
Electron transfer bridge inducing polarization of nitrogen molecules for enhanced photocatalytic nitrogen fixation†
Huiyi
Li‡
a,
Jiongrong
Wang‡
b,
Zhoushilin
Ruan‡
a,
Pengfei
Nan
d,
Binghui
Ge
 d,
Ming
Cheng
a,
Lan
Yang
a,
Xiaohong
Li
a,
Qilong
Liu
c,
Bicai
Pan
a,
Qun
Zhang
*a,
Chong
Xiao
*ac and
Yi
Xie
*ac
d,
Ming
Cheng
a,
Lan
Yang
a,
Xiaohong
Li
a,
Qilong
Liu
c,
Bicai
Pan
a,
Qun
Zhang
*a,
Chong
Xiao
*ac and
Yi
Xie
*ac
aHefei National Research Center for Physical Sciences at the Microscale, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), University of Science and Technology of China, Hefei, Anhui 230026, China
bKey laboratory of Strongy-Coupled Quantum Matter Physics, Department of Physics University of Science and Technology of China, Hefei, Anhui 230026, People's Republic of China
cInstitute of Energy, Hefei Comprehensive National Science Center, Hefei, Anhui 230031, China
dKey Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China
First published on 22nd August 2023
Abstract
Ammonia (NH3) plays a crucial role in the production of fertilizers, medicines, fibers, etc., which are closely relevant to the development of human society. However, the inert and nonpolar properties of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N seriously hinder artificial nitrogen fixation under mild conditions. Herein, we introduce a novel strategy to enhance the photocatalytic efficiency of N2 fixation through the directional polarization of N2 by rare earth metal atoms, which act as a local “electron transfer bridge.” This bridge facilitates the transfer of delocalized electrons to the distal N atom and redirects the polarization of adsorbed N2 molecules. Taking cerium doped BiOCl (Ce–BiOCl) as an example, our results reveal that the electrons transfer to the distal N atom through the cerium atom, resulting in absorbed nitrogen molecular polarization. Consequently, the polarized nitrogen molecules exhibit an easier trend for NN cleavage and the subsequent hydrogenation process, and exhibit a greatly enhanced photocatalytic ammonia production rate of 46.7 μmol g−1 h−1 in cerium doped BiOCl, nearly 4 times higher than that of pure BiOCl. The original concept of directional polarization of N2 presented in this work not only deepens our understanding of the N2 molecular activation mechanism but also broadens our horizons for designing highly efficient catalysts for N2 fixation.
N seriously hinder artificial nitrogen fixation under mild conditions. Herein, we introduce a novel strategy to enhance the photocatalytic efficiency of N2 fixation through the directional polarization of N2 by rare earth metal atoms, which act as a local “electron transfer bridge.” This bridge facilitates the transfer of delocalized electrons to the distal N atom and redirects the polarization of adsorbed N2 molecules. Taking cerium doped BiOCl (Ce–BiOCl) as an example, our results reveal that the electrons transfer to the distal N atom through the cerium atom, resulting in absorbed nitrogen molecular polarization. Consequently, the polarized nitrogen molecules exhibit an easier trend for NN cleavage and the subsequent hydrogenation process, and exhibit a greatly enhanced photocatalytic ammonia production rate of 46.7 μmol g−1 h−1 in cerium doped BiOCl, nearly 4 times higher than that of pure BiOCl. The original concept of directional polarization of N2 presented in this work not only deepens our understanding of the N2 molecular activation mechanism but also broadens our horizons for designing highly efficient catalysts for N2 fixation.
 Yi Xie | Since our initial publication in 2014, we have been joyfully observing the growth and advancement of Materials Horizons. As the journal celebrates its 10th anniversary, we wish for its sustained triumph and prosperity. It is with great excitement that we introduce our latest article, which uncovers captivating revelations within extensively researched materials. Here's to a wonderful 10th anniversary and the continued flourishing of Materials Horizons for years to come! |
New conceptsPhotocatalytic nitrogen fixation is emerging as a promising approach to efficiently activate the inert and nonpolar properties of NN, enabling the conversion of nitrogen molecules into activated products. In this study, we propose a novel approach to enhance the polarization of nitrogen molecules by constructing an electron transfer bridge. By employing this new concept of directional polarization of nitrogen, we achieved significantly enhanced photocatalytic efficiency in N2 fixation, offering immense potential to mitigate the greenhouse effect. By utilizing the electron transfer bridge, electrons can be transferred efficiently and directionally, which can effectively overcome the limitations posed by the inert and nonpolar nature of nitrogen molecules. This strategy opens up exciting opportunities for the design of advanced catalysts and contributes to our broader knowledge of nitrogen activation processes.
|
Nitrogen, as a kind of activated nitrogen building block, plays an important role in agriculture (fertilizer production) and the chemical industry. At present, the annual output of ammonia in the world is as high as 200 million tons.1 The industrial synthesis of NH3 mainly relies on the traditional Haber–Bosch process, which uses heterogeneous iron-based catalysts at high temperatures (300–500 °C) and high pressures (150–300 atm) under harsh reaction conditions. Statistics show that its average annual energy consumption exceeds 1–2% of the world's total energy consumption, accompanied by about 1.4% of global carbon dioxide emissions each year.2–4 Undoubtedly, this has indulged the current energy crisis and environmental pollution problems. Therefore, it is difficult, yet urgent, to seek and develop green NH3 synthesis methods. Compared with the Haber–Bosch method, the photocatalytic N2 reduction reaction for NH3 production uses solar energy as the only energy source and uses cheap and easily available water and nitrogen as reactants.5–7 However, the photocatalytic activation of N2 is extraordinarily difficult due to the weak adsorption of inert dinitrogen molecules on the surface of catalysts and the high ionization energy of nonpolar N
N covalent triple bonds toward dissociation.
