
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon transport on self-assembled block copolymer electrolytes with different side chain chemistries†
Mario V.
Ramos-Garcés‡
 a,
Dodangodage Ishara
Senadheera
b,
Karthik
Arunagiri
a,
Polyxeni P.
Angelopoulou
c,
Georgios
Sakellariou
c,
Ke
Li§
b,
Bryan D.
Vogt
a,
Revati
Kumar
*b and
Christopher G.
Arges
*a
a,
Dodangodage Ishara
Senadheera
b,
Karthik
Arunagiri
a,
Polyxeni P.
Angelopoulou
c,
Georgios
Sakellariou
c,
Ke
Li§
b,
Bryan D.
Vogt
a,
Revati
Kumar
*b and
Christopher G.
Arges
*a
aDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: chris.arges@psu.edu
bDepartment of Chemistry, Louisiana State University, Baton Rouge, LA 70803, USA. E-mail: revatik@lsu.edu
cDepartment of Chemistry, National and Kapodistrian University of Athens, 15771 Athens, Greece
First published on 11th January 2023
Abstract
Ion-exchange membranes (IEMs) are used in electrochemical systems for a wide variety of applications, including water purification, mineral recovery, and energy storage and conversion. These materials often dictate the ohmic overpotential drop in electrochemical systems and can have a profound impact on process efficiency. Central to the rationale design of ion-conducting polymers is a fundamental understanding as to how chemical composition and macromolecular architecture influence ion and water transport. Herein, we report the preparation of three microphase separated block copolymer electrolytes (BCEs) with long-range order that have different side chain chemistries in the ionic domain. The side chain variants are alkyl and alkoxy pendants and a zwitterionic group. The side chain chemistries were installed post-assembly of crosslinked block copolymers. Unexpectedly, we observed that more hydrophobic alkyl side chain yields about an order of magnitude greater ionic conductivity when compared to alkoxy and zwitterionic side chains despite similar IEC values and water uptake. Molecular dynamics simulations reveal that the hydrophilic alkoxy moieties and zwitterion structure coordinate with water making less free water available for mediating ionic conductivity. Conversely, the hydrophobic alkyl side chains give rise to large, interconnected water clusters that promote ionic conduction of the counterion.
Introduction
Numerous electrochemical systems, such as water purification units, electrolyzers, and fuel cells, deploy ion-exchange membranes (IEMs) as separators and ionomer materials as electrode binders.1–4 These materials are required to transport ionic species from one electrode to another, or one compartment to another, through a charged polymer network that prevents bulk fluid crossover. In electrochemical separations, IEMs and ionomer binders that selectively promote the permeability of one ion over others are critical for selective ionic separations – which is important to critical materials and mineral recovery5,6 and organic acid capture from processed biomass.7,8The ionic conductivity of IEMs and ionomer binders is a key bulk material property that governs the ohmic overpotential drop in electrochemical cells. Depending on the cell current density, as well as other sources of overpotential, the ohmic overpotential can have a large influence on the energy efficiency of electrochemical systems as well as their sizing for a given application.9,10
Block copolymer electrolytes (BCEs) are a subset of IEMs and ionomer materials. BCEs consist of tethered, repeating ionic moieties in one polymer block, while other block(s) feature non-ionic repeat units that are often glassy and endow the material with mechanical integrity.11,12 Additionally, phase separation of the blocks via self-assembly allows the formation of controlled microstructures with long-range order, making them model systems that allow researchers to explore how mesoscale morphology and periodic feature sizes govern transport properties as well as ion-partitioning.13,14 Furthermore, perfect alignment of ionic grains from one electrode to another eliminates structure defects that often obfuscate interpretation as to how chemical composition alters transport and partitioning properties.15
There are previous reports demonstrating that side chain chemistry in polymeric IEMs can alter ionic conductivity. For example, flexible alkoxy side chains in anion exchange membranes (AEMs) were shown to promote ionic conductivity over rigid alkyl side chains.16 Short-side chain perfluorosulfonic acid proton exchange membranes enhance proton conductivity under lower relative humidity conditions when benchmarked against longer-side chain variants like Nafion.17
Herein, the role of side chemistry (alkoxy, alkyl, and zwitterionic) on ionic conductivity of BCEs was assessed as thin films on interdigitated electrode arrays and confirmed using molecular dynamics. This work builds off our previous reports that prepared BCE thin films with long-range order by alkylating the pyridine in self-assembled poly(styrene-block-2-vinyl pyridine) (PSbP2VP) with iodomethane to form poly(styrene-block-2-vinyl pyridine/pyridinium iodide) (PSbP2VP/NMP+I−).18,19 We study this class of BCEs as thin films as opposed to bulk membranes as it allows us to control the microstructure over large areas – which is difficult to achieve in bulk, free standing membranes. Controlling the microstructure is vital for drawing definitive conclusions about how the composition impacts ion transport and other material properties. This information can play an important role in the rational design of bulk IEM separators. This work reports the functionalization of the pyridine group in self-assembled PSbP2VP with bromo-terminated alkoxy, alkyl, and sulfonated molecules as opposed to iodomethane. To perform these Menshutkin reactions post PSbP2VP self-assembly, it was necessary to crosslink the polystyrene block with UV light. Crosslinking allowed the Menshutkin reaction to proceed without disrupting the self-assembled microstructure. Thin film ionic conductivity measurements demonstrate that the BCE with alkyl side chains have the highest ionic conductivity across different relative humidity values. BCEs featuring alkoxy and zwitterionic side chains had comparable ionic conductivity when fully hydrated; the BCE with a zwitterionic installed group had almost no ionic conductivity measured under relative humidity conditions. Molecular dynamics (MD) simulations were performed on the BCEs with different side chain chemistries. These simulations showed that water coordination to the ether groups in the alkoxy and the zwitterionic group reduced the amount of uncoordinated water available to mediate ionic conduction via both translational diffusion and anion hopping.
Methods
Materials
Poly(styrene-block-2-vinyl pyridine) (PSbP2VP) with Mn value of 40k–44k (PDI: 1.10) was purchased from Polymer Source Inc and was used as received. Silicon wafers with and without thermally grown oxide layers of 1 μm were acquired from Pure Wafer Inc. The silicon wafers with thermally grown oxide layers were used for fabricating interdigitated electrodes (IDEs). Silicon wafers without thermally grown oxide layers were used for atomic force microscopy (AFM) and X-ray photoelectron spectroscopy (XPS). Toluene, N,N-dimethylformamide (DMF), ethanol 200 proof, acetone, N-methylpyrrolidone (NMP), isopropyl alcohol, 1-bromododecane (97%), and sodium 2-bromoethanesulfonate (98%) were acquired from VWR or Sigma-Aldrich and used as received. n-Hexane (95+% extra pure) was acquired from Acros Organics. The polyethylene glycol (PEG) linkers bromo-PEG3-bromo (98%) and 1-Br-PEG4 (98%) were acquired from BroadPharm®.IDE fabrication
IDEs were fabricated at Penn State's Nanofabrication Laboratory using a similar procedure previously reported by Arges et al.19 The IDE fabrication process commenced with the application of a lift-off resist (LOR™ SA) to a silicon wafer with a thermally grown oxide layer of 1 μm via spin coating at 4000 rpm for 45 seconds and subsequently baking the coated wafer at 185 °C for 3 minutes. In the next step, a positive photoresist (SPR™ 3012) was spin coated on top of the lift-off resist optical at 4000 rpm for 45 seconds and subsequently baked at 80 °C. An MA/BA Gen4-serie mask and bond aligner tool was usedto expose the photoresist (60 mJ cm−2), followed by the developing step. This developing step consisted of submerging the already exposed sample into a Microdeposit® MF® CD-26 developer for 1 min, followed by a thorough rinsed with DI water. The last two steps were the evaporation of 15 nm of titanium (Ti) and 135 nm of gold (Au) by electron beam evaporation (Temescal FC-2000) and lift-off of the resist with NMP at 70 °C. Each IDE consisted of 22 teeth that were 0.45 cm long and 0.1 mm wide. The separation between the teeth was also 0.1 mm.Self-assembly of PSbP2VP on wafer substrates
The formation of PSbP2VP perpendicular lamellae required a grafted random copolymer brush (mono-hydroxy terminated PSrP2VP (HO-PSrP2VP, 80% PS)) on the silicon wafer to provide a non-preferential surface. This brush was synthesized via nitroxide mediated polymerization (NMP).20 A 1 wt% solution of HO-PSrP2VP in toluene was spin coated onto silicon wafers at 4000 rpm for 45 seconds. This RCP was grafted onto the surface of the substrate by thermal treatment at 200 °C for 10 min in a N2 filled glovebox. Then, a 1.5 wt% solution of the PSbP2VP 40k–44k block copolymer in DMF was spin coated onto the substrate with the grafted polymer brush. The phase separation of the BCP was performed by solvent annealing with acetone for 2 h at a flow rate of 45 sccm and 5 sccm of N2 as the carrier gas.BCE formation with different side chain chemistries
The BCEs with different side chain chemistries were prepared by alkylating the P2VP domain via a Menshutkin reaction with bromo-terminated reactants. Before the Menshutkin reactions, the PS domain in self-assembled PSbP2VP was crosslinked using a XL-1500 UV crosslinker (Spectroline®). The sample was placed in a vacuum sealed chamber with a transparent quartz window (0.5 cm thickness) and then was loaded into the XL-1500 UV crosslinker equipment. The sample was crosslinked at 254 nm with an exposure energy of 800 mJ cm−2. The crosslinking step was necessary to preserve the nanostructures after the Menshutkin reaction for all the BCEs prepared. For the alkyl side chain BCE, the wafer with the crosslinked PSbP2VP (xLPSbP2VP) was submerged in a 10% v/v 1-bromododecane solution in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 hexane:ethanol mixture. The reaction was performed for 24 h at 50 °C under constant stirring. The other two BCEs were prepared in a similar fashion with some minor differences in procedure. The alkoxy BCE was prepared by submerging the xLPSbP2VP in a 5% v/v solution of bromo-PEG3-bromo in pure ethanol solvent. This reaction was performed for 24 h at room temperature without stirring. Finally, the zwitterionic BCE was prepared by submerging the xLPSbP2VP in a 0.1 M solution of 2-bromoethanesulfonate in DI water. The reaction was performed for 48 h at 50 °C under constant stirring. Fig. S1 (ESI†) presents a flow diagram depicting the preparation of the BCEs reported in this work.
