
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of F-doped materials and applications in catalysis and rechargeable batteries
Jiale
Huo
 ab,
Yaofang
Zhang
*ab,
Weimin
Kang
ac,
Yan
Shen
ab,
Xiang
Li
ab,
Zirui
Yan
ab,
Yingwen
Pan
ab and
Wei
Sun
ab
ab,
Yaofang
Zhang
*ab,
Weimin
Kang
ac,
Yan
Shen
ab,
Xiang
Li
ab,
Zirui
Yan
ab,
Yingwen
Pan
ab and
Wei
Sun
ab
aState Key Laboratory of Separation Membranes and Membrane Processes, Tiangong University, Tianjin 300387, PR China. E-mail: zhangyaofang@tiangong.edu.cn
bSchool of Physical Science and Technology, Tiangong University, Tianjin 300387, PR China
cSchool of Textile Science and Engineering, Tiangong University, Tianjin 300387, China
First published on 8th May 2023
Abstract
Elemental doping is one of the most essential techniques for material modification. It is well known that fluorine is considered to be a highly efficient and inexpensive dopant in the field of materials. Fluorine is one of the most reactive elements with the highest electronegativity (χ = 3.98). Compared to cationic doping, anionic doping is another valuable method for improving the properties of materials. Many materials have physicochemical limitations that affect their practical application in the field of catalysis and rechargeable ion batteries. Many researchers have demonstrated that F-doping can significantly improve the performance of materials for practical applications. This paper reviews the applications of various F-doped materials in photocatalysis, electrocatalysis, lithium-ion batteries, and sodium-ion batteries, as well as briefly introducing their preparation methods and mechanisms to provide researchers with more ideas and options for material modification.
1 Introduction
Since the 21st century, environmental issues have become the primary problems hindering sustainable development. Over the past 20 years, many methods have been used to remove toxic pollutants, such as coagulation, membrane treatment, adsorption, ion exchange, and photocatalysis.1–3 Since Fujishima and Honda first demonstrated photocatalytic hydrolysis at TiO2 electrodes under UV light in 1972,4 catalytic technology has been applied in various fields to solve the increasingly serious environmental pollution problems. At present, semiconductor photocatalysis is attracting much attention from researchers because of its promising applications in degrading organic pollutants to clean up air and water pollution. The principle of photocatalysis is based on the redox ability of photocatalysts in the presence of light to degrade pollutants.5 Photocatalysis, although varying in detail in terms of reactions and mechanisms, can be described in terms of four crucial steps (as shown in Fig. 1a): (I) absorption of light to generate electron–hole pairs; (II) separation of the excitation charge; (III) transfer of electrons and holes to the surface of the photocatalyst; and (IV) redox reactions using the charge on the surface. Some organic pollutants such as phenol,6–8 toluene,9–11 methyl orange,12 methylene blue,13,14 rhodamine B,15,16 diclofenac,17–19 atrazine,20,21 and perfluorooctanoic acid22,23 enter the atmosphere and water bodies, seriously endangering environmental safety and threatening the health of living organisms. Common photocatalysts such as titanium dioxide, zinc oxide, tin oxide, zirconium dioxide, and cadmium sulfide have shown good photocatalytic performance.24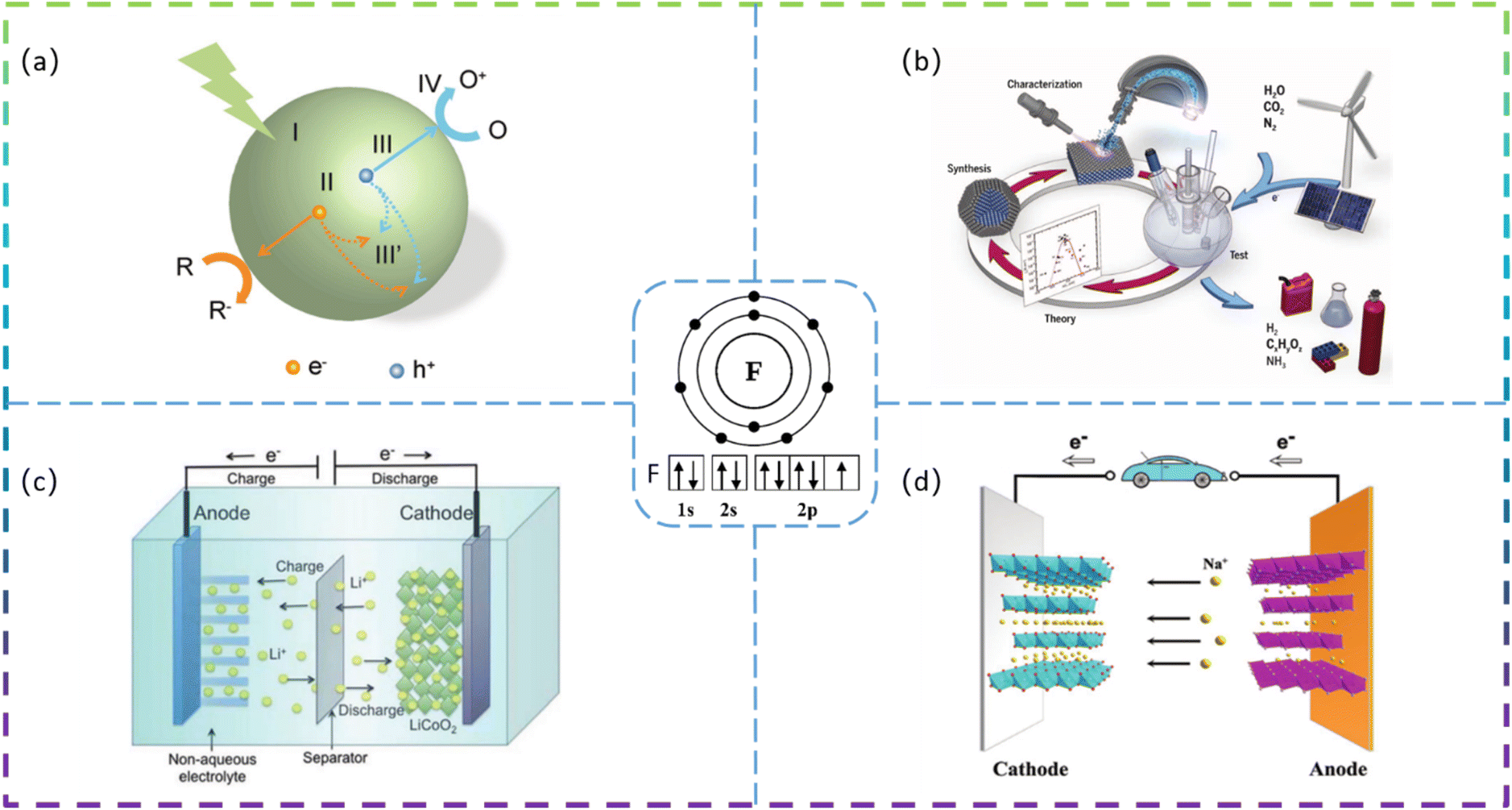 | ||
| Fig. 1 (a) Steps in the photocatalytic reaction process. (b) Schematic showing electrochemical conversion of water, carbon dioxide, and nitrogen into value-added products (e.g., hydrogen, hydrocarbons, oxygenates, and ammonia), using energy from renewable sources. (c) Schematic diagram of the lithium intercalation–deintercalation reaction mechanism in a rechargeable lithium-ion battery. (d) Schematic illustration of SIBs (the inset shows the electron configuration of the F atom). | ||
Developing sustainable, environmentally friendly pathways to produce globally significant fuels and chemicals can play an important role in reducing carbon emissions while providing the raw materials necessary for human survival.25–27 One promising goal is to use electrochemical conversion processes to convert atmospheric molecules (such as water, carbon dioxide, and nitrogen) into higher-value products (such as hydrogen, hydrocarbons, oxygenates, and ammonia) by combining them with renewable energy sources (Fig. 1b).28 Electrocatalysts play a key role in these energy conversion technologies as they increase the rate, efficiency, and selectivity of the chemical conversions involved. However, conventional electrocatalysts are not sufficient. The greatest challenge is to develop advanced electrocatalysts and improve their performance to achieve widespread access to clean energy technologies. Electrocatalysis is a catalytic action that causes charge transfer at the electrode and electrolyte interface to accelerate a reaction. Electrocatalysis is widely used for the electrolytic reduction of carbon dioxide29–31 and nitrogen,32–35 organic degradation,36 hydrogen evolution reaction,37 and oxygen reduction reactions.38 The rational selection of electrode materials is of great importance for the economical and efficient operation of electrodes. Excellent electrode materials should not only be effective in degrading pollutants, but also have stable electrochemical properties and be inexpensive.
Lithium-ion batteries (LIBs) have received a lot of attention because of their high energy density, high power density, long cycle life, and low memory effect.39 LIBs have been under development as a power source for portable electronic devices, electric vehicles, and energy storage systems. The development of next-generation LIBs with high energy/power density and long life is now necessary to address the rapidly growing energy demand in our daily lives. Typically, LIBs consist mainly of a positive electrode, a negative electrode, a separator, and a liquid electrolyte (Fig. 1c).40 The electrode material is the key to developing high-energy, high-power density lithium-ion batteries. In recent years, sodium-ion batteries (SIBs) have attracted extensive research attention due to their abundant and cheap sodium resources. A schematic diagram of the operating principle of a sodium ion battery is shown in Fig. 1d.41
Researchers modify materials to achieve more outstanding properties through various techniques such as doping, coating, controlling the structure of the material, and pre-treatment. Among the various dopants, fluorine is a very common and efficient dopant because of its abundant and cheap source. It is well known that F-doped materials have a wide range of applications, and we have searched the Web of Science for articles on F-doped material applications over the last decade and found that they are highly represented in photocatalytic, electrocatalytic, LIB and SIB applications. Fig. 2 summarises the number of SCI papers published on F-doped materials in photocatalysis, electrocatalysis, and batteries in the last decade (up to September 2022). It is clear that F-doped materials are receiving increasing attention in these applications.
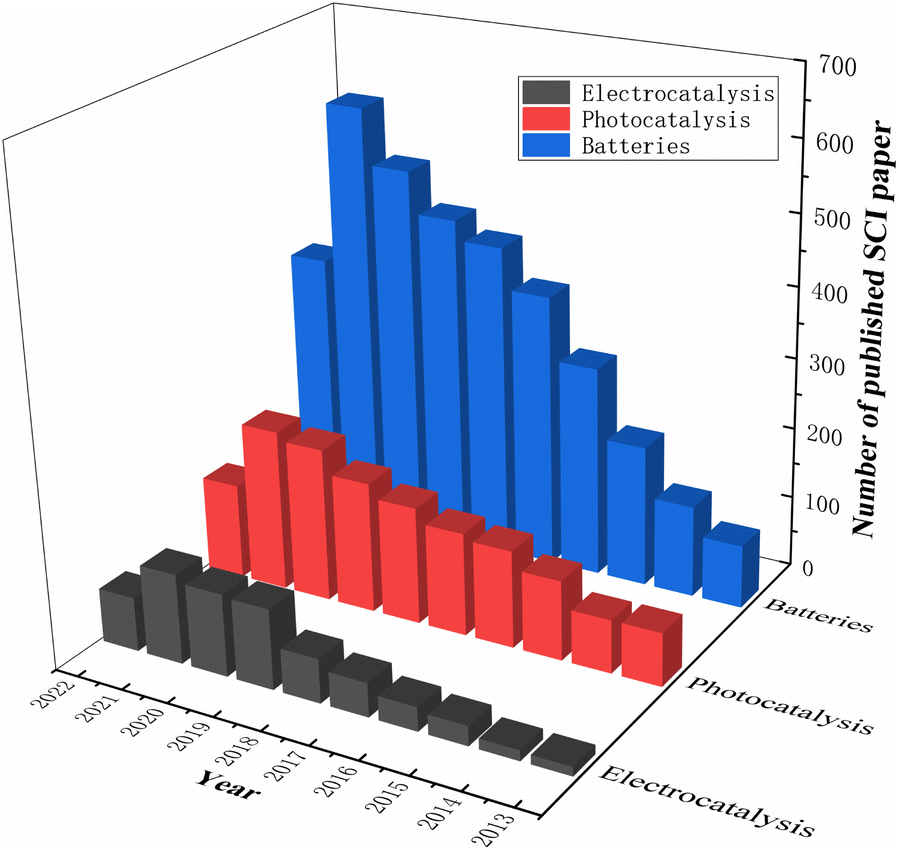 | ||
| Fig. 2 Statistics of F-doped materials' core publications in photocatalysis, electrocatalysis and batteries. | ||
2 Applications and synthetic strategies for F-doped materials in catalysis
2.1 Photocatalysis
Yu et al. fabricated square-shaped TiO2 nanocrystals doped with different F contents via the sonochemical method. The doping of the optimal content of F (1.3 mol%) gives a 5.3 times increase in the phenol degradation rate (Fig. 3a). The high photocatalytic degradation activity of the doped TiO2 is due to the factor that the F doping increases the number of hydroxyl groups on the TiO2 surface, which effectively reduces the recombination rate of photogenerated electron/hole pairs and thus generates more ·OH radicals to decompose phenol molecules.44
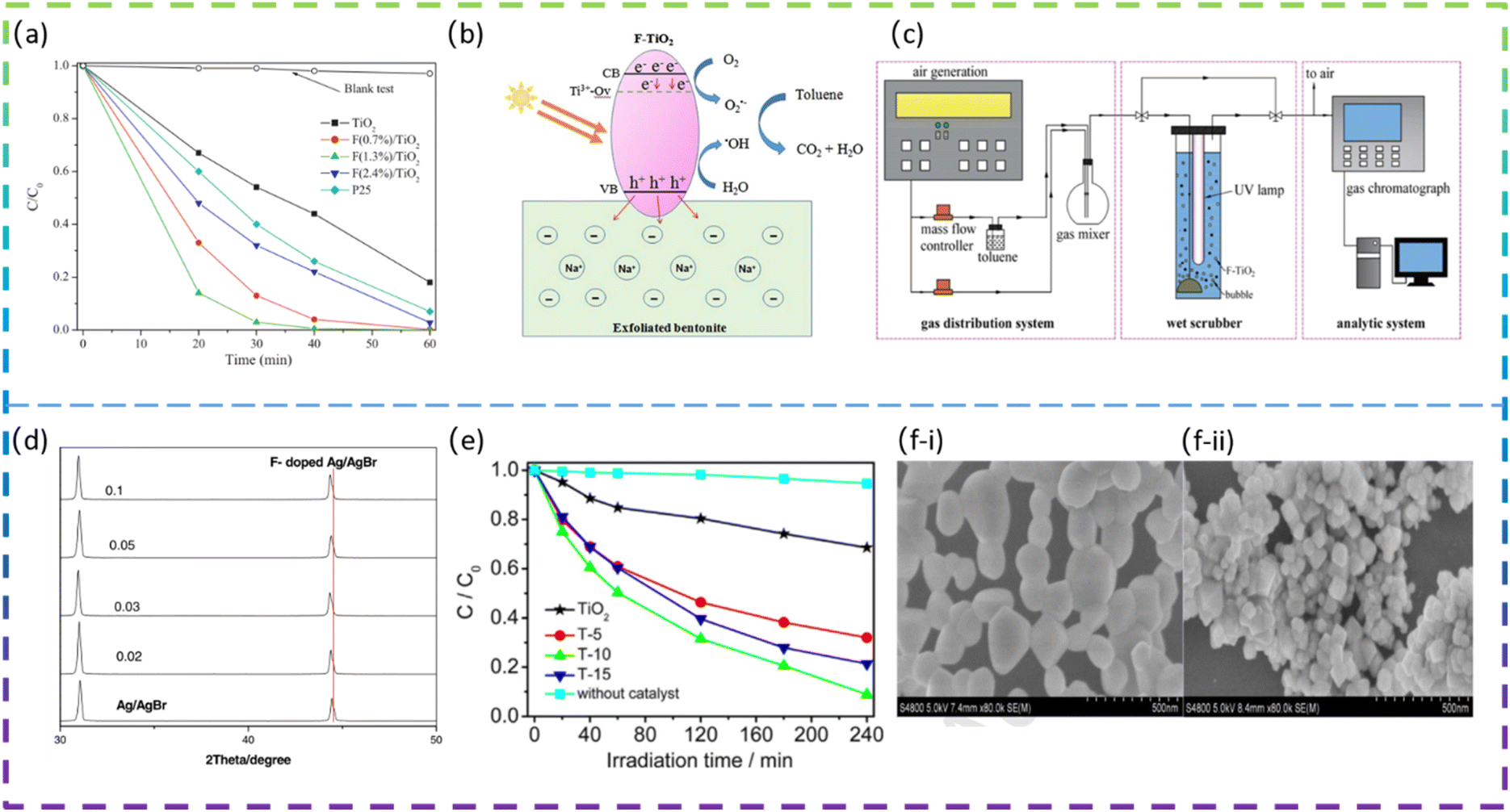 | ||
| Fig. 3 (a) Concentration changes of the phenol as a function of light irradiation time. (b) Proposed mechanism for the photocatalytic degradation of toluene on the TF5-400/EB composite. (c) Schematic of the experimental setup. (d) Partially enlarged XRD patterns of undoped and F-doped Ag/AgBr. (e) Photocatalytic degradation of methylene blue by TiO2, T-5, T-10, and T-15 under visible light irradiation. SEM images of the as-prepared CeO2 nanoparticles (f-i) and F(1/3)-CeO2 nanoparticles (f-ii). | ||
Wang et al. prepared an F-doped TiO2/exfoliated bentonite (TF/EB) composite by dispersing F-doped TiO2 (TF) nanoparticles on an exfoliated bentonite (EB) using a facile sol–gel method. The presence of F− ions increases the percentage of exposed reactive facets on TiO2. In addition, F− ions doped in the TiO2 lattice create oxygen vacancies and Ti3+ defects, which affect the transfer and migration of photocatalytic carriers and induce visible light photocatalytic activity. Fig. 3b shows the proposed mechanism for the improved photocatalytic performance of the TF5-400/EB (F/Ti atomic ratio was 5% and calcination was done at 400 °C) photocatalyst.10 Xie et al. synthesized F-doped TiO2 and employed it in wet-scrubbing coupled with the photocatalytic oxidation (WPCO) process. The experimental setup is shown in Fig. 3c. Toluene removal efficiency reached 80% over F-TiO2, while it was only 25% over TiO2 in the WPCO process. In addition, its corresponding mineralization capacity is significantly higher, with CO2 from the oxidation of toluene in the coupling reaction being approximately 92 ppmv. The F-doping produces more mobile ·OH, which is responsible for the excellent toluene degradation and mineralization photocatalytic activity.11
