
MOF catalysis meets biochemistry: molecular insights from the hydrolytic activity of MOFs towards biomolecules
Charlotte
Simms†
 ,
Angelo
Mullaliu†
,
Siene
Swinnen†
,
Francisco
de Azambuja
* and
Tatjana N.
Parac-Vogt
*
,
Angelo
Mullaliu†
,
Siene
Swinnen†
,
Francisco
de Azambuja
* and
Tatjana N.
Parac-Vogt
*
Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: francisco.deazambuja@kuleuven.be; tatjana.vogt@kuleuven.be
First published on 20th December 2022
Abstract
Performing reactions under physiologically relevant conditions often challenges the catalysts' robustness, reactivity and recyclability. Widely regarded as stable and versatile materials, metal–organic frameworks (MOFs) are an emerging platform for the development of materials with enzyme-like characteristics (i.e., nanozymes), whose applications in bioanalytical devices, biomolecule study, and therapeutics is attracting increasing attention. Despite these promising prospects, developing MOF-based nanozymes that operate in aqueous medium over a broad pH range, and in the presence of a high concentration of salts is frequently challenged by MOFs' low stability in water, unreliable reactivity, and favorable adsorption of substrates. In this minireview, we share detailed molecular insights on the reactivity of MOFs as nanozymes for hydrolysis reactions. Specifically, we discuss key aspects of MOF structure/activity relationship based on our recent work developing Zr-based MOF nanozymes for the hydrolysis of peptides and proteins. Further, an overview of recent works targeting the hydrolysis of other biomolecules highlighting current limitations, and promising research directions for improving the applicability of MOFs in biochemical contexts complement this analysis.
 Charlotte Simms | Charlotte Simms studied Biochemistry and Biological Chemistry at the University of Nottingham, UK and then completed her MSc thesis in the Laboratory of Bioinorganic Chemistry, KU Leuven, focusing on the structure and reactivity of bimetallic metal–organic frameworks under the supervision of Prof. Parac-Vogt. As an FWO PhD fellow, she is now investigating the relationship between synthesis, structure and reactivity of bimetallic MOFs for use as artificial nanozyme catalysts. |
 Angelo Mullaliu | Angelo Mullaliu obtained his Ph.D. at the University of Bologna, Italy, working on battery materials and synchrotron-based characterization methods. Following his PhD, he joined the Helmholtz Institute Ulm (HIU), Germany, for a postdoc focused on Li- and Na-ion batteries and X-ray techniques. After obtaining an FWO fellowship, he moved to KU Leuven, Belgium, to study the interaction of MOFs with biomolecules, particularly with X-ray absorption fine structure (XAFS) spectroscopy. |
 Siene Swinnen | Siene Swinnen completed her studies in Biochemistry at KU Leuven. At present, she is an FWO PhD fellow in the Laboratory of Bioinorganic Chemistry, led by Prof Parac-Vogt, and develops metal–organic frameworks as artificial enzymes for protein hydrolysis in the field of proteomics. |
 Francisco de Azambuja | Francisco de Azambuja is an organic chemist educated in Brazil (PhD 2015, State University of Campinas), Germany and USA. Broadly interested in catalysis and catalytic materials, he aims to develop novel reactions and earth-abundant metal catalysts as alternatives to sensitive late transition metal catalysts. |
 Tatjana N. Parac-Vogt | Tatjana N. Parac-Vogt is a full professor of chemistry at KU Leuven, Belgium. She studied chemistry at the University of Belgrade and obtained her PhD from Iowa State University (Ames, USA). Following her PhD, she enjoyed postdoctoral stays at the group of Professor Ken Raymond (University of California, Berkeley, USA), and in Germany as an Alexander von Humboldt fellow. At KU Leuven, she leads an interdisciplinary team performing research at the interface of inorganic chemistry, biochemistry, materials science and catalysis. Tatjana is a member of AcademiaNet, a global portal of outstanding female scientists, and is currently the Vice-President of the European Rare-Earth and Actinide Society. She is a fellow of the Royal Society of Chemistry and has been elected as a Chemistry Europe fellow (Class 2020/2021), the highest award given by an association of European chemical societies. |
Design, System, ApplicationThe use of metal–organic frameworks (MOFs) in aqueous conditions for carrying out biologically relevant hydrolytic reactions is a promising avenue of research for the development of robust bioassays with varied applications. However, intrinsic limitations of MOFs' properties, and reactivity under physiologically relevant conditions largely hinder their technological development within this context. In this article, we present an overview of the development of Zr-based MOFs as useful materials for the hydrolysis of peptides, and proteins. After introducing key initial findings, and reaction mechanism, a critical discussion on the structure–activity relationship of the MOF materials used in these reactions is presented. Further on, the first attempts to optimize MOF structures through modifications on the organic linker, metal node, and synthesis protocols is discussed, keeping it in perspective with the development of MOF catalysts in other areas of science. |
1. Introduction
Natural enzymes are widely used both in academic research and industry for a variety of purposes. For example, the use of trypsin for protein hydrolysis,1 horse-radish peroxidase for chemiluminescent oxidation,2 and deoxyribonuclease I for DNA hydrolysis3 are some of the most prominent examples. However, natural enzymes are generally substrate specific, highly sensitive to the reaction conditions, and easily become denatured in response to high temperature or non-physiological pH. Moreover, they can be quite expensive, especially if they are not readily available.4,5 On the other hand, nanosized materials such as MOFs have emerged as promising pH, temperature and chemically resistant mimics of natural enzymes, widening both the substrate scope and the range of reaction environments in which they can be used. These so-called nanozymes are usually prepared from cheap and readily available materials, have a long ‘shelf-life’ and can often be reused several times.6,7 In this context, the Lewis acidity, water stability and tunable hydrophobicity of metal–organic frameworks (MOFs) prompted their development as heterogeneous nanozymes for a large range of biomolecules,8,9 and their exploration for sensing,10,11 hydrolysis,12,13 and oxidation14 of biomolecules.The growing popularity of MOFs as robust nanozymes for varied applications also arises from their key advantages over other types of porous materials. Formed of metallic nodes (often referred to as secondary building units, SBU) joined by organic linker groups, these hybrid inorganic–organic materials have infinite and precise 2D or 3D structures characterized by a high crystallinity and porosity, which result in very large accessible surface areas.15–17 Their intrinsic modular nature provides a unique opportunity for structure design, featuring atomic-level control through linker-SBU combination. Together with straightforward synthetic protocols (or post-synthetic modifications),18–20 this simplicity prompted the development of thousands of unique MOF structures, with vast potential as catalysts,21–24 absorbents,25 filters,26 structural hosts for biological substances,27 energy storage materials28 and sensors.29 In comparison, the structural diversity of fully inorganic aluminosilicate zeolites (M2/xO·Al2O3·ySiO2·zH2O, x = valence, y = 2–200, z = confined water), which share many structural and chemical properties with MOFs,15,17,30,31 relies on a template-dependent synthesis – be that solvent or surface – to ensure structural uniformity, and to prevent fracturing of the material during transfers. On the other hand, fully organic covalent organic frameworks (COFs), which sustain the beneficial structural parameters of MOFs such as surface area, pore size, tunability, with robust covalent connectivity,32 are challenged by the compromise between crystallinity, stability and complexity.33 As such, design and tunning of COFs and zeolites' structure is not as straightforward as for the MOFs.
In this minireview, we share our perspective on the challenges and pitfalls of designing MOF-based nanozymes for the transformation of biologically relevant substrates in reactions performed in physiologically-related aqueous conditions. Although of high potential for many areas of biosciences, this emerging field has received far less attention than other MOF applications in catalysis. The unique properties of these types of substrates present challenges of a different nature to MOF catalysts (e.g., strong adsorption, reactions in aqueous systems of diverse acidity and composition, etc.). Accordingly, this minireview discusses key aspects of the structure–activity relationship of a set of MOFs used for hydrolysis of biomolecules in order to put findings in perspective with other areas of MOF catalysis, and point out fertile research directions to further rationally design these materials for applications in crucial areas such as the biochemistry, biomedical, and sensing fields. To this end, the discussion presented here is largely based on our recent work developing Zr-based MOFs (Zr-MOFs) as protease mimics for the (selective) hydrolysis of peptides, and proteins.34 We critically re-assess key recent findings (since 2018), highlight the main challenges faced so far, and identify possible routes to overcome current shortcomings towards improved MOF-based nanozymes. Specifically, we discuss the relationship between different MOF structures and resulting reactivity, highlight the proposed reaction mechanisms, and how the MOF structure can be modified through synthesis to tune reactivity and substrate interactions. To put these findings in a broader perspective, we then highlight recent advances encompassing other biologically relevant substrates such as nucleic acids, sugars, and lipids, and conclude by outlining key future research directions to bring MOFs to the next level in this area.
2. Zr-MOFs for the hydrolysis of peptides and proteins: structure vs. reactivity
Due to the inertness of the peptide bond35 and the need to overcome the drawbacks of proteolytic enzymes such as high cost, sensitivity to the reaction conditions, and self-digestion, MOFs have drawn increasing attention as more robust and inexpensive alternatives for peptide bond cleavage.34,36 The high Lewis acidity of MOFs formed from group (IV) metal cations, especially zirconium, has paved the path for their use as efficient heterogeneous catalysts for the hydrolysis of several peptides,37–41 and the selective cleavage of proteins, e.g., horse heart myoglobin (Mb)42 and hen egg white lysozyme (HEWL).37–39,43 The fragmentation of HEWL by Zr-based MOF-808, for instance, can be detected within 1 hour of incubation, while 55% of the protein is hydrolyzed after 25 hours.37 Interestingly, the cleavage of proteins generates only a few medium-sized protein fragments (3–15 kDa), suggesting the hydrolysis proceeds in a selective manner. The cleavage sites could be extrapolated by estimating the molecular weights of the protein fragments observed after the reaction, and the results suggest Asp-X/X-Asp are preferably cleaved.44 However, unambiguous detection of cleavage sites is still missing.42At the molecular level, the hydrolysis of peptide bonds by Zr-MOFs likely happens at the Zr6O8 cluster node of the MOF structures. Early theoretical modelling of peptide bond hydrolysis using MOF-808 and glycylglycine (Gly-Gly) as a model system was carried out to understand the mechanism of peptide bond hydrolysis.45 The reaction was predicted to proceed in a linker-free face of the Zr6O8 cluster involving a pair of Zr centers counterbalanced by labile water/hydroxyl groups as ligands. Glycylglycine promptly replaces the water/hydroxyl pair, and binds by bridging the Zr pair with its carboxylate group. This most stable binding mode, however, is not the one leading to the lowest energy reaction pathway. A bidentate NH2/C(O)NH coordination leaving the terminal COO− group free to deprotonate the attacking water molecule has been identified as the most probable reactive intermediate (Scheme 1). Nucleophilic attack by an external molecule of water is most likely the rate-limiting step in this mechanism, and the assistance of carboxylate group is key to decrease the energy barrier of this step. Importantly, the preliminary understanding of this mechanism also inspired the development of other important reactions. One clear example was the formation of amide bonds, which explored the same binding equilibrium to push the reaction in the opposite direction to hydrolysis.46,47
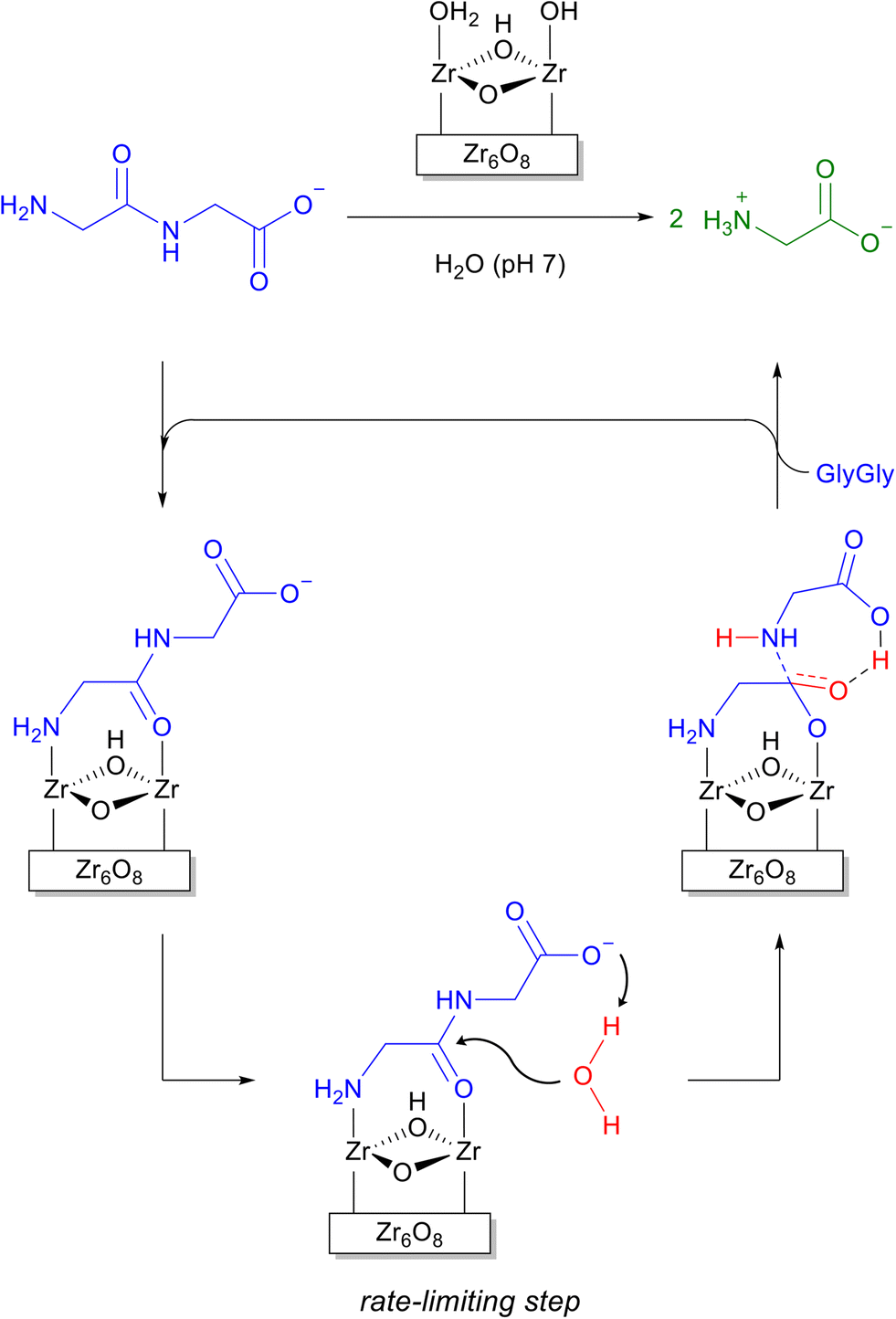 | ||
| Scheme 1 General mechanism of peptide bond hydrolysis at the Zr6O8 cluster nodes of Zr-MOFs. | ||
In proteins, hydrolysis happens at internal peptide bonds, and the reaction mechanism likely involves basic functional groups of the amino acids' side chains.44,48 No studies on the mechanism of protein hydrolysis by Zr-MOFs have been reported yet. However, it is plausible that Zr-MOFs cleave the peptide bonds in proteins through a similar mechanism as predicted for Zr-POMs,48 since both types of catalysts hydrolyze peptides by a very similar mechanism.49 In general, hydrolysis of internal peptide bonds requires the assistance of a side-chain group to deprotonate the water nucleophile, as the terminal COO− group that assists the reaction in dipeptides is not available. Notably, the aspartate's side-chain COO− group has been found particularly suitable to accelerate the hydrolysis of internal peptide bonds.34,44,48 In accordance with this hypothesis, the similar molecular weight of fragments generated by Zr-MOFs and Zr-polyoxometalates (Zr-POMs) during the hydrolysis of hen egg white lysozyme (HEWL) and myoglobin (Mb) strongly suggests that similar principles are governing the hydrolysis of proteins by both materials. However, more studies are definitely needed to understand the mechanistic details, especially on how MOF-protein interactions influence the cleavage selectivity.
In addition to these observations related to the mechanism at the molecular level, other results strongly suggest that peptide bond hydrolysis by Zr-MOFs is largely influenced by mass transport phenomena. Mechanistically, the substrates need to be adsorbed by the MOF's external and/or internal surface before undergoing the actual cleavage reaction. Likewise, the products need to be efficiently released after hydrolysis has happened. In the case of Zr-MOFs, the affinity of substrates and products can vary significantly depending on the conditions, MOF structure, and substrate nature. In the case of peptides, one of the few systematic studies addressing their adsorption by Zr-MOFs identified many different driving forces for the adsorption to occur.50 More specifically, the Zr6O8 cluster node has been found to exert a double role by strongly coordinating the carboxylate groups, but also repelling positively charged protonated N-terminal groups. These effects are so important that the distance between C-, and N-terminal ends in a dipeptide was found to affect the extent of adsorption. In addition, the hydrophobic interactions between linkers and substrates are also highly relevant – direct relationships between linker hydrophobicity and adsorption were clear for extreme cases like NU-1000 and MOF-808 materials. In the case of proteins, their adsorption by porous materials is a matter of great interest for the development of robust biocatalysts (enzyme immobilization), and selective biosensors. Thus, many more studies are available.51 However, similar studies prompting an understanding of Zr-MOF/protein interaction, especially under hydrolytically relevant conditions, are still lacking.
Considering the great influence of MOF structure on the reactivity observed, and its direct relationship with key mechanistic aspects of the reaction, gaining insight into the structure/activity relationship is key to advancing the field of nanozymatic catalysis. For this reason, the following sections revisit MOFs' structural features in detail and attempts to associate it with some aspects of their catalytic activity. In this case, while all Zr-based MOFs studied so far (MOF-808, MIP-201, UiO-66, and NU-1000) possess {Zr6O8} clusters as metallic nodes, they have different organic linkers. Furthermore, formal linker connectivity varies considerably depending on the MOF, and interesting reactivity trends indicate that both catalytic cores and linkers can be used to tune the material's physicochemical properties and modulate catalytic performance.
2.1. Inorganic catalytic cluster
A three-dimensional network of inorganic catalytic nodes linked together via organic molecules forms the complex MOFs' hybrid structure. Network nodes (SBUs) are metallic ions or metal oxo clusters, most commonly lanthanide ions or early transition metals due to their high coordination numbers and Lewis acidity, although di- and trivalent metals are also used.17,52 These nodes are directly connected through n organic linkers, where n designates the node connectivity. In the case of Zr-MOFs, such node is a zirconium oxo cluster, whose skeleton consists of six Zr atoms placed at the vertices of an octahedron with eight triangular faces (Fig. 1a). In a model structure, a μ3-oxygen atom or μ3-hydroxyl ligand lies on the top of each face of the Zr octahedron, bridging the Zr vertices of that face (cf.Fig. 1b and c). Thus, each vertex is indirectly connected to four adjacent Zr atoms through bridging μ3-ligands, and μ3-oxygens fill half of Zr′s coordination sites. In the pristine state, the presence of four μ3-O and four μ3-OH for the building block of UiO-66, results in a [Zr6(μ3-OH)4(μ3-O)4] core. After heat exposure (250–300 °C), dehydroxylation occurs, and two hydroxyl ligands leave the cluster together with the hydrogen atoms of the remaining two μ3-OH, giving rise to a fully dehydrated {Zr6O8} inorganic core.53 The structural rearrangement frees one coordination site per Zr (coordination number (CN) = 8 → 7) enhancing the Lewis acidity of the {Zr6O8} node. Thus, interactions with substrates, and subsequently the polarization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, is enhanced. This facilitates water nucleophilic addition to the carbonyl group, resulting in an overall increase in the catalytic activity (Scheme 1).
O bond, is enhanced. This facilitates water nucleophilic addition to the carbonyl group, resulting in an overall increase in the catalytic activity (Scheme 1).
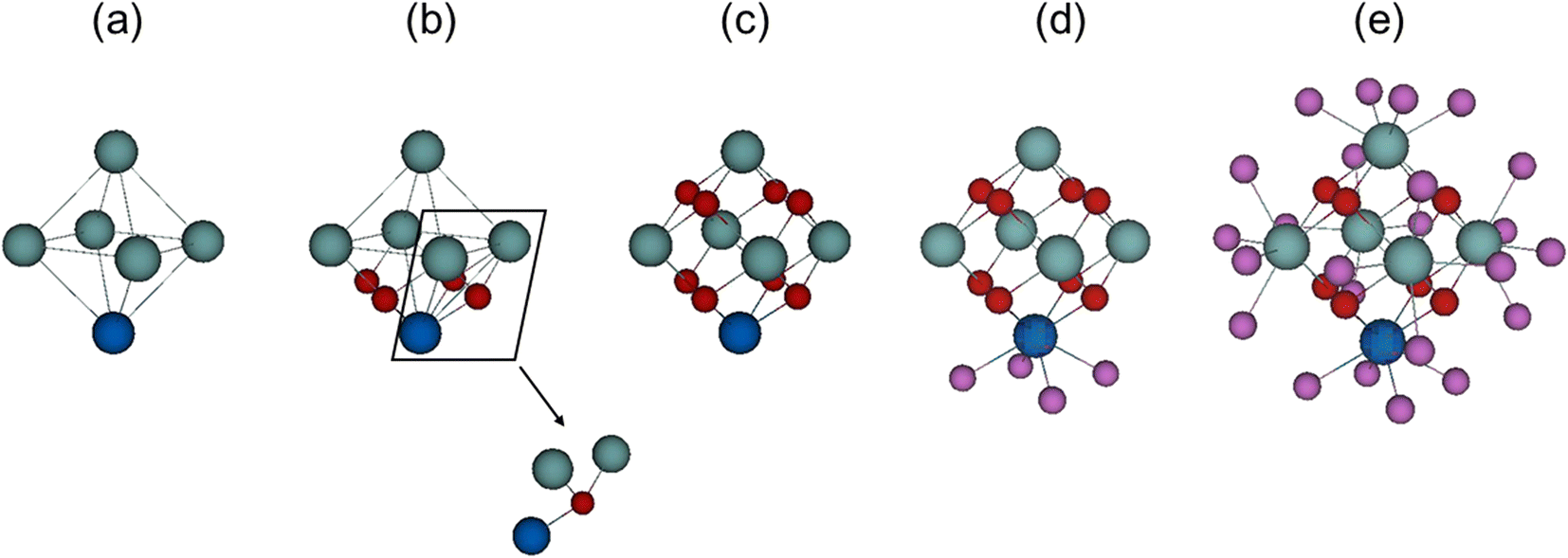 | ||
| Fig. 1 Structure of the Zr6O8 cluster node: (a) inorganic catalytic clusters are made of six Zr atoms (cyan and blue spheres) placed at the vertices of an octahedron. (b) Each Zr atom, for instance, the blue-colored one, is surrounded by four μ3-O/OH (red spheres). Each μ3-O/OH is shared with other two Zr atoms placed on the same triangular face (detail); panel (c) shows the complete {Zr6O8} catalytic cluster, with the Zr coordination sites half-filled. (d) The Zr coordination environment is completed by other four oxygen atoms (violet) deriving from the linker, modulator, or water, forming with the previous oxygens a square antiprismatic structure. In panel (e), all Zr atoms have a full coordination sphere of oxygens. | ||
Besides μ3-oxygens, Zr′s coordination sphere, typically having a coordination number of 8 and arranged in a square antiprismatic geometry, is filled with four other oxygen atoms (μ-O) belonging to the linker, modulator, or water (cf.Fig. 1d and e). Interatomic distances between Zr and μ-O ligands (∼2.2 Å) are slightly longer than Zr–μ3-O (∼2.1 Å), while each Zr is placed ∼3.5 Å away from the four adjacent Zr atoms, and ∼5.0 Å distant from the opposite Zr vertex.53–55 While μ3-O can be affected by heat treatment in the case of dehydroxylation, as mentioned above, they are generally not altered during the catalytic reaction. μ-O are more labile and can be exchanged with substrates or other ligands. For example, facile post-synthetic ligand exchange has been reported in many instances,56–58 together with the removal of coordinating modulator ligands to free Zr(IV) coordination sites and enhance MOF's catalytic activity.59
Furthermore, by replacing Zr with another metal M, interatomic M–O and M–M distances and related bond strength can be modulated, and these changes have been reported to directly influence MOFs' properties, including the catalytic activity. Essentially, three main situations can be considered in this case: i) M < Zr; ii) M ∼ Zr; iii) M > Zr. By post-synthetic exchange of Zr by Ti, which is smaller than Zr (M < Zr), changes of M–M/Zr interatomic distances and overall structure result in smaller pores' sizes, which enhances the adsorption of CO2.60 Considering the case when M ∼ Zr, the metal substitution does not affect structural distances significantly, but the properties of the new metal can still affect reactivity. For example, Hf4+ ion is known as the “elemental twin” of Zr4+. Despite the substantial difference in atomic number, they have very similar ionic radii (Zr4+ = 0.84 Å and Hf4+ = 0.83 Å for CN = 8) because of the lanthanide contraction.61,62 Thus, {Zr6O8} and {Hf6O8} clusters are highly similar in terms of geometrical distances.63 However, their M–O bond strengths are slightly different (dissociation enthalpies for Zr–O and Hf–O bonds are 776 and 802 kJ mol−1, respectively),64 most likely due to the presence of 4f orbitals in Hf, which could hybridize with the oxygen 2p states. This small difference renders the {Hf6O8} clusters more reactive, and Hf-NU-1000 provides higher yields in the formation of cyclic carbonates from CO2 and epoxides.64,65 Finally, an example of M > Zr is the replacement of Zr with Ce(IV). Ce4+ is minimally affected by the lanthanide contraction, being at the beginning of the series; therefore, its ionic radius is larger (1.00 Å (ref. 66)) than Zr(IV), resulting in longer M–O (and M–M/Zr) interatomic distances in a {Ce6O8} cluster.63,67 This certainly affects the lability of M–μ-O bonds, and consequently may affect reaction outcome (e.g., by influencing the adsorption/desorption of substrates and products). Moreover, Ce(IV) is able to expand its coordination sphere thanks to its 4f orbitals mixing with the orbitals of the incoming substrate.68 This has been suggested as the underlying reason for the greater hydrolysis rate of phosphate substrate by Ce-UiO-66 in comparison with Zr-UiO-66.69 These changes imparted by the presence of Ce are also consistent with the effect of a partial Zr replacement by Ce in a series of mixed Zr/Ce UiO-66 MOFs, which showed superior catalytic activity compared to a pure Zr-UiO-66 MOF.41
In light of these results, the extrapolation of interatomic distances, bond angles, and coordination environment could be pivotal in correlating structure and catalytic activity. The interatomic M–O, M–M′ (M′ = M or M′ ≠ M), or M–C (deriving from the linker or modulator) distances can be obtained by using X-ray absorption spectroscopy (XAS) at the metal M K-edge. As an element-selective technique, XAS probes the transitions from inner core electrons (1s shell in the case of K-edge spectroscopy) to unoccupied energy states and has been used to study the structural coordination environment of several metal-oxo compounds, including discrete clusters55,70 and MOFs.54,67 Indeed, an accurate description of the local structure around the photo absorber can be achieved by interpreting the modulation of the XAS signal in the extended region of the spectrum (EXAFS) arising from the interaction of the ejected core electron (photo electron) with the potential described by the neighboring atoms. However, more studies will be required to evaluate how the cluster's structure affects binding to the substrate and vice versa. Moreover, other techniques need to be employed to investigate the clusters' connectivity and derived properties, which significantly impact catalytic performance, as will be discussed below.