:50 hexane:ethanol mixture. The reaction was performed for 24 h at 50 °C under constant stirring. The other two BCEs were prepared in a similar fashion with some minor differences in procedure. The alkoxy BCE was prepared by submerging the xLPSbP2VP in a 5% v/v solution of bromo-PEG3-bromo in pure ethanol solvent. This reaction was performed for 24 h at room temperature without stirring. Finally, the zwitterionic BCE was prepared by submerging the xLPSbP2VP in a 0.1 M solution of 2-bromoethanesulfonate in DI water. The reaction was performed for 48 h at 50 °C under constant stirring. Fig. S1 (ESI†) presents a flow diagram depicting the preparation of the BCEs reported in this work.
Physical and chemical characterization of BCEs
Topography images of the three BCEs with different side chain moieties were obtained with atomic force microscopy (AFM; Horiba SmartSPM 1000). The AFM used uncoated Si cantilevers (ACTA-SS, k = 37 N m−1, 125 μm length) operating at a resonant frequency of 287 kHz and a free amplitude of ≈30 nm. The thickness of the thin films and polymer brushes was determined with a Woollam M-2000F Focused Beam Spectroscopic Ellipsometer and the data was analyzed using the CompleteEASE software. X-ray photoelectron spectroscopy (XPS) was performed on a Scienta Omicron ECSA 2SR X-ray Photoelectron Spectroscope. All spectra were calibrated to the adventitious carbon 1s peak at 284.8 eV and fitted using a Shirley background. Pyridine conversion to pyridinium was calculated using the relative peak area ratio of the two N 1s peaks corresponding to the tertiary and quaternary nitrogen atoms, respectively, using the CasaXPS software. The IEC values of the BCE thin films were calculated using the ratio of pyridinium to pyridine as determined by XPS (see eqns (S1)–(S6) in the ESI†).The BCE film resistance was determined using electrochemical impedance spectroscopy (EIS) (Gamry Reference 3000 Potentiostat/Galvanostat) carried out in galvanostatic mode. The frequency range for EIS was set from 100000–0.1 Hz with a 1 × 10−6 A amplitude. The electrode pad areas of the IDE substrate were cleaned with a cotton Q-tip after spin coating the polymer brush to remove the film for electrical connections and cleaned a second time after spin coating the BCP. The measurements of the ionic conductivity under relative humidity were performed inside a temperature and humidity controlled chamber from ESPEC North America, Inc. (model BTX-475) at 30 °C at the following relative humidity percentages: 95%, 75%, and 50%. To measure the conductivity when the films are interfaced with DI water, a drop of DI water was place on top of the IDEs used as substrates for the preparation of the BCEs. The ionic conductivity (κ) was calculated using eqn (1) below:21
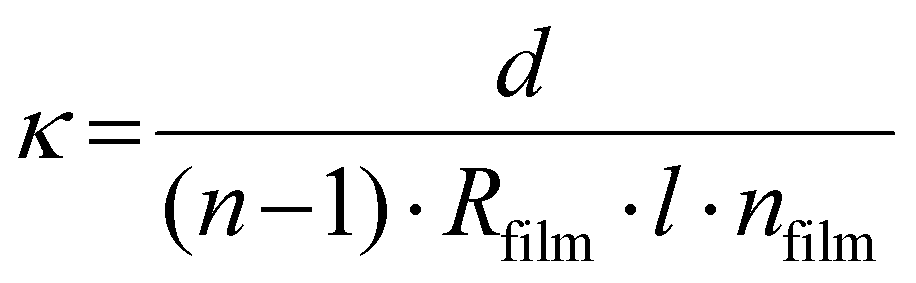 | (1) |
Water uptake in the films was determined using a quartz crystal microbalance (QCM) with dissipation (QCM-D, Q-Sense E1). All measurements were performed at 30 °C. The clean, uncoated sensors were measured in air to provide a baseline. The coated sensors were first measured in dry air to obtain the approximate thickness of the coating. The thickness was estimated from the change in frequency between the uncoated crystal and the coated crystal. This calculation used the Sauerbrey expression to obtain the areal mass of the film. The thickness was estimated by assuming a density of 1 g cm−3 for the copolymer coating. Subsequently, the crystals were exposed to humidified air, corresponding to approximately 55% relative humidity, and then liquid type I water. Multiple overtones (3rd–11th) were analyzed using the Sauerbrey expression22 to determine the masses under these conditions. For the liquid water, the response of the crystal to the bulk liquid was subtracted by considering the kinematic viscosity of water at 30 °C and intrinsic properties of quartz.23 The mass reported to be associated with the sorbed liquid water includes the surface associated water.
Molecular dynamic simulations
The relationship between the microstructures, the solvation of the charge transport ion as well as transport properties for the three block copolymers (consisting of alkoxy side chains, alkyl side chains, and zwitterionic side chains) were investigated systematically by performing all-atom molecular dynamics simulations. In each system, there are 30 polymers. Each block copolymer is a 20-mer with a block of 10 styrene monomers followed by an equal length segment having alternating charged (2-vinyl pyridinium) and uncharged (2-vinyl pyridine) monomers. The positively charged pyridinium moieties comprised the respective side chains (alkoxy, alkyl, zwitterion). Fig. S2 (ESI†) provides the structure of each model BCE used for MD simulation study. The reason for including non-alkylated pyridine in the ionic domain of the model BCE is to account for the fact that 100% alkylation of the pyridine N is not achievable experimentally. A bromide counterion and ten water molecules were introduced to each positively charged pyridinium moiety in each BCE system. The water amount was based on the previous simulation work of BCE with methylated pyridinium domains, which was six water molecules per pyridinium moiety and was decided by solution uptake experiments.24 Here an approximate value of 10 was considered to account for the side chains. The systems were simulated using the OPLSAA25 force field along with the SPC/E26 model for water using the LAMMP27,28 software package. Detailed information on the simulation setup is provided in the ESI.† The solvation structures were calculated from the replica exchange simulation trajectories at 300 K. The translational dynamics of the system were studied using the trajectories from the canonical MD simulations at a constant temperature. Finally, non-equilibrium simulations in the presence of an electric field were carried out to determine the ionic conductivities.24,28 The simulation setup is discussed in detail in the ESI.†Results and discussion
PSbP2VP BCEs with different side chain chemistries and long-range order necessitates that the fixed charge groups be added post self-assembly. Block copolymers and BCEs with high enthalpic interaction between the different blocks often cause preferential wetting of one block to the substrate or free surface during deposition to a substrate and annealing. This prevents perpendicular alignment. Probing the long-range order of BCEs and discerning their microstructure via top-down imaging and X-ray scattering techniques necessitates perpendicular alignment. Fig. S3 (ESI†) gives an AFM image of PSbP2VP with perpendicular lamellae morphology.The conversion of self-assembled PSbP2VP to a BCE post self-assembly without disrupting the nanostructure previously occurred by infiltrating iodomethane vapor into the polymer film.18 Iodomethane is fairly volatile at room temperature making the vapor infiltration reaction possible. However, the reactants used to prepare different side chain chemistry to PSbP2VP are not suitable for vapor infiltration because the bromo-terminated reactants are not volatile and have low saturated vapor pressure values. Hence, the PSbP2VP starting materials were submerged in solutions of the corresponding bromo-terminated alkoxy, alkyl, and sulfonate reactants. Numerous attempts to optimize the reactant concentration and solvent type for the Menshutkin reaction always resulted in loss of the nanostructure of the self-assembled PSbP2VP or removal of the PSbP2VP thin film (see Fig. S4, ESI†). To circumvent this problem, the polystyrene domain in the self-assembled PSbP2VP was crosslinked by exposing the thin film to UV light. The resiliency of the self-assembled crosslinked PSbP2VP (xLPSbP2VP) to ethanol was evaluated by immersing the samples in the solvent at different temperatures (20 °C, 40 °C, and 50 °C) for 3 hours. The AFM micrographs presented in Fig. S5 (ESI†) demonstrate that non-crosslinked PSbP2VP loses its perpendicular lamellae structure when immersing the sample in ethanol at temperatures of 40 °C and 50 °C. Conversely, xLPSbP2VP shows retention of its self-assembled morphology after being submerged in ethanol for 3 h at all temperatures studied (20 °C, 40 °C, and 50 °C).