Yu et al. successfully synthesized F-doped TiO2 (F-TiO2) using a modified sol–gel method. The results showed that the photocatalytic degradation of methylene blue by F-TiO2 was enhanced, with a maximum degradation rate of 91% for F-TiO2 and 32% for pure TiO2 under visible light irradiation. The doping of F in TiO2 increases the absorption of visible light, promotes the separation of photogenerated electrons and holes, and enhances the photocatalytic oxide species; the doping of F also leads to enhanced surface acidity and promotes the adsorption of reactant molecules, thus improving the photocatalytic activity of TiO2.47 Miao et al. synthesized pure CeO2 and fluorine-doped CeO2 (F-doped CeO2) using a low-temperature solution combustion method and subsequent heat treatment in air. The prepared samples doped with different nominal mass ratios of sodium fluoride were denoted as F(x)-CeO2, where x is 1/10, 3/10, 1/3, and 2/5, respectively. It is found that F-doping makes the CeO2 nanoparticles smaller in size and the CeO2 nanocubes formed exposed a higher proportion of the reaction surface. The SEM images of pure CeO2 and F-doped CeO2 are shown in Fig. 3f-i and f-ii. It can be clearly seen that pure CeO2 (Fig. 3f-i) has an irregular spherical morphology with an average diameter of about 100 nm, with some of the particles agglomerated into larger ones. In contrast, F(1/3)-CeO2 (Fig. 3f-ii) has a size of about 50 nm. The band gap is estimated to be 3.16 eV and 2.88 eV, for pure CeO2 and F-doped CeO2 nanocubes, respectively. Interestingly, the F-doped CeO2 nanoparticles exhibited excellent photocatalytic activity for the degradation of organic pollutants under UV and visible light irradiation. The apparent reaction rate constant k for methylene blue decomposition on the optimized F-doped CeO2 nanocubes is 9.5 times higher than that on pure CeO2 and 2.2 times higher than that on commercial TiO2, respectively.48
Rueda-Salaya et al. prepared zinc oxide catalysts doped with different amounts of fluoride by the sol–gel technique. Compared to undoped ZnO, F-doped ZnO has better photocatalytic performance for DCF mineralization under simulated solar radiation conditions. The DCF of fluorine-doped ZnO degradation in solar radiation is shown in Fig. 4a. Under the optimized experimental conditions (1 g L−1 ZnO-F20 (ZnO semiconductor modified by the incorporation of 10 wt% fluorine) catalyst, DCF solution pH maintained at 6.5), the DCF was completely degraded, the chloride ions were completely released and the energy accumulated during drug degradation was 400 kJ m−2 with a mineralization rate of about 90%. The results show that as the amount of fluoride-doping increases, the crystal size decreases, the band gap value decreases slightly, and the specific surface area increases. F− substituent addition to the ZnO network reduces the rate of complexation of e−/h+ pairs and improves photocatalytic activity under simulated solar irradiation.49 Vitiello et al. synthesized fluorine-doped ZnO nano- and meso-crystals by a hydrothermal approach and investigated the following F atomic concentrations: 0 at% (bare, ZnO:F0), 0.25 at% (ZnO:F0.25), 0.5 at% (ZnO:F0.5) and 1 at% (ZnO:F1). Surprisingly, the sample with the lowest fluorine content (ZnO:F0.25) had the best photocatalytic efficiency. As the amount of F doping increases, there is a tendency for the defect concentration to increase. It is argued that the excess dopant increases the extent of defects in the ZnO crystal structure and becomes recombination centers for photogenerated carriers, leading to the deterioration of photocatalytic activity. In fact, the introduction of F− ions into the ZnO lattice can increase the concentration of free electrons in the conduction band and increase the n-type characteristics of the sample. ZnO:F0.25 has a higher hydrophilicity, which means a higher rate of hydroxyl radical production under UV irradiation. All doped samples exhibited high rates of DCF degradation and mineralization, mainly due to their high efficiency in generating hydroxyl radicals (·OH). The ZnO:F synthesis method and the mechanism of photocatalytic degradation of DCF are shown in Fig. 4b.50
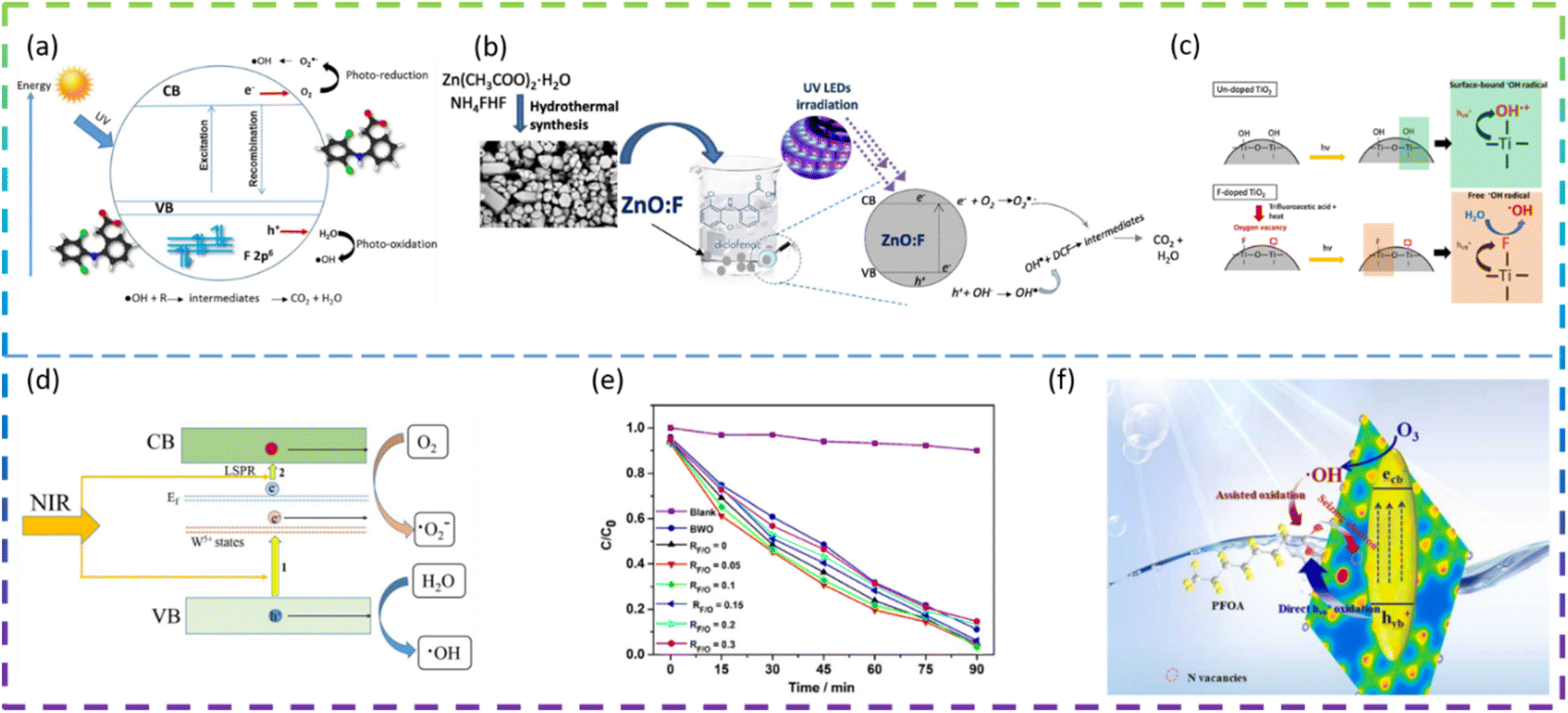 | ||
| Fig. 4 (a) Schematic representation of DCF degradation using fluorine-doped zinc oxide under solar irradiation. (b) ZnO:F synthesis method and photocatalytic degradation mechanism of DCF. (c) Schematic diagram showing formation of surface-bound and free hydroxyl radicals on TiO2. (d) Schematic illustration of the NIR photocatalytic mechanism of F-doped (NH4)0.33WO3. (e) Photocatalytic degradation of RhB solution by BWO and the F-doped rGO-BWO samples with different RF/O values under visible light (λ > 400 nm) illumination. (f) Schematic diagram of F-doped g-C3N4 photocatalytic degradation of PFOA. | ||
Samsudin et al. prepared mesoporous nanocrystalline F-doped TiO2 for the photocatalytic degradation of atrazine using trifluoroacetic acid (TFA) as a fluorine precursor using a modified sol–gel method. The photocatalytic activity of F-doped TiO2 is enhanced under UV light irradiation compared to undoped TiO2. The F-doped TiO2 also exhibits visible photocatalytic activity, which is due to the exogenous light absorption triggered by the oxygen vacancies and Ti3+. The excellent photocatalytic activity under UV irradiation is due to the effective electron and hole separation and the enhanced absorption intensity in the UV region. In addition, upon photoexcitation, free radicals are generated on the surface of F-doped TiO2, while less reactive hydroxyl radicals are generated on the surface of undoped TiO2 (Fig. 4c). The higher fluorine doping in TiO2 does not necessarily control the enhanced photocatalytic activity due to the excess oxygen vacancies acting as carrier traps. This suggests that an optimum doping condition exists for F-doped TiO2 to maximize photocatalytic activity.51
Kang et al. synthesized F-doped (NH4)0.33WO3 as a novel near-infrared (NIR) photocatalyst. The NIR degradation of RhB by F-doped (NH4)0.33WO3 was 83% within 180 min. The best F-doped (NH4)0.33WO3 showed a NIR degradation rate of 0.0102 min−1 for RhB, which is about 8.5 times higher than that of (NH4)0.33WO3. The enhanced NIR photocatalytic performance of F-doped (NH4)0.33WO3 can be attributed to the remarkably enhanced generation and separation of NIR localized surface plasmon resonance (LSPR) induced electron–hole pairs (Fig. 4d).52 Yu et al. synthesized reduced graphene oxide-Bi2WO6 (rGO-BWO) photocatalysts with the different RF/O values (molar ratio of the F molar mass and the O molar mass of Bi2WO6) via a one-step hydrothermal method. The photocatalytic activity of the F-doped rGO-BWO with RF/O = 0.05 was superior to that of rGO-BWO and other F-doped rGO-BWO under 500 W Xe lamp irradiation. F−-ion doping enhances surface acidity, generates oxygen vacancies, increases active centers, and effectively inhibits electron–hole pair complexation through the rapid migration of photogenerated carriers.53
Chen et al. regulated N vacancies, hydrophobic sites, and the electron-rich zone by doping F into g-C3N4 (CN) to accelerate photocatalytic ozonation of PFOA. F-CN activity was better than CN activity, with F-CN/Vis/O3 removal of PFOA at 74.3% and CN/Vis/O3 removal of PFOA at only 57.1%. The doping of F narrows the Eg of F-CN and shifts the VB potential downwards, which facilitates the generation of more Ecb− (photogenerated electron)–Hvb+ (photogenerated hole) and improves the oxidation of Hvb+. In addition, the doping of F leads to more N vacancies on F-CN, which facilitates the Hvb+ oxidation of PFOA. Hydrophobic sites and electron-rich regions induced by F atoms can facilitate the mass transfer of O3 and PFOA, promote the reaction of O3 with Ecb− and inhibit the binding of Ecb−–Hvb+. The schematic diagram of the F-doped g-C3N4 photocatalytic degradation of PFOA is shown in Fig. 4f.54
Zhang et al. prepared Br-doped Bi2O2CO3 nanosheets via a one-step hydrothermal reaction. Under simulated solar irradiation, the sample with a trace Br content of 1.17 wt% showed the highest degradation efficiency for RhB, with a rate constant k that was 3.6 times higher than that of pure Bi2O2CO3. According to crystal structure and Density Functional Theory (DFT) calculations, the interstitial doping of Br from cetyltrimethylammonium bromide (CTAB) into the Bi2O2CO3 lattice as a dopant changes the band gap position of Bi2O2CO3, generating delocalized impurity states at the Fermi energy level. The improved electronic structure gives Br-doped Bi2O2CO3 a broader light-harvesting and accelerated charge migration.56
Zai et al. prepared rose-like I-doped Bi2O2CO3 microspheres via a hydrothermal process. Under visible light (λ > 400 nm), RhB is completely degraded within 6 min and about 90% of Cr(VI) is reduced after 25 min. According to the crystal structure of Bi2O2CO3 and DFT calculations, I− can partially replace CO32− in Bi2O2CO3, narrowing its band gap and enhancing its visible light absorption.57
Ding et al. designed P-doped CeVO4 nanobelts by electrospinning process. In consecutive levofloxacin photocatalytic experiments, the P-doped CeVO4 isomeric nanoribbons exhibited excellent photocatalytic activity, high long-term stability, and excellent reusability. The partial phase transition from tetragonal zircon (t-CeVO4) to monoclinic monazite (m-CeVO4) was induced by P-doping, and such a homogeneous junction resulted in a fully optimized t-CeVO4/m-CeVO4 interface with enhanced visible light capture and electron–hole transfer separation, contributing to effective visible light-induced photocatalysis.59
Shen et al. prepared original CTF-1 (original covalent triazine frameworks) and a series of CTFSx doped with different contents of S. The S-doped CTF exhibited excellent photocatalytic activity and cyclic stability compared to the original CTF-1. CTFS5 (covalent triazine frameworks doped with a mass of 0.02 g S) showed the best photocatalytic performance. The visible photocatalytic reaction showed a degradation rate of 53.2% for methyl orange and 84.7% for bisphenol A within 6 h. The degradation rate constants were 0.145 h−1 and 0.29 h−1, which were 3.6 and 5.8 times higher than those of CTF-1, respectively. Further studies have shown that enhanced visible light absorption, reduced free carrier recombination, rapid separation and transfer of photogenerated electrons and holes, and increased –OH oxidation capacity are important factors in the significant enhancement of their photocatalytic activity.60
In order to better demonstrate the synthesis and photocatalytic applications of F-doped materials/other element-doped materials, their preparation methods and photocatalytic properties are summarised in Table 1.