As illustrated in Scheme 1, there are two distinct mechanisms for nitrogen reduction to ammonia under the action of a catalyst, namely dissociative and associative. In the dissociation mechanism, the NN bond is destroyed before the hydrogenation reaction, however, an extremely formidable dissociation energy of 941 kJ mol−1 made it difficult. Besides, the associative mechanism contains two different pathways, namely, the alternating pathway and the distal pathway.8 The associative alternating pathway postulates that H+ is alternately attached to two N atoms. In other words, this nitrogen activation pathway involves sequential functionalization of both the proximal nitrogen (Np) and distal nitrogen (Nd) atoms. Unfortunately, the two N atoms suffer from various obstacles. On one hand, due to the close electron interaction between the adsorption center and the Np atom in M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NpNd complexes, the Np atom usually exhibits a higher electron density.9,10 Despite this, the steric hindrance of Np and the excessively strong Np-adsorbent interaction still preclude reactivity.11 On the other hand, the distal Nd atoms cannot be activated due to the depressed electron delivery efficiency. The above two obvious problems put the alternating pathway in a dilemma. In the associative distal pathway, generally, hydrogenation occurs preferentially on the nitrogen atom farthest from the surface, generating an equivalent of NH3 and leaving an adsorbed nitrogen atom on the catalyst surface, which is subsequently hydrogenated to give a second equivalent of NH3. Though the distal and flexible Nd atom provides more opportunities for progressive H+ functionalization, the lack of electrons on Nd still constricts nitrogen activation.12 Therefore, how to increase the electron density of the Nd atom, namely, the directional polarization of the nitrogen molecule, has become the most effective and practical demand to enhance the activation of the nitrogen molecule.
NpNd complexes, the Np atom usually exhibits a higher electron density.9,10 Despite this, the steric hindrance of Np and the excessively strong Np-adsorbent interaction still preclude reactivity.11 On the other hand, the distal Nd atoms cannot be activated due to the depressed electron delivery efficiency. The above two obvious problems put the alternating pathway in a dilemma. In the associative distal pathway, generally, hydrogenation occurs preferentially on the nitrogen atom farthest from the surface, generating an equivalent of NH3 and leaving an adsorbed nitrogen atom on the catalyst surface, which is subsequently hydrogenated to give a second equivalent of NH3. Though the distal and flexible Nd atom provides more opportunities for progressive H+ functionalization, the lack of electrons on Nd still constricts nitrogen activation.12 Therefore, how to increase the electron density of the Nd atom, namely, the directional polarization of the nitrogen molecule, has become the most effective and practical demand to enhance the activation of the nitrogen molecule.
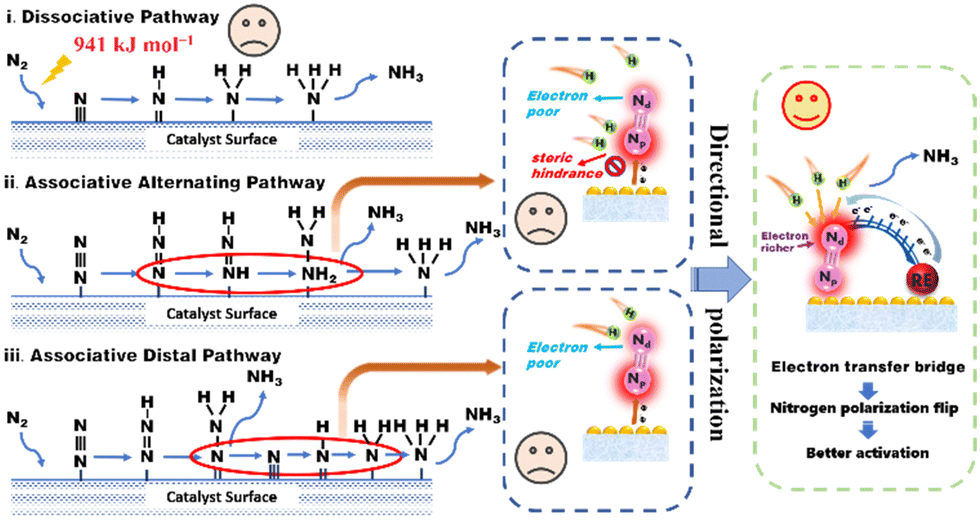 | ||
| Scheme 1 Schematic diagram of the nitrogen activation process and our designed directional polarization. The common nitrogen activation mechanisms include the dissociative pathway, the alternating associative pathway, and the distal associative pathway. The scheme points out the existing problems in the above pathways. To circumvent these issues, after the electron transfer bridge was formed by rare earth atoms, the electrons are delivered through the catalyst to the distal N atom, resulting in nitrogen molecular polarization. | ||
As a typical type of layered two-dimensional material, bismuth oxyhalide (BiOX) has been widely researched as a modern nitrogen photoreduction catalyst.13 The structure is mainly composed of the [Bi2O2]2+ layer in the middle and the [X]− layer on both sides.14 The widespread surface oxygen vacancies (OVs) behave as the N2 reduction sites by the photoexcited conduction band electrons.15 The charge delivered from the OVs to the adsorbed N2 leads to electron accumulation on the Np atom rather than the more flexible Nd atom. Blame to above-mentioned complications, the nitrogen photoreduction efficiency of this traditional photocatalyst remains at a standstill. Recently, owing to the abundant coupling of the metal and ligand orbitals in the support and its changes in the d-band center (εd) of the metal atoms, traditional transition metals, precious metals, and rare earth metals have led to a series of successful design of catalysts.16–18 Among these metal elements, the 4f electrons of rare earth elements are initially highly delocalized compared to the valence 5d and 6s electrons, leading to their chemical inertness, especially when stabilized through coordination.19,20 However, the quantum nature of d–f exchange interactions results in strong coupling between the 3d, 4f, and 5d orbitals. While the 4f energy levels in RE oxides cause the 4f electrons to be strongly localized, hybridization with lighter elements (e.g., oxygen via the 2p orbital) can induce delocalization and covalent interactions.21,22 These delocalized electrons can then be transferred to the adsorbed reaction molecules through the “electron transfer bridge”.23 The electron transfer bridge enables rapid migration of photogenerated electrons to active sites, preventing recombination with holes. This efficient charge separation enhances the photocatalytic efficiency and promotes desirable redox reactions, leading to improved overall performance in various photocatalytic processes.24–26 These findings encourage us to dig into the possibility of the N2 molecule's directional polarization for more efficient activation of NN triple bonds by the introduction of rare earth metals.