Fig. 1 shows the AFM images of the materials after the alkylation reactions via the above-mentioned scheme using xLPSbP2VP submerged in solution containing the bromo-terminated reactants. From the AFM images, the nanostructures of the microphase separated block copolymers were still intact post the Menshutkin reactions as the crosslinked polystyrene locked the structure in place. Nonetheless, the use of a mono-halogenated PEG reactant, 1-Br-PEG4, still causes the loss of the nanostructures even when using xLPSbP2VP (see Fig. S6, ESI†). For this reason, a di-bromo PEG reactant was used, Br-PEG3-Br. The di-bromo moiety was used to crosslink the P2VP domain as observed in other studies that have used di-bromo butane (DBB).29 By using the Br-PEG3-Br, we were able to retain the nanostructures as seen in Fig. 1. The micrographs of the BCEs show perpendicular lamellae structures with a periodic spacing (L0) of 44 nm. These morphologies and periodic spacing are identical to the parent self-assembled BCP and indicate that the alkylation reaction conditions does not affect the self-assembled structure (Fig. S3, ESI†).
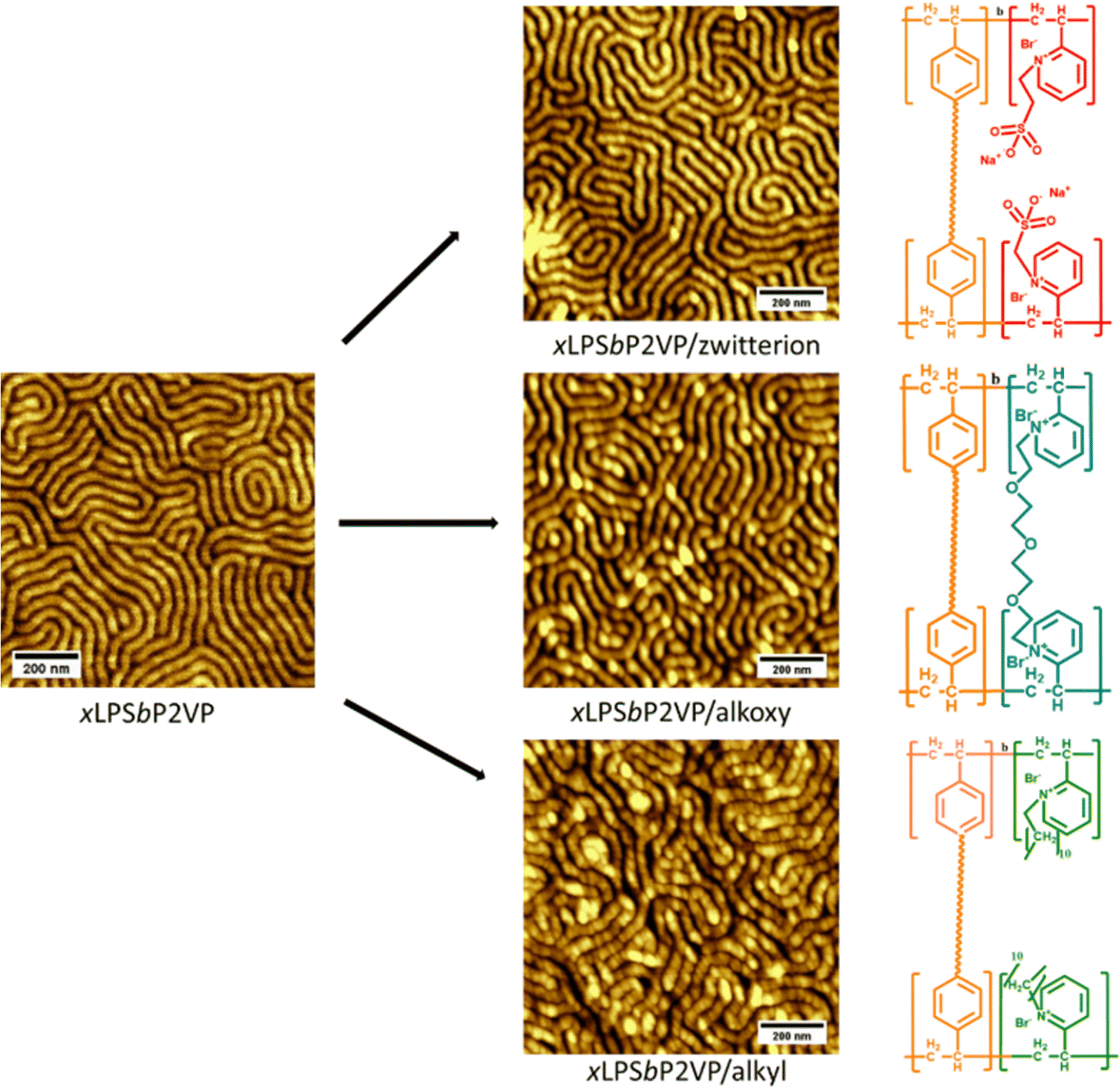 | ||
| Fig. 1 AFM micrographs of xLPSbP2VP (left), xLPSbP2VP/zwitterion (top), xLPSbP2VP/alkoxy (middle), and xLPSbP2VP/alkyl (bottom) BCEs. The chemical structures of the BCEs are represented on the right side of their corresponding AFM image. | ||
After devising a scheme to retain nanostructures post Menshutkin reactions, XPS was performed to confirm the presence of the ionic moieties in the BCEs. The xLPSbP2VP material shows only one peak corresponding to the tertiary nitrogen and it appears at 399.1 eV (Fig. S7, ESI†). The Menshutkin reaction converts pyridine into pyridinium – fixed charge with a bromide counterion. The nitrogen in the pyridinium has a higher N 1s binding energy. Fig. 2 shows the XPS spectra for the xLPSbP2VP materials after the Menshutkin reaction with the three different bromo-terminated reactant agents (bromo-PEG3-bromo, 2-bromoethanesulfonate, and 1-bromododecane). The high-resolution N 1s spectrum show two peaks that arise from pyridine and pyridinium. The N 1s binding energy for pyridinium in xLPSbP2VP/alkoxy (Fig. 2b) occurred at 408.07 eV – which is 9 eV higher compared to the pyridine N (399.1 eV). The higher binding energy for the N 1s in pyridinium arises from its electron deficiency as it is positively charged. The N 1s peaks for pyridinium in the xLPSbP2VP/zwitterion and xLPSbP2VP/alkyl BCEs had peaks at binding energy values that were close to the pyridine N 1s peak. Deconvolution of the N 1s peak was necessary to accurately determine the binding energy of both types of nitrogen atoms. The binding energy difference for these two BCEs are in the range of 1.6–1.8 eV.
