
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical recycling of poly(ethylene terephthalate) via sequential glycolysis, oleoyl chloride esterification and vulcanization to yield durable composites†
Claudia V.
Lopez
and
Rhett C.
Smith
 *
*
Department of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: rhett@clemson.edu
First published on 2nd June 2023
Abstract
Herein we report a method for the chemical recycling of poly(ethylene terephthalate) (PET) by a three-stage process employing sustainably-sourced organic materials and industrial byproduct sulfur. In this protocol, PET was subject to glycolysis with diethylene glycol to yield low molecular weight oligomers with hydroxyl end groups. The glycolyzed PET (GPET) was then reacted with oleoyl chloride to yield esterified PET (EPET) containing vulcanizable olefin units. The oligomers constituting GPET and EPET were elucidated by MALDI-TOF spectrometry. EPET underwent inverse vulcanization with elemental sulfur (90 wt%) for 35 min or 24 h to yield xPES or mPES, respectively. The composition, thermal, morphological, thermal and mechanical properties were characterized. The composites exhibited good to excellent mechanical properties that were improved significantly by extending the reaction time from 35 min used to prepare xPES (compressive strength = 10.5 MPa, flexural strength = 2.7 MPa) to 24 h used to prepare mPES (compressive strength = 26.9 MPa, flexural strength = 7.7 MPa). Notably, the compressive and flexural strengths of mPES represent 158% and 208% of the values required for residential building foundations made from traditional materials such as ordinary Portland cement. The three-stage approach delineated herein thus represents a way to mediate chemical recycling of waste plastic with green coreagents to yield composites having mechanical properties competitive with existing commercial structural materials.
Introduction
The global production of plastics has increased precipitously over the last half-century.1 Synthesis of plastic from non-renewable sources like petroleum and natural gas derivatives precludes eternal propagation of this practice. Post-consumer plastics from single-use applications such as food/beverage packaging and diapers, as well as from end-of-use durable goods such as appliances, building materials and furniture persist in the environment to epidemic levels.2–5 One of the most commonly-used polymers is polyethylene terephthalate (PET). PET is a semi-crystalline polyester produced from the esterification reaction between ethylene glycol and terephthalic acid. Attractive properties of virgin PET such as high tensile and flexural strength and transparency make it one of the most widely used materials in food and beverage packaging and in the production of synthetic fibres. Of the >100 billion PET bottles produced annually, only ∼35 billion are recovered, and as few as ∼9 billion of these are ultimately recycled.1Various mechanical and chemical recycling methods have been widely investigated to reclaim PET,6–11 most commonly through bottle-to-bottle thermal reprocessing or chemical recycling to yield monomers or fuels. Some of the most common chemical recycling routes involve depolymerization of PET by hydrolysis, aminolysis, transesterification, or glycolysis reactions.12–19 During the glycolysis reaction, for example, PET is depolymerized by heating with glycols such as ethylene glycol or diethylene glycol (Scheme 2), sometimes with the addition of a catalyst, to yield low molecular weight derivatives useful in other processes. The glycolysis reaction of PET with ethylene glycol, for example, can give bis(hydroxyethyl)terephthalate in good yield. This is in turn used in the production of virgin PET.1
Esterification reactions are also effective for depolymerizing PET. In one study, new ecological polyester was prepared from PET waste bottles, diethylene glycol and oleic acid.20 The recycled unsaturated polyester showed enhanced thermal and mechanical properties including an increase of 14.6% in the hardness of the recycled product versus PET. This effort to recycle PET using oleic acid resulted in the formation of a rigid 3-D crosslinked polyester-styrene system. Unsaturated fatty acids and high fatty acid-content fat/oil products having low nutritional value represent an abundant, low-cost feedstock that can be used in reactions that aim to yield value-added products.21–25
Ordinary Portland cement (OPC) production accounts for about 30% of global materials utilization with concomitant emission of >8% of anthropogenic CO2,26 contributing significantly to increases in global temperatures.27 Carbon dioxide emission from cement production is only predicted to increase in the next several decades, highlighting the essential but sometimes overlooked role that new structural materials should play in a holistic approach to climate change attenuation.28 Historically, the monetary cost of cement production was relatively low and the climate cost was not fully understood, so alternatives to OPC were not widely sought outside of some efforts to develop specialty products for applications requiring properties unattainable with OPC. Sulfur cements were one of the few early classes of potential OPC surrogates.29–34 Such sulfur cements can exhibit high mechanical strength typical of OPC but with greater environmental resilience to acidic conditions and weather-induced degradation.30–32,35–47
Most high sulfur content materials (HSMs) reported to date have been prepared by inverse vulcanization (Scheme 1),48 a reaction analogous to Goodyear's vulcanization,49 but generally using a greater proportion of sulfur. During inverse vulcanization reactions, sulfur – an underutilized by-product of fossil fuel refining – is heated to temperatures above 159 °C to generate sulfur radicals that react with olefins to generate chemically stable materials with enhanced properties.
 | ||
| Scheme 1 General inverse vulcanization of an olefin with sulfur. | ||
Until the green renaissance, sulfur cements were prepared by reacting elemental sulfur with petrochemical olefins. More recently, HSMs have been prepared using a wide range of bioderived comonomers such as polysaccharide derivatives,50–54 lignin derivatives55,56 such as guaiacol57 or chlorolignin,58 lignocellulosic biomass,59–61 triglyceride sources,62 terpenoids,63,64 and others.65–68 More recently, novel strategies have been implemented for catalytic inverse vulcanization, mechanochemical synthesis,69 and lowering reaction temperatures. Concomitant improvements in processing/recycling HSMs have also been developed including use of room-temperature S–S metathesis or combinations of heat/pressure,1,70–73 Demonstrated application spaces for HSMs67,74–77 include their use in IR transparent lenses,78 lithium–sulfur batteries,79 water purification,79–96 and in adhesives.97
Although numerous bio-olefin-derived HSMs represent promising routes to repurpose biowaste sources into value-added goods, recycled-PET could also be considered a as potential comonomer to be used in the inverse vulcanization process. Such approach could be a valuable addition to current chemical recycling strategies to reclaim waste plastic. In the current context, it was thus of interest to (1) depolymerize PET waste using glycols to generate lower molecular weight oligomers. (2) Perform the esterification reaction of the glycolyzed-PET using an unsaturated fatty acid derivative to incorporate olefinic units into the PET polymer chain and (3) Synthesize chemically stable composites with interesting thermal and mechanical properties using the PET-esterified material as the comonomer in the atom economical inverse vulcanization reaction.
Herein, we report the synthesis of HSMs using recycled PET directly as a comonomer in the chemical reaction. This chemical modification was achieved in three steps (Scheme 2): first, the depolymerization of waste PET by diethylene glycol and zinc acetate as the catalyst to yield glycolyzed-PET (GPET). Then GPET reacted with oleoyl to chloride to produce esterified-PET (EPET), and finally, the reaction of 10 wt% EPET with 90 wt% sulfur via inverse vulcanization to yield xPES and mPES, where x indicates an “express” synthesis with just 35 min of heating and m indicates more heating over 24 h. The chemical composition, thermal and mechanical properties of the materials will be discussed.
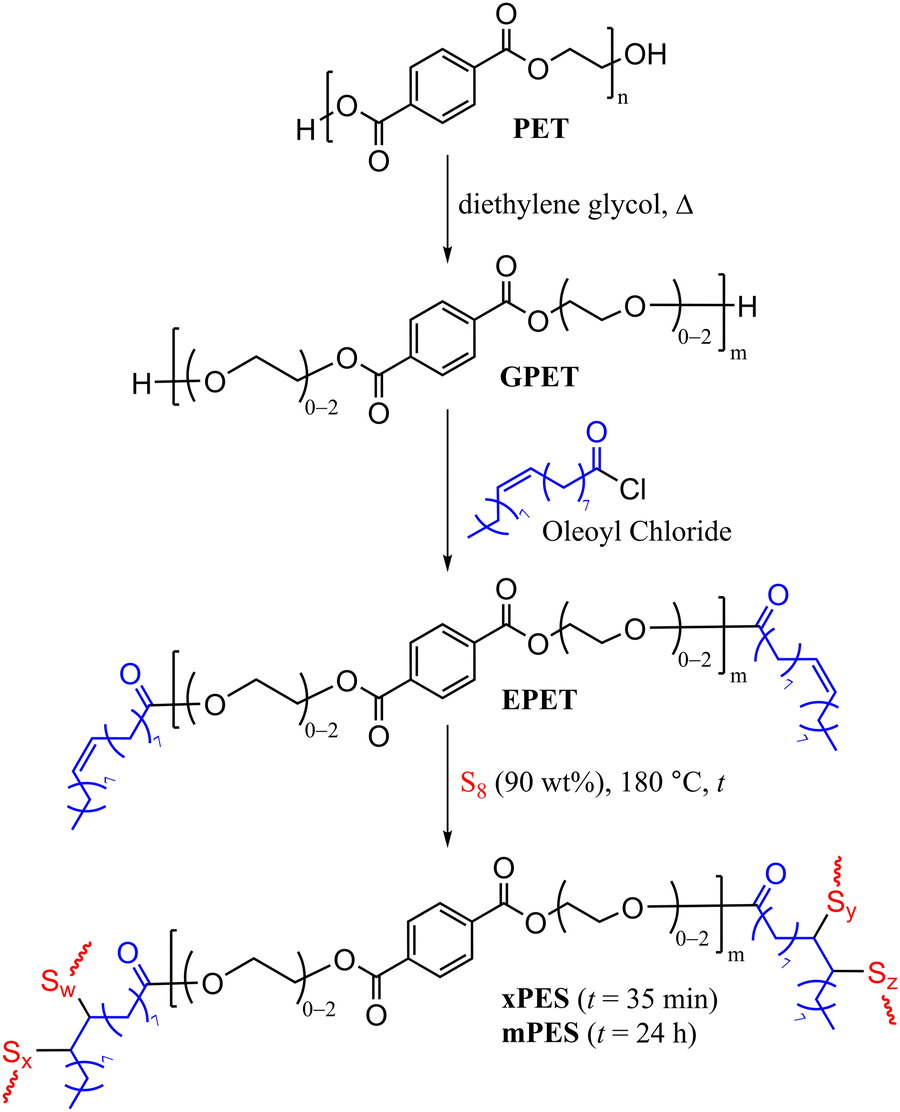 | ||
| Scheme 2 Sequential glycolysis, esterification, and vulcanization of PET. MALDI-TOF analysis showed that m ranges from 2–6. | ||
Results and discussion
Depolymerization and esterification of PET
Clear, colourless, postconsumer PET water bottles (Mn = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw = 82000, PDI = 1.9) were cleaned and cut into small pieces. These PET pieces were rinsed with ethanol and DI water, then dried in a vacuum oven at 40 °C for 24 h. The PET so prepared was subjected to glycolysis by its reaction with diethylene glycol and zinc acetate for 10 hours at 185 °C according to the reported procedure (Scheme 2, characterization is detailed in the ESI,† Fig. S1–S4 and Table S1),20 to give GPET. MALDI-TOF spectrometry revealed that GPET is a mixture of oligomers comprising 2–6 terephthalate units (Scheme 2).
000, Mw = 82000, PDI = 1.9) were cleaned and cut into small pieces. These PET pieces were rinsed with ethanol and DI water, then dried in a vacuum oven at 40 °C for 24 h. The PET so prepared was subjected to glycolysis by its reaction with diethylene glycol and zinc acetate for 10 hours at 185 °C according to the reported procedure (Scheme 2, characterization is detailed in the ESI,† Fig. S1–S4 and Table S1),20 to give GPET. MALDI-TOF spectrometry revealed that GPET is a mixture of oligomers comprising 2–6 terephthalate units (Scheme 2).
Oleoyl chloride was selected as the esterification reagent because it is readily prepared from bio-derived oleic acid and provides the required olefins for crosslinking via vulcanization in the next stage. GPET was therefore reacted with oleoyl chloride under an atmosphere of dry N2 for 24 h at room temperature to yield the esterified product EPET. EPET was analysed by 1H NMR spectrometry and FT-IR spectroscopy (full spectra along with those of oleoyl chloride for comparison are provided in Fig. S5–S7 in the ESI†). Proton NMR analysis revealed resonances for EPET consistent with the structure shown in Scheme 2. The monomers and oligomers present in EPET were further elucidated by MALDI-TOF spectrometry, which further confirmed the esterification of terminal alcohol moieties in GPET with oleoyl units (MALDI-TOF data and structures of oligomers are provided in Fig. S8 and Table S2 in the ESI†).
The TGA thermogram for EPET (Fig. S4, ESI†) shows a first decomposition step at around 220 °C. This step is attributed to the loss of low molecular fragments via cleavage of ester bonds present between the PET backbone and the oleoyl chloride chain. Similarly, a second degradation step between 360–380 °C represents the cleavage of the polymeric chains in PET.
The total unsaturation content of EPET was quantified by integrating the olefinic proton resonance from oleoyl units (∼5.4 ppm) versus the aldehydic proton resonance (∼10.2 ppm) of internal standard 2,3,4,5,6-pentafluorobenzaldehyde (full spectrum provided in Fig. S6 in the ESI†). This analysis revealed that EPET has 3.6 mmol g−1 of olefin moieties that could undergo a vulcanization reaction.
Synthesis and chemical characterization of composites
In the third step of the process shown in Scheme 2, the reaction of 90 wt% sulfur and 10 wt% EPET at 185 °C was undertaken. Prior work has noted that monomer composition, heating time and conditions can significantly impact the properties of HSMs prepared by inverse vulcanization.98 To explore the limits of reaction time influence, the vulcanization of EPET was undertaken under identical conditions for either 35 min to yield xPES, or 24 h to yield mPES. After the designated heating time, each reaction mixture was cooled to room temperature, giving a quantitative yield of either xPES as a light-brown solid or mPES as a black solid (Fig. 1). Both the composites are remeltable solids, allowing them to be readily poured into moulds in the heated liquid form and then cooled to give samples shapes such as cylinders (Fig. 1(A) and (C)) or dog-bones (Fig. 1(B) and (D)) appropriate for compressive strength or tensile strength testing, respectively. Previous work has demonstrated a correlation between sulfur catenate length (and thus crosslink density or sulfur rank) and the colour of the material, with a lighter colour generally indicative of shorter catenates.99,100 The less-crosslinked nature of light brown-coloured xPES was supported by fractionation studies and mechanical tests (vide infra). | ||
| Fig. 1 Samples of xPES (A), (B) and mPES (C), (D) shaped for compressive (A) and (C) or tensile (B) and (D) strength testing. | ||
The FT-IR spectra for xPES and mPES (Fig. S9, ESI†) confirm the loss of intensity for bands attributable to sp2 C–H stretches for cis-alkenes that had been present in EPET at around 3005 cm−1 with concomitant emergence of a peak at 664 cm−1 attributed to C–S stretches from the bonds formed during the inverse vulcanization process.
Notably, the inverse vulcanization process is 100% atom economical in this case. In some inverse vulcanization reactions, the atom economy falls below 100% due to H2S formation, typically via sulfur radical-mediated abstraction of benzylic or allylic protons or loss of sublimed sulfur. In the current case the vessel was covered with a watch glass to preclude sublimation loss and no side reaction-derived mass loss was observable: a 100% yield of the target composites was accomplished within the error limits of mass measurement.
HSMs prepared by inverse vulcanization are generally best described as composites because there is a small percentage of added sulfur that is not covalently incorporated into the organic-sulfur network. Several techniques have been reported to quantify the free versus bound sulfur in HSMs.101 Free sulfur (S8) is completely soluble in CS2, while sulfur that is covalently crosslinked in a densely-crosslinked sulfur/olefin network is not soluble in CS2. This solubility difference allowed us to use fractionation studies using CS2 to quantify free versus covalently incorporated sulfur in xPES and mPES composites. In the current study, CS2 extractions followed by elemental analysis of the fractions revealed that most of the sulfur present in the composites was effectively incorporated as covalently-bound catenates as indicated in Scheme 1. Composite xPES had only 10 wt% of extractable free sulfur, while mPES had 20 wt% of CS2-extractable material. The heating time had a notable impact on the chemical composition of the extractable portions of the composites. Elemental microanalysis of the CS2-extractable fraction of mPES was found to be 98% elemental sulfur, indicating that essentially all of the organic molecules from EPET have been effectively sequestered into the insoluble network solid through the formation of C–S bonds over 24 h of heating in the vulcanization reaction. In sharp contrast, the soluble fraction of xPES was 93% sulfur, with 7% of the mass attributable to extractable organic material (Table S2, ESI†). These data suggest that lower molecular-mass fragments not comprising part of the network solid are still present after the abbreviated 35 min heating time. These data, and the mechanical properties of the composites (vide infra) reinforce the sensitive nature of HSMs to heating time. Given that essentially all of the organic comonomer comprises part of the network polymer in mPES and the amount of sulfur effectively incorporated into the network is also known, the average length of the oligosulfur catenates linking organic units (known as a material's sulfur rank) was calculated (details are in the ESI†). A wide range of average sulfur ranks, from 1 to >500, have been reported for other vulcanized materials depending on the olefin content and the proportion of sulfur employed for their synthesis. For mPES the sulfur rank was thus found to be 62. This compares well to other composites prepared by inverse vulcanization with 90 wt% sulfur, such as CanBG9025 and OSS90,53 which have similar sulfur ranks to that in mPES, of 60 and 69, respectively. The organic crosslinker for CanBG90 was 1:1 (mass/mass) brown grease (a high fatty acid-content animal fat) and sunflower oil, while for OSS90 the organic crosslinker was octenyl succinate-modified cellulose.
Thermal, morphological and mechanical properties of composites
TGA of composites xPES and mPES (Fig. S4, ESI†) revealed decomposition temperatures (Td) of 217 °C and 215 °C, respectively (Table 1). These Td values lie within the range of other composites comprising similarly high proportions of sulfur, wherein thermally-induced mass loss is attributable to degradation of the polysulfide domains.| Materials | T d /°C | T m /°C | T g,DSC /°C | Cold crystallization peaks/°C | ΔHm J g−1 | ΔHcc J g−1 | Percent crystallinityd | Percent insoluble fractione |
|---|---|---|---|---|---|---|---|---|
| a The temperature at which the 5% mass loss was observed. b The temperature at the peak maximum of the endothermic melting from the third heating cycle. c Glass transition temperature. d The reduction of percent crystallinity of each sample was calculated with respect to sulfur (normalized to 100%). e Percent of non-extractable sulfur in each sample after CS2 extractions. | ||||||||
| xPES | 217 | 117.7 | −36.8 | 15.0 | 33.9 | −22.8 | 25 | 90 |
| mPES | 215 | 116.8 | −36.0 | 3.7 | 33.6 | −18.2 | 34 | 80 |
| GPET | 176 | NA | NA | NA | NA | NA | NA | NA |
| EPET | 221 | NA | NA | NA | NA | NA | NA | NA |
| S8 | 229 | 118.5 | NA | NAb | 44.8 | NA | 100 | 0 |
Differential scanning calorimetry (DSC, Table 1 and Fig. S10 of the ESI†) was used to investigate the composites’ thermal/morphological transitions over the range of −60 to 140 °C. Both xPES and mPES exhibited the characteristic melting peak of orthorhombic sulfur at temperatures between 115 and 118 °C. In the second heating cycle, xPES and mPES showed glass transitions (Tg) at −37 °C, diagnostic for the presence of polymeric sulfur catenates in the organosulfur crosslinked network. Cold crystallization peaks were also observed in xPES and mPES at 15 and 3.7 °C, respectively. These cold crystallization peaks reflect the partial organization of the polymer chains above the glass transition temperature, which is commonly observed in network-stabilized polymeric sulfur domains.
The percent crystallinity of xPES and mPES relative to crystalline orthorhombic sulfur was calculated from the integration of melting and cold crystallization enthalpies observed in the DSC spectra (the equation used for the calculations is provided in the Experimental section). From these integrations, it can be concluded that both composites have amorphous and crystalline regions, with percent crystallinities between 25–34% (Table 1).
Scanning electron microscopy with elemental mapping by energy-dispersive X-ray analysis (SEM-EDX) confirmed that xPES and mPES form microscopically homogeneous materials (Fig. S13 in ESI†). In both cases, the composites had largely uniform distributions of carbon, oxygen and sulfur. These data provide further evidence of the successful chemical reaction between EPET and elemental sulfur to yield homogeneous composites.
Cylinders, dog-bones, and rectangular prisms of the composites for compressive, tensile, and flexural strength testing, respectively, were readily prepared by melting the xPES and mPES composites at 160 °C and pouring the liquid into silicon moulds (examples are shown in Fig. 1). The composites were allowed to stand at room temperature for four days before any mechanical testing to allow direct compare their mechanical properties to those of reported HSMs that are likewise aged for four days prior to measurement.25,62,63
Mechanical test stand analysis was used to measure the compressive strength of composites (Fig. 2(A) and Table 2; stress–strain plots are provided in Fig. S11 in the ESI†). The compressive strength of xPES was 10.5 ± 0.1 MPa, representing 62% of the compressive strength of ordinary Portland cement (OPC). The compressive strength of mPES, however, was 26.9 ± 0.6 MPa, which is 158% of the compressive strength of OPC. The significantly lower compressive strength of xPES versus that of mPES may be attributed to a lower extent of crosslinking in xPES as a result of the shorter reaction time. This hypothesis is supported by incomplete incorporation of organics into the network structure that was observed in fractionation studies (vide supra, Table S1 in ESI†). Composites comprise vulcanized aromatic/fatty acid components, so a comparison of their properties to those of other vulcanized composites comprising fatty acid or aromatic subunits with 90 wt% sulfur is also of interest. Composite mPES, for example exhibits a lower compressive strength than that of CanBG90,25 which has significant fatty acid and triglyceride content but no aromatic moieties. On the other hand, the compressive strength of mPES is significantly higher than that of ZOS90 (19.4 MPa) that employed oleic acid as the organic comonomer, or PS90 (21.3 MPa)59,60 containing lignocellulosic biomass (peanut shells having only ∼1 wt% triglyceride content).
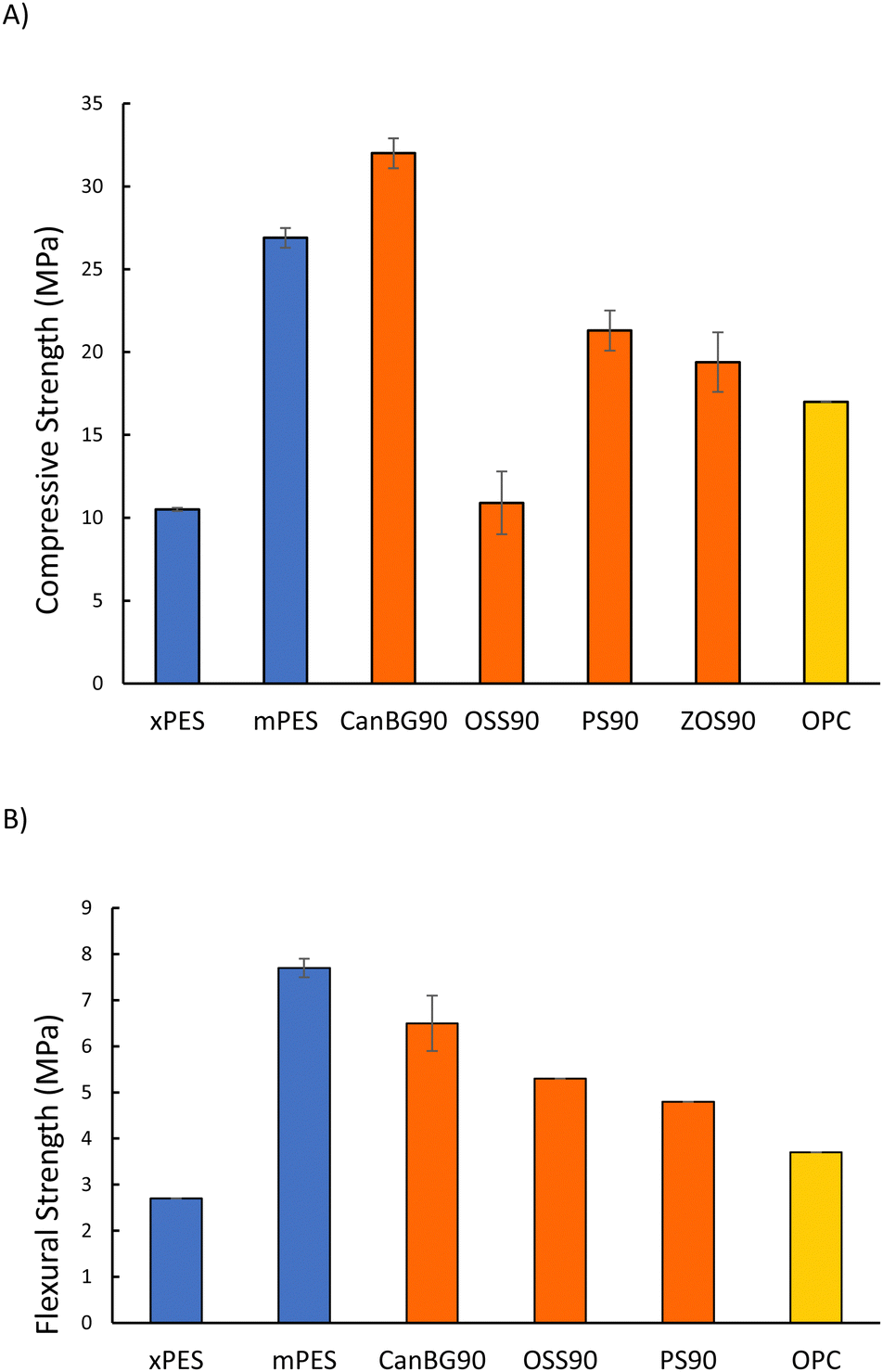 | ||
| Fig. 2 Compressive strength (A) and flexural strength (B) for composites xPES and,PES compared to other high sulfur-content materials and the familiar structural product ordinary Portland cement (OPC). | ||
| Materials | Compressive strength (MPa) | Flexural strength/modulus (MPa) | Ultimate tensile strength at break (MPa) | Elongation at break (%) | Sulfur rank | Ref. |
|---|---|---|---|---|---|---|
| xPES | 10.5 ± 0.1 | 2.7/100 | 0.27 ± 0.02 | <1 | ND | This work |
| mPES | 26.9 ± 0.6 | 7.7/320 | 0.21 ± 0.04 | <1 | 62 | This work |
| CanBG90 | 32.0 ± 0.9 | 6.5/420 | ND | ND | 60 | 23 |
| OSS90 | 10.9 ± 1.9 | 5.3/660 | ND | ND | 69 | 51 |
| PS90 | 21.3 ± 1.2 | 4.8/950 | ND | ND | 257 | 57,58 |
| ZOS90 | 19.4 ± 1.8 | ND | ND | ND | ND | 22 |
| GS80 | ND | ND | 2.32 ± 0.02 | 10.9 ± 2.1 | ND | 55 |
| GS90 | ND | ND | 1.16 ± 0.53 | 4.3 ± 1.8 | ND | 55 |
| I-BPA80 | ND | ND | 1.04 ± 0.16 | 89 ± 9 | ND | 68 |
| Portland cement | 17.0 | 3.7/580 | ND | ND | NA | — |
PES composites were also subjected to dynamic mechanical analysis (DMA) at room temperature in single cantilever mode to assess their flexural strengths/moduli (Fig. 2(B) and Table 2; stress–strain plots are provided in Fig. S12 in the ESI†). As with the compressive tests, mPES exhibited significantly higher flexural strength and moduli than that of xPES. The flexural strength of mPES is higher than those reported for CanBG90 (6.5 MPa), PS90 (4.8 MPa), or OSS90 (5.3 MPa).
The ultimate tensile strength at break (UTS) of mPES (0.21 MPa), was similar to that of xPES (0.27 MPa), with both materials showing relatively poor tensile performance and low elongation at break (<1%) as compared to previously reported composites having similar sulfur content and aromatic organic crosslinking groups. Composites GS80 (UTS = 2.32 MPa, elongation at break = 4%) and GS90 (UTS = 1.16 MPa, elongation at break = 11%), for example, were prepared by crosslinking guaiacol and 80 or 90 wt% sulfur, respectively.57 Composite I-BPA80, prepared by inverse vulcanization of (O,O′-diallyl-2,2′,6,6′-tetrabromo bisphenol A and 80 wt% sulfur) exhibited a similar UTS (1.04 MPa) to GS90 with a significantly greater elongation at break of 89 ± 9%.70
Conclusions
The chemical recycling of poly(ethylene terephthalate) was achieved in a process that involved the glycolysis of waste PET by diethylene glycol followed by its esterification reaction using fatty acid derivatives to yield esterified-PET (EPET). EPET was then directly used as the organic comonomer in the inverse vulcanization reaction with elemental sulfur and a variation in the reaction time (35 min or 24 h) yielded composites xPES and mPES, respectively. The influence of extending the reaction times in the chemical and mechanical properties of the composites was evaluated. The longer reaction time led to significantly higher compressive and flexural strength for mPES than for xPES. This improvement in the mechanical properties of mPES can be attributed to a more complete crosslinking of the organic domains in the composite after 24 h of reaction, as confirmed by fractionation studies. The synthetic approach to repurposing PET reported herein represents a promising way to directly incorporate waste PET as the organic comonomer in the atom economical inverse vulcanization reaction to yield sustainable building materials with mechanical properties that exceed that of commercial structural materials.Experimental
General considerations and instrumentation
Matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) data were recorded on a MicrofleX LRF equipped with a 337 nm nitrogen laser in the positive reflectron ion mode. The scanning mass-to-charge (m/z) range for the experiments was between 600–1600 using α-cyano-4-hydroxycinnamic acid as the matrix.Fourier transform infrared spectra were obtained using an IR instrument (Shimadzu IRAffinity-1S) with an ATR (attenuated total reflectance) attachment. Scans were collected over the range 400–4000 cm−1 at ambient temperature with a resolution of 8.
SEM was acquired on a Schottky Field Emission Scanning Electron Microscope SU5000 operating in variable pressure mode with an accelerating voltage of 15 keV.
TGA data were recorded using a Mettler Toledo TGA 2 STARe System operating over the range 20–800 °C with a heating rate of 10 °C min−1 under a flow of N2 (100 mL min−1). Each measurement was acquired in triplicate and presented results represent an average value.
DSC data were acquired using a Mettler Toledo DSC 3 STARe System operating over the range of −60 to 140 °C with a heating rate of 10 °C min−1 under a flow of N2 (200 mL min−1). Each DSC measurement was carried out over three heat–cool cycles.
DMA was performed using a Mettler Toledo DMA 1 STARe System in single cantilever mode. DMA samples were cast from silicone resin moulds made using a commercial Smooth-On Oomoo® 30 tin-cure kit. Samples were manually sanded to ensure uniform dimensions of approximately 15 × 8 × 1.5 mm. Sample dimensions were measured using a digital calliper with 0.01 mm resolution. The force was varied from 0 to 10 N with a ramp rate of 0.2 N min−1 measured isothermally at 25 °C.
Carbon disulfide extractions were performed by suspending 0.3 g of finely ground material (measured to 0.0001 g accuracy) in 20 mL of CS2, allowing the solid to settle for 30 minutes, pipetting off the supernatant into a separate vial, and adding another 20 mL of CS2. This process was repeated an additional three times so that a total of 5 washes were performed and constant residual mass was confirmed. The residual CS2 was evaporated under a flow of N2, and each vial was weighed to determine the fraction that was soluble (collected as supernatant) or insoluble (remained in the initial vial). This process was performed in duplicate, yielding identical results within balance error both times for each material analysed.
Compressional analysis was performed on a Mark-10 ES30 test stand equipped with an M3-200 force gauge (1 kN maximum force with 1 N resolution) with an applied force rate of 3–4 N s−1. Compression cylinders were cast from silicone resin moulds (Smooth-On Oomoo 30 tin-cure) with diameters of approximately 6 mm and heights of approximately 10 mm. Samples were manually sanded to ensure uniform dimensions and measured with a digital calliper with 0.01 mm resolution. Compressional analysis was performed in triplicate and results were averaged.
Proton NMR spectra were acquired on a Bruker NEO-300 MHz at room temperature and data was processed with TopSpin 4.0.6 software. All spectra reported were calibrated to the residual solvent signal.
Materials
Diethylene glycol (VWR), zinc acetate (Sigma Aldrich), oleoyl chloride (Sigma Aldrich), and sulfur (Dugas Diesel) were used without further purification. PET-sulfur composite materials were aged for 4 d prior to any mechanical testing.Synthesis
CAUTION: Heating elemental sulfur with organics can result in the formation of H2S gas. H2S is toxic, foul-smelling, and corrosive. Although we did not observe any mass loss attributable to gas generation, the temperature must be carefully controlled to prevent thermal spikes, which contribute to the potential for H2S evolution. Rapid stirring shortened heating times, and very slow addition of reagents can help prevent unforeseen temperature spikes.Synthesis of xPES
Preparation of xPES involved the reaction of EPET (1.0 g, 10 wt%) and sulfur (9.0 g, 0.03 mol, 90 wt%). Sulfur was first melted in an oil bath at 160 °C with rapid mechanical stirring. Then, the temperature was heated further to 185 °C, where sulfur exists primarily as polymeric diradicals. Once the temperature was stable, EPET was slowly added to the sulfur while stirring. The reaction mixture was stirred for 35 min at 185 °C. Within 35 min of the reaction time, the desired product, a homogeneous solution was produced. Upon cooling to room temperature, the material solidified to a light-brown solid composite in quantitative yield (10 g). ELEM. ANAL calcd: C 4.00, H 1.00, S 90.00; found: C 7.61, H 1.11, S 89.28.Synthesis of mPES
Preparation of mPES involved the reaction of EPET (1.0 g, 10 wt%) and sulfur (9.0 g, 0.03 mol, 90 wt%). Sulfur was first melted in an oil bath at 160 °C with rapid mechanical stirring. Then, the temperature was heated further to 185 °C, where sulfur exists primarily as polymeric diradicals. Once the temperature was stable, EPET was slowly added to the sulfur while stirring. The reaction mixture was stirred for 24 h at 185 °C. Within 24 h of the reaction time, the desired product, a homogeneous solution was produced. Upon cooling to room temperature, the material solidified to a black solid composite in quantitative yield (10 g). ELEM. ANAL calcd: C 4.00, H 1.00, S 90.00; found: C 8.22, H 0.50, S 90.48.Author contributions
Claudia Lopez: data curation, formal analysis, investigation, validation, roles/writing – original draft. Rhett C. Smith: conceptualization, funding acquisition, methodology, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the National Science Foundation grant number CHE-2203669.Notes and references
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- J. Jeong and J. Choi, Chemosphere, 2019, 231, 249–255 CrossRef CAS PubMed.
- M. Cole, P. Lindeque, C. Halsband and T. S. Galloway, Mar. Pollut. Bull., 2011, 62, 2588–2597 CrossRef CAS PubMed.
- V. Hidalgo-Ruz, L. Gutow, R. C. Thompson and M. Thiel, Environ. Sci. Technol., 2012, 46, 3060–3075 CrossRef CAS PubMed.
- I. Gestoso, E. Cacabelos, P. Ramalhosa and J. Canning-Clode, Sci. Total Environ, 2019, 687, 413–415 CrossRef CAS PubMed.
- B. S. Maddodi, L. U. A, S. Devesh, A. U. Rao, G. B. Shenoy, H. T. Wijerathne, N. Sooriyaperkasam and P. K. M, Eng. Sci., 2022, 18, 329–336 CAS.
- D. Pan, F. Su, H. Liu, C. Liu, A. Umar, L. C. Castañeda, H. Algadi, C. Wang and Z. Guo, ES Mater. Manuf., 2021, 11, 3–15 CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- K. M. Akato, N. A. Nguyen, P. V. Bonnesen, D. P. Harper and A. K. Naskar, ACS Omega, 2018, 3, 10709–10715 CrossRef CAS PubMed.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- E. Langer, S. Waśkiewicz, M. Lenartowicz-Klik and K. Bortel, Polym. Degrad. Stab., 2015, 119, 105–112 CrossRef CAS.
- D. J. Suh, O. O. Park and K. H. Yoon, Polymer, 2000, 41, 461–466 CrossRef CAS.
- B. Liu, X. Lu, Z. Ju, P. Sun, J. Xin, X. Yao, Q. Zhou and S. Zhang, Ind. Eng. Chem. Res., 2018, 57, 16239–16245 CrossRef CAS.
- K. Troev, G. Grancharov, R. Tsevi and I. Gitsov, J. Appl. Polym. Sci., 2003, 90, 1148–1152 CAS.
- Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 340–348 CrossRef CAS.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Polym. Degrad. Stab., 2011, 96, 399–403 CrossRef CAS.
- O. Mecit and A. Akar, Macromol. Mater. Eng., 2001, 286, 513–515 CrossRef CAS.
- H. M. Naguib and X. H. Zhang, Polym. Test., 2018, 69, 450–455 CrossRef CAS.
- A. D. Smith, T. Thiounn, E. W. Lyles, E. K. Kibler, R. C. Smith and A. G. Tennyson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1704–1710 CrossRef CAS.
- A. D. Smith, C. D. McMillen, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 CrossRef CAS.
- A. D. Smith, C. D. McMillin, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 CrossRef CAS.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustainable Chem. Pharm., 2020, 16, 100249 CrossRef.
- C. V. Lopez, A. D. Smith and R. C. Smith, RSC Adv., 2022, 12, 1535–1542 RSC.
- K. L. Scrivener, V. M. John and E. M. Gartner, Cem. Concr. Res., 2018, 114, 2–26 CrossRef CAS.
- P. A. Arias, N. Bellouin, E. Coppola, R. G. Jones, G. Krinner, J. Marotzke, V. Naik, M. D. Palmer, G.-K. Plattner, J. Rogelj, M. Rojas, J. Sillmann, T. Storelvmo, P. W. Thorne, B. Trewin, K. A. Rao, B. Adhikary, R. P. Allan, K. Armour, G. Bala, R. Barimalala, S. Berger, J. G. Canadell, C. Cassou, A. Cherchi, W. Collins, W. D. Collins, S. L. Connors, S. Corti, F. Cruz, F. J. Dentener, C. Dereczynski, A. D. Luca, A. D. Niang, F. J. Doblas-Reyes, A. Dosio, H. Douville, F. Engelbrecht, V. Eyring, E. Fischer, P. Forster, B. Fox-Kemper, J. S. Fuglestvedt, J. C. Fyfe, N. P. Gillett, L. Goldfarb, I. Gorodetskaya, J. M. Gutierrez, R. Hamdi, E. Hawkins, H. T. Hewitt, P. Hope, A. S. Islam, C. Jones, D. S. Kaufman, R. E. Kopp, Y. Kosaka, J. Kossin, S. Krakovska, J.-Y. Lee, T. M. J. Li, T. K. Maycock, M. Meinshausen, S.-K. Min, P. M. S. Monteiro, T. Ngo-Duc, F. Otto, I. Pinto, A. Pirani, K. Raghavan, R. Ranasinghe, A. C. Ruane, L. Ruiz, J.-B. Sallée, B. H. Samset, S. Sathyendranath, S. I. Seneviratne, A. A. Sörensson, S. Szopa, I. Takayabu, A.-M. Treguier, B. V. D. Hurk, R. Vautard, K. V. Schuckmann, S. Zaehle, X. Zhang and K. Zickfeld, in Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Cambridge University Press, 2021 Search PubMed.
- R. M. Andrew, Earth Syst. Sci. Data, 2018, 10, 195–217 CrossRef.
- A.-M. O. Mohamed and M. E. Gamal, Sulfur concrete for the construction industry, J. Ross Publishing, Fort Lauderdale, FL, 2010 Search PubMed.
- Journal, 1978.
- M. Al-Ansary, E. Masad and D. Strickland, Adv. Gas Process., 2010, 2, 121–130 CAS.
- A. Taylor, N. Tran, R. May, D. Timm, M. Robbins and B. Powell, J. Assoc. Asphalt Paving Technol., 2010, 79, 403–441 CAS.
- Z.-g Zuo, G.-s Cao and H.-l Song, J. Qingdao Technol. Univ., 2011, 32, 26–29 CAS.
- X.-D. Hu, Y.-M. Gao, L.-R. Lin, S. Zhong and X.-W. Dai, J. Wuhan Univ. Technol., 2013, 35, 10–13 CAS.
- M. Dehestani, E. Teimortashlu, M. Molaei, M. Ghomian, S. Firoozi and S. Aghili, Data Brief, 2017, 13, 137–144 CrossRef CAS PubMed.
- E. D. Weil, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 59, 325–340 CrossRef CAS.
- WO2004-EP53357, 2005.
- I. Deme, Adv. Chem. Ser., 1978, 165, 172–189 CrossRef CAS.
- J. E. Gillott, I. J. Jordaan, R. E. Loov, N. G. Shrive and M. A. Ward, Adv. Chem. Ser., 1978, 165, 98–112 CrossRef CAS.
- G. J. Kennepohl and L. J. Miller, Adv. Chem. Ser., 1978, 165, 113–134 CrossRef CAS.
- D.-Y. Lee, Prod. R&D, 1975, 14, 171–177 Search PubMed.
- S. Gwon, E. Ahn and M. Shin, Composites, Part B, 2019, 162, 469–483 CrossRef CAS.
- S. Gwon, S.-Y. Oh and M. Shin, Constr. Build. Mater., 2018, 181, 276–286 CrossRef CAS.
- S. Gwon and M. Shin, Constr. Build. Mater., 2019, 228, 116784 CrossRef CAS.
- P. Szajerski, A. Bogobowicz and A. Gasiorowski, J. Hazard. Mater., 2020, 381, 121180 CrossRef CAS PubMed.
- M. Lewandowski and R. Kotynia, MATEC Web Conf., 2018, 219, 3006 CrossRef CAS.
- S. Mohammed and V. Poornima, Mater. Today: Proc., 2018, 5, 23888–23897 CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- US3633A, Goodyear Charles, 1844.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- M. K. Lauer and R. C. Smith, Compr. Rev. Food Sci. Food Saf., 2020, 19, 1–53, DOI:10.1111/1541-4337.12627.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, Mater. Adv., 2021, 2, 2391–2397 RSC.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, Mater. Adv., 2022, 3, 4186–4193 RSC.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734–748 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer and R. C. Smith, J. Mater. Chem. A, 2020, 8, 20318–20322 RSC.
- M. S. Karunarathna, A. G. Tennyson and R. C. Smith, J. Mater. Chem. A, 2020, 8, 548–553 RSC.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 590–594 RSC.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 2271–2278 RSC.
- M. K. Lauer, Z. E. Sanders, A. D. Smith and R. C. Smith, Mater. Adv., 2021, 2, 7413–7422 RSC.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 CrossRef CAS.
- C. P. Maladeniya, M. S. Karunarathna, M. K. Lauer, C. V. Lopez, T. Thiounn and R. C. Smith, Mater. Adv., 2020, 1, 1665–1674 RSC.
- C. P. Maladeniya and R. C. Smith, J. Compos. Sci., 2021, 5, 257 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer, A. G. Tennyson and R. C. Smith, Polym. Chem., 2020, 11, 1621–1628 RSC.
- T. Thiounn, M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Sustainable Chem., 2020, 1, 183–197 CrossRef.
- T. Thiounn, M. K. Lauer, M. S. Bedford, R. C. Smith and A. G. Tennyson, RSC Adv, 2018, 8, 39074–39082 RSC.
- T. Thiounn, A. G. Tennyson and R. C. Smith, RSC Adv., 2019, 9, 31460–31465 RSC.
- P. Yan, W. Zhao, F. McBride, D. Cai, J. Dale, V. Hanna and T. Hasell, Nat. Commun., 2022, 13, 4824 CrossRef CAS PubMed.
- T. Thiounn, M. S. Karunarathna, L. M. Slann, M. K. Lauer and R. C. Smith, J. Polym. Sci., 2020, 58, 2943–2950 CrossRef CAS.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044 CrossRef CAS PubMed.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361 CrossRef CAS.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675 RSC.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- C. R. Westerman and C. L. Jenkins, Macromolecules, 2018, 51, 7233–7238 CrossRef CAS.
- C. R. Westerman, P. M. Walker and C. L. Jenkins, J. Visualized Exp., 2019, e59620, DOI:10.3791/59620.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. MacKay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- C. V. Lopez, C. P. Maladeniya and R. C. Smith, Electrochem., 2020, 1, 226–259 CrossRef.
- M. L. Eder, C. B. Call and C. L. Jenkins, ACS Appl. Polym. Mater., 2022, 4, 1110–1116 CrossRef CAS.
- J. H. Worthington Max, L. Kucera Renata, T. Gibson Christopher, A. Sibley, D. Slattery Ashley, A. Campbell Jonathan, F. K. Alboaiji Salah, J. Young, N. Adamson, R. Gascooke Jason, A. Lewis David, S. Quinton Jamie, V. Ellis Amanda, M. Chalker Justin, S. Albuquerque Ines, J. L. Bernardes Goncalo, A. Muller Katherine, A. Johs, D. Jampaiah, M. Sabri Ylias, K. Bhargava Suresh and J. Ippolito Samuel, Chemistry, 2017, 23, 16219–16230 CrossRef CAS PubMed.
- L. J. Esdaile and J. M. Chalker, Chemistry, 2018, 24, 6905–6916 CrossRef CAS PubMed.
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 CrossRef.
- Y. Zhang, T. S. Kleine, K. J. Carothers, D. D. Phan, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, Polym. Chem., 2018, 9, 2290–2294 RSC.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- P. T. Dirlam, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1635–1668 CrossRef CAS.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. LaVilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156 CrossRef CAS PubMed.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722 RSC.
- J. J. Griebel, G. Li, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 173–177 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS PubMed.
- V. P. Oleshko, J. Kim, J. L. Schaefer, S. D. Hudson, C. L. Soles, A. G. Simmonds, J. J. Griebel, R. S. Glass, K. Char and J. Pyun, MRS Commun., 2015, 5, 353–364 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, A. V. Astashkin, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258–1261 CrossRef CAS PubMed.
- S. Namnabat, J. J. Gabriel, J. Pyun and R. A. Norwood, Proc. SPIE, 2014, 8983, 89830D CrossRef.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS PubMed.
- M. Mann, E. Kruger Jessica, F. Andari, J. McErlean, R. Gascooke Jason, A. Smith Jessica, J. H. Worthington Max, C. C. McKinley Cheylan, A. Campbell Jonathan, A. Lewis David, T. Hasell, V. Perkins Michael and M. Chalker Justin, Org. Biomol. Chem., 2019, 17, 1929–1936 RSC.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- C. Herrera, K. J. Ysinga and C. L. Jenkins, ACS Appl. Mater. Interfaces, 2019, 11, 35312–35318 CrossRef CAS PubMed.
- J. A. Smith, X. Wu, N. G. Berry and T. Hasell, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1777–1781 CrossRef CAS PubMed.
- C. B. Meyer, T. Stroyer-Hansen, D. Jensen and T. V. Oommen, J. Am. Chem. Soc., 1971, 93, 1034–1035 CrossRef CAS.
- B. Meyer, Chem. Rev., 1964, 64, 429–451 CrossRef CAS.
- J. J. Dale, S. Petcher and T. Hasell, ACS Appl. Polym. Mater., 2022, 4, 3169–3173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic and characterization including IR spectra, NMR Spectra, TGA and DSC data, details on mechanical testing. See DOI: https://doi.org/10.1039/d2ma00986b |
| This journal is © The Royal Society of Chemistry 2023 |