
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Factors influencing the catalytic activity of metal-dependent histidine-rich peptides: sequence, conformation, stereochemistry, self-assembly or their interplay?†
Patrizia
Janković
 a,
Marko
Babić
a,
Marko
Perčić
bcd,
Ana S.
Pina
e and
Daniela
Kalafatovic
*ad
a,
Marko
Babić
a,
Marko
Perčić
bcd,
Ana S.
Pina
e and
Daniela
Kalafatovic
*ad
aUniversity of Rijeka, Department of Biotechnology, Radmile Matejčić 2, 51000 Rijeka, Croatia. E-mail: daniela.kalafatovic@uniri.hr
bUniversity of Rijeka, Faculty of Engineering, Vukovarska 58, 51000 Rijeka, Croatia
cUniversity of Rijeka, Centre for Micro- and Nanosciences and Technologies, 51000 Rijeka, Croatia
dUniversity of Rijeka, Center for Artificial Intelligence and Cybersecurity, 51000 Rijeka, Croatia
eInstituto de Tecnologia Química e Biológica António Xavier (ITQB), Universidade Nova de Lisboa, Av. da República, 2780-157, Oeiras, Portugal
First published on 13th September 2023
Abstract
The sequence-to-function relationship of peptide-based catalysts remains a challenge, as even subtle modifications at the sequence level can alternate their catalytic activity. A set of linear and cyclic histidine-rich peptides was synthesized to assess the impact of amino acid disposition, cyclization, and incorporation of D-amino acids on their ability to self-assemble, coordinate Zn2+ ions, and show intrinsic hydrolase-like activity. Self-assembly into β-sheets was confirmed for both linear peptides and one cyclic analogue (cy-hh) by FTIR, ThT binding, CD, and AFM. Interestingly, only peptide A demonstrated efficient ester hydrolysis of p-NPA, p-NPB and p-NPO substrates, indicative of its effective Zn2+ coordination. Our findings highlight that increased rigidity of the peptide can hinder metal ion coordination by limiting the necessary conformational adjustments for optimal Zn2+ binding. These insights into the structural changes underlying the function of short peptides offer valuable knowledge for the design of metal-dependent peptide-based catalysts.
Design, System, ApplicationWe investigated the impact of modifications at the sequence level, including amino acid disposition, cyclization and chirality of key residues involved in catalysis on the functionality and self-assembly propensity of Ac-IHIHINI-Am and its analogues. Our research sought to uncover the molecular principles governing catalytic activity in metal-dependent peptides. While the introduction of rigidity in cyclic peptides hindered the coordination of metal ions due to strict geometric requirements, the conformationally heterogeneous linear analogue exhibited more favorable metal coordination, proving to be the critical factor for achieving catalytic activity, surpassing the importance of self-assembly propensity. This work contributes to a deeper comprehension of catalytic activity in histidine-rich peptides and holds great promise for the future exploration of novel metal-dependent catalytic peptides. Understanding the intricate relationship between peptide sequence and function allows for the optimization of their catalytic performance and opens up diverse possibilities for their application in various industrial processes and therapeutic settings. |
1 Introduction
Nanozymes are a class of nanoscale materials that exhibit intrinsic catalytic activity, mimicking the functions of natural enzymes.1,2 They show unique properties that make them attractive for applications ranging from medical biosensing,3 treatment of bacterial infections,4 to environmental remediation.5 Nanozymes are typically composed of inorganic, metal, or metal oxide nanoparticles that resemble catalytic functions, including peroxidase, oxidase, catalase, and other activities.3 They offer several advantages over traditional enzymes, including versatility of design, ease of synthesis and functionalization.6Peptides have gained attention as a promising class of molecules able to catalyze biochemical transformations.7–11 Their significance comes from the fact that amino acids responsible for various catalytic mechanisms can be incorporated into a variety of designs and also from their ability to self-assemble in supramolecular architectures with emerging properties. In addition, peptides have a plausible evolutionary role as promiscuous precursors to complex and well-organized enzyme structures with extraordinary levels of substrate specificity and catalytic efficiency.12 A deeper understanding of their evolutionary journey could provide valuable insights into the fundamental principles underlying enzyme catalysis and pave the way for the design of novel catalytic sequences and biomimetic systems.8,12–14
Peptide-based catalysts can be designed and engineered precisely to mimic the active sites of enzymes, allowing the development of highly specific and efficient catalytic systems.15 Peptides can be modified with various functional groups or metals to further enhance their catalytic performance.16,17 Furthermore, they offer excellent biocompatibility and low immunogenicity, making them suitable for biomedical applications.18,19 Compared to enzymes, catalytic peptides offer several advantages, such as their smaller size, simpler structures, modularity, and ease of production.20,21 Using peptides as catalysts may enable access to a chemical space larger than that of traditional small-molecule catalysts, leading to new possibilities for synthetic chemistry and biotechnology.
Catalytic peptides often display lower efficiencies compared to enzymes because of the lack of complex three-dimensional structures present in enzymes. To bridge this gap between simplicity and functionality, self-assembly has emerged as a promising approach.15,19 Peptide self-assembly is the spontaneous organization of peptides into well-defined structures that display unique physical and chemical properties, different from those of their individual constituent monomers.22,23 By organizing peptides into specific architectures, it is possible to create catalytic sites with enhanced activity and selectivity.20,21,24 In addition, peptides are susceptible to proteolytic degradation, have reduced stability in plasma, and limited oral bioavailability.18 Nevertheless, recent advances in peptide synthesis and modification techniques have helped overcome some of these limitations. For example, N-acetylation25,26 or cyclization27,28 are effective strategies for increasing the stability and structural diversity of peptides, leading to improved bioactivity and pharmacokinetics.29
Several design strategies have been developed to identify effective catalytic peptides, including mimicking natural enzymes, incorporating catalytic motifs or cofactors, and using directed evolution or combinatorial chemistry.21,30,31 The most prominent strategies to mimic natural enzymes include metal coordination and incorporation of catalytic triad residues into short peptide sequences with self-assembly propensity.32 The coordination of metal ions within peptides is often accomplished through specific amino acid residues, such as histidine, cysteine, aspartic acid, and glutamic acid, that can act as ligands for metal ions and consequently stabilize them within the peptide structure. Such stable and active metal–peptide complexes have been used to catalyze oxidation, reduction, hydrolysis, and isomerization.16,33 One of the designs studied most extensively involves alternating hydrophobic amino acids with histidines, which can coordinate Zn2+ ions.34 The optimal amino acid combination consists of an interplay of isoleucines, histidines, and a single glutamine. To evaluate the effect of sequence composition on self-assembly and catalytic activity, multiple analogs, including substitution of tyrosine–glutamine, were proposed that showed increased binding affinity to Zn2+ and the potential to form photo-induced cross-links via dityrosine.35 In general, short peptides capped through acetylation at the N-terminus and amidated at the C-terminus exhibit higher activity, whereas uncapped peptides and substitutions leading to increased net charge such as isoleucine-aspartic acid mutation lead to a reduction in activity. The sequence-dependent activity of the Ac-IHIHIQI-Am, Ac-IHIHIRI-Am, and Ac-IHIHIYI-Am heptapeptides assuming cross-β amyloid fibrillar morphologies showed that a flexibility–rigidity balance is required to efficiently mimic the active sites of the enzyme and resulted in higher esterase activity in fibers with increased flexibility.36
Despite the development of numerous catalytic peptides, fundamental understanding of the key factors that influence their performance and the sequence–activity relationship remains challenging. This is due to the complex interplay between their intrinsic structure and the extrinsic environment. The conformational heterogeneity of peptides could be one of the reasons why the catalytic performance of peptides is lower than that of enzymes. To address this issue, we propose the introduction of rigidity through cyclization to assess whether the reduced flexibility of short peptides can have an impact on their catalytic activity. The present study involved the design and synthesis of two linear and two cyclic histidine-rich peptides that shared the same set of amino acids, isoleucine, histidine, and asparagine. The impact of amino acid disposition within the sequence, cyclization, and incorporation of D-amino acids in cyclic analogues was evaluated to determine their effect on peptide self-assembly propensity, ability to coordinate zinc ions and the intrinsic hydrolase-like activity (Fig. 1). Our findings demonstrate that in addition to the well-established design principles of catalytic peptides, such as sequence motifs for metal coordination and self-assembly propensity, the flexibility of the peptide sequence plays a crucial role in metal coordination, which ultimately impacts the catalytic activity.