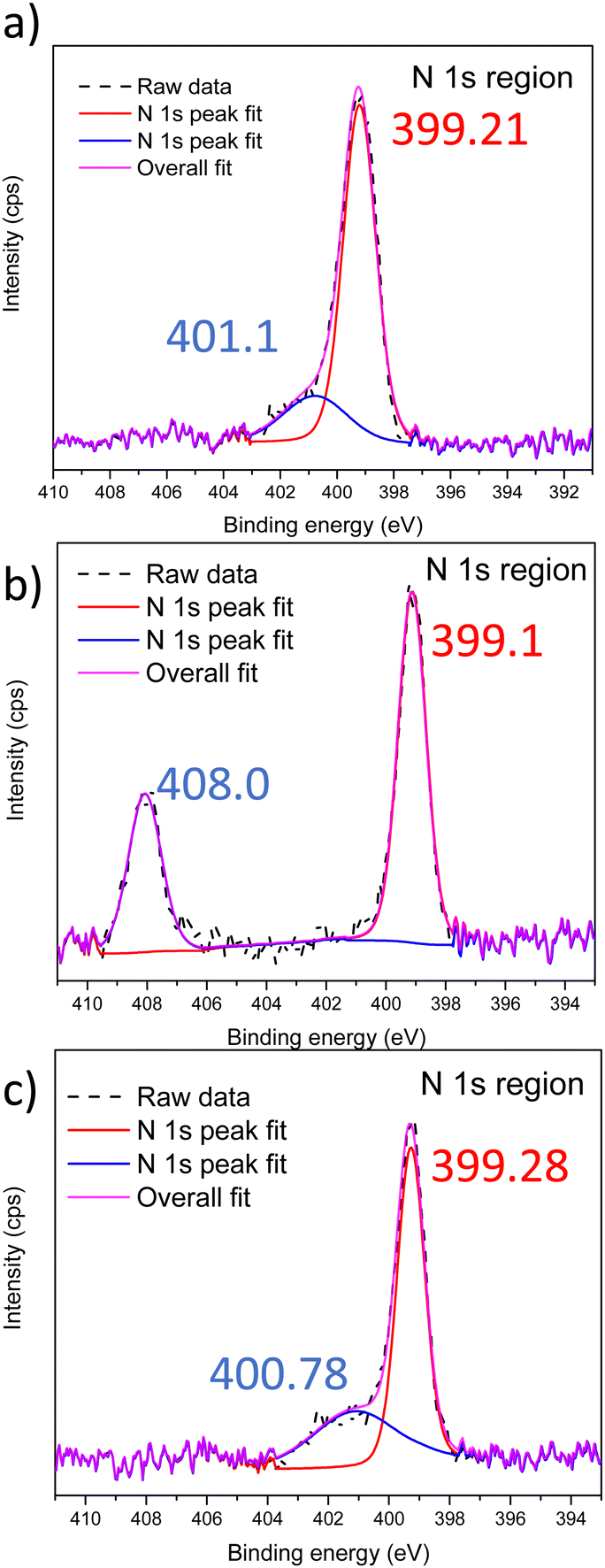 | ||
| Fig. 2 XPS spectra in the N 1s region of (a) xLPSbP2VP/zwitterion, (b) xLPSbP2VP/alkoxy and (c) xLPSbP2VP/alkyl BCEs. | ||
The higher N 1s binding energy for pyridinium in xLPSbP2VP/alkoxy BCE versus xLPSbP2VP/alkyl BCEs is ascribed to the alkyl side chain variant being more electron donating than the alkoxy side chain. However, it is unclear why the N 1s binding energy for pyridinium in xLPSbP2VP/alkoxy is higher than xLPSbP2VP/zwitterion and why the N 1s binding energy for pyridinium for xLPSbP2VP/zwitterion is similar to xLPSbP2VP/alkyl because the sulfonate moiety in xLPSbP2VP/zwitterion is electron withdrawing and in close proximity to the pyridinium group. There could be other factors at play beyond effects of electron donating and withdrawing characteristics of adjacent moieties. For instance, pyridinium screening by the bromide counterion could impact the binding energy value of the N 1s of pyridinium. Furthermore, interactions between sulfonate group oxygen atoms and nitrogen might also affect N 1s binding energy values. Future work will investigate this effect by performing near-ambient pressure XPS at National Lab User Facilities with different partial pressure values of water that can dictate the extent of ionic charge dissociation.
The percentage of pyridine groups to pyridinium groups was determined from the peak integration of the tertiary and quaternary signals. While XPS is a surface sensitive technique (ca. 5 nm), previous works have shown that XPS gives the same trend and similar results as FTIR (a bulk probing technique).18 This suggests that alkylation takes places within the film with a reasonable degree of homogeneity and XPS results can be seen as a representation of the thin film's chemical state over its thickness (∼20 to 30 nm). For the xLPSbP2VP/zwitterion, xLPSbP2VP/alkoxy, and xLPSbP2VP/alkyl BCEs, the pyridine to pyridinium conversion was 21%, 27%, and 24%, respectively (Table 1). These conversion values were used to calculate the ion-exchange capacity (IEC) values of the thin film BCEs using the eqns (S1)–(S6) in the ESI.† The IEC values for the samples varied by less than 30%. Additionally, the counterion was also probed with XPS by performing high resolution scans in the bromine (Br) 3d region (Fig. S8, ESI†). These scans show that bromine is present in all three BCEs with two peaks observed corresponding to the Br 3d5/2 and 3d3/2 orbitals, further confirming the successful alkylation of pyridine groups in the BCP. In Fig. S9 (ESI†), the high-resolution scans of the Na 1s and S 2p regions for the xLPSbP2VP/zwitterion BCE are shown. These high-resolution scans confirm the presence of tethered sulfonate and sodium counterions in xLPSbP2VP/zwitterion.
| Fraction of pyridinium repeat units in the P2VP block | IEC (mmol g−1) | |
|---|---|---|
| xLPSbP2VP/alkyl | 0.24 | 0.92 |
| xLPSbP2VP/alkoxy | 0.27 | 1.10 |
| xLPSbP2VP/zwitterion | 0.21 | 0.85 |
The ionic conductivity of the different BCEs was measured with EIS by determining the film resistance extracted from the equivalent model fit of Nyquist data. Herein, we used a model that only consists of resistors (R) and constant phase elements (CPE); both having physical manifestations of charge transport in IDE configurations. This model has been previously used to determine the film resistance of BCEs based on the PSbP2VP BCP.15 Alternative electric circuit equivalent models have been used for thin film Nafion, PFSA, and lithium salt imbibed PEO conductivity.21,30–32Fig. 3a shows the Nyquist plot obtained from EIS measurements for the xLPSbP2VP/alkoxy BCE when interfaced with DI water along with the multivariable equivalent model circuit. In this figure, Rfilm is the resistance and CPEfilm the dielectric behavior of the polymeric film between the electrodes; CPEdl models the ion depletion double-layer at the polymer–electrode interface (accumulation of charge at the electrode); Re models the electrode resistance (e.g., electrical contact).
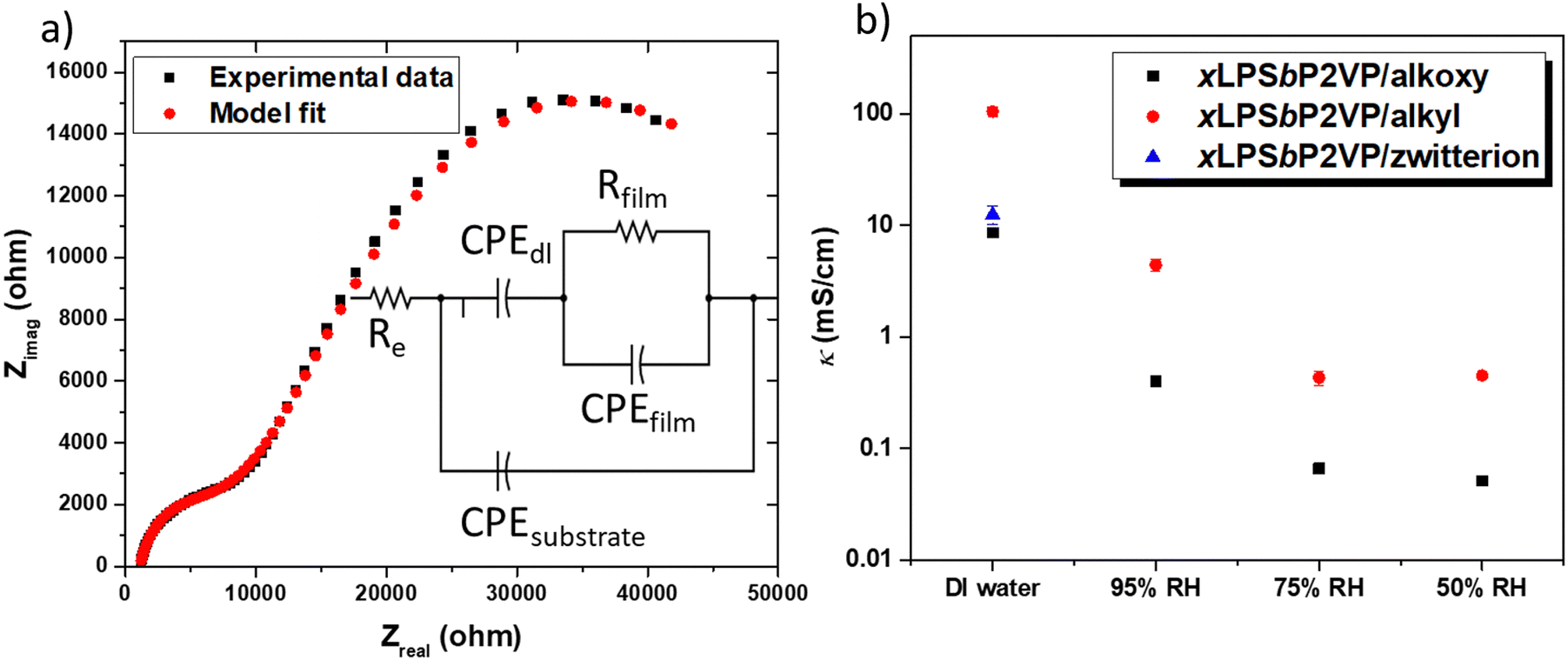 | ||
| Fig. 3 (a) Representative electric circuit model fit to Nyquist data of thin film xLPSbP2VP/alkoxy BCE interfaced with DI water. (b) Ionic conductivity of the three BCEs samples when interfaced with DI water and at different relative humidity percentages (95%, 75%, and 50%). All measurements were performed on IDEs substrates at room temperature for DI water measurements and 30 °C for RH measurements. Note: xLPSbP2VP/zwitterion showed ∼0 mS cm−1 conductivity under RH environments. | ||
The ionic conductivity of the three BCEs showed marked differences as a function of side chain chemistry. Fig. 3b shows the ionic conductivity values for the three BCEs interfaced with liquid DI water and under three different relative humidity conditions: 95%, 75%, and 50%. All BCEs show their highest conductivity when they were interfaced with liquid DI water. Furthermore, the BCEs gave higher ionic conductivity values at larger RH values. Larger partial pressures of water result in greater hydration of the thin film BCEs. Water is central to dissociating ion charge pairs and promotes counterion transport under electrochemical potential gradients.14 Notably, there is over an order magnitude drop in ionic conductivity when moving from the BCE interfaced with DI water to 95% RH (water vapor). Similar observations have been observed with sulfonated ionomers and the differences in ionic conductivity between 100% RH and DI liquid water is known as Schroeder's paradox.33
From the three BCEs studied herein, xLPSbP2VP/alkyl gave highest bromide ion conductivity at 105 ± 10 mS cm−1 in deionized water. The alkoxy and zwitterionic BCEs demonstrated a bromide conductivity (8.6 ± 0.7 and 12.5 ± 2.3 mS cm−1, respectively) of roughly one order of magnitude less than the alkyl BCE when these are interfaced with liquid DI water. Bromide ion conductivity over 100 mS cm−1 at room temperature for xLPSbP2VP/alkyl represents a very high value. The result was unexpected because the alkyl side chain is more hydrophobic when compared to the alkoxy and zwitterionic side chains. It was initially hypothesized that the ether groups in the alkoxy side chain would aid in counterion dissociation from pyridinium because the electron rich ethers would repel the electron rich halide counterion. Plus, the alkoxy side chains are more hydrophilic in nature and it was anticipated they would adsorb more water for a given external partial pressure of water. Additional water within the BCE should typically favor higher ionic conductivity. As it will be shown later from MD simulations, the alkoxy side chains in xLPSbP2VP/alkoxy coordinate water and reduce the water's ability to assist with ionic conductivity.
In the case of the of the zwitterionic BCE, we hypothesized that the additional ionic group, tethered sulfonate with a sodium counterion, would increase the dielectric constant, as reported in zwitterionic polymer electrolytes for batteries,34,35 while also providing a second counterion that can diffuse and carry current through the BCE thin film. However, xLPSbP2VP/zwitterion showed the second lowest conductivity in deionized water and had no ionic conductivity when exposed to humidified environments at 30 °C. This observation suggested that the 50 to 95% RH did not provide enough water to xLPSbP2VP/zwitterion to dissociate the ion charge pairs for ionic conductivity.
QCM measured the water uptake of the different BCE thin film variants at 55% RH and when interfaced with liquid DI water (Table 2). All the BCE thin films adsorbed water. The data in Table 2 demonstrate that the water uptake is a function of film thickness and without precise control of thickness, it is difficult to infer how the different sidechain chemistries affect water uptake.
| Material | Film thicknessa (nm) | Water uptake @ ∼55% RH (μg cm−2) | Liquid water uptake (μg cm−2) |
|---|---|---|---|
| a Assumes density of the coating is 1 g cm−3. Film thickness value on quartz crystal. b Maximum uptake. Some extraction from film occurs. c Surface did not fully wet and unable to determine uptake as some air remained trapped at the film surface. | |||
| xLPSbP2VP/alkyl sample 1 | 31.3 ± 2.4 | 0.102 ± 0.013 (3.1 wt%) | 1.78 ± 0.32 (36.2 wt%) |
| xLPSbP2VP/alkyl sample 2 | 40.2 ± 2.1 | 0.259 ± 0.035 (6.1 wt%) | 1.43 ± 0.13b (26.2 wt%) |
| xLPSbP2VP/alkoxy sample 1 | 25.7 ± 0.2 | 0.477 ± 0.012 (15.6 wt%) | 0.801 ± 0.33b (23.7 wt%) |
| xLPSbP2VP/alkoxy sample 2 | 13.7 ± 0.3 | 0.014 ± 0.001 (1.0 wt%) | 0.102 ± 0.03b (6.9 wt%) |
| xLPSbP2VP/zwitterion | 117.5 ± 5.3 | 0.832 ± 0.670 (6.6 wt%) | —c |
The IEC and water uptake data for the three variant BCE films cannot explain why the xLPSbP2VP/alkyl sample had about an order of magnitude higher ionic conductivity when compared to xLPSbP2VP/alkoxy and xLPSbP2VP/zwitterion in liquid water and under different relative humidity values. Hence, these observations motivated molecular dynamics simulations to examine the state of water within the BCEs and how they may affect ionic conductivity.
To gain more insight into this observed experimental behavior, molecular dynamics simulations were performed. These simulations assisted in examining the molecular level differences between the microstructure, the ion association, and transport properties within these newly designed BCE systems. First, the solvation environment was examined around the charged moieties. To that end, radial distribution (g(r)) functions (Fig. 4) were calculated between corresponding atoms to compare the differences in the cation/anion (pyridinium/bromide) association, as well as the level of hydration around the ions in all three BCE cases. The corresponding radial distribution (g(r)) function between the C atom of the first methylene group of the side chain attached to the pyridinium N atom and bromide ions/water molecules provides a direct measurement of the ion pairing and solvation around each pyridinium cation. The results reveal that ion-charge pairs (bromide ion and positively charged pyridinium) in the BCEs associate weakly, and the water present strongly solvates the bromide ions in the system, which is confirmed by the sharp and higher peaks for g(r) calculated between the bromide ion and water O atoms (Fig. 4c) compared to the corresponding broad peaks of g(r) in Fig. 4a. When considering the ion-charge pairs, the alkyl side chains show a higher number of bromides associated with the pyridinium cations (red curve in Fig. 4a) as there are no other sites for bromide ions to interact with, owing to the hydrophobic nature of alkyl side chains. In the alkoxy side chain BCE system, the bromide ions interact with the oxygens of the hydrophilic side chain, leaving fewer ions to interact with the pyridinium site (black curve in Fig. 4a). This is confirmed by the g(r) calculated between the bromide ions and oxygen of alkoxy side chains provided in Fig. S10a (ESI†). The g(r) between the methylene C and bromide ion for the zwitterionic side chain BCE system shows that ions interact very weakly with the pyridinium cation (blue curve in Fig. 4a). This is because the sodium counterions in the xLPSbP2VP/zwitterion system associate very strongly with the bromide ion, which is confirmed by the g(r) calculated between sodium and bromide ions in Fig. S10b (ESI†). This strong association is also the reason for the smaller number of water molecules associated with bromide ion (blue curve in Fig. 4c). Fig. 4b confirms that there is no strong association between the water molecules and the pyridinium cations in all three cases as the corresponding g(r) curves are comparatively broad and less structured.
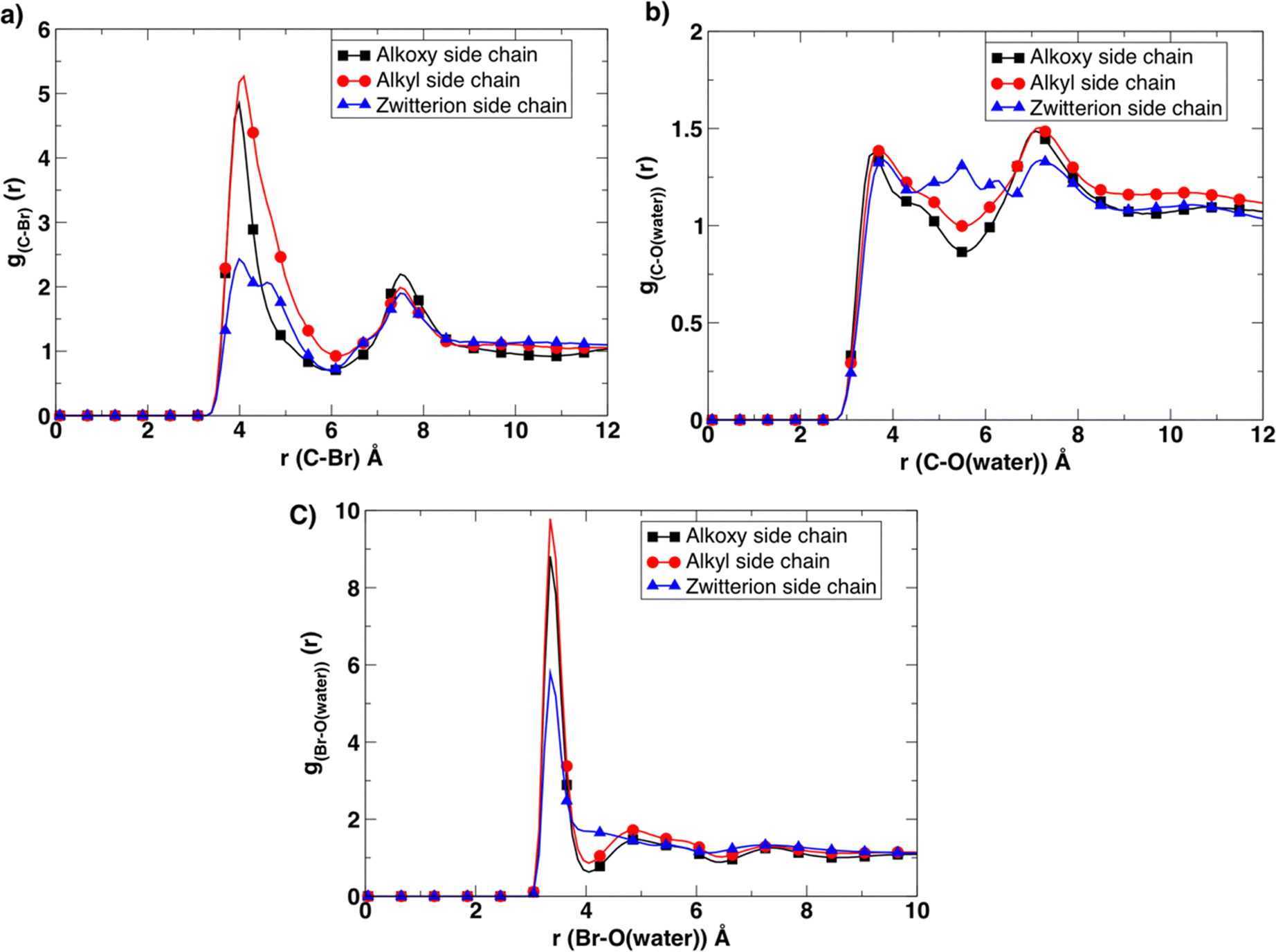 | ||
| Fig. 4 The radial distribution function g(r) between (a) the C atom (the first methylene group of the side chain attached to the pyridinium N atom) and the bromide atom. (b) The C atom (the first methylene group of the side chain attached to the pyridinium N atom) and water O atom. (c) The radial distribution function g(r) calculated between the bromide atom and water O atom. | ||
Furthermore, the BCE with alkyl side chains showed distinct water-rich interconnected hydrophilic regions and water-poor hydrophobic regions (i.e., where the aggregation of styrene and alkyl side chains takes place), while water in BCE with alkoxy side chains and zwitterionic side chains is more distributed as is clear from the representative simulation snapshots and the probability distribution of the largest interconnected water clusters in Fig. 5a. The curves represent the distribution of the largest water clusters as a function of the number of water molecules in the cluster for each system. The largest water cluster of hydrogen-bonded water molecules was calculated for each frame in each simulation by considering the intermolecular O–H length between two water molecules. A water molecule is hydrogen bonded to another if an intermolecular O–H distance between them is less than 2.5 Å.36 The distribution for xLPSbP2VP/alkoxy and xLPSbP2VP/zwitterion systems shows narrow and higher peaks between N = 50 and 100. In contrast, the distribution of the largest water cluster for xLPSbP2VP/alkyl systems is broad, with a peak occurring around N = 150. These observations reveal that hydrophilic alkoxy and zwitterionic side chains have disturbed the formation of larger hydrogen-bonded water clusters within the alkoxy and zwitterionic BCE systems. In comparison, hydrophobic alkyl side chains have contributed to the formation of larger interconnected water clusters.
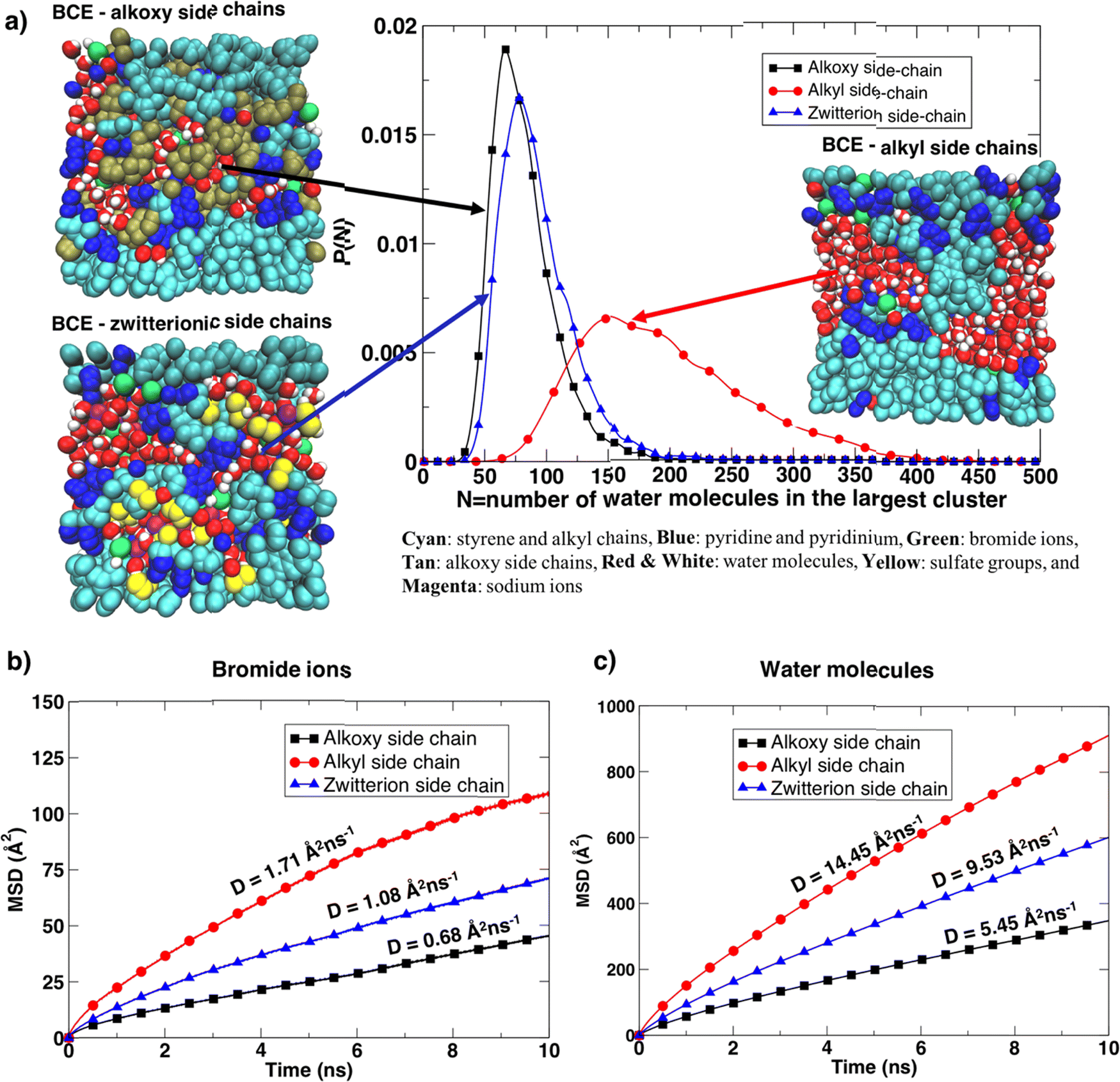 | ||
| Fig. 5 (a) Probability distribution P(n) of the largest water clusters as a function of the number of water molecules (n) in the clusters of BCEs with different side chains. Snapshots obtained from each system represent the interconnected and distributed water regions. (b) Mean square displacement (MSD) of bromide ion as a function of time and (c) mean square displacement (MSD) of water molecules as a function of time. Note the corresponding diffusion constants (D) are given by 1/6 of the slope of the corresponding MSD vs. time. | ||
The translational dynamics of the bromide ion and water are faster for the BCE with alkyl side chains, as is clear from the slopes of the mean square displacement (MSD) of the ion/water versus time graphs provided in Fig. 5b and c for the three systems. The bromide ion and water self-diffusion constant (D) values calculated from the slope of the MSD as a function of time (D ≈ 1/6 of the slope) are highest for alkyl side chain systems, and lowest for the system with alkoxy side chains. The diffusion coefficient values for the xLPSbP2VP/zwitterion system show intermediate properties. The diffusion constant (D) for the sodium ions was also calculated for the zwitterionic case (Fig. S10d, ESI†) and the value (1.70 Å2 ns−1) is similar to the value of the bromide ion diffusion constant in the xLPSbP2VP/alkyl system, which is 1.73 Å2 ns−1. This shows that sodium cations diffuse faster than the bromide anions in the zwitterionic system. Sodium cations also show a stronger association with water molecules than bromide ions in the xLPSbP2VP/zwitterion system, which is confirmed by the higher peak of the g(r) calculated between sodium ion and water O atoms (Fig. S10c, ESI†).