Herein, inspired by the above-mentioned advantages, conceptually directional polarization of N2, which receives electrons through an electron transfer bridge formed by rare earth atoms, was first presented as an effective way to promote photocatalytic N2 fixation. Taking Ce-doped BiOCl (Ce–BiOCl) as a representative, the insights gained from the experimental and theoretical analysis results indicate that the surface electron distribution dramatically changed to form an efficient electron transfer bridge, which transfers electrons to the Nd atom and results in polarization of the NN triple bond. As a result, the redirected polarized N2 accelerates the hydrogenation process for photocatalytic N2 reduction in Ce–BiOCl nanosheets with an ammonia production rate of 46.7 μmol g−1 h−1, nearly 4 times compared to pure BiOCl. This study sheds light on the rational design of nitrogen polarization for N2 photoreduction and other important chemical transformations.
All Bragg diffraction peaks in the X-ray diffraction (XRD) pattern (Fig. 1a) for BiOCl and Ce–BiOCl were well-indexed to BiOCl (P4/nmm JCPDS 06-0249). No diffraction peaks of Ce were observed in the XRD patterns of Ce–BiOCl nanosheets, which indicated the absence of impurity phases. X-ray photoelectron spectroscopy (XPS) analysis was used to verify the chemical state of the samples. Two peaks located at 904.2 and 884.7 eV were assigned to the Ce 3d3/2 and Ce 3d5/2 signals of Ce3+, respectively (Fig. 1b).27–29 For BiOCl, the Bi 4f XPS spectra were attributed to Bi 4f7/2 and Bi 4f5/2 of Bi3+, respectively. The high-resolution O 1s spectra (Fig. S1b, ESI†) can be indexed into two distinctive binding energy peaks located at 529.9 and 531.8 eV, belonging to the lattice oxygen and adsorbed oxygen on the BiOCl surface, respectively.30 Specifically, the XPS peak representing lattice oxygen (lattice O) experienced a red shift of 0.332 eV, while the peak representing oxygen vacancy (OV) showed a red shift of 0.243 eV. Additionally, the XPS peaks of Bi exhibited a double peak shift of 0.353 eV. The decrease of the binding energy of Bi 4f and O 1s demonstrated the weakening of the electron screening effect due to the increased electron density, and thus the increase of binding energy means an increased electron density.31 It is possible that Bi atoms attract additional electrons from neighboring Ce atoms, influencing the charge environment around Bi and affecting the Bi–O bond length or bond strength, consequently affecting the XPS peaks associated with Bi–O bonds more significantly than those associated with oxygen vacancies. In addition, the immobile XPS peak location of Cl 2p manifests that the chemical environment of Cl may not be disturbed by the introduction of Ce (Fig. S1c, ESI†). Thus, the shift of the binding energy is ascribed to the electronic interaction and charge transfer between Ce and the [Bi2O2]+ layer.
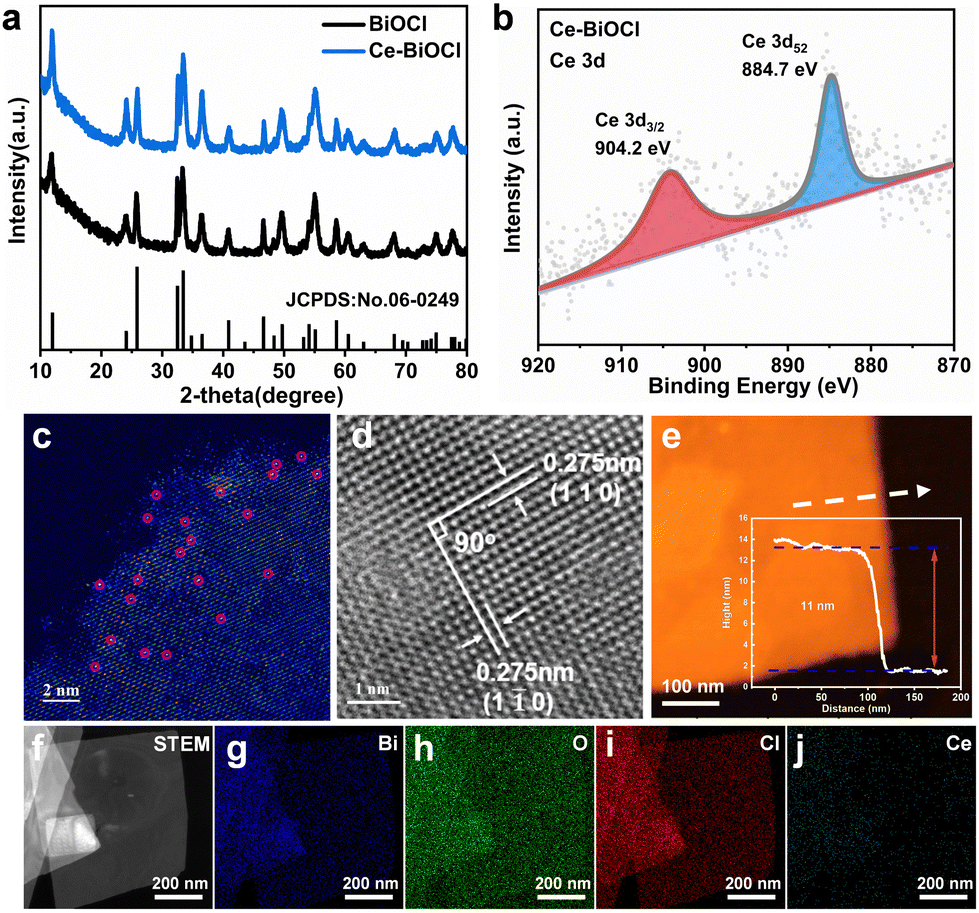 | ||
| Fig. 1 Structural characterization. (a) XRD patterns of BiOCl and Ce–BiOCl. (b) High-resolution Ce 3d XPS spectra of Ce–BiOCl. Atomic resolution HAADF-STEM images of (c) Ce–BiOCl. (d) HRTEM image of Ce–BiOCl. (e) AFM images and corresponding height profiles of Ce–BiOCl. (f) STEM image of Ce–BiOCl and (g)–(j) the corresponding elemental distribution of Bi, O, Cl and Ce. | ||
The as-prepared BiOCl samples all show sheet-like structures with horizontal sizes ranging around 500 nm and no obvious morphology changes were observed between BiOCl and others as shown in the transmission electron microscopy (TEM) images in Fig. S2 and S3 (ESI†). In order to prove the existing form of lanthanide from the atomic level, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) more intuitively with aberration correction was used to detect the surface of two-dimensional Ce–BiOCl nanosheets. In the HAADF-STEM image, irregularly distributed bright spots are located on the lattice sites, representing the combined information of BiOCl and Ce. This combined presence of BiOCl and Ce atoms contributes to the highest scattering intensity, resulting in the brightest appearance in the image. (Fig. 1c). This finding further proved that the lanthanide atoms are loaded onto the substrate, which is consistent with the above-mentioned XRD and XPS results. As shown in the high-resolution transmission electron microscopy (HRTEM) image (Fig. 1d), the observed lattice spacing of 0.275 nm has an even distribution, corresponding to the (110) or (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) crystal plane spacing, revealing that the dominant exposed crystal plane of Ce–BiOCl nanosheets is the (001) crystal plane.32 Due to the difference in atomic radii, with Ce having a radius of 1.65 and Bi having a radius of 1.46, the interplanar spacing in Ce–BiOCl is measured at 0.275 nm, whereas in BiOCl, it is observed at 0.271 nm (Fig. S4, ESI†). The AFM characterization indicates that the thickness of the nanosheet is about 11 nm (Fig. 1e and Fig. S5, ESI†). Furthermore, we performed energy-dispersive X-ray spectroscopy (EDS) mapping on Ce–BiOCl, as revealed in Fig. 1f–j. The distribution of Bi, O, Cl, and Ce elements corresponding to the STEM image manifests the uniform distribution. In summary, it can be considered that Ce has been successfully introduced to two-dimensional BiOCl nanosheets.