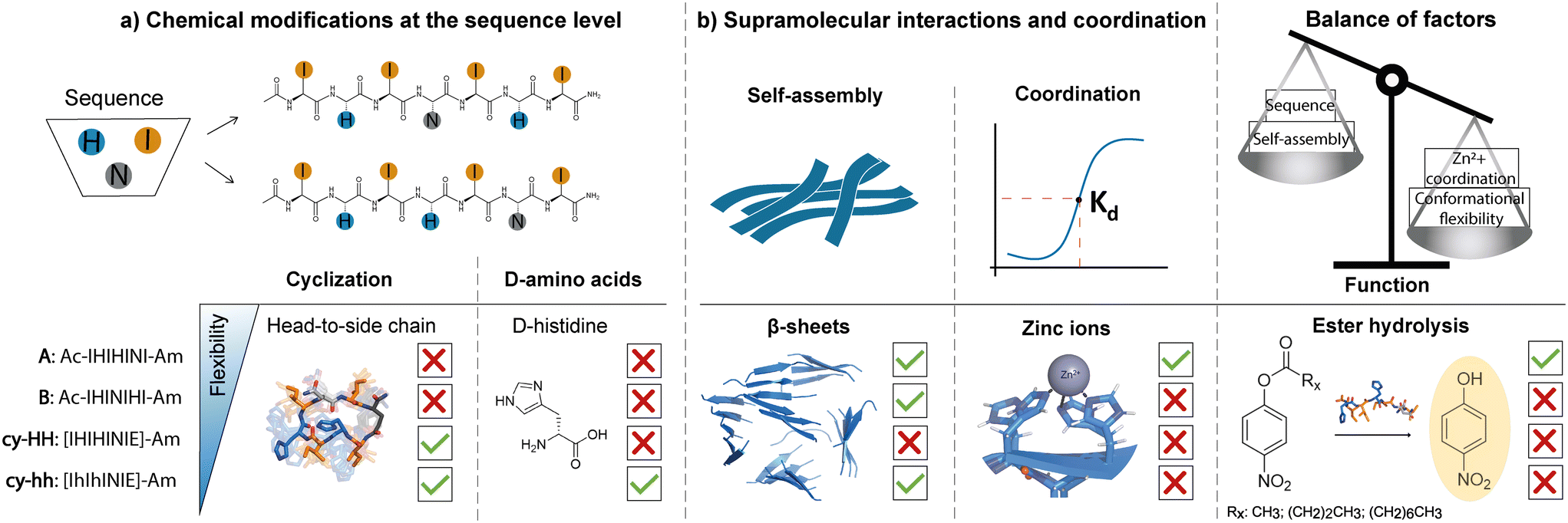 | ||
| Fig. 1 Schematic representation of a) sequence-level modifications, including head-to-side chain cyclization and incorporation of D-histidines; and b) the consequent effects on self-assembly propensity, zinc ion coordination and catalytic activity in histidine-rich short peptides. We proposed two linear peptides (A and B) with different amino acid arrangements at the sequence level, where histidine residues, responsible for catalysis, were placed at different locations in the sequence. Both sequences self-assembled into β-sheets, however, only peptide A that efficiently coordinated zinc (Kd determined) showed catalytic activity. Modifications at the sequence level affecting peptide flexibility, through cyclization (cy-HH, cy-hh) and incorporation of D-histidines (cy-hh) in peptide A resulted in β-sheets for cy-hh and the absence of supramolecular structures for cy-HH, highlighting the influence of chirality on the self-assembly behavior of cyclic peptides. However, both modifications led to a lack of metal coordination resulting in a loss of esterase activity, showing that the interplay of various factors shapes the functionality of catalytic peptides. | ||
2 Results and discussion
A set of two linear and two cyclic histidine-rich peptides was evaluated in terms of their aggregation propensity and catalytic activity toward ester hydrolysis in the presence of Zn2+ ions (Table 1). Peptides were prepared using Fmoc-solid phase peptide synthesis on Rink amide resin, whereby head-to-side chain cyclization was performed on-resin through the formation of an amide bond between the N-terminus and the glutamic acid (E) side chain, after deprotection of the 2-phenylisopropyl (O-2-PhiPr) group. The linear peptides, Ac-IHIHINI-Am (Fig. S1†) and Ac-IHINIHI-Am (Fig. S2†), were N-terminal acetylated and C-terminal amidated to minimize the effect of terminal charges on catalytic efficiency.34 For the same reason, the cyclic peptides, [IHIHINIE]-Am (Fig. S3†) and [IhIhINIE]-Am (Fig. S4†), were C-terminal amidated while the N-terminus was modified through cyclization. The E residue was added to the cyclic peptides to allow the head-to-side chain cyclization which was chosen to avoid the oligomerization that often arises with the backbone cyclization and to achieve higher purity by performing the cyclization on resin rather than in solution.37 The aggregation behavior of the obtained peptides was assessed through the ThT assay, IR and AFM, which confirmed the propensity to form fibrillar β-sheet assemblies (Fig. 2). Furthermore, the secondary structure of the aggregates was determined using circular dichroism (CD) (Fig. 2) and modeled with molecular dynamics (MD) simulations (Fig. S13†), which confirmed their predominant β-sheet nature.| Sequence | Name | Cyclization | Sequence modification | Catalytic activity (p-NPA) | CAC (M) |
|---|---|---|---|---|---|
| Ac-IHIHINI-Am | A | — | Introduction of N | Yes | 67 |
| Ac-IHINIHI-Am | B | — | N to H position substitution | — | 67 |
| [IHIHINIE]-Am | cy-HH | Head-to-side chain | Addition of E for cyclization | — | — |
| [IhIhINIE]-Am | cy-hh | Head-to-side chain | Replacement of both H with h | — | 54 |
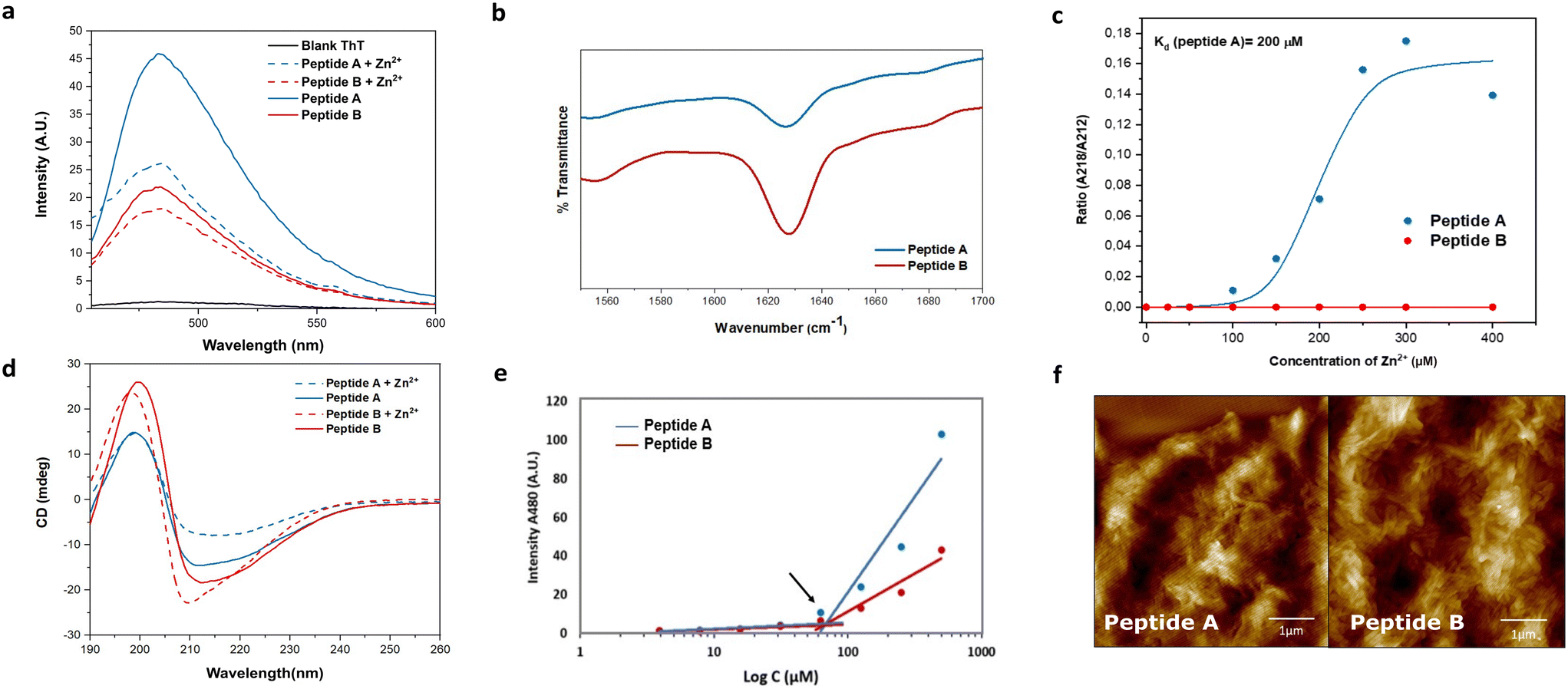 | ||
| Fig. 2 Characterization of linear peptides: (a) ThT assay for peptides A (blue) and B (red) with Zn2+ (dashed line) and without Zn2+ (solid line) performed at 250 μM peptide concentration. (b) ATR-FTIR spectra of peptide A and peptide B showing a peak at 1623 cm−1 indicating the presence of β-sheets. (c) Zn2+ titration curve of peptide A (blue) and peptide B (red), demonstrating the differential affinity for the metal. Peptide A exhibits strong metal binding with a binding constant (Kd) of 200 μM, while peptide B shows no affinity. (d) CD spectra of peptide A (25 μM) and peptide B (25 μM) with and without Zn2+, indicating the presence of β-sheet assemblies in both peptides. (e) CAC in MiliQ water, plotted for different concentrations of both peptides as a function of ThT fluorescence emission at 480 nm. (f) AFM images showing fibrillar morphologies for peptide A (left) and peptide B (right), recorded at a concentration of 250 μM (scale bar: 1 μm). | ||
2.1 Impact of side chain length and amino acid disposition on the self-assembly propensity of linear peptides
The role of alternating hydrophilic and hydrophobic amino acids, particularly isoleucines and histidines, in the design of promising catalytic peptide candidates has been well established in previous studies.34–36 Histidine, being a common component of active sites in esterases, is frequently incorporated in the design of catalytic peptides.38,39 Motivated by the Ac-IHIHIQI-Am sequence,34 we made a specific amino acid modification by substituting glutamine with asparagine in Ac-IHIHINI-Am (peptide A). This substitution involves a minor difference of only one –CH2 group in the side chain of the amino acid. Our hypothesis was that this slight alteration in the peptide sequence could potentially enhance self-assembly by promoting the formation of asparagine ladders.40 These ladder-like structures feature repetitive patterns of asparagine residues that contribute to the stabilization of the supramolecular arrangement. Moreover, we examined the impact of the amino acid disposition within the sequence in Ac-IHINIHI-Am (peptide B). In this case, the asparagine residue was positioned at the center of the sequence, while maintaining the alternating pattern of hydrophobic and hydrophilic residues.Circular dichroism (CD) and infrared (IR) spectroscopies were used to confirm the adoption of a supramolecular β-sheet conformation by peptides A and B. In the IR spectra (Fig. 2b), both peptides exhibited a distinct peak at 1628 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the amide bond, a characteristic feature of peptide self-assembly.41 Under metal-free conditions, peptides A and B showed similar CD spectra, characterized by a minimum around 212 nm and a maximum at 197 nm, indicative of β-sheet-like assemblies (Fig. 2d). Upon the addition of Zn2+, peptide A displayed a red shift of the minimum from 212 to 218 nm, indicating a more ordered cross-β configuration. In contrast, peptide B exhibited a blue shift in the minimum toward 210 nm, consistent with the formation of a less organized β-sheet-like arrangement, as previously reported.34 These observations validate the hypothesis that substituting glutamine (Q) for asparagine (N) in peptide A improves the stability of its supramolecular β-sheet arrangement.
O stretching vibration of the amide bond, a characteristic feature of peptide self-assembly.41 Under metal-free conditions, peptides A and B showed similar CD spectra, characterized by a minimum around 212 nm and a maximum at 197 nm, indicative of β-sheet-like assemblies (Fig. 2d). Upon the addition of Zn2+, peptide A displayed a red shift of the minimum from 212 to 218 nm, indicating a more ordered cross-β configuration. In contrast, peptide B exhibited a blue shift in the minimum toward 210 nm, consistent with the formation of a less organized β-sheet-like arrangement, as previously reported.34 These observations validate the hypothesis that substituting glutamine (Q) for asparagine (N) in peptide A improves the stability of its supramolecular β-sheet arrangement.
To explore the interaction between the peptides and the metal cofactor, a Zn2+ titration was conducted using CD, enabling the monitoring of structural changes as reflected by shifts in the minimum across a range of Zn2+ concentrations.42 Peptide A exhibited a strong affinity for the metal cofactor with a binding constant (Kd) of 200 μM. On the contrary, peptide B showed a loss of affinity for Zn2+ (Fig. 2c) that could be due to different supramolecular arrangements for A and B. Peptide A is expected to assemble similarly to Ac-IHIHIQI-Am, with Ile residues constituting the central hydrophobic core and His residues projected outwards, facilitating metal ion coordination.34,43 However, placement of Asn in the center of the peptide sequence (peptide B) could cause differential positioning of histidines within supramolecular assemblies or steric hindrance for efficient coordination, resulting in loss of binding affinity for Zn2+ ions. At enzyme active sites, Zn2+ is typically coordinated in a trigonal bipyramidal or tetrahedral geometry44 that might be difficult to achieve for peptides with increased distances between His residues (B). Furthermore, MD simulations provided insights into the conformational flexibility of peptide B, revealing significant fluctuations of its His residues over time, likely contributing to the lack of binding affinity to the metal cofactor (Fig. S12†).
Fibril formation was investigated using the thioflavin T (ThT) binding assay and atomic force microscopy (AFM). Peptide A demonstrated a higher enhancement in ThT fluorescence compared to peptide B, indicating a greater abundance of β-sheet-like assemblies (Fig. 2a). Interestingly, upon the addition of Zn2+, the ThT signal was quenched, possibly due to coordination of Zn2+ with the ThT anilino group, leading to signal attenuation.35 Importantly, the reduction in signal intensity was not uniform for both peptides, confirming distinct interactions with the metal. The critical aggregation concentration (CAC) of both peptides was determined at 67 μM, obtained by measuring the fluorescence intensity of ThT as a function of the peptide concentration (Fig. 2e). Furthermore, the AFM images provided visual evidence of bundles of fibers for both peptides (Fig. 2f).
To further confirm β-sheet formation, all-atom MD simulations were performed by simulating 24 instances of the same peptide molecule for each of the two linear peptides in 0.08 M Tris buffer. Both GROMACS simulations showed self-assembly in several shorter β-sheets (Fig. S13†), in contrast to the uniform β-sheet bilayer reported for the Ac-IHIHIQI-Am sequence.34,36 The solvent accessible surface area (SASA) analysis of peptides A and B shows convergence to a stable surface in less than 50 ns for both molecules (Fig. S14†), with Ac-IHINIHI-Am having a slightly lower average SASA (150.25 nm2) than Ac-IHIHINI-Am (140.13 nm2). This indicates that both peptides exhibit rapid aggregation and self-assembly, forming stable nanostructures in solution.
2.2 Amino acid disposition within the sequence affects the functionality of linear peptides
The esterase activity of linear peptides was assessed using the widely employed p-NPA substrate. Peptide A demonstrated a moderate catalytic efficiency (kcat/KM) of 4.19 M−1 s−1 (Fig. 3a) at pH 7.4. To reflect the effect of the positional mutation of Q with N, the p-NPA assay was also performed at pH 8 and showed an improved catalytic performance for peptide A compared to the reported Ac-IHIHIQI-Am system,34 yielding a kcat/KM value of 83.21 M−1 s−1 (Fig. S7†). On the contrary, peptide B showed a loss of activity in both the presence and absence of the metal cofactor. These results highlight the importance of selecting amino acids capable of coordinating zinc (Zn2+) and arranging them appropriately within the peptide sequence to maintain catalytic activity. Our findings suggest that the self-assembly ability of histidine-rich peptides alone may not be sufficient to ensure the catalytic performance of Ac-IHIHINI-Am analogues, as opposed to the ability to control the catalytic efficiency in previously reported Ac-IHIHIQI-Am analogues where the β-sheet propensity could be correlated with catalytic activity.34 To confirm the essential role of zinc for the catalytic function of peptide A, we performed a control experiment in which the catalytic activity of peptide A was assessed in the absence of zinc ions (Fig. S8†), resulting in lack of esterase activity. Furthermore, we evaluated the activity of peptide A on substrates with longer alkyl chains (Table 2). Our results showed improved activity for para-nitrophenyl butyrate (p-NPB) and reduced activity for para-nitrophenyl octanoate (p-NPO) (Fig. 3a and S9†). The enhanced activity for p-NPB indicates a higher affinity for the substrate, as reflected in the changes in KM rather than kcat, which may be attributed to the hydrophobic interactions between the Ile residues in the peptide and the alkyl chain of the substrate.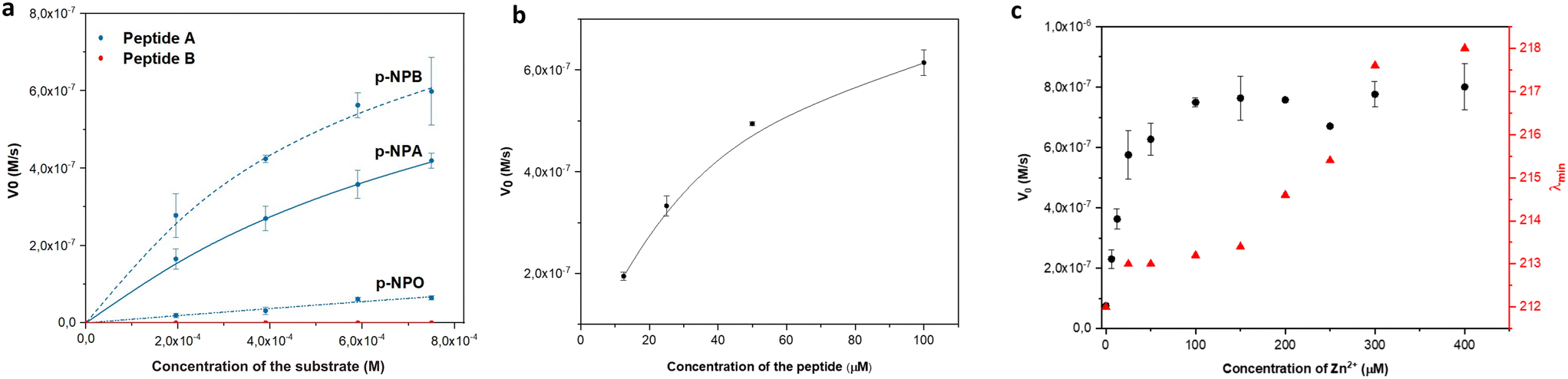 | ||
| Fig. 3 Catalytic activity of linear peptides: (a) Michaelis–Menten plot showing the catalytic activity of peptides A (blue) and B (red) towards p-NPA, p-NPB, p-NPO. (b) Dependence of the p-NPA hydrolysis rate on the concentration of peptide A. (c) The activity of peptide A (black) and the shift in the minima (red) in CD as a function of Zn2+ concentration. | ||
| Substrate | k cat (10−3 s−1) | K M (10−3 M) | V max (10−6 M s−1) | k cat/Km (M−1 s−1) |
|---|---|---|---|---|
| p-NPA | 4.00 ± 0.01 | 0.98 ± 0.15 | 1.00 ± 0.01 | 4.19 ± 0.65 |
| p-NPB | 4.36 ± 0.36 | 0.55 ± 0.17 | 1.06 ± 0.09 | 7.83 ± 1.6 |
| p-NPO | 1.17 ± 0.17 | 12 ± 1.21 | 1.15 ± 0.041 | 0.41 ± 1.06 |
To better understand the relationship between peptide concentration and the corresponding catalytic activity, we performed p-NPA assays using varying concentrations of peptide A. The activity exhibited a positive correlation with the increase in peptide concentration, indicating that self-assembly contributes to the enhancement of catalytic activity. In particular, our findings showed first-order kinetics within the concentration range of 12.5–50 μM, below the CAC (Fig. 3b), indicating that the rate of the catalytic reaction is directly proportional to the peptide concentration. However, when the concentration exceeded the CAC (100 μM), a slight saturation of the curve was observed. This suggests that the system approached maximum velocity, reaching a point where the catalytic activity became independent of the peptide concentration, following zero-order kinetics. Changes in behavior can be attributed to various factors, including changes in surface area, confinement effects, and the accessibility of active sites within assemblies. Additionally, to satisfy the requirements of the Michaelis–Menten kinetics, the concentration of the substrate needs to be at least one order of magnitude higher than the concentration of the peptide catalyst. When the concentration of peptide A is above its CAC (100 μM), it approaches the substrate concentration limited by the poor solubility of p-NPA. Therefore, the observed activity at peptide concentrations above CAC might follow different kinetics. Although the esterase activity of peptide B was too low to accurately determine the parameters for the Michaelis–Menten kinetics, we observed a linear increase in activity with increasing concentrations of the peptide (Fig. S6†).
We performed zinc titrations to explore the influence of metal concentration on the catalytic activity of peptide A and to establish a correlation with conformational changes, as determined by CD spectroscopy. Our findings showed that the peak activity coincided with the transition of the assemblies towards a more ordered structure (Fig. 3c). This indicates that a minimum of 4 eq of zinc is necessary to attain a catalytically competent species, which aligns with the initiation of the formation of an ordered supramolecular structure.
2.3 The effect of cyclization and chirality on catalytic activity and self-assembly propensity of peptide A derivatives
Cyclic peptides offer several advantages over linear ones, such as increased stability against proteases due to the constrained conformation provided by their cyclic structure, which leads to longer half-lives and improved bioavailability.29 Additionally, cyclization can result in enhanced binding affinity and specificity for target molecules.45,46 However, our understanding of cyclic catalytic peptides capable of coordinating metal atoms is limited. In this study, we aim to address this gap by proposing a cyclic analogue of the catalytically active peptide A (cy-HH). To achieve this, we incorporated a glutamic acid residue into the peptide sequence, which served as a linker and formed an amide bond with the N-terminus. The cyclization neutralized the side chain, eliminating any charge interference with the catalytic reaction. The cyclic analogue exhibited improved solubility compared to its linear counterpart, which presented challenges in determining the critical aggregation concentration (CAC). Furthermore, analysis using ATR-FTIR, ThT binding, and CD (Fig. 4a–c) did not provide any evidence of β-sheet formation.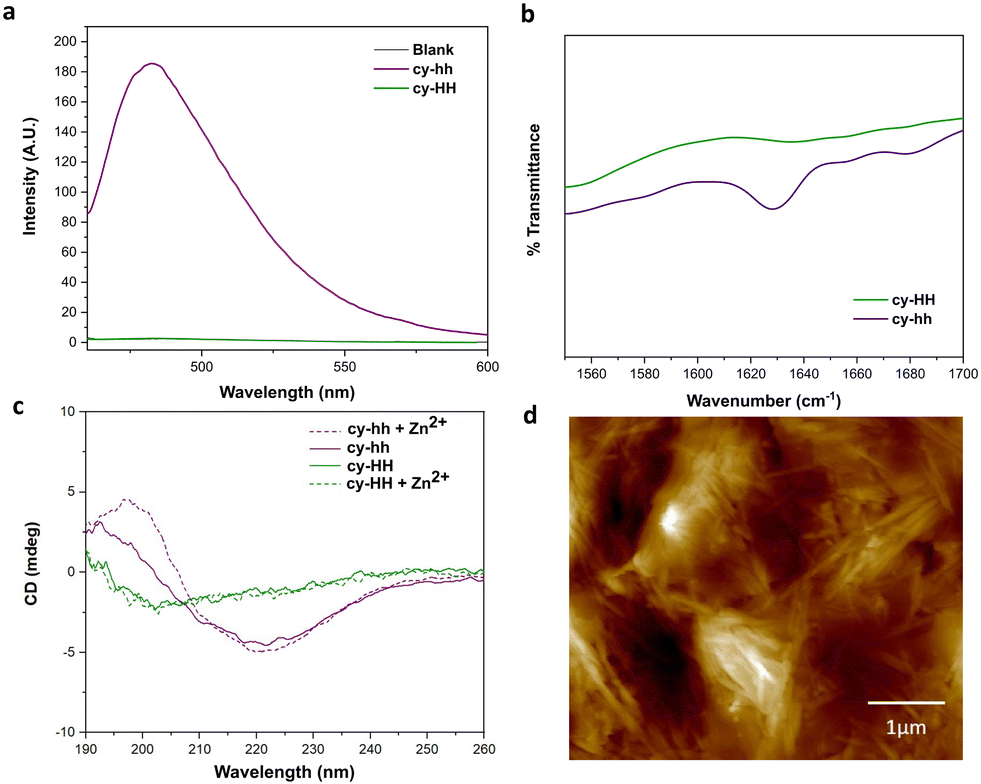 | ||
| Fig. 4 Characterization of cyclic cy-HH (green) and cy-hh (purple) peptides: (a) ThT assay performed at 250 μM of peptide in presence (dashed line) and absence (solid line) of Zn2+. (b) IR spectra showing a peak at 1630 cm−1 indicating β-sheets only for cy-hh. (c) CD spectra of cy-HH (25 μM) and cy-hh (25 μM) in presence and absence of Zn2+ showing β-sheets only for cy-hh. (d) AFM image showing elongated fibrils of cy-hh (250 μM). | ||
A more subtle way of controlling conformational constrains is through alternating the chirality of amino acid residues, which can influence the packing arrangements in self-assembled cyclic peptides. Alternating D- and L-amino acids promotes the formation of assemblies due to a growing number of intermolecular interactions.12 By incorporating D-histidines into the cyclic peptide sequence (cy-hh), we restored a self-assembly pattern similar to that of its linear counterpart. Cy-hh exhibited an increase in ThT fluorescence intensity, indicating β-sheet formation (Fig. 4a) and displayed a CAC of 54 μM (Fig. S5†), comparable to linear sequences (67 μM). The fibrillar morphology of the nanostructures was confirmed by AFM (Fig. 4d). The self-assembly of cy-hh into β-sheets was further validated by the presence of a band at 1630 cm−1 in the IR spectrum and characteristic CD spectral features, including a minimum at 220 nm and a maximum at 197 nm (Fig. 4b and c). In contrast to linear peptides where zinc affects the arrangement of the supramolecular assemblies, cyclic peptides do not sense the addition of Zn2+ ions as no shifts in the minimum are observed in the CD spectra, suggesting a lack of interaction with the cofactor, which consequently results in the absence of catalytic function (Fig. 4c).
To investigate the potential relationship between conformational rigidity and coordination deficiency, we conducted MD simulations. RMSD measurements revealed reduced backbone fluctuations (Fig. S11†) for the cyclic peptides, and cluster analysis showed a decreased number of clusters (Fig. S10†), indicating their greater rigidity compared to their linear counterparts (Table 3).
