. Mathews, Nanoscale, 2019, 11, 12370–12380 RSC.
- S. Bera, R. K. Behera and N. Pradhan, J. Am. Chem. Soc., 2020, 142, 20865–20874 CrossRef CAS PubMed.
- Y. Yang, J. T. Lee, T. Liyanage and R. Sardar, J. Am. Chem. Soc., 2019, 141, 1526–1536 CrossRef CAS.
- J. Hao, X. Qu, L. Qiu, G. Li, Y. Wei, G. Xing, H. Wang, C. Yan, H. S. Jang, Z. Cheng and J. Lin, Adv. Opt. Mater., 2018, 4, 1801323 CrossRef.
- S. Wang, J. Yu, M. Zhang, D. Chen, C. Li, R. Chen, G. Jia, A. L. Rogach and X. Yang, Nano Lett., 2019, 19, 6315–6322 CrossRef CAS PubMed.
- D. Yang, X. Li, Y. Li, B. Cai, L. Su, S. Zhang, Z. Juan, C. Meng, D. Geng, J. Chen, F. Xu and H. Zeng, Nano Res., 2022, 15, 502–509 CrossRef CAS.
- D. Yang, X. Li, W. Zhou, S. Zhang, C. Meng, Y. Wu, Y. Wang and H. Zeng, Adv. Mater., 2019, 31, 1900767 CrossRef.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS.
- R. Grisorio, E. Fanizza, M. Striccoli, D. Altamura, C. Giannini, I. Allegretta, R. Terzano and G. P. Suranna, ChemNanoMat, 2020, 6, 356–361 CrossRef CAS.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- D. Yang, X. Li, Y. Wu, C. Wei, Z. Qin, C. Zhang, Z. Sun, Y. Li, Y. Wang and H. Zeng, Adv. Opt. Mater., 2019, 7, 1900276 CrossRef.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427–2433 CrossRef CAS.
- J. Zhang, H. Zhang, W. Cao, Z. Pang, J. Li, Y. Shu, C. Zhu, X. Kong, L. Wang and X. Peng, J. Am. Chem. Soc., 2019, 141, 15675–15683 CrossRef CAS.
- J. De Roo, M. Ibanez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Koyalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS.
- X. G. Peng, Adv. Mater., 2003, 15, 459–463 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS.
- M. I. Bodnarchuk, S. C. Boehme, S. ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef CAS.
- B. Zhang, L. Goldoni, J. Zito, Z. Dang, G. Almeida, F. Zaccaria, J. de Wit, I. Infante, L. De Trizio and L. Manna, Chem. Mater., 2019, 31, 9140–9147 CrossRef CAS.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819–824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, absorbance and PL spectra, XRD patterns, TEM images, FTIR spectra, and XPS spectra. See DOI: https://doi.org/10.1039/d2nr06208a |
| This journal is © The Royal Society of Chemistry 2023 |