| No. | Materials | Preparation | Fluorine source/other doping element sources | Object | Light source | Photocatalytic efficiency | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | F-doped TiO2 | Sonochemical method | NH4F | 0.050 g L−1 phenol | 365 nm UV lamp | 90% in 60 min | 44 |
| 2 | F-doped TiO2/exfoliated bentonite | Sol–gel method | NH4F | 2000 ppb toluene | UV | 71.6% in 80 min | 10 |
| 3 | F-doped TiO2 | One-step hydrothermal method | NH2HF2 | 30 ppmv toluene | UV | 80% in 120 min | 11 |
| 4 | F-doped Ag/AgBr | Ion exchange method | NaF | 20 mg L−1 methyl orange | Visible-light | 91.9% in 12 min | 12 |
| 5 | F-doped TiO2 | Modified sol–gel method | NH4F | 10 mg L−1 methylene blue | Visible-light | 91% in 240 min | 47 |
| 6 | F-doped CeO2 | Solution combustion method | NaF | 10 mg L−1 methylene blue | UV | 92.1% in 6 min | 48 |
| 7 | F-doped ZnO | Sol–gel method | NH4F | 10 mg L−1 DCF | Simulated solar radiation | 88.4% | 49 |
| 8 | F-doped ZnO | Hydrothermal approach | NH4FHF | 10 mg L−1 DCF | UV | 85% in 30 min | 50 |
| 9 | F-doped TiO2 | Modified sol–gel method | Trifluoroacetic acid | 0.5 mg L−1 atrazine | UV and visible light | 53.9 and 36.6% in 180 min | 51 |
| 10 | F-doped (NH4)0.33WO3 | HF | 20 mg L−1 RhB | Near-infrared | 83% in 180 min | 52 | |
| 11 | F-doped reduced graphene oxide-Bi2WO6 | One-step hydrothermal method | NaF | 5 mg L−1 RhB | Visible-light | About 95% in 90 min | 53 |
| 12 | F-doped g-C3N4 | — | NH4F | 10 mg L−1 PFOA | Visible-light | 74.3% in 4 h | 54 |
| 13 | Cl-doped Bi2MoO6 | Hydrothermal approach | NaCl | 200 mL 10−5 M RhB | Visible-light | 99.5% in 160 min | 55 |
| 14 | Br-doped Bi2O2CO3 | Hydrothermal approach | Cetyltrimethylammonium bromide | 10−5 M RhB | UV-vis | 100% in 30 min | 56 |
| 15 | I-doped Bi2O2CO3 | Hydrothermal approach | NaI | 10−5 M RhB | Visible-light | 100% in 6 min | 57 |
| 16 | N-doped TiO2 | Hydrothermal method | Urea | 10 mg L−1 methylene blue | Visible-light | 87.9% in 180 min | 58 |
| 17 | P-doped CeVO4 | Electrospinning process | (NH4)2HPO4 | 20 mg L−1 levofloxacin | Simulated sunlight | 92.9% in 140 min | 59 |
| 18 | S-doped covalent triazine | — | S | 1 mg L−1 methyl orange | Visible-light | 53.2% in 6 h | 60 |
2.2 Electrocatalysis
Ni et al. synthesized fluorine-doped caged porous carbon (F-CPC) as an efficient catalyst for CO2RR by using a polymer-derived method. Fig. 5a shows the synthesis process of the F-CPC sample. The optimized F-CPC has a large specific surface area, medium mesopores, and abundant micropores as well as a high electrical conductivity. When used as a catalyst for the CO2RR, F-CPC exhibits faradaic efficiency (FE) of 88.3% for CO at −1.0 V vs. RHE with a current density of 37.5 mA cm−2. Fluorine doping can adjust the electronic structure by generating a positive charge density, thus enhancing the adsorption of *COOH and inhibiting the hydrogen evolution reaction (HER).30 Han et al. fabricated a fluorine-tuned single-atom catalyst with an ultrathin nanosheet morphology (only ∼1.25 nm) and high Ni content of 5.92 wt% by a polymer-assisted pyrolysis approach (Fig. 5b). As a result, this catalyst with unique structural features exhibits a remarkable electrocatalytic performance for CO2-to-CO conversion with FE over 95% in a wide potential range and an outstanding CO evolution rate of 1146 mmol gcat−1 h−1 at −0.97 V vs. RHE. F-doping alters the electronic configuration of the central Ni–N4 site, thereby lowering the energy barrier to CO2 activation, which facilitates the generation of key *COOH intermediates.31 Chen et al. reported the facile construction of F-doped Fe-N-C single-site catalysts (SSC) and the electron-withdrawing character of fluorine could stabilize Fe3+ sites, which promotes the CO faradaic efficiency (FECO) from the volcano-like highest value (88.2% @ −0.40 V) to the high plateau (>88.5% @ −0.40 to −0.60 V), with a much-increased partial current (JCO) (from 3.24 to 11.23 mA cm−2). The fluorine doping stabilizes the Fe3+ sites and enriches the defects, thus promoting the CO2RR thermodynamically, inhibiting competitive hydrogen evolution reactions, and increasing the electroactive surface area kinetically, improving charge transfer. The Fe-FN-C was synthesized by a convenient adsorption–pyrolysis route, as shown in Fig. 5c.29
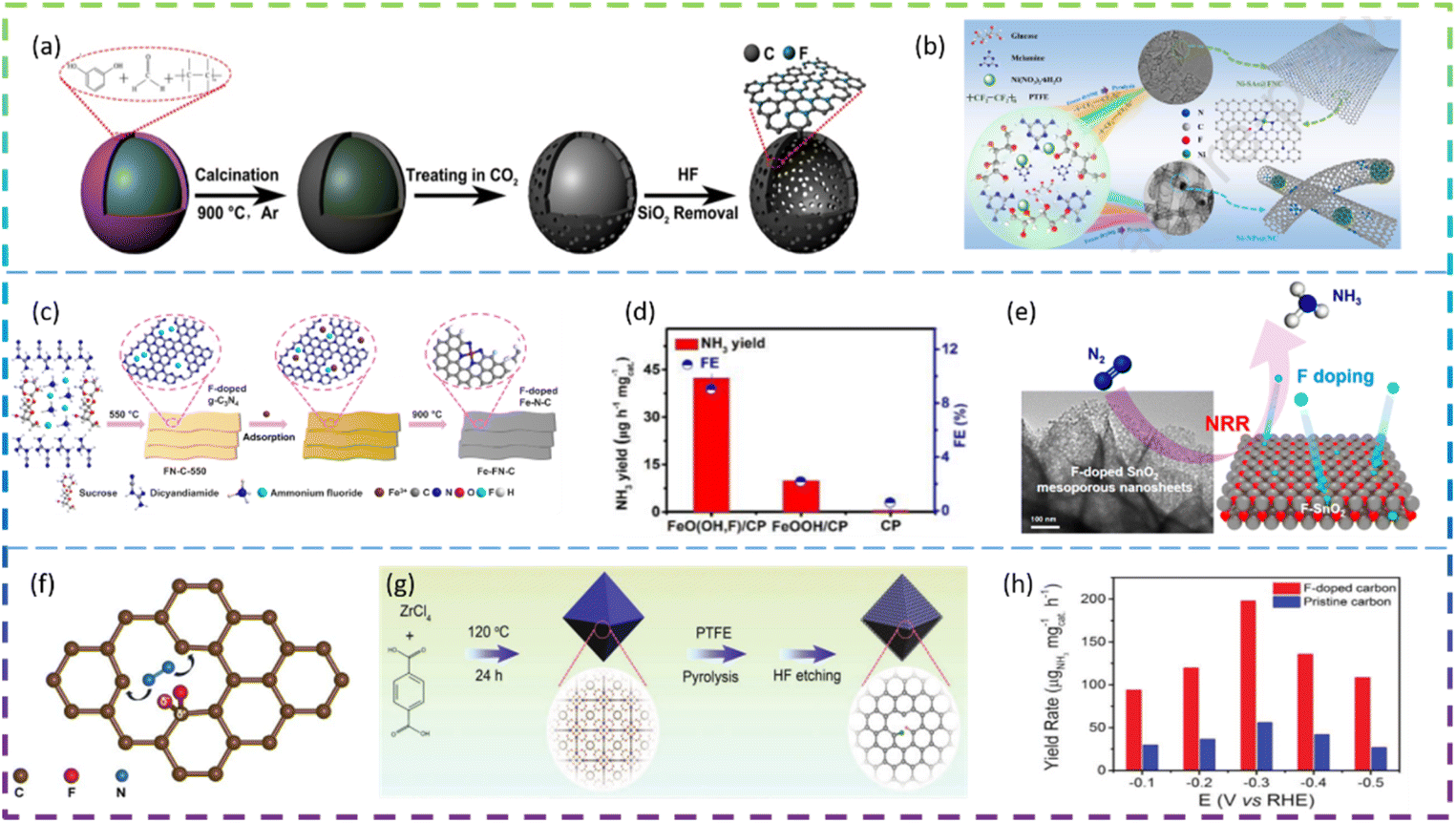 | ||
| Fig. 5 (a) Schematic illustration to the formation process of the fluorine-doped cagelike porous carbon (F-CPC). (b) Schematic illustration for the formation and structures of Ni-SAs@FNC and Ni-NPs@NC. (c) Schematic diagram for the synthesis of Fe-FN-C. (d) NH3 yields and FEs of different electrodes at −0.60 V after electrolysis for 2 h. (e) Schematic diagram of electrocatalytic reduction of N2 on fluorine-doped SnO2 porous nanosheets. (f) Schematic of the atomic structure of fluorine-doped carbon. (g) Schematic of the synthetic procedure for F-doped carbon. (h) Yield rate for NH3 of F-doped carbon and pristine carbon at different applied potentials. | ||
Zhu et al. reported that the NRR activity of β-FeOOH nanorods can be greatly improved by simple F-doping. When tested at −0.60 V vs. the reversible hydrogen electrode (RHE) in 0.5 M LiClO4, these β-FeO(OH, F) nanorods obtain optimal NH3 yield (42.38 μg h−1 mgcat−1) and FE (9.02%), much higher than those of pristine β-FeOOH (10.01 μg h−1 mgcat−1, 2.16%) (Fig. 5d). DFT calculations show that the reaction energy barrier for β-FeO(OH, F) (0.24 eV) is lower than that for β-FeOOH (0.59 eV), resulting in an enhanced activity. Overall, their theoretical calculations show that the performance of the NRR can be significantly improved by the substitution of OH groups by F atoms and therefore β-FeO(OH, F) is a promising electrocatalyst for the NRR, which agrees well with the experimental electrochemical results.33 Liu et al. developed fluorine (F)-doped SnO2 mesoporous nanosheets on carbon cloth (F-SnO2/CC) as an efficient NRR electrocatalyst. In 0.1 M Na2SO4, F-SnO2/CC exhibited an NH3 yield of 19.3 μg h−1 mg−1 and an FE of 8.6% at −0.45 V. It has high durability and is superior to most of the reported NRR catalysts. Specifically, the DFT results reveal that F-doping increases conductivity and increases the density of positive charges on the active Sn sites, thereby reducing the reaction energy barrier and enhancing NRR activity. The schematic diagram of the electrocatalytic reduction of N2 on fluorine-doped SnO2 porous nanosheets is shown in Fig. 5e.34 Yuan et al. reported that a non-metallic fluorine doped carbon improved the efficiency of the electrochemical NRR. The fluorine-doped carbon exhibits better NRR performance than the undoped carbon, with a peak ammonia production rate (6.9 μg h−1 cm−2) and a corresponding high faradaic efficiency (12.1%) at −0.55 V versus the reversible hydrogen electrode (RHE). The doping of fluorine allows the electron distribution of the carbon to be adjusted, which provides positively charged carbon sites that are more likely to adsorb N2 rather than H+ under acidic water conditions. Doping of the carbon skeleton with F atoms can effectively lead to the positive polarisation of adjacent carbon atoms. These strongly polarised carbon atom positions can serve as effective locations for the adsorption of protons by the N2 molecule (Fig. 5f).35 Liu et al. introduced F atoms into a 3D porous carbon framework (F-doped carbon) for N2 electroreduction and developed a highly efficient metal-free catalyst. Fig. 5g exhibits the synthetic procedure of F-doped carbon. At −0.2 V versus the reversible hydrogen electrode (RHE), the F-doped carbon achieves the highest FE of 54.8% for NH3, which is 3.0 times as high as that (18.3%) of pristine carbon frameworks. It is worth noting that at −0.3 V versus RHE, the yield rate of F-doped carbon for NH3 reaches 197.7 μgNH3 mgcat−1 h−1 (Fig. 5h). Mechanistic studies have shown that the improved electroreduction performance of F-doped N2 is mainly due to the enhanced N2 binding strength and easier dissociation of N2 into *N2H. Due to the difference in electronegativity between F and C atoms, F bonds with C atoms to form Lewis acid sites. As a result, the repulsion between the Lewis acid site and the proton H inhibits the activity of the H2 evolution reaction, thus improving the selectivity of the electroreduction of N2 to NH3.32
2.2.3.1 2-Electron pathway reactions. Hydrogen peroxide (H2O2) is an important environmentally friendly chemical that is widely used in the chemical industry, sanitation, and environmental remediation. The current industrial synthesis of hydrogen peroxide is carried out by the anthraquinone process, which has the disadvantages of fragmented production, high energy consumption, the generation of large amounts of organic by-products, and the need to transport the resulting H2O2 to the point of use.70,71 Therefore, the electrochemical synthesis of H2O2via the two-electron pathway of the oxygen reduction reaction (ORR) becomes an attractive alternative method.
Zhao et al. prepared F-doped porous carbon with excellent performance for electrochemical synthesis of H2O2 by carbonization of MIL-53 (Al). The F content has an influence on the production of H2O2, and the best catalytic activity for H2O2 electrosynthesis is achieved when the F content is 3.41 at%. The resultant F-doped porous carbon (FPC) catalysts exhibited good H2O2 selectivity of 97.5–83.0% and the H2O2 production rate could reach 112.6–792.6 mmol h−1 g−1 over the potential range of 0.2 V to −0.3 vs. RHE (pH 1). The addition of CF2,3 facilitates the adsorption of O2 and the desorption of OOH, resulting in a high selectivity for the two-electron reduction reaction of oxygen. The synthetic process for the fabrication of F-doped porous carbon is shown in Fig. 6a.72 Wang et al. used F to modify carbon nanotubes (CNT) and H2O2 was produced with a gas diffusion electrode (GDE) based on the F-doped CNT catalyst as cathode. The results show that F-doping can improve the oxygen reduction activity and the selectivity of H2O2, which in turn improves the yield of H2O2. The F-CNT prepared with 0.6 M HF (CNT-F-0.6) showed significantly higher hydrogen peroxide production (47.6 mg L−1) and current efficiency (89.5%) than carbon nanotubes (29.6 mg L−1, 70.1%) at a bias voltage of −1.3 V (vs. SCE) and pH 7, and still maintained high catalytic activity for five consecutive reaction cycles. The improved performance of the F-modified CNT-GDE can be attributed to the CF2 and CF3 on the F-doped CNT catalyst (Fig. 6b).73 Zeng et al. synthesized an F-doped carbon electrocatalyst in an F2 atmosphere. The fluorine-doped carbon was synthesized in a sealed fluidized bed, and the device is shown in Fig. 6c. H2O2 was generated via a two-electron pathway using F-doped carbon as the cathode. 3 h of electrolysis resulted in a H2O2 concentration of 2837.5 mg L−1 and a selectivity of 95.7%. And 12 cycles (3 h each) of H2O2 concentration remained constant, indicating good stability of the catalyst. Fluorine has high electronegativity and induces polarisation of adjacent carbon, creating active centers and increasing the force between oxygen and carbon. The X-ray photoelectron spectra (XPS) and DFT data confirm that the CF2 bond type facilitates the adsorption of O2 and the desorption of OOH, thereby increasing the ability of the catalyst to produce H2O2.74
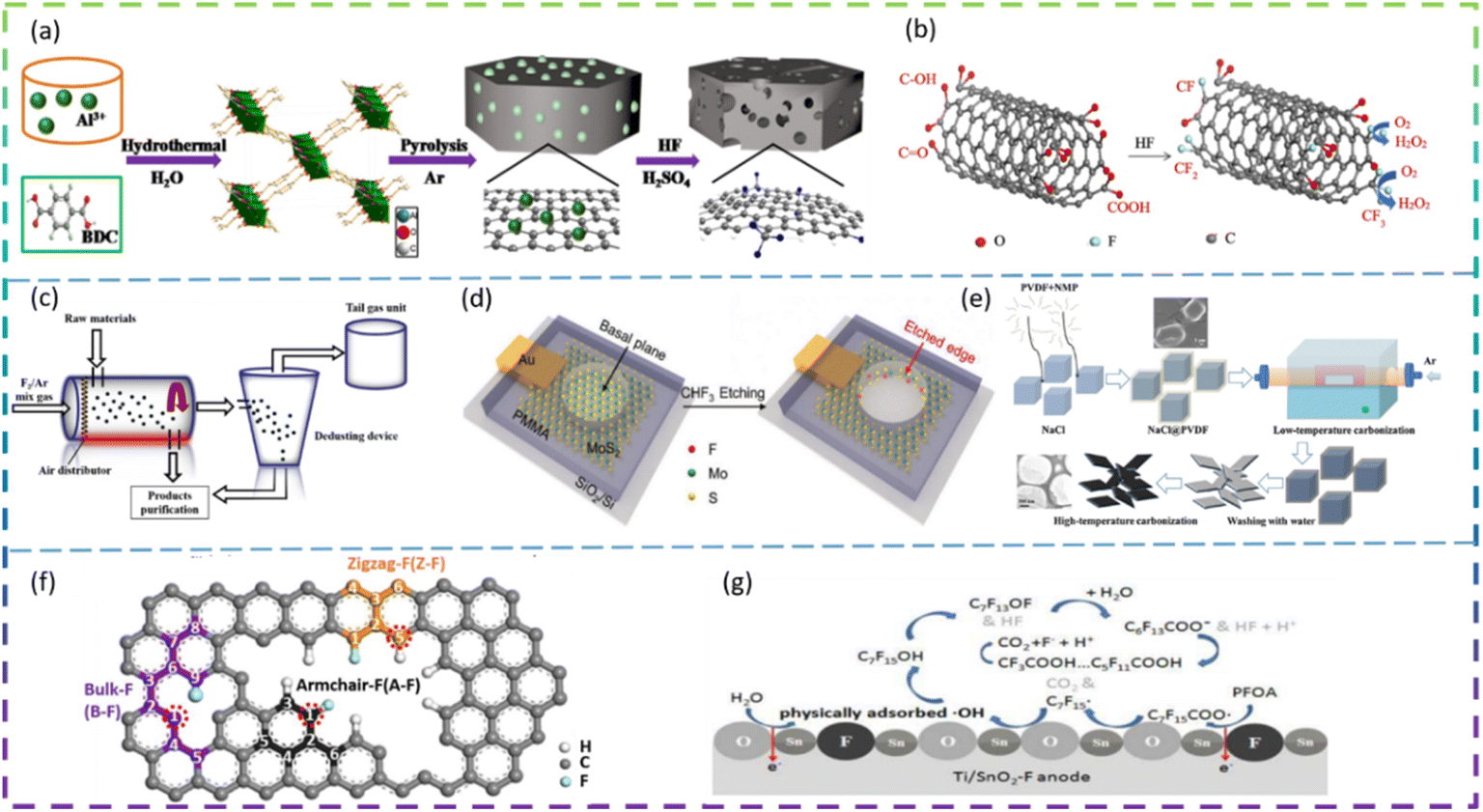 | ||
| Fig. 6 (a) Procedure for the synthesis of F-doped hierarchically porous carbon catalysts. (b) Enhanced two-electron oxygen reduction process by F-CNT. (c) Schematic diagram of the synthesis device of F-doped carbon. (d) Schematic illustration of in situ creation and doping of etched edges on MoS2 by CHF3 plasma. (e) Illustration of the synthesis procedure for FC. (f) Schematic depicting the structures of A-F, B-F and Z-F. (g) Schematic diagram of PFOA degradation on the Ti/SnO2-F electrode. | ||
2.2.3.2 4-Electron pathway reactions. The ORR is a key reaction in rechargeable metal–air cells and fuel cells, and has been extensively studied due to the pollution problems caused by the use of fossil fuels.75,76 Chang et al. firstly produced 2D porous F-doped carbon nanosheets by using polyvinylidene fluoride as precursor and NaCl as the template (Fig. 6e). FC-900 (pyrolyzed at 900 °C) shows an impressive ORR due to the large specific surface area (1031 m2 g−1) and the highly active sites. The FC nanosheets exhibit high ORR activity as Pt/C catalysts under alkaline conditions due to the synergistic effect of defects and appropriate F dopants producing highly active sites, thus ensuring high-performance zinc–air batteries. When the samples were further calcined at 900 °C for different times, the ORR activity of the samples obtained improved slightly, indicating that the defects have much less influence on the ORR performance than the fluorine doping. The ORR activity on F-doped graphene was studied by DFT. In total, three kinds of F doping patterns are modeled for DFT calculation, namely, the F doped on bulk (denoted as B-F), armchair edge (denoted as A-F), and zigzag edge (denoted as Z-F), with their structures shown in Fig. 6f.38
Zhang et al. used the CHF3 plasma etching to create F-doped MoS2 edge sites with enhanced HER performance and stability. The doping of the more electronegative fluorine atoms at the MoS2 edge positions resulted in a fivefold enhancement in activity compared to the original edge and was attributed to a more moderate binding energy of the hydrogen species (Fig. 6d). The study provides two-dimensional materials as a platform for understanding edge site doping effects at the atomic level and offers a new avenue for the design of efficient catalysts.37
Yang et al. prepared an F-doped Ti/SnO2 (Ti/SnO2-F) electrode using tin tetrafluoride as a novel single-source precursor, which provided a promising anode material for wastewater treatment containing highly stable PFOA. The Ti/SnO2-F electrode has higher oxidation activity and longer service life than the Ti/SnO2–X (X = Cl, Br, I, Sb) electrode, decomposing over 99% of the PFOA (50 mL 100 mg L−1) in 30 min of electrolysis. The similar ionic radii of F and O as well as the strong electronegativity of F lead to high electrochemical stability and a smooth surface, generating weakly adsorbed ·OH. Details of the mechanism of PFOA degradation on the Ti/SnO2-F electrode are shown in Fig. 6g.36
Ji et al. developed P-doped Ag nanoparticles embedded in N-doped carbon nanoflakes (denoted as P-Ag@NC) for effective hydrogen evolution electrocatalysis. P-Ag@NC exhibits stronger catalytic activity than undoped Ag@NC, requiring an overpotential of 78 mV to drive 10 mA cm−2 in 0.5 M H2SO4, which is 198 mV lower than for Ag@NC. In addition, the catalyst exhibits strong long-term electrochemical durability. DFT calculations show that P doping optimizes the hydrogen adsorption free energy to a more thermoneutral value.82
Li et al. demonstrated sulfur-doped graphene (S-G) can be utilized to stabilize ultrafine (sub-2 nm) Au25(PET)18 clusters to enable stable NRR without significant structural degradation. The Au25@S-G exhibits an ammonia yield rate of 27.5 μgNH3 mgAu−1·h−1 at −0.5 V with faradaic efficiency of 2.3%. Sulfur dopants play a key role in stabilizing the chemical state and coordination environment of the Au atomic clusters.83
In order to better demonstrate the synthesis and electrocatalytic applications of F-doped materials/other element-doped materials, their preparation methods and electrocatalytic properties are summarised in Table 2.
