
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D printed SrNbO2N photocatalyst for degradation of organic pollutants in water†
Antonio
Iborra-Torres
 a,
Matej
Huš
bcd,
Kiem
Nguyen
e,
Antonis
Vamvakeros
fg,
Muhammad Tariq
Sajjad
e,
Steven
Dunn
e,
Myrjam
Mertens
h,
Simon
Jacques
f,
Andrew M.
Beale
fgi,
Blaž
Likozar
b,
Geoffrey
Hyett
a,
Suela
Kellici
e and
Vesna
Middelkoop
*h
a,
Matej
Huš
bcd,
Kiem
Nguyen
e,
Antonis
Vamvakeros
fg,
Muhammad Tariq
Sajjad
e,
Steven
Dunn
e,
Myrjam
Mertens
h,
Simon
Jacques
f,
Andrew M.
Beale
fgi,
Blaž
Likozar
b,
Geoffrey
Hyett
a,
Suela
Kellici
e and
Vesna
Middelkoop
*h
aDepartment of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
bDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, SI-1001 Ljubljana, Slovenia
cAssociation for Technical Culture of Slovenia, Zaloška 65, SI-1001 Ljubljana, Slovenia
dResearch Institute, Conservation Centre, Institute for the Protection of Cultural Heritage of Slovenia, Poljanska 40, SI-1000 Ljubljana, Slovenia
eLondon Centre for Energy Engineering, School of Engineering, London South Bank University, London SE1 0AA, UK
fFinden, Building R71, Harwell Campus, Oxfordshire, OX11 0QX, UK
gDepartment of Chemistry, University College London, London WC1H 0AJ, UK
hSustainable Materials Management, Flemish Institute for Technological Research (VITO), B-2400 Mol, Belgium. E-mail: vesna.middelkoop@vito.be
iResearch Complex at Harwell, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxon OX11 0FA, UK
First published on 13th March 2023
Abstract
Organic pollutants in water are a major concern for the environment and human health, and require urgent attention. Here, we developed for the first time monolithic structures by 3D printing of perovskite metal oxynitride, SrNbO2N, for photocatalytic degradation of organic pollutant in water. Advanced, synchrotron-based XRD-CT measurements were employed to gain structural insight into photocatalyst formulation and assess the fidelity of design in terms of both the chemical and physical form of the photocatalysts to be imaged. Our 3D printed material showed excellent photocatalytic activity, degrading 100% of methylene blue (MB) as well as good stability for three cycle operations. This is due to high adsorption of the 3D printed oxynitride towards MB which enhanced its photoredox reactivity. It is also evident from the excellent charge transfer demonstrating a charge transfer rate of (1.5 ± 0.2) × 108 s−1. We performed Time-Dependent Density Functional Theory (TD-DFT) calculations to understand the photocatalyst structure and degradation pathways. Our calculated band gap (at Γ) of 1.88 eV is in good agreement with the experimental values. We found that the highest valence bands were contributed by N p orbitals and the lowest conduction bands corresponded to Nb d orbitals offering avenues for fine-tuning the band gap. Hence, the ability to tailor photocatalyst monoliths by 3D printing renders their water treatment application more facile compared to their powder suspension counterparts.
1 Introduction
Metal oxynitrides with the general formula ABO2N (where A = Sr, Ba, Ca; B = Nb, Ta) are promising materials that combine the advantages of oxides and nitrides, leading to small band gaps that make them suitable as visible light photocatalysts.The mechanism of photocatalytic degradation typically involves the generation of electron–hole pairs by absorbing photons from light, followed by the transfer of these charges to the surface of the photocatalyst, where they can participate in redox reactions. Perovskite oxynitrides have been shown to have a wide band gap and can be excited by UV light, leading to the generation of electron–hole pairs. Fig. 1 illustrates how these charges, when reacting with water molecules, lead to the production of reactive oxygen species (ROS), such as hydroxyl radicals (˙OH). The ROS have the ability to oxidize and degrade organic compounds, including dyes.1 These photocatalytic properties (including a visible light absorption band) make perovskite oxynitrides (for example Sr, Nb and Ti based) promising materials for water splitting2 and the degradation of organic pollutants and biological contaminants in water3–5 including degradation of organic compounds such as acetone (LaTi(O,N)3),6 methyl orange (CaTaO2N;7 CaNbO2N and SrNbO2N)8 and methylene blue,9 among others.10
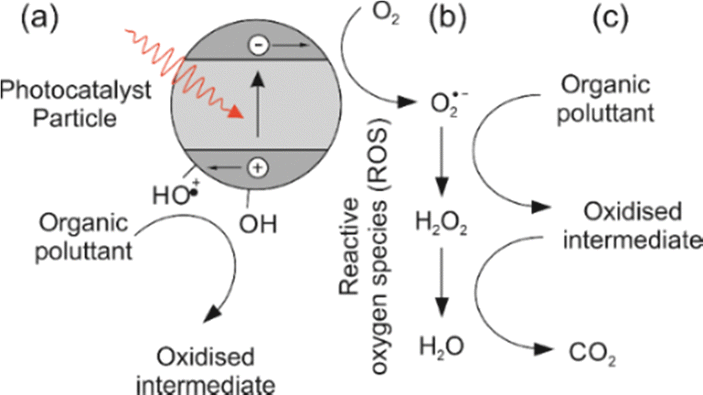 | ||
| Fig. 1 Generation of reactive oxygen species (ROS) and process of photo-oxidation of an organic pollutant. Step (a) is absorption of a photon to generate an excited electron–hole pair which can then migrate to the surface of the photocatalyst particle, where (b) the electron can then reduce atmospheric oxygen to the more labile superoxide radical. In step (c) the superoxide radical and subsequent ROS can oxidise an organic pollutant to CO2. The holes can form surface bound hydroxyl radicals which can also directly oxidise organic molecules.1 | ||
Previously we reported8 seven tantalum and niobium oxynitrides ABON2 (where A = Ca, Sr, Ba; B = Nb or Ta) for self-cleaning reactions using dichloroindophenol (in the presence of a sacrificial electron donor) and stearic acid as test pollutants. Although all of the oxynitride samples showed the ability to degrade the test pollutant, they did so poorly with at best 1.5% of the dye being degraded after 3 hours. However, after decorating the oxynitride with co-catalyst particles of cobalt oxide, a significantly higher activity was observed. All of the oxynitrides were active, with the most active being SrNbO2N that degraded 25% of the dye after 3 hours – a twenty fold increase compared to the sample without the co-catalyst.
In recent years, there have been a number of examples of 3D structured photocatalytic reactors using TiO2 deposited on a solid foam.11,12 No examples of 3D printed oxynitride-based photocatalysts have been reported to date, only a very limited number of other photocatalyst compositions.13–16 The photocatalytic microreactors based on microfluidic technology that use a catalyst dispersed in powder form, have been extensively reported.17 However, it requires an additional step to separate the catalyst from the products, thus leading to an increase in the experimental cost and time. 3D printed photocatalytic structures can resolve the challenges associated with the immobilisation of the photocatalyst as well as its recovery and ease of product separation. In addition, 3D printing allows complex shapes and optimised geometries to be easily tailored, thereby facilitating the flow patterns and efficient light management. One of the major advantages of the orderly-structured micro-channel catalytic or adsorbent beds over randomly powder packed beds is an increased mass transfer, reduced pressure drop and higher selectivity per cubic metre of reactor volume, by their controlled porosity and controlled product distribution.18–21 Examples of the development and characterisation of the 3D printed microchannel catalysts can be found in our previous work.22–25
This work describes the development, characterisation and modelling of oxynitride SrNbO2N material that was 3D printed and employed for the photodegradation of a simulant pollutant (methylene blue dye in aqueous solution) under simulated solar light irradiation. For that, a printing ink containing SrNbO2N was first formulated followed by the deposition of stacked fibres using direct ink writing (DIW) to form a monolithic photocatalyst. This monolith was evaluated for its efficiency to generate reactive oxygen species and to degrade pollutant (MB) in aqueous solution. It should be noted that relevant scientific literature suggest that MB is a suitable standard to test the photocatalytic performance of materials.26 The 3D printed monolith showed excellent photocatalytic activity with 100% degradation of pollutant within just 3.5 hours. The photophysical study shows excellent charge transfer between the 3D printed perovskite (SrNbO2N oxynitride) structure and MB. The DFT calculations of photocatalytic structure and degradation pathways show good agreement with experimental data.
2 Materials and methods
2.1 Synthesis of SrNbO2N powder
Perovskite metal oxynitride SrNbO2N was synthesised by ammonolysis of its respective oxide precursor. The oxide precursor was synthesised via the polymeric precursor method (PPM). For this, NbCl5 (0.89 mmol, Sigma-Aldrich, 99%) was dissolved in 6.5 mL of methanol. Then, SrCO3 (0.89 mmol, Sigma-Aldrich, 99.99%) was added and stirred until complete dissolution. Finally, citric acid (19.86 mmol, Fisher) and propylene glycol (76.8 mmol, Sigma-Aldrich, 99.5%) were added to the Sr–Nb-containing solution while keeping the ratio of metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid:propylene glycol at 1:11:43.
:citric acid:propylene glycol at 1:11:43.