0) crystal plane spacing, revealing that the dominant exposed crystal plane of Ce–BiOCl nanosheets is the (001) crystal plane.32 Due to the difference in atomic radii, with Ce having a radius of 1.65 and Bi having a radius of 1.46, the interplanar spacing in Ce–BiOCl is measured at 0.275 nm, whereas in BiOCl, it is observed at 0.271 nm (Fig. S4, ESI†). The AFM characterization indicates that the thickness of the nanosheet is about 11 nm (Fig. 1e and Fig. S5, ESI†). Furthermore, we performed energy-dispersive X-ray spectroscopy (EDS) mapping on Ce–BiOCl, as revealed in Fig. 1f–j. The distribution of Bi, O, Cl, and Ce elements corresponding to the STEM image manifests the uniform distribution. In summary, it can be considered that Ce has been successfully introduced to two-dimensional BiOCl nanosheets.
To evaluate the photocatalytic nitrogen reduction performance, the gas–liquid experiments were performed in an N2-saturated water solution under simulated solar-light irradiation detected by ion chromatography (IC) (Fig. S6 and S7, ESI†). Before nitrogen reduction, the catalyst powder was dispersed in 40 mL of water and then irradiated under a 300 W Xe lamp for 5 h to eliminate any surface organic contaminants during the catalyst preparation process. As shown in Fig. S8 (ESI†), N 1s XPS confirmed that the surface organic contaminants were eliminated by this photocatalytic oxidation process. The pristine BiOCl nanosheets obtained relatively low NH3 production rates of 11.4 μmol g−1 h−1. With different amounts of Ce doping, the catalysts all exhibited better catalytic performance compared with the pure BiOCl (Fig. 2a). Among those, Ce–BiOCl-3 (denoted as Ce–BiOCl) exhibited an increase in the NH3 production rate of up to nearly 46.7 μmol g−1 h−1, 4 times higher than that of pure BiOCl. However, we noticed that excessive Ce doping induced a negative effect on the photocatalytic process, resulting in a reduced rate of N2 fixation, owing to possible agglomeration. We further carry out time-dependent measurements with continuous light irradiation for up to 3 h over BiOCl and Ce–BiOCl. As shown in Fig. 2b, the detected amount of ammonia was gradually increased by prolonging the reaction time, corroborating the photocatalytic N2 fixation to ammonia. In addition, no evident inactivation has been detected during the cycling stability test of Ce–BiOCl indicating excellent stability and durability (Fig. 2c). It can be observed that there is no obvious structure and morphology change based on the XRD pattern and the TEM image after the photocatalytic reaction, respectively (Fig. S9, ESI†). Moreover, wavelength-dependent apparent quantum efficiency (AQE) experiments, an important parameter to evaluate catalyst photoelectric conversion performance, were performed under monochromatic light irradiation on Ce–BiOCl. The AQE values are about 0.39%, 0.33%, 0.20%, 0.14%, 0.06%, and 0.02% at 254, 295, 320, 380, 500, and 600 nm, respectively (Fig. 2d). We also conducted a series of control experiments for comparison, as shown in Table S1 (ESI†). The Ce–BiOCl samples did not show the activity of photocatalytic NH3 production when using Ar/He as the reaction gas. To verify the exact origin of the generated ammonia, a 15N labeling experiment was carried out and examined by 1H nuclear magnetic resonance (1H-NMR) spectroscopy. As shown in Fig. 2e, the doublet pattern with a coupling constant of JN–H = 72 Hz corresponded to 15NH4+ in DMSO-d6. Meanwhile, when performed in a 14N2 atmosphere, the resulting spectrum manifested a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet pattern with a coupling constant of JN–H = 52 Hz, which agreed with the 14NH4+ signal.33 The above results revelaed that the synthesized NH3 completely came from photocatalytic N2 reduction.
:1:1 triplet pattern with a coupling constant of JN–H = 52 Hz, which agreed with the 14NH4+ signal.33 The above results revelaed that the synthesized NH3 completely came from photocatalytic N2 reduction.