The bromide ion hopping rate (from equilibrium simulations) along the pyridinium backbone and the conductivity from non-equilibrium simulations were calculated and are provided in Table 3. As in previous work, hopping is considered to take place when the bromide ion goes from the solvation shell of one pyridinium cation to another.24 Both values for the BCE system with alkyl side chains are higher compared to the BCE systems containing alkoxy and zwitterionic side chains. This agrees with the experimental ionic conductivity results when interfaced with liquid DI water. The significantly higher bromide hopping rate from simulations seen for the alkyl side chain case compared to the alkoxy results in a much higher conductivity value (factor of 3.6) in contrast to what one would predict based on bromide diffusion constants (factor of 2.5).
| BCE-alkyl | BCE-alkoxy | BCE-zwitterionic | |
|---|---|---|---|
| Bromide ion hopping rate along pyridinium backbone | 5.7 ns−1 | 1.9 ns−1 | 2.6 ns−1 |
| Conductivity from non-equilibrium simulations | 101.0 mS cm−1 | 6.58 mS cm−1 | 3.97 mS cm−1 |
The fraction of bound and unbound water was also determined to further understand the role of the side chain chemistry in water mobility in the systems. The coordination number as a function of the distance, n(r) is given by the equation 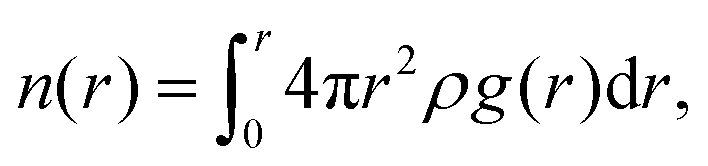 and provides a direct measurement of water coordinated at different sites in the polymer. The values of n(r) at the first minimum of g(O(ether)–O(water))(r) which is 0.83, and g(O(sulfate)–O(water))(r) which is 1.54, (see Fig. S11a and b, ESI†) provide the average number of water molecules coordinated to an ether oxygen in the alkoxy BCE and a sulfate oxygen in the zwitterionic BCE, respectively. To calculate the total number of water molecules, this average number is multiplied by the number of oxygen atoms at the side chain. The alkoxy and zwitterionic BCEs both have 3 oxygens in the side chain attached to a pyridinium cation. In addition, the n(r) calculated at r = 4 Å for each g(r) provided in Fig. 4b gives the average number of water molecules coordinated to the pyridinium cation in each system. For the MD simulation study, 10 water molecules were added per pyridinium moiety consisting of each side chain in all three systems. Table 4 provides a summary of the water coordination at different sites (side chain and pyridinium cation) and the fractions of bound and unbound water in each system. The values again confirm that a higher fraction of uncoordinated water is available in the alkyl BCE to promote ion transport while in the alkoxy and zwitterionic BCEs a lower fraction of unbound water is available to mediate ion conduction.
and provides a direct measurement of water coordinated at different sites in the polymer. The values of n(r) at the first minimum of g(O(ether)–O(water))(r) which is 0.83, and g(O(sulfate)–O(water))(r) which is 1.54, (see Fig. S11a and b, ESI†) provide the average number of water molecules coordinated to an ether oxygen in the alkoxy BCE and a sulfate oxygen in the zwitterionic BCE, respectively. To calculate the total number of water molecules, this average number is multiplied by the number of oxygen atoms at the side chain. The alkoxy and zwitterionic BCEs both have 3 oxygens in the side chain attached to a pyridinium cation. In addition, the n(r) calculated at r = 4 Å for each g(r) provided in Fig. 4b gives the average number of water molecules coordinated to the pyridinium cation in each system. For the MD simulation study, 10 water molecules were added per pyridinium moiety consisting of each side chain in all three systems. Table 4 provides a summary of the water coordination at different sites (side chain and pyridinium cation) and the fractions of bound and unbound water in each system. The values again confirm that a higher fraction of uncoordinated water is available in the alkyl BCE to promote ion transport while in the alkoxy and zwitterionic BCEs a lower fraction of unbound water is available to mediate ion conduction.
| System | No. of water molecules coordinated to the side chain | No. of water molecules coordinated to the pyridinium cation | Total number of water molecules coordinated per pyridinium moiety | Fraction of bound water | Fraction of unbound water |
|---|---|---|---|---|---|
| BCE-alkoxy | 2.49 | 1.34 | 3.83 | 0.38 | 0.62 |
| BCE-zwitterionic | 4.62 | 1.39 | 6.01 | 0.60 | 0.40 |
| BCE-alkyl | N/A | 1.24 | 1.24 | 0.12 | 0.88 |
The experimental and computational results elucidate the ion transport behavior as a function of side chain chemistry for the three BCEs studied herein. The lower ionic conductivity of the alkoxy material results from the interactions between the water molecules and the oxygens atoms on the PEG side chain through hydrogen bonding, leading to the lower water and bromide diffusion coefficients observed on the MD simulations. As mentioned previously, this ether O–water interaction results in fewer water molecules around the bromide counterion as well as a more distributed water arrangement. Additionally, the XPS results show that the nitrogen in the pyridinium group has a larger positive character, possibly holding stronger the counterion via electrostatic forces. In the case of xLPSbP2VP/zwitterion, the interactions between the sulfate oxygen atoms with the pyridinium cation, as well as the sodium ion with the bromide counterion, also prevent a larger water cluster around the counterion. Additionally, the interaction between the two ions in the zwitterionic moiety significantly hinders the ion transport as seen from the experimental results under humidity-controlled conditions. However, the xLPSbP2VP/alkyl BCE allows the agglomeration of larger water clusters due to the hydrophobic nature of the side chain and a lower fraction of bound water. The larger water clusters in this case increase the water diffusion constant as well as the aqueous solvation of the bromide ion. This, in turn, facilitates the transport of the bromide ion, both from diffusion and hopping along the ionic domain of the polymer electrolyte, giving rise to high ionic conductivity values.
Conclusions
The xLPSbP2VP architecture allowed us to examine ionic conductivity with different side chain group chemistries while controlling for backbone type and mesoscale morphology. Thin film ionic conductivity measurements and molecular dynamics revealed that the alkyl side chain variant promoted the highest ionic conductivity under fully hydrated and partially hydrated (50% RH) conditions when compared to alkoxy and zwitterionic side chains. This was unexpected as the alkoxy and zwitterionic side chains are more hydrophilic and were expected to adsorb more water when the BCE film was exposed to external environments with lower RH values. Molecular dynamics simulations revealed that the oxygen atoms of the PEG side chain, hinders ionic conductivity due to coordination with water molecules and creating a larger distribution of them, reducing the agglomeration of water molecules around the ions. Additionally, it was observed that for the zwitterionic BCE, interactions between the two counterions (bromide and sodium) and the sulfate and the pyridinium hinders ionic transport under applied electric fields. These observations explain why xLPSbP2VP/zwitterion under RH conditions have no ionic conductivity (i.e., the films are starved of water needed to break the ionic charge pairs). With an alkyl side chain chemistry, a BCE with a very high bromide conductivity of 105 mS cm−1 was measured. We assert that the hydrophobic alkyl side chain steers water toward the tethered pyridinium and bromide counterion. Directing the water to the ion aides in dissociation and mediates ionic conductivity. Systematic tuning of BCE chemistry while retaining control of the mesostructure represent effective tools for probing the molecular interactions that govern ionic transport in polymer electrolytes. Using the reaction schemes reported here, future work will study how the different side chain chemistries affect ionic activity coefficients and partitioning. Furthermore, we plan to determine how these side chain chemistries influence ionic conductivity and ionic activity in non-phase separated systems (e.g., random copolymer electrolytes). The ionic conductivity of the BCEs will also be improved by increasing the IEC values in these materials.Author contributions
M. V. R.-G. performed all experiments, D. I. S., K. L. and R. K. performed MD simulations, B. V. performed QCM experiments, analyzed QCM data, and assisted with thin film preparation on quartz crystals, K. A. prepared BCE thin films on quartz crystals, G. S. and P. A. synthesized random copolymer brushes, M. V. R.-G. and C. G. A. wrote the manuscript, all authors reviewed and edited the manuscript.Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences program under Award Number DE-SC0022304. This research used resources of the LSU Shared Instrumentation and Nanofabrication Facilities, as well as from The Pennsylvania State University Material Characterization and Nanofabrication Laboratories. R. K and D. I. S. are grateful for computer time on the LSU-High Performance Computing (LSU-HPC) clusters as well as the clusters of the Louisiana Optical Network Initiative (LONI). The authors are also grateful to Deepra Bhattacharya for helpful discussions and software help.References
- H. Strathmann, A. Grabowski and G. Eigenberger, Ind. Eng. Chem. Res., 2013, 52, 10364–10379 CrossRef CAS.
- J. Kamcev and B. D. Freeman, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 111–133 CrossRef CAS PubMed.
- S. Jiang, H. Sun, H. Wang, B. P. Ladewig and Z. Yao, Chemosphere, 2021, 282, 130817 CrossRef CAS PubMed.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS.
- M. A. Alkhadra, X. Su, M. E. Suss, H. Tian, E. N. Guyes, A. N. Shocron, K. M. Conforti, J. P. de Souza, N. Kim, M. Tedesco, K. Khoiruddin, I. G. Wenten, J. G. Santiago, T. A. Hatton and M. Z. Bazant, Chem. Rev., 2022, 122, 13547–13635 CrossRef CAS PubMed.
- X. Su, Electrochem. Soc. Interface, 2020, 29, 55–61 CrossRef CAS.
- M. L. Jordan, T. Kulkarni, D. I. Senadheera, R. Kumar, Y. J. Lin and C. G. Arges, J. Electrochem. Soc., 2022, 169, 043511 CrossRef.
- N. Kim, J. Jeon, R. Chen and X. Su, Chem. Eng. Res. Des., 2022, 178, 267–288 CrossRef CAS.
- V. M. Palakkal, J. E. Rubio, Y. J. Lin and C. G. Arges, ACS Sustainable Chem. Eng., 2018, 6, 13778–13786 CrossRef CAS.
- V. M. Palakkal, L. Valentino, Q. Lei, S. Kole, Y. J. Lin and C. G. Arges, npj Clean Water, 2020, 3, 5 CrossRef CAS.
- Y. Kambe, C. G. Arges, S. N. Patel, M. P. Stoykovich and P. F. Nealey, Electrochem. Soc. Interface, 2017, 26, 61–67 CrossRef CAS.
- M. J. Park and N. P. Balsara, Macromolecules, 2008, 41, 3678–3687 CrossRef CAS.
- M. J. Park and N. P. Balsara, Macromolecules, 2010, 43, 292–298 CrossRef CAS.
- Q. Lei, K. Li, D. Bhattacharya, J. Xiao, S. Kole, Q. Zhang, J. Strzalka, J. Lawrence, R. Kumar and C. G. Arges, J. Mater. Chem. A, 2020, 8, 15962–15975 RSC.
- Y. Kambe, C. G. Arges, D. A. Czaplewski, M. Dolejsi, S. Krishnan, M. P. Stoykovich, J. J. De Pablo and P. F. Nealey, Nano Lett., 2019, 19, 4684–4691 CrossRef CAS PubMed.
- Y. Zhu, L. Ding, X. Liang, M. A. Shehzad, L. Wang, X. Ge, Y. He, L. Wu, J. R. Varcoe and T. Xu, Energy Environ. Sci., 2018, 11, 3472–3479 RSC.
- M. Yandrasits, M. Lindell, M. Schaberg and M. Kurkowski, Electrochem. Soc. Interface, 2017, 26, 49–53 CrossRef CAS.
- C. G. Arges, Y. Kambe, H. S. Suh, L. E. Ocola and P. F. Nealey, Chem. Mater., 2016, 28, 1377–1389 CrossRef CAS.
- C. G. Arges, Y. Kambe, M. Dolejsi, G.-P. Wu, T. Segal-Pertz, J. Ren, C. Cao, G. S. W. Craig and P. F. Nealey, J. Mater. Chem. A, 2017, 5, 5619–5629 RSC.
- D. Bhattacharya, S. Kole, O. Kizilkaya, J. Strzalka, P. P. Angelopoulou, G. Sakellariou, D. Cao and C. G. Arges, Small, 2021, 17, 2100437 CrossRef CAS PubMed.
- D. Sharon, P. Benningto, C. Liu, Y. Kambe, B. X. Dong, V. F. Burnett, M. Dolejsi, G. Grocke, S. N. Patel and P. F. Nealey, J. Electrochem. Soc., 2018, 165, H1028–H1039 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- K. K. Kanazawa, Chemical Sensors Iv, Proceedings of the Symposium, 1999, 99, 306–313 Search PubMed.
- M. V. Ramos-Garcés, K. Li, Q. Lei, D. Bhattacharya, S. Kole, Q. Zhang, J. Strzalka, P. P. Angelopoulou, G. Sakellariou and R. Kumar, RSC Adv., 2021, 11, 15078–15084 RSC.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem. C, 1987, 91, 6269–6271 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- K. Li, V. Subasinghege Don, C. S. Gupta, R. David and R. Kumar, J. Chem. Phys., 2021, 154, 184505 CrossRef CAS PubMed.
- Y. Kang, J. J. Walish, T. Gorishnyy and E. L. Thomas, Nat. Mater., 2007, 6, 957–960 CrossRef CAS PubMed.
- S. Farzin, A. Sarella, M. A. Yandrasits and S. K. Dishari, J. Phys. Chem. C, 2019, 123, 30871–30884 CrossRef CAS.
- D. K. Paul, R. McCreery and K. Karan, J. Electrochem. Soc., 2014, 161, F1395–F1402 CrossRef CAS.
- S. Chatterjee, E. Zamani, S. Farzin, I. Evazzade, O. A. Obewhere, T. J. Johnson, V. Alexandrov and S. K. Dishari, JACS Au, 2022, 2, 1144–1159 CrossRef CAS PubMed.
- X. C. Chen, D. T. Wong, S. Yakovlev, K. M. Beers, K. H. Downing and N. P. Balsara, Nano Lett., 2014, 14, 4058–4064 CrossRef CAS PubMed.
- W. Mei, A. J. Rothenberger, J. E. Bostwick, J. M. Rinehart, R. J. Hickey and R. H. Colby, Phys. Rev. Lett., 2021, 127, 228001 CrossRef CAS PubMed.
- S. D. Jones, H. Nguyen, P. M. Richardson, Y.-Q. Chen, K. E. Wyckoff, C. J. Hawker, R. J. Clément, G. H. Fredrickson and R. A. Segalman, ACS Cent. Sci., 2022, 8, 169–175 CrossRef CAS PubMed.
- R. Kumar, J. R. Schmidt and J. L. Skinner, J. Chem. Phys., 2007, 126, 204107 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods. Schematic flow diagram of BCE preparation. AFM images of PSbP2VP, PSbP2VP after Menshutkin reactions, PSbP2VP and xLPSbP2VP after being submerged in ethanol. XPS spectra of deconvoluted N peak of xLPSbP2VP, deconvoluted Br, Na, and S peaks of the different BCEs. MD simulation, radial distribution function, mean square displacement, diffusion constant, and coordination number. Equations for calculating the IEC values of the thin film BCEs. See DOI: https://doi.org/10.1039/d2ma00919f |
| ‡ Present address: Ecolectro, Inc., 61 Brown Rd STE 103 Ithaca, NY 14850. |
| § Present address: Agency for Science, Technology and Research, Singapore. |
| This journal is © The Royal Society of Chemistry 2023 |