| Peptide | Average RMSD (Å) | N clusters | 1st cluster (%) | 2nd cluster (%) | 3rd cluster (%) | His-His avg distance (Å) |
|---|---|---|---|---|---|---|
| A | 4.71 | 296 | 1.33% | 0.66% | 0.66% | 11.03 |
| B | 4.62 | 295 | 1.00% | 1.00% | 0.66% | 13.61 |
| cy-HH | 2.28 | 76 | 18.60% | 14.95% | 5.32% | 9.42 |
| cy-hh | 1.75 | 30 | 52.16% | 10.93% | 7.95% | 9.51 |
3 Conclusions
In conclusion, our study highlights the crucial role of amino acid disposition within the sequence of linear peptides for their esterase activity. We demonstrated that selecting amino acids capable of coordinating Zn2+ and arranging them appropriately within the peptide sequence is essential for maintaining catalytic activity. Cyclization of linear peptides can impair their catalytic function by increasing rigidity, which hinders the coordination of metal ions. This rigidity limits the necessary sequence adjustments to optimize the conformation required for effective Zn2+ coordination. This conformational optimization process is more feasible in flexible and conformationally heterogeneous linear peptides. Furthermore, these results suggest that chirality can play a crucial role in the self-assembly propensity of cyclic peptides. Therefore, the design and optimization of cyclic peptides with improved catalytic activity and self-assembly properties require careful consideration of various factors, including rigidity, chirality, and coordination chemistry. Further structural modifications are necessary to restore catalytic activity in the cyclic analogues cy-HH and cy-hh. These findings offer new prospects for the development of metal-dependent peptide-based catalysts in which the amino acids and their disposition within the sequence need to be carefully selected. We envision that the extension of this study to different cyclization strategies and other amino acid permutations will lead to the optimization of the geometry of the active residues and the discovery of next-generation histidine-rich peptide catalysts. With the application of this knowledge, we hope in the future to develop more efficient and selective peptide-based catalysts for ester hydrolysis.4 Experimental
4.1 Materials
Lyophilized linear peptides (95.0%), N-terminal acetylated and C-terminal amidated, were custom synthesized by Genecust (Boynes, France). All p-nitrophenyl esters (95.0%), including p-nitrophenyl acetate (p-NPA), p-nitrophenyl butyrate (p-NPB) and p-nitrophenyl octanoate (p-NPO), Fmoc-amino acids, Novabiochem® resin (Rink Amide AM), ThT, DIPEA, HBTU, TIS, TFA and piperidine (99.0%) were purchased from Sigma-Aldrich (St. Louis, Missouri, SAD). ZnCl2 was purchased from Gram mol (Zagreb, Croatia). All other chemicals and reagents are analytical grade and are available from commercial sources.4.2 Peptide synthesis
Cyclic peptides were synthesized by fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide synthesis on the Novabiochem Rink Amide AM resin (100–200 mesh) (loading of 0.78 mmol g−1) using L- and D-amino acids. The resin was swollen for 30 min in DMF. The removal of Fmoc was performed with 20% piperidine in DMF. Chain growth was carried out using a three-fold excess of amino acids over the resin in DMF, using N,N-diisopropylethylamine (DIPEA) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) as coupling and activating agents in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio relative to amino acids, respectively. The coupling and deprotection steps were separated by DMF (3 × 30 s) and DCM washes (3 × 30 s). Intramolecular (amide bond) cyclization was performed between the side chain of the C-terminal Glu (E) and the N-terminus. The Glu residue involved in intramolecular cyclization was protected with the 2-phenylisopropyl (O-2-PhiPr) group. After complete elongation of the peptide chain, the resin was treated with 1% (v/v) trifluoroacetic acid (TFA) in DCM (4 × 10 min) for selective removal of O-2-PhiPr, followed by neutralization with 5% DIPEA in DCM (4 × 5 min). Cyclization was performed on resin with PyBOP/Hobt/DiPEA (5, 5 and 10 equiv) in DMF for 3 h. Mini-cleavage with ∼5 mg resin was performed to check product yield after cyclization. The cleavage of the cyclic peptide from the resin was achieved using TFA:triisopropylsilane (TIS):H2O = 95:2.5:2.5. The crude peptide was precipitated and washed in cold diethyl ether, centrifuged (3 × 10 min) at 4 °C, dissolved in 20% acetonitrile (ACN) in MiliQ water, and lyophilized.
:1 ratio relative to amino acids, respectively. The coupling and deprotection steps were separated by DMF (3 × 30 s) and DCM washes (3 × 30 s). Intramolecular (amide bond) cyclization was performed between the side chain of the C-terminal Glu (E) and the N-terminus. The Glu residue involved in intramolecular cyclization was protected with the 2-phenylisopropyl (O-2-PhiPr) group. After complete elongation of the peptide chain, the resin was treated with 1% (v/v) trifluoroacetic acid (TFA) in DCM (4 × 10 min) for selective removal of O-2-PhiPr, followed by neutralization with 5% DIPEA in DCM (4 × 5 min). Cyclization was performed on resin with PyBOP/Hobt/DiPEA (5, 5 and 10 equiv) in DMF for 3 h. Mini-cleavage with ∼5 mg resin was performed to check product yield after cyclization. The cleavage of the cyclic peptide from the resin was achieved using TFA:triisopropylsilane (TIS):H2O = 95:2.5:2.5. The crude peptide was precipitated and washed in cold diethyl ether, centrifuged (3 × 10 min) at 4 °C, dissolved in 20% acetonitrile (ACN) in MiliQ water, and lyophilized.
4.3 Peptide analysis and purification
The crude peptides were analyzed by HPLC-MS on the XSELECT CSH C18 column (4.6 × 150 mm, 3.5 μm, Waters, Milford, MA, USA) with an Agilent 1260 series HPLC chromatograph coupled to an Agilent 6460 triple quadrupole mass spectrometer system (Agilent Technologies, Santa Clara, CA, USA). The elution was carried out with linear gradients of solvent B (0.1% v/v TFA in ACN) into A (0.1% v/v TFA in H2O) over 20 min. The peptides were purified by preparative HPLC on the Aeris PEPTIDE XB-C18 column (10 × 250 mm, 5 μm, Phenomenex) with an Agilent Infinity 1260 semipreparative HPLC instrument using a flow rate of 5 ml min−1 and linear gradients of solvent B (0.1% v/v TFA in ACN) in A (0.1% v/v TFA in H2O) for 30 min with detection at 214 nm on a DAD detector. The purity of the collected fractions was analyzed and those with satisfactory purity (>90%) were merged and lyophilized. The purity of the final products was analyzed by HPLC-MS on the XSELECT CSH C18 column (4.6 × 150 mm,3.5 μm, Waters, Milford, MA, USA) in an Agilent 1260 series HPLC chromatograph coupled to an Agilent 6460 triple quadrupole mass spectrometer system (Agilent Technologies, Santa Clara, CA, USA) eluting with linear gradients of solvent B (0.1% v/v TFA in ACN) into A (0.1% v/v TFA in H2O) for 20 min at 1 mL min−1 flow rate.4.4 Preparation of peptide solutions
The 1.1 mM stock solution of purified lyophilized peptides was prepared by dissolving the peptides in 10 mM HCl. The working solution at pH 7.4 was prepared by mixing 200 μL of the stock solution with 1.8 ml of buffer solution (25 mM Tris, 1 mM ZnCl2).4.5 Catalytic activity assessment
The catalytic activity assay was performed on a Hidex Sense microplate reader to monitor the absorbance of the product (p-nitrophenol) for 30 minutes (22 °C) in 96-well plates at 405 nm. Stock solutions of 0.1 M of p-nitrophenyl esters (p-nitrophenyl acetate, p-nitrophenyl butyrate, p-nitrophenyl octanoate) (Sigma-Aldrich, United States) were used to prepare substrate solutions in 25 mM Tris (pH 7.4), 1 mM ZnCl2. 150 μL of freshly prepared substrate solutions (to the final substrate concentration of 0.195–0.75 mM) were added to 50 L of buffered (25 mM Tris, 1 mM ZnCl2) peptide solutions at pH 7.4 (to a final peptide concentration of 25 μM). The calibration curve was experimentally obtained from a standard 4-nitrophenol (p-NP) solution (Sigma-Aldrich, United States) in the concentration range of 0–100 μM. The reported results correspond to the average of at least two independent measurements. Kinetic parameters were obtained by fitting the data to the Michaelis–Menten equation V0 = kcat·[E]0·[S]0/(KM + [S]0) using Origin 2018 software.4.6 Circular dichroism
The far-UV CD spectra (190–300 nm) were acquired with a Jasco J-815 spectropolarimeter with a Peltier temperature control system at 20 °C, in a 1 mm quartz cuvette under nitrogen flow and 2 nm bandwidth. Each spectrum represents an average of three scans. The peptide concentrations were maintained at 25 μM and the measurements were performed in 5 mM Tris (pH 7.4) or 5 mM Tris (pH 7.4) containing 0.5 mM ZnCl2.4.7 Zn2+ titrations
For the p-NPA assay analysis, 11 different samples were prepared on a 96-well plate. Each sample consisted of 50 μL of the peptide working solution, 100 μL of 25 mM pH 7.4 Tris buffer with various concentrations of ZnCl2 (0, 6.25, 12.5, 25, 50, 100, 150, 200, 250, 300, 400 μM). Subsequently, 50 μL of p-NPA solution in 25 mM pH 7.4 Tris buffer (without additional zinc chloride) was added to each well, resulting in a final substrate concentration of 750 μM. For the CD measurements, nine different samples were prepared in Eppendorf tubes. Each sample contained 12.5 μL of the peptide stock solution, 500 μL of 5 mM pH 7.4 Tris buffer with various concentrations of ZnCl2 (0, 25, 50, 100, 150, 200, 250, 300, 400 μM). Dissociation constants KD (μM) were determined in OriginPro9 by fitting the data to the Hill equation δd = [Peptidefree]n/(KnD + [Peptidefree]) where KD is the dissociation constant and n is the Hill coefficient that describes peptide cooperativity.4.8 Thioflavin-T (ThT) binding assay and critical aggregation concentration (CAC) determination
Fluorescence measurements were performed using an Agilent Cary Eclipse fluorescence spectrophotometer. ThT binding assay was performed using a range of buffered (25 mM Tris, pH 7.4) peptide concentrations (0.0039–0.500 mM) with and without 1 mM ZnCl2, incubated with ThT stock solution prepared in methanol to a final concentration of 25 μM for 15 minutes. The samples were excited at 450 nm and the emission spectra were recorded in the range of 460–600 nm on the spectrophotometer. The excitation and emission bandwidths were both set to 5 nm. The CAC was obtained by plotting the intensity of the emission peak at 480 nm against the peptide concentration.4.9 Fourier-transform infrared spectroscopy (FTIR)
Hydrogen bonding patterns characteristic for peptide self-assembly were investigated using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR). ATR-FTIR spectra were recorded for the peptide solutions in D2O (Sigma-Aldrich, United States), using an Agilent Technologies Cary 630 FTIR in the range 650–4000 cm−1 with a resolution of 16 cm−1.4.10 Atomic force microscopy (AFM)
The surface topology scans of the analyzed samples are obtained by using the Bruker Dimension Icon atomic force microscope in tapping and ScanAsyst modes. Tapping mode was used to obtain a high resolution scan of surface detail by using a Bruker SNL-10 type D (low stiffness) silicon nitride cantilever with a silicon tip of 2 nm tip radius and ScanAsyst silicon nitride cantilevers. The dynamic properties of the cantilever such as natural frequency and normal stiffness are obtained using the thermal tuning method, where the thermal noise spectra of the cantilever were measured and fitted to a Lorentzian harmonic oscillator model in air which corresponded to 22 kHz for SNL10-D and 75 kHz for the ScanAsyst cantilever. The calibration obtained allows the use of a minimal contact force for the measurement that has minimal impact on the surface, so the average force used was 2 pN ± 5%. The scans were performed with scan sizes of 5 and 10 μm2 selected from a large 90 μm2 sweep scan with 512 scan lines each with 512 data points acquired per line. The obtained data was consequently processed in order to provide tilt and bow corrections using the proprietary Bruker Nanoscope Analysis software.4.11 Molecular dynamics simulations
Molecular dynamics simulations were performed for linear (Ac-IHIHINI-Am, Ac-IHINIHI-Am) and head-to-side chain cyclized ([IHIHINIE]-Am, [IhIhINIE]-Am) peptides using the GROMACS simulation package (version 2023.00-TunePME) and the CHARMM36m force field.47 Peptide molecules were built in Pymol48 using the “fab” command and modified in the Avogadro software49 to introduce head-to-side-chain cyclization and D-oriented histidines when needed. The Avogadro-generated MOL2 files were then uploaded to CHARMM-GUI's ligand reader and modeler (https://www.charmm-gui.org/?doc=input/ligandrm)50,51 to create topologies and parameter files for such modified molecules. First, single peptide molecules were simulated to assess their flexibility, predominant conformation, and histidine distances. For this purpose, the peptides were modeled to have histidines with protonated ε-nitrogen atoms. Each solution box contained 0.08 M Tris buffer, which was also generated using ligand reader and modeler for the tris(hydroxymethyl)aminomethane molecule. The generated files were placed into the CHARMM-GUI's multicomponent assembler, prior to simulations in GROMACS. All simulations were performed using the TIP3P water model, using a temperature of 298.15 K, in a square water box 45 Å in side length. Energy minimization was limited to 50000 steps with a convergence cutoff of 100.0 kJ mol−1 nm−1 using a steepest descent algorithm, which always ended before the step limitation was reached. The system was then equilibrated during 200 ps in an NPT ensemble using a Nose–Hoover thermostat. The simulations were run through cluster analysis using the GROMOS clustering system52,53 in GROMACS. Whole peptide molecules (including the side chain and hydrogen atoms) were used for clustering, with a 0.1 nm RMSD cutoff.
To assess the aggregation propensity of linear peptides, and to estimate their aggregation time, multipeptide simulations were set up using the CHARMM-GUI's PDB reader and modeler54,55 in building topology and parameter files for the multicomponent assembler. Ac-IHIHINI-Am and Ac-IHINIHI-Am were used to make two separate systems containing 24 instances of the same peptide, dissolved in a 60 Å water box and 0.025 M Tris buffer. The peptides were modeled to have histidines with protonated ε nitrogen in His at position 2 and protonated δ nitrogen in His at position 4. These systems were simulated for 500 ns. The same minimization and equilibration processes were used as with the first set of peptide simulations. A solvent accessible surface area (SASA) analysis56 was performed on these multipeptide systems to evaluate their aggregation time.
All the simulations were run on Bura supercomputer, on 5 nodes for each individual simulation. The nodes contain 2 Xeon E5 processors with 64 GB memory, and the system runs on a Red Hat Enterprise Linux 7 and Slurm Workload Manager operating system.
Author contributions
Patrizia Janković: conceptualization, methodology, investigation, writing – original draft preparation. Marko Babić: methodology, formal analysis, writing – original draft preparation. Marko Perčić: methodology, visualization. Ana S. Pina: methodology, visualization. Daniela Kalafatovic: conceptualization, investigation, supervision, writing – reviewing and editing, funding acquisition. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work utilized resources of the Bura supercomputer facility at the University of Rijeka, Center for Advanced Computing and Modeling. This work was supported by the Croatian Science Foundation [grant numbers UIP-2019-04-7999, DOK-2021-02-3496] and the University of Rijeka [grant number uniri-mladi-intpo-22-32790].Notes and references
- X. Ren, D. Chen, Y. Wang, H. Li, Y. Zhang, H. Chen, X. Li and M. Huo, J. Nanobiotechnol., 2022, 20, 92 CrossRef CAS PubMed.
- R. Mandal, A. Ghosh, N. K. Rout, M. Prasad, B. Hazra, S. Sar, S. Das, A. Datta and P. K. Tarafdar, Org. Biomol. Chem., 2023, 21, 4473–4481 RSC.
- A. Shamsabadi, T. Haghighi, S. Carvalho, L. C. Frenette and M. M. Stevens, Adv. Mater., 2023, 2300184 CrossRef PubMed.
- J. Yi, Q. Deng, Z. Liu, H. Wang, X. Liu, J. Ren and X. Qu, Small, 2023, 2301096 CrossRef CAS PubMed.
- P. Makam, S. S. Yamijala, V. S. Bhadram, L. J. Shimon, B. M. Wong and E. Gazit, Nat. Commun., 2022, 13, 1505 CrossRef CAS PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 RSC.
- T. Schnitzer, J. W. Rackl and H. Wennemers, Chem. Sci., 2022, 13, 8963–8967 RSC.
- A. S. Pina, L. Morgado, K. L. Duncan, S. Carvalho, H. F. Carvalho, A. J. Barbosa, B. D. P. Mariz, I. P. Moreira, D. Kalafatovic and B. M. M. Faustino, et al. , Chem. Sci., 2022, 13, 210–217 RSC.
- X. Liu, R. Waters, H. E. Gilbert, G. T. Barroso, K. M. Boyle and L. S. Witus, RSC Adv., 2021, 11, 23714–23718 RSC.
- M. A. Beasley, A. D. Dunkelberger, M. D. Thum, E. S. Ryland, K. P. Fears, A. B. Grafton, J. C. Owrutsky, J. G. Lundin and C. R. So, J. Mater. Chem. B, 2022, 10, 9400–9412 RSC.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- V. Alva and A. N. Lupas, Curr. Opin. Struct. Biol., 2018, 48, 103–109 CrossRef CAS PubMed.
- J. Han, H. Gong, X. Ren and X. Yan, Nano Today, 2021, 41, 101295 CrossRef CAS.
- Y. Lou, B. Zhang, X. Ye and Z.-G. Wang, Mater. Today Nano, 2023, 100302 CrossRef CAS.
- P. Dowari, M. K. Baroi, T. Das, B. K. Das, S. Das, S. Chowdhuri, A. Garg, A. Debnath and D. Das, J. Colloid Interface Sci., 2022, 618, 98–110 CrossRef CAS PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. Bernardes, E. Gazit and T. P. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- O. Zozulia, M. Dolan and I. Korendovych, Chem. Soc. Rev., 2018, 47, 3621–3639 RSC.
- S. Carvalho, D. Q. Peralta Reis, S. V. Pereira, D. Kalafatovic and A. S. Pina, Isr. J. Chem., 2022, 62, e202200029 CrossRef CAS.
- A. Lampel, R. Ulijn and T. Tuttle, Chem. Soc. Rev., 2018, 47, 3737–3758 RSC.
- P. Janković, I. Šantek, A. S. Pina and D. Kalafatovic, Front. Chem., 2021, 9, 723473 CrossRef PubMed.
- Z. Li, S. Y. Joshi, Y. Wang, S. A. Deshmukh and J. B. Matson, Angew. Chem., Int. Ed., 2023, e202303755 CAS.
- R. Ree, S. Varland and T. Arnesen, Exp. Mol. Med., 2018, 50, 1–13 CrossRef CAS PubMed.
- Y. Marciano, N. Nayeem, D. Dave, R. V. Ulijn and M. Contel, ACS Biomater. Sci. Eng., 2023, 9, 3379–3389 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- R. Jwad, D. Weissberger and L. Hunter, Chem. Rev., 2020, 120, 9743–9789 CrossRef CAS PubMed.
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- Y. Maeda, O. V. Makhlynets, H. Matsui and I. V. Korendovych, Annu. Rev. Biomed. Eng., 2016, 18, 311–328 CrossRef CAS PubMed.
- K. L. Duncan and R. V. Ulijn, Biocatalysis, 2015, 1, 67–81 Search PubMed.
- P. Janković, E. Otović, G. Mauša and D. Kalafatovic, Data Brief, 2023, 109290 CrossRef PubMed.
- S. Lai, D. Yang, Y. Wang, X. Ju, W. Liu, H. Li, D. Wang, Y. Zhao, J. Wang and H. Xu, Colloids Surf., A, 2023, 665, 131257 CrossRef CAS.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stohr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- Z. Al-Garawi, B. McIntosh, D. Neill-Hall, A. Hatimy, S. Sweet, M. Bagley and L. Serpell, Nanoscale, 2017, 9, 10773–10783 RSC.
- R. Song, X. Wu, B. Xue, Y. Yang, W. Huang, G. Zeng, J. Wang, W. Li, Y. Cao and W. Wang, et al. , J. Am. Chem. Soc., 2018, 141, 223–231 CrossRef PubMed.
- L. Gentilucci, R. De Marco and L. Cerisoli, Curr. Pharm. Des., 2010, 16, 3185–3203 CrossRef CAS PubMed.
- M. Babić, P. Janković, S. Marchesan, G. Mauša and D. Kalafatovic, J. Chem. Inf. Model., 2022, 62, 6398–6410 CrossRef PubMed.
- M. Belieres, N. Chouini-Lalanne and C. Dejugnat, RSC Adv., 2015, 5, 35830–35842 RSC.
- T. D. Do, N. E. De Almeida, N. E. LaPointe, A. Chamas, S. C. Feinstein and M. T. Bowers, Anal. Chem., 2016, 88, 868–876 CrossRef CAS PubMed.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- A. Rodger and M. A. Ismail, Spectrophotometry and Spectrofluorimetry: A Practical Approach, Oxford University Press, 2000 Search PubMed.
- M. Lee, T. Wang, O. V. Makhlynets, Y. Wu, N. F. Polizzi, H. Wu, P. M. Gosavi, J. Stohr, I. V. Korendovych and W. F. DeGrado, et al. , Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6191–6196 CrossRef CAS PubMed.
- K. A. McCall, C. Chin Huang and C. A. Fierke, J. Nutr., 2000, 130, 1437S–1446S CrossRef CAS PubMed.
- K. Patel, L. J. Walport, J. L. Walshe, P. D. Solomon, J. K. Low, D. H. Tran, K. S. Mouradian, A. P. Silva, L. Wilkinson-White and A. Norman, et al. , Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26728–26738 CrossRef CAS PubMed.
- S. H. Joo, Biomol. Ther., 2012, 20, 19 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmuller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- L. Schrodinger and W. DeLano, PyMOL, http://www.pymol.org/pymol Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- S. Kim, J. Lee, S. Jo, C. L. Brooks III, H. S. Lee and W. Im, J. Comput. Chem., 2017, 38, 1879–1886 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- H. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- S. Jo, X. Cheng, S. M. Islam, L. Huang, H. Rui, A. Zhu, H. S. Lee, Y. Qi, W. Han, K. Vanommeslaeghe, A. D. MacKerell, B. Roux and W. Im, Biomolecular Modelling and Simulations, Academic Press, 2014, vol. 96, pp. 235–265 Search PubMed.
- S.-J. Park, N. Kern, T. Brown, J. Lee and W. Im, J. Mol. Biol., 2023, 167995 CrossRef CAS PubMed.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00117b |
| This journal is © The Royal Society of Chemistry 2023 |