| No. | Materials | Preparation | Fluorine source/other doping element sources | Application | Performance | Ref. |
|---|---|---|---|---|---|---|
| 1 | F-doped cage like porous carbon | Polymer-derived method | HF | CO2 reduction reaction | FE of 88.3% for CO at −1.0 V vs. RHE with a current density of 37.5 mA cm−2 | 30 |
| 2 | F-doped single Ni atoms supported on graphitized carbon | Polymer-assisted pyrolysis approach | PTFE | CO2 reduction reaction | FE over 95% in a wide potential range and an outstanding CO evolution rate of 1146 mmol gcat−1 h−1 at −0.97 V vs. RHE | 31 |
| 3 | F-doped Fe-N-C | Convenient adsorption–pyrolysis route | NH4F | CO2 reduction reaction | FECO (>88.5% @ −0.40 to −0.60 V), JCO (11.23 mA cm−2) | 29 |
| 4 | F-doped β-FeOOH | NaF | N2 reduction reaction | NH3 yield (42.38 μg h−1 mgcat−1) and FE (9.02%) | 33 | |
| 5 | F-doped SnO2/CC | Facile solvothermal approach | NaF | N2 reduction reaction | NH3 yield of 19.3 μg h−1 mg−1 and a faradaic efficiency of 8.6% at −0.45 V (vs. RHE) | 34 |
| 6 | F-doped carbon | — | PTFE | N2 reduction reaction | Peak ammonia production rate (6.9 μg h−1 cm−2) and a corresponding high faradaic efficiency (12.1%) at −0.55 V vs. RHE | 35 |
| 7 | F-doped porous carbon | Polymer-assisted pyrolysis approach | PTFE | N2 reduction reaction | FE of 54.8% for the NH3 product at −0.2 V vs. RHE, the yield rate for NH3 reached 197.7 μgNH3 mgcat−1 h−1 | 32 |
| 8 | F-doped hierarchically porous carbon | — | HF | Oxygen reduction reaction | H2O2 selectivity of 97.5–83.0% and the H2O2 production rate reached 112.6–792.6 mmol h−1 g−1 in the potential range of 0.2 V to −0.3 V vs. RHE (pH 1) | 72 |
| 9 | F-doped modified carbon nanotubes | — | HF | Oxygen reduction reaction | H2O2 production (47.6 mg L−1) and current efficiency (89.5%) at a bias voltage of −1.3 V (vs. SCE) and pH 7 | 67 |
| 10 | F-doped carbon | — | F2 | Oxygen reduction reaction | The H2O2 concentration and selectivity of the catalyst can reach 2837.5 mg L−1 and 95.7% after electrolysis for three hours | 73 |
| 11 | F-doped MoS2 edge electrodes | Plasma etching strategy | CHF3 | Hydrogen evolution reactions | Overpotential of 160 mV (j = −0.02 nA μm−1), Tafel slope of the etched edge: 136 mV dec−1 | 37 |
| 12 | F-doped Ti/SnO2 | — | SnF4 | Degradation of PFOA | Decomposed over 99% of PFOA (50 mL, 100 mg L −1) within 30 min of electrolysis | 36 |
| 13 | Cl-doped ultrathin graphdiyne | Corrosion strategy | Cl2 | N2 reduction reaction | NH3 production rate of 10.7 μg h−1 cm−2 and a high FE of 8.7% at −0.45 V and −0.4 V, respectively | 79 |
| 14 | S,N dual-doped graphene-like carbon | Polymer-assisted pyrolysis | Dibenzyl sulfide and melamine | Oxygen reduction reaction | — | 81 |
| 15 | P-doped Ag | — | NaH2PO2 | Hydrogen evolution reactions | Overpotential of 78 mV to drive 10 mA cm−2 in 0.5 M H2SO4 | 82 |
| 16 | S-doped graphene | Solution-phase method | — | N2 reduction reaction | Ammonia production rate of 27.5 μgNH3 mgAu−1 h−1 with a FE of 2.3% at −0.5 V | 83 |
3 Applications and synthetic strategies for F-doped materials in rechargeable ion batteries
3.1 Lithium-ion batteries
Fluorine doping is used extensively in electrodes to optimize their electrochemical properties. For example, Bai et al. synthesized an F-doped Li4Ti5O12 anode material via the solid-state reaction. The F doping had no effect on the crystal structure of Li4Ti5O12 and the particle size of the samples remained at a uniform size of about 1 μm. A comparison of Fig. 7a left and right images indicates that the molecular structure of Li4Ti5O12 has not changed after F doping and also that it belongs to the Fd3m space group, except that the unit-cell contains one F atom at 32e sites replacing one O atom (Fig. 7a). In addition, the best performing material Li4Ti5O11.9F0.1 delivers superior rate capacities of 165, 162.1, 160.0, 142.3, 125.1, and 99.2 mA h g−1, at 0.2C, 0.5C, 1C, 3C, 5C, and 10C, respectively, which are higher than those of Li4Ti5O12 (150.0, 135.0, 123.5, 93.7, 76.4, and 56.8 mA h g−1 at the same C rates), and the cycling retention is 82.7% after 150 cycles at 5C. The Li4Ti5O11.9F0.1 electrode has higher charge transfer kinetics (Fig. 7b), lower electrode polarisation, and a higher lithium-ion diffusion coefficient than other electrodes.90
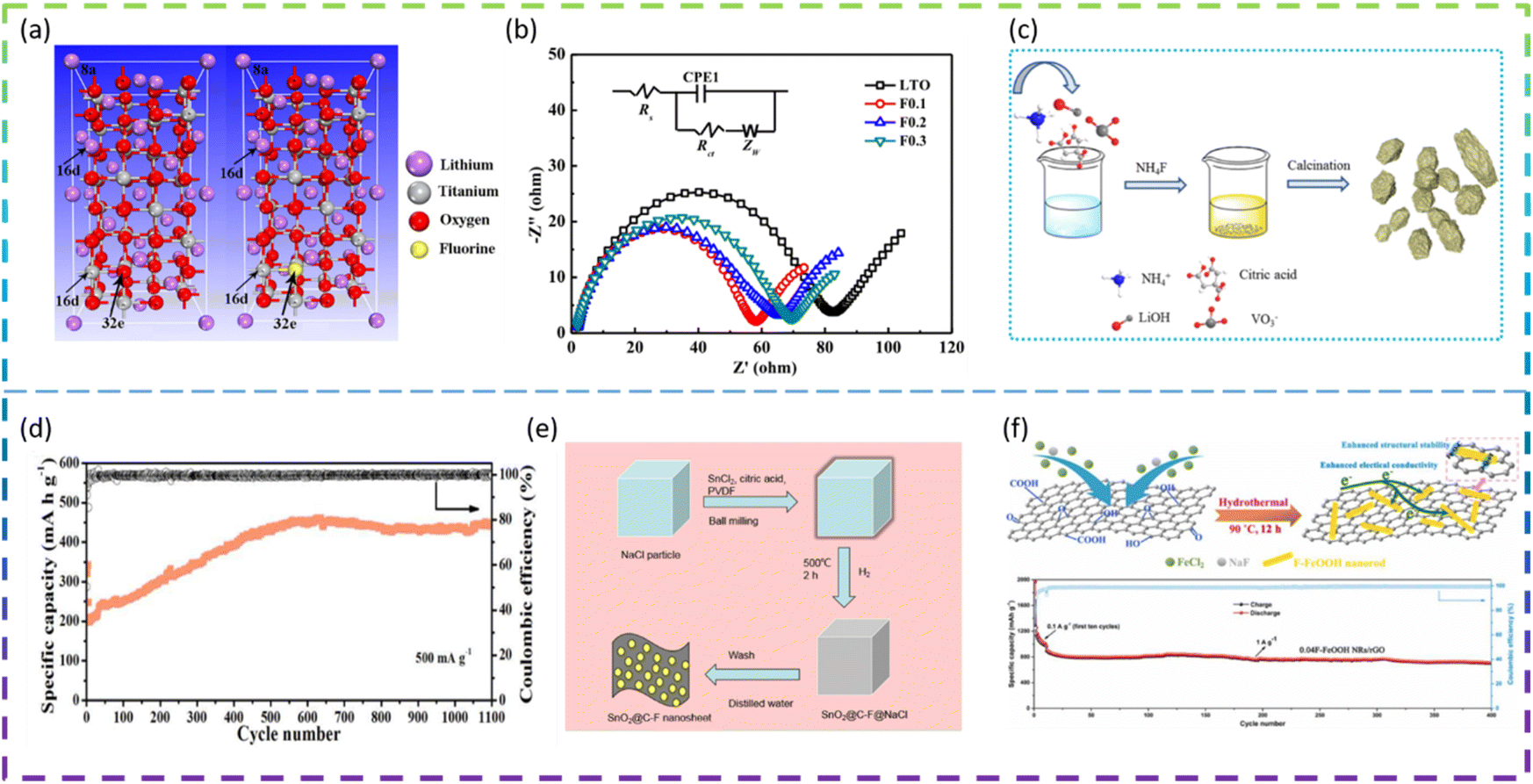 | ||
| Fig. 7 (a) The crystal structures of Li21Ti27O64 and Li21Ti27O63F1. (b) EIS curves (Nyquist plots) of all samples. (c) Preparation scheme for F-doped Li3VO4. (d) Long-term cycling (at 500 mA g−1 for 1100 cycles except for the first five cycles at 100 mA g−1) for 5 at% F-doping. (e) Schematic of the synthetized porous SnO2@C-F mesoporous nanosheets. (f) Diagram of the F-FeOOH NRs/rGO synthesis process and long-term cycling performance of 0.04F-FeOOH NRs/rGO at 1 A g−1. | ||
Liu et al. synthesized a series of F-doped Li3VO4 samples as anode materials for LIBs by a modified sol–gel method for the first time (Fig. 7c). The results show that the 5 at% F-doped sample has a long cycle life (1100 cycles at 500 mA g−1, except for the first five 100 mA g−1 cycles) (Fig. 7d) and an optimized rate performance (255 mA h g−1 at 1000 mA g−1), which is better than for the undoped sample. The doping of F leads to the appearance of oxygen vacancies and a reduction of the band gap, which effectively improves the electronic conductivity, the reduction of the electron and ion transport energy barriers, the complete rearrangement of the structure, the optimization of the SEI layer and the appearance of nanosheets.91
Feng et al. developed novel fluorine-doped porous SnO2@C (SnO2@C-F) nanosheets as an anode material for LIBs by a facile ball milling method with the NaCl template (Fig. 7e). The porous SnO2@C-F nanosheet samples exhibited excellent electrochemical capacity (821 mA h g−1 at 200 mA g−1 after 100 cycles), excellent rate capacity (816.62, 684.46, 579.2, 476.08, 410.94, 361.64, 324.1 and 734.0 mA h g−1, respectively) and long-term cycling stability (419.4 mA h g−1 at 2.0 A g−1 after 800 cycles). The formation of metal fluorides (Sn–F bond) and the strong electronic coupling between fluorine-doped carbon and SnO2 nanoparticles can greatly improve the structural stability and electronic conductivity of SnO2 negative electrode materials.92
Jin et al. synthesized F-doped Ni3V2O8 nanorods as an anode material for LIBs via a molten-salt method. The F-doped Ni3V2O8 nanorods have an initial coulombic efficiency of 90.8%, better cycling performance (983 mA h g−1 after 100 cycles at 200 mA g−1), and excellent long-term stability (616 mA h g−1 after 500 cycles at 2000 mA g−1). The results show that the one-dimensional F-doped oxygen-vacancy containing Ni3V2O8 nanorods accelerate the migration of Li+ ions and improve the electrochemical reaction kinetics.93
Zhu et al. synthesized novel hybrids of fluorine-doped FeOOH nanorods/reduced graphene oxide (F-FeOOH NRs/rGO) as anode materials for LIBs via a simple one-step hydrothermal method (Fig. 7f). Various amounts of NaF (1, 2 and 5 mM) were used to fabricate the hybrids with different F contents, which were denoted as 0.02F-FeOOH NRs/rGO, 0.04F-FeOOH NRs/rGO and 0.1FFeOOH NRs/rGO, respectively. As a result, the 0.04F-FeOOH NRs/rGO hybrid shows excellent lithium storage performance of 1207.4 mA h g−1 after 160 cycles at 0.1 A g−1. In addition, it exhibits impressive rate capability and long-term recyclability (701.4 mA h g−1 at 1 A g−1 after 400 cycles) (Fig. 7f). The doping of F not only improves the electrical conductivity of FeOOH but also contributes to the formation of a large number of fine nanoparticles that shorten the transport distance of Li+ and provide more electroactive sites for the storage of Li+.94
Zhao et al. fabricated fluorine-doped ZnFe2O4 (ZFO-F) as an anode material for LIBs via a quick quenching method. The results show that the sample with the optimum amount of F doping has the smallest charge transfer resistance, the highest specific capacity of 950 mA h g−1 after stabilization at 0.5 A g−1, and excellent rate performance, up to 312 mA h g−1 at a high current density of 5 A g−1 while it is only 200 mA h g−1 for pure ZnFe2O4. It was shown that the successful doping of the element F into ZFO brought about an abundance of active centers and improved electronic conductivity.95
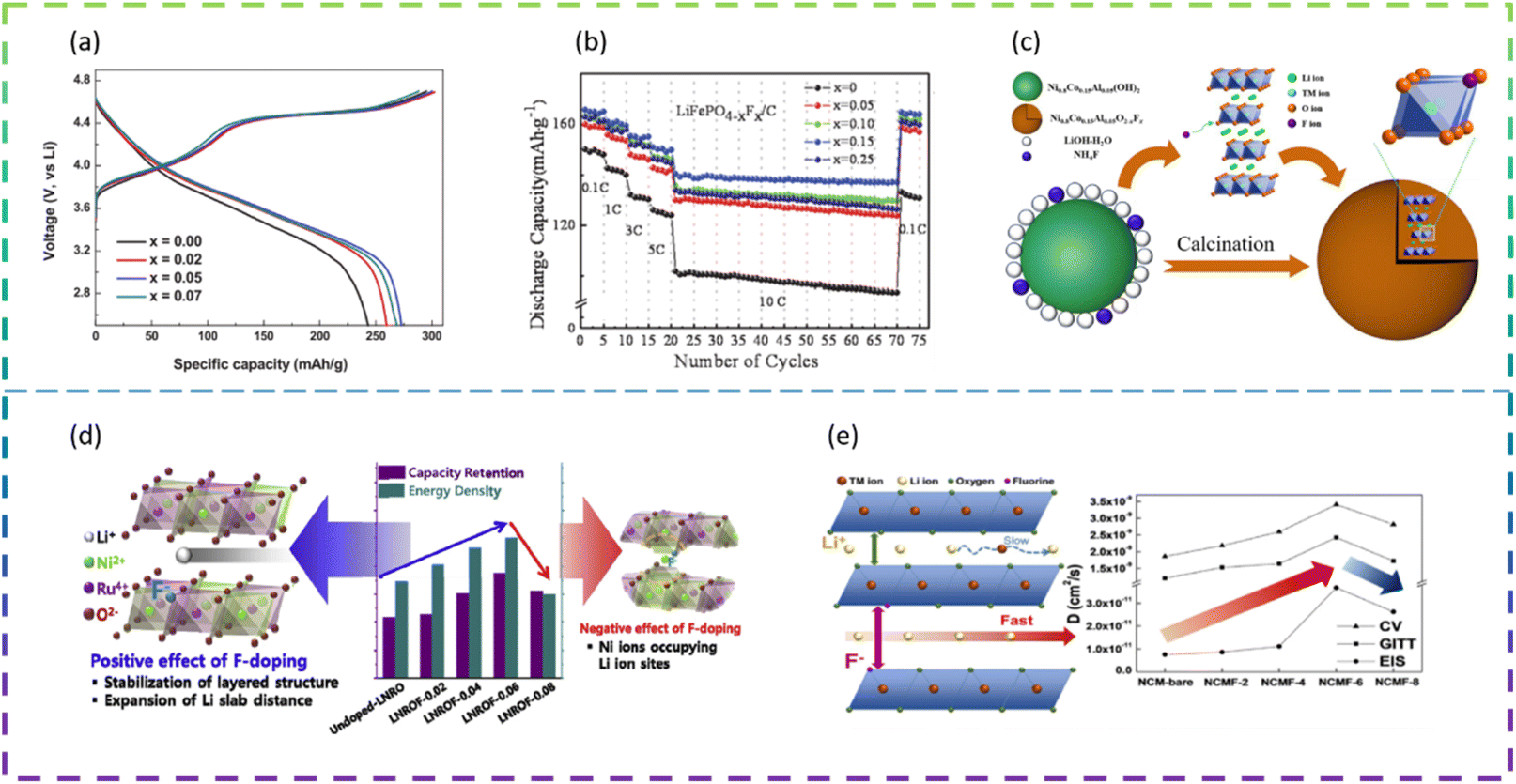 | ||
| Fig. 8 (a) Initial charge/discharge curves. (b) Rate and cycle performances of undoped and F-doped LiFePO4/C samples. (c) Schematic illustration of the in situ synthesis route for the F-doped LiNi0.8Co0.15Al0.05O2−xFx (0 ≤ x ≤ 0.1) composite powders. (d) Positive and negative effects of F-doped Li1.15Ni0.275Ru0.575O2. (e) Comparison of Li-ion diffusion coefficients of the samples measured by CV, GITT, and EIS analysis. | ||
Wang et al. employed F doping in improving the electrochemical performance of the Li2MnSiO4 cathode material. The results show that Li2MnSiO3.97F0.03/C with the best electrochemical performance provides an initial specific discharge capacity of 279 mA h g−1 at a current density of 25 mA g−1 from 1.5 V to 4.8 V. In addition, it maintains a higher capacity (201 mA h g−1) after 50 cycles than Li2MnSiO4 without F (145 mA h g−1). Firstly, F doping not only broadens the lattice parameters but also reduces the particle size, which synergistically improves the Li+-ion diffusion of Li2MnSiO4. Secondly, F doping improves the electronic conductivity of Li2MnSiO4/C by inhibiting the formation of C–O bonds in the carbon layer. At the same time, F doping improves the crystallinity of Li2MnSiO4 and stabilizes the crystal structure.98
Choi et al. synthesized F-doped Li1.15Ni0.275Ru0.575O2 cathode structures (LNROF-x, 0 < x < 0.1) using a facile solid-state reaction. The long life cycle and rate performance of the F-doped Li1.15Ni0.275Ru0.575O2 material can be attributed to the stronger structural stability of the metal–F bond than the metal–O bond and the increased diffusive movement of the lithium ions due to the increased Li sheet spacing. Nevertheless, excessive F doping causes the performance of LNROF-0.08 to deteriorate, with increased resistance to Li+-ion movement and Li/Ni anti-location defects leading to a lower diffusion coefficient (Fig. 8d).99
Wang et al. synthesized F− doped LiNi0.8Co0.15Al0.05O2 (LiNi0.8Co0.15Al0.05O2−xFx (0 ≤ x ≤ 0.1)) as LIB cathode materials by an in situ modified method (Fig. 8c). The reversible discharge specific capacity of the half-cell assembled with LiNi0.8Co0.15Al0.05O1.96F0.04 composite electrodes was 157.8 mA h g−1, with a capacity retention of 98.3% after 100 cycles at 2.0C and 25 °C. Even at a high temperature of 60 °C and a high current density of 5.0C, it was still possible to reach a discharge specific capacity of 142.6 mA h g−1 with a capacity retention of 89.1%. The significant improvement in lithium storage performance is mainly attributed to the introduction of F− ions into the lattice, replacing the metal–O bond with a stronger metal–F bond, which not only effectively stabilizes the main structure and maintains structural integrity, but also prevents the erosion of HF and inhibits the increase in polarisation during continuous cycling.100
Kim et al. prepared Ni-rich ternary cathode materials (NCM) doped with various amounts of F dopant by a solid-state reaction method. The F-doped cathode material exhibited better cycling and rate performance compared to the undoped NCM samples, due to the relatively strong bond between the transition metal and F and the improved Li+ ion transport behavior. However, when the F doping exceeds the optimum value, the performance of LIBs deteriorates due to the deterioration of Li+ ion transport caused by Li/Ni anti-substitution defects (Fig. 8e).101
Kim et al. synthesized Br-doped Li4Ti5O12−xBrx (x = 0, 0.1, 0.3, 0.5, and 0.7) (LTOBrx) anode materials for LIBs by a conventional solid-state reaction technique. The LTOBr0.5 sample had a maximum capacity of 125 mA h g−1 at 1C compared to 115 mA h g−1 for pure LTO. However, the highly Br-doped sample (LTOBr0.7) showed a slight decrease in capacity. Most of the Br ions are located on the surface/interface of the agglomerated particles, rather than in the bulk lattice. As a result, electrons and lithium ions form narrow conduction paths on their surfaces/interfaces. The presence of these narrow surface conductive tubes improves the rate capability of the LTOBr sample during the charge/discharge process.103
Wang et al. synthesized Br-doped Li4Ti5O12 microspheres (LTOS) as anode materials for LIBs using a cetyltrimethylammonium hydroxide-assisted hydrothermal method and further calcination. Among the doped samples, 0.2Br-LTOS has excellent rate performance and cycling capability. Even at 50C and 60C, its specific capacities are still as high as 101 and 96 mA h g−1, respectively. Moreover, its capacity retention ratio is about 87.8% after 1000 cycles at 10C, which is higher than the 76.5% of pure LTOS. The spherical morphology and spinel-type microstructure of LTOS remain unchanged after proper Br doping, but the relative content of Ti3+ increases, which facilitates the rapid transport of electrons and ions.104
3.2 Sodium-ion batteries
Chen et al. synthesized F-doped Na2Ti3O7 nanorods by a solid-phase method as an anode for SIBs. In Fig. 9a and b, the cell with NTOF0.3 electrodes showed the best electrochemical performance. The specific capacity of the F-doped Na2Ti3O7 sample is 30% higher than that of pure Na2Ti3O7, due to the increased Na+ diffusion coefficient. The F doping improves the charge transfer resistance of the material and the resulting material also exhibits better rate properties and cycling for more than 800 cycles.115
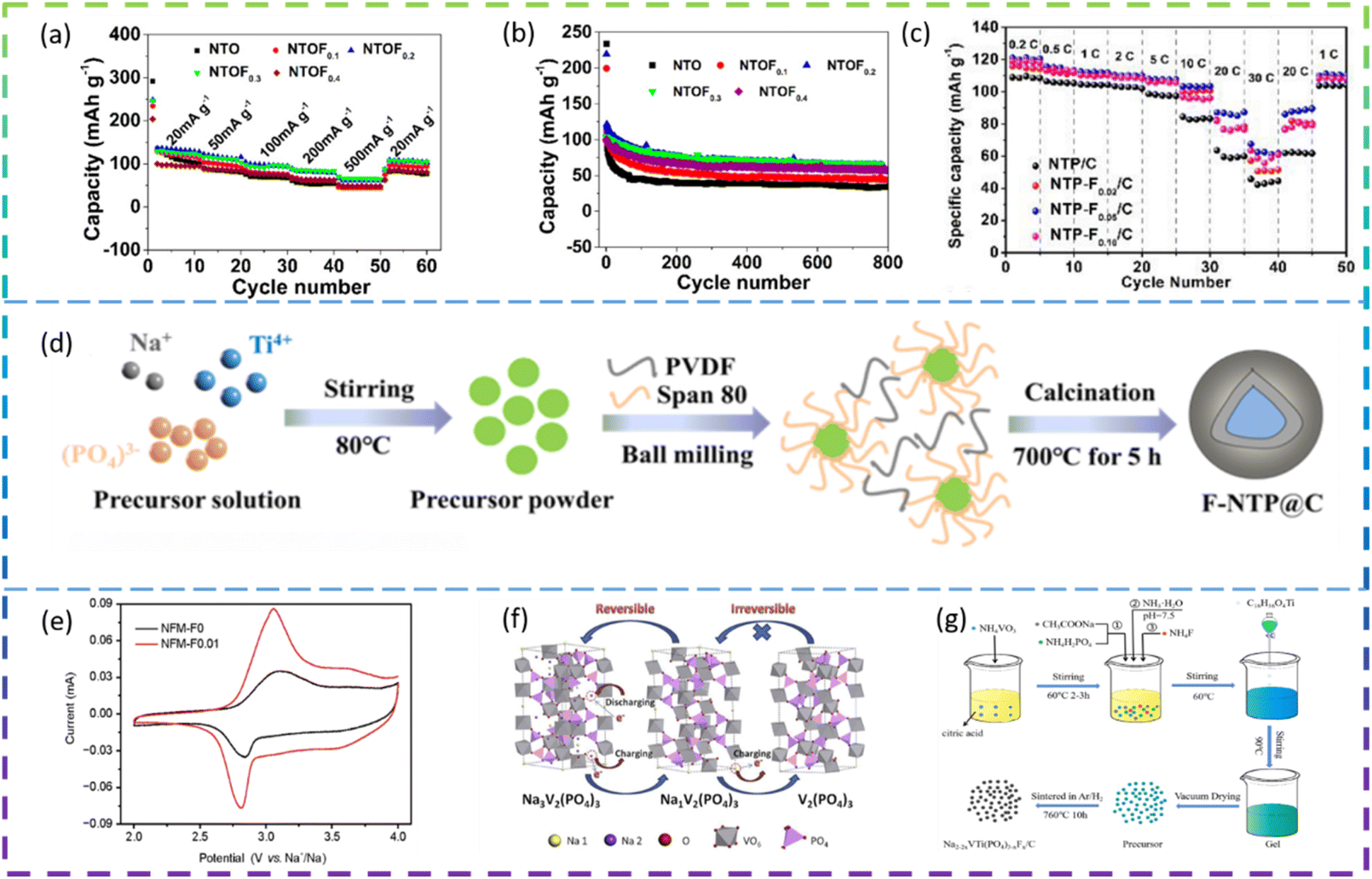 | ||
| Fig. 9 (a) The specific capacity performance of NTO and NTOFx at different current densities. (b) The cycling properties of NTO and NTOFx at a current density of 100 mA g−1. (c) Rate performance of Na1−2xTi2(PO4)3−xFx/C from 0.2C to 30C. (d) Schematic illustration of the synthetic route of F-NTP@C. (e) The first CV curves of NFM-F0 and NFM-F0.01 at a scan rate of 0.1 mV s−1. (f) Schematic of the structural degradation of Na3V2(PO4)3. (g) Schematic illustration of the preparation process of Na2−2xVTi(PO4)3−xFx/C. | ||
Wei et al. synthesized F-doped Na1−2xTi2(PO4)3−xFx (denoted as NTP-Fx/C, x = 0, 0.02, 0.05, 0.10) as anode materials for SIBs by a sol–gel method. Among the F-doped composites, NTP-F0.05/C shows the best electrochemical performance: the specific capacity decreases from 121 mA h g−1 to 85.8 mA h g−1 as the current density increases from 0.2C to 20C. It offers a specific discharge capacity of 62.5 mA h g−1 even at 30C, demonstrating superior rate capability (Fig. 9c). NTP-F0.05/C also exhibits a high reversible capacity of 102.5 mA h g−1 at 10C and a retention capacity of 90% after 1000 cycles, demonstrating excellent cycling performance and structural stability. The results of the electrical analysis show that fluorine doping significantly enhances the diffusion kinetics of Na+. Meanwhile, density functional theory calculations indicate that the F-doped nanotubes have excellent electrochemical properties due to their enhanced intrinsic ion/electron conductivity.116
Deng et al. prepared fluorine-doped NaTi2(PO4)3@C composite (F-NTP@C) anode materials for SIBs by a sol–gel method followed by a high temperature solid state method (Fig. 9d). The modified F-NTP@C material with a 3D porous structure exhibits excellent rate performance (108.7 mA h g−1 at 50C) and long cycle life (75.5% capacity retention after 2000 cycles at 10C). It also shows temperature adaptability over a wide range from 0 to 50 °C. By combining experimental data with theoretical calculations, it can be concluded that the enhanced electronic conductivity and fast Na+ kinetics are attributed to the doping of F− ions and the design of the 3D porous structure.117
Zhao et al. prepared N/P co-doped soft carbon nanoboxes (NPSC) as anodes for SIBs using petroleum pitch as raw material. At a high current density of 1 A g−1, the NPSC can provide a high reversible capacity of 162 mA h g−1 for 3000 cycles. DFT calculations show that the N and P co-doped carbon has more electrons near the Fermi level, thus facilitating electron transport.119
Chen et al. synthesized an F-doped Na3V2(PO4)2.93F0.07/C (F-0.07-NVP/C) composite as the cathode for SIBs by a solid-state reaction method. Compared to the undoped NVP/C sample, the F-0.07-NVP/C composite has a discharge capacity of 113 mA h g−1 at 10 mA g−1, which is very close to the theoretical capacity (117 mA h g−1). In terms of cycling performance, a reversible capacity of 97.8 mA h g−1 can be obtained and 86% of the capacity is maintained after 1000 cycles at 200 mA g−1. F doping can inhibit the structural degradation of Na3V2(PO4)3 to V2(PO4)3 and improve the structural stability (Fig. 9f). At the same time, it can reduce the particle size to decrease the pathway for Na+ diffusion, thus effectively increasing the ionic conductivity.121
Song et al. synthesized F-doped Na2−2xVTi(PO4)3−xFx/C (denoted as NVTP-Fx, x = 0.01, 0.03 and 0.05) as cathodes for SIBs through a simple sol–gel method (Fig. 9g). Among the F-doped materials, the NVTP-F0.03 electrode exhibits the best electrochemical performance. When the current density is increased from 0.1C to 10C, the specific capacity decreases from 134.3 to 75.4 m A h g−1, and even when the applied current density is increased to 20C, a specific discharge capacity of 59.5 mA h g−1 is provided, which is much higher than that of the original sample. In addition, the NVTP-F0.03 sample exhibits excellent cycling stability. After 100 cycles at 0.5C a capacity retention of 96.5% was achieved, and after 500 cycles at higher currents (2C) a capacity retention of 81.3% was still achieved. The results of the electrical analysis showed that the Na+ diffusion kinetics were greatly improved after fluorine doping and that the carbon coating effectively facilitated charge transfer at the electrode.122
In order to better demonstrate the synthesis and application of F-doped materials/other element-doped materials in electrode materials, the preparation methods and cell performance are summarised in Table 3.
| No. | Materials | Preparation | Fluorine source/other doping element sources | Application | Specific capacity (mA h g−1) | Cycling number | Current rate (mA g−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | F-doped Li4Ti5O12 | Solid-state reaction | NH4F | LIB anode | 106.4 | 150 | — | 90 |
| 2 | F-doped Li3VO4 | Sol–gel method | NH4F | LIB anode | 450 | 1100 | 500 | 91 |
| 3 | F-doped porous SnO2@C | Ball milling method | PVDF | LIB anode | 419.4 | 800 | 2000 | 92 |
| 4 | F-doped Ni3V2O8 | Molten-salt method | NaF | LIB anode | 616 | 500 | 2000 | 93 |
| 5 | F-doped FeOOH | One-step hydrothermal method | NaF | LIB anode | 701.4 | 400 | 1000 | 94 |
| 6 | F-doped ZnFe2O4 | Quick quenching method | NH4F | LIB anode | 850 | 500 | 500 | 95 |
| 7 | Li[Li1/6Ni1/6Co1/6Mn1/2]O2−xFx | Solid state reaction method | LiF | LIB cathode | ∼650 | 300 | — | 96 |
| 8 | F-doped LiFePO4/C | Co-precipitation method | HF | LIB cathode | 129.8 | 50 | 3400 | 97 |
| 9 | F-doped Li2MnSiO4/C | Solid-state reaction | LiF | LIB cathode | 201 | 50 | 25 | 98 |
| 10 | F-doped Li1.15Ni0.275Ru0.575O2 | Solid-state reaction | NH4F | LIB cathode | 150 | 100 | 20 | 99 |
| 11 | F-doped LiNi0.8Co0.15Al0.05O2 | Co-precipitation process | NH4F | LIB cathode | 149.9 | 100 | — | 100 |
| 12 | F-doped Na2Ti3O7 | Solid-phase method | NaF | SIB anode | ∼85 | 800 | 100 | 115 |
| 13 | F-doped NaTi2(PO4)3/C | Sol–gel method | NaF | SIB anode | 67.3 | 1000 | 1330 | 116 |
| 14 | F-doped NaTi2(PO4)3@C | Sol–gel method | PVDF | SIB anode | 84.5 | 2000 | 722 | 117 |
| 15 | F-doped O3-type NaNi1/3Fe1/3Mn1/3O2 | Solid-state reaction | NaF | SIB cathode | ∼110 | 70 | 150 | 120 |
| 16 | F-doped Na3V2(PO4)3/C | Solid-state reaction | NaF | SIB cathode | 97.8 | 1000 | 200 | 121 |
| 17 | F-doped Na2VTi(PO4)3 | Sol–gel method | NH4F | SIB cathode | ∼80 | 500 | 270 | 122 |
| 18 | Cl-doped LiFePO4/C | High-temperature solid phase method | NH4Cl | LIB cathode | 105.3 | 500 | — | 102 |
| 19 | Br-doped Li4Ti5O12 | Solid-state reaction | LiBr | LIB anode | ∼122 | 300 | — | 103 |
| 20 | Br-doped Li4Ti5O12 | CTAOH-assisted hydrothermal treatment | LiBr | LIB anode | 106.2 | 1000 | 1750 | 104 |
| 21 | S-doped graphene | Modified thermal process | — | SIB anode | 217 | 1000 | 3200 | 118 |
| 22 | N/P co-doped soft carbon | — | (NH4)2HPO4 | SIB anode | 162 | 3000 | 1000 | 119 |
4 Conclusions
This paper reviews the progress of research on the synthesis of F-doped materials and their applications in batteries and catalysts. In recent years, F has gained a lot of attention in the field of materials as a cheap and efficient dopant. F-doping has been applied by researchers in photocatalysis, electrocatalysis, and rechargeable batteries. Firstly, F as a dopant in photocatalytic degradation of organic matter can inhibit photogenerated electron/hole recombination, increase the amount of –OH on the surface of the photocatalyst and increase the concentration of free electrons, and also make the particle size smaller, increase the specific surface area of the catalyst and improve the photocatalyst activity. Secondly, the introduction of F in electrocatalysis can change the electronic structure of the electrocatalyst and lower the reaction energy potential barrier, while increasing the electrical conductivity and improving the electrocatalytic selectivity and Faraday efficiency. Finally, doping F in the positive and negative electrode materials of LIBs and SIBs can improve the electronic conductivity of the materials, reduce the electrode polarisation, stabilize the crystal structure and accelerate the diffusion rate of lithium or sodium ions, thus improving the electrochemical performance of the batteries.It is worth noting that, despite the good progress made in these areas with F-doped materials, their future development still faces challenges. F-doping is actually a relatively common topic in materials research, as it can alter the physical and chemical properties of materials and has important implications for a number of applications such as photocatalysts, electrocatalysts, and anode and cathode materials for rechargeable batteries, etc. However, the following may be among the reasons for the relative paucity of research on F doping: firstly, F doping is not possible for all materials and only some specific materials can be effectively doped with F ions. Therefore, this may limit the application and research of F doping in some materials. Secondly, the preparation method of F-doping is complicated and the amount of F ions doped is difficult to control, which makes the experimental study of F-doping difficult and requires more effort and time to optimise the preparation process. Thirdly, the doping of F ions in the material may affect the stability of the material, which may limit the use of F doping in some applications. Therefore, when conducting F-doping studies, the stability of the material needs to be fully considered, which adds to the difficulty of the study. Finally, there may be relatively few theoretical studies of F-doping compared to experimental studies, and this may be one of the reasons for the relative paucity of F-doping studies. Theoretical studies can be done to investigate the effect of F-doping on material properties through computational simulations and other methods, providing a theoretical basis and guidance for experimental studies.
F-doping has many potential applications, such as enhancing the optical, electrical and magnetic properties of materials and improving the performance of semiconductor devices. The following are the development prospects and research trends in F-doping: for example, F doping can be used to improve the performance of semiconductor materials, such as increasing the electrical conductivity of the material, reducing the particle size, increasing the specific surface area of the material, etc. F doping can also be used to develop new types of semiconductor materials, such as fluoride semiconductor materials, which have high electrical and optical properties and can be used in infrared detectors, laser diodes, etc. F doping is used to develop new types of catalysts, for example, doping F into titanium dioxide catalysts can improve their catalytic performance and stability. F doping can also be used to improve the performance of rechargeable batteries, accelerate ion conduction rates, stabilise the structure of anode and cathode materials and extend battery life.
In other words, F-doped materials have excellent and unique physicochemical properties and have promising applications in catalysis and energy. We believe that in the future, with the joint efforts of researchers, F-doped materials will receive more attention and be widely used in more fields, showing the uniqueness of F-doped materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (61904123, 51973157) and the Tianjin Technical and Engineering Center of Nonwovens and State Key Laboratory of Membrane and Membrane Separation, Tiangong University (KF202103).References
- X. Jin, L. Ye, H. Xie and G. Chen, Coord. Chem. Rev., 2017, 349, 84–101 CrossRef CAS.
- S. R. Mishra and M. Ahmaruzzaman, Nanoscale, 2022, 14, 1566–1605 RSC.
- J. Sun, X. Li, Y. Quan, Y. Yin and S. Zheng, Chemosphere, 2015, 136, 181–189 CrossRef CAS PubMed.
- J. Wen, X. Li, W. Liu, Y. Fang, J. Xie and Y. Xu, Chin. J. Catal., 2015, 36, 2049–2070 CrossRef CAS.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- M. Moradi, Y. Vasseghian, A. Khataee, M. Harati and H. Arfaeinia, Sep. Purif. Technol., 2021, 261, 118274 CrossRef CAS.
- O. G. Sas, P. B. Sánchez, B. González and Á. Domínguez, Sep. Purif. Technol., 2020, 236, 116310 CrossRef CAS.
- W. Raza, J. Lee, N. Raza, Y. Luo, K.-H. Kim and J. Yang, J. Ind. Eng. Chem., 2019, 71, 1–18 CrossRef CAS.
- T. D. Vo, C. Lin, C. E. Weng, C. S. Yuan, C. W. Lee, C. H. Hung, X. T. Bui, K. C. Lo and J. X. Lin, J. Environ. Manage., 2018, 217, 327–336 CrossRef CAS PubMed.
- Q. Wang, B. Rhimi, H. Wang and C. Wang, Appl. Surf. Sci., 2020, 530, 147286 CrossRef CAS.
- R. Xie, D. Lei, Y. Zhan, B. Liu, C. H. A. Tsang, Y. Zeng, K. Li, D. Y. Leung and H. Huang, Chem. Eng. J., 2020, 386, 121025 CrossRef CAS.
- T. Bao, L. Song and S. Zhang, Appl. Organomet. Chem., 2018, 32, e4349 CrossRef.
- H. Koyuncu and A. R. Kul, Appl. Water Sci., 2020, 10, 1–14 CrossRef.
- S. Parakala, S. Moulik and S. Sridhar, Chem. Eng. J., 2019, 375, 122015 CrossRef CAS.
- A. A. Al-Gheethi, Q. M. Azhar, P. Senthil Kumar, A. A. Yusuf, A. K. Al-Buriahi, R. M. S. Radin Mohamed and M. M. Al-Shaibani, Chemosphere, 2022, 287, 132080 CrossRef CAS PubMed.
- S. A. Bhat, N. Rashid, M. A. Rather, S. A. Bhat, P. P. Ingole and M. A. Bhat, Chem. Phys. Lett., 2020, 754, 137724 CrossRef CAS.
- P. Sathishkumar, R. A. A. Meena, T. Palanisami, V. Ashokkumar, T. Palvannan and F. L. Gu, Sci. Total Environ., 2020, 698, 134057 CrossRef CAS PubMed.
- B. M. Sharma, J. Becanova, M. Scheringer, A. Sharma, G. K. Bharat, P. G. Whitehead, J. Klanova and L. Nizzetto, Sci. Total Environ., 2019, 646, 1459–1467 CrossRef CAS PubMed.
- Y. Li, L. Zhang, X. Liu and J. Ding, Sci. Total Environ., 2019, 658, 333–342 CrossRef CAS PubMed.
- K. Poonia, V. Hasija, P. Singh, A. A. Parwaz Khan, S. Thakur, V. K. Thakur, S. Mukherjee, T. Ahamad, S. M. Alshehri and P. Raizada, J. Cleaner Prod., 2022, 367, 133087 CrossRef CAS.
- A. Fareed, A. Hussain, M. Nawaz, M. Imran, Z. Ali and S. U. Haq, Environ. Technol. Innovation, 2021, 24, 101840 CrossRef CAS.
- R. Chen, Y. Zhuang, Y. Yu and B. Shi, Water Res., 2021, 190, 116660 CrossRef CAS PubMed.
- S. Poothong, E. Papadopoulou, J. A. Padilla-Sanchez, C. Thomsen and L. S. Haug, Environ. Int., 2020, 134, 105244 CrossRef CAS PubMed.
- C. Sun, J. Yang, M. Xu, Y. Cui, W. Ren, J. Zhang, H. Zhao and B. Liang, Chem. Eng. J., 2022, 427, 131564 CrossRef CAS.
- B. Wu, H. Meng, D. M. Morales, F. Zeng, J. Zhu, B. Wang, M. Risch, Z. J. Xu and T. Petit, Adv. Funct. Mater., 2022, 32, 2204137 CrossRef CAS.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H. Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio Jr, L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruna, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Chen, G. Li, Y. Zeng, L. Yan, X. Wang, L. Yang, Q. Wu and Z. Hu, Nano Res., 2022, 15, 7896–7902 CrossRef CAS.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y. M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed.
- S.-G. Han, D.-D. Ma, S.-H. Zhou, K. Zhang, W.-B. Wei, Y. Du, X.-T. Wu, Q. Xu, R. Zou and Q.-L. Zhu, Appl. Catal., B, 2021, 283, 119591 CrossRef CAS.
- Y. Liu, Q. Li, X. Guo, X. Kong, J. Ke, M. Chi, Q. Li, Z. Geng and J. Zeng, Adv. Mater., 2020, 32, e1907690 CrossRef PubMed.
- X. Zhu, Z. Liu, H. Wang, R. Zhao, H. Chen, T. Wang, F. Wang, Y. Luo, Y. Wu and X. Sun, Chem. Commun., 2019, 55, 3987–3990 RSC.
- Y. P. Liu, Y. B. Li, H. Zhang and K. Chu, Inorg. Chem., 2019, 58, 10424–10431 CrossRef CAS PubMed.
- D. Yuan, Z. Wei, P. Han, C. Yang, L. Huang, Z. Gu, Y. Ding, J. Ma and G. Zheng, J. Mater. Chem. A, 2019, 7, 16979–16983 RSC.
- B. Yang, C. Jiang, G. Yu, Q. Zhuo, S. Deng, J. Wu and H. Zhang, J. Hazard. Mater., 2015, 299, 417–424 CrossRef CAS PubMed.
- R. Zhang, M. Zhang, H. Yang, G. Li, S. Xing, M. Li, Y. Xu, Q. Zhang, S. Hu, H. Liao and Y. Cao, Small Methods, 2021, 5, e2100612 CrossRef PubMed.
- Y. Chang, J. Chen, J. Jia, X. Hu, H. Yang, M. Jia and Z. Wen, Appl. Catal., B, 2021, 284, 119721 CrossRef CAS.
- J. Neumann, M. Petranikova, M. Meeus, J. D. Gamarra, R. Younesi, M. Winter and S. Nowak, Adv. Energy Mater., 2022, 12, 2102917 CrossRef CAS.
- C. F. J. Francis, I. L. Kyratzis and A. S. Best, Adv. Mater., 2020, 32, e1904205 CrossRef PubMed.
- Z. Hu, Q. Liu, S. L. Chou and S. X. Dou, Adv. Mater., 2017, 29, 1700606 CrossRef PubMed.
- V. Vaiano, M. Matarangolo, J. J. Murcia, H. Rojas, J. A. Navío and M. C. Hidalgo, Appl. Catal., B, 2018, 225, 197–206 CrossRef CAS.
- N. Villota, J. M. Lomas and L. M. Camarero, Ultrason. Sonochem., 2017, 39, 439–445 CrossRef CAS PubMed.
- C. Yu, Q. Fan, Y. Xie, J. Chen, Q. Shu and J. C. Yu, J. Hazard. Mater., 2012, 237–238, 38–45 CrossRef CAS.
- N. Imanaka, T. Masui and K. Yasuda, Chem. Lett., 2011, 40, 780–785 CrossRef CAS.
- B. Belaissaoui, Y. Le Moullec and E. Favre, Energy, 2016, 95, 291–302 CrossRef CAS.
- W. Yu, X. Liu, L. Pan, J. Li, J. Liu, J. Zhang, P. Li, C. Chen and Z. Sun, Appl. Surf. Sci., 2014, 319, 107–112 CrossRef CAS.
- H. Miao, G.-F. Huang, J.-H. Liu, B.-X. Zhou, A. Pan, W.-Q. Huang and G.-F. Huang, Appl. Surf. Sci., 2016, 370, 427–432 CrossRef CAS.
- L. Rueda-Salaya, A. Hernández-Ramírez, L. Hinojosa-Reyes, J. Guzmán-Mar, M. Villanueva-Rodríguez and E. Sánchez-Cervantes, J. Photochem. Photobiol., A, 2020, 391, 112364 CrossRef CAS.
- G. Vitiello, G. Iervolino, C. Imparato, I. Rea, F. Borbone, L. De Stefano, A. Aronne and V. Vaiano, Sci. Total Environ., 2021, 762, 143066 CrossRef CAS PubMed.
- E. M. Samsudin, S. B. A. Hamid, J. C. Juan, W. J. Basirun, A. E. Kandjani and S. K. Bhargava, Appl. Surf. Sci., 2016, 365, 57–68 CrossRef CAS.
- Y. Kang, X. Wu and Q. Gao, ACS Sustainable Chem. Eng., 2019, 7, 4210–4219 CrossRef CAS.
- H. Yu, C. Chu and X. An, Appl. Organomet. Chem., 2019, 33, e4682 CrossRef.
- Z. Chen, W. Chen, G. Liao, X. Li, J. Wang, Y. Tang and L. Li, J. Hazard. Mater., 2022, 428, 128222 CrossRef CAS PubMed.
- A. Phuruangrat, P. Dumrongrojthanath, B. Kuntalue, S. Thongtem and T. Thongtem, Mater. Lett., 2017, 196, 256–259 CrossRef CAS.
- G.-Y. Zhang, J.-J. Wang, X.-Q. Shen, J.-J. Wang, B.-Y. Wang and D.-Z. Gao, Appl. Surf. Sci., 2019, 470, 63–73 CrossRef CAS.
- J. Zai, F. Cao, N. Liang, K. Yu, Y. Tian, H. Sun and X. Qian, J. Hazard. Mater., 2017, 321, 464–472 CrossRef CAS PubMed.
- C. Liu, L. Zhang, R. Liu, Z. Gao, X. Yang, Z. Tu, F. Yang, Z. Ye, L. Cui, C. Xu and Y. Li, J. Alloys Compd., 2016, 656, 24–32 CrossRef CAS.
- W. Ding, X. Zhang, X. Liu, Q. Lu, M. Wei and Y. Pang, J. Environ. Chem. Eng., 2021, 9, 105985 CrossRef CAS.
- Y. Shen, J.-y. Hu, L. Lu, C. Zhu, Q.-l. Fang and S. Song, J. Zhejiang Univ. Sci. A, 2022, 23, 988–997 CrossRef CAS.
- T. N. Do, C. You and J. Kim, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- N. Li, X. Z. Chen, W. J. Ong, D. R. MacFarlane, X. J. Zhao, A. K. Cheetham and C. H. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS.
- J. Gu, F. Heroguel, J. Luterbacher and X. Hu, Angew. Chem., Int. Ed. Engl., 2018, 57, 2943–2947 CrossRef CAS.
- T. Wu, M. M. Melander and K. Honkala, ACS Catal., 2022, 12, 2505–2512 CrossRef CAS.
- X. Li, Y. Tian, X. Wang, Y. Guo and K. Chu, Sustainable Energy Fuels, 2021, 5, 4277–4283 RSC.
- W. Cai, Y. Han, Y. Pan, X. Zhang, J. Xu, Y. Zhang, Y. Sun, S. Li, J. Lai and L. Wang, J. Mater. Chem. A, 2021, 9, 13483–13489 RSC.
- S. Zhao, X. Lu, L. Wang, J. Gale and R. Amal, Adv. Mater., 2019, 31, e1805367 CrossRef PubMed.
- F. Wang, Y. p. Liu, H. Zhang and K. Chu, ChemCatChem, 2019, 11, 1441–1447 CrossRef CAS.
- C. Zuo, L. Li, W. Chen and Z. Zhang, Appl. Surf. Sci., 2021, 554, 149546 CrossRef CAS.
- Y. Wang, G. I. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2020, 11, 2003323 CrossRef.
- K. Zhao, Y. Su, X. Quan, Y. Liu, S. Chen and H. Yu, J. Catal., 2018, 357, 118–126 CrossRef.
- W. Wang, X. Lu, P. Su, Y. Li, J. Cai, Q. Zhang, M. Zhou and O. Arotiba, Chemosphere, 2020, 259, 127423 CrossRef CAS.
- S. Zeng, S. Wang, H. Zhuang, B. Lu, C. Li, Y. Wang and G. Wang, Electrochim. Acta, 2022, 420, 140460 CrossRef CAS.
- X. Zheng, X. Cao, Z. Sun, K. Zeng, J. Yan, P. Strasser, X. Chen, S. Sun and R. Yang, Appl. Catal., B, 2020, 272, 118967 CrossRef CAS.
- Q. Yang, Y. Jia, F. Wei, L. Zhuang, D. Yang, J. Liu, X. Wang, S. Lin, P. Yuan and X. Yao, Angew. Chem., Int. Ed. Engl., 2020, 59, 6122–6127 CrossRef CAS PubMed.
- M. Nemiwal, V. Gosu, T. C. Zhang and D. Kumar, Int. J. Hydrogen Energy, 2021, 46, 10216–10238 CrossRef CAS.
- R. Moradi and K. M. Groth, Int. J. Hydrogen Energy, 2019, 44, 12254–12269 CrossRef CAS.
- H. Zou, W. Rong, B. Long, Y. Ji and L. Duan, ACS Catal., 2019, 9, 10649–10655 CrossRef CAS.
- H. Luo, X. Wang, C. Wan, L. Xie, M. Song and P. Qian, Nanomaterials, 2022, 12, 1081 CrossRef CAS PubMed.
- J. Li, Y. Zhang, X. Zhang, J. Huang, J. Han, Z. Zhang, X. Han, P. Xu and B. Song, ACS Appl. Mater. Interfaces, 2017, 9, 398–405 CrossRef CAS PubMed.
- X. Ji, B. Liu, X. Ren, X. Shi, A. M. Asiri and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 4499–4503 CrossRef CAS.
- M. Li, B. Zhang, T. Cheng, S. Yu, S. Louisia, C. Chen, S. Chen, S. Cestellos-Blanco, W. A. Goddard and P. Yang, Nano Res., 2021, 14, 3509–3513 CrossRef CAS.
- J. Li, Y. Cai, H. Wu, Z. Yu, X. Yan, Q. Zhang, T. Z. Gao, K. Liu, X. Jia and Z. Bao, Adv. Energy Mater., 2021, 11, 2003239 CrossRef CAS.
- T. Weigel, F. Schipper, E. M. Erickson, F. A. Susai, B. Markovsky and D. Aurbach, ACS Energy Lett., 2019, 4, 508–516 CrossRef CAS.
- C. Yang, X. Ou, X. Xiong, F. Zheng, R. Hu, Y. Chen, M. Liu and K. Huang, Energy Environ. Sci., 2017, 10, 107–113 RSC.
- G. Li, S. Cao, L. Fu, S. Wan and Q. Liu, Electrochim. Acta, 2021, 386, 138470 CrossRef CAS.
- M. Cabello, E. Gucciardi, A. Herran, D. Carriazo, A. Villaverde and T. Rojo, Molecules, 2020, 25, 2494 CrossRef CAS PubMed.
- H. Wang, Q. Pan, Q. Wu, X. Zhang, Y. Huang, A. Lushington, Q. Li and X. Sun, J. Mater. Chem. A, 2017, 5, 4576–4582 RSC.
- X. Bai, W. Li, A. Wei, Q. Chang, L. Zhang and Z. Liu, Solid State Ionics, 2018, 324, 13–19 CrossRef CAS.
- X. Liu, G. Li, D. Zhang, L. Meng, B. Li and L. Li, Electrochim. Acta, 2020, 354, 136655 CrossRef CAS.
- Y. Feng, C. Bai, K. Wu, H. Dong, J. Ke, X. Huang, D. Xiong and M. He, J. Alloys Compd., 2020, 843, 156085 CrossRef CAS.
- R. Jin, R. Li and H. Xu, Ceram. Int., 2021, 47, 24625–24631 CrossRef CAS.
- S. Zhu, B. Liang, X. Mou, X. Liang, H. Huang, D. Huang, W. Zhou, S. Xu and J. Guo, J. Alloys Compd., 2022, 905, 164142 CrossRef CAS.
- Q. Zhao, P. Peng, P. Zhu, G. Yang, X. Sun, R. Ding, P. Gao and E. Liu, New J. Chem., 2022, 46, 9612–9617 RSC.
- J. H. Song, A. Kapylou, H. S. Choi, B. Y. Yu, E. Matulevich and S. H. Kang, J. Power Sources, 2016, 313, 65–72 CrossRef CAS.
- C. Gao, J. Zhou, G. Liu and L. Wang, J. Alloys Compd., 2017, 727, 501–513 CrossRef CAS.
- C. Wang, Y. Xu, X. Sun, B. Zhang, Y. Chen and S. He, J. Power Sources, 2018, 378, 345–352 CrossRef CAS.
- S. Choi, M.-C. Kim, S.-H. Moon, H. Kim and K.-W. Park, Electrochim. Acta, 2019, 326, 135015 CrossRef CAS.
- J. Wang, C. Liu, G. Xu, C. Miao, M. Wen, M. Xu, C. Wang and W. Xiao, Chem. Eng. J., 2022, 438, 135537 CrossRef CAS.
- S.-B. Kim, H. Kim, D.-H. Park, J.-H. Kim, J.-H. Shin, J.-S. Jang, S.-H. Moon, J.-H. Choi and K.-W. Park, J. Power Sources, 2021, 506, 230219 CrossRef CAS.
- H. Liu, S.-h. Luo, S.-x. Yan, Y.-f. Wang, Q. Wang, M.-q. Li and Y.-h. Zhang, J. Electroanal. Chem., 2019, 850, 113434 CrossRef CAS.
- J. B. Kim, S. G. Lee, S.-Y. Choi, J. Kim and S. O. Kim, Ceram. Int., 2019, 45, 17574–17579 CrossRef.
- J. Wang, S. Zhao, J. Xie, D. Ruan and F. Zhao, J. Solid State Chem., 2021, 303, 122479 CrossRef CAS.
- W. Weng, J. Xu, C. Lai, Z. Xu, Y. Du, J. Lin and X. Zhou, J. Alloys Compd., 2020, 817, 152732 CrossRef CAS.
- Q. Deng, F. Zheng, W. Zhong, Q. Pan, Y. Liu, Y. Li, G. Chen, Y. Li, C. Yang and M. Liu, Chem. Eng. J., 2020, 392, 123735 CrossRef CAS.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed. Engl., 2018, 57, 102–120 CrossRef CAS PubMed.
- Z. Y. Gu, J. Z. Guo, X. X. Zhao, X. T. Wang, D. Xie, Z. H. Sun, C. D. Zhao, H. J. Liang, W. H. Li and X. L. Wu, InfoMat, 2021, 3, 694–704 CrossRef CAS.
- Z. Zhang, Y. Du, Q. C. Wang, J. Xu, Y. N. Zhou, J. Bao, J. Shen and X. Zhou, Angew. Chem., Int. Ed. Engl., 2020, 59, 17504–17510 CrossRef CAS PubMed.
- G. Zheng, Q. Lin, J. Ma, J. Zhang, Y. B. He, X. Tang, F. Kang, W. Lv and Q. H. Yang, InfoMat, 2021, 3, 1445–1454 CrossRef CAS.
- Z. Yan, Q.-W. Yang, Q. Wang and J. Ma, Chin. Chem. Lett., 2020, 31, 583–588 CrossRef CAS.
- M. Wu, W. Ni, J. Hu and J. Ma, Nano-Micro Lett., 2019, 11, 44 CrossRef CAS PubMed.
- M. Song, C. Wang, D. Du, F. Li and J. Chen, Sci. China: Chem., 2019, 62, 616–621 CrossRef CAS.
- Y. Qi, W. Gao, H. Wang, D. Liu, J. Deng, B. Guo, S. Bao and M. Xu, Chem. Eng. J., 2020, 402, 126118 CrossRef CAS.
- Z. Chen, L. Lu, Y. Gao, Q. Zhang, C. Zhang, C. Sun and X. Chen, Materials, 2018, 11, 2206 CrossRef PubMed.
- P. Wei, Y. Liu, Y. Su, L. Miao, Y. Huang, Y. Liu, Y. Qiu, Y. Li, X. Zhang, Y. Xu, X. Sun, C. Fang, Q. Li, J. Han and Y. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 3116–3124 CrossRef CAS PubMed.
- Q. Deng, Q. Cheng, X. Liu, C. Chen, Q. Huang, J. Li, W. Zhong, Y. Li, J. Hu, H. Wang, L. Wu and C. Yang, Chem. Eng. J., 2022, 430, 132710 CrossRef CAS.
- B. Quan, A. Jin, S. H. Yu, S. M. Kang, J. Jeong, H. D. Abruna, L. Jin, Y. Piao and Y. E. Sung, Adv. Sci., 2018, 5, 1700880 CrossRef PubMed.
- Y. Zhao, Y. Cong, H. Ning, X. Fei, C. Wu, H. Wang, Z. He, Y. Wang, Q. Zhao and M. Wu, J. Alloys Compd., 2022, 918, 165691 CrossRef CAS.
- Q. Zhang, Y. Huang, Y. Liu, S. Sun, K. Wang, Y. Li, X. Li, J. Han and Y. Huang, Sci. China Mater., 2017, 60, 629–636 CrossRef CAS.
- Y. Chen, Y. Xu, X. Sun, B. Zhang, S. He, L. Li and C. Wang, J. Power Sources, 2018, 378, 423–432 CrossRef CAS.
- K. Song, Y. Zhu, Y. Jiang, X. Wang, J. Ma, C. Dai and L. Zhao, J. Electroanal. Chem., 2021, 900, 115597 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |