
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePore size and electronic tuning in cerium-doped CoFe-LDH for the oxygen evolution reaction†
Parul
Aggarwal
 ,
Bhupendra
Singh
and
Amit
Paul
*
,
Bhupendra
Singh
and
Amit
Paul
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhopal, M.P–462066, India. E-mail: apaul@iiserb.ac.in; Tel: +91-755-2691343
First published on 8th August 2023
Abstract
A series of cerium-doped CoFe-layered double hydroxide (LDH) materials were synthesized using a co-precipitation method, and they were utilized for the oxygen evolution reaction. Among all the materials, CoFeCe2, having the highest cerium doping, exhibited a remarkable mass activity of 294.15 A g−1, turnover frequency (TOF) of 1.82 s−1, and TOFEIS of 126.76 s−1, and these activity parameters were 6.5–8 times higher than those of undoped CoFe-LDH (CoFeCe0). Electrochemically active surface area (ECSA) measurements suggested a massive increase in roughness factor (21 times) for CoFeCe2 compared to undoped CoFeCe0. BET results revealed that cerium doping resulted in mostly a narrow mesoporous distribution, which helped to increase the surface area and pore volume. Employing the mechanism of LDH formation reported earlier, we proposed that the cerium doping minimized the interparticle electrostatic repulsion during LDH synthesis and hence, an ordered LDH nanosheet with shallow pore distribution (mostly narrow mesopores) was observed for CoFeCe2, while the pore distribution was very wide for undoped LDH (CoFeCe0) due to the formation of disordered LDH nanosheets. XPS results provided evidence that the cerium doping resulted in electron transfer from Co2+ to Ce4+ to generate more Co3+ in the synthesized LDH material, which acted as active sites for water oxidation and helped to enhance water oxidation. The high ionic radius of the cerium ion also resulted in lattice defects that decreased the material's crystallinity and increased the catalytic reactivity by providing more active sites. Furthermore, during water oxidation, the unsymmetrical electron occupancy in the t2g orbitals of Co2+ leads to a dynamic Jahn–Teller distortion, which results in the lengthening of the Co–O bonds, and further facilitates the formation of O–O bonds during the OER process.
Introduction
Since the 19th century, non-renewable energy resources have been utilized as the main energy supply to support human activities.1 However, the massive consumption of fossil fuels caused by the rapidly increasing world population has resulted in an increasing energy crisis and environmental pollution. These concomitant environmental problems have motivated scientists to explore green and sustainable energy systems to reduce carbon footprints for long-term development.2–7 In this regard, electrochemical water splitting is a promising and fascinating solution for the renewable energy conversion of water to clean fuel such as hydrogen.8,9 The water splitting reaction consists of two half-cell reactions: the cathodic hydrogen evolution reaction, HER: 2H+ + 2e− → H2 and the anodic oxygen evolution reaction, OER: 2H2O → O2 + 4H+ + 4e−.10 Among the two reactions, the OER is the bottleneck in water splitting since it involves a 4e−/4H+ proton-coupled electron transfer (PCET) reaction and is, therefore, kinetically sluggish in nature.11 The thermodynamic potential for the OER is 1.23 V vs. normal hydrogen electrode (NHE);12 however, the reaction requires a significantly higher potential due to a large kinetic barrier, and this additional potential is known as the overpotential. Thus, one of the major hotspots of research is the reduction of this kinetic barrier for the OER by interposing highly efficient catalytic materials.13 In the past, Ru/Ir-oxides have been widely regarded as excellent electrocatalysts for the OER. However, low abundance and high cost have hindered their commercial utilization.14–16 Thus, first-row transition metals, particularly cobalt, nickel, and iron-based oxide materials, have drawn attention as electrocatalysts for the OER due to their low cost, reliability, high activity, and long-term stability in highly alkaline solutions.17–30In this regard, layered double hydroxide (LDH) has drawn attention due to its exposed active sites, which facilitates electron transport properties towards electrocatalysis.31,32 LDH is a class of hydrotalcite-like inorganic layered material made up of positively charged brucite-like host layers, and charge-balancing anions are incorporated inside the interlayer spaces.33,34 The basic structure of the LDH can be described by the general formula [M1−xII·MxIII·(OH)2]x+·Ax/nn−·yH2O, wherein MII and MIII, and An− are divalent metal ions, trivalent metal ions, and guest anions, respectively.35 LDH materials have wide application in the areas of catalysis,31 fluorescence and luminescence processes,35,36 separation processes,37 drug delivery, etc.38 Among them, the LDHs that can promote the OER include nickel- and cobalt-based LDHs, such as NiFe-LDH,39,40 NiAl-LDH,41 CoAl-LDH,42,43 CoMn-LDH,44–46 CoFe-LDH,47–49 and so on. However, the pore sizes of LDHs are usually broad (ranging from 1–80 nm), which limits the number of active sites for catalysis and thus decreases the OER.50–54 For nanomaterial applications in catalysis or charge storage, pore size plays an important role since narrow pore size can significantly increase the number of active sites.9 For example, Gogotsi and co-workers highlighted that for supercapacitor applications, the presence of ultramicropores (<1 nm) in nanomaterials results in a massive increase of charge storage capacity due to vast enhancement of electrolyte accessible surface area.55,56 However, for electrocatalysis, it can be envisioned that a somewhat wider pore size will be ideal since product accumulation inside micropores may result in pore bursting. Indeed, for the oxygen reduction reaction (ORR), a narrow mesoporous diameter of 3.2 nm was found to be most suitable.57 Unfortunately, investigations on the role of pore size or control of pore size for LDH-based materials in heterogeneous electrocatalysis are rare.54,58 However, literature reports suggest that doping of other elements in LDH can significantly improve the catalytic performance of the OER.43 It has been argued that heteroatom doping could modify the electronic structure of the catalyst, which may optimize the adsorption energy of reaction intermediates.59–62 The incorporated atoms could act as the active centres toward electrocatalytic reactions, thereby synergistically contributing towards the improvement of the electrocatalytic performance.63 It is also evidenced that the introduction of heteroatoms may also induce lattice distortion, which improves the number of active sites and enhances the surface-active areas.64,65 In this regard, we envisioned that heteroatom doping in LDH-based materials could not only tune the electronics of the materials but also alter the pore distribution as well, a phenomenon which has been overlooked so far. Considering the literature report on the mechanism of LDH formation,66 we conceptualized that large heteroatom doping, such as cerium, could help to achieve narrow pore size distribution by minimizing interparticle electrostatic repulsion in LDH-based materials and thus may significantly enhance catalytic activity.66–68
Herein, a series of Ce-doped CoFe-layered double hydroxide (LDH) complexes have been synthesized by the co-precipitation method, wherein doping of cerium (Ce) into cobalt-iron layered double hydroxide (LDH) has been investigated. We studied the electrocatalytic performance of CoFe LDH doped with different concentrations of rare earth elements (e.g., Ce). In the following few sections, we discuss the synthesis and characterization of these LDHs. Furthermore, utilizing electrochemical techniques such as cyclic voltammetry, potential-dependent electrochemical impedance spectroscopy, etc., we explain their reactivity in terms of pore size distribution and electronics of the synthesized nanomaterials, which resulted in differences in physical processes towards oxygen evolution.
Experimental
Chemicals
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) and ammonium cerium(IV) sulfate dihydrate ((NH4)4Ce(SO4)4·2H2O) were purchased from Sigma-Aldrich. Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) was purchased from Rankem. Sodium hydroxide (NaOH) was purchased from Emplura. Nafion perfluorinated resin solution was purchased from Sigma-Aldrich. Isopropyl alcohol and ethanol were purchased from Rankem Pvt. Ltd. Milli-Q water (resistivity ∼18.2 MΩ cm) was used throughout the study. The chemicals were used without any further purification.Electrode materials
The platinum (Pt) wire was purchased from Alfa Aesar. The Saturated Calomel Electrode (SCE) and Glassy Carbon (GC) electrode were purchased from CH Instruments Inc. TX, USA.Synthesis of Ce-doped CoFe-layered double hydroxide (LDH)
The Ce-doped CoFe-LDHs were prepared under a N2 atmosphere by the co-precipitation method69,70 (Scheme 1). Co(NO3)2·6H2O (6 mmol), Fe(NO3)3·9H2O (3 mmol), and various molar ratios of cerium (Ce(NH4)4(SO4)4·2H2O (x = 0, 0.5, 1, 2 mmol)) were dissolved in 15 mL Milli-Q water. After that, they were mixed by magnetic stirring for 20 min to form a clear solution. In another flask, 0.7 M NaOH was prepared. Then, these two solutions were added dropwise in a round bottom flask, maintaining pH ∼9 in the N2 environment condition over 2 h (Scheme 1). The solution mixture was aged at 40 °C for 1 h. The precipitate was maintained in the same state for 24 h. After that, the precipitate was washed by centrifugation using a mixture of ethanol and Milli-Q water to remove the loosely attached surface products, and then dried in air at 60 °C for 12 h. The collected materials are denoted depending on mmol of Ce salt added (CoFeCe0: 0 mmole Ce, CoFeCe0.5: 0.5 mmole Ce, CoFeCe1: 1 mmole Ce, CoFeCe2: 2 mmole Ce, and CoFeCe3: 3 mmole Ce).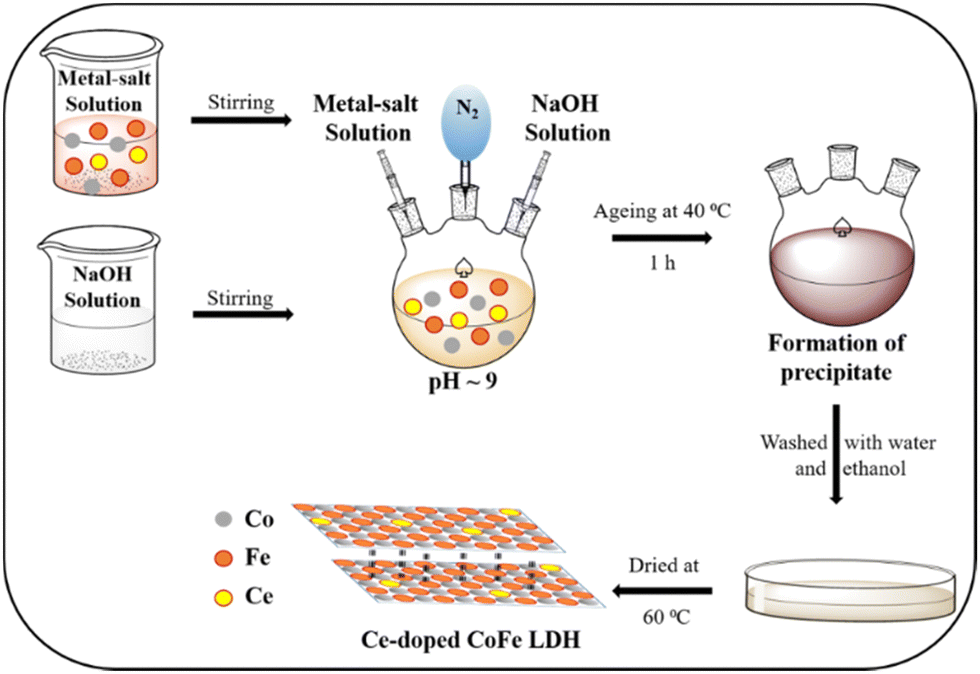 | ||
| Scheme 1 The synthesis methodology for Ce-doped CoFe LDHs. | ||
Characterization
Powder X-ray diffraction (PXRD) patterns were collected to confirm the structural pattern of the synthesized materials and also to study the crystallinity of the prepared sample by PANalytical Empyrean XRD using Cu Kα radiation (1.54 Å) in a 2θ range of 10–80°. Functional groups in Ce-doped CoFe-LDHs were identified using FT-IR spectroscopy. Spectra were collected using a PerkinElmer Model UATR Spectrum Two Instrument in the range of 500–4000 cm−1. The three-dimensional morphology of the prepared samples was analyzed using scanning electron microscopy (SEM). The dried samples were directly spread on conductive carbon tape and sputter-coated with gold for 2 min to improve the conductivity. Experiments were performed using a high-resolution field emission scanning electron microscope (HRFESEM) ZEISS of model ULTRA Plus at a working voltage of 20 kV. The internal structure of the prepared materials was analyzed using a transmission electron microscope (TEM FEI TALOS 200SS) operated at an accelerated voltage of 200 kV. The samples were prepared by drop-casting the suspension of the sample in isopropanol (0.5 mg in 15 mL) on the surface of a carbon-coated copper grid, and the solvent was removed by desiccating it overnight. Percentage elemental composition was calculated and analysed by a fully automated PC controlled elemental analyser. X-ray photoelectron spectroscopy (XPS) studies were performed using a K-Alpha ESCA System (Thermo Scientific) with an Al Kα (1486.6 eV) X-ray source. N2 adsorption/desorption experiments were carried out using a Quantachrome Autosorb QUA21011 instrument. Prior to the adsorption experiments, the samples were degassed under vacuum at 60 °C for 12 h. TGA experiments were performed using a PerkinElmer thermogravimetric analyser in the temperature range of 25–800 °C at a scan rate of 5 °C min−1. The content of Co, Fe, and Ce in all the prepared samples was determined by digesting a very small quantity of these samples in 25 mL savillex Teflon pressure decomposition vessels by dissolving in HCl:HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solutions. After overnight digestion, it was diluted 10 times prior to high resolution inductively coupled plasma optical emission spectrometry (ICP-OES) (Agilent Technologies Model: 725).
:1 v/v) solutions. After overnight digestion, it was diluted 10 times prior to high resolution inductively coupled plasma optical emission spectrometry (ICP-OES) (Agilent Technologies Model: 725).
Electrochemical analysis
For electrochemical studies, 1.2 mg of the catalyst was dispersed in a mixture of 12.5 μL Nafion perfluorinated resin solution and 250 μL 3:1 (v/v) water–isopropyl alcohol. From this, a 1 μL aliquot was drop-cast to deposit the catalyst on a cleaned and polished 3 mm diameter glassy carbon electrode (GC), leading to a catalyst loading of 0.065 mg cm−2. Then, the electrode was dried at 70 °C for 30 min prior to use for the water oxidation study. The electrochemical experiments were carried out in 1 M aq. KOH solution (pH 13.9) consisting of the GC, saturated calomel electrode (SCE), and Pt wire as the working, reference, and counter electrodes, respectively, at room temperature (25 °C). CH Instruments, Austin, TX (CHI 760D), and BioLogic (SP300 and SP240) potentiostats were used for all the electrochemical measurements. Cyclic voltammetry (CV) experiments were performed at a scan rate of 5 mV s−1, while the solution was stirred at 1600 rpm to avoid O2 bubble accumulation near the electrode surface. The electrochemical results mentioned in this work were iR compensation corrected and potentials are reported with respect to the reversible hydrogen electrode (RHE) following the relation E (V vs. RHE) = E (vs. SCE) + 0.242 + 0.059 × pH.71 Electrochemical impedance spectroscopy (EIS) experiments were measured at different potentials over a frequency range from 105 to 10−1 s−1 with an amplitude of 5 mV. For the long-term stability test, the potential of the electrode was held at 1.54 V vs. RHE for 24 h. Electrochemically active surface areas (ECSAs) were determined by collecting CVs in the non-faradaic region (+0.15 to +0.25 V vs. SCE) without stirring the solution at different scan rates.
Turnover frequency (TOF), mass activity, and specific activity of all the catalysts were evaluated utilizing eqn (1)–(3).72,73
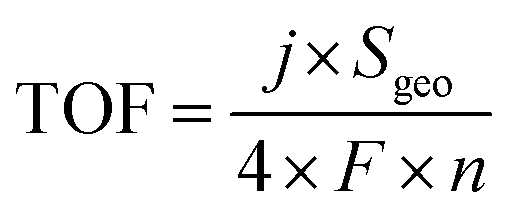 | (1) |
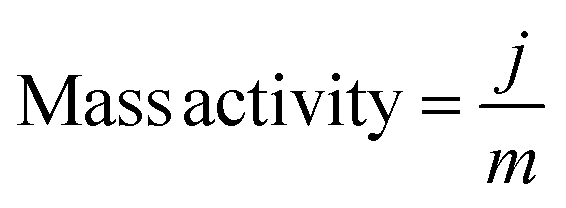 | (2) |
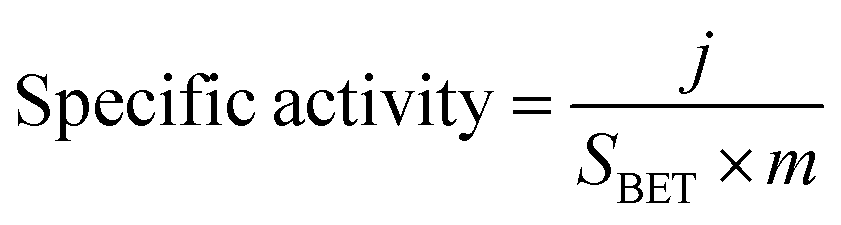 | (3) |
In these equations, j, Sgeo, SBET, F, m and n represent the current density at 350 mV overpotential (mA cm−2), geometrical surface area of the electrode (cm2), BET surface area (m2 g−1) from N2 sorption analysis, Faraday constant (96485 C mol−1), mass density (mg cm−2), and moles of the catalyst, respectively. Moles of catalysts were determined using ICP-OES.
Oxygen-detection experiments
A HI 764080 digital polarographic dissolved oxygen probe was used to measure the change of dissolved oxygen concentration in 1 M KOH solution kept in a four-mouth sealed electrode cell during controlled potential electrolysis (CPE). The experiment was performed by holding the GC electrode potential at 1.54 V vs. RHE for 30 min. Before performing the chronoamperometry, the sensor was calibrated using two points against solution and air while 1 M KOH solution was purged with Ar gas until the probe sensor showed zero O2 concentration.Results and discussion
The synthesized LDHs were characterized by various advanced techniques. First, in order to understand the crystal structure, the synthesized materials were analyzed by PXRD. Fig. 1 shows the PXRD patterns of the prepared Ce-doped CoFe-LDH powders, exhibiting the typical characteristic diffraction peaks of the hydrotalcite-like structure of LDH. The observed diffraction peaks located at 11°, 23°, 34°, 38°, 46°, 59°, and 60° can be indexed to the (003), (006), (012), (015), (018), (110), and (113) planes of the Ce-doped CoFe-LDH structure, respectively (PDF#79–1744) (Fig. 1).74–76 It was observed that the varying molar ratio of cerium ions in the CoFe-LDH influenced the crystallinity of Ce-doped CoFe-LDH. The crystallinity of the material decreased with increasing the cerium concentration in the CoFe-LDH layers, evidenced by the broadening of the peaks (Fig. 1). The CoFeCe0.5 sample exhibited the best crystallinity, signifying a regular arrangement of cerium ions on the brucite-like layer of CoFe-LDH. However, a further increase of cerium ion doping led to lattice defects in LDH, and resulted in declining crystallinity (Fig. 1). Lattice defects are introduced in the crystal lattice due to the higher ionic radius of Ce, which occupies a larger space in the lattice and distorts the lattice structure. Hence, Ce-doping affects the crystallization process of CoFe-LDH, which induces local defects, and results in lattice distortion. Besides, impurity peaks were observed which can be attributed to Ce- and NH4-based salts such as ammonium cerium(IV) sulfate dihydrate used for cerium doping.16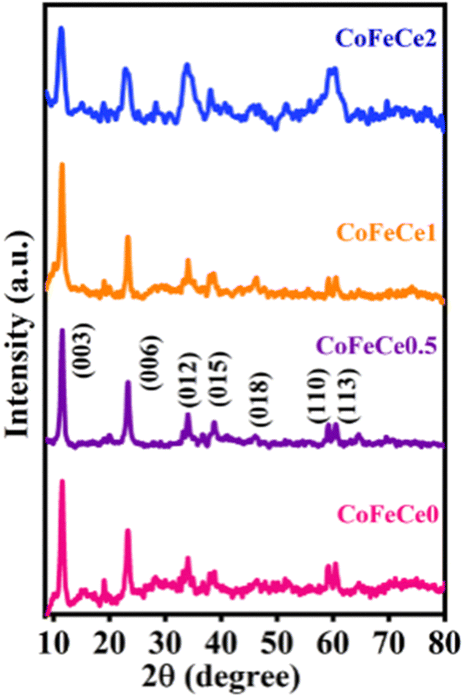 | ||
| Fig. 1 PXRD patterns of Ce-doped CoFe LDHs. | ||
Thereafter, the functional group analysis was performed using FTIR spectroscopy (Fig. S1 and Table S1, ESI†). The band near ∼3400 cm−1 was due to the stretching vibration of hydroxyl groups in the interlayer, and the band observed at ∼1600 cm−1 is assigned to the bending vibration of water molecules69 (Fig. S1 and Table S1, ESI†). IR bands at 755 and 470 cm−1 were due to stretching vibration of M–O and M–O–M (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Co, Fe, Ce), respectively (Fig. S1 and Table S1, ESI†). After doping of cerium ions into the LDH structure, a shift to a lower wavenumber for the M–O band (614 cm−1) was observed, suggesting a new type of M–O bond formation in the LDH structure when the cerium concentration was at a maximum (Fig. S1 and Table S1, ESI†).69,77 Also, the intensity of the NO3− peak decreased with a subsequent increase in the SO42− peak due to the use of sulphate salt during synthesis (Fig. S1 and Table S1, ESI†).
Co, Fe, Ce), respectively (Fig. S1 and Table S1, ESI†). After doping of cerium ions into the LDH structure, a shift to a lower wavenumber for the M–O band (614 cm−1) was observed, suggesting a new type of M–O bond formation in the LDH structure when the cerium concentration was at a maximum (Fig. S1 and Table S1, ESI†).69,77 Also, the intensity of the NO3− peak decreased with a subsequent increase in the SO42− peak due to the use of sulphate salt during synthesis (Fig. S1 and Table S1, ESI†).
The thermal behavior of the Ce-doped CoFe-LDHs was studied using TGA (Fig. S2, ESI†). In the temperature range of 50 to 200 °C, the weight loss of the catalysts was nearly 6%, which can be attributed to the desorption of H2O molecules attached to the catalysts through adsorption or hydration to the surface (Fig. S2, ESI†). In the temperature range of 700 to 780 °C, weight loss of 1% was observed for the CoFeCe0, CoFeCe0.5 and CoFeCe1 (Fig. S2, ESI†). However, in the case of CoFeCe2-LDH, the weight loss in the temperature range 700 to 780 °C was 5% (Fig. S2, ESI†), which can be attributed to the structural disorder in the crystal structure of CoFeCe2, which was also evident from the PXRD patterns (vide supra).77
The surface morphology of the Ce-doped CoFe-LDH samples was investigated by SEM. The SEM image of CoFeCe0.5 showed a clear hexagonal nanosheet-like morphology (Fig. 2b), which supports the PXRD pattern results that the material had the highest crystallinity (vide supra). With increasing concentration of the cerium ion in the CoFe-LDH layers, SEM images revealed an increase in morphological disorder which can be correlated with somewhat decline of crystallinity of the materials with increased cerium doping (Fig. 2a–d). In order to understand the morphology of the material better, the TEM analysis of the Ce-doped CoFe LDHs was carried out. TEM images revealed abundant ultrathin hexagonal nanosheets with varying diameters (Fig. 2e and Fig. S3, ESI†), which is consistent with the results obtained by SEM (vide supra). These hexagonal ultrathin nanosheets are stacked together to form a layered structure. The SAED patterns show the formation of the clear diffraction ring for CoFeCe0.5, which is credited to the highest crystallinity of the LDH, and the fuzzy dispersed diffraction ring indicated a decline of crystallinity with increase in cerium doping (Fig. S4, ESI†). Furthermore, lattice fringes with spacing ranging around 0.268–0.281 nm have been observed utilizing the HRTEM image of the prepared LDH samples (Fig. S5, ESI†), which further confirmed the formation of the LDH. Energy dispersive X-ray (EDX) spectroscopy and elemental mapping confirmed the presence of Co, Fe, and Ce elements with uniform distribution (Fig. 2g–i and Fig. S3, ESI†). These results firmly established successful doping of Ce within the LDH lattice.
 | ||
| Fig. 2 Morphological characterization of Ce-doped CoFe-LDHs: (a)–(d) SEM image, (e) a low-resolution TEM micrograph of CoFeCe2 LDH, and (f) SEM image of CoFeCe2 LDH taken for elemental mapping. Elemental mapping for (g) Co, (h) Fe and (i) Ce from the selected area of the SEM image as shown in (f). | ||
X-ray photoelectron spectroscopy (XPS) was performed to study the surface chemical states of Ce-doped CoFe-LDHs. The survey XPS spectra identified the presence of Co, Fe, Ce, C, and O atoms (Fig. 3a and Fig. S6a, S6c, S6e, ESI†). The O1s spectra revealed peaks at 531.4 and 532.6 eV corresponding to the M–O network and surface –OH groups, respectively (Fig. 3b).22,78,79 The Fe2p spectrum has been fitted into two pairs of peaks, corresponding to Fe3+ (710.6 and 724.5 eV) and two satellite peaks at 717.5 and 734.5 eV (Fig. 3c).80,81 Co2p spectra revealed two peaks at 780.5 and 781.8 eV for Co3+ 2p3/2 and Co2+ 2p3/2 respectively (Fig. 3d).21,58 The spin–orbit coupling peaks were located at 796.3 and 797.7 eV for Co3+ 2p1/2 and Co2+ 2p1/2, respectively (Fig. 3d).21 Two small peaks at 786.3 and 804.8 eV were observed for satellite peaks of Co (Fig. 3d).21,78,82,83 A sequential increase in the ratio of Co3+/Co2+ was observed with the increase in the concentration of cerium (Fig. S6 and Table S2, ESI†). This observation was due to the fact that doping of Ce4+ results in oxidation of Co2+ to Co3+ through electron acceptance from Ce4+ to form stable Ce3+, and hence more Co2+ can be oxidized to Co3+, which is considered as the active site for electrocatalytic water oxidation.84 The Ce 3d XPS spectrum was deconvoluted into two series of subpeaks according to the Ce 3d spin–orbit states. The two peaks at 885.5 and 904.7 eV were attributed to Ce3+ 3d5/2 and Ce3+ 3d3/2, respectively (Fig. 3e).69 Additionally, the peaks at 882.3 and 898.3 eV were attributed to the oxidation states of Ce4+ 3d5/2 and Ce4+ 3d3/2, respectively which indicates that Ce exists in the mixed valence state of Ce3+ and Ce4+ (Fig. 3e).69,85
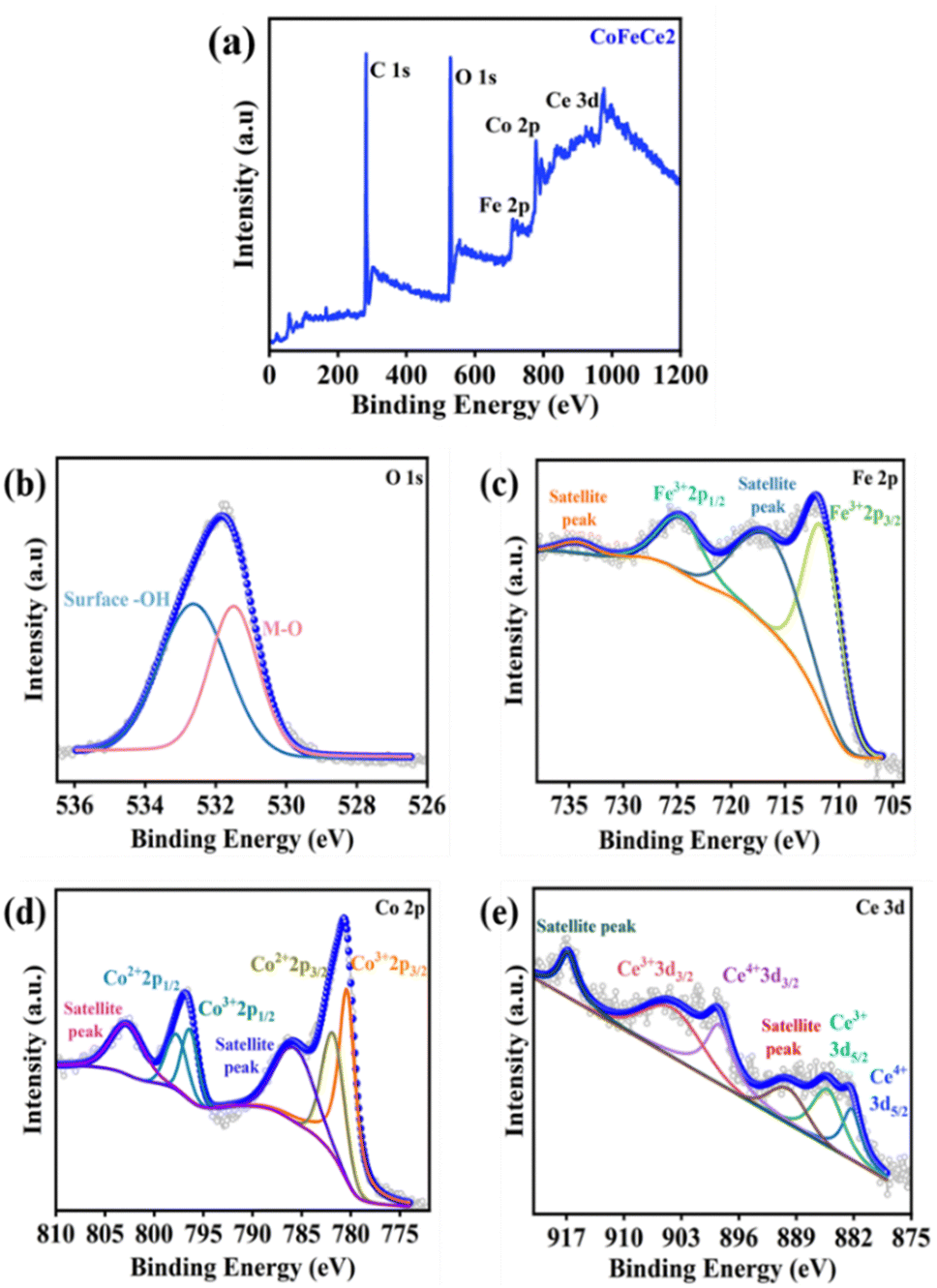 | ||
| Fig. 3 XPS spectra of CoFeCe2. (a) XPS survey scan, (b) deconvoluted O1s spectra, (c) deconvoluted Fe2p spectra, (d) deconvoluted Co2p spectra, and (e) deconvoluted Ce3d spectra. | ||
Once the chemical nature of the LDHs was confirmed, BET N2 sorption analyses of the LDHs were performed to understand the porous nature of the synthesized materials. The isotherms were closely related to type III isotherm profiles according to the IUPAC classification revealing weak adsorbate–adsorbent interaction (Fig. 4a).86 The BET surface area of CoFeCe2 was 195.7 m2 g−1, which was significantly higher compared to the other LDHs (Table 1). The hysteresis loop located in the pressure range P/P0 (0.6–0.9) indicated that the LDHs were mesoporous in nature87,88 (Fig. 4a). Cumulative pore volumes were calculated at P/P0 of 0.98 and are listed in Table 1. Cumulative pore volumes vs. pore diameter of these nanomaterials are shown in Fig. 4a insets. These results revealed significantly narrow pore size distribution for CoFeCe2 (0–35 nm) compared to other LDHs (CoFeCe0 (0–150 nm), CoFeCe0.5 (0–70 nm) and CoFeCe1 (0–70 nm)) (Fig. 4b and Fig. S7, ESI†). The pore diameter distribution revealed major peaks at 2.4, 2.3 and 2.2 for CoFeCe0, CoFeCe0.5 and CoFeCe1, respectively, while the most intense peak was located at 3.8 nm for CoFeCe2 (Fig. 4b). The pore diameter distribution for CoFeCe2 was concentrated in the narrow mesoporous (0–35 nm) region, while the pore distribution was significantly wider for other LDHs (Fig. 4b and Table S3, ESI†). Specifically, in the case of LDH without Ce-doping (CoFeCe0) the pore distribution was very wide with a very shallow peak at 2.36 nm (Fig. 4b inset). These results suggest that cerium doping resulted in a narrower pore distribution for the synthesized LDHs compared to undoped LDH, with the most narrow pore distribution for CoFeCe2. The highest pore volume in the narrow mesoporous range was also obtained for CoFeCe2 (Table S3, ESI†), which was beneficial for increasing the catalytic activity (vide infra). The doping of cerium ions resulted in an initial decrease in surface area and pore volume presumably due to increased crystallinity as evident from PXRD and SEM analysis (vide supra), but thereafter a systematic increase in surface area and pore volume of LDH was observed with increasing cerium doping, suggesting an important role of cerium in LDH pore formation, which led to increased surface areas, narrow pore distribution, and increased pore volume (Table 1, Fig. 4a, b and Fig. S7, ESI†).89 It is interesting to observe that CoFeCe2 had a significantly higher narrow mesoporous pore volume (2–5 nm), compared to other LDHs (Table S3, ESI†).
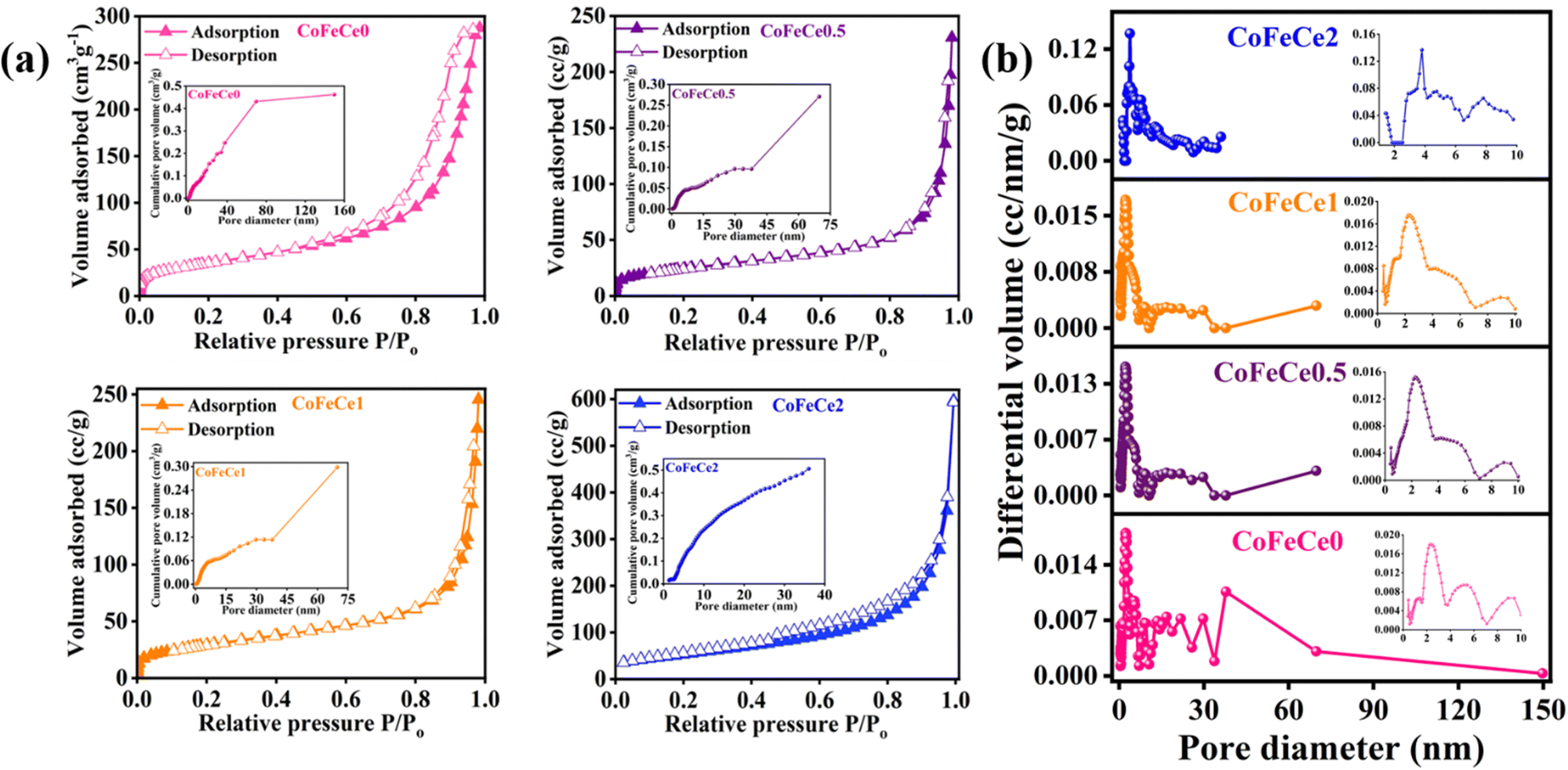 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherm profile at 77 K. Insets show the cumulative pore volume of the respective samples, and (b) pore size distribution (inset shows small pore region) of Ce-doped CoFe LDHs. | ||
| Sample | BET surface area (m2 g−1) | Pore distribution/peak (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| CoFeCe0 | 126.6 | 2.36 | 0.44 |
| CoFeCe0.5 | 86.4 | 2.25 | 0.36 |
| CoFeCe1 | 106.0 | 2.24 | 0.38 |
| CoFeCe2 | 195.7 | 3.79 | 0.51 |
HRTEM images of LDHs revealed the presence of hexagonal nucleates in all LDHs, as shown in Fig. 5a–d, marked by yellow dotted lines. Employing these images, the average diameter of hexagonal nucleates has been calculated by taking 10 samples and it revealed a systematic increase in the diameter of hexagonal nucleates from 8.85 to 13.31 nm from CoFeCe0 to CoFeCe2 (Fig. 5e and Table S4, ESI†).
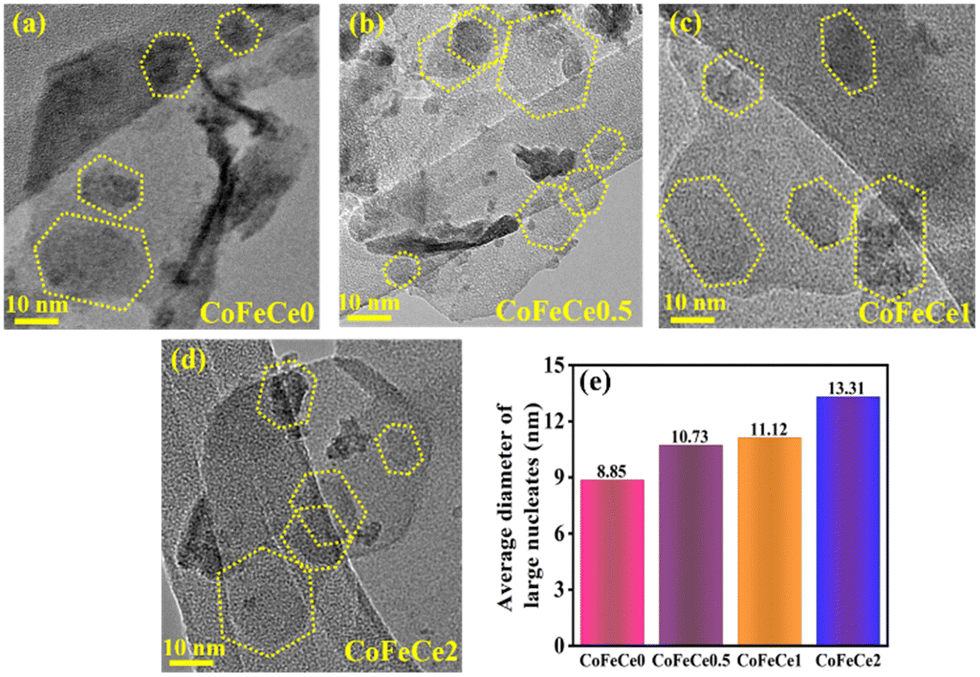 | ||
| Fig. 5 (a)–(d) HRTEM images of different Ce-doped CoFe LDHs showing large nucleates (dotted yellow line), and (e) bar graph showing the trend in the average diameter of large nucleates in different LDHs. | ||
The obtained BET results and TEM results can be understood by considering the LDH formation mechanism proposed by Alexei A. Lapkin and co-workers (Scheme 2).66 The LDH crystallization process involves three steps, and they are nucleation, surface growth, and aggregation.66 Firstly, the nucleation of different ions results in the formation of small hexagonal nucleates. In the second step, these small nucleates grow along the plane with the simultaneous attachment of hydroxyl or hydroxyl-cerium on the edges66 to increase the size, resulting in the formation of large hexagonal nucleates and this process is called surface growth. In the last step, these large hexagonal nucleates aggregate to form the final arrangements of LDH nanosheets. The interparticle distance between these large hexagonal nucleates dictates the pore distribution of LDH nanomaterials. In the absence of cerium ions, these large hexagonal nucleates experience higher electrostatic repulsive forces due to high charge concentration on the hydroxyl ion present on the edges of the hexagonal nucleates. This strong repulsive force results in a large interparticle distance, while the nucleates do not face such repulsive force due to the absence of hydroxyl ions, resulting in a shorter interparticle distance, and hence a wide pore distribution was observed for CoFeCe0 (vide supra) (Scheme 2).
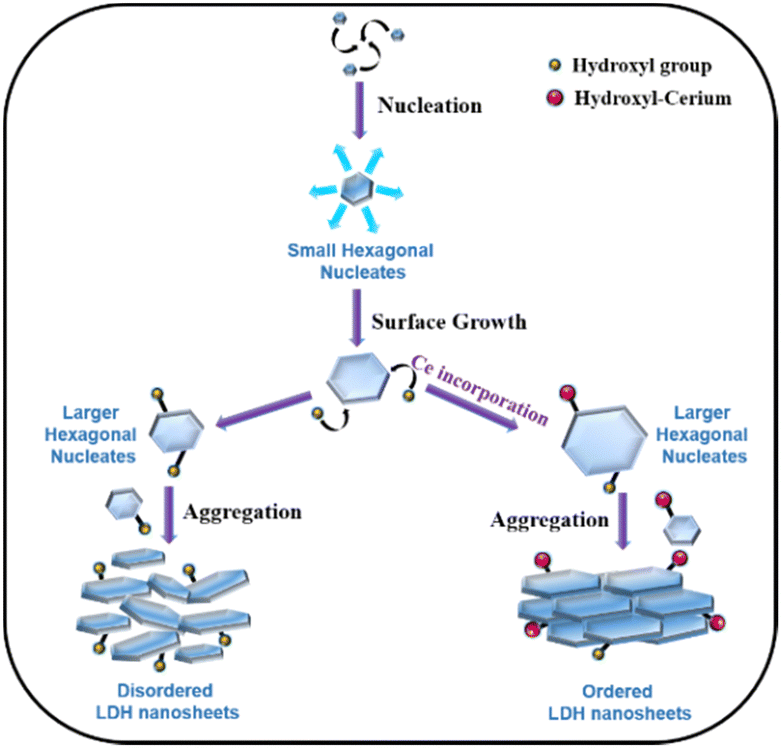 | ||
| Scheme 2 Schematic of the crystallization mechanism for LDH formation. | ||
We propose that for nanomaterials having cerium doping, cerium will be attached to the hydroxyl group at the edge-attached hydroxyl groups due to chemisorption.66 Due to the large atomic size of cerium, the concentration of charge decreased on the hexagonal nucleates, which results in a decrease in electrostatic repulsive force between the different nucleates (Scheme 2). Hence, the interparticle distance decreases, which results in a narrower pore distribution, and hence ordered LDH nanosheets are obtained (vide supra). This decrease in repulsive force also results in an increase in the average diameter of the nucleates with an increase in cerium doping (vide supra).
Water oxidation by Ce-doped CoFe LDHs
The LDHs were employed for electrochemical OER in 1 M KOH solution at room temperature in a three-electrode system. Cyclic voltammograms (CVs) were collected for all the materials, and CVs revealed increasing catalytic activity evident from increasing catalytic current density at higher overpotential with increasing Ce doping in LDHs, i.e., the trend was CoFeCe2 > CoFeCe1 > CoFeCe0.5 > CoFeCe0 (Fig. 6a). Overpotential is an important parameter to judge the potential of a water oxidation catalyst, which is defined as the excess potential over the equilibrium potential required to derive a significant current, and in water oxidation studies, 10 mA cm−2 current density has been taken as the reference point considering its relevance in solar fuel.90 Among all the LDHs, CoFeCe2 showed the lowest overpotential of 320 ± 7 mV (Fig. 6c). However, further increase of Ce doping (CoFeCe3 LDH) resulted in increased overpotential (367 mV) and Tafel slope (64 mV dec−1), which could be due to structural changes at higher cerium concentration (Fig. S8, ESI†).39 At this point, it is also important to emphasize that the overpotential depends on catalyst loading on the electrode surface, electrode configuration, etc.; hence literature comparisons based on overpotential should be avoided.53 The kinetics of water oxidation can be related to Tafel slopes, i.e. the smaller the value of the Tafel slope, the better the electrocatalytic performance. With increasing cerium doping, the Tafel slope decreased from 77 to 47 mV dec−1 (Fig. 6b).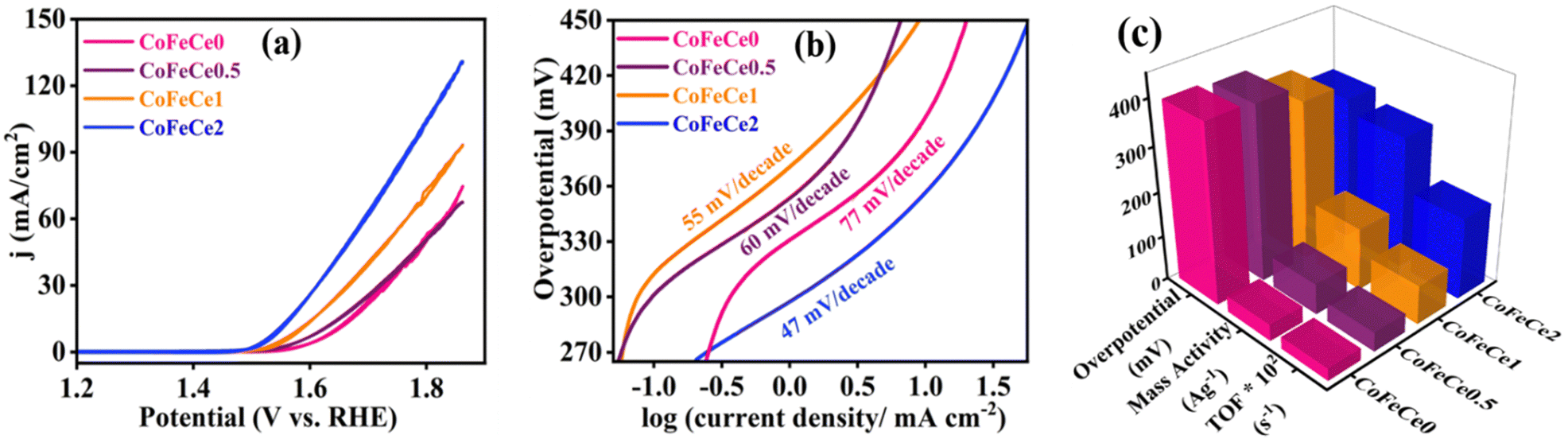 | ||
| Fig. 6 Electrochemical results of LDHs. (a) CVs at a scan rate of 5 mV s−1, (b) Tafel plots of Ce-doped CoFe LDHs during water oxidation and (c) overpotentials, mass activities, and TOFs of the Ce-doped CoFe LDHs during water oxidation. Overpotentials were measured at 10 mA cm−2 current density. Mass activities and TOFs were calculated at 350 mV overpotential. | ||
Mass activities and TOFs are listed as important parameters to judge the activity of a catalyst.21,22,53 TOF quantifies the reactivity of a catalyst per active site of the material deposited on the electrode at the specified reaction conditions.91 Mass activities and TOFs were calculated at 350 mV overpotential. CoFeCe2 showed a remarkable mass activity of 294.15 A g−1, which is 2.1, 4.2 and 8.0 times higher than that of CoFeCe1 (140.15 A g−1), CoFeCe0.5 (69.59 A g−1) and CoFeCe0 (37.11 A g−1), respectively (Fig. 6c, Table 2). A similar trend was observed for TOFs wherein CoFeCe2 showed the best performance with a TOF value of 1.823 s−1, which was higher than that of CoFeCe1 (0.873 s−1), CoFeCe0.5 (0.432 s−1) and CoFeCe0 (0.277 s−1), respectively (Fig. 6c, Table 2). The mass activities reported by Jakhad and coworkers92 for NiFe LDH, and Liu and coworker93 for (FeCoNiCrMn)3O4-400 were 141.2 and 67.3 A g−1, respectively. Besides, the TOFs reported by Ye and coworkers54 for Co3Fe1 LDH, Jakhad and coworkers92 for NiFe LDH, and Liu and coworkers93 for (FeCoNiCrMn)3O4-400 were 0.31, 0.25, and 0.16 s−1, respectively. This comparison suggests significant improvement of reactivity presented herein compared to previous reports of similar materials. Mass activity and TOF for CoFeCe2 are compared with more recent literature reports of similar LDH-based materials (Table S5, ESI†). Specific activities calculated at 350 mV overpotential also showed increasing catalytic reactivity with increased cerium doping (Table 2).
| Sample | TOF (s−1) | Mass activity (A g−1)@η = 350 mV | Specific activity (mA cm−2)@η = 350 mV |
|---|---|---|---|
| CoFeCe0 | 0.277 | 37.11 | 0.03 |
| CoFeCe0.5 | 0.432 | 69.59 | 0.08 |
| CoFeCe1 | 0.873 | 140.12 | 0.13 |
| CoFeCe2 | 1.823 | 294.15 | 0.15 |
The electrochemically active surface area (ECSA) of the catalysts is used to understand the electrolyte accessibility inside the surface of the nanomaterials, which governs the catalytic performances. Larger ECSA suggests more exposed active sites for the OER reaction. However, ECSA depends on the surface area of the electrode used, and hence the roughness factor (RF) is more appropriate for comparison, which is calculated by dividing the ECSA by the geometrical surface area. RFs were calculated by performing CV experiments in a non-faradaic zone (potential region: +0.15 to 0.25 V vs., SCE, ESI† for calculation methodology).9 Representative CVs revealed that the charging currents trend as CoFeCe2 > CoFeCe1 > CoFeCe0.5 > CoFeCe0 (Fig. 7a and Fig. S9a–c, ESI†). Charging currents for anodic and cathodic components at 0.2 V vs. scan rate are plotted for the calculation of ECSAs (Fig. 7b and Fig. S9d–f, ESI†), and the values demonstrated that ECSA for CoFeCe2 was nearly twenty-one-fold higher than that of CoFeCe0 (Table S6, ESI†). RFs for LDHs have been plotted in Fig. 7c, which shows a significant increase of RF value with increasing cerium doping and a massive RF value of 432.4 was obtained for CoeFeCe2, signifying nearly 433 times increase of electrolyte accessible surface area compared to the geometrical surface area of the electrode (Fig. 7c and Table S6, ESI†). It is important to mention that this RF value is significantly higher than the RF values (23–60) for the OER previously reported.9,22
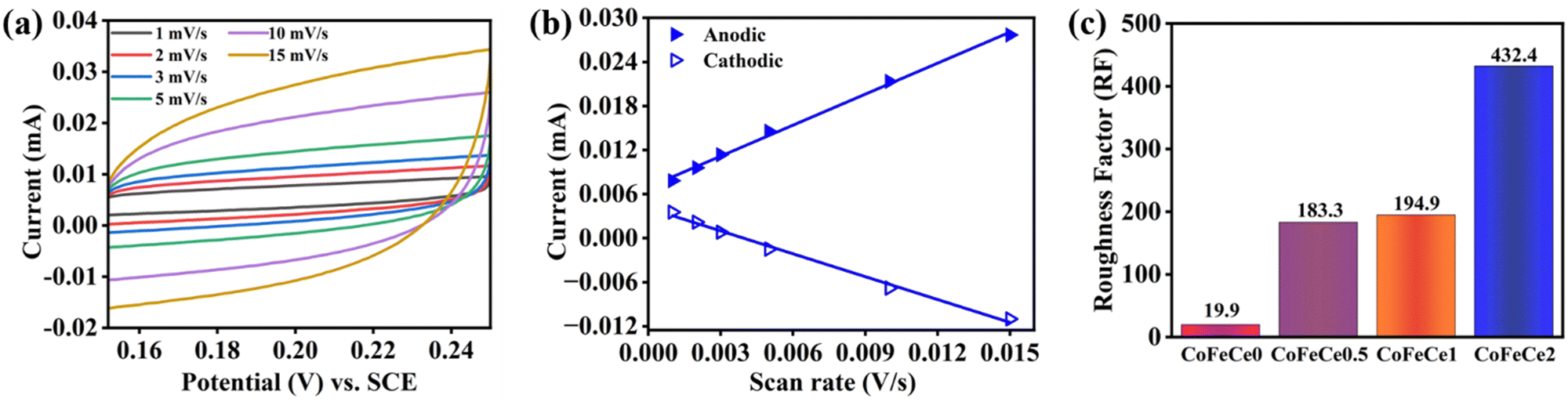 | ||
| Fig. 7 (a) CVs at different scan rate in a non-faradaic potential region (0.15 to 0.25 V vs. SCE). (b) Plot of anodic and cathodic charging currents at 0.2 V (vs. SCE) vs. scan rate of voltammetry of CoFeCe2 LDH. (c) Roughness factor of Ce-doped CoFe LDHs. | ||
In order to understand the improved reactivity of CoFeCe2 LDH, electrochemical impedance spectroscopy (EIS) was performed (Fig. 8). The Nyquist diagram (a plot of imaginary vs. real components of impedance) collected at 1.54 vs. RHE showed two semicircles. The diameter of the first small semicircle is associated with charge transfer resistance (Rct) at the electrode–electrolyte interface, which occurs at a faster time scale, i.e., in the high-frequency region (Fig. 8a), and the larger second semicircle in the lower frequency range is associated with the reaction steps related to water oxidation, and charge relaxation of the surface adsorbed intermediate species (Fig. 8a).94–96 The Rct for CoFeCe2 was estimated to be 3.9 Ω, which was significantly lower than that of CoFeCe1 (7.4 Ω), CoFeCe0.5 (7.3 Ω) and CoFeCe0 (8.1 Ω), which were obtained by fitting the EIS results using an equivalent electrical circuit (Fig. S10 and Table S7, ESI†).97 The pseudoresistance (Rp), which represents resistance due to electron transfer between ions and surface adsorbed species to convert O2 from water, decreased significantly from CoFeCe0 to CoFeCe2 (74.6 to 45.5 Ω) (Table S7, ESI†).14 Bode plots showed that the maximum phase angle in the mid-frequency region was lowest for CoFeCe2, suggesting fast charge propagation, which is also in agreement with the Tafel slope value (Fig. 8b).22
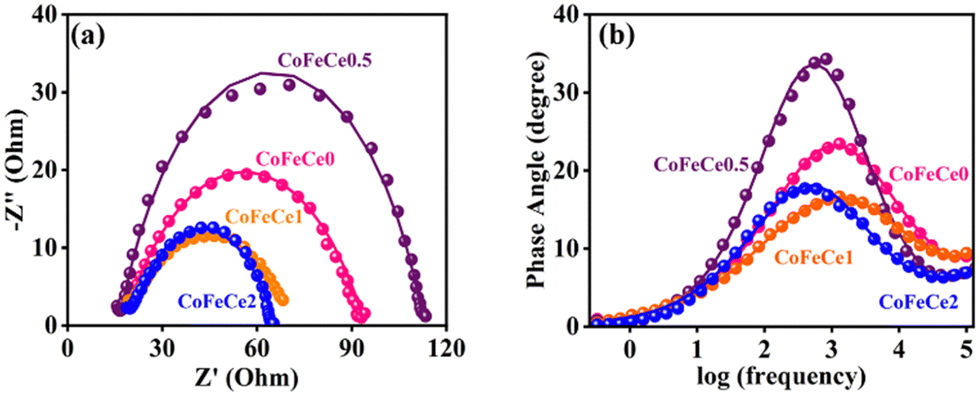 | ||
| Fig. 8 (a) Nyquist plots, and (b) Bode plots at a bias potential of 1.54 V vs. RHE in the frequency range from 105 to 10−1 s−1 with 0.005 V amplitude for different LDHs. The experimental results are represented by discrete points and equivalent circuit fitted results are represented by solid lines. | ||
At this point, it is important to emphasize that in the evaluation of TOF using potential-sweep measurements such as CV, different resistive components become additive, such as resistance of the catalyst, charge transfer resistance and mass-transport resistance.91,98 Thus, TOF obtained from CV provides a lower-limit value for catalytic activity.99 However, EIS is a stationary technique which minimizes the contribution of different resistive components and hence, it can provide an upper limit of TOF, which is termed TOFEIS. TOFEIS can be calculated using eqn 4, wherein n, τ, Rct, and Cμ are the number of transferred electrons per catalytic site (4 for OER), time constant (s) for a charge transfer process at the catalyst–solution interface (e.g., water oxidation), charge-transfer resistance at the catalyst/solution interface, and the catalyst's chemical capacitance.
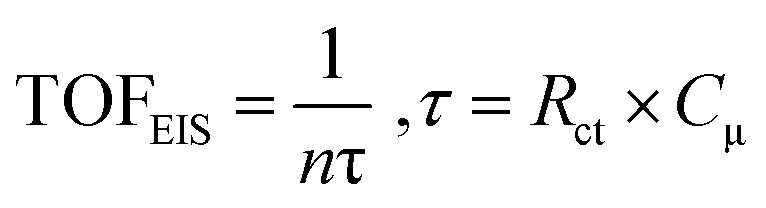 | (4) |
TOFEIS was 126.76 s−1 for CoFeCe2, which was nearly 10 fold higher than the value reported by Idan Hod and coworkers.98
For catalytic application, the long-term stability of the catalyst is another important parameter which needs to be evaluated. A long-term chronoamperometric experiment was performed for CoFeCe2 and CoFeCe1 LDH by applying a potential of 1.63 V vs. RHE (400 mV overpotential) for 24 h. The current densities were stabilized after 12 h, and a higher current was observed for CoFeCe2 (10 mA cm−2) compared to CoFeCe1 (7.5 mA cm−2) (Fig. S11a, ESI†), which can be attributed to the increased activity due to enhanced Ce-doping (vide supra). A slow decline in the current density was observed beyond 12 h, which could be due to the blockage of pores due to oxygen evolution.
Product determination and post-catalysis characterization
In order to confirm O2 as the product of the water oxidation reaction, controlled potential electrolysis was performed by holding the potential of a GC electrode coated with CoFeCe2 LDH for 30 min at 1.54 V vs. RHE in 1 M KOH. 8.5 μmol of O2 was detected at the end of the experiment, while electrochemistry experiments suggested a maximum of 9.24 μmol of O2 could have evolved. Hence, a faradaic efficiency of 92% was obtained. The amount of evolved O2 at different time intervals is plotted (dark cyan triangles) and compared with the calculated amount of O2 from electrochemistry (red line) (Fig. S11b, ESI†). A long-term electrolysis experiment was performed to understand the stability of CoFeCe2. The SEM image after electrolysis revealed that the morphology of the catalyst remained unaltered (Fig. S12, ESI† and Fig. 2d). EDS analysis revealed a slight increase in oxygen percentage and a slight decrease in cobalt, iron, and cerium percentage after the electrolysis, which could be due to surface-adsorbed oxygen during electrolysis (Fig. S13, ESI†). Besides, the elemental mapping before and after electrolysis suggested the presence of similar surface functionality (Fig. S14, ESI†). Fig. S15a, ESI† shows the XRD of CoFeCe2 before and after electrolysis and it revealed that the characteristic peaks of LDH were retained after electrocatalysis. To find the chemical nature and valence state of the CoFeCe2 LDH after long-term electrolysis, XPS was performed. XPS spectra after electrolysis indicated that O1s, Fe2p, Co2p, and Ce3d retained their characteristic peaks in the same region (Fig. S15b–e, ESI† and Fig. 3). Hence, all these results suggest that the chemical nature of the CoFeCe2 remained the same after electrolysis.Discussion about enhanced OER activity by Cerium-doped CoFe-LDH
The enhanced reactivity of cerium-doped LDHs presumably has two origins, and they are electronic and morphological. Iron doping also helps in the enhancement of catalytic activity by modulating the crystal and electronic structure of cobalt; besides, Fe3+ also acts as catalytic sites for the OER.100,101 Since the concentration of iron was kept the same in all the samples, the synergistic role of iron will be similar for all the LDHs. During water oxidation, the electrons are transferred from OH− to the cobalt center, which results in the formation of CoII-OH from CoIII (Scheme 3a, step 1). In the next step, the reduced CoII-OH would undergo a deprotonation step to release a water molecule and form CoII-O (Scheme 3a, step 2). The next electron transfer step is the rate-determining step (RDS), which involves the formation of CoII-OOH (Scheme 3a, step 3). In the final step, the evolution of oxygen occurs, and the catalyst is regenerated as CoIII (Scheme 3a, Step 4).102 Co2+ is a d7 system, which exists as a high spin complex with a weak ligand OH−; hence the electronic configuration of Co2+ is t52g e2g. The unsymmetrical electron occupancy in the t2g orbitals of Co2+ leads to a dynamic Jahn–Teller distortion, which results in the lengthening of the Co–O bonds,103,104 and further facilitates the formation of O–O bonds during the OER process. Water oxidation by LDHs occurs at CoIII centres, and cerium doping systematically enhances the concentration of CoIII in the nanomaterials during the OER due to electron transfer from CoII to CeIV, which results in the formation of CoIII and CeIII, respectively. This hypothesis is supported by XPS results, which revealed a systematic increase of Co3+ concentration with increasing cerium concentration (Table S2, ESI†), as depicted in Scheme 3b. Due to enhanced CoIII ions in the nanomaterials, water oxidation is favored with increasing cerium concentration, and hence, the highest reactivity was obtained for CoFeCe2.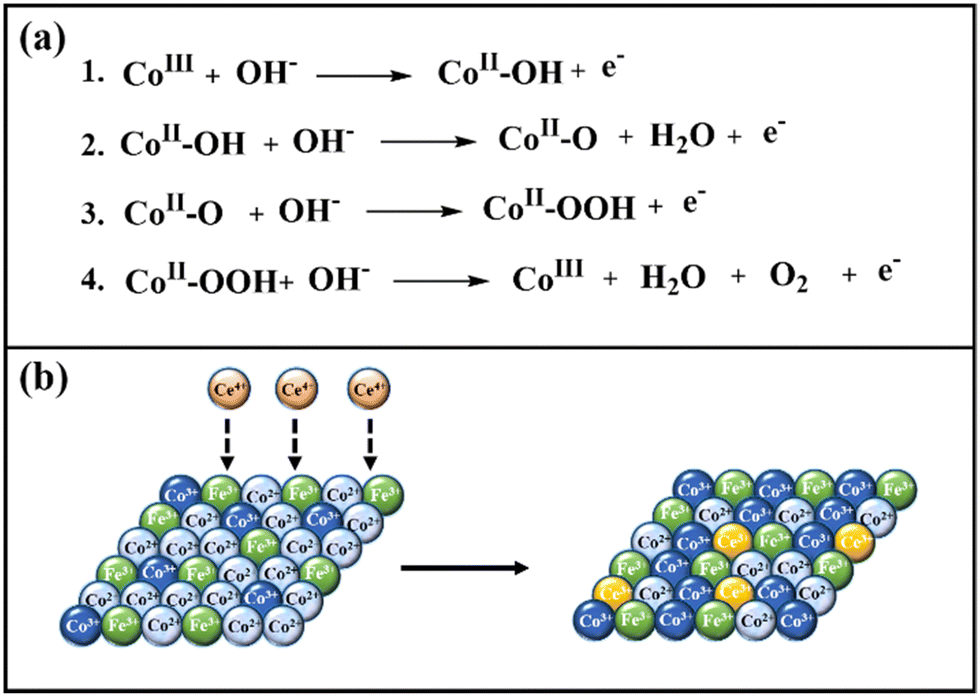 | ||
| Scheme 3 (a) OER mechanism, and (b) role of cerium doping in increasing the concentration of active sites (Co3+). | ||
We further propose that the enhanced reactivity of cerium-doped LDHs has a morphological origin as well. BET and TEM results revealed a narrow pore size distribution with increasing cerium doping, which resulted in a massive and systematic increase in ECSA and RF values (vide supra). These results suggest that the electrolyte accessibility inside the nanomaterials was systematically improved with cerium doping. From the mechanistic understanding of LDH formation, it was evident that cerium doping minimized the electrostatic repulsion between large hexagonal nucleates, which led to uniform interparticle distance. Hence, an ordered regular arrangement with narrow mesoporous distribution was observed for CoFeCe2, while the pore distribution was very wide for undoped LDH (CoFeCe0) (Scheme 2) (vide supra). It is also important to emphasize that the large atomic radius of cerium ions caused lattice distortion, which resulted in a decrease in the crystallinity of cerium-doped LDH, and this phenomenon also helped to improve the active sites for catalysis.
Conclusions
In conclusion, we have demonstrated excellent water oxidation by synthesizing Ce-doped CoFe-LDH prepared by the co-precipitation method, in which the catalytic activity of the LDH increased by increasing the cerium doping. Among all the materials, CoFeCe2 having the highest cerium doping, exhibited a remarkable mass activity of 294.15 A g−1, TOF of 1.82 s−1, and TOFEIS of 35.35 s−1. These values were significantly higher than the previously reported literature values54,92,93 (Mass activity = 67.3–141.2 A g−1 and TOF = 0.16–0.31 s−1). Moreover, a massive roughness factor value of 433 was obtained, suggesting 433 times enhancement of electrolyte accessibility compared to the geometrical surface area of the electrode. This outstanding reactivity of CoFeCe2 LDH can be attributed to:i. The mostly narrow mesoporous pore distribution of CoFeCe2 increased electrolyte accessibility inside the nanomaterials and hence increased the catalytic activity. We proposed that the cerium doping minimized the electrostatic repulsion between large hexagonal nucleates formed after nucleation and surface growth steps. In consequence, ordered LDH nanosheets were formed, which resulted in narrow pore distribution for CoFeCe2, while in the absence of cerium doping, high electrostatic repulsion between hydroxyl groups provided very wide pore distribution for undoped LDH (CoFeCe0) due to disordered LDH nanosheet formation.
ii. Furthermore, cerium doping helped to increase Co3+ ions in the material by electron donation from Co2+ to Ce4+ to form stable Ce3+. These Co3+ centres acted as the active sites for the OER.
iii. The high ionic radius of cerium ions resulted in lattice defects, which decreased the crystallinity of the material. This phenomenon improved the active sites for catalysis and helped to enhance the OER.
iv. Moreover, during OER reaction steps, the unsymmetrical electron occupancy in the t2g orbitals of Co2+ leads to a dynamic Jahn–Teller distortion, which results in the lengthening of the Co–O bonds and further facilitated the formation of O–O bonds during the OER process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.P. acknowledges financial support from DST-SERB New Delhi (CRG/2020/002493), infrastructural support from IISER Bhopal, and the FIST supported TEM facility to the Dept. of Chemistry, IISER Bhopal. P. A. acknowledges DST-INSPIRE for the fellowship.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- A. Morozan, H. Johnson, C. Roiron, G. Genay, D. Aldakov, A. Ghedjatti, C. T. Nguyen, P. D. Tran, S. Kinge and V. Artero, ACS Catal., 2020, 10, 14336–14348 CrossRef CAS.
- X. Shi, H.-J. Peng, T. J. P. Hersbach, Y. Jiang, Y. Zeng, J. Baek, K. T. Winther, D. Sokaras, X. Zheng and M. Bajdich, ACS Energy Lett., 2022, 7, 2228–2235 CrossRef CAS.
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC.
- M. A. Mushtaq, M. Arif, G. Yasin, M. Tabish, A. Kumar, S. Ibraheem, W. Ye, S. Ajmal, J. Zhao, P. Li, J. Liu, A. Saad, X. Fang, X. Cai, S. Ji and D. Yan, Renewable Sustainable Energy Rev., 2023, 176, 113197 CrossRef CAS.
- W. Ye, Y. Yang, M. Arif, S. Yang, X. Fang, M. A. Mushtaq, X. Chen and D. Yan, ACS Sustainable Chem. Eng., 2020, 8, 15946–15952 CrossRef CAS.
- Y. Jiang, X. Li, T. Wang and C. Wang, Nanoscale, 2016, 8, 9667–9675 RSC.
- J. Saha, D. R. Chowdhury, P. Jash and A. Paul, Chem. – Eur. J., 2017, 23, 12519–12526 CrossRef CAS PubMed.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS PubMed.
- M. T. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- G. S. Hutchings, Y. Zhang, J. Li, B. T. Yonemoto, X. Zhou, K. Zhu and F. Jiao, J. Am. Chem. Soc., 2015, 137, 4223–4229 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem. Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- P. Bothra and S. K. Pati, ACS Energy Lett., 2016, 1, 858–862 CrossRef CAS.
- K. Nejati, A. R. Akbari, S. Davari, K. Asadpour-Zeynali and Z. Rezvani, New J. Chem., 2018, 42, 2889–2895 RSC.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 RSC.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- W. Hua, H. Liu, J. G. Wang and B. Wei, Nanomaterials, 2017, 7, 433 CrossRef PubMed.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed.
- P. Jash, P. Srivastava and A. Paul, Chem. Commun., 2019, 55, 2230–2233 RSC.
- P. Jash, V. Aravind and A. Paul, New J. Chem., 2019, 43, 6540–6548 RSC.
- S. Ibraheem, G. Yasin, A. Kumar, M. A. Mushtaq, S. Ibrahim, R. Iqbal, M. Tabish, S. Ali and A. Saad, Appl. Catal., B, 2022, 304, 120987 CrossRef CAS.
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS.
- R. Gao, J. Zhu and D. Yan, Nanoscale, 2021, 13, 13593–13603 RSC.
- Z. Guo, W. Ye, X. Fang, J. Wan, Y. Ye, Y. Dong, D. Cao and D. Yan, Inorg. Chem. Front., 2019, 6, 687–693 RSC.
- C. Huang, Y. Zhong, J. Chen, J. Li, W. Zhang, J. Zhou, Y. Zhang, L. Yu and Y. Yu, Chem. Eng. J., 2021, 403, 126304 CrossRef CAS.
- R.-Y. Fan, J.-Y. Xie, H.-J. Liu, H.-Y. Wang, M.-X. Li, N. Yu, R.-N. Luan, Y.-M. Chai and B. Dong, Chem. Eng. J., 2022, 431, 134040 CrossRef CAS.
- H.-J. Liu, R.-N. Luan, L.-Y. Li, R.-Q. Lv, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 461, 141714 CrossRef CAS.
- W.-K. Gao, M. Yang, J.-Q. Chi, X.-Y. Zhang, J.-Y. Xie, B.-Y. Guo, L. Wang, Y.-M. Chai and B. Dong, Sci. China Mater., 2019, 62, 1285–1296 CrossRef CAS.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- B. Zhang, J. Zhang, X. Tan, D. Tan, J. Shi, F. Zhang, L. Liu, Z. Su, B. Han, L. Zheng and J. Zhang, Chem. Commun., 2018, 54, 4045–4048 RSC.
- R. Ma, Z. Liu, K. Takada, N. Iyi, Y. Bando and T. Sasaki, J. Am. Chem. Soc., 2007, 129, 5257–5263 CrossRef CAS PubMed.
- M. Kurmoo, Chem. Mater., 1999, 11, 3370–3378 CrossRef CAS.
- H. Ma, R. Gao, D. Yan, J. Zhao and M. Wei, J. Mater. Chem. C, 2013, 1, 4128–4137 RSC.
- Y. Zhao, H. Lin, M. Chen and D. Yan, Ind. Eng. Chem. Res., 2014, 53, 3140–3147 CrossRef CAS.
- A. M. Fogg, J. S. Dunn, S.-G. Shyu, D. R. Cary and D. O’Hare, Chem. Mater., 1998, 10, 351–355 CrossRef CAS.
- K. Zou, H. Zhang and X. Duan, Chem. Eng. Sci., 2007, 62, 2022–2031 CrossRef CAS.
- L. Wu, L. Yu, F. Zhang, D. Wang, D. Luo, S. Song, C. Yuan, A. Karim, S. Chen and Z. Ren, J. Mater. Chem. A, 2020, 8, 8096–8103 RSC.
- Y. Wang, S. Tao, H. Lin, G. Wang, K. Zhao, R. Cai, K. Tao, C. Zhang, M. Sun, J. Hu, B. Huang and S. Yang, Nano Energy, 2021, 81, 105606 CrossRef CAS.
- L. Feng, Y. Du, J. Huang, L. Cao, L. Feng, Y. Feng, Q. Liu, D. Yang and K. Kajiyoshi, Sustain. Energy Fuels, 2020, 4, 2850–2858 RSC.
- J. Ping, Y. Wang, Q. Lu, B. Chen, J. Chen, Y. Huang, Q. Ma, C. Tan, J. Yang, X. Cao, Z. Wang, J. Wu, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 7640–7645 CrossRef CAS PubMed.
- M. Rong, F. Yang, C. Yu, S. Wang, H. Zhong and Z. Cao, Colloids Surf., A, 2020, 607, 125419 CrossRef CAS.
- B. Liu, M. Zhang, Y. Wang, Z. Chen and K. Yan, J. Alloys Compd., 2021, 852, 156949 CrossRef CAS.
- Z. Wang, Y. Hu, W. Liu, L. Xu, M. Guan, Y. Zhao, J. Bao and H. Li, Chem. – Eur. J., 2020, 26, 9382–9388 CrossRef CAS PubMed.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- S. Nagappan, A. Karmakar, R. Madhu, S. S. Selvasundarasekar, S. Kumaravel, K. Bera, H. N. Dhandapani, D. Sarkar, S. M. Yusuf and S. Kundu, ACS Appl. Energy Mater., 2022, 5, 11483–11497 CrossRef CAS.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W.-X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- F. Shi, K. Zhu, X. Li, E. Wang, X. Zhu and W. Yang, J. Energy Chem., 2021, 61, 327–335 CrossRef CAS.
- A. A. Ensafi, A. Nabiyan, M. Jafari-Asl, M. Dinari, H. Farrokhpour and B. Rezaei, Energy, 2016, 116, 1087–1096 CrossRef CAS.
- B. Rezaei, H. Khosropour, A. A. Ensafi, M. Dinari and A. Nabiyan, RSC Adv., 2015, 5, 75756–75765 RSC.
- C. Li, M. Wei, D. G. Evans and X. Duan, Small, 2014, 10, 4469–4486 CrossRef CAS PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- C. Ye, M.-Q. Wang, S.-J. Bao and C. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 30887–30893 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- G. A. Ferrero, K. Preuss, A. B. Fuertes, M. Sevilla and M. M. Titirici, J. Mater. Chem. A, 2016, 4, 2581–2589 RSC.
- J. Long, J. Zhang, X. Xu and F. Wang, Mater. Chem. Phys., 2020, 254, 123496 CrossRef CAS.
- Y. Wu, M. Tariq, W. Q. Zaman, W. Sun, Z. Zhou and J. Yang, ACS Omega, 2020, 5, 7342–7347 CrossRef CAS PubMed.
- L. Guo, X. Bai, H. Xue, J. Sun, T. Song, S. Zhang, L. Qin, K. Huang, F. He and Q. Wang, Chem. Commun., 2020, 56, 7702–7705 RSC.
- Y. Liu, S. Liu, Y. Wang, Q. Zhang, L. Gu, S. Zhao, D. Xu, Y. Li, J. Bao and Z. Dai, J. Am. Chem. Soc., 2018, 140, 2731–2734 CrossRef CAS PubMed.
- C. Zhou, H. Yuan, P. Hu and H. Wang, Chem. Commun., 2020, 56, 15201–15204 RSC.
- S. E. Balaghi, C. A. Triana and G. R. Patzke, ACS Catal., 2020, 10, 2074–2087 CrossRef CAS.
- M. Gong, Z. Deng, D. Xiao, L. Han, T. Zhao, Y. Lu, T. Shen, X. Liu, R. Lin, T. Huang, G. Zhou, H. Xin and D. Wang, ACS Catal., 2019, 9, 4488–4494 CrossRef CAS.
- C. Wang, H. Xu, H. Shang, L. Jin, C. Chen, Y. Wang, M. Yuan and Y. Du, Inorg. Chem., 2020, 59, 3321–3329 CrossRef CAS PubMed.
- N. A. Jose, H. C. Zeng and A. A. Lapkin, Nat. Commun., 2018, 9, 4913 CrossRef PubMed.
- H. Xu, H. Shang, C. Wang, L. Jin, C. Chen and Y. Du, Nanoscale, 2020, 12, 2126–2132 RSC.
- L. Su, Y. Zhao, F. Yang, T. Wu, G. Cheng and W. Luo, J. Mater. Chem. A, 2020, 8, 11923–11927 RSC.
- M. Rong, H. Zhong, S. Wang, X. Ma and Z. Cao, Colloids Surf., A, 2021, 625, 126896 CrossRef CAS.
- S. Nagappan, A. Karmakar, R. Madhu, H. N. Dhandapani, K. Bera, A. De and S. Kundu, ACS Appl. Energy Mater., 2022, 5, 12768–12781 CrossRef CAS.
- L. Lorencova, T. Bertok, E. Dosekova, A. Holazova, D. Paprckova, A. Vikartovska, V. Sasinkova, J. Filip, P. Kasak, M. Jerigova, D. Velic, K. A. Mahmoud and J. Tkac, Electrochim. Acta, 2017, 235, 471–479 CrossRef CAS PubMed.
- X. Liu, P. Su, Y. Chen, B. Zhu, S. Zhang and W. Huang, New J. Chem., 2018, 42, 9449–9454 RSC.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- Z. Ba, Y. Han, D. Qiao, D. Feng and G. Huang, Ind. Eng. Chem. Res., 2018, 57, 15225–15233 CAS.
- W. Zhang, Y. Li, L. Zhou, Q. Zheng, F. Xie, K. H. Lam and D. Lin, Electrochim. Acta, 2019, 323, 134595 CrossRef CAS.
- A. Karmakar, K. Karthick, S. Kumaravel, S. S. Sankar and S. Kundu, Inorg. Chem., 2021, 60, 2023–2036 CrossRef CAS PubMed.
- M. Dinari, H. Allami and M. M. Momeni, J. Electroanal. Chem., 2020, 877, 114643 CrossRef CAS.
- W. Guo, Y. E. L. Gao, L. Fan and S. Yang, Chem. Commun., 2010, 46, 1290–1292 RSC.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- K. Li, B. Zhao, J. Bai, H. Ma, Z. Fang, X. Zhu and Y. Sun, Small, 2020, 16, 2001974 CrossRef CAS PubMed.
- C. Ye, J. Liu, Q. Zhang, X. Jin, Y. Zhao, Z. Pan, G. Chen, Y. Qiu, D. Ye, L. Gu, G. I. N. Waterhouse, L. Guo and S. Yang, J. Am. Chem. Soc., 2021, 143, 14169–14177 CrossRef CAS PubMed.
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X.-Q. Yang, H. Wang and X. Sun, Adv. Energy Mater., 2018, 8, 1701694 CrossRef.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, Chem. Sci., 2017, 8, 3484–3488 RSC.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- N. Yan, L. Hu, Y. Li, Y. Wang, H. Zhong, X. Hu, X. Kong and Q. Chen, J. Phys. Chem. C, 2012, 116, 7227–7235 CrossRef CAS.
- M. Xu, F. Wang, M. Zhao, S. Yang and X. Song, Electrochim. Acta, 2011, 56, 4876–4881 CrossRef CAS.
- N. Baliarsingh, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2013, 1, 4236–4243 RSC.
- A. S. Batchellor and S. W. Boettcher, ACS Catal., 2015, 5, 6680–6689 CrossRef CAS.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- J. Saha, A. Kumar, A. Pm and V. Jakhad, Chem. Commun., 2020, 56, 8770–8773 RSC.
- C. Duan, X. Li, D. Wang, Z. Wang, H. Sun, R. Zheng and Y. Liu, Sustainable Energy Fuels, 2022, 6, 1479–1488 RSC.
- A. Q. Mugheri, A. Tahira, U. Aftab, M. I. Abro, S. R. Chaudhry, L. Amaral and Z. H. Ibupoto, RSC Adv., 2019, 9, 5701–5710 RSC.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CrossRef CAS.
- M. Arunkumar and A. Paul, ACS Omega, 2017, 2, 8039–8050 CrossRef PubMed.
- G. Wu, N. Li, D. Zhou, K. Mitsuo and B. Xu, Solid State Chem., 2004, 177, 3682–3692 CrossRef CAS.
- C. Singh, I. Liberman, R. Shimoni, R. Ifraemov and I. Hod, J. Phys. Chem. Lett., 2019, 10, 3630–3636 CrossRef CAS PubMed.
- A. Fan, C. Qin, X. Zhang, X. Dai, Z. Dong, C. Luan, L. Yu, J. Ge and F. Gao, ACS Sustainable Chem. Eng., 2019, 7, 2285–2295 CrossRef CAS.
- Y. Zheng, R. Gao, Y. Qiu, L. Zheng, Z. Hu and X. Liu, Inorg. Chem., 2021, 60, 5252–5263 CrossRef CAS PubMed.
- Q. Wang and D. O’Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- K. Bera, A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, Inorg. Chem., 2021, 60, 19429–19439 CrossRef CAS PubMed.
- D. A. T. Gary and L. Miessler, Inorganic Chemistry, Prentice Hall, Pearson, 1999 Search PubMed.
- J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhi, Inorganic Chemistry: Principles of Structure and Reactivity, Pearson Education, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental details, schemes for synthesis, structural characterization and catalytic activity of the catalysts, figures and tables. See DOI: https://doi.org/10.1039/d3ma00324h |
| This journal is © The Royal Society of Chemistry 2023 |