The solution was then heated at 120 °C for 90 minutes to allow the complete evaporation of the methanol, resulting in formation of a transparent resin. The gel containing the metal cations was placed into an alumina crucible and pre-calcined in air for 1 h at 350 °C, followed by 1 h calcination at 650 °C and finally 2 h at 800 °C at a constant heating rate of 10 °C min−1 at each step. The heating cycle converted the gel into a white metal oxide powder. The resultant oxide powder in the alumina crucible was heated (3 °C min−1 ramp rate) and nitrided inside a tube furnace under an ammonia flow (of 250 mL min−1 anhydrous, BOC, 99.98%) for 15 h at 850 °C. The SrNbO2N product was cooled down under flowing ammonia to avoid its re-oxidation.
2.2 3D printing of SrNbO2N powder
For the design of the photocatalyst monoliths, matrix-like structures were produced using a rapid prototyping/3D printing process commonly referred to as Direct Ink Writing (DIW). The monolith manufacturing steps are represented in Fig. 2. The photocatalyst monolith was deposited layer-by-layer by moving the nozzle in x–y–z and laying down the ink in the desired pattern. The printing ink was prepared from the active 5 wt% SrNbO2N material pre-mixed with 95 wt% Puralox alumina (Sasol TM100/150 UF) by adjusting the target ratios of 44 wt% of an organic binder component (3.5% aqueous solution of methylcellulose) and 7.7 wt% of 1 mol L−1 HNO3(aq) solution in the mixture. | ||
| Fig. 2 (left) SrNbO2N-containing printing ink prepared by mixing SrNbO2N/Al2O3 power, organic binder and acid solution to obtain a printable viscosity; resulting 3D printed monolith freshly printed, dried and thermally treated (viewed from left to right). | ||
The 3D printed photocatalysts were subsequently subjected to a heat treatment and ammonolysis to remove the organic binder component and obtain the final desired structure and photocatalytic properties. For the removal of the organic binder component, the 3D printed monolith was heated under a flow of air (250 mL min−1) at 650 °C for 2 hours (heating rate of 10 °C min−1).
For the deposition of the Cobalt oxide co-catalyst, the 3D printed monolith was immersed in 1.12 mL of a 0.009 M Co(NO3)2 (Sigma-Aldrich, 98%) water solution aiming at 2% wt loading. The suspension was treated for 5 minutes in an ultrasound bath and heated to promote the evaporation of the water. The resulting monolith was treated under flowing ammonia (250 mL min−1) for 15 hours at 850 °C (heating rate of 3 °C min−1). Finally, the co-catalyst was activated via mild oxidation in air at 200 °C for 1 h.
2.3 X-ray diffraction computed tomography (XRD-CT)
XRD-CT measurements were performed at beamline ID15A27 of the ESRF using a 100keV monochromatic X-ray beam focused to have a spot size of ca. 40 ×20μm (horizontal × vertical). 2D powder diffraction patterns were acquired using the Pilatus3 X CdTe 2M hybrid photon counting area detector. The 3D printed photocatalyst was mounted onto a goniometer which was placed on a rotation stage itself mounted on top of a translation stage. The sample-to-detector distance was approximately 0.8 m. The XRD-CT scans were measured by performing a series of zigzag line scans.28 An exposure time of 10 ms and an angular range of 0–180° with 300 projections in total were used for the XRD-CT dataset. The fast axis was the translation axis (fly scanning) and slow axis was the rotation axis. A translation step size of 100 microns was applied; in total 330 translation steps were made per line scan corresponding to a covered area of 3.3 ×3.3 cm2. The detector calibration was performed using a CeO2 standard. Every 2D diffraction image was converted to 1D powder diffraction pattern using the nDTomo and PyFAI software packages with implemented trimmed mean filter (10%) to remove the artefacts due to hot spots of (single) crystalline material.29–32 The data integration was performed with fast GPU processing. The reconstructed images were obtained using the filtered back projection (FBP) algorithm. The collected data were corrected in respect to synchrotron current. The integrated diffraction patterns were reshaped into sinograms and centered; the air scatter signal was subtracted from the data. The voxel size in the reconstructed images (330 × 330 pixels) corresponded to 100 × 100 × 20 μm3.
The analysis of diffraction data was performed with the TOPAS v7 software33 on a voxel-by-voxel basis. A pseudo-Voigt peak shape function calculated after the analysis of the CeO2 calibrant pattern was used for diffraction data. The data were also analysed using the Direct Least-Squares Reconstruction (DLSR) approach34 which overcomes the parallax artefact. Due to the memory expensive requirements of the DLSR approach, the data had to be downscaled by a factor of two to 165 × 165 pixels. This means that the pixel size in the reconstructed DLSR maps corresponds to 200 microns. The downscaled sinograms (containing parallax artefact) were used to reconstruct images using the conventional approach and perform analysis using the Rietveld method. To stabilize the DLSR refinements, the SrNbO2N scale factor map obtained from the conventional analysis was used as an input to the DLSR. A mask was created based on the SrNbO2N scale factor map which allowed us to decrease the number of structures needed to be refined. A 3XS Data Science Workstation C264X2 with 2× Intel Xeon Silver 4216 and 350 GB RAM was used to perform the refinements for both the conventional approach (FBP + Rietveld) and the DLSR approach.
2.4 Conventional characterisation
Conventional characterisation of the photocatalyst monolith was carried out using SEM, EDX, XRD. SEM and EDX images were recorded on a FEI Nova Nano SEM 450 instrument equipped with a Bruker Quantax 200 system featuring the XFlash 5030 detector and operating at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) analysis of the starting SrNbO2N powder was performed using a JEOL JEM 1010 instrument operated at an acceleration voltage of 100 kV. High resolution transmission electron microscopy (HRTEM) analysis of the 3D printed photocatalyst monolith after the degradation tests was obtained using a JEOL JEM 2100 instrument at an acceleration voltage of 200 kV. Powder XRD patterns were measured on a PANalytical Empyrean diffractometer equipped with a Co-tube and operated at 40 keV and 45 mA.2.5 Photocatalytic experiments
The photocatalytic activity of the 3D printed photocatalyst was investigated by the degradation of methylene blue (MB) in water employing a solar-simulator setup consisting of a quartz tungsten halogen lamp (Osram 1 kW R7s 22000) cooled by an electric fan.
Characteristically, the 3D printed photocatalyst (2.29 g) was suspended in MB aqueous standard solution (0.04 mM, 0.4 L) in a photocatalytic reactor (glass beaker, Corning Pyrex Griffin). To ensure the establishment of an adsorption–desorption equilibrium, the reaction mixture was agitated in the dark for 30 minutes. It was kept at a distance (27 cm) from the light source and constantly agitated throughout the photocatalytic process. The sample was irradiated for 210 minutes. During the irradiation procedure, aliquots were taken at 30 minutes intervals, and analysed using UV-vis spectrophotometry (Shimadzu UV-1800) to determine MB photodegradation (by changes observed at λabs = 665 nm). A calibrated correlation between the measured absorbance and its concentration was used for the quantitative determination of MB. The rate of direct degradation of MB was measured in the absence of the catalyst and was found to be negligible. The adsorption coefficient, qt (the adsorbed mass (mg) of MB per mass (g) of catalyst, at time (t) was calculated according to the following equation):35
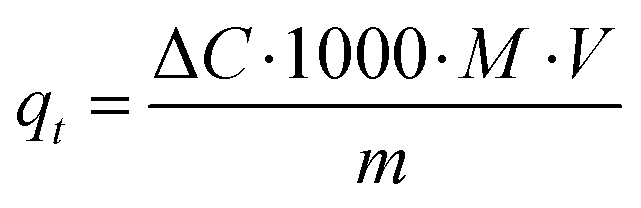 | (1) |
To assess the catalyst's efficiency over time, the 3D printed monolith was recycled and then re-exposed to irradiation, using the same testing approach as previously described. The sample was recovered after each run by periodically washing it with ethanol followed by drying. To investigate the photocatalytic degradation of MB by the 3D photocatalytic monolith the following parameters were considered:, the initial contaminant (dye) concentration, initial pH solution, the presence of oxidants and electron acceptors, photocatalyst concentration, photocatalyst particle size, reactor/structured matrix dimensions, reaction temperature as well as light intensity.
2.6 Transient photoluminescence spectroscopy
Time-resolved photoluminescence (TRPL) decays were measured using Edinburgh Instrument FLS 1000 by exciting the samples at 375 nm using picosecond laser (EPL-375) and detecting the emission at 725 nm.2.7 Theoretical calculations
For ab initio calculations of electronic properties of MB and its degradation productions with the LCAO method, Gaussian 16 was used. For ground state calculations, a density functional theory (DFT) approach with a hybrid functional M06-2X36 and People's37–40 basis set 6-311+G was taken. The Minnesota functionals are known to describe thermochemistry of the main groups well.41 Solvation was accounted for implicitly, using the SMD variation of the integral equation formalism (IEFPCM) and using the dielectric constant for water.42 For excited molecules on account of photoactivation, only the first excited state was considered according to Kasha's rule. Time-dependent (TD) DFT was used.43 Using this approach, we calculate the first excited state to lie 1.95 eV above the ground state, corresponding to an adsorption band at 636 nm, which is in good agreement with the experimentally measured absorption peak around 665 nm.44Intermediates were geometrically optimized until the forces on all atoms dropped below 1.5 × 10−5 hartree bohr−1. Vibrational analysis was performed to confirm that stable stationary points were found. Photocatalytic degradation of MB was modelled as an attack of the OH* radical on the ground state and excited MB molecule in solution.
For catalyst structure calculations, plane-wave DFT as implemented in VASP 5.4.1 was used.45,46 Preliminary calculations, including geometry optimizations, were performed with a GGA functional (PBE) with the projector-augmented wave method (PAW).47,48 To account for d electrons of niobium, a Hubbard value of 3 eV was used in the DFT+U approach,49 although the effect of this value is small.50 For the plane waves expansion, a cut-off of 500 eV was employed. Dispersion interaction was described with the Grimme D3 correction. For band structure and band gap evaluation, single point calculations with a hybrid functional (HSE06) were performed on the PBE-level optimized structures.51 A gamma centered Monkhorst–Pack mesh 11 × 11 × 7 was sufficient to describe a 5.81 × 5.81 × 8.18 Å unit cell, which agrees well with the experimental determination (5.71 × 5.71 × 8.10 Å).50,52,53
3. Results and discussion
3D printed SrNbO2N monolith was characterised using UV-vis spectrophotometry, transient photoluminescence spectroscopy, XRD-CT, SEM-EDX and hybrid-DFT calculations. The photocatalytic performance of the 3D printed SrNbO2N monolith was investigated using methylene blue as pollutant dye, under simulated solar light irradiation.3.1 XRD-CT characterisation
The reconstructed images (Fig. 3) for the 3D printed photocatalyst were investigated with the aim to provide the physicochemical information and distribution of the active SrNbO2N phase. The results were obtained from the Rietveld analysis of the XRD-CT data for the SrNbO2N phase using both the conventional and DLSR approaches.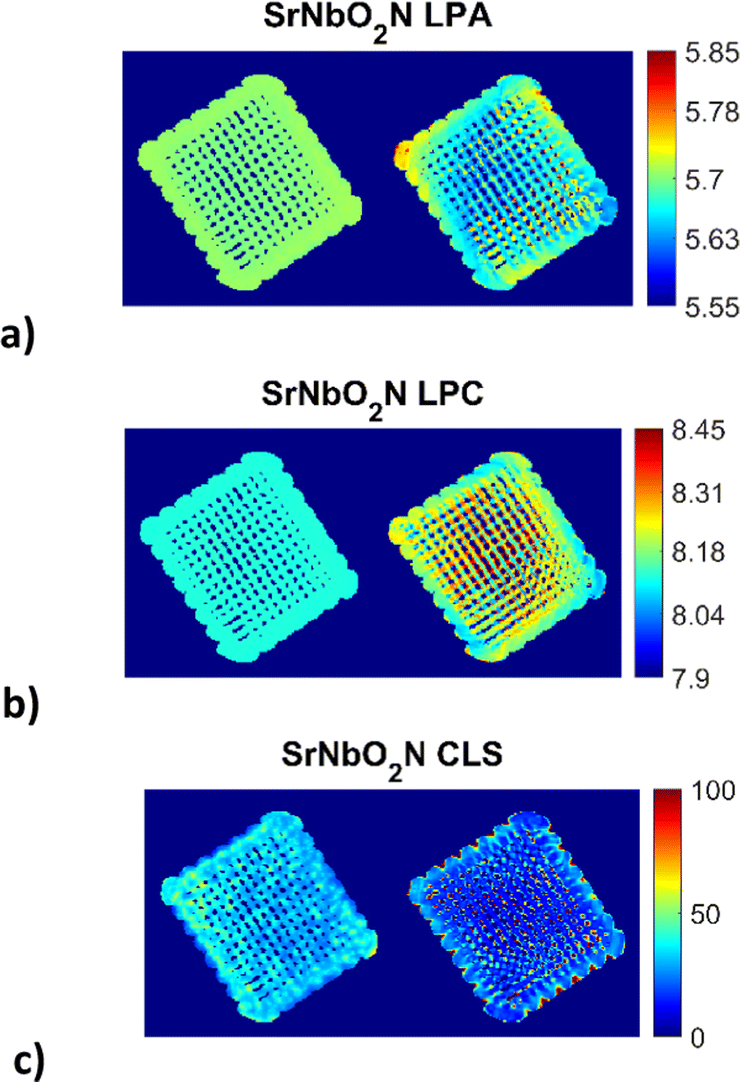 | ||
| Fig. 3 Reconstructed XRD-CT images of the 3D printed photocatalyst based on the Rietveld analysis of four isolated SrNbO2N peaks (single phase model) using the conventional approach (left) and the DLSR approach (right) shown in each of the three sub-figures: (a) lattice parameter a (LPA) maps (colour bar axis in Å), (b) lattice parameter c (LPC) maps (colour bar axis in Å), (c) crystallite (CLS) maps (colour bar axis in nm). Every pixel in the reconstructed XRD-CT image corresponds to a single diffraction pattern. The pixel size in the reconstructed DLSR maps corresponds to 200 μm as the data had to be downscaled by a factor of two to 165 × 165 pixels. Accompanying figures can be found in the ESI† (see Fig. S1 and S2). | ||
The XRD-CT data on the left-hand side of each sub-figure was obtained using the conventional approach that presents the reconstructed images based on the FBP algorithm and the analysis of the local diffraction patterns which is based on the Rietveld method. This type of analysis can lead to accurate scale factor and weight fraction maps for the various crystalline components in the sample despite the parallax artefacts present in the data. One global scale factor was refined for the SrNbO2N phase, i.e., the distribution of the component was forced to be the one obtained from the conventional approach. However, a scale factor was applied to this map which was refined during the DLSR analysis (presented on the right hand-side of each sub-figure). The reconstructed maps of the lattice parameters and crystallite size of the SrNbO2N phase were refined spatially. For both the conventional FBP-Rietveld and DLSR approaches, a selected region of the diffraction patterns was used: Q = 2.01–2.3, 3.58–3.93, 4.74–5 and 5.6–5.9 Å−1. These regions contained isolated SrNbO2N diffraction peaks (including the (112), (204), (420), (116), (316) and (512) reflections) which allowed to avoid using the difficult-to-model Al2O3 support phase in the refinement process. The weighted profile R-factor (Rwp) is ca. 8–9% for most pixels. The overall Rwp for the global DLSR refinement is similar too, at 8.8%. ensuring that the comparison of the reconstruction algorithms was not biased.
It can be observed that for the conventional approach to XRD-CT, image analysis leads to more uniform and underestimated values for the lattice parameters while local inhomogeneities can be observed across the sample by the DLSR approach. The DLSR approach accounts for the loss of physico-chemical information associated with diffraction peak shape and position and directly yields the varying lattice parameter and crystallite size throughout the scanned cross section. The values of the lattice parameters a and c vary from 5.60–5.80 and 8.04–8.32 Å respectively. These values are in agreement with lattice parameters results obtained from the bulk sample by the conventional XRD measurements (presented in ESI,† Fig. S3 and S4): 5.71 and 8.12 Å for a and c respectively. Both the conventional and XRD-CT diffraction patterns were indexed with the space group I4/mcm.54 The DLSR approach yields lower and more uniform values for the crystallite size. Fig. S2 in the ESI† shows a histogram of the crystallite size distribution obtained from the DLSR CLS map.
3.2 Conventional characterisation
In addition to the crystallite (CLS) maps Fig. S6–S8 in the ESI† provide complementary micron-scale SEM and EDX maps of the microstructure and distribution of active SrNb phase across the alumina support of the 3D printed photocatalyst. TEM and HRTEM images (Fig. S9 and S10, ESI†) show the photocatalyst nanoparticles of the starting SrNbO2N powder and the 3D printed monolith material after the degradation tests, respectively.3.3 Photocatalytic experiments
The 3D printed monolith was tested for photodegradation of MB under a solar light simulator. The conversion of MB (C = final concentration to C0 = initial concentration, (C/C0 × 100)) was recorded during 210 min solar light irradiation period (see Fig. 4). The photocatalytic reaction rate was calculated using first-order kinetics (ln(C/C0) = −kt), where ln(C/C0) vs. irradiation time was linear in all cases. The rate constant, k, was determined from the slope of the best fit line. The photo-response data are summarised in Table 1.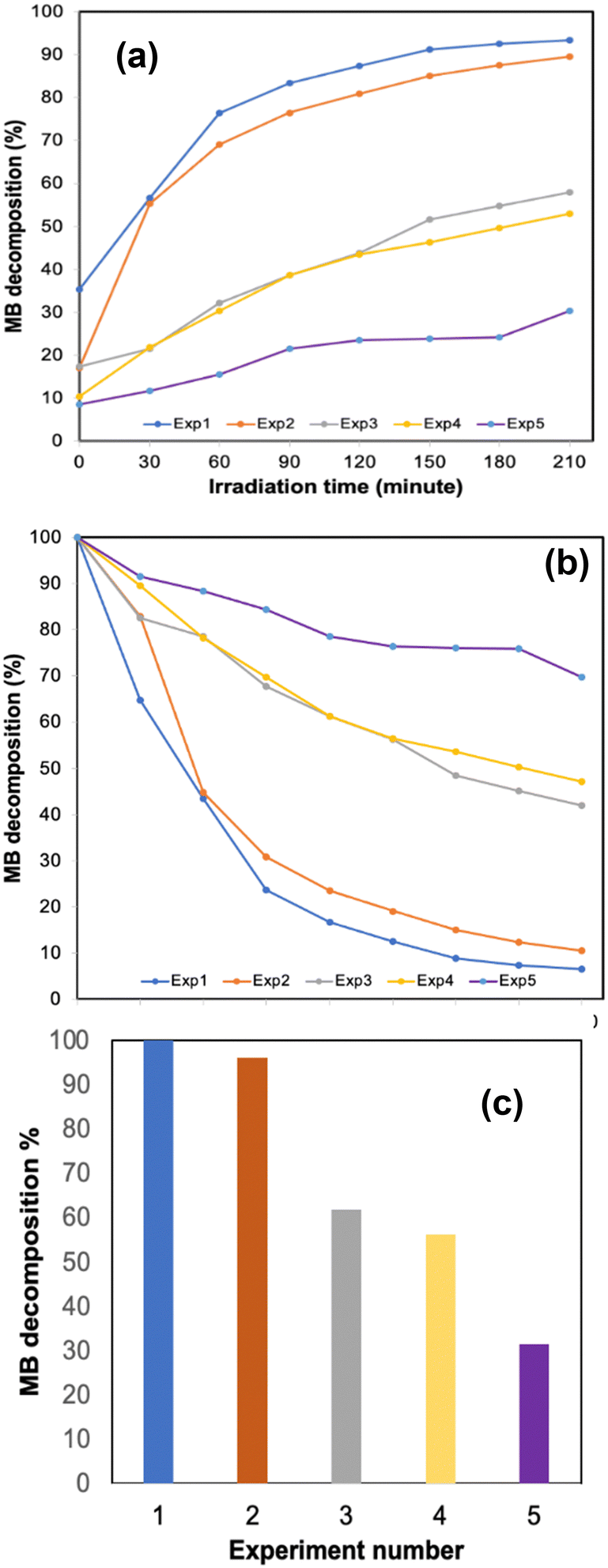 | ||
| Fig. 4 Methylene blue decomposition % (C/C0 ×100) over time (a) after post-adsorption equilibrium (MB/3D monolith mixture was kept 30 min in the dark; this was assigned as t = 0) and (b) catalytic activity for the entire duration of the experiment (including the adsorption time) (c) the final MB decomposition % with catalyst cycling runs. | ||
| Experiment | Rate constant (1014 s−1) | MB photodegradation (%) | q t |
|---|---|---|---|
| 1st photocatalytic activity | 4 | 100 | 2.23 |
| 2nd photocatalytic activity | 3 | 96 | 2.14 |
| 3rd photocatalytic activity | 0.6 | 62 | 1.38 |
| 4th photocatalytic activity | 0.5 | 56 | 1.26 |
| 5th photocatalytic activity | 0.2 | 32 | 0.70 |
The 3D printed monolith demonstrated excellent photocatalytic activity, with ca. 100% degradation of MB in the allotted 210 minutes time period and a significant two-fold enhancement when compared to our previously reported work, in which we tested photodegradation of blue DCIP dye in the presence of a sacrificial electron donor.8 The 3D printed oxynitride also exhibited adsorption toward MB, allowing it to effectively react with photogenerated active species on the surface of the 3D printed oxynitride, enhancing photoredox activity. It should be noted that, a control experiment carried out under the same conditions but without the addition of photocatalyst, provided an assessment of the extent of direct photochemical reaction of MB which was proved to be negligible when compared with that in the presence of the catalyst (see Fig. S11 in the ESI;† Fig. S12, ESI† shows the absorption spectra of MB degradation in the absence of the catalyst). Irradiance curve (irradiance readings as a function of wavelength) for the light source used in the experiments shown Fig. 4–6 can be found in ESI,† Fig. S13. The Tauc plot for the SrNbO2N photocatalyst used as a conventional method for extracting the band gap of a material is presented in ESI,† Fig. S14.
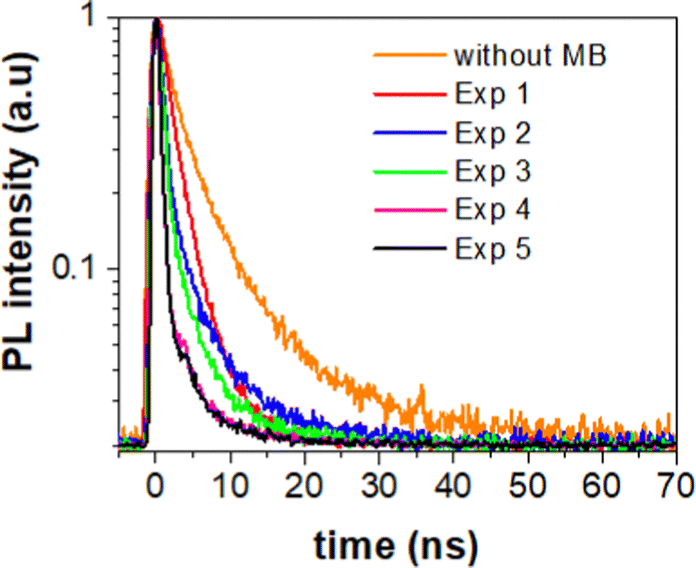 | ||
| Fig. 5 PL decay spectra of 3D monolith without (orange line) and with MB (red, blue, green, magenta and black lines). Red line corresponds to PL decay measured after the first photocatalytic activity run, blue line is PL decay of 3D monolith measured after 2nd photocatalytic activity run, green line represents PL decay of 3D monolith measured after 3rd photocatalytic activity experiment, magenta line represents PL decay of 3D monolith measured after 4th photocatalytic activity experiment and black line represents PL decay of 3D monolith measured after 5th photocatalytic activity experiment. | ||
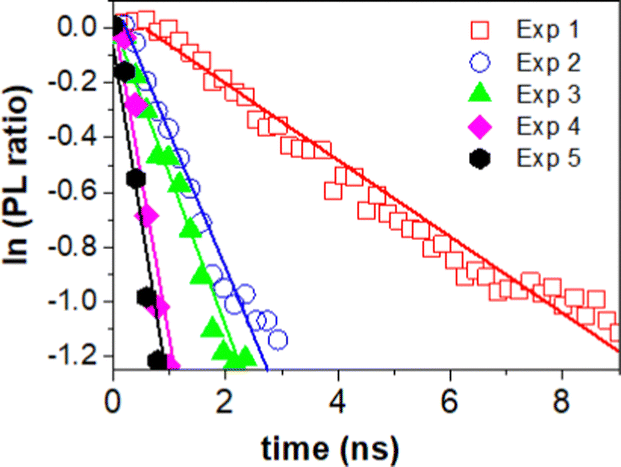 | ||
| Fig. 6 Natural logarithm of the PL ratio of 3D monolith with and without MB. Red dots – PL ratio with MB after first photocatalytic activity. Blue dots – PL ratio with MB after 2nd photocatalytic activity experiment. Green dots – PL ratio with MB after 3rd photocatalytic activity experiment. Magenta dots – PL ratio with MB after 4th photocatalytic activity experiment. Black dots – PL ratio with MB after 5th photocatalytic activity experiment. | ||
One important feature associated with the usage of photocatalysts is long term stability and their cyclic use. To evaluate the efficiency over time, a series of experiments were conducted where the 3D printed monolith was recycled and subjected to photocatalytic testing under solar light. The change in methylene blue concentration is shown in Table 1. The 3D printed monolith exhibited good stability for 3 cycle operations, reaching an adsorption plateau for cycles 2 and 3, while remaining efficient in terms of photocatalytic degradation of MB.
It should be noted that cobalt oxide, when used (even in low concentrations) as a co-catalyst with perovskite oxynitrides, can enhance the photocatalytic activity of the perovskite oxynitride by acting as a promoter and providing additional active sites for the adsorption and activation of reactants. The CoOx species (shown in Fig. S8, ESI†) can also facilitate the transfer of electrons and holes between the perovskite oxynitride and dye molecules, leading to more efficient degradation as previously described by the authors.8
3.4 Transient photoluminescence spectroscopy characterisation
To extract the information about the charge transfer, we performed time-resolved photoluminescence (PL) spectroscopy. For that, we first measured natural PL decay of 3D printed monolith in the absence of methylene blue (MB) and used it as a reference (black line). We then measured PL decay in the presence of MB after the photocatalytic activity experiment (red line) as shown in Fig. 5. The PL decays in the presence of MB gets faster due to the charge transfer between 3D printed SrNbO2N and MB.We determined the lifetimes by fitting the data with multi-exponentials. The equation and parameters are provided in the ESI† (see eqn (S1) and Table S1).
The charge transfer can be quantitatively determined from the additional rate of decay in the presence of MB (quencher).55,56 We determine this rate of quenching by taking the differentiation or the slope of the natural logarithms of the PL ratio of 3D monolith with MB and without MB (Fig. 6) using:
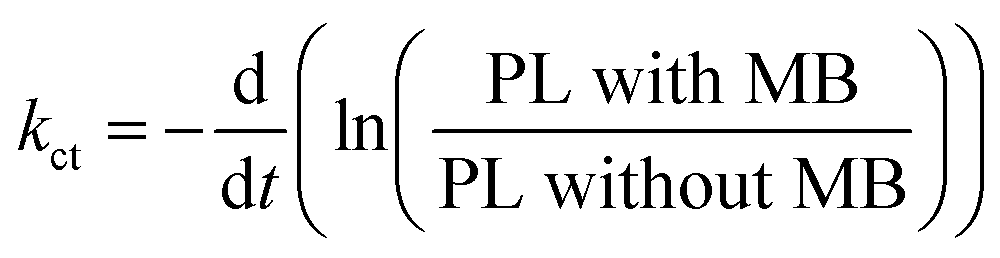 | (2) |
The resulting charge transfer rate of (1.5 ± 0.2) × 108 s−1 after first photocatalytic activity, (5.0 ± 0.5) × 108 s−1 after 2nd photocatalytic activity, (5.6 ± 0.6) × 108 s−1 after 3rd photocatalytic activity experiment, (1.2 ± 0.2) × 109 s−1 after 4th photocatalytic activity experiment and (1.3 ± 0.1) × 109 s−1 after 5th photocatalytic activity experiment.
3.5 Regioselectivity in the methylene blue molecule
There are nine distinct sites in methylene blue, which are susceptible to a radical attack: six aromatic carbon atoms (numbered 1, 1a, 2, 3, 4, 4a), the nitrogen (5) and sulphur atoms (6) in the aromatic ring and the amine nitrogen atom (7), as shown in Fig. 7.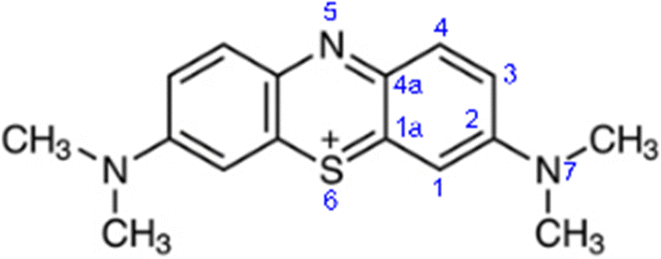 | ||
| Fig. 7 Numbering of the heavy atoms in MB. | ||
In studying photodegradation, we are most interested in the changes in reactivity upon the excitation of methylene blue. From the natural bond order (NBO) analysis, a charge redistribution is noticeable. In the ground state, the S6 atom is positively charged +0.485e, while N5 and N7 are negatively charged with −0.366e and −0.421e, respectively. As shown in Fig. 8, the N5 atom is the most electropositive part of the molecule. In the excited state, however, the electrons accumulate in the hetero-ring, increasing the charge of S6 to +0.395e and N5 to −0.515e. On the other hand, the charge density on the amine group and outer aromatic rings slightly decreases.
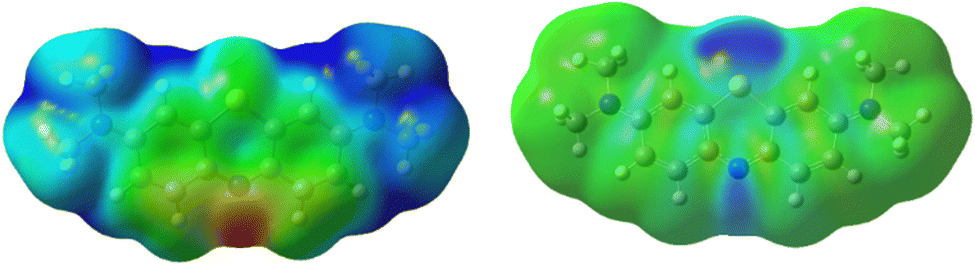 | ||
| Fig. 8 (left) Electrostatic potential of methylene blue in ground state shows and (right) differential electron density between the excited and ground state. | ||
While listed numerically in Table 2, this is more easily shown as a differential electron density between the excited and ground state in Fig. 8. It demonstrates that as electrons flow towards the excited molecule, the N5 becomes more reactive.
| Site | Atom charge in atomic units | |
|---|---|---|
| Ground state | First excited state | |
| 1 | −0.301 (−0.048) | −0.245 (−0.007) |
| 1a | −0.157 | −0.207 |
| 2 | +0.281 | +0.247 |
| 3 | −0.268 (−0.018) | −0.231 (+0.019) |
| 4 | −0.125 (+0.123) | −0.172 (+0.075) |
| 4a | +0.093 | +0.196 |
| 5 | −0.366 | −0.515 |
| 6 | +0.485 | +0.395 |
| 7 | −0.421 | −0.367 |
A more quantitative measure of the regioselectivity are the Fukui functions, which indicate the change in electron density when the number of electrons in the molecule changes. The Fukui function for the nucleophilic attack in the ground state shows the highest regioselectivity at N5 and N7 and moderate regioselectivity at C2 and C4a. Conversely, the electrophilic attack is most likely at C1 and C3. The radical attack will most likely proceed on N5, C4a, C1 and C3.
In the excited state, the electron density is perturbed (see Fig. 8), which also affects the regioselectivity. The nucleophilic attack is still most likely at N5, but C3 and N7 also show non-negligible reactivity, while the electrophilic attack is centred on N5 and S6. The excited molecule is susceptible to a radical attack on N5, S6, C1, C1a, C2 and C4. The Fukui functions are plotted in Fig. 9.
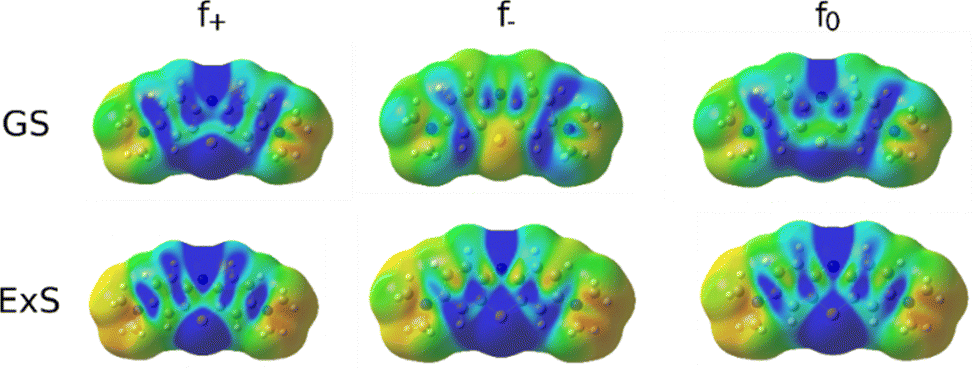 | ||
| Fig. 9 Fukui functions of MB for the nucleophilic (f+), electrophilic (f−) and radical attack (f0) in the ground state (GS) and the first excited state (ExS). | ||
3.6 Degradation routes of methylene blue
While the full mechanism of photochemical degradation of methylene blue is complex and unwieldly to detail with first-principle methods, a useful and common approximation is to study the reaction of MB with hydroxyl radicals (OH*).57 Under photoactive conditions, there is an abundance of OH*, which attack the MB molecule. The activation barriers for such reactions are low, usually only slightly above the diffusion limit, and depend on the catalyst surface and irradiation. As a proxy quantity, we studied the stability of different intermediates between OH* and methylene blue because the activation barriers of analogous reactions correlate with the energy differences, which is known as the BEP correlation. Additionally, abstraction of hydrogen from the aromatic ring (sites 1, 3, 4) or methyl group is also studied.As shown in Table 3, there is considerable difference in the activity of different sites in MB. OH* will most favourably abstract a hydrogen atom from the methyl group, which activates the cascade of N-demethylation. Other (aromatic) hydrogen atoms are not likely to be abstracted (152–160 kJ mol−1). Alternatively, OH* can bind to carbon atom, most favourably to C1, followed by C3 (relative energy +51 kJ mol−1 with respect to C1-intermediate), C4a (+63 kJ mol−1) and C4 (+81 kJ mol−1). The heteroatoms (N5, S6 and N7) are least likely to be attacked by the OH* radical. This shows that in the ground state, the degradation by the OH* radical begins with N-demethylation, while the aromatic ring structure is attacked at the C1 or C3 site. However, this changes upon excitation.
| Site | Energy (relative to the most stable intermediate) in kJ mol−1 | |
|---|---|---|
| Ground state | First excited state | |
| 1-OH | +51 | +171 |
| 1a-OH | +98 | +207 |
| 2-OH | +102 | +99 |
| 3-OH | +63 | +129 |
| 4-OH | +81 | +289 |
| 4a-OH | +74 | +160 |
| 5-OH | +115 | +65 |
| 6-OH | +137 | +163 |
| 7-OH | +141 | +231 |
| 1-H abstraction | +153 | +198 |
| 3-H abstraction | +152 | +206 |
| 4-H abstraction | +160 | +206 |
| Methyl-H abstraction | 0 | 0 |
While MB is flat in the ground and excited state, its intermediate with OH* is planar in the ground state but assumes a bent geometry in the first excited state (see Fig. 10). This has consequences for the degradation pathways, as well. While the attack on the methyl group with hydrogen abstraction remains most favourable, the reactivity of ring sites is changed. For instance, while poorly reactive in the ground state, N5 is preferentially attacked by OH* when MB is in the first excited state. The reactivity of the aromatic carbon atoms does not change as drastically with C3, C4a and C4 remaining most reactive (but less than the aromatic nitrogen atom N5). Experimentally, it was confirmed previously that N-demethylation proceeds concomitantly with the degradation of the phenothiazine structure of MB,58 which is in line with our calculation of similar energies for the OH attack on C1 and hydrogen cleave from N-methyl (difference of 13 kcal mol−1), as shown in Table 3.
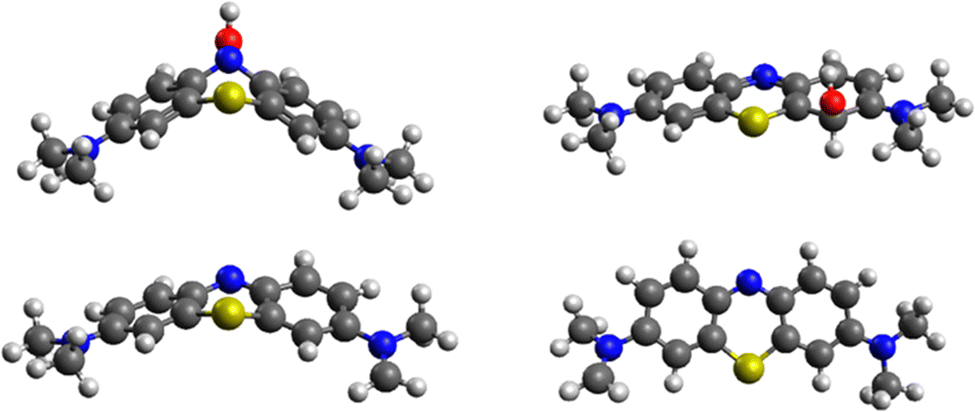 | ||
| Fig. 10 Structures of most stable (top) MB-OH* intermediates (left) in the excited state (site 5) and (right) the ground state (site 1) and (bottom) MB structures after hydrogen abstraction from the methyl group (left) in the excited and ground state (right). | ||
Once a hydrogen atom is cleaved from the methyl group, N-demethylation rapidly follows, yielding azure B, azure A, azure C and thionine with three, two, one and no methyl groups, respectively.59 Since the mechanism of radical N-demethylation is analogous for all methyl groups, we present the calculations for one cascade, only.
We model hydrogen atoms to be cleaved off by an impacting OH* radical, yielding H2O in the process. The results show that C–H bond cleavages are more exothermic than C–N bond cleavage, meaning that N-demethylation proceeds stepwise through –NR-CH3, –NR-CH2*, –NR-CH, –NR-C*, –NR*, and –NRH intermediates. In the last step, the –NR* radical does not abstract a hydrogen atom from a water molecule (this reaction would be endothermic) but instead reacts with a new MB molecule in a new exothermic reaction, propagating the reaction chain by converting it to –NR-CH2*. The mechanism and energetics in the ground and excited states are similar (see Table 4 and Fig. 11).
| Reaction | Energy in kJ mol−1 | |
|---|---|---|
| Ground state | First excited state | |
| Ar–NCH3CH3 + OH* → Ar–NCH3CH2* + H2O* | −179 | −201 |
| Ar–NCH3CH3 + OH* → Ar–NCH3* + CH3OH | −66 | −134 |
| Ar–NCH3CH2* + OH* → Ar–NCH3CH + H2O* | −98 | −55 |
| Ar–NCH3CH2* + OH* → Ar–NCH3 + CH2OH* | +21 | −25 |
| Ar–NCH3CH + OH* → Ar–NCH3C* + H2O* | −88 | −284 |
| Ar–NCH3CH + OH* → Ar–NCH3* + CHOH | −43 | −132 |
| Ar–NCH3C* + OH* → Ar–NCH3* + COH | −128 | −22 |
| Ar–NCH3* + H2O → Ar–NHCH3+ OH* | +100 | +175 |
| Ar–NCH3* + Ar–NCH3CH3 → Ar–NHCH3+ Ar–NCH3CH2* | −78 | −26 |
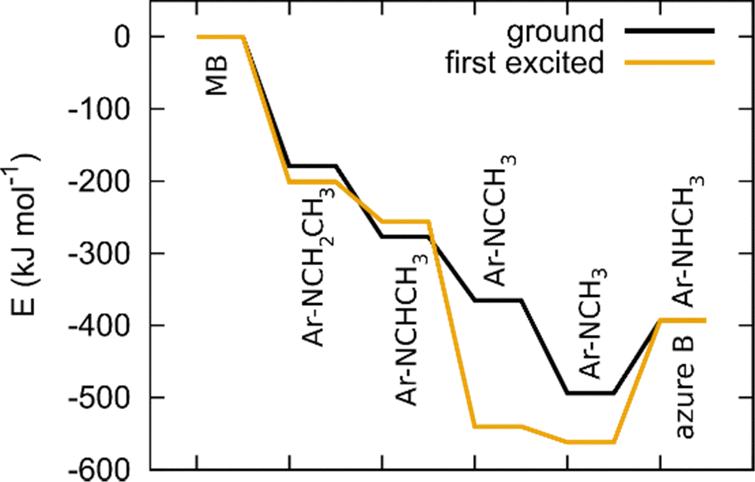 | ||
| Fig. 11 Energetics of N-demethylation of MB to azure B in the ground state and first excited state. | ||
3.7 Electronic structure of SrNbO2N
SrNbO2N is a promising semiconductor due to its low band gap, which was experimentally measured at 1.8 eV.59 Consequently, it has a characteristic bluish hue with a high absorption tail above 650 nm. This makes it well suited for methylene blue photodegradation, because MB is experimentally known to absorb at 665 nm,44 corresponding to 1.85 eV. Its applicability for photodegradation of different organic compounds can be tailored by changing its O:N ratio and surface substitutions. As shown by Aschauer et al.,60 the catalytic performance of the material changes with a N-by-O substitution, changing the overpotential for ORR, the band gap etc.
In SrNbO2N (and other perovskite oxynitrides), niobium atoms are in octahedral sites surrounded by nitride and oxide anions. Sr atoms are at the A sites, Nb atoms are at the B sites (ABO3). The position of nitrogen and oxygen anions can vary and several isomers are possible,61 which has already been extensively studied with first-principle methods.50,62–65 Our results show that nitrogen atoms assume the equatorial cis positions, which is consistent with the findings of Stotskyi et al.50
For an additional insight into the origin of its electronic properties, we calculated the band structure (and band gap), which is shown in Fig. 12. The calculated band gap (at Γ) was 1.88 eV, which is consistent with the experimentally determined values. The highest valence bands are contributed by N p orbitals, showing that its band gap can be fine-tuned by nitrogen doping. The lowest conduction bands correspond mostly to Nb d orbitals, which offer another avenue of band gap manipulation. Oxygen has a smaller effect on the band gap position.
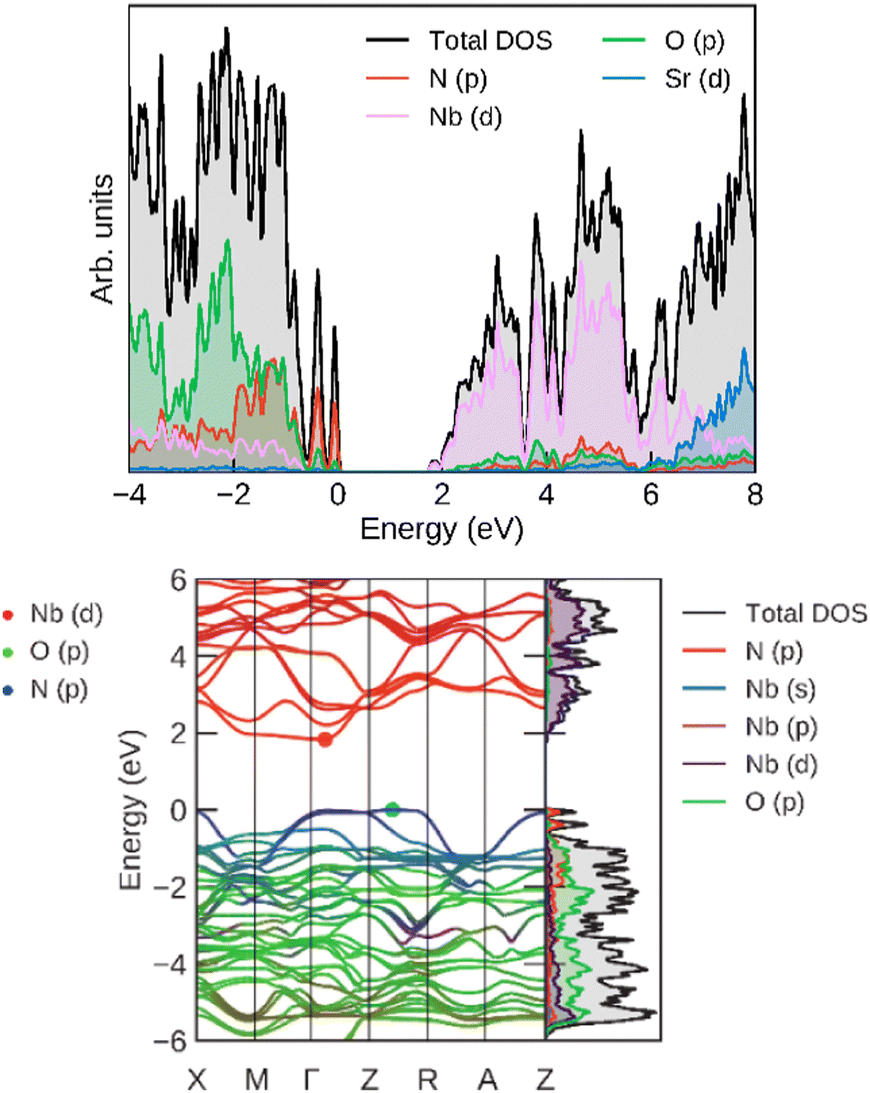 | ||
| Fig. 12 (left) Total and projected densities of states and (right) band structure for SrNbO2N, calculated at the HSE06 level. | ||
4. Conclusions
SrNbO2N perovskite oxynitride mixed with an alumina support powder was 3D printed and tested for degradation of organic pollutant in water. We observed that our 3D structure exhibited excellent photocatalytic properties and can degrade a high concentration of 100% organic pollutant (MB) within just a few hours. Further, it demonstrates good stability with more than 60% degradation efficiency after 3 cycle operations. This is due to our perovskite materials’ excellent photophysical properties, which include visible light harvesting, long excited state lifetime, and fast charge transfer, as well as their high adsorption toward MB. Theoretical calculations revealed that the aromatic nitrogen atom in MB becomes most reactive as the electron density flows towards it. This was confirmed by TD-DFT calculations of the stability of MB–OH* intermediates. The most stable intermediate in the excited state is formed with the attack upon the aromatic nitrogen, showing the most probable site for the photodegradation of MB. In addition, Electronic structure calculation shows a bandgap of 1.88 eV. The decomposition to atom types shows that the valence band is populated by N p electrons, while the conduction band is contributed by Nb d orbitals. While Sr serves as the scaffolding, the exchange of O by N and their relative position in the octahedra around Nb allows for fine-tuning of the bandgap.The reconstructed DLSR maps provide a wealth of new detail on the fidelity of the 3D printed photocatalyst design and its physico-chemical characteristics accounting for the varying lattice parameters and crystallite sizes of the active SrNbO2N phase throughout the monolith. The results of the XRD-CT characterisation efforts serve to guide the design and development of the novel tailor-made geometries and composition.
Hence, this work demonstrates that DIW is a viable approach for developing structured photocatalyst for water treatment. The resultant structured form of the photocatalyst offers the advantage of facile catalyst recovery (when compared to a powder suspension) by avoiding additional steps in product separation. We believe that 3D printing has the potential to revolutionise the field of catalysis and semiconductor photocatalysis by facilitating the controlled manufacture of both supports and catalysts at the micron level, allowing photocatalyst structures to be formed with tailored strength, porosity and functionality.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.AIT, GH, MM and VM designed and implemented the material synthesis and 3D printing procedures. KN, MTS, SD and SK were responsible for aspects of photocatalytic testing (PL spectroscopy) and advised on the analysis of the photocatalytic properties while MH and BL performed and interpreted the theoretical calculations of the photodegradation mechanism. AV, VM, SDMJ and AMB designed the XRD-CT experiments, acquired the XRD-CT data and subsequently performed the Rietveld analysis of the data. VM, SK, AV, GH and BL directed the research. MTS, SD, MH, AV, AIT, GH, SK and VM are responsible for writing and editing the manuscript with feedback given by all authors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Antonio Iborra-Torres thanks the EPSRC for provision of funding for his doctorate. Kiem Nguyen thanks LSBU for the PhD scholarship. Muhammad Tariq Sajjad thanks LSBU and Edinburgh Instrument for financial support. Matej Huš and Blaž Likozar were funded by the Slovenian Research Agency (M. H.: project J1-3020, core funding P2-0421, infrastructure funding I0-0039; B. L.: project N2-0135, core funding P2-0152). We would like to thank ESRF for beamtime and Marco di Michiel (ID15A, ESRF) for preparing beamline instrumentation and setup and for his help with the experimental XRD-CT data acquisition. We particularly thank Raymond Kemps for his assistance with electron microscopy. The authors appreciate the financial and research support provided by their respective institutions.References
- J. C. Colmenares and R. Luque, Heterogeneous photocatalytic nanomaterials: prospects and challenges in selective transformations of biomass-derived compounds, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, Synthesis and Photocatalytic Activity of Perovskite Niobium Oxynitrides with Wide Visible-Light Absorption Bands, ChemSusChem, 2011, 4(1), 74–78 CrossRef CAS PubMed.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang, P. Huo and X. Li, A review on heterogeneous photocatalysis for environmental remediation: From semiconductors to modification strategies, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS.
- N. Das and S. Kandimalla, Application of perovskites towards remediation of environmental pollutants: an overview, Int. J. Environ. Sci. Technol., 2017, 14, 1559–1572 CrossRef CAS.
- Y. Li, X. Li, F. Li, M. Li, Z. Li and X. Li, A Review of Co-Catalysts for Perovskite Oxynitrides in Photocatalytic Water Treatment, Front. Chem., 2021, 9, 640265 Search PubMed.
- R. Aguiar, A. Kalytta, A. Reller, A. Weidenkaff and S. G. Ebbinghaus, Photocatalytic decomposition of acetone using LaTi(O,N)3 nanoparticles under visible light irradiation, J. Mater. Chem., 2008, 18, 4260–4265 RSC.
- F. Oehler, R. Naumann, R. Koferstein, D. Hesseb and S. G. Ebbinghaus, Photocatalytic activity of CaTaO2N nanocrystals obtained from a hydrothermally synthesized oxide precursor, Mater. Res. Bull., 2016, 73, 276–283 CrossRef CAS.
- F. Oehler and S. G. Ebbinghaus, Photocatalytic properties of CoOx-loaded nano-crystalline perovskite oxynitrides ABO2N (A = Ca, Sr, Ba, La; B = Nb, Ta), Solid State Sci., 2016, 54, 43–48 CrossRef CAS.
- V. B. R. Boppana, D. J. Doren and R. F. Lobo, A Spinel Oxynitride with Visible-Light Photocatalytic Activity, ChemSusChem, 2010, 3(7), 814–817 CrossRef CAS PubMed.
- A. Iborra-Torres, A. N. Kulak, R. G. Palgrave and G. Hyett, Demonstration of visible light-activated photocatalytic self-cleaning by thin films of perovskite Tantalum and Niobium Oxynitrides, ACS Appl. Mater. Interfaces, 2020, 12(30), 33603–33612 CrossRef CAS PubMed.
- S. Hajiesmaili, S. Josset, D. Begin, C. Pham-Huu, N. Keller and V. Keller, 3D solid carbon foam-based photocatalytic materials for vapor phase flow-through structured photoreactors, Appl. Catal., A, 2010, 382(1), 122–130 CrossRef CAS.
- S. Josset, S. Hajiesmaili, D. Begin, D. Edouard, C. Pham-Huu, M.-C. Lett, N. Keller and V. Keller, UV-A photocatalytic treatment of Legionella pneumophila bacteria contaminated airflows through three-dimensional solid foam structured photocatalytic reactors, J. Hazard. Mater., 2010, 175(1–3), 372–381 CrossRef CAS PubMed.
- Y. Li, B. Li and L. Li, Fabrication of 3D photocatalytic α-Fe2O3 structure using Direct Ink Writing Method, Mod. Phys. Lett. B, 2014, 28, 1450051 CrossRef.
- M. R. Skorski, J. M. Esenther, Z. Ahmed, A. E. Miller and M. R. Hartings, The chemical, mechanical, and physical properties of 3D-printed materials composed of TiO2-ABS nanocomposites, Sci. Technol. Adv. Mater., 2016, 17, 89–97 CrossRef PubMed.
- H. Mei, W. Huang, H. Liu, L. Pan and L. Cheng, 3D printed carbon-ceramic structures for enhancing photocatalytic properties, Ceram. Int., 2019, 45(12), 15223–15229 CrossRef CAS.
- S. Lawson, L. Xin, H. Thakkar, A. A. Rownaghi and F. Rezaei, Recent advances in 3D printing of structured materials for adsorption and catalysis applications, Chem. Rev., 2021, 121(10), 6246–6291 CrossRef CAS PubMed.
- S. Das and V. C. Srivastava, Microfluidic-based photocatalytic microreactor for environmental application: a review of fabrication substrates and techniques, and operating parameters, Photochem. Photobiol. Sci., 2016, 15, 714–730 CrossRef CAS PubMed.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Chemistry in Microstructured Reactors, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- F. Rezaei and P. Webley, Optimum structured adsorbents for gas separation processes, Chem. Eng. Sci., 2009, 64, 5182–5191 CrossRef CAS.
- R. Kumar Patel, A. Kumar Chawla, P. Loulergue, B. Teychene and J. K. Pandey, 3D-printed Microchannel loaded with hematite nanoadsorbent for fluoride removal from water, Mater. Lett., 2019, 254, 190–193 CrossRef CAS.
- S. García, S. Poulston, D. Modeshia, P. Stavarek, M. Ujcic, F. Lali, M. A. Alves, J. D. Araújo, M. Krusche, F. Ullrich and D. Maier, Continuous production of Squalane using 3D printed catalytic supports, Johnson Matthey Technol. Rev., 2019, 63(3), 191–204 CrossRef.
- C. Jacquot, V. Middelkoop, A. Köckritz, A. Pohar, R. Bienert, S. Kellici, I.-A. Baragau, B. Venezia, A. Gavriilidis, B. Likozar and A. M. Beale, 3D printed catalytic reactors for aerobic selective oxidation of benzyl alcohol into benzaldehyde in continuous multiphase flow, Sustainable Mater. Technol., 2021, 30, e00329 CrossRef CAS.
- V. Middelkoop, T. Slater, M. Florea, F. Neat
![[u with combining cedilla]](https://www.rsc.org/images/entities/char_0075_0327.gif) , S. Danaci, V. Onyenkeadi, K. Boonen, B. Saha, I. A. Baragau and S. Kellici, Next frontiers in cleaner synthesis: 3D printed graphene-supported CeZrLa mixed-oxide nanocatalyst for CO2 utilisation and direct propylene carbonate production, J. Cleaner Prod., 2019, 214, 606–614 CrossRef CAS.
, S. Danaci, V. Onyenkeadi, K. Boonen, B. Saha, I. A. Baragau and S. Kellici, Next frontiers in cleaner synthesis: 3D printed graphene-supported CeZrLa mixed-oxide nanocatalyst for CO2 utilisation and direct propylene carbonate production, J. Cleaner Prod., 2019, 214, 606–614 CrossRef CAS. - V. Middelkoop, A. Vamvakeros, D. De Wit, S. Jacques, S. Danaci, C. Jacquot, Y. De Vos, D. Matras, S. Price and A. Beale, 3D printed Ni/Al2O3 based catalysts for CO2 methanation: a comparative and operando XRDCT study, J. CO2 Util., 2019, 33, 478–487 CrossRef CAS.
- T. Karsten, V. Middelkoop, D. Matras, A. Vamvakeros, S. Poulston, V. Grosjean, B. Rollins, F. Gallucci, H. R. Godini, S. D. M. Jacques, A. M. Beale and J.-U. Repke, Multi-scale studies of 3D printed MnNaW/SiO2 catalyst for oxidative coupling of methane, Catalysts, 2021, 11(3), 290 CrossRef CAS.
- K. Thompson, et al., Screening tests for the evaluation of nanoparticle titania photocatalysts, J. Chem. Technol. Biotechnol., 2009, 84(11), 1717–1725 CrossRef CAS.
- G. Vaughan, R. Baker, R. Barret, J. Bonnefoy, T. Buslaps, S. Checchia, D. Duran, F. Fihman, P. Got, J. Kieffer, S. A. J. Kimber, K. Martel, C. Morawe, D. Mottin, E. Papillon, S. Petitdemange, A. Vamvakeros, J. P. Vieux and M. Di Michiel, ID15A at the ESRF – a beamline for high-speed operando X-ray diffraction, diffraction tomography and total scattering, J. Synchrotron Radiat., 2020, 27, 515–528 CrossRef CAS PubMed.
- A. Vamvakeros, S. D. M. Jacques, M. Di Michiel, P. Senecal, V. Middelkoop, R. J. Cernik and A. M. Beale, Interlaced X-ray diffraction computed tomography, J. Appl. Crystallogr., 2016, 49, 485–496 CrossRef CAS PubMed.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, The fast azimuthal integration Python library: pyFAI, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed.
- A. Vamvakeros, S. D. M. Jacques, M. Di Michiel, V. Middelkoop, C. K. Egan, R. J. Cernik and A. M. Beale, Removing multiple outliers and single-crystal artefacts from X-ray diffraction computed tomography data, J. Appl. Crystallogr., 2015, 48, 1943–1955 CrossRef CAS.
- J. Kieffer, S. Petitdemange and T. J. Vincent, Real-time diffraction computed tomography data reduction, Synchrotron Radiat., 2018, 25, 612–617 CrossRef CAS PubMed.
- A. Vamvakeros, nDTomo Software Suite, 2018, Available online: https://github.com/antonyvam/nDTomo Search PubMed.
- A. A. Coelho, TOPAS and TOPAS-Academic: an optimization program integrating computer algebra and crystallographic objects written in C++, J. Appl. Crystallogr., 2018, 51(1), 210–218 CrossRef CAS.
- A. Vamvakeros, A. A. Coelho, D. Matras, H. Dong, Y. Odarchenko, S. W. T. Price, K. T. Butler, O. Gutowski, A. C. Dippel, M. Zimmerman, I. Martens, J. Drnec, A. M. Beale and S. D. M. Jacques, DLSR: A solution to the parallax artefact in X-ray diffraction computed tomography data, J. Appl. Crystallogr., 2020, 53(6), 1531–1541 CrossRef CAS.
- T. D. Nguyen-Phan, et al., The role of graphene oxide content on the adsorption-enhanced photocatalysis of titanium dioxide/graphene oxide composites, Chem. Eng. J., 2011, 170, 226–232 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino and B. G. Janeskoet al., Gaussian 16, revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, 72(10), 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. Von Ragué Schleyer, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F, J. Comput. Chem., 1983, 4(3), 294–301 CrossRef CAS.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions, J. Phys. Chem. A, 2013, 117(47), 12590–12600 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42, 845 RSC.
- D. Melgoza, A. Hernández-Ramírez and J. M. Peralta-Hernández, Comparative efficiencies of the decolourisation of Methylene Blue using Fenton's and photo-Fenton's reactions, Photochem. Photobiol. Sci., 2009, 8(5), 596–599 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57(3), 1505–1509 CrossRef CAS.
- V. Stotskyi and U. Aschauer, Water oxidation catalysis on the nitrogen-deficient SrNbO2N(001) Surface, J. Phys. Chem. C, 2021, 125, 2424–2430 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- H. Ouhbi and U. Aschauer, Nitrogen loss and oxygen evolution reaction activity of perovskite oxynitrides, ACS Mater. Lett., 2019, 1, 52–57 CrossRef CAS.
- Y.-I. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Characterization of the structural, optical, and dielectric properties of oxynitride perovskites AMO2N (A) (Ba, Sr, Ca; M) (Ta, Nb), Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- Y. I. Kim, P. M. Woodward, K. Z. Baba Kishi and C. W. Tai, Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- D. Aldakov, M. T. Sajjad, V. Ivanova, A. K. Bansal, J. Park, P. Reiss and I. D. W. Samuel, Mercaptophosphonic acids as efficient linkers in quantum dot sensitized solar cells, J. Mater. Chem. A, 2015, 3, 19050–19060 RSC.
- J. Park, M. T. Sajjad, P.-H. Jouneau, A. Ruseckas, J. Faure-Vincent, I. D. Samuel, P. Reiss and D. Aldakov, Efficient eco-friendly inverted quantum dot sensitized solar cells, J. Mater. Chem. A, 2016, 4, 827–837 RSC.
- B. A. Marinho, L. Suhadolnik, B. Likozar, M. Huš, Ž. Marinko and M. Čeh, Photocatalytic, electrocatalytic and photoelectrocatalytic degradation of pharmaceuticals in aqueous media: Analytical methods, mechanisms, simulations, catalysts and reactors, J. Cleaner Prod., 2022, 343, 131061 CrossRef CAS.
- T. Zhang, T. Oyamaa, A. Aoshimaa, H. Hidakaa, J. Zhao and N. Serpone, Photooxidative N-demethylation of methylene blue in aqueous TiO2 dispersions under UV irradiation, J. Photochem. Photobiol., A, 2001, 140(2), 163–172 CrossRef CAS.
- X. Sun, G. Liu and X. Xu, Defects management and efficient photocatalytic water oxidation reactions over Mg modified SrNbO2N, J. Mater. Chem. A, 2018, 6, 10947–10957 RSC.
- H. Ouhbi and U. Aschauer, Nitrogen loss and oxygen evolution reaction activity of perovskite oxynitrides, ACS Mater. Lett., 2019, 1(1), 52–57 CrossRef CAS.
- X. Xu and H. Jiang, Anion order in perovskite oxynitrides AMO2N (A = Ba, Sr, Ca; M = Ta, Nb): a first-principles based investigation, RSC Adv., 2020, 10, 24410–24418 RSC.
- L. Yang, J. Yuab, Q. Fu, L. Kong and X. Xu, Mesoporous single-crystalline SrNbO2N: Expediting charge transportation to advance solar water splitting, Nano Energy, 2022, 95, 107059 CrossRef CAS.
- N. Umezawa and A. Janotti, Controlling the Electronic Structures of Perovskite Oxynitrides and their Solid Solutions for Photocatalysis, ChemSusChem, 2016, 9, 9 CrossRef PubMed.
- Ph Borse, H. G. Kim and J. S. Lee, Difference in electronic structure between tetragonal and cubic SrNbO2N, J. Appl. Phys., 2005, 98, 043706 CrossRef.
- J. S. Gelves-Badillo, A. H. Romero and A. C. Garcia-Castro, Unveiling the mechanisms behind the ferroelectric response in the Sr(Nb,Ta)O2N oxynitrides, Phys. Chem. Chem. Phys., 2021, 23, 17142–17149 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma01076c |
| This journal is © The Royal Society of Chemistry 2023 |