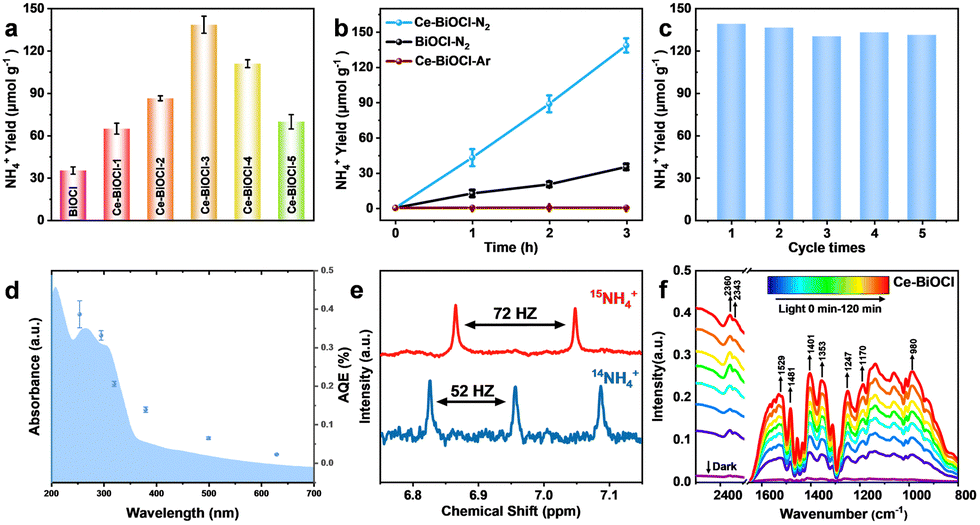 | ||
| Fig. 2 Photocatalytic activity under a 300 W xenon lamp, 15N isotope tracing results and in situ-FTIR results. (a) Photocatalytic NH4+ yields of BiOCl, Ce–BiOCl-1, Ce–BiOCl-2, Ce–BiOCl-3, Ce–BiOCl-4, and Ce–BiOCl-5. (b) Photocatalytic NH4+ yields for 3 h under Ar and N2 atmospheres. (c) Cycling stability in the first three hours for the cycling stability test of Ce–BiOCl. (d) AQE of Ce–BiOCl. (e) 1H NMR spectra of 15NH4+ and 14NH4+ products after photocatalytic reactions using 15N2 and 15N2 gases as the nitrogen source, respectively. (f) The in situ-DRIFTS of Ce–BiOCl under illumination. | ||
Regarding the distinction between the associative alternating pathway and associative distal pathway for N2 activation, the distinctive intermediate N2H4 of the associative alternating pathway was not detected, which indicated that nitrogen is activated in the associative distal pathway (Fig. S10, ESI†).34 In order to further reveal the photocatalytic process of N2 fixation, in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) was performed (Fig. 2f). With the light irradiation time prolonging, these vibration peaks grew rapidly indicating efficient N2 adsorption, activation, and conversion processes on Ce–BiOCl. The bands at 1402 and 1354 cm−1 are ascribed to the in-plane bending vibration of the activated O–H bond. Notably, the bands at 1248 and 1529 cm−1 represent the wagging vibration absorption peaks of the –NH2 group. The gradually increased peaks of 1354, 1187 and 1058 cm−1 could be attributed to σ(N–H) derived from intermediates and NH3 species, respectively.35 The bands at 1247, 1134 and 980 cm−1 could be assigned to weakly adsorbed gas-phase NH3 generated through the process. Moreover, the 1308 and 1187 cm−1 bands arose from the characteristic absorption of the final product NH4+.36 These results confirmed the reaction energy diagram of the nitrogen fixation pathway of *N2 → *NNH → *NNH2 → *NNH3 → *N + NH3 → *NH → *NH2 → *NH3 → NH3. However, the N2 adsorption signals and all the vibration signals were weakened in pure BiOCl (Fig. S11, ESI†). The measurement confirmed that Ce significantly enhances the adsorption of N2 and accelerates the conversion process of N2 to NH3.
To deeply understand the relaxation processes of photogenerated carriers, femtosecond time-resolved transient absorption (fs-TA) spectroscopy using a pump–probe configuration was employed, which is a powerful tool for tracking the photogenerated carrier relaxation process. In TA measurements, a short, usually femtosecond (fs), duration optical pulse (the pump) excites the sample and, after a temporal delay, a less powerful short pulse (the probe) is used to monitor the excited population of charge carriers. In photocatalytic systems, photogenerated charge carriers (electron–hole pairs) are created upon light absorption. These charge carriers can either be effectively separated and utilized for desired reactions, or they may recombine, leading to a loss of photocatalytic efficiency.37 The use of a 320 nm pump was enough to promote the electrons from the valence band to the conduction band, and a time delayed broadband white light in the wavelength range of 450–770 nm was used for probing. As shown in Fig. 3a and b, the fs-TA spectral profiles of the two samples manifested similar broad photo-induced bleaching signals (negative value), further verifying that the introduction of Ce3+ had limited influence on the crystal structure of BiOCl. Remarkably, however, the signal minimum of Ce–BiOCl was about twice that of BiOCl, suggesting that Ce3+ could open a new electron transfer channel that leads to the rapid transfer of photogenerated electrons and hence effective spatial charge separation. In order to glean information about the carrier lifetime of interest, we analyzed the fs-TA kinetics through a bi-exponential global fitting in a representative probing region of 655–705 nm, as shown in Fig. 3c and d. In photocatalytic nitrogen fixation, the hundred-picosecond-scale lifetime observed in our Ce–BiOCl system is likely due to charge recombination.38,39 Charge recombination refers to the process where photogenerated electron–hole pairs recombine before participating in desired photocatalytic reactions. This recombination pathway competes with the desired charge separation and utilization for nitrogen fixation reactions, leading to a loss of photocatalytic efficiency.40,41 Apparently, the average carrier lifetime of Ce–BiOCl (i.e., 262 ± 4 ps) was prolonged by ∼43% relative to that of BiOCl (i.e., 183 ± 6 ps), indicating that the photogenerated carriers in the Ce–BiOCl case are prone to participating in the photocatalytic reaction due to the slowing down of charge recombination therein. Moreover, as shown in Fig. S12a (ESI†), the transient photocurrent response of Ce–BiOCl was significantly enhanced compared to that of pure BiOCl, indicating that the Ce–BiOCl sample has an enhanced photo-induced charge transfer capability. As presented in Fig. S12b (ESI†), the EIS results reveal that the arc radius of Nyquist plots for the Ce–BiOCl sample is smaller than that for BiOCl in Na2SO4 solution. The BiOCl samples showed an absorption edge of about 390 nm, which is the inherent absorption characteristic of BiOCl materials.
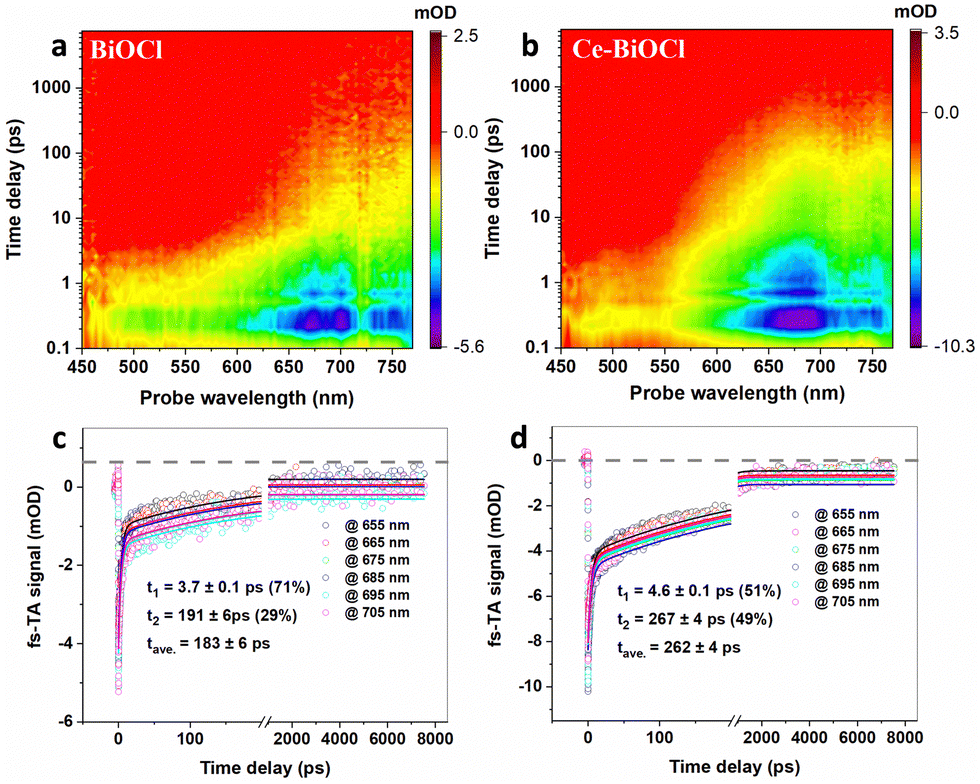 | ||
| Fig. 3 Photogenerated carrier dynamics tracked by fs-TA spectroscopy. Pseudocolor fs-TA spectra of (a) BiOCl and (b) Ce–BiOCl. Representative fs-TA kinetic traces (probe in the wavelength range of 655–705 nm) and the bi-exponential global fitting results of (c) BiOCl and (d) Ce–BiOCl. | ||
In order to analyze the adsorption form and adsorption capacity of N2 molecules on the surface of Ce-doped BiOCl, we conducted the N2 temperature programmed desorption (N2-TPD) test. The desorption peaks at 363.0 and 556.7 °C were attributed to the physical and chemical adsorption of N2 on Ce–BiOCl, respectively. The desorption temperature of BiOCl are 361.0 °C and 547.9 °C (Fig. S13, ESI†). This indicates that the binding strength of N2 with Ce–BiOCl is stronger than that with BiOCl, manifesting that Ce–BiOCl can facilitate N2 adsorption. Raman spectroscopy can provide information about the polarizability of gas molecules, which can be related to their polarity. As shown in Fig. 4a, the band observed at 2330 cm−1 in Raman spectra was assigned to the Q-branch of the vibration–rotation band of gaseous N2, while no signal was detected when the gas was changed to Ar.42 A slight shift in the Raman peak position of N2 was observed on the surface of both BiOCl and Ce–BiOCl compared to pure N2, which can be attributed to the polarization change of nitrogen molecules.43 To further clarify the promotion effect of Ce–BiOCl on N2 activation, quasi-in situ XANES spectra of the N K-edge were performed.
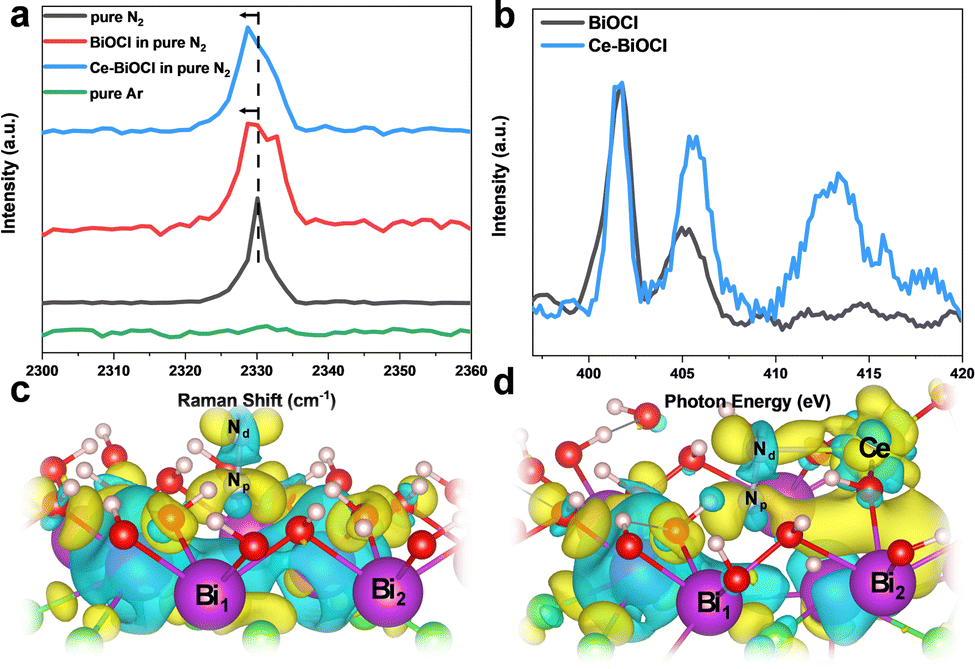 | ||
| Fig. 4 (a) Raman spectra of as-prepared different samples. (b) Quasi-in situ XANES spectra of the N K-edge of BiOCl and Ce–BiOCl after N2 treatment. Theoretical simulation results of the charge density difference. Bader charge analysis of (c) BiOCl and (d) Ce–BiOCl, where the yellow and cyan isosurfaces correspond to an increase in the number of electrons and the depletion zone, respectively. | ||
As shown in Fig. 4b, the peaks in the 399–408 eV range were identified as multi-electron excitations related to the notable N 1s → π* transition, while the peak at approximately 414 eV was referred to as σ* shaped resonance.44,45 The energy position and intensity of σ* resonance are highly sensitive to the internuclear distance or bond length, which means that a longer bond length would lead to a lower position and higher intensity of σ* resonance for first-row diatomics.46,47 The σ* resonance of adsorbed N2 for Ce–BiOCl demonstrated a distinct shift in position and increased intensity compared to that of BiOCl, indicating that the NN triple bond is unevenly stretched and effectively polarized over Ce–BiOCl.
In order to obtain insights into the origin of the improvement of ammonia production, we conducted first-principles calculations using the generalized gradient approximation of DFT using VASP. The cleavage of the NN bond is the rate-determining key to the N2 fixation reaction.48 According to the Bader charge in Fig. 4c and d, after the generation of an OV on the (001) surface, the electrons are mainly localized on the two nearest Bi atoms and are transferred to the end-on adsorbed N2 after its adsorption.49 Notably, the introduction of Ce broke its nearby charge symmetry, and the two vicinal Bi1/Bi2 charge ratios surrounding the OV changed from 13.27/13.26 to 13.20/14.17. Upon N2 access, the charge on two N atoms had been redistributed. In other words, the distal Nd atom could be particularly electron richer than the proximal Np atom. The charge ratio of Nd/Np changed from 5.09/5.26 to 5.35/5.17 (Table S2, ESI†). After the N2 adsorption obstacle was conquered, there were two main rate-determining steps in the process, the first H adsorption onto the N2 molecule and the desorption of the product NH3. Since the distal N atom is less sterically encumbered, the electron crossover on the two N atoms will effectively promote the first hydrogenation on the distal N and the product desorption.50 Consequently, the calculated N2 adsorption energy (Eads) is 0.206 eV in BiOCl, and in sharp contrast, the Eads value significantly decreased to −0.05 eV (Fig. S14, ESI†). Thus, we believe that Ce is anchored by BiOCl which not only can result in a change in the density of the surrounding electron cloud but also can better help to absorb N2 molecules.
Conclusions
In summary, we propose a novel strategy to redirect the polarization of adsorbed N2 molecules through the “electron transfer bridge” of rare earth atoms for optimization of photocatalytic N2 fixation. Our experimental and theoretical analysis results demonstrate that the rare earth atom can construct an electron transfer pathway, leading to fast charge carrier transfer and an increase in electron density for the Nd atom, ultimately causing the polarization of N2 molecules. As a result, the less sterically encumbered Nd with engorged electrons can better engage in the subsequent hydrogen activation process. Taking Ce-BiOCl as an example, our results show that the Ce–BiOCl nanosheet displays significantly enhanced photocatalytic N2 reduction performance with an ammonia production rate of 46.7 μmol g−1 h−1, nearly 4 times higher than that of pure BiOCl. Given the unique electron configurations and various types of rare earth metal elements, it is reasonable to expect that more efficient nitrogen fixation catalysts can be developed by introducing rare earth elements with different electron delivery capabilities. The findings of this study not only offer a highly efficient catalytic system for nitrogen photofixation but also provide valuable insights into the activation of inert molecular polarization.Author contributions
C. X. conceived the idea. C. X. and Y. X. supervised this project. H. Y. L. designed and performed the sample synthesis, characterization, and photocatalytic activity measurements. J. R. W. and B. C. P. conducted the theoretical analysis. Z. S. L. R. and Q. Z. conducted the fs-TA measurements and analyses. P. F. N and B. H. G conducted the HAADF measurements and analyses. M. C., L. Y., X. H. L. and Q. L. L. assisted in performing photocatalytic activity experiments and initial analysis of the data. H. Y. L., C. X. and Y. X. co-wrote the manuscript. All authors participated in the overall scientific interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36030300), the Youth Innovation Promotion Association CAS (Y202092), the National Natural Science Foundation of China (21890750 and 22173090), the National Key Research and Development Program of China (2018YFA0208702), the Fundamental Research Funds for the Central University (WK2340000094), the Innovation Program for Quantum Science and Technology (2021ZD0303303), and the Anhui Initiative in Quantum Information Technologies (AHY090200). The computational center of USTC and Hefei Advanced Computing Center are acknowledged for computational support. We thank the Catalysis and Surface Science Endstation at the BL11U beamline and the Infrared Spectroscopy and Microspectroscopy at the BL01B beamline in the National Synchrotron Radiation Laboratory (NSRL) for help in the characterization.Notes and references
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- T. L. Root, J. T. Price, K. R. Hall, S. H. Schneider, C. Rosenzweig and J. A. Pounds, Nature, 2003, 421, 57–60 CrossRef CAS PubMed.
- X. Chen, N. Li, Z. Kong, W. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- M. Cheng, C. Xiao and Y. Xie, J. Mater. Chem. A, 2019, 7, 19616–19633 RSC.
- M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- B. Huang, N. Li, W. Ong and N. Zhou, J. Mater. Chem. A, 2019, 7, 27620–27631 RSC.
- S. Hu, X. Chen, Q. Li, F. Li, Z. Fan, H. Wang, Y. Wang, B. Zheng and G. Wu, Appl. Catal., B, 2017, 201, 58–69 CrossRef CAS.
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS.
- T. M. Buscagan and D. C. Rees, Joule, 2019, 3, 2662–2678 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- Q. Hao, C. Liu, G. Jia, Y. Wang, H. Arandiyan, W. Wei and B. Ni, Mater. Horiz., 2020, 7, 1014–1029 RSC.
- Y. Shiraishi, M. Hashimoto, K. Chishiro, K. Moriyama, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2020, 142, 7574–7583 CrossRef CAS PubMed.
- R. Gao, J. Wang, Z. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang, C. Shi, J. Lim and J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zboril, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- H. C. Herper, O. Y. Vekilova, S. I. Simak, I. Di Marco and O. Eriksson, J. Phys.: Condens.Matter, 2020, 32, 215502 CrossRef CAS PubMed.
- H. Wang, X. Gong, Y. Guo, Y. Guo, G. Lu, P. Hu and H. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- M. Alaydrus, M. Sakaue and H. Kasai, Phys. Chem. Chem. Phys., 2016, 18, 12938–12946 RSC.
- Z. Chen and J. Yang, J. Chem. Phys., 2022, 156, 211101 CrossRef CAS PubMed.
- H. Ma, J. Liu, Y. Wang, S. Zuo, Y. Yu and B. Li, Mater. Adv., 2022, 3, 7546–7558 RSC.
- X. Wang, Y. Zhu, H. Li, J. Lee, Y. Tang and G. Fu, Small Methods, 2022, 6, 2200413 CrossRef CAS PubMed.
- Q. Chen, G. Gao, Y. Zhang, Y. Li, H. Zhu, P. Zhu, Y. Qu, G. Wang and W. Qin, J. Mater. Chem. A, 2021, 9, 15820–15826 RSC.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, e202218460 CrossRef CAS PubMed.
- X. Wang, F. You, Q. Yao, K. Wang, Y. Liao, G. Tong, X. Wang, T. Wu and W. Wu, Mater. Horiz., 2023, 10, 2677–2690 RSC.
- Z. Dai, F. Qin, H. Zhao, J. Ding, Y. Liu and R. Chen, ACS Catal., 2016, 6, 3180–3192 CrossRef CAS.
- W. Wang, J. Chen, Y. Tao, S. Zhu, Y. Zhang and X. Wu, ACS Catal., 2019, 9, 3498–3510 CrossRef CAS.
- Y. Mi, L. Wen, Z. Wang, D. Cao, R. Xu, Y. Fang, Y. Zhou and Y. Lei, Nano Energy, 2016, 30, 109–117 CrossRef CAS.
- Y. Shi, G. Zhan, H. Li, X. Wang, X. Liu, L. Shi, K. Wei, C. Ling, Z. Li, H. Wang, C. Mao, X. Liu and L. Zhang, Adv. Mater., 2021, 33, 2100143 CrossRef CAS PubMed.
- J. Yang, H. Bai, Y. Guo, H. Zhang, R. Jiang, B. Yang, J. Wang and J. C. Yu, Angew. Chem., Int. Ed., 2021, 60, 927–936 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- X. Dong, Z. Cui, X. Shi, P. Yan, Z. Wang, A. C. Co and F. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200937 CrossRef CAS PubMed.
- Z. Wu, B. Jiang, Y. Liu, H. Wang and R. Jin, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS PubMed.
- S. Yang, C. Wang, J. Li, N. Yan, L. Ma and H. Chang, Appl. Catal., B, 2011, 110, 71–80 CrossRef CAS.
- D. A. Wheeler and J. Z. Zhang, Adv. Mater., 2013, 25, 2878–2896 CrossRef CAS PubMed.
- Y. Shi, G. Zhan, H. Li, X. Wang, X. Liu, L. Shi, K. Wei, C. Ling, Z. Li, H. Wang, C. Mao, X. Liu and L. Zhang, Adv. Mater., 2021, 33, 2100143 CrossRef CAS PubMed.
- B. Wang, W. Zhang, G. Liu, H. Chen, Y. Weng, H. Li, P. K. Chu and J. Xia, Adv. Funct. Mater., 2022, 32, 2202885 CrossRef CAS.
- P. Zhang, T. Wang, X. Chang and J. Gong, Acc. Chem. Res., 2016, 49, 911–921 CrossRef CAS PubMed.
- Y. Yao, X. Bao, Y. Zhu, X. Sui, A. Hu, P. Bai, S. Wang, H. Yang, X. Liu and Y. Gao, Nano. Res, 2023, 16, 10420–10428 CrossRef CAS.
- A. Koo, U. D. Lanke, F. Budde, S. Granville, H. J. Trodahl, A. Bittar, J. B. Metson, V. J. Kennedy, A. Markwitz and B. J. Ruck, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235202 CrossRef.
- D. Dill, S. Wallace and J. L. Dehmer, Phys. Rev. Lett., 1979, 43, 1005–1008 CrossRef.
- Y. Zhang, T. Hou, Q. Xu, Q. Wang, Y. Bai, S. Yang, D. Rao, L. Wu, H. Pan, J. Chen, G. Wang, J. Zhu, T. Yao and X. Zheng, Adv. Sci., 2021, 8, 2100302 CrossRef CAS.
- F. Sette, J. Stöhr and A. P. Hitchcock, J. Chem. Phys., 1984, 81, 4906–4914 CrossRef CAS.
- A. P. Hitchcock and J. Stöhr, J. Chem. Phys., 1987, 87, 3253–3255 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- H. Li, J. Shang, J. Shi, K. Zhao and L. Zhang, Nanoscale, 2016, 8, 1986–1993 RSC.
- X. Zhang, C. Fan, Y. Wang, Y. Wang, Z. Liang and P. Han, Comput. Mater. Sci., 2013, 71, 135–145 CrossRef CAS.
- Q. Hao, C. Liu, G. Jia, Y. Wang, H. Arandiyan, W. Wei and B. Ni, Mater. Horiz., 2020, 7, 1014–1029 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01041d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |