
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanotubes from bacteriophage tail sheath proteins: internalisation by cancer cells and macrophages†
Dovydas
Gabrielaitis‡
 a,
Vilmante
Zitkute‡
b,
Lina
Saveikyte
a,
Greta
Labutyte
c,
Martynas
Skapas
d,
Rolandas
Meskys
c,
Vida
Casaite
c,
Ausra
Sasnauskiene§
*b and
Urte
Neniskyte§
*ae
a,
Vilmante
Zitkute‡
b,
Lina
Saveikyte
a,
Greta
Labutyte
c,
Martynas
Skapas
d,
Rolandas
Meskys
c,
Vida
Casaite
c,
Ausra
Sasnauskiene§
*b and
Urte
Neniskyte§
*ae
aDepartment of Neurobiology and Biophysics, Institute of Biosciences, Life Sciences Center, Vilnius University, Vilnius, Lithuania. E-mail: urte.neniskyte@gmc.vu.lt
bDepartment of Biochemistry and Molecular Biology, Institute of Biosciences, Life Sciences Center, Vilnius University, Vilnius, Lithuania. E-mail: ausra.sadauskaite@gf.vu.lt
cDepartment of Molecular Microbiology and Biotechnology, Institute of Biochemistry, Life Sciences Center, Vilnius University, Vilnius, Lithuania
dInstitute of Biotechnology, Vilnius University, Vilnius, Lithuania
eVU-EMBL Partnership Institute, Vilnius University, Vilnius, Lithuania
First published on 7th June 2023
Abstract
Bionanoparticles comprised of naturally occurring monomers are gaining interest in the development of novel drug transportation systems. Here we report on the stabilisation, cellular uptake, and macrophage clearance of nanotubes formed from the self-assembling gp053 tail sheath protein of the vB_EcoM_FV3 bacteriophage. To evaluate the potential of the bacteriophage protein-based nanotubes as therapeutic nanocarriers, we investigated their internalisation into colorectal cancer cell lines and professional macrophages that may hinder therapeutic applications by clearing nanotube carriers. We fused the bacteriophage protein with a SNAP-tag self-labelling enzyme and demonstrated that its activity is retained in assembled nanotubes, indicating that such carriers can be applied to deliver therapeutic biomolecules. Under physiological conditions, the stabilisation of the nanotubes by PEGylation was required to prevent aggregation and yield a stable solution with uniform nano-sized structures. Colorectal carcinoma cells from primary and metastatic tumours internalized SNAP-tag-carrying nanotubes with different efficiencies. The nanotubes entered HCT116 cells via dynamin-dependent and SW480 cells – via dynamin- and clathrin-dependent pathways and were accumulated in lysosomes. Meanwhile, peritoneal macrophages phagocytosed the nanotubes in a highly efficient manner through actin-dependent mechanisms. Macrophage clearance of nanotubes was enhanced by inflammatory activation but was dampened in macrophages isolated from aged animals. Altogether, our results demonstrate that gp053 nanotubes retained the cargo's enzymatic activity post-assembly and had the capacity to enter cancer cells. Furthermore, we emphasise the importance of evaluating the nanocarrier clearance by immune cells under conditions mimicking a cancerous environment.
Introduction
Complex protein nanostructures, due to their unique physical and biological properties, provide numerous possibilities for novel targeted drug delivery systems1,2 and are exceptionally popular because they are easy to manufacture, modify, and can self-assemble after expression.3–6 Naturally occurring proteins that self-assemble into higher-order complexes can offer a valuable source of biocompatible building blocks for nanostructure development. Bacteriophages and other viruses are examples of biological structures that are comprised of monomeric proteins, which orderly assemble into homogeneous nanometre-sized super-structures. Such naturally occurring nanostructures have controllable size and orientation and can be applied in nanobiotechnology as filamentous particles (from M13 bacteriophage),7,8 polysheaths (from T4 bacteriophage),9 or spheric capsids (from Qβ bacteriophage).10 The geometry and surface modifications of protein-based nanostructures can easily be altered genetically; therefore, protein-based self-assembling nanoparticles arise as a sought-after competitor, compared to conventional inorganic nanostructures.Importantly, nanostructures composed of proteins can potentially be applied in drug targeting and delivery. Compared to metal or polymer-based nanoparticles, nanocarriers composed of proteins are attracting increasing interest,11 because of their biocompatibility, efficient biodegradability, and controllable drug release.12 Conventional pharmacology requires the use of high doses of bioactive molecules, which inevitably cause side effects by interacting with cells that are not the target of interest.13 To minimize unwanted side effects of systemic therapy, the drug of interest should be shielded from degradation and released from the carrier only in cancerous tissue in a dosage-controlled manner.14
One of the rapidly growing areas of the application of nanocarriers as drug transport systems is targeted chemotherapy for oncological pathologies.15 Cancerous tissue possesses morphologically abnormal vasculature, which can be passively penetrated by nanosized structures, whereas in healthy vasculature, the nanoparticles cannot penetrate the vessels. Prior to the accumulation in cancerous tissue, while traveling from the point of admission (intravenous, intramuscular, intradermal injections, or orally) the nanoparticles can be cleared by resident macrophages, competing with the uptake by cancer cells.16 To minimise macrophage clearance, surface modifications can be applied to the nanocarrier like PEGylation17 or encapsulation in a cell membrane containing CD47.18,19 After successfully accumulating in pathological tissue, the nanoparticles can be used not only as a drug carrier but also as an imaging marker, as most possess specific photo-physical capabilities.20 When in cancerous tissue, the drug of interest needs to be internalised into the cells to enable the treatment, yet some pharmaceuticals are cell impermeable. Therefore, packing them into nanosized carriers that can permeate the plasma membrane can increase the drug uptake, and decrease the required dosage.21 Nanoparticle uptake mechanisms vary depending on particle size,22 surface modifications,23 and the ability to self-opsonise by serum proteins or form a protein layer around the nanoparticle.24
Here, we present the application of the chimeric self-assembling nanotubes made of the tail sheath protein gp053 of the bacteriophage vB_EcoM_FV3.25 To determine if the fusion of an enzymatic protein to the C-terminus of gp053, which is oriented towards the inner cavity of the nanotube, sustains its enzymatic activity, the self-labelling SNAP-tag protein was used to obtain 053SNAP nanotubes. Labelling of the chimeric nanotubes with fluorescent SNAP-tag substrates proved that the enzymes fused to gp053 retained the enzymatic activity and were suitable to track the intracellular localisation of the nanotubes. To demonstrate the potential of 053SNAP nanotubes to target cancer cells, we evaluated the internalisation mechanism of 053SNAP by colorectal cancer cells from primary and metastatic tumours. In addition, we assessed the clearance of 053SNAP nanotubes by peritoneal macrophages and evaluated how it is modulated by the age of the host and macrophage inflammatory state.
Results and discussion
Characterisation of self-assembled nanotubes from chimeric bacteriophage tail sheath protein
To characterise the nanostructures formed by the gp053 protein, we fused a SNAP-tag to the C-terminus of gp053,26 allowing us to estimate their structure in solution and intracellular localisation in in vitro studies via fluorescence microscopy. Although the sequences of the bacteriophage tail sheath proteins are diverse, their structural and modelling data indicate that they share a conserved core and are assembled according to the same structural architecture, with both termini of the protein pointing towards the inside of the tube.27,28 This structural homology, together with the AlphaFold 2 (ref. 29) modelling and assembly visualization on Chimera 1.13.1,30 suggests that the C-terminus of the gp053 protein is located inside the tube, directing the fused SNAP-tag to the inside of the tube (Fig. S1†). The transmission electron microscopy (TEM) and high-performance liquid chromatography (HPLC) analysis of the purified protein structures revealed that SNAP-tag modification did not block the protein's ability to form polymeric tubular structures (Fig. 1A and S2†). In addition, the SNAP-tag allowed the nanotubes to be observed under a fluorescence microscope after labelling with a fluorescent tag consisting of SNAP-specific benzylguanine-derived substrate conjugated with Alexa Fluor 488 fluorescent dye (Fig. 1B). The successful labelling of the SNAP-tag proved that after the folding into a tubular structure, the enzymatic activity of the fused protein is retained, suggesting that after the nanoparticles are purified, they can be used to carry intact and active enzymes. Our recent study has shown that the fusion of an amidohydrolase YqfB to a nanotube forming monomer 041Δ200 resulted in a 3-fold decrease in enzymatic activity.31 Therefore, at least a partial impairment in the activity of SNAP-tag within the nanotubes can be anticipated, but there was sufficient binding of the fluorescent enzyme substrate to observe the nanotubes within the cells.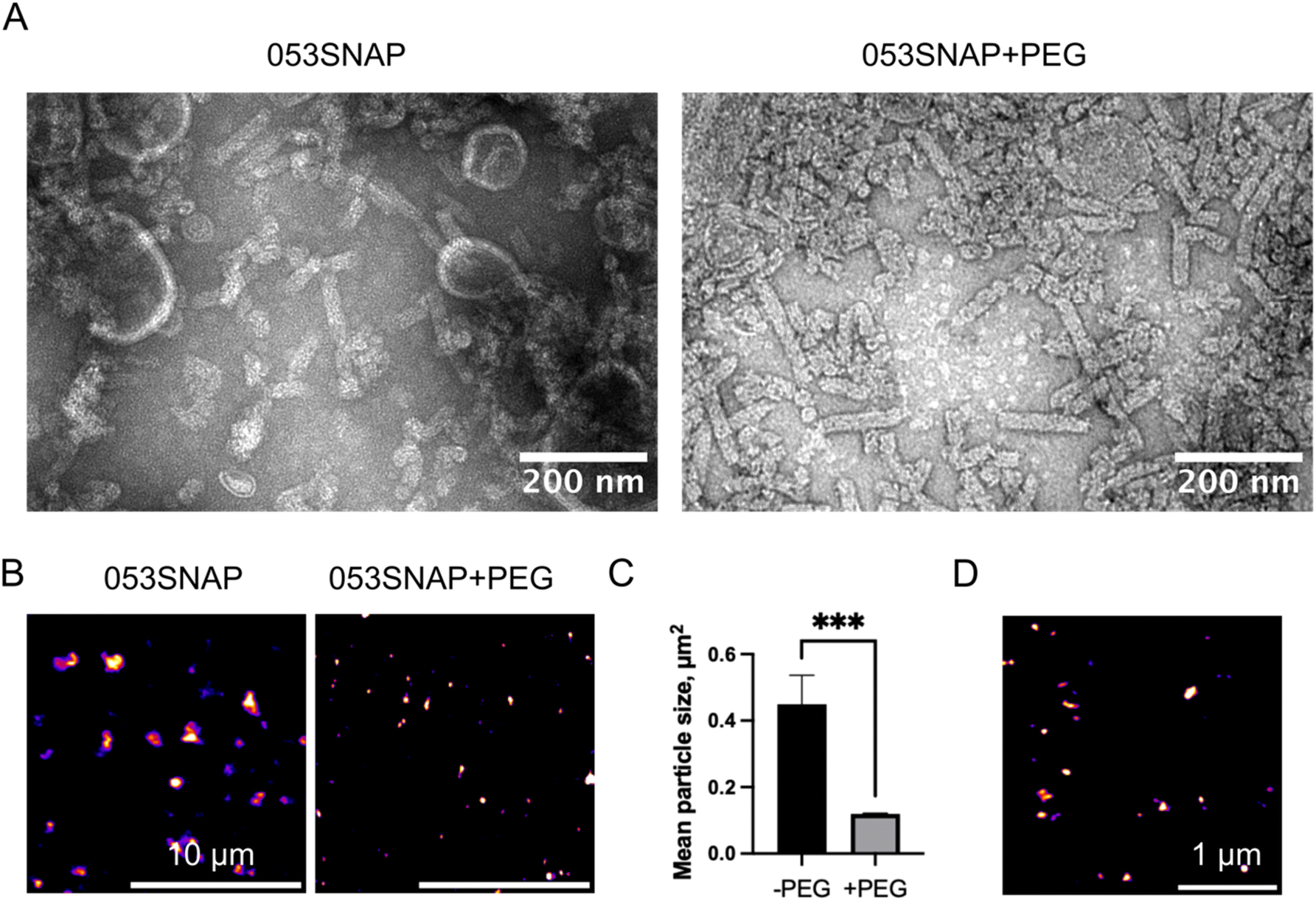 | ||
| Fig. 1 Size evaluation of 053SNAP nanotubes. (A) Transmission electron microscopy images of 053SNAP self-assembled structures without (left) and with (right) PEG functionalisation; scale bar 200 nm. (B) Confocal images of 053SSNAP particles without (left) and with (right) PEG functionalisation; scale bar 10 μm. (C) The size of 053SNAP particles with and without PEG functionalisation, measured in confocal images. (D) Stimulated emission depletion (STED) micrograph of PEG-stabilised 053SNAP particles conjugated with SNAP-Cell® 647-SiR; scale bar 1 μm. Data are represented as means ± sem; ***p < 0.001. | ||
The retention of enzymatic activity in nanostructures formed by fused recombinant proteins was also demonstrated in our previous study with a bacteriophage tail sheath protein derived from the vB_KleM-RaK2 bacteriophage,31 confirming that proteinaceous nanotubes are a suitable carrier for enzymes.
To test if the self-assembled nanotubes can be applied to eukaryotic cells under physiological conditions, the stability of the nanotubes was evaluated in a common cell culture medium (Dulbecco's Modified Eagle Medium (DMEM) with 10% of fetal bovine serum). The nanotubes were found to aggregate when diluted in cell culture medium (Fig. 1B). To limit this aggregation, the nanotubes were stabilised by PEGylation. PEGylation significantly reduced the aggregation of nanotubes diluted in cell culture medium, as demonstrated by confocal microscopy image analysis, showing a 4-fold decrease in nanostructures' area (Fig. 1B and C). As the size of nanotubes was expected to be beyond the diffraction limit of light microscopy, we then used super-resolution imaging to measure the size of the resulting nanostructures. The 053SNAP particles were conjugated with the SNAP-Cell 647-SiR fluorescent probe and imaged with a stimulated emission depletion (STED) system (Fig. 1D). The particles' size estimated from the STED images was 131 ± 11 nm in diameter, indicating that PEGylated nanotubes retained their distinct size under physiological conditions.
The ultrastructure of the purified 053SNAP nanotubes and the effect of PEGylation were further evaluated using TEM. We have previously found that the unmodified gp053 protein forms polysheaths of up to 1000 nm in length.25 Interestingly, we found that the fusion of the SNAP-tag resulted in the formation of up to 10-fold shorter tubular structures, significantly reducing their size (Fig. 1A). TEM analysis showed that gp053 protein fused to SNAP-tag formed nanotubes ranging from 35 to 126 nm in length, averaging 68 ± 26 nm in length and 27 ± 1 nm in width, which is almost 2-fold smaller compared to the size obtained by STED (Fig. 1D), suggesting that PEGylation reduced nanotube aggregation, but did not abrogate it completely. The decrease in length caused by the fusion of a protein was also reported in our previous work, where the size of the nanotubes formed by gp39 sheath protein from the vB_EcoS_NBD2 bacteriophage with an EGFP molecule fused to its C-terminus were 3-fold shorter than the nanotubes formed by the wild-type protein.32 Importantly, in the TEM micrographs, 053SNAP particles that were PEGylated did not differ from non-modified ones, indicating that PEGylation did not affect the self-assembly of the nanotubes (Fig. 1A). To evaluate the size and structure in solution more thoroughly, single molecule localisation microscopy (STORM, PALM) or AFM could be applied in the future.
The internalisation of 053SNAP particles by colorectal cancer cells
Following the examination of the structure and stability of 053SNAP chimeric protein nanotubes, we aimed to evaluate their feasibility as nanocarriers for delivery into cancer cells. The ability of nanotubes to enter cancer cells was studied using colorectal cancer cell lines HCT116, SW480, and SW620. In this study, the SW480 and SW620 paired cell lines were used for the comparison of nanotubes' entry into cells derived from the primary tumour (SW480) and lymph node metastasis (SW620), both derived from the same patient. In addition, HCT116 cells from a different patient were used, as they have different cancer-driver genes' mutation status than SW480 and SW620 cells. Although all three cell lines possess mutated KRAS, only SW480 and SW620 cells have mutant TP53, while HCT116 cells have mutant PIK3CA.33 The protein encoded by PIK3CA is directly involved in endocytic processes34 and may therefore modify the uptake of nanoparticles. Moreover, recent findings suggest that endocytic trafficking also can be influenced by mutant p53.35 Therefore, to assess 053SNAP nanotube uptake, we have chosen cell lines having distinct origins and genetic features that may lead to differences in endocytic activity.To evaluate nanotubes' ability to enter cancer cells, the PEGylated nanotubes composed of 053SNAP proteins labelled with Alexa 488 were added to the cell culture medium at a concentration of 50 μg ml−1. We first investigated the uptake of nanotubes into HCT116 cells. The fluorescence specific to nanotubes was registered using confocal microscopy after 1, 2, 4, 6, and 20 h of incubation with nanotubes. Nanotubes' internalisation was first noticed after 4 h incubation with 053SNAP. The internalized nanotubes accumulated with time, and after 20 h of incubation the median of internalised 053SNAP fluorescence was almost 5-fold higher compared to the 4 h timepoint (p < 0.0001) (Fig. 2B). After 20 h of incubation, the nanotubes colocalised with acidic organelles (lysosomes) stained with LysoTracker, indicating the uptake of nanotubes into the endolysosomal system. The median of Pearson's coefficient after 4 h incubation was 0.042 and after 20 h of incubation, it increased to 0.348 (p < 0.001) (Fig. 2C), implying that the longer incubation with nanotubes favours their accumulation into lysosomes.
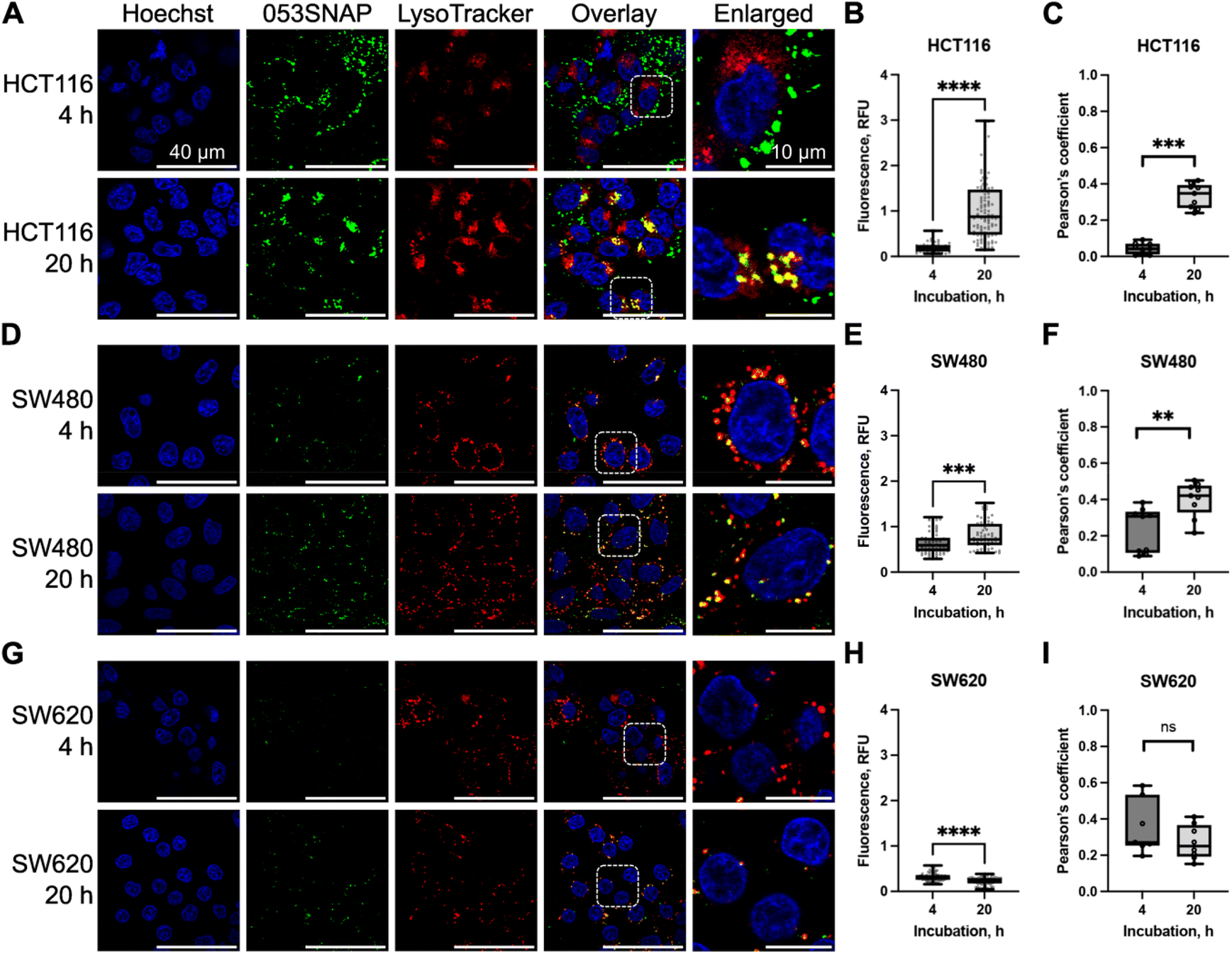 | ||
| Fig. 2 The internalisation of 053SNAP and its colocalization with lysosomes in human colorectal cancer cell lines: HCT116 (A–C), SW480 (D–F), and SW620 (G–I). (A, D and G) Fluorescence microscopy images representing nuclei (blue, Hoechst33342), 053SNAP (green), and lysosomes (red, LysoTracker). Scale bar 40 and 10 μm for enlarged images. (B, E and H) 053SNAP-specific integrated fluorescence intensity. (C, F and I) Pearson's coefficient measuring 053SNAP colocalization with lysosomes. Data are represented as the minimum value, first quartile, median, third quartile, and the maximum value, every dot presents one cell (B, E and H) or one image (C, F and I). **/***/****p < 0.01/0.001/0.0001; ns – non-significant. | ||
We then compared the internalisation of nanotubes into HCT116 and SW480 cells, both of which are derived from primary tumours. The fluorescence signal of nanotubes within SW480 cells was higher after 4 h of incubation with 053SNAP but lower after 20 h compared to HCT116 cells. The median of fluorescence was 0.59 at 4 h after incubation and increased to 0.718 at 20 h (p < 0.001) (Fig. 2D and E). SW480 cells efficiently targeted nanotubes to the lysosomal compartment, as the median of Pearson's coefficient for 053SNAP colocalization with lysosomes was 0.307 after 4 h and increased up to 0.421 after 20 h of incubation (p < 0.01) (Fig. 2F). Despite differences in the mutations in the cancer-driver genes TP53 and PIK3CA, which can affect endocytic activity, HCT116 and SW480 cell lines accumulated nanoparticles at similar levels.
Finally, we assessed the accumulation of 053SNAP in metastatic SW620 cells. In contrast to HCT116 and SW480 cells, nanotubes did not accumulate over time in SW620 cells. The median of fluorescence after 4 and 20 h of incubation were 0.298 and 0.246, respectively (Fig. 2G and H). Furthermore, the colocalization of nanotubes with lysosomes in the SW620 cell line was lower than in other cell lines and did not change after a longer incubation period: the median of the Pearson's coefficient after 4 and 20 h of incubation was 0.271 and 0.25, respectively (Fig. 2I). Reduced nanotube uptake by SW260 cells could be explained by the lower endocytic activity of metastatic cells compared to primary tumour cells (SW480 or HCT116). Despite the identical genotype, the SW620 cell line internalized a lower amount of 053SNAP than the SW480 cell line. Differences between SW620 and SW480 cells were identified at the epigenetic level,33 which may suggest the cause of difference in 053SNAP internalisation. Endocytosis is tightly linked to cell migration which is crucial for metastatic processes36 and the cells derived from metastases can bear changes in endocytic processes. These results highlight the importance of evaluating potential drug carriers in both primary and metastatic tumour cells to assess their potential to be used in targeted therapy. The incubation with 053SNAP did not change cell viability up to 48 h of incubation (Fig. S4†). In addition, even high doses (up to 200 μg ml−1) of 053SNAP did not induce any changes in cell shape or nucleus morphology (Fig. S5†). This suggests that the nanotubes do not exhibit any short-term cytotoxic effect on cancer cells, and thus should not cause any unwanted side-effects.
Clathrin and dynamin-2 are required for nanotubes' internalisation into colorectal cancer cells
To define the molecular pathways involved in the internalisation of 053SNAP particles by colorectal cancer cells, we have investigated the importance of clathrin and dynamin for the endocytosis of nanotubes. HCT116 and SW480 cells were chosen for this analysis since nanotubes efficiently accumulated only in these two cell lines. To interfere with clathrin-dependent endocytosis in HCT116 cells, we downregulated clathrin heavy chain (CLHC) and dynamin-2 (DNM) proteins using specific siRNAs, by 60% and 70%, respectively (Fig. S5A†). The downregulation of CLHC did not change the accumulation of nanotubes in HCT116 cells (Fig. 3A and B). The median of integrated fluorescence density using non-targeting (NT) siRNA was 0.230 and the median after CLHC silencing was 0.185. In contrast, the reduction of DNM almost completely prevented the internalisation of nanotubes in HCT116 cells (Fig. 3A and B). The median of integrated fluorescence density after DNM silencing was reduced to 0.015 (p < 0.0001), indicating that HCT116 cells internalize 053SNAP nanotubes in a clathrin-independent, but dynamin-dependent manner.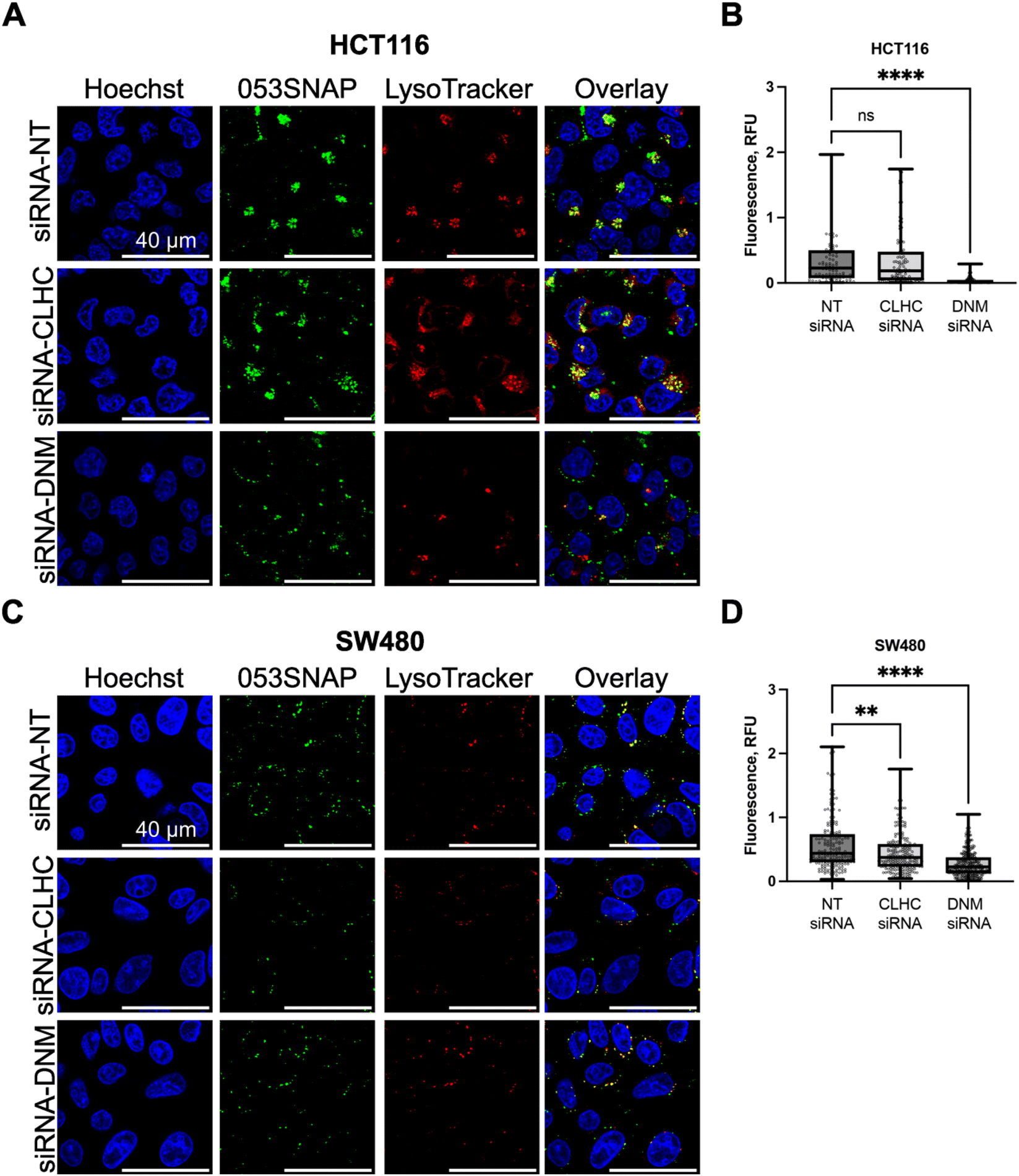 | ||
| Fig. 3 The internalisation of 053SNAP after the silencing of clathrin heavy chain (CLHC) and dynamin-2 (DNM) with specific siRNAs in HCT116 (A and B) and SW480 (C and D) cells. (A and C) Fluorescence microscopy images representing nuclei (blue, Hoechst33342), 053SNAP (green) and lysosomes (red, LysoTracker); scale bar 40 μM. (B and D) 053SNAP-specific integrated fluorescence intensity. Data are represented as the minimum value, first quartile, median, third quartile, and the maximum value, every dot presents integrated fluorescence density in one cell. **/****p < 0.01/0.0001; ns – non-significant. | ||
The silencing of CLHC or DNM by specific siRNAs in SW480 cells reduced their protein levels by 90% and 80%, respectively (Fig. S5B†). CLHC or DNM silencing downregulated 053SNAP nanotube internalisation into SW480 cells (Fig. 3C and D). The median of integrated fluorescence density using NT siRNA was 0.439 and it was significantly reduced to 0.373 after CLHC silencing (p < 0.001) and up to 0.227 after DNM silencing (p < 0.0001) (Fig. 3C and D). These results indicated that uptake of nanotubes by SW480 cells relied on clathrin–dynamin-dependent endocytosis as well as another, likely passive, mechanism.
Interestingly, while both HCT116 and SW480 cells internalised 053SNAP in a dynamin-dependent manner, only SW480 cell uptake of 053SNAP was hindered by clathrin silencing. Even though clathrin-mediated endocytosis is one of the main entry mechanisms of various nanoparticles,37,38 there are several clathrin-independent and dynamin-dependent endocytic pathways: caveolae-mediated endocytosis, Rho/Rac pathway, and fast endophilin mediated endocytosis (FEME).39 Pinocytosis could also be used for the 053SNAP entry into HCT116 cells, as it can be dynamin-dependent and several studies report the uptake of various nanoparticles by pinocytosis.40,41
Clearance of 053SNAP nanotubes by peritoneal macrophages
The therapeutic efficacy of targeted carriers depends not only on the efficient uptake by target cells but also on competing clearance by various immune cells in the body. One of the major phagocytes contributing to the clearance of drug carriers for intraperitoneal organs are peritoneal macrophages. Peritoneal macrophages are immune cells, which reside in the peritoneal cavity and clear apoptotic and necrotic cells, cancerous cells, and foreign bodies.42,43 To estimate the interference of the macrophage with the nanotube therapy in the peritoneal cavity, we evaluated whether and how 053SNAP nanotubes are eliminated by primary peritoneal macrophages isolated from mice. Macrophages isolated from young animals (2–4 months old) efficiently internalised 053SNAP (Fig. 4A). As the internalisation of nanoparticles may cause cytotoxic side-effects,44 we evaluated whether internalised 053SNAP lead to macrophage apoptosis and/or necrosis. Cells that internalised 053SNAP did not show any changes in cellular viability (Fig. S6†), indicating that the clearance of nanocarriers would not decrease the number of macrophages in the peritoneal cavity.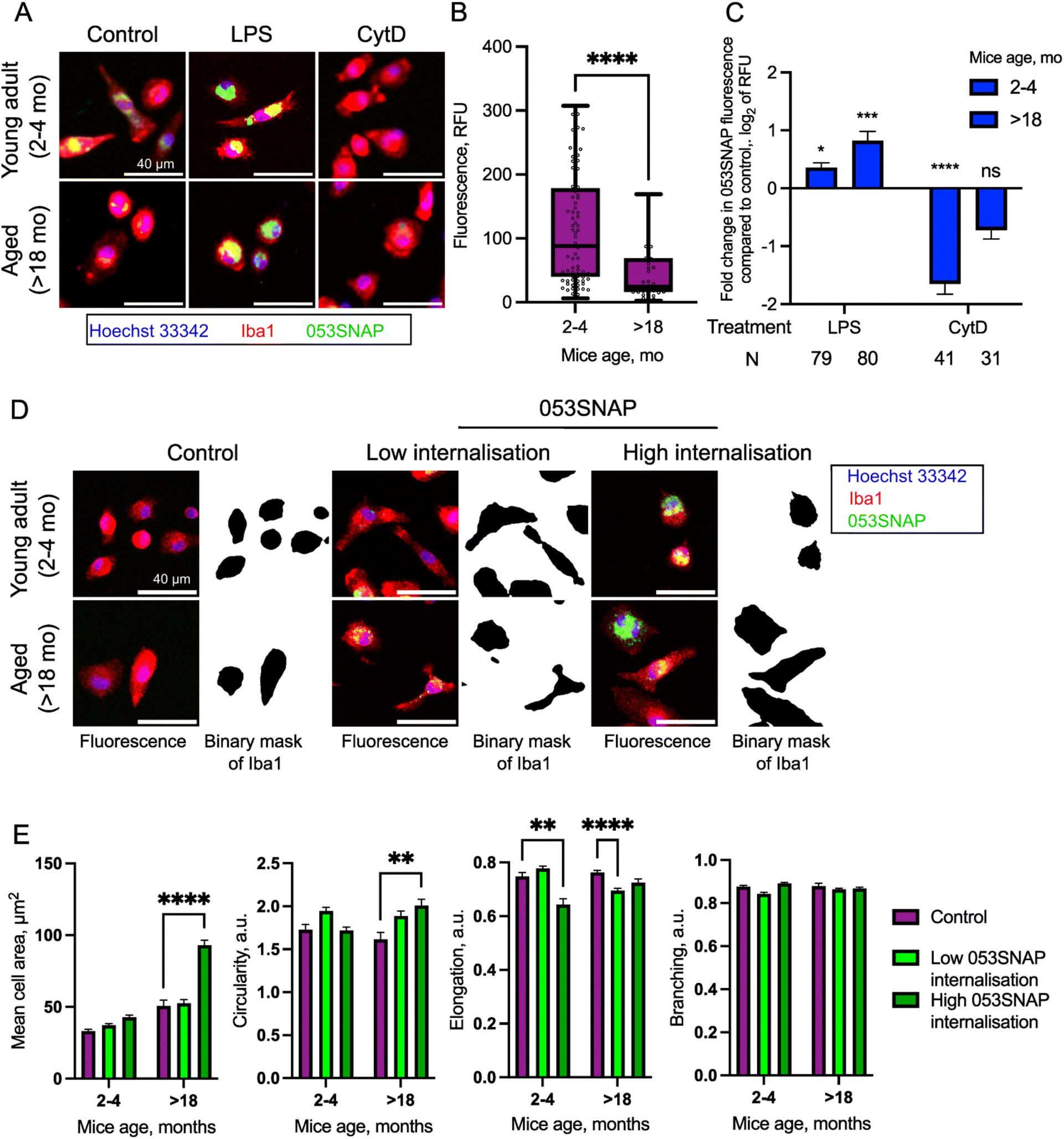 | ||
| Fig. 4 Internalisation of 053SNAP nanotubes into primary mouse peritoneal macrophages. (A) Fluorescence images of primary mouse peritoneal macrophages that were incubated with 053SNAP nanotubes with or without pre-treatment with either LPS or CytD (blue – Hoechst3342, red – anti-Iba1, green – 053SNAP); scale bar 40 μm. (B) Quantification of 053SNAP nanotube internalization into primary peritoneal macrophages isolated from young adult and aged mice. (C) Fold change of 053SNAP nanotubes internalised by primary peritoneal macrophages isolated from young adult and aged mice and treated with either LPS or CytD. (D) Fluorescence images of Iba1-labelled primary macrophages and their segmented masks representing the cell shape. Macrophages were isolated from young adult and aged mice and incubated with or without 053SNAP. Low internalization and high internalization cells were defined based on their internalized 053SNAP fluorescence; scale bar 40 μm. (E) Cell size, circularity, elongation and branching of primary peritoneal macrophages isolated from young adult and aged mice, with and without the incubation with 053SNAP. Data are represented as the minimum value, first quartile, median, third quartile, maximum value, every dot presents average integrated fluorescence intensity in one image. Data are represented as means ± sem; */**/***/****p < 0.05/0.01/0.001/0.0001. | ||
Oncological pathologies are closely related to aging,45 which also modulates the immune response of tissue macrophages. Therefore, we evaluated how aging affects the clearance of 053SNAP nanotubes in peritoneal macrophages isolated from aged mice (>18 months old). Interestingly, we noticed that the uptake of 053SNAP was 2-fold lower in cells isolated from aged animals (p < 0.0001) (Fig. 4B), which is in line with a study that demonstrated a 2-fold decrease of phagocytic capacity of peritoneal macrophages isolated from aged mice compared to those isolated from young animals.46 This suggests that the clearance of nanotubes could be decreased in aged patients thus ensuring high efficacy in the primary demographic for cancer treatment.
We then assessed if the uptake of 053SNAP nanotubes by the peritoneal macrophages was active (actin-dependent) or passive (actin-independent). To address this question we pre-treated the cells with cytochalasin D (CytD), which impedes actin polymerisation and hinders all actin-dependent uptake, including endocytosis and phagocytosis.47 CytD pre-treatment reduced internalised 053SNAP in cells from young animals (p < 0.0001) (Fig. 4A and C), suggesting an active actin-dependent internalisation mechanism. Interestingly, the blocking of actin polymerisation did not have a statistically significant decrease in uptake by aged animal cells (p = 0.23) (Fig. 4C), indicating that 053SNAP may also enter peritoneal macrophages passively in an actin-independent manner. Previous studies have reported that the inhibition of actin polymerization by CytD does not hinder pulmonary macrophage uptake of ultrafine (<0.1 and 0.2 μm) nanoparticles, but decreases the intracellular localisation of larger (1 μm) particles.48
Cancerous tissue presents inflammatory milieu and the inflammation is known to modulate macrophage phagocytic capacity, therefore we investigated how the uptake of 053SNAP is affected by inflammatory activation of macrophages. To elicit inflammatory response, we pre-treated macrophages with bacterial cell wall lipopolysaccharide (LPS).49 The activation with LPS increased 053SNAP uptake by macrophages isolated from both young animals (p < 0.05) and aged animals (p < 0.001, Fig. 4A and C). Importantly, in response to inflammatory activation the phagocytosis was enhanced almost twice as high in macrophages isolated from aged animals compared to those from young mice. The increase in clearance by activated macrophages is not favourable, as most macrophages within and around cancerous tissue will be in the activated state. To tackle this, it may be necessary to combine nanocarrier therapy with anti-inflammatory drugs to limit macrophages' clearance. Even though the macrophages isolated from aged animals had an increased uptake of 053SNAP45 after activation with LPS, the average recorded fluorescence intensity in LPS activated macrophages isolated from aged animals was 2-fold lower compared to macrophages from young animals. These results suggest that the therapeutic effects may not be hindered in aged groups that are at the center of interest for cancer drug development.
Some nanostructures themselves can cause local inflammation,50,51 therefore we evaluated whether the internalisation of 053SNAP stimulates inflammatory response in the peritoneal macrophages. To see if the macrophages that internalized 053SNAP showed morphological changes that can be associated with inflammatory response,52 we evaluated their size, circularity, elongation, and branching (Fig. 4D, E and S7†). It has been demonstrated that an increase in cell area and circularity as well as a decrease in elongation and branching is associated with inflammatory response.53,54 To specifically assess the effect of internalised 053SNAP rather than macrophage response to 053SNAP in the medium due to surface receptor activation, the cells were divided into two groups. Low internalisation cells were defined as those that contained less than the median of 053SNAP fluorescence intensity, while high internalisation cells were those that exhibited higher than the median of 053SNAP fluorescence intensity. High internalisation macrophages from young animals showed a statistically significant decrease in elongation (Fig. 4E), suggesting that the uptake of 053SNAP might have activated the macrophages. Meanwhile, high internalisation macrophages isolated from aged animals did not demonstrate any changes in elongation but had a significant increase in size and circularity (Fig. 4E), both of which also may indicate inflammatory activation. Notably, low internalisation macrophages from aged animals also had a decreased elongation (together with a statistically insignificant increase in circularity, p = 0.12) (Fig. 4E) which suggests inflammatory activation by low quantities of internalised 053SNAP or to the exposure to 053SNAP in the medium. Alternatively, the increase of the size of high internalization macrophages can be caused by phagocytosis itself that results in the expansion of the cell enveloping phagocytic targets.55 Importantly, none of the cells showed changes in branching, which indicates that the cells did not present the morphologies related to strong inflammatory activation. Therefore, further investigation, preferentially evaluation of cytokine expression, is required to establish whether nanotubes derived from bacteriophage sheath proteins elicit an immune response in macrophages that may hinder their application in therapy.
While conventional metal nanoparticles are known to be inert and do not cause any inflammatory responses,56 the interaction of the immune system with biomolecule-based nanoparticles is still under investigation. For example, the particles assembled from the T4 phage head protein do not elevate cytokines and reactive oxygen species production in an in vivo model or human blood cells,57 in contrast to our 053SNAP particles that might cause inflammatory activation of macrophages. Favourably, it has been reported that PEGylation, which we used to stabilise 053SNAP, not only stabilises nanostructures, but it also interferes with the opsonisation of the nanoparticles and thus hinders the formation of the protein rich layer, known as the “protein corona”.17 The protein corona formed around nanocarriers is crucial for their longevity in the bloodstream, as it modulates the internalisation of nanoparticles into cancer cells and their clearance by macrophages.24,58 Without the protein corona, macrophages cannot recognise the particles as phagocytic targets, thus reducing their clearance.59,60 Recent reports have shown that the protein corona can still assemble on PEGylated particles, albeit less efficiently compared to non-coated particles. Therefore, other surface modification methods might be required to further reduce 053SNAP clearance by macrophages, such as polyglycerol, which has demonstrated promising results.61
In summary, our findings indicate that 053SNAP self-assemble into sub-micron sized nanostructures that retain the enzymatic activity of the fused SNAP-tag protein, which faces the inner cavity of the nanotube. PEGylated nanotubes were stable under physiological conditions and were actively internalised in primary and metastatic cancer cells. The internalisation into cancer cells increased with time and did not cause direct cytotoxicity. The evaluation of the internalisation mechanism revealed that HCT116 and SW480 cells internalised 053SNAP in a dynamin-dependent manner, while SW480 cells internalised the particles in a both dynamin- and clathrin-dependent mechanism. We also found that peritoneal macrophages actively internalised 053SNAP, which may interfere with the application of these nanocarriers to therapy. Notably, the clearance of nanotubes by macrophages isolated from aged animals was reduced, thus suggesting that the efficacy of nanocarriers based on the gp053 phage may be age-dependent.
Conclusions
In this study we presented 053SNAP self-assembling chimeric proteinaceous nanotubes and demonstrated that they can be used as a basis for the development of novel enzyme-nanocarriers for the delivery into cancer cells. The fusion of the SNAP-tag self-labelling enzyme to the C-terminus of the gp053 protein yielded sub-micron sized nanotubes with retained enzymatic activity, indicating that these nanostructures can carry intact enzymatic cargo. Primary and metastatic colorectal cancer cells actively internalized 053SNAP nanotubes, indicating their potential as a therapeutic delivery agent. However, primary macrophages readily cleared of the nanotubes and might therefore hinder their application in therapy. Interestingly, the ability of macrophages to clear the nanotubes decreased with age, indicating that protein-based nanotubes may be a suitable platform for developing cancer treatment for older patients – the dominant cancer demographic. Ultimately, our results demonstrated that nanotubes that self-assemble from bacteriophage sheath proteins are a suitable platform for future developments of novel nanocarriers.Experimental section
Gene construction
The full-length 053 gene (GeneID: 14011712) was amplified from phage FV3 DNA by using primers 053NcoF and 053SalR
and 053SalR  The obtained DNA fragment was digested with restriction endonucleases NcoI and SalI and inserted into the NcoI–SalI sites of pET21d vector. SNAP-tag with the linker sequence was cut from the pET21b_SNAP_gb plasmid (a gift from Paul Heppenstall (Dhandapani et al., 2018)) and inserted into SalI and XhoI restriction endonuclease sites of the pET21d_053 plasmid. The resulting plasmid pET21_053SNAP was amplified in E. coli DH5α, verified by DNA sequencing, and used for protein synthesis.
The obtained DNA fragment was digested with restriction endonucleases NcoI and SalI and inserted into the NcoI–SalI sites of pET21d vector. SNAP-tag with the linker sequence was cut from the pET21b_SNAP_gb plasmid (a gift from Paul Heppenstall (Dhandapani et al., 2018)) and inserted into SalI and XhoI restriction endonuclease sites of the pET21d_053 plasmid. The resulting plasmid pET21_053SNAP was amplified in E. coli DH5α, verified by DNA sequencing, and used for protein synthesis.
Expression and purification of 053SNAP
BL21(DE3) cells transformed with pET21_053SNAP were grown in 50 ml of LB medium at 30 °C to an OD (600) 0.8. Protein synthesis was induced by 0.1 mM IPTG. The cells were grown for 20 h at 20 °C, collected by centrifugation, and suspended in 5 ml of 10 mM potassium phosphate buffer solution pH 7.5. The cells were disrupted by sonication and the soluble protein fraction was purified as described previously.31Modification of 053SNAP
The purified 053SNAP protein was labelled with SNAP-Surface Alexa Fluor 488 (50 nmol) according to the manufacturer's recommendations (New England BioLabs). MS(PEG)12 methyl–PEG–NHS–ester reagent was used for PEGylation according to the manufacturer's recommendations (Thermo Fisher Scientific). After PEGylation, proteins were dialyzed in 10 mM potassium phosphate, pH 7.5 buffer, and filtered through 0.22 μm filters. The protein size was analysed by SDS–PAGE (14% separating and 4.0% stacking gels) according to Laemmli.62 The concentration of protein was determined using the Pierce™ Coomassie Plus (Bradford) Assay Reagent by the standard microplate protocol.Transmission electron microscopy (TEM)
TEM analysis was performed as described previously.31 The particle size was estimated using ImageJ (FIJI) software63 choosing the sample size n = 30.HPLC analysis
Soluble cell-free extract (25 μl) was labelled with SNAP-Surface Alexa Fluor 488, diluted 10×, and subjected to high-performance liquid chromatography (HPLC) with a UV-vis and fluorescence detection system (Shimadzu, Kyoto, Japan). A TSKgel G4000SWXL column was used for analysis as described previously,31 equilibrated in 10 mM Tris–HCl, pH 8.0.Evaluation of nanotube aggregation
To evaluate the aggregation of 053SNAP nanotubes conjugated with either SNAP-Surface® Alexa Fluor® 488 or SNAP-Cell® 647-SiR (New England Biolabs) in cell culture medium, the nanotube solution was diluted in Dulbecco's modified Eagle's medium with Nutrient Mixture F-12 (DMEM/F12-10, Gibco) supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS, Gibco) for a final concentration of 50 μg ml−1 and incubated for 4 h in 37 °C, 5% CO2. The solution was then imaged at 1 μm above the glass surface using a Leica TCS SP8 system with a 63×/1.4 NA oil-immersion objective for confocal imaging and a 100×/1.40 NA oil-immersion objective for stimulated emission depletion (STED) imaging. For STED imaging 775 nm depletion laser was used. Particle size from the micrographs was evaluated on ImageJ (FIJI) software.63Cell lines
Colorectal carcinoma cells HCT116 (MSI, TP53 proficient, K-Ras-mutated; CCL-247; ATCC), colorectal adenocarcinoma cells SW480 (the primary tumour, MSS, TP53, and K-Ras mutated; CLL3227; ATCC), and SW620 (lymph node metastasis, MSS, TP53, and K-Ras mutated; CLL-228; ATCC) were used for experiments. The HCT116 cell line was cultivated in Roswell Park Memorial Institute 1640 medium (RPMI 1640, Gibco), SW480, and SW620 cell lines – in Dulbecco's modified Eagle's medium (DMEM, Gibco). Both cell culture media were supplemented with 10% FBS (Gibco), stable L-glutamine (Gibco), and penicillin and streptomycin solution (Gibco). Cells were grown in a humidified atmosphere containing 5% CO2 at 37 °C.siRNA transfection
siRNA transfection was performed in 8-well plates for microscopy (C8-1-N; Cellvis) and in 12-well plates for western blot analysis (Thermo Fisher Scientific). Cells were seeded in a cell culture medium without antibiotics at the density of 2 × 104 cells per cm2. Cells were transfected with siRNAs at 24 h after cell seeding. siRNA transfection was performed following the manufacturer's recommendations. Transfection reagent Lipofectamine™ RNAiMAX (Thermo Fisher Scientific) and siRNAs were diluted in an Opti-MEM™ medium (Thermo Fisher Scientific). AllStars non-targeting siRNA mix for human transcripts (1027281; Qiagen) was used as a negative control. Specific siRNAs targeting the transcripts of clathrin heavy chain (TAATCCAATTCGAAGACCAAT; SI00299880; Qiagen) and dynamin-2 (CTCATACGTGGCCATCATCAA; SI04224591; Qiagen) were used for gene silencing. The final concentration of siRNAs was 6 nM. The cell culture medium was changed at 24 h after transfection. To evaluate protein downregulation, cells were harvested for western blot analysis 48 h after transfection. To investigate nanotube uptake, 053SNAP (50 μg ml−1) was added to the cell culture medium 24 h after the transfection.053SNAP uptake assay in cancer cells
HCT116, SW480, and SW620 cells were seeded to 8-well plates (C8-1-N; Cellvis) at the density of 2 × 104 cells per cm2. 053SNAP nanotubes (50 μg ml−1) were added to the medium 48 h after cell seeding and incubated at 37 °C, 5% CO2 for 1, 2, 4, 6 and 20 h. LysoTracker™ Deep Red (50 nM, Thermo Fisher Scientific) was used for lysosome staining, and Hoechst33342 (10 μg ml−1, Sigma) for nuclei staining. Cells were incubated with lysosome dye for 40 min and with nuclei dye for 15 min at 37 °C in 5% CO2. Before imaging, cells were washed with PBS and covered with fresh cell culture medium. Cells were imaged on a Leica TCS SP5 confocal system with a 63×/1.4 NA oil objective. The wavelengths of excitation and emission for nuclei (Hoechst33342): λex/λem = 405 nm/420–480 nm; 053SNAP nanotubes labelled with Alexa Fluor 488: λex/λem = 488 nm/500–550 nm; lysosomes labelled with LysoTracker™ Deep Red: λex/λem = 633 nm/660–710 nm. Images were analysed using ImageJ (FIJI) software.63 JACoP plugin was used to evaluate colocalisation.64Crystal violet assay
A crystal violet assay was used to evaluate cell viability after incubation with 053SNAP. HCT116, SW480, and SW620 cells were seeded to a 12-well plate at density 2 × 104 cells per cm2 and cultivated for 48 h. Afterwards, 053SNAP (50 μg ml−1) was added to the cell culture medium, and cells were incubated for 24 h or 48 h. A crystal violet assay was performed as previously described.65Western blot analysis
Cells were collected and lysed for 15 min on ice in radioimmunoprecipitation assay buffer (RIPA, Thermo Fisher Scientific) with the Protease Inhibitor Cocktail for General Use (Sigma-Aldrich). Then, the cell lysates were centrifuged for 15 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g at 4 °C. The supernatants were collected, and the protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). For each sample, 50 μg of total protein was used for electrophoresis. Protein samples were concentrated by 4% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) using 80 V and separated by 10% SDS–PAGE using 120 V. Proteins were transferred to a nitrocellulose membrane using a semi-dry blotter. Blots were probed with anti-clathrin heavy chain antibody (Ma1-065; Invitrogen), anti-dynamin-2 antibody (PA-1661; Invitrogen), or anti-β-actin antibody (MA5-15739; Thermo Fisher Scientific). The membrane-bound primary antibody of dynamin-2 was detected by horseradish peroxidase-conjugated (HRP) secondary anti-rabbit antibody (31460; Thermo Fisher Scientific). The membrane-bound antibodies of clathrin heavy chain and β-actin were detected by HRP secondary anti-mouse antibody (31430; Thermo Fisher Scientific). The immunoreactive bands were developed using Pierce® ECL Western Blotting Substrate (Thermo Fisher Scientific). Densitometric analysis was performed using ImageJ (FIJI) software.63 Only the signals falling within the linear range were used for densitometric analysis. The levels of clathrin heavy chain and dynamin-2 were normalised to the level of β-actin that was used as a loading control.
000 × g at 4 °C. The supernatants were collected, and the protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). For each sample, 50 μg of total protein was used for electrophoresis. Protein samples were concentrated by 4% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) using 80 V and separated by 10% SDS–PAGE using 120 V. Proteins were transferred to a nitrocellulose membrane using a semi-dry blotter. Blots were probed with anti-clathrin heavy chain antibody (Ma1-065; Invitrogen), anti-dynamin-2 antibody (PA-1661; Invitrogen), or anti-β-actin antibody (MA5-15739; Thermo Fisher Scientific). The membrane-bound primary antibody of dynamin-2 was detected by horseradish peroxidase-conjugated (HRP) secondary anti-rabbit antibody (31460; Thermo Fisher Scientific). The membrane-bound antibodies of clathrin heavy chain and β-actin were detected by HRP secondary anti-mouse antibody (31430; Thermo Fisher Scientific). The immunoreactive bands were developed using Pierce® ECL Western Blotting Substrate (Thermo Fisher Scientific). Densitometric analysis was performed using ImageJ (FIJI) software.63 Only the signals falling within the linear range were used for densitometric analysis. The levels of clathrin heavy chain and dynamin-2 were normalised to the level of β-actin that was used as a loading control.
Primary cell isolation
C57BL/6J mice were obtained from the local Vilnius University Life Sciences Center colony. Animal studies were conducted in accordance with Directive 2010/63/EU requirements and were approved by the Lithuanian State Food and Veterinary Service (B6-(1.9)-2653). Young adult (2–4 months old) and aged (>18 months old) C57BL/6J mice were anesthetised subcutaneously with ketamine (100 mg kg−1, Bioketan) and xylazine (10 mg kg−1, Sedaxylan). Peritoneal macrophages were isolated as previously described.66 Instead of a 20 G needle, we used a 1000 μl pipette tip to aspirate peritoneal fluid. Isolated cells were seeded at a 35 cells per mm2 density onto 8-well plates coated with 0.001% poly-L-lysine (Sigma) and in DMEM/F12 medium supplemented with heat inactivated 10% (v/v) FBS. Macrophages were allowed to adhere for 1 h at 37 °C, 5% CO2, and nonadherent cells were removed by washing three times with warm Dulbecco's phosphate-buffered saline without calcium and magnesium (DPBS, Gibco). Remaining adherent macrophages were then maintained in DMEM/F12 medium supplemented with heat inactivated 10% (v/v) FBS. Cell culture medium and all solutions (except DPBS) were supplemented with 50 U per ml penicillin/streptomycin (Gibco).053SNAP uptake assay in macrophages
For the evaluation of nanotube uptake in primary peritoneal macrophages, after 24 h in vitro, the medium was changed to either fresh medium or fresh medium with LPS (100 ng ml−1, L6511, Sigma) and incubated for 18 h at 37 °C, 5% CO2. For treatment with cytochalasin D (1 μM, C8273, Sigma), cells were pre-treated for 1 h at 37 °C, 5% CO2 prior to adding the nanotubes. Afterwards, peritoneal macrophages were incubated with 053SNAP nanotubes (50 μg ml−1) for 2.5 h at 37 °C, 5% CO2. Then cell nuclei were stained with Hoechst33342 (10 μg ml−1, Sigma) alone or together with propidium iodide (1 μg ml−1, Sigma) diluted in fresh medium for 15 min at 37 °C, 5% CO2. Labelled cells were washed with cell culture medium and fixed with 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature (RT). For immunolabelling, cells were permeabilised with 0.1% Triton X-100 in PBS for 15 min at RT and blocked with 5% donkey serum (Gibco) in PBS for 1 h, at RT. Iba1 was immunodetected by incubation with rabbit anti-Iba1 primary antibodies (1:1000, 01919741, FUJIFILM Wako) for 1 h at RT followed by donkey anti-rabbit Alexa Fluor™ 594 secondary antibody (1:500, A11012, Thermo Fisher Scientific) incubation in PBS with 2.5% normal donkey serum for 1 h at RT. Cells were then imaged on a Leica TCS SP8 confocal system with a 20×/0.75 NA dry objective. The wavelengths of excitation and emission for nuclei (Hoechst33342): λex/λem = 405 nm/415–489 nm; 053SNAP nanotubes labelled with Alexa Fluor 488: λex/λem = 499 nm/508–584 nm; anti-Iba1 labelled with Alexa Fluor 594: λex/λem = 598 nm/608–750 nm. Confocal microscopy images of primary peritoneal macrophages were analysed using CellProfiler software.67 The Iba1 signal was used to define cellular contour, in which the integrated intensity of 053SNAP-specific fluorescence was quantified for every cell.
Peritoneal macrophage viability evaluation
The viability of peritoneal macrophages was defined in live cells based on their labelling with Hoechst33342 (10 μg ml−1) and propidium iodide (1 μg ml−1). Cells were labelled for 15 min at 37 °C, 5% CO2, washed with prewarmed DMEM/F12 without phenol red (Gibco), and then imaged on EVOS® FL Cell Imaging System using a 20× objective. Images of the nuclei, 053SNAP, and propidium iodide were acquired using EVOS® DAPI (λex/λem = 335–379 nm/417–477 nm), GFP (λex/λem = 467–494 nm/500–548 nm) and RFP (λex/λem = 542–552 nm/573–633 nm) LED cubes. Cell necrosis was identified as a propidium iodide positive cell nucleus. Cell apoptosis was identified as a propidium iodide negative, condensed cell nucleus without distinguishable nucleoli.Statistical analysis
To evaluate the particle size of the control and PEGylated 053SNAP, 10 images were acquired for each sample, and Welch's t-test was used to compare particle size. For cancer cell experiments at least 3 independent experiments were performed, and the unpaired two-samples Wilcoxon test was used for statistical comparison. For peritoneal macrophage experiments, at least three independent experiments were performed for each group and a two-way ANOVA was used with Tukey's multiple comparison post hoc test. Normality was tested using the Shapiro–Wilk test. The difference between samples/groups was considered statistically significant at a p-value of <0.05. Statistical analysis was performed on GraphPad Prism 9.0 (GraphPad Software, Inc.)Author contributions
DG: investigation, visualisation, writing – original draft. VZ: investigation, visualisation, writing – original draft. LS: investigation, visualisation. GL: resources. MS: investigation, visualisation. RM: resources, supervision. VC: funding acquisition, investigation, resources, writing – review & editing. AS: investigation, supervision, visualization, writing – review & editing. UN: supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Research Council of Lithuania, grant number S-SEN-20-4. UN was supported by the European Regional Development Fund under grant agreement no. 01.2.2-CPVA-V-716-01-0001 with the Central Project Management Agency (CPVA), Lithuania.Notes and references
- V. Gupta, A. Aseh, C. N. Ríos, B. B. Aggarwal and A. B. Mathur, Int. J. Nanomed., 2009, 115(4), 115–122 CrossRef PubMed.
- A. G. Cheetham, P. Zhang, Y. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- K. Karami, N. Jamshidian, A. Hajiaghasi and Z. Amirghofran, New J. Chem., 2020, 44, 4394–4405 RSC.
- J. J. Milligan, S. Saha, I. C. Jenkins and A. Chilkoti, Curr. Opin. Biotechnol., 2022, 74, 146–153 CrossRef CAS PubMed.
- X. Hu, M. Liao, H. Gong, L. Zhang, H. Cox, T. A. Waigh and J. R. Lu, Curr. Opin. Colloid Interface Sci., 2020, 45, 1–13 CrossRef CAS.
- S. Lee, T. H. T. Trinh, M. Yoo, J. Shin, H. Lee, J. Kim, E. Hwang, Y. Lim and C. Ryou, Int. J. Mol. Sci., 2019, 20, 5850 CrossRef CAS PubMed.
- J.-H. Lee, J. H. Lee, J. Xiao, M. S. Desai, X. Zhang and S.-W. Lee, Nano Lett., 2019, 19, 2661–2667 CrossRef CAS PubMed.
- T. Sawada and T. Serizawa, Bull. Chem. Soc. Jpn., 2018, 91, 455–466 CrossRef CAS.
- B. F. Poglazov, A. V. Efimov, S. Marco, J. Carrascosa, T. A. Kuznetsova, L. G. Aijrich, L. P. Kurochkina and V. V. Mesyanzhinov, J. Struct. Biol., 1999, 127, 224–230 CrossRef CAS PubMed.
- D. Lauster, S. Klenk, K. Ludwig, S. Nojoumi, S. Behren, L. Adam, M. Stadtmüller, S. Saenger, S. Zimmler, K. Hönzke, L. Yao, U. Hoffmann, M. Bardua, A. Hamann, M. Witzenrath, L. E. Sander, T. Wolff, A. C. Hocke, S. Hippenstiel, S. De Carlo, J. Neudecker, K. Osterrieder, N. Budisa, R. R. Netz, C. Böttcher, S. Liese, A. Herrmann and C. P. R. Hackenberger, Nat. Nanotechnol., 2020, 15, 373–379 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- S. Hong, D. W. Choi, H. N. Kim, C. G. Park, W. Lee and H. H. Park, Pharmaceutics, 2020, 12, 604 CrossRef CAS PubMed.
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Adv. Drug Delivery Rev., 2018, 127, 46–57 CrossRef CAS PubMed.
- S. Yadav, A. K. Sharma and P. Kumar, Front. Bioeng. Biotechnol., 2020, 8, 127 CrossRef PubMed.
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- W. P. Caron, J. C. Lay, A. M. Fong, N. M. La-Beck, P. Kumar, S. E. Newman, H. Zhou, J. H. Monaco, D. L. Clarke-Pearson, W. R. Brewster, L. Van Le, V. L. Bae-Jump, P. A. Gehrig and W. C. Zamboni, J. Pharmacol. Exp. Ther., 2013, 347, 599–606 CrossRef CAS PubMed.
- L. Sanchez, Y. Yi and Y. Yu, Nanoscale, 2017, 9, 288–297 RSC.
- Y. Qie, H. Yuan, C. A. von Roemeling, Y. Chen, X. Liu, K. D. Shih, J. A. Knight, H. W. Tun, R. E. Wharen, W. Jiang and B. Y. S. Kim, Sci. Rep., 2016, 6, 26269 CrossRef CAS PubMed.
- Z. Liu, X. Zhou, Q. Li, Y. Shen, T. Zhou and X. Liu, Acta Pharm. Sin. B, 2022, 13(1), 327–343 CrossRef PubMed.
- Y. H. Chung, H. Cai and N. F. Steinmetz, Adv. Drug Delivery Rev., 2020, 156, 214–235 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, in Site-Specific Protein Labeling, ed. A. Gautier and M. J. Hinner, Humana, New York, USA, 1st edn, 2015, ch. 3, pp. 29–53 Search PubMed.
- A. M. Bannunah, D. Vllasaliu, J. Lord and S. Stolnik, Mol. Pharm., 2014, 11, 4363–4373 CrossRef CAS PubMed.
- G. Sanità, B. Carrese and A. Lamberti, Front. Mol. Biosci., 2020, 7, 587012 CrossRef PubMed.
- E. Voronovic, A. Skripka, G. Jarockyte, M. Ger, D. Kuciauskas, A. Kaupinis, M. Valius, R. Rotomskis, F. Vetrone and V. Karabanovas, ACS Appl. Mater. Interfaces, 2021, 13, 39076–39087 CrossRef CAS PubMed.
- E. Šimoliūnas, L. Truncaitė, R. Rutkienė, S. Povilonienė, K. Goda, A. Kaupinis, M. Valius and R. Meškys, Viruses, 2019, 11, 50 CrossRef PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- P. Evseev, M. Shneider and K. Miroshnikov, Viruses, 2022, 14, 1148 CrossRef CAS PubMed.
- A. A. Aksyuk, P. G. Leiman, L. P. Kurochkina, M. M. Shneider, V. A. Kostyuchenko, V. V. Mesyanzhinov and M. G. Rossmann, EMBO J., 2009, 28, 821–829 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- G. Labutytė, S. Povilonienė, E. Šimoliūnas, D. Gabrielaitis, M. Skapas, A. Noreika, R. Meškys and V. Časaitė, Nanomaterials, 2021, 11, 3031 CrossRef PubMed.
- A. Špakova, I. Dalgėdienė, R. Insodaitė, A. Sasnauskienė, A. Žvirblienė and R. Petraitytė-Burneikienė, Virus Res., 2020, 290, 198194 CrossRef PubMed.
- D. Ahmed, P. W. Eide, I. A. Eilertsen, S. A. Danielsen, M. Eknæs, M. Hektoen, G. E. Lind and R. A. Lothe, Oncogenesis, 2013, 2, e71 CrossRef CAS PubMed.
- C. Basquin and N. Sauvonnet, Commun. Integr. Biol., 2013, 6, e24243 CrossRef PubMed.
- A. M. Lakoduk, C.-F. Lee and P.-H. Chen, Int. J. Biochem. Cell Biol., 2021, 131, 105905 CrossRef CAS PubMed.
- I. Khan and P. S. Steeg, Br. J. Cancer, 2021, 124, 66–75 CrossRef CAS PubMed.
- M. S. de Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC.
- Z. Hu, Y. Pan, J. Wang, J. Chen, J. Li and L. Ren, Biomed. Pharmacother., 2009, 63, 155–164 CrossRef CAS PubMed.
- K. Sandvig, S. Kavaliauskiene and T. Skotland, Histochem. Cell Biol., 2018, 150, 107–118 CrossRef CAS PubMed.
- H. C. Verdera, J. J. Gitz-Francois, R. M. Schiffelers and P. Vader, J. Controlled Release, 2017, 266, 100–108 CrossRef PubMed.
- M. C. B. Lira-Nogueira, V. P. Gibson, V. Nicolas, N. S. Santos-Magalhães and C. Vauthier, Pharm. Res., 2022, 39, 1135–1150 CrossRef CAS PubMed.
- T. Liu, F. Liu, L.-W. Peng, L. Chang and Y.-M. Jiang, Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics, 2018, 26, 817–826 CrossRef PubMed.
- Y. Li, Z. Wang, X. Ma, B. Shao, X. Gao, B. Zhang, G. Xu and Y. Wei, Pathog. Dis., 2014, 72(2), 111–123 CAS.
- Y.-M. Kwon, Z. Xia, S. Glyn-Jones, D. Beard, H. S. Gill and D. W. Murray, Biomed. Mater., 2009, 4, 025018 CrossRef PubMed.
- M. C. White, D. M. Holman, J. E. Boehm, L. A. Peipins, M. Grossman and S. Jane Henley, Am. J. Prev. Med., 2014, 46, S7–S15 CrossRef PubMed.
- E. Linehan, Y. Dombrowski, R. Snoddy, P. G. Fallon, A. Kissenpfennig and D. C. Fitzgerald, Aging Cell, 2014, 13, 699–708 CrossRef CAS PubMed.
- J. Magae, T. Nagi, K. Takaku, T. Kataoka, H. Koshino, M. Uramoto and K. Nagai, Biosci., Biotechnol., Biochem., 1994, 58, 104–107 CrossRef CAS PubMed.
- M. Geiser, B. Rothen-Rutishauser, N. Kapp, S. Schürch, W. Kreyling, H. Schulz, M. Semmler, V. I. Hof, J. Heyder and P. Gehr, Environ. Health Perspect., 2005, 113, 1555–1560 CrossRef PubMed.
- S. Soudi, A. Zavaran-Hosseini, Z. M. Hassan, M. Soleimani, F. J. Adegani and S. M. Hashemi, Cell J., 2013, 15, 45–54 CAS.
- H. M. Braakhuis, M. V. Park, I. Gosens, W. H. De Jong and F. R. Cassee, Part. Fibre Toxicol., 2014, 11, 18 CrossRef PubMed.
- P. Pedata, C. Petrarca, E. M. Garzillo and M. Di Gioacchino, Int. J. Immunopathol. Pharmacol., 2016, 29, 343–353 CrossRef PubMed.
- C. Chi, X. Jiang, L. Su, Z. Shen and X. Yang, J. Cell. Mol. Med., 2015, 19, 2741–2750 CrossRef CAS PubMed.
- S. Schütze, S. Ribes, A. Kaufmann, A. Manig, J. Scheffel, S. Redlich, S. Bunkowski, U.-K. Hanisch, W. Brück and R. Nau, Oncotarget, 2014, 5, 12573–12592 CrossRef PubMed.
- F. Y. McWhorter, C. T. Davis and W. F. Liu, Cell. Mol. Life Sci., 2015, 72, 1303–1316 CrossRef CAS PubMed.
- C. S. Zent and M. R. Elliott, FEBS J., 2017, 284, 1021–1039 CrossRef CAS PubMed.
- B. J. Swartzwelter, A. Verde, L. Rehak, M. Madej, V. F. Puntes, A. C. De Luca, D. Boraschi and P. Italiani, Nanomaterials, 2021, 11, 207 CrossRef CAS PubMed.
- P. Miernikiewicz, K. Dąbrowska, A. Piotrowicz, B. Owczarek, J. Wojas-Turek, J. Kicielińska, J. Rossowska, E. Pajtasz-Piasecka, K. Hodyra, K. Macegoniuk, K. Rzewucka, A. Kopciuch, T. Majka, A. Letarov, E. Kulikov, H. Maciejewski and A. Górski, PLoS One, 2013, 8, e71036 CrossRef CAS PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- R. Rampado, S. Crotti, P. Caliceti, S. Pucciarelli and M. Agostini, Front. Bioeng. Biotechnol., 2020, 8, 116 CrossRef PubMed.
- P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, J. Controlled Release, 2003, 90, 323–334 CrossRef CAS PubMed.
- Y. Zou, S. Ito, F. Yoshino, Y. Suzuki, L. Zhao and N. Komatsu, ACS Nano, 2020, 14, 7216–7226 CrossRef CAS PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- D. Dabkeviciene, A. Sasnauskiene, E. Leman, R. Kvietkauskaite and V. Kirveliene, Anticancer Res., 2014, 34, 5295–5302 CAS.
- X. Zhang, R. Goncalves and D. M. Mosser, Curr. Protoc. Immunol., 2008, 83, 14.1.1–14.1.14 Search PubMed.
- D. R. Stirling, M. J. Swain-Bowden, A. M. Lucas, A. E. Carpenter, B. A. Cimini and A. Goodman, BMC Bioinform., 2021, 22, 433 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00166k |
| ‡ Equally contributed to the first authorship. |
| § Equally contributed to the last authorship. |
| This journal is © The Royal Society of Chemistry 2023 |