2.2. From clusters to MOFs: tuning reactivity via linkers
The organic linkers connecting the inorganic catalytic cores have a large influence on MOF structure, properties, and reactivity. Linkers range from a simple benzene-dicarboxylic acid to complex polyaromatic systems containing several binding groups, allowing for control over the size of pores and surface area of the MOF through linker choice. Rigid linkers are preferred to preserve crystalline porous structures, but they are not crucial.16,71 Linkers can also be functionalized with groups such as –NH2, –NO2, or –SO4 to modify the surface without disrupting the original framework.72 Furthermore, the linkers have a direct relationship with the cluster connectivity, and their properties highly impact selectivity and hydrolytic activity. Thus, by carefully selecting the ligand at the synthesis stage, the MOF can have several aspects of their reactivity designed ahead of their use.The nature of the linker directly correlates with the interactions and reactivity observed with peptides. The four basic Zr-MOF structures used for peptide hydrolysis so far, i.e., UiO-66, MOF-808, NU-1000, and MIP-201 are synthesized by adopting different carboxylates as the linkers (see Table 1). More precisely, UiO-66 is built using the anionic form of 1,4-benzendicarboxylic acid (bdc), while MOF-808, NU-1000, and MIP-201 use the deprotonated forms of 1,3,5-benzenetricarboxylic acid (btc), 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy), and 3,3,5,5′-tetracarboxydiphenylmethane (H4mdip), respectively. As shown in Table 1, the linkers differ in the number of coordination sites: bdc is a ditopic linker, meaning that it can coordinate at two coordination sites, btc is tritopic (three coordination sites), while H4TBAPy and H4mdip are tetratopic (four coordination sites). The different number of sites contributes to determining the Zr6 cluster connectivity and MOF's pore size and shape (cf.Table 1), while the nature of the linker affects other properties, such as hydrophobicity and affinity to certain substrates. For the MOF-808, UiO-66, and NU-1000 triad, it has been observed that lower connectivity, thus a higher number of available Zr(IV) sites, correlates with enhanced glycylglycine (Gly-Gly) adsorption.50 When considering a wide selection of dipeptides, increased hydrophilicity of the peptide's side chain further boosts adsorption in the case of MOF-808. On the other side, the hydrophobicity of H4TBAPy, used as the linker for NU-1000, negatively affects the adsorption of Gly-Gly although NU-1000 features very large pores (cf.Table 1). However, the same MOF promotes hydrophobic interactions, cation–π, or π–π interactions with hydrophobic and/or aromatic functional groups in other dipeptides.
| MOF | Linker | Space group | Connectivitya | Pore size | MOF's graphical representation |
|---|---|---|---|---|---|
| a Formal connectivity. The actual structure may contain missing linker defects depending on the synthesis conditions (see DeStefano et al. for a discussion73). | |||||
| UiO-66 |
 bdc
bdc
|
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) m (225) |
12 | 11.7 Å |
|
| a = 20.7 Å | |||||
| MOF-808 |
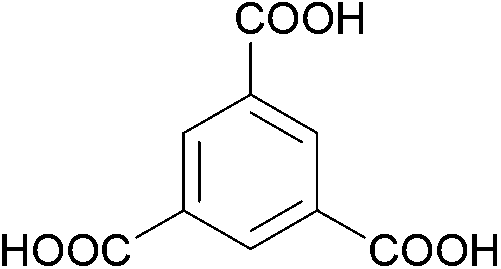 btc
btc
|
Fdm (227) |
6 | 7–10 Å (tetrahedral pore) |
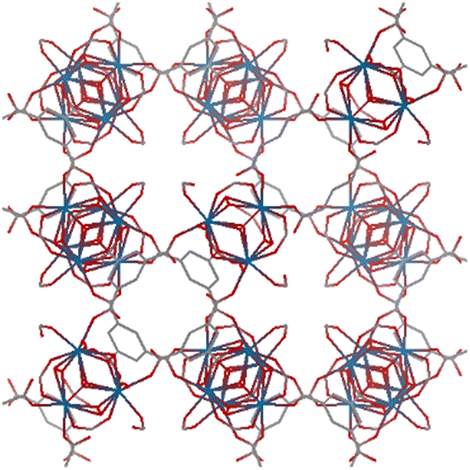
|
| a = 35.1 Å | 18.4 Å (octahedral pore) | ||||
| NU-1000 |
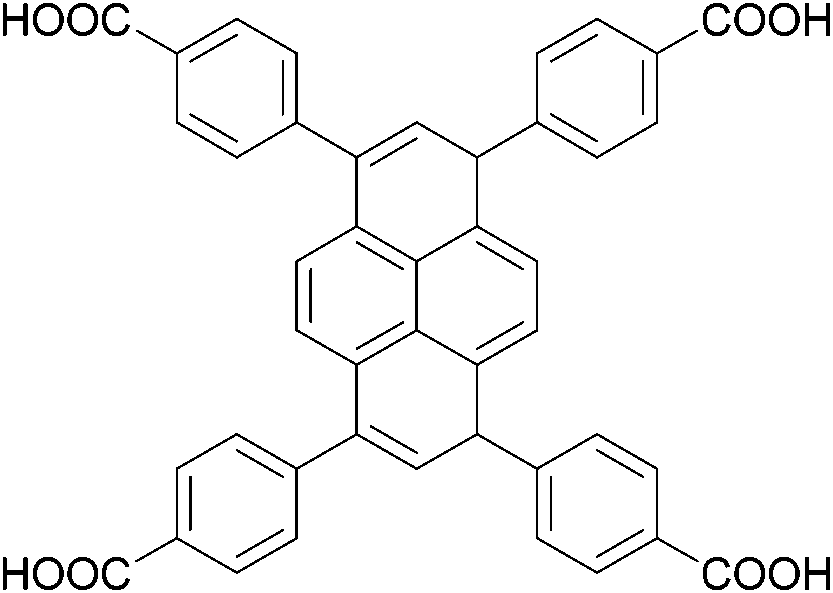 H
4
TBAPy
H
4
TBAPy
|
P6/mmm (191) | 8 | 12 Å (triangular pore) |
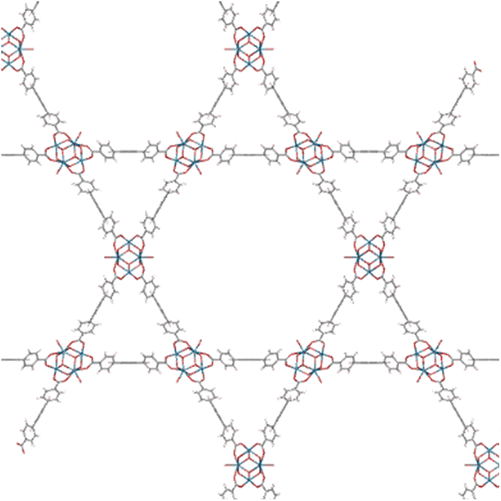
|
| a = 39.3 Å | 31 Å (hexagonal pore) | ||||
| c = 16.6 Å | |||||
| MIP-201 |
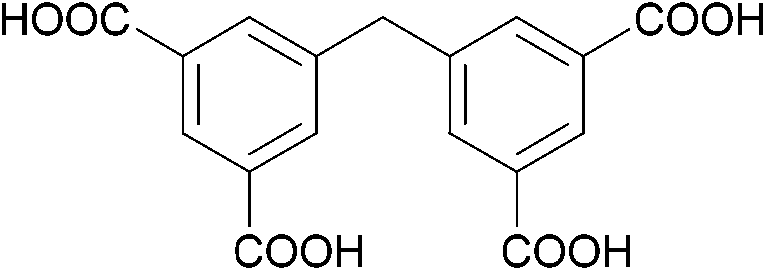 H
4
mdip
H
4
mdip
|
Im (204) |
6 | 7 Å, 10.5 Å |
|
| a = 24.6 Å | |||||
Besides affecting adsorption, linker connectivity also inversely correlates with hydrolytic reactivity, and MOFs with a higher connectivity promoted a slower reaction, as encountered in the literature for other reactions.74,75 As seen in Table 2, the rate of Gly-Gly hydrolysis at 60 °C and pD = 7.4 strongly correlated with the MOF's connectivity, with 6-connected MOF-808 and MIP-201 displaying the highest hydrolysis rates (k = 2.69 × 10−4 s−1 and 1.85 × 10−5 s−1, respectively), followed by the 8-connected NU-1000 (k = 1.61 × 10−6 s−1) and the formally 12-connected UiO-66 (k =7.9 × 10−7 s−1). The substantial dissimilarity in the MOFs' hydrolytic performance is even more clear from the corresponding half-lives of 0.72 hours, 10.4 hours, five days, and ten days, respectively.
A striking difference in the transition state energies, particularly in the entropy of activation ΔS‡, experimentally determined according to the Eyring equation, strongly suggests the MOF topology resulting from the choice of linkers also directly impacts the observed reactivity, presumably by influencing the overall mass transport across the material (Table 2). Despite some variations in the enthalpy of activation ΔH‡, the ΔS‡ term associated with the transition state for NU-1000 is significantly different. While both MOF-808 (space group (s.g.) Fdm, no. 227) and UiO-66 (s.g. Fmm, no. 225) feature a cubic structure, NU-1000 crystallizes in the hexagonal P6/mmm lattice (s.g. no. 191) and can be pictured as a Kagome lattice,76i.e., a two-dimensional pattern with large hexagonal mesoporous channels (31 Å in diameter). Presumably, a lower degree of freedom in the transport for NU-1000 compared to a cubic MOF with 3D channels, results in the more negative entropic parameter for NU-1000 and negatively affects the reaction rate.12 However, in a similar structure with 2D mesoporous channels interconnected via 3D cavities, i.e., a nanoporous carbon employed as a supercapacitor and adsorbent, an increase in mass transport was observed in the material's bulk.77 Thus, a 3D-interconnected Kagome lattice might experience an increase in the transport of substrates to the catalytic sites in the material's bulk and, consequently, improve catalytic activity. Overall, this experimental evidence suggests that more effort has to be directed to researching the structure/activity interrelation to design more performant hybrid materials by fine-tuning structural features. Although the {Zr6O8} inorganic cores catalyze the hydrolysis, it is evident that linkers and, more in general, MOF's architecture play a crucial role in influencing catalytic performance.
3. Modifications to the MOF structure, and its impact on the reactivity
Due to the general ease of MOF synthesis and immense variability achieved through use of different organic linkers and metal cluster configurations, there is much evidence showing how the structure of the MOF influences its reactivity. It is possible to take this further, by changing or fine-tuning structural features within a chosen MOF to enhance reactivity. One of the most explored areas of structural modulation is referred to as ‘Defect-Engineering’, i.e., creating more ‘open-metal sites’ (OMS) on the clusters to increase the possibility of substrate-active site interactions, and diffusion in the pores, resulting in increased catalytic activity.52,78,79 Other strategies include controlling the size of the particle to increase the accessible surface area,80 increasing the pore area or adding functional groups to linkers to change the character of the pore (pore engineering), or modifying the surface to enhance substrate–MOF interactions.19,81 Finally, as referred to in section 2.1, it is also possible to insert a second metal to alter the reactivity of the MOF, whilst preserving the global structure of the starting MOF.82,83 In this context, initial systematic studies attempting to apply some of these strategies to the hydrolysis of peptides and proteins have been reported. In essence, three main strategies (creating more defects in the structure, tuning the pore environment, and modifying the synthesis procedure to control the particle size, and other MOF features) have been applied using UiO-66 and MOF-808 Zr-MOFs. The main findings arising from this work are discussed in the following paragraphs.3.1. Structural modifications
As noted in section 2.2, the connectivity of the cluster also has a strong role in determining the reactivity of the MOF through the presence of OMS, with the 6 connected MOF-808 being substantially more active towards peptide bond hydrolysis than the 12 connected UiO-66.37,39 Further evidence of the relationship between cluster connectivity, and reaction rates was observed by inducing defects in the MOF structure. Specifically, hydrolysis rates of UiO-66 could be boosted in a straightforward manner by increasing the amount of missing linkers defects, which enables additional open-metal sites formally decreasing the cluster connectivity while maintaining the general architecture of UiO-66.84 In this case, the synthesis of UiO-66 in the presence of trifluoroacetic acid (TFA) promoted the formation of a more defective structure (UiO-66-TFA), increasing the availability of free catalytic sites and improving the hydrolysis rate constant (k = 7.97 × 10−6 s−1, half life = 24.2 h) in comparison to the pristine UiO-66 (k = 7.9 × 10−7 s−1, half-life = 240 h). The greater accessibility of catalytic sites was further confirmed by the higher surface area of UiO-66-TFA (985 m2 g−1) in comparison with the ‘pristine’ UiO-66 (861 m2 g−1) used in this work, giving the similar particle size observed for both MOFs.38A distinct approach to enhance the number of accessible catalytic sites by enlarging the external surface area was demonstrated using MOF-808, indicating that the majority of hydrolytic activity occurs on the external surface of the MOF rather than within the pores.43 Although well known for having large BET surface areas, often in the range of 1000–2000 m2 g−1, the external surface area of MOF-808 particles could be improved by tuning the synthesis conditions. Specifically, MOF-808 was prepared using a two step approach, in which Zr6O8 clusters were preformed in isopropanol and acetic acid before reacting with the benzene-tricarboxylic acid linker in a water/formic acid mixture at room temperature. This much greener and safer synthesis than previous standard solvothermal reactions in DMF/formic acid at elevated temperatures also allowed for much greater control over key structural features. By changing the BTC linker concentration used in this two step synthesis of MOF-808, the particle size could be carefully controlled, resulting in control over the internal and external surface areas of the MOF. The other structural features such as the total BET surface area and number of defects of the MOF were largely unchanged, but as the particle size was reduced from 850 nm to 35 nm, the external surface area increased 16-fold. When used for peptide bond hydrolysis, the hydrolysis rate was 223% higher for the 35 nm MOF compared to the largest particle size of 850 nm. Therefore, the external accessible metal sites on MOF-808 surface are major contributors for the hydrolytic activity, while the porous structure seems to be much less relevant in this case.
Additionally, when incubated with the small (14.4 kDa) monomeric protein hen egg white lysozyme (HEWL), MOF-808 with different particle sizes also hydrolysed the protein into several smaller fragments.37,43 In this case, while the molecular weight of the fragments observed was similar to the original MOF-808 report, the concentration of fragments released in solution was heavily influenced by the particle size, with small MOF particles allowing for the detection of more fragments. This was probably because the greater external surface area provided more external sites for protein–MOF interaction and subsequent hydrolysis. In addition, with a reduced internal surface area, protein fragments are less likely to become trapped within the pores of the MOF, resulting in greater substrate recovery in the conditions used for hydrolysis (60 °C, pH 7, water).
Interestingly, a comparison of the hydrolytic activity discussed above with the original report on MOF-808 nanozymatic activity, hints that the synthesis method has a great effect on the reactivity of the MOF. This topic remains rarely addressed despite the many publications probing the effects of synthesis on the structure. In the first report, MOF-808 was synthesized from H3BTC and ZrOCl2·8H2O in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMF and formic acid at 130 °C for 48 h, and this material hydrolysed the dipeptide Gly-Gly with a rate of 2.69 × 10−4 s−1 (half-life = 0.72 h), giving almost complete conversion to glycine in 5 h.37 On the other hand, the two-step synthesis of MOF-808 discussed above hydrolysed Gly-Gly with a maximum rate of 3.87 × 10−5 s−1 (half-life = 4.98 h) for the smallest particle.43 Although a clear understanding about this discrepancy is still elusive, an intriguing initial study addressing this gap probed the effect of using different Zr precursors for the material synthesis in the reactivity of MOF-808.85
:1 mixture of DMF and formic acid at 130 °C for 48 h, and this material hydrolysed the dipeptide Gly-Gly with a rate of 2.69 × 10−4 s−1 (half-life = 0.72 h), giving almost complete conversion to glycine in 5 h.37 On the other hand, the two-step synthesis of MOF-808 discussed above hydrolysed Gly-Gly with a maximum rate of 3.87 × 10−5 s−1 (half-life = 4.98 h) for the smallest particle.43 Although a clear understanding about this discrepancy is still elusive, an intriguing initial study addressing this gap probed the effect of using different Zr precursors for the material synthesis in the reactivity of MOF-808.85
Analysis of the reactivity towards Gly-Gly hydrolysis observed for MOF-808 materials prepared from different Zr precursors suggests the precursor has a lasting effect on the reactivity, at times surpassing those arising from reaction time or synthesis temperature (Table 3). This is demonstrated by the 130-20 series (synthesized at 130 °C for 20 minutes in a mixture of HCOOH and DMF in a 2.5:1 ratio), with ZrO(NO3)2 and ZrOCl2 producing equally active MOFs (glycine yield of ∼33%), and almost identical surface areas, even though they contain a very different number of missing linker defects. On the other hand, the effect of synthesis duration on structural features and reactivity of the MOF was comparatively less pronounced as exemplified by the MOFs prepared from ZrCl4 at 130 °C for 20 min (130-20-E) and 24 h (130-24-E). In this case, the BET surface area is not affected by the synthesis duration, but number of missing linkers almost doubles, as does the reactivity. Although clear chemical reasons behind these differences in reactivity, and principles for designing synthesis conditions have yet to be elucidated, this initial work highlights that factors frequently overlooked might be highly relevant for the reactivity of MOF catalysts, at least towards polar/charged biomolecules like peptides. Key structural features, and reactivity data of MOF-808 synthesized from a range of different metal precursors, and synthesis conditions are outlined in Table 3.
| MOF | Precursor | Synthesis duration (h) | BET surface area (m2 g−1) | Missing linkers | Glycine formed after 5 h (%) |
|---|---|---|---|---|---|
| 130-20-A | Zr(OiPr)4 | 0.33 | 1060 | 1.59 | 23 |
| 130-20-B | ZrO(NO3)2·xH2O | 0.33 | 2285 | 1.18 | 33 |
| 130-20-D | ZrOCl2·8H2O | 0.33 | 2290 | 0.58 | 34 |
| 130-20-E | ZrCl4·4H2O | 0.33 | 1500 | 1.34 | 16 |
| 130-24-E | ZrCl4·4H2O | 24 | 1610 | 2.50 | 32 |
3.2. Chemical modifications
In addition to altering the structure of the native MOF to create more open metal sites, the hydrolytic activity of UiO-66 could also be modulated by functionalization of the linker. For the bdc linker, electronic effects can be tuned by functionalizing the benzene ring with an electron-withdrawing nitro group (–NO2), inducing an increased Lewis acidity of the {Zr6O8} catalytic sites, as suggested by theoretical calculations as well as experimental evidence.86,87 The direct consequence and advantage of an enhanced Lewis acidity was readily observed in the hydrolysis of Gly-Gly.38 Using a UiO-66 MOF material prepared with –NO2 functionalized bdc linker (UiO-66-NO2), a ten-fold increase in the hydrolysis rate (k = 8.36 × 10−6, half-life = 23 h) compared to the non-functionalized UiO-66 (k = 7.9 × 10−7, half-life = 240 h) was observed. Interestingly, the UiO-66 linker functionalization with an amino group (UiO-66-NH2) yields a similar reactivity (k = 8.97 × 10−6, half-life = 21.5 h) as UiO-66-NO2, despite the opposite effect of the two functional groups (NH2 is an electron-donating substituent).Changes to the MOF structure, and to the chemical environment around the catalytic sites might have contributed to unexpectedly high activity of UiO-66-NH2. The similar reactivity of the two expectedly different MOFs was rationalized by considering, on one side, the much higher BET surface and lower linker connectivity of UiO-66-NH2 caused by the larger number of defects present. On the other, functionalization of the linker by a NH2 group provides additional opportunities for H-bonding of peptide substrate nearby the metal active sites (H2O adsorption differences are likely minimal),88 thereby enhancing the reactivity.89,90 Similar scenario has been proposed for an aldol condensation catalysed by UiO-66, where UiO-66 functionalization with a NH2 group was found to enhance substrate adsorption, and subsequently reactivity, in comparison with a non-functionalized material.91 Put into perspective, these findings underline the relevance of tuning the interactions between polar/charged substrates like peptides and linkers for achieving a desired reactivity, in line with the observed for adsorption trends mentioned previously.50 However, systematic studies on these effects have yet to be reported.
The surface chemistry of the MOF can also be modified by using surfactants to tune the MOF–substrate interactions, without having to resort to linker modifications. Comparing the hydrolysis of horse heart myoglobin (HHM) by MOF-808 in the absence,92 and in the presence of different surfactants reveals that different fragmentations are obtained due to the altered MOF–protein interactions (Fig. 2). With the anionic surfactant sodium dodecyl sulfate (SDS), HHM is fully denatured by the surfactant as determined by Trp-fluoresence, and HHM is fully adsorbed onto the MOF. Unfolding of the protein improves MOF–HHM interaction, resulting in cleavage sites being more accessible for interacting with the OMS on the MOF, resulting in more hydrolysis fragments than when no surfactant was used (Fig. 2). The neutral surfactant TX-100 also resulted in unfolding of HHM, and detection of two additional hydrolysis fragments. Unlike SDS, TX-100 was also adsorbed onto the MOF without the protein being present, as was the zwitterionic CHAPS. However, CHAPS hindered hydrolysis of HHM through blocking the active sites of the MOF. A second zwitterionic surfactant Zw3-12 also hindered HHM hydrolysis. However, Zw3-12 did not interact well with the MOF on its own, and likely blocked HHM–MOF interaction by forming micelles on the surface of the MOF. Considering the challenging synthesis of some MOF structures containing functionalized linkers, these results clearly show that this unconventional approach can become an interesting alternative to tune MOF reactivity toward biomolecules in the future.
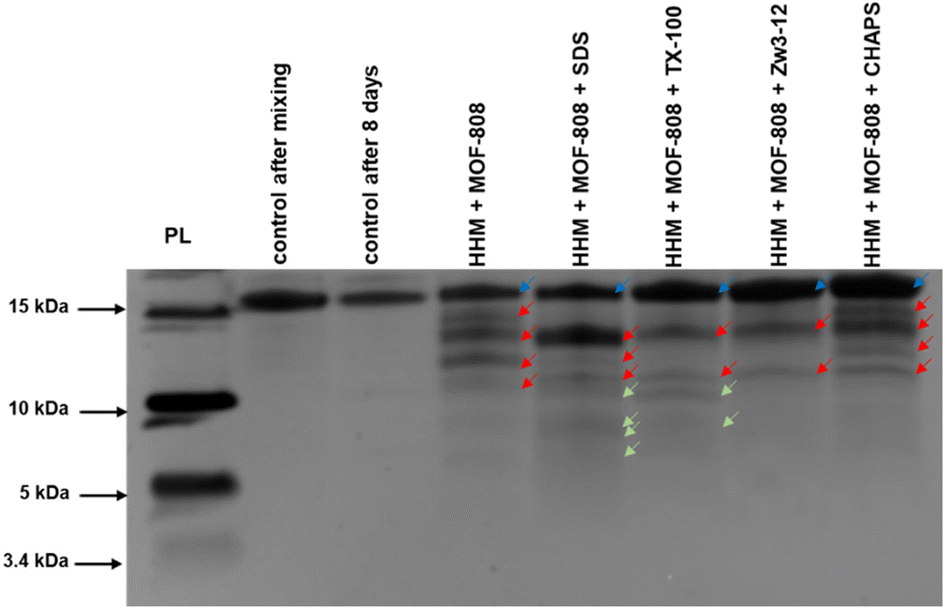 | ||
| Fig. 2 Hydrolysis of myoglobin by MOF-808 in absence and presence of surfactants, analysed with silver stained SDS-PAGE. Conditions: HHM (20 μM), MOF-808 (2 μmol), SDS, Zw3-12, CHAPS, TX-100 (0.5%), after 8 days of incubation at 60 °C in water at pH 7.4. PL stands for Mw reference. The dark blue and red arrows indicate the intact protein and the produced fragments respectively, and green arrows indicate the additional peptide fragments formed in the presence of surfactants (reproduced with permission from John Wiley and Sons from ref. 92). | ||
3.3. Cluster modifications
Another modification to the MOF structure can be achieved by changing the metals in the cluster itself, directly impacting the intrinsic reactivity of the active sites.82,83 For the hydrolysis of peptides, MOFs containing hafnium and cerium have been recently disclosed. By using HfIV in place of ZrIV in NU-1000, the MOF receives an interesting balance of Lewis and Brønsted acidity, which affects substrate interaction and subsequent reactivity, without changing the topology of the MOF.40 For example, it was found that due to the slight reduction in Lewis acidity the adsorption of HEWL was lower in Hf-NU-1000 in comparison with Zr-NU-1000. Detailed analysis of the thermodynamics of Gly-Gly hydrolysis by both forms of NU-1000 revealed that the enthalpy of activation with Hf-NU-1000 was greater than for Zr-NU-1000 (ΔH‡ = 60 kJ mol vs. 49 60 kJ mol respectively) most likely due to the greater strength of the Hf–O bond compared to the Zr–O bond. This hinders substrate exchange kinetics, and results in a slower rate of Gly-Gly hydrolysis with the Hf-MOF. On the other hand, an approach involving mixed metal MOFs was used to circumvent the low aqueous stability of CeIV-MOFs,93 and incorporate cerium to the clusters of UiO-66. By combining cerium(IV) ammonium nitrate and zirconium(IV) dinitrate oxide hydrate salts in a solvothermal synthesis, a series of bimetallic Zr/Ce UiO-66 was synthesized, containing varying percentages of CeIV in place of ZrIV.94 When Gly-Gly was hydrolysed by a series of Zr/Ce-UiO-66 MOFs, the presence of CeIV increased the reactivity of the MOF, in line with an early report on the superiority of CeIV salts for peptide bond hydrolysis.95 As percentage of Ce increased from 28–61%, the reaction rate doubles, accompanied by an increase in BET surface area from 1394 to 1723 m2 g−1.414. Reactivity of MOFs with other types of biomolecules
Unlike the systematic studies on the intrinsic hydrolytic activity of MOFs for the cleavage of peptide bonds, the hydrolysis of other biomacromolecules (e.g. nucleic acids, sugars or lipids) by MOFs is not as thoroughly investigated. However, in some cases the hydrolytic activity of MOFs towards compounds having similar types of hydrolysable bonds has been extensively investigated in other contexts. Thus, the discussion below focus only on the aspects that are closely related to the reactivity found in biomolecules. For details on the use of MOFs in these related areas, readers are kindly referred to the reviews cited along the text. Importantly, beside using MOF's intrinsic reactivity, the hydrolysis of several biomacromolecules was achieved using MOFs as a platform for the immobilization of other types of catalysts such as enzymes,51 and polyoxometalates (POMs).96 For example, enzyme immobilization onto MOFs enabled the hydrolysis of adenosine triphosphate (ATP),97 and cellulose.98 Alternatively, embedding polyoxometalates onto MOFs was utilized for the hydrolytic cleavage of the DNA model substrate bis(4-nitrophenyl)phosphate,99 the acidolysis of soybean oil100 and the hydrolysis of cellulose.101Among the hydrolysable bonds present in biomacromolecules other than a peptide bond, the hydrolysis of phosphoester bonds is probably the most investigated reaction using MOF's intrinsic reactivity, especially when considering Zr-MOFs. However, most research so far concerned the capture, and destruction of chemical warfare agents (CWA) and pesticides.102–104 The Zr-MOF assisted hydrolysis of phosphoester bonds of CWA and pesticides (Fig. 3), which relates to the chemical reactivity of those present in nucleic acids, has been proposed to proceed through a simple addition–elimination type of mechanism, that is, an initial nucleophilic addition of a hydroxide ligand to the phosphoester bond affords a yet elusive pentavalent phosphorus intermediate, which then eliminates the alcohol moiety, reforming the PO bond, and resulting into the final hydrolyzed product (Fig. 4).
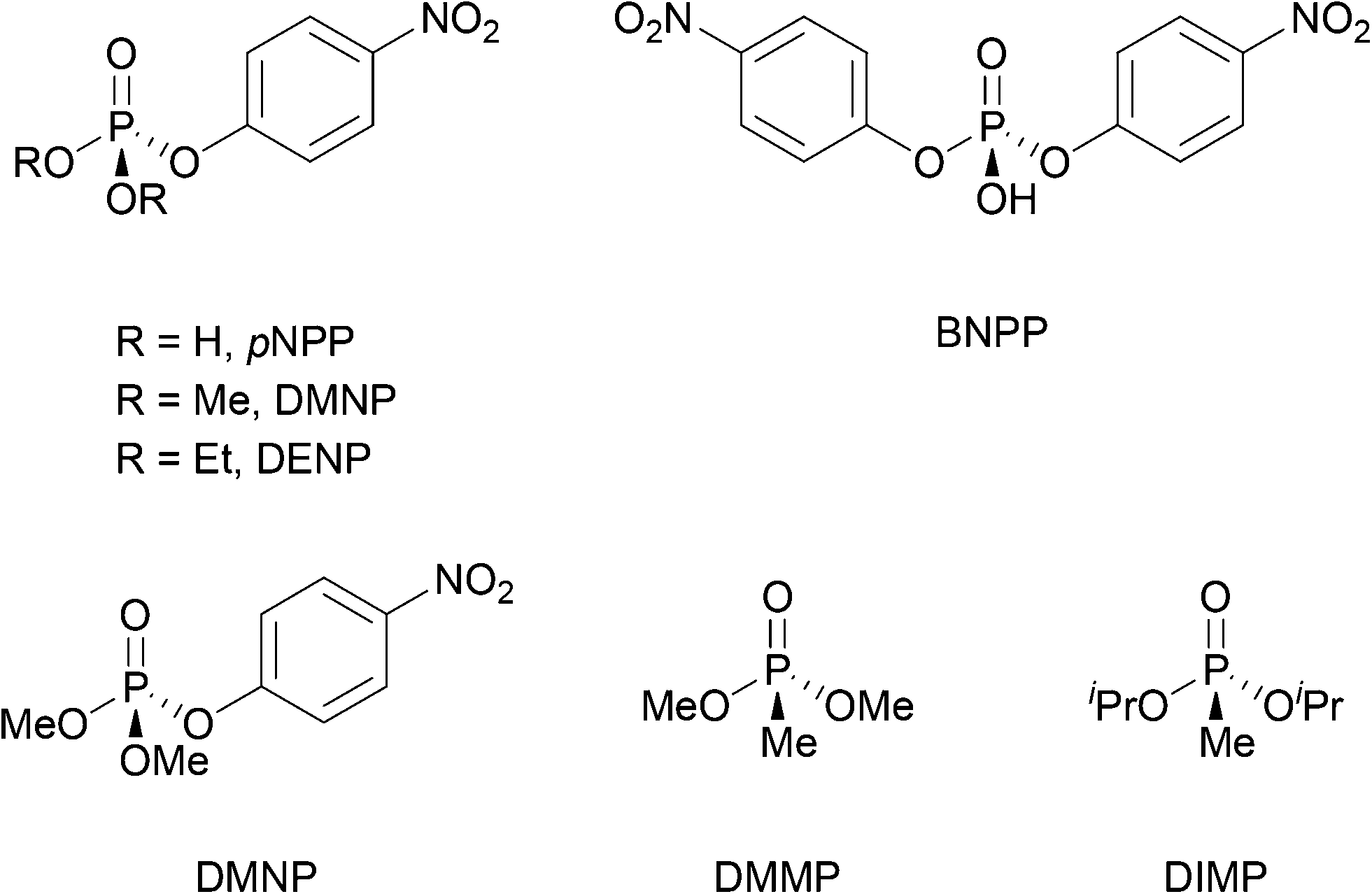 | ||
| Fig. 3 CWA and pesticide model substrates containing a phosphoester bond. | ||
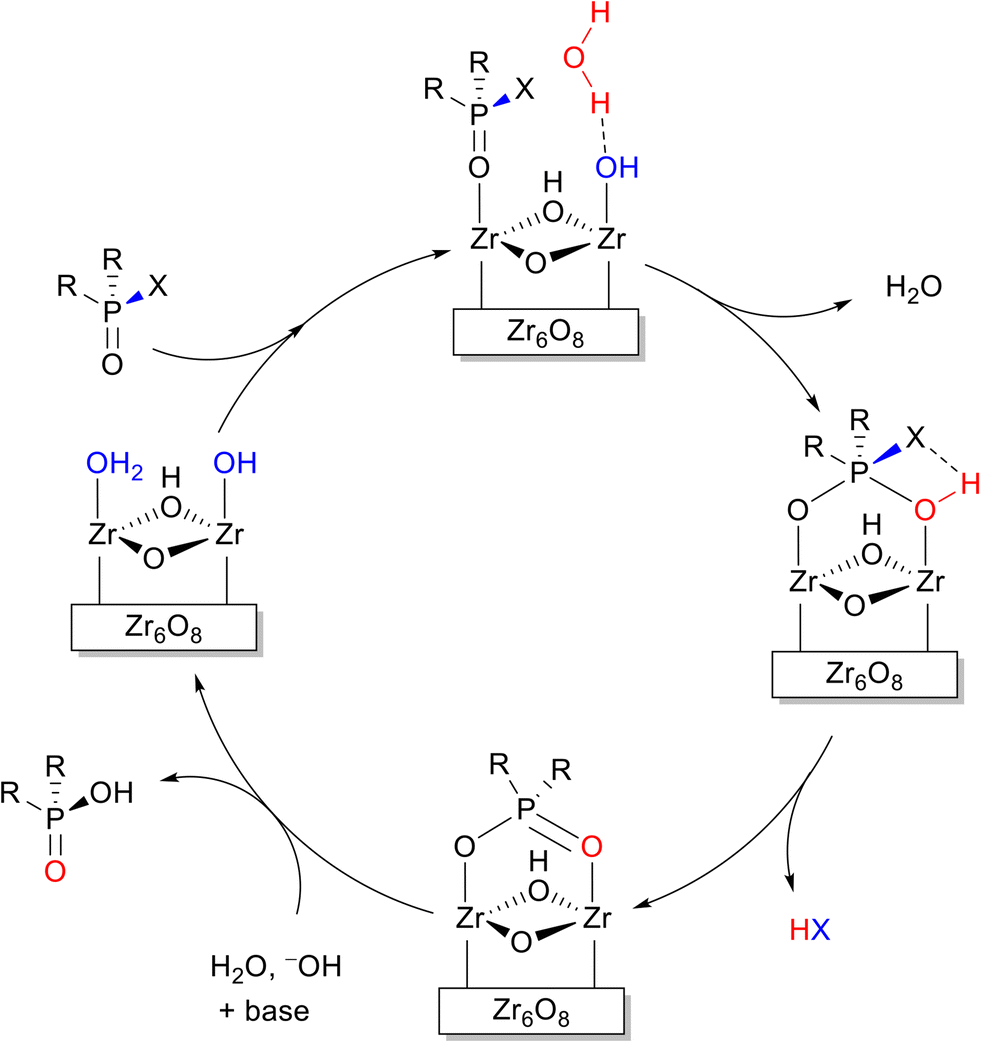 | ||
| Fig. 4 Reaction mechanism proposed for the hydrolysis of phosphoester bonds by Zr-MOFs. | ||
In addition to CWA and pesticides, bis(4-nitrophenyl)phosphate (BNPP) or para-4-nitrophenylphosphate (pNPP) have been extensively used as model substrates to explore phosphatase-like activity of MOFs, also because the reaction can be conveniently followed by UV-vis or 31P-NMR spectroscopy. Different groups have recently employed these substrates to investigate phosphatase biomimicry of Zr-, Ce-, and Hf-MOFs, exploring different aspects of this reactivity.
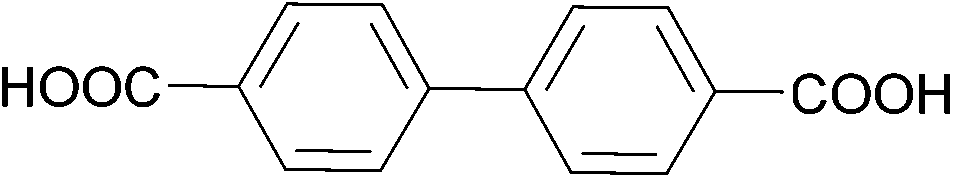
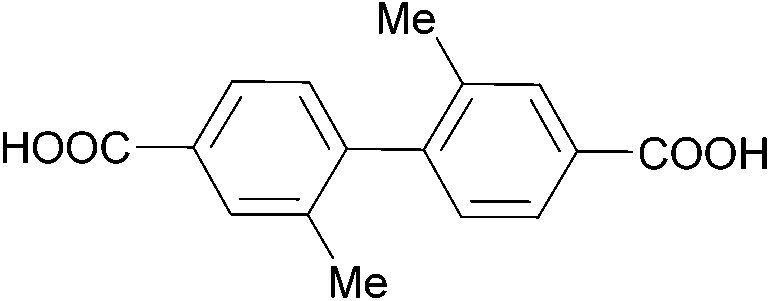
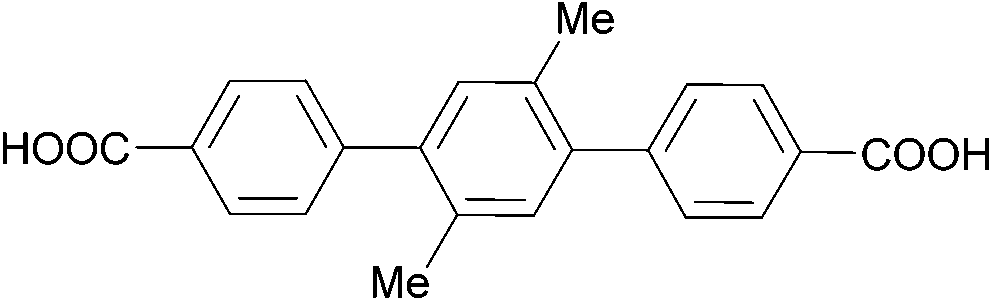
The effect of acidity was extensively evaluated for Zr-based MOFs. In 2019, Xu et al. have shown that MOFs with different topology and cluster connectivity, namely UiO-66 and UiO-67 with fcu topology, and PCN-700, PCN-701 and PCN-703 with bcu topology (Table 4), could hydrolyse pNPP in HEPES buffer solutions (100 mM, pH = 8) at 37 °C.105 The open Zr sites are suggested to be the responsible catalytically active sites, since a positive correlation was observed between the cluster connectivity and reaction rates. Using UiO-66 as a representative material, MOF stability under the reaction conditions and subsequent recyclability without significant changes were demonstrated, although adsorption of p-nitrophenol could be observed. Furthermore, different pH conditions were explored for the activation of Zr-UiO-66 by soaking UiO-66 in HEPES buffer solutions with pH values ranging from 2 to 8. This treatment did not affect the overall number of missing-linker defects, but by removing different amounts of monocarboxylates bound to the Zr6O8 cluster, it modified the number of open metal sites across the MOF series – in other words, it activated the MOF. Using the original reaction conditions (HEPES buffer 100 mM, pH = 8, 37 °C), a strong linear correlation between the catalytic activity and the number of open metal sites was again observed. However, a high activity was intringuingly observed for MOFs treated at both low and high pH values. Further evaluation of the influence of the reaction mixture acidity showed the rates for UiO-66, UiO-67, and PCN-700 peak at pH values around 8, and are largely affected by even small deviations from this pH. Using DFT calculations, which were consistent with ex situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis, authors suggest a low energy pathway becomes accessible when the Zr–O–Zr moiety is saturated with a hydroxyl group and a water molecule, but the oxygens bridging the Zr atoms atom remain deprotonated. This is an intriguing feature, as according to the DFT results the bridging oxygens do not directly participate in the reaction. In addition, the results of this study show that this feature can be modulated by controlling the pH when the reactions are conducted in aqueous solutions, which is an interesting alternative to the extreme heating approaches discussed in section 2.1. Moreover, similar effects might also be relevant for other types of biomolecules, but such detailed studies have yet to be reported.
Despite the similarity with model substrates, the first report on the MOF-assisted hydrolysis of adenosine triphosphate (ATP) was only reported in 2020, when Ce-UiO-66 was shown to dephosphorylate ATP.106 Ce-UiO-66 was able to hydrolyse ATP and ADP at 37 °C in HEPES buffer (pH 7.4, in D2O), generating adenosine diphosphate (ADP) at first, and adenosine monophosphate (AMP) as a final product. No pyrophosphate peak was detected, suggesting a sequential hydrolysis, and no dephosphorylation of AMP was reported. Unlike the enzyme apyrase, responsible for ATP hydrolysis, Ce-UiO-66 was able to almost instantly convert ATP in AMP, presumably due to the proximity of catalytic sites in the periodic MOF structure. This probably helped to abate the accumulation of ADP during the reaction progress. Furthermore, the presence of Ca2+ cations, essential for apyrase activity, had no significant influence on the performance of Ce-UiO-66. Although Ce-UiO-66 also functioned optimally around pH 7.4, its reactivity could be increased with temperature elevation up to 60 °C unlike the apyrase enzyme. More importantly, negligible activity towards ATP was observed under these conditions for MOFs based on others metals (Zr, Hf, Zn, Co, and Cr), which have been successfully used previously for the hydrolysis of phosphate bonds in model compounds. This showcases the discrepancy one may encounter between model substrates, and actual biomolecules. To study more the unique reactivity of cerium, X-ray photoelectron spectroscopy (XPS) was used to probe the MOF surface under the assumption the ATP molecules are too large to penetrate pores, and reaction must occur on the external surface. The study showed that the Ce6O8 clusters are composed of Ce(III) and Ce(IV) centers. However, control experiments strongly suggest only Ce(IV) is able to activate the phosphate group for hydrolysis, although the nearby Ce(III) center might still assist the reaction by enhancing the nucleophilicity of a water molecule coordinated to it.106
Later on, the same group showed the same reactivity could be used to hydrolyse phosphodiester linkages in various DNA molecules.107 An 88-mer single-stranded DNA (ssDNA) was fully cleaved after 6 h at pH 7.4 and 60 °C using Ce-UiO-66. It was possible to carry out the reaction under physiological conditions, but the yield increased with raising the temperature to 60 °C, suggesting an activation energy barrier for the reaction. The MOF remained catalytically active after pre-treatments with pH ranging from 3 to 9. Next, the authors were able to hydrolytically cleave double-stranded DNA (dsDNA), a supercoiled plasmid pBR322 consisting of 4361 base pairs, in 24 h.
Other Ce-MOFs have also been reported to hydrolyse phosphoester bonds. For example, Ce6O8-based Ce-FMA MOF featuring fumaric acid as a linker afforded a 12-connected MOF structure with the metal clusters arranged in cubic close packing.9 This MOF has been reported to hydrolyze a plethora of biological compounds: ATP, ADP, AMP, and β-glycerophosphate (β-GP) in neutral environment (HEPES buffer pH 7.0 and 7.5), though plasmid DNA was not cleaved, most likely due to steric hindrance. Finally, an example of Ce(III)-based MOF with a phosphatase-like activity has also been reported. Ce-TCPE MOF structure consist of tetrakis(4-carboxyphenyl)ethylene (TCPE) linkers connecting Ce(III) centers. However, the reactivity observed was much lower than the one observed for Ce(IV) centers lodged in Ce6O8 clusters.108
MOFs containing two types of metals in their structure have also been probed as phosphatase mimics, and are an interesting alternative to improve materials' versatility. Here, we highlight two recent examples, one leading to more robust structures, which could circumvent to some extent the common pitfall of low stability of MOFs in water and buffer solutions under biologically inspired reaction conditions,109–112 and another illustrating how a second metal imparts the material with an additional type of reactivity, and enables multienzymatic activity mimics.
Dong et al. synthesized two new Hf6O8-based MOFs using isonicotinic acid (or 3-aminoisonicotinic acid) as a linker (Hf–Ni and Hf–Ni–NH2, respectively).113 The pyridine ring on these linkers was coordinated to Ni2+ centers, assembling a network of [Hf48Ni6] cubic nanocages, whose catalytic sites were assumed to be the Hf–OH–Hf motifs. The Hf–Ni exhibited the best performance for the hydrolysis of pNPP in HEPES buffer at pH 7.4 and room temperature, while Hf UiO-66 was not active under these conditions. Furthermore, these MOFs selectively catalyzed hydrolysis of P–O bonds in the presence of substrates containing S–O or C–O bonds. Additionally, a ten times higher reaction rate was observed for the dephosphorylation of pNPP compared to BNPP, which was attributed to steric hindrance. This study contributes to the design of biomimetic materials with controllable selectivity and possibly therapeutic MOFs in the future.
Another example of a bimetallic MOF was FePCN MOF.114 This MOF contains, beside the typical zirconium oxo clusters nodes, iron porphyrin as the organic linker. FePCN was able to not only hydrolyze the ATP, but also exert oxidase and peroxidase activity towards the substrates 1,4-dihydropyridine and H2O2. These reactions were all carried out in HEPES buffer of pH 7.2, at different temperatures. To screen for oxidase and peroxidase activity, the reactions were set up for about 30 minutes at 37 °C, while for the phosphatase activity the temperature of 65 °C was used and the reaction was set up for an hour. The MOF could be recycled four times and still delivered high yields of 86%, 97% and 99% for oxidase-, peroxidase- and phosphatase-mimicking reactions, respectively. The catalytic active sites for oxidase and peroxidase-like activity was attributed to the iron located in the linkers of the MOF, while the phosphatase-like activity originated from the Zr nodes. This work is interesting for the development of nanomaterials with intrinsic multienzyme-mimicking activities.
Intrinsic glycosidase and lipase-like activity of MOFs are even more rarely studied in contrast to biomimicry of phosphatases. In the previously mentioned study,9 the cleavage of the glycosidic bond of two model substrates, 2-nitrophenyl-β-D-galactopyranoside and 4-nitrophenyl-N-acetyl-β-D-glucosamine, catalyzed by Ce-FMA MOF was investigated. The cleavage was successful, however when the same experiment with Ce-FMA was repeated with maltose and lactose to cleave the α-1,4 and β-1,4 glycosidic bonds respectively, no monosaccharides were detected. These results suggest that the reactions of the model substrates benefited from a good leaving group. Therefore, the polysaccharide carboxymethyl chitosan was chosen for another attempt in alkaline (pH 8.0) environments at 37 °C and 60 °C. This time, successful cleavage was obtained and the reusability of the Ce-FMA MOF was investigated, although its activity already decreased in the second and third usage cycles. Using the same Ce-FMA MOF, an attempt to hydrolyze the lipid ubiquitously present in living organisms cephalin failed. In this case, authors proposed that steric hindrance inhibited cleavage.
The reactivity discussed above intersects with a number of possible biological applications. For example, the application of MOFs as biosensors for ATP10,115–117 hydrolysis or other phosphatase activity118 is widely investigated for metabolic alteration therapy in cancer research.119,120 Further, MOFs have also been employed for the enrichment of phospho- and/or glycopeptides and glycopeptide profiling needed in proteomics.121–124 In the field of gene editing, MOFs have proven to be a carrier for genetic material, able to form Zr–O–P bonds and even to be of assistance in CRISPR technology.117,125–129 In other words, it can be concluded that more systematic research investigations are needed for the optimization of reactions with biomacromolecules that utilize the intrinsic catalytic activity of MOFs. Nevertheless, there is an enormous variety of possible applications for these reactions, which certainly underlines the potential of this research field.
5. Outlook
In summary, MOF catalysis for the transformation of biomolecules is a field of growing interest, and great potential. The recent work highlighted here shows that the catalytic activity of MOF materials towards biomolecules follows similar trends to those observed with more conventional organic substrates, and many of the tools developed for the design and synthesis of MOFs can definitely be used to tune the reactivity of MOF catalysts towards biomolecules. However, the charged and polar nature of biomolecules like peptides and proteins poses unique challenges to translate this reactivity into applied systems (e.g., the strong adsorption of substrates and products to the MOF). Moreover, biomacromolecules like proteins and nucleic acids solubility and stability are dependent on buffers, and the presence of salts, which may compromise the stability of several MOF structures.By far, the most attractive characteristic of MOF materials is the tunability of their structure. From there to practical applications, it will be crucial to identify structural features that enhance performance without compromising reactivity and selectivity. Therefore, advances in this area will likely require developing alternatives beyond the traditional tools of MOF chemistry such as controlling defects, pore engineering, etc. In addition, better understanding of the MOF surface chemistry in aqueous conditions will be essential to modulate substrate/product adsorption, and control its effects on the overall reactivity. In this context, the initial work discussed indicates many research directions could contribute for the rationale design of MOFs in this field. For example:
1. Different metals can be used as secondary building blocks, as many different metals have been shown catalytic competent in the cleavage of peptide bonds and proteins.130,131 Initial evidence on the promising reactivity of MOF based on Hf (ref. 40) oxo clusters indicates that other tetravalent metals also render MOFs stable enough for the hydrolysis of proteins under physiological-like conditions. Naturally, metals other than tetravalent ones can also be explored. One example is copper, which was featured in an early report of the protease-like activity of the Cu-BTC MOF (HKUST-1).36 Finally, bimetallic MOF structures combining two metal in the SBU,41 or in the MOF as a whole,113,114 may prove interesting to enhance and diversify MOF performance.
2. Improving the stability of materials under physiological-like conditions, which needs to consider the presence of buffers, and relatively high ionic strengths are to be addressed. Systematic studies of the stability of prototypic MOFs such as the UiO-66 (ref. 112 and 132) have been reported, but the effect of defects, MOF composition, and topology is largely unknown.
3. Devising alternatives to reverse the adsorption of substrates and products are also needed, especially if enhanced recyclability is envisioned.
Stability, tunability, intrinsic hydrolysis activity, recyclability and control over substrate interactions confirms the potential for MOFs to be used as artificial nanozymes for the hydrolysis of proteins and other biomolecules, providing a promising alternative to circumvent limitations of traditional enzymatic methods such as protein digestion with trypsin for use in middle-down proteomics. Clearly, major gaps still remain in the reaction selectivity, especially for more complex biomolecules. However, recent knowledge gained in the tuning of the structure of MOFs towards a certain reaction lights the way to enhance the control over MOF-protein interactions, and site-selective cleavage within protein sequences.34 Although efficient ways of achieving such control have yet to be identified, further structural insights will be key to continuing to expand the range of proteins and other biomolecules hydrolysable by MOFs. Together, these challenges also highlight the potential of MOFs as nanozymes. Once current limitations are addressed, MOF materials that are more suitable to deal with challenges uniquely associated with biomolecules can be rationally designed, quickly boosting the development of highly specialized MOF-based nanozymes, whose unique advantages would be key to develop disruptive technologies in biochemistry, and biomedical, analytical and environmental sciences, among others.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank KU Leuven and Research Foundation – Flanders (FWO) for funding. C. S. (68090/11C9320N), A. M. (1228622N), S. S. (1S61322N), F. d. A. (195931/1281921N) thank the FWO for fellowships.References
- E. Vandermarliere, M. Mueller and L. Martens, Getting intimate with trypsin, the leading protease in proteomics, Mass Spectrom. Rev., 2013, 32, 453–465, DOI:10.1002/mas.21376.
- N. C. Veitch, Horseradish peroxidase: a modern view of a classic enzyme, Phytochemistry, 2004, 65, 249–259, DOI:10.1016/j.phytochem.2003.10.022.
- L. Lauková, B. Konečná, Ľ. Janovičová, B. Vlková and P. Celec, Deoxyribonucleases and Their Applications in Biomedicine, Biomolecules, 2020, 10, 1036 CrossRef PubMed , https://www.mdpi.com/2218-<?pdb_no 273X?>273X<?pdb END?>/10/7/1036.
- K. M. Koeller and C.-H. Wong, Enzymes for chemical synthesis, Nature, 2001, 409, 232–240, DOI:10.1038/35051706.
- P. V. Iyer and L. Ananthanarayan, Enzyme stability and stabilization—Aqueous and non-aqueous environment, Process Biochem., 2008, 43, 1019–1032, DOI:10.1016/j.procbio.2008.06.004.
- H. Wei and E. Wang, Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes, Chem. Soc. Rev., 2013, 42, 6060–6093, 10.1039/C3CS35486E.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes (II), Chem. Soc. Rev., 2019, 48, 1004–1076, 10.1039/C8CS00457A.
- X. Huang, S. Zhang, Y. Tang, X. Zhang, Y. Bai and H. Pang, Advances in metal–organic framework-based nanozymes and their applications, Coord. Chem. Rev., 2021, 449, 214216 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0010854521004902.
- S. Li, Z. Zhou, Z. Tie, B. Wang, M. Ye, L. Du, R. Cui, W. Liu, C. Wan, Q. Liu, S. Zhao, Q. Wang, Y. Zhang, S. Zhang, H. Zhang, Y. Du and H. Wei, Data-informed discovery of hydrolytic nanozymes, Nat. Commun., 2022, 13, 827, DOI:10.1038/s41467-022-28344-2.
- L. Qin, X. Wang, Y. Liu and H. Wei, 2D-Metal–Organic-Framework-Nanozyme Sensor Arrays for Probing Phosphates and Their Enzymatic Hydrolysis, Anal. Chem., 2018, 90, 9983–9989, DOI:10.1021/acs.analchem.8b02428.
- M. Daniel, G. Mathew, M. Anpo and B. Neppolian, MOF based electrochemical sensors for the detection of physiologically relevant biomolecules: An overview, Coord. Chem. Rev., 2022, 468, 214627 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0010854522002223.
- R. Fang and J. Liu, Cleaving DNA by nanozymes, J. Mater. Chem. B, 2020, 8, 7135–7142, 10.1039/D0TB01274B.
- L. Gabrielli, L. J. Prins, F. Rastrelli, F. Mancin and P. Scrimin, Hydrolytic Nanozymes, Eur. J. Org. Chem., 2020, 2020, 5044–5055, DOI:10.1002/ejoc.202000356.
- M. Bilal, M. Adeel, T. Rasheed and H. M. N. Iqbal, Multifunctional metal–organic frameworks-based biocatalytic platforms: recent developments and future prospects, J. Mater. Res. Technol., 2019, 8, 2359–2371 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2238785418308093.
- J. Caro, Are MOF membranes better in gas separation than those made of zeolites?, Curr. Opin. Chem. Eng., 2011, 1, 77–83 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2211339811000128.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed , https://science.sciencemag.org/content/sci/341/6149/1230444.full.pdf.
- N. Rangnekar, N. Mittal, B. Elyassi, J. Caro and M. Tsapatsis, Zeolite membranes – a review and comparison with MOFs, Chem. Soc. Rev., 2015, 44, 7128–7154, 10.1039/C5CS00292C.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Metal–organic frameworks: versatile heterogeneous catalysts for efficient catalytic organic transformations, Chem. Soc. Rev., 2015, 44, 6804–6849, 10.1039/C4CS00395K.
- L. Feng, G. S. Day, K.-Y. Wang, S. Yuan and H.-C. Zhou, Strategies for Pore Engineering in Zirconium Metal-Organic Frameworks, Chem, 2020, 6, 2902–2923, DOI:10.1016/j.chempr.2020.09.010.
- L. Jiao, J. Wang and H.-L. Jiang, Microenvironment Modulation in Metal–Organic Framework-Based Catalysis, Acc. Mater. Res., 2021, 2, 327–339, DOI:10.1021/accountsmr.1c00009.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Metal–organic framework materials as catalysts, Chem. Soc. Rev., 2009, 38, 1450–1459, 10.1039/B807080F.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172, 10.1039/C8CS00256H.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120, 8468–8535, DOI:10.1021/acs.chemrev.9b00685.
- A. Ahmad, S. Khan, S. Tariq, R. Luque and F. Verpoort, Self-sacrifice MOFs for heterogeneous catalysis: Synthesis mechanisms and future perspectives, Mater. Today, 2022, 55, 137–169, DOI:10.1016/j.mattod.2022.04.002.
- D. Wu, P.-F. Zhang, G.-P. Yang, L. Hou, W.-Y. Zhang, Y.-F. Han, P. Liu and Y.-Y. Wang, Supramolecular control of MOF pore properties for the tailored guest adsorption/separation applications, Coord. Chem. Rev., 2021, 434, 213709 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0010854520308833.
- S. K. Firooz and D. W. Armstrong, Metal-organic frameworks in separations: A review, Anal. Chim. Acta, 2022, 340208, DOI:10.1016/j.aca.2022.340208.
- X. Wang, P. C. Lan and S. Ma, Metal–Organic Frameworks for Enzyme Immobilization: Beyond Host Matrix Materials, ACS Cent. Sci., 2020, 6, 1497–1506, DOI:10.1021/acscentsci.0c00687.
- I. Hussain, S. Iqbal, C. Lamiel, A. Alfantazi and K. Zhang, Recent advances in oriented metal–organic frameworks for supercapacitive energy storage, J. Mater. Chem. A, 2022, 10, 4475–4488, 10.1039/D1TA10213C.
- P. Kumar, A. Deep and K.-H. Kim, Metal organic frameworks for sensing applications, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0165993615001090.
- A. Khaleque, M. M. Alam, M. Hoque, S. Mondal, J. B. Haider, B. Xu, M. A. H. Johir, A. K. Karmakar, J. L. Zhou, M. B. Ahmed and M. A. Moni, Zeolite synthesis from low-cost materials and environmental applications: A review, Environ. Adv., 2020, 2, 100019 CrossRef , https://www.sciencedirect.com/science/article/pii/S2666765720300193.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Confinement in a Zeolite and Zeolite Catalysis, Acc. Chem. Res., 2021, 54, 2894–2904, DOI:10.1021/acs.accounts.1c00274.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Gościańska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. Diercks and S. Wuttke, The Current Status of MOF and COF Applications, Angew. Chem., Int. Ed., 2021, 60, 23975–24001, DOI:10.1002/anie.202106259.
- F. Haase and B. V. Lotsch, Solving the COF trilemma: towards crystalline, stable and functional covalent organic frameworks, Chem. Soc. Rev., 2020, 49, 8469–8500, 10.1039/D0CS01027H.
- F. de Azambuja, J. Moons and T. N. Parac-Vogt, The Dawn of Metal-Oxo Clusters as Artificial Proteases: From Discovery to the Present and Beyond, Acc. Chem. Res., 2021, 54, 1673–1684, DOI:10.1021/acs.accounts.0c00666.
- A. Radzicka and R. Wolfenden, Rates of Uncatalyzed Peptide Bond Hydrolysis in Neutral Solution and the Transition State Affinities of Proteases, J. Am. Chem. Soc., 1996, 118, 6105–6109, DOI:10.1021/ja954077c.
- B. Li, D. Chen, J. Wang, Z. Yan, L. Jiang, D. Deliang, J. He, Z. Luo, J. Zhang and F. Yuan, MOFzyme: Intrinsic protease-like activity of Cu-MOF, Sci. Rep., 2014, 4, 6759, DOI:10.1038/srep06759.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. De Vos and T. N. Parac-Vogt, Superactivity of MOF-808 toward Peptide Bond Hydrolysis, J. Am. Chem. Soc., 2018, 140, 6325–6335, DOI:10.1021/jacs.8b01902.
- H. G. T. Ly, G. Fu, F. de Azambuja, D. E. De Vos and T. N. Parac-Vogt, Nanozymatic Activity of UiO-66 Metal-Organic Frameworks: Tuning the Nanopore Environment Enhances Hydrolytic Activity toward Peptide Bonds, ACS Appl. Nano Mater., 2020, 3, 8931–8938, DOI:10.1021/acsanm.0c01688.
- A. Loosen, F. de Azambuja, S. Smolders, J. Moons, C. Simms, D. De Vos and T. N. Parac-Vogt, Interplay between structural parameters and reactivity of Zr6-based MOFs as artificial proteases, Chem. Sci., 2020, 11, 6662–6669, 10.1039/D0SC02136A.
- J. Moons, A. Loosen, C. Simms, F. de Azambuja and T. Parac-Vogt, Heterogeneous nanozymatic activity of Hf oxo-clusters embedded in a metal-organic framework towards peptide bond hydrolysis, Nanoscale, 2021, 13, 12298–12305, 10.1039/D1NR01790J.
- A. Loosen, C. Simms, S. Smolders, D. E. De Vos and T. N. Parac-Vogt, Bimetallic Ce/Zr UiO-66 Metal–Organic Framework Nanostructures as Peptidase and Oxidase Nanozymes, ACS Appl. Nano Mater., 2021, 4, 5748–5757 CrossRef CAS , https://pubs.acs.org/doi/abs/10.1021/acsanm.1c00546.
- S. Wang, H. G. T. Ly, M. Wahiduzzaman, C. Simms, I. Dovgaliuk, A. Tissot, G. Maurin, T. N. Parac-Vogt and C. Serre, A zirconium metal-organic framework with SOC topological net for catalytic peptide bond hydrolysis, Nat. Commun., 2022, 13, 1284, DOI:10.1038/s41467-022-28886-5.
- S. Dai, C. Simms, I. Dovgaliuk, G. Patriarche, A. Tissot, T. N. Parac-Vogt and C. Serre, Monodispersed MOF-808 Nanocrystals Synthesized via a Scalable Room-Temperature Approach for Efficient Heterogeneous Peptide Bond Hydrolysis, Chem. Mater., 2021, 33, 7057–7066, DOI:10.1021/acs.chemmater.1c02174.
- J. Moons, F. de Azambuja, J. Mihailovic, K. Kozma, K. Smiljanic, M. Amiri, T. Cirkovic Velickovic, M. Nyman and T. N. Parac-Vogt, Discrete Hf18 Metal-oxo Cluster as a Heterogeneous Nanozyme for Site-Specific Proteolysis, Angew. Chem., Int. Ed., 2020, 59, 9094–9101, DOI:10.1002/anie.202001036.
- D. Conic, K. Pierloot, T. N. Parac-Vogt and J. N. Harvey, Mechanism of the highly effective peptide bond hydrolysis by MOF-808 catalyst under biologically relevant conditions, Phys. Chem. Chem. Phys., 2020, 22, 25136–25145, 10.1039/D0CP04775A.
- F. de Azambuja, A. Loosen, D. Conic, M. van den Besselaar, J. N. Harvey and T. N. Parac-Vogt, En Route to a Heterogeneous Catalytic Direct Peptide Bond Formation by Zr-Based Metal–Organic Framework Catalysts, ACS Catal., 2021, 11, 7647–7658, DOI:10.1021/acscatal.1c01782.
- I. Abánades Lázaro, R. S. Forgan and F. G. Cirujano, MOF nanoparticles as heterogeneous catalysts for direct amide bond formations, Dalton Trans., 2022, 51, 8368–8376, 10.1039/D2DT00369D.
- H. G. T. Ly, T. T. Mihaylov, P. Proost, K. Pierloot, J. N. Harvey and T. N. Parac-Vogt, Chemical Mimics of Aspartate-Directed Proteases: Predictive and Strictly Specific Hydrolysis of a Globular Protein at Asp−X Sequence Promoted by Polyoxometalate Complexes Rationalized by a Combined Experimental and Theoretical Approach, Chem. – Eur. J., 2019, 25, 14370–14381, DOI:10.1002/chem.201902675.
- T. T. Mihaylov, H. G. T. Ly, K. Pierloot and T. N. Parac-Vogt, Molecular Insight from DFT Computations and Kinetic Measurements into the Steric Factors Influencing Peptide Bond Hydrolysis Catalyzed by a Dimeric Zr(IV)-Substituted Keggin Type Polyoxometalate, Inorg. Chem., 2016, 55, 9316–9328, DOI:10.1021/acs.inorgchem.6b01461.
- A. Loosen, F. de Azambuja and T. N. Parac-Vogt, Which factors govern the adsorption of peptides to Zr(IV)-based metal-organic frameworks?, Mater. Adv., 2022, 3, 2475–2487, 10.1039/D1MA01027A.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Metal–Organic Framework-Based Enzyme Biocomposites, Chem. Rev., 2021, 121, 1077–1129, DOI:10.1021/acs.chemrev.0c01029.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Defect-Engineered Metal–Organic Frameworks, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed , https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201411540.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130, 13850–13851, DOI:10.1021/ja8057953.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory, Chem. Mater., 2011, 23, 1700–1718, DOI:10.1021/cm1022882.
- C. Hennig, S. Weiss, W. Kraus, J. Kretzschmar and A. C. Scheinost, Solution Species and Crystal Structure of Zr(IV) Acetate, Inorg. Chem., 2017, 56, 2473–2480, DOI:10.1021/acs.inorgchem.6b01624.
- Z. Hu, S. Faucher, Y. Zhuo, Y. Sun, S. Wang and D. Zhao, Combination of Optimization and Metalated-Ligand Exchange: An Effective Approach to Functionalize UiO-66(Zr) MOFs for CO2 Separation, Chem. – Eur. J., 2015, 21, 17246–17255, DOI:10.1002/chem.201503078.
- M. Taddei, R. J. Wakeham, A. Koutsianos, E. Andreoli and A. R. Barron, Post-Synthetic Ligand Exchange in Zirconium-Based Metal–Organic Frameworks: Beware of The Defects!, Angew. Chem., Int. Ed., 2018, 57, 11706–11710, DOI:10.1002/anie.201806910.
- X. Zhao, Z. Zhang, X. Cai, B. Ding, C. Sun, G. Liu, C. Hu, S. Shao and M. Pang, Postsynthetic Ligand Exchange of Metal–Organic Framework for Photodynamic Therapy, ACS Appl. Mater. Interfaces, 2019, 11, 7884–7892, DOI:10.1021/acsami.9b00740.
- G. Fu, B. Bueken and D. De Vos, Zr-Metal-Organic Framework Catalysts for Oxidative Desulfurization and Their Improvement by Postsynthetic Ligand Exchange, Small Methods, 2018, 2, 1800203, DOI:10.1002/smtd.201800203.
- C. Hon Lau, R. Babarao and M. R. Hill, A route to drastic increase of CO2 uptake in Zr metal organic framework UiO-66, Chem. Commun., 2013, 49, 3634–3636, 10.1039/C3CC40470F.
- H. Huang, Y. Niu, Z. Zhao, H. Hei and D. Zhu, On the enigma of Nb-Ta and Zr-Hf fractionation—A critical review, J. Earth Sci., 2011, 22, 52–66, DOI:10.1007/s12583-011-0157-x.
- Y. Niu, Earth processes cause Zr–Hf and Nb–Ta fractionations, but why and how?, RSC Adv., 2012, 2, 3587–3591, 10.1039/C2RA00384H.
- S. Waitschat, D. Fröhlich, H. Reinsch, H. Terraschke, K. A. Lomachenko, C. Lamberti, H. Kummer, T. Helling, M. Baumgartner, S. Henninger and N. Stock, Synthesis of M-UiO-66 (M = Zr, Ce or Hf) employing 2,5-pyridinedicarboxylic acid as a linker: defect chemistry, framework hydrophilisation and sorption properties, Dalton Trans., 2018, 47, 1062–1070, 10.1039/C7DT03641H.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, A Hafnium-Based Metal–Organic Framework as an Efficient and Multifunctional Catalyst for Facile CO2 Fixation and Regioselective and Enantioretentive Epoxide Activation, J. Am. Chem. Soc., 2014, 136, 15861–15864, DOI:10.1021/ja508626n.
- P. García-García and A. Corma, Hf-based Metal-Organic Frameworks in Heterogeneous Catalysis, Isr. J. Chem., 2018, 58, 1062–1074, DOI:10.1002/ijch.201800099.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767, DOI:10.1107/S0567739476001551.
- K. A. Lomachenko, J. Jacobsen, A. L. Bugaev, C. Atzori, F. Bonino, S. Bordiga, N. Stock and C. Lamberti, Exact Stoichiometry of CexZr6–x Cornerstones in Mixed-Metal UiO-66 Metal–Organic Frameworks Revealed by Extended X-ray Absorption Fine Structure Spectroscopy, J. Am. Chem. Soc., 2018, 140, 17379–17383, DOI:10.1021/jacs.8b10343.
- H. Shigekawa, M. Ishida, K. Miyake, R. Shioda, Y. Iijima, T. Imai, H. Takahashi, J. Sumaoka and M. Komiyama, Extended x-ray absorption fine structure study on the cerium(IV)-induced DNA hydrolysis: Implication to the roles of 4f orbitals in the catalysis, Appl. Phys. Lett., 1999, 74, 460–462 CrossRef CAS , https://aip.scitation.org/doi/abs/10.1063/1.123036.
- T. Islamoglu, A. Atilgan, S.-Y. Moon, G. W. Peterson, J. B. DeCoste, M. Hall, J. T. Hupp and O. K. Farha, Cerium(IV) vs Zirconium(IV) Based Metal–Organic Frameworks for Detoxification of a Nerve Agent, Chem. Mater., 2017, 29, 2672–2675, DOI:10.1021/acs.chemmater.6b04835.
- G. Kickelbick, M. P. Feth, H. Bertagnolli, B. Moraru, G. Trimmel and U. Schubert, EXAFS Investigations on Nanocomposites Composed of Surface-Modified Zirconium and Zirconium/Titanium Mixed Metal Oxo Clusters and Organic Polymers, Monatsh. Chem., 2002, 133, 919–929, DOI:10.1007/s007060200062.
- G. Férey, Hybrid porous solids: past, present, future, Chem. Soc. Rev., 2008, 37, 191–214, 10.1039/B618320B.
- S. Ali Akbar Razavi and A. Morsali, Linker functionalized metal-organic frameworks, Coord. Chem. Rev., 2019, 399, 213023, DOI:10.1016/j.ccr.2019.213023.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Room-Temperature Synthesis of UiO-66 and Thermal Modulation of Densities of Defect Sites, Chem. Mater., 2017, 29, 1357–1361, DOI:10.1021/acs.chemmater.6b05115.
- K. Epp, A. L. Semrau, M. Cokoja and R. A. Fischer, Dual Site Lewis-Acid Metal-Organic Framework Catalysts for CO2 Fixation: Counteracting Effects of Node Connectivity, Defects and Linker Metalation, ChemCatChem, 2018, 10, 3506–3512, DOI:10.1002/cctc.201800336.
- A. H. Valekar, M. Lee, J. W. Yoon, J. Kwak, D.-Y. Hong, K.-R. Oh, G.-Y. Cha, Y.-U. Kwon, J. Jung, J.-S. Chang and Y. K. Hwang, Catalytic Transfer Hydrogenation of Furfural to Furfuryl Alcohol under Mild Conditions over Zr-MOFs: Exploring the Role of Metal Node Coordination and Modification, ACS Catal., 2020, 10, 3720–3732, DOI:10.1021/acscatal.9b05085.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Scalable synthesis and post-modification of a mesoporous metal-organic framework called NU-1000, Nat. Protoc., 2016, 11, 149–162, DOI:10.1038/nprot.2016.001.
- Y. Liang, Z. Li, R. Fu and D. Wu, Nanoporous carbons with a 3D nanonetwork-interconnected 2D ordered mesoporous structure for rapid mass transport, J. Mater. Chem. A, 2013, 1, 3768–3773, 10.1039/C3TA01307C.
- R. Hardian, S. Dissegna, A. Ullrich, P. L. Llewellyn, M.-V. Coulet and R. A. Fischer, Tuning the Properties of MOF-808 via Defect Engineering and Metal Nanoparticle Encapsulation, Chem. – Eur. J., 2021, 27, 6804–6814, DOI:10.1002/chem.202005050.
- X. Feng, H. S. Jena, C. Krishnaraj, K. Leus, G. Wang, H. Chen, C. Jia and P. Van Der Voort, Generating Catalytic Sites in UiO-66 through Defect Engineering, ACS Appl. Mater. Interfaces, 2021, 13, 60715–60735, DOI:10.1021/acsami.1c13525.
- M. B. Majewski, H. Noh, T. Islamoglu and O. K. Farha, NanoMOFs: little crystallites for substantial applications, J. Mater. Chem. A, 2018, 6, 7338–7350, 10.1039/C8TA02132E.
- Y. Kim and S. Huh, Pore engineering of metal–organic frameworks: introduction of chemically accessible Lewis basic sites inside MOF channels, CrystEngComm, 2016, 18, 3524–3550, 10.1039/C6CE00612D.
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Mixed-metal metal–organic frameworks, Chem. Soc. Rev., 2019, 48, 2535–2565, 10.1039/C8CS00337H.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Mixed-Metal MOFs: Unique Opportunities in Metal–Organic Framework (MOF) Functionality and Design, Angew. Chem., Int. Ed., 2019, 58, 15188–15205, DOI:10.1002/anie.201902229.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, Synthesis Modulation as a Tool To Increase the Catalytic Activity of Metal–Organic Frameworks: The Unique Case of UiO-66(Zr), J. Am. Chem. Soc., 2013, 135, 11465–11468, DOI:10.1021/ja405078u.
- C. Simms, F. de Azambuja and T. N. Parac-Vogt, Enhancing the Catalytic Activity of MOF-808 Towards Peptide Bond Hydrolysis through Synthetic Modulations, Chem. – Eur. J., 2021, 27, 17230–17239, DOI:10.1002/chem.202103102.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Electronic Effects of Linker Substitution on Lewis Acid Catalysis with Metal–Organic Frameworks, Angew. Chem., Int. Ed., 2012, 51, 4887–4890, DOI:10.1002/anie.201108565.
- S. Li, W. Wang, S. Lei and J.-Z. Cui, Boosting Catalytic Efficiency of Metal-Organic Frameworks with Electron-Withdrawing Effect for Lewis-Acid Catalysis, ChemistrySelect, 2021, 6, 7732–7735, DOI:10.1002/slct.202101471.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Tuning the Adsorption Properties of UiO-66 via Ligand Functionalization, Langmuir, 2012, 28, 15606–15613, DOI:10.1021/la3035352.
- G. Fu, F. G. Cirujano, A. Krajnc, G. Mali, M. Henrion, S. Smolders and D. E. De Vos, Unexpected linker-dependent Brønsted acidity in the (Zr)UiO-66 metal organic framework and application to biomass valorization, Catal. Sci. Technol., 2020, 10, 4002–4009, 10.1039/D0CY00638F.
- T. Islamoglu, M. A. Ortuño, E. Proussaloglou, A. J. Howarth, N. A. Vermeulen, A. Atilgan, A. M. Asiri, C. J. Cramer and O. K. Farha, Presence versus Proximity: The Role of Pendant Amines in the Catalytic Hydrolysis of a Nerve Agent Simulant, Angew. Chem., Int. Ed., 2018, 57, 1949–1953, DOI:10.1002/anie.201712645.
- J. Hajek, M. Vandichel, B. Van de Voorde, B. Bueken, D. De Vos, M. Waroquier and V. Van Speybroeck, Mechanistic studies of aldol condensations in UiO-66 and UiO-66-NH2 metal organic frameworks, J. Catal., 2015, 331, 1–12, DOI:10.1016/j.jcat.2015.08.015.
- C. Simms, N. Savić, K. De Winter and T. N. Parac-Vogt, Understanding the role of surfactants in the interaction and hydrolysis of myoglobin by Zr-MOF-808, Eur. J. Inorg. Chem., 2022, e202200145, DOI:10.1002/ejic.202200145.
- M. Lammert, C. Glißmann, H. Reinsch and N. Stock, Synthesis and Characterization of New Ce(IV)-MOFs Exhibiting Various Framework Topologies, Cryst. Growth Des., 2017, 17, 1125–1131, DOI:10.1021/acs.cgd.6b01512.
- M. Lammert, C. Glißmann and N. Stock, Tuning the stability of bimetallic Ce(iv)/Zr(iv)-based MOFs with UiO-66 and MOF-808 structures, Dalton Trans., 2017, 46, 2425–2429, 10.1039/C7DT00259A.
- T. Takarada, M. Yashiro and M. Komiyama, Catalytic Hydrolysis of Peptides by Cerium(IV), Chem. – Eur. J., 2000, 6, 3906–3913, DOI:10.1002/1521-3765(20001103)6:21<3906::AID-CHEM3906>3.0.CO;2-J.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Heterogenisation of polyoxometalates and other metal-based complexes in metal–organic frameworks: from synthesis to characterisation and applications in catalysis, Chem. Soc. Rev., 2021, 50, 6152–6220, 10.1039/D0CS00323A.
- P. Ji, T.-Y. Wang, G.-F. Luo, W.-H. Chen and X.-Z. Zhang, A tumor-cell biomimetic nanoplatform embedding biological enzymes for enhanced metabolic therapy, Chem. Commun., 2021, 57, 9398–9401, 10.1039/D1CC03494D.
- L. Wang, W. Zhi, J. Wan, J. Han, C. Li and Y. Wang, Recyclable β-Glucosidase by One-Pot Encapsulation with Cu-MOFs for Enhanced Hydrolysis of Cellulose to Glucose, ACS Sustainable Chem. Eng., 2019, 7, 3339–3348, DOI:10.1021/acssuschemeng.8b05489.
- Q. Han, L. Zhang, C. He, J. Niu and C. Duan, Metal–Organic Frameworks with Phosphotungstate Incorporated for Hydrolytic Cleavage of a DNA-Model Phosphodiester, Inorg. Chem., 2012, 51, 5118–5127, DOI:10.1021/ic202685e.
- W. Xie, X. Yang and P. Hu, Cs2.5H0.5PW12O40 Encapsulated in Metal–Organic Framework UiO-66 as Heterogeneous Catalysts for Acidolysis of Soybean Oil, Catal. Lett., 2017, 147, 2772–2782, DOI:10.1007/s10562-017-2189-z.
- J. Han, Y. Wang, J. Wan and Y. Ma, Catalytic hydrolysis of cellulose by phosphotungstic acid–supported functionalized metal-organic frameworks with different electronegative groups, Environ. Sci. Pollut. Res., 2019, 26, 15345–15353, DOI:10.1007/s11356-019-04923-7.
- T. G. Grissom, A. M. Plonka, C. H. Sharp, A. M. Ebrahim, Y. Tian, D. L. Collins-Wildman, A. L. Kaledin, H. J. Siegal, D. Troya, C. L. Hill, A. I. Frenkel, D. G. Musaev, W. O. Gordon, C. J. Karwacki, M. B. Mitchell and J. R. Morris, Metal–Organic Framework- and Polyoxometalate-Based Sorbents for the Uptake and Destruction of Chemical Warfare Agents, ACS Appl. Mater. Interfaces, 2020, 12, 14641–14661, DOI:10.1021/acsami.9b20833.
- K. O. Kirlikovali, Z. Chen, T. Islamoglu, J. T. Hupp and O. K. Farha, Zirconium-Based Metal–Organic Frameworks for the Catalytic Hydrolysis of Organophosphorus Nerve Agents, ACS Appl. Mater. Interfaces, 2020, 12, 14702–14720, DOI:10.1021/acsami.9b20154.
- S. Rojas, A. Rodríguez-Diéguez and P. Horcajada, Metal–Organic Frameworks in Agriculture, ACS Appl. Mater. Interfaces, 2022, 14, 16983–17007, DOI:10.1021/acsami.2c00615.
- M. Xu, L. Feng, L.-N. Yan, S.-S. Meng, S. Yuan, M.-J. He, H. Liang, X.-Y. Chen, H.-Y. Wei, Z.-Y. Gu and H.-C. Zhou, Discovery of precise pH-controlled biomimetic catalysts: defective zirconium metal–organic frameworks as alkaline phosphatase mimics, Nanoscale, 2019, 11, 11270–11278, 10.1039/C9NR02962A.
- J. Yang, K. Li, C. Li and J. Gu, Intrinsic Apyrase-Like Activity of Cerium-Based Metal–Organic Frameworks (MOFs): Dephosphorylation of Adenosine Tri- and Diphosphate, Angew. Chem., Int. Ed., 2020, 59, 22952–22956, DOI:10.1002/anie.202008259.
- J. Yang, K. Li and J. Gu, Hierarchically Macro-Microporous Ce-Based MOFs for the Cleavage of DNA, ACS Mater. Lett., 2022, 4, 385–391, DOI:10.1021/acsmaterialslett.1c00797.
- Y. Dou, L. Yang, L. Qin, Y. Dong, Z. Zhou and D. Zhang, Efficient hydrolytic cleavage of phosphodiester with a lanthanide-based metal-organic framework, J. Solid State Chem., 2021, 293, 121820 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0022459620306514.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Metal–organic frameworks as heterogeneous catalysts for oxidation reactions, Catal. Sci. Technol., 2011, 1, 856–867, 10.1039/C1CY00068C.
- N. C. Burtch, H. Jasuja and K. S. Walton, Water Stability and Adsorption in Metal–Organic Frameworks, Chem. Rev., 2014, 114, 10575–10612, DOI:10.1021/cr5002589.
- H. Bunzen, Chemical Stability of Metal-organic Frameworks for Applications in Drug Delivery, ChemNanoMat, 2021, 7, 998–1007, DOI:10.1002/cnma.202100226.
- D. Bůžek, S. Adamec, K. Lang and J. Demel, Metal–organic frameworks vs. buffers: case study of UiO-66 stability, Inorg. Chem. Front., 2021, 8, 720–734, 10.1039/D0QI00973C.
- J. Dong, H.-D. An, Z.-K. Yue, S.-L. Hou, Y. Chen, Z.-J. Zhang, P. Cheng, Q. Peng and B. Zhao, Dual-Selective Catalysis in Dephosphorylation Tuned by Hf6-Containing Metal–Organic Frameworks Mimicking Phosphatase, ACS Cent. Sci., 2021, 7, 831–840, DOI:10.1021/acscentsci.0c01581.
- C. Yang, Z. Jiang, Q. Wu, C. Hu, C. Huang, Y. Li and S. Zhen, One-component nano-metal-organic frameworks with superior multienzyme-mimic activities for 1,4-dihydropyridine metabolism, J. Colloid Interface Sci., 2022, 605, 214–222 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0021979721011693.
- X. Zhao, J. Fang, Y. Li and C. Huang, Highly selective recognition of adenosine 5′-triphosphate against other nucleosides triphosphate with a luminescent metal-organic framework of [Zn(BDC)(H2O)2]n(BDC = 1,4-benzenedicarboxylate), Sci. China: Chem., 2013, 56, 1651–1657, DOI:10.1007/s11426-013-4905-x.
- X.-M. Hai, N. Li, K. Wang, Z.-Q. Zhang, J. Zhang and F.-Q. Dang, A fluorescence aptasensor based on two-dimensional sheet metal-organic frameworks for monitoring adenosine triphosphate, Anal. Chim. Acta, 2018, 998, 60–66 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S000326701731187X.
- K. Yu, T. Wei, Z. Li, J. Li, Z. Wang and Z. Dai, Construction of Molecular Sensing and Logic Systems Based on Site-Occupying Effect-Modulated MOF–DNA Interaction, J. Am. Chem. Soc., 2020, 142, 21267–21271, DOI:10.1021/jacs.0c10442.
- X.-Y. Yao, Q. Wang, Q. Liu, M. Pang, X.-M. Du, B. Zhao, Y. Li and W.-J. Ruan, Ultrasensitive Assay of Alkaline Phosphatase Based on the Fluorescent Response Difference of the Metal–Organic Framework Sensor, ACS Omega, 2020, 5, 712–717, DOI:10.1021/acsomega.9b03337.
- H.-S. Wang, Metal–organic frameworks for biosensing and bioimaging applications, Coord. Chem. Rev., 2017, 349, 139–155 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S001085451730348X.
- W. J. Moon and J. Liu, Interfacing Catalytic DNA with Nanomaterials, Adv. Mater. Interfaces, 2020, 7, 2001017, DOI:10.1002/admi.202001017.
- J. Chen, K. Li, J. Yang and J. Gu, Bimetallic Ordered Large-Pore MesoMOFs for Simultaneous Enrichment and Dephosphorylation of Phosphopeptides, ACS Appl. Mater. Interfaces, 2021, 13, 60173–60181, DOI:10.1021/acsami.1c18201.
- H. Xu, M. Liu, X. Huang, Q. Min and J.-J. Zhu, Multiplexed Quantitative MALDI MS Approach for Assessing Activity and Inhibition of Protein Kinases Based on Postenrichment Dephosphorylation of Phosphopeptides by Metal–Organic Framework-Templated Porous CeO2, Anal. Chem., 2018, 90, 9859–9867, DOI:10.1021/acs.analchem.8b01938.
- M. Zhao, T. Chen and C. Deng, Porous anatase TiO2 derived from a titanium metal–organic framework as a multifunctional phospho-oriented nanoreactor integrating accelerated digestion of proteins and in situ enrichment, RSC Adv., 2016, 6, 51670–51674, 10.1039/C6RA03837A.
- Y. Ji, Z. Xiong, G. Huang, J. Liu, Z. Zhang, Z. Liu, J. Ou, M. Ye and H. Zou, Efficient enrichment of glycopeptides using metal–organic frameworks by hydrophilic interaction chromatography, Analyst, 2014, 139, 4987–4993, 10.1039/C4AN00971A.
- Y. Wang, Z. Liu, H. Zhang, J. Liu, H. Dai, T. Ji, F. Liu, P. Cao, J. Zou, S. Wang, L. Wang and Z. Wang, MOF effectively deliver CRISPR and enhance gene-editing efficiency via MOF's hydrolytic activity of phosphate ester bonds, Chem. Eng. J., 2022, 439, 134992 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S1385894722004983.
- S. Chand, O. Alahmed, W. S. Baslyman, A. Dey, S. Qutub, R. Saha, Y. Hijikata, M. Alaamery and N. M. Khashab, DNA-Mimicking Metal–Organic Frameworks with Accessible Adenine Faces for Complementary Base Pairing, JACS Au, 2022, 2, 623–630, DOI:10.1021/jacsau.1c00516.
- J. Peng, Y. Hu, H. Zhang, L. Wan, L. Wang, Z. Liang, L. Zhang and R. A. Wu, High Anti-Interfering Profiling of Endogenous Glycopeptides for Human Plasma by the Dual-Hydrophilic Metal–Organic Framework, Anal. Chem., 2019, 91, 4852–4859, DOI:10.1021/acs.analchem.9b00542.
- C. Pu, H. Zhao, Y. Hong, Q. Zhan and M. Lan, Facile Preparation of Hydrophilic Mesoporous Metal–Organic Framework via Synergistic Etching and Surface Functionalization for Glycopeptides Analysis, Anal. Chem., 2020, 92, 1940–1947, DOI:10.1021/acs.analchem.9b04236.
- Q. Liu, C.-H. Deng and N. Sun, Hydrophilic tripeptide-functionalized magnetic metal–organic frameworks for the highly efficient enrichment of N-linked glycopeptides, Nanoscale, 2018, 10, 12149–12155, 10.1039/C8NR03174F.
- N. E. Wezynfeld, T. Frączyk and W. Bal, Metal assisted peptide bond hydrolysis: Chemistry, biotechnology and toxicological implications, Coord. Chem. Rev., 2016, 327–328, 166–187 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0010854516300169.
- B. G. Kathryn and K. Miki, Major Advances in the Hydrolysis of Peptides and Proteins by Metal Ions and Complexes, Curr. Org. Chem., 2006, 10, 1035–1049 CrossRef , https://www.eurekaselect.com/node/56182/article.
- Y. Wang, J. Yin, W. Cao, Y. Fu and X. Kong, The instability of a stable metal-organic framework in amino acid solutions, Nano Res., 2022, 15, 6607–6612, DOI:10.1007/s12274-022-4346-y.
Footnote |
| † Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |