
Mechanistic investigation of a Ni-catalyzed electrochemical reductive cleavage of the α-O-4 bond in the lignin model compound benzyl phenyl ether†
Fang
Lin
ab,
Predrag V.
Petrović
 bc,
Ho-Yin
Tse
bc,
Hanno C.
Erythropel
bcd,
Jason Chun-Ho
Lam
*e and
Paul T.
Anastas
*abcdf
bc,
Ho-Yin
Tse
bc,
Hanno C.
Erythropel
bcd,
Jason Chun-Ho
Lam
*e and
Paul T.
Anastas
*abcdf
aDepartment of Chemistry, Yale University, 225 Prospect St, New Haven, CT, USA. E-mail: paul.anastas@yale.edu
bCenter for Green Chemistry & Green Engineering at Yale, 370 Prospect St, New Haven, CT, USA
cYale School of the Environment, 195 Prospect St, New Haven, CT, USA
dDepartment of Chemicals and Environmental Engineering, Yale University, 17 Hillhouse Ave, New Haven, CT, USA
eCity University of Hong Kong, School of Energy and Environment, Kowloon Tong, Hong Kong. E-mail: jason.lam@cityu.edu.hk
fYale School of Public Health, 60 College St, New Haven, CT, USA
First published on 13th October 2023
Abstract
The electrochemical reductive cleavage of the C–O bond in the lignin α-O-4 model compound benzyl phenyl ether (BPE) at room temperature was investigated using earth-abundant nickel as a catalyst in methanol. Experiments using a divided cell setup using either NiCl2·6H2O salt or pre-deposited Ni on a carbon paper cathode (Ni/CP) under an inert atmosphere revealed the essential role of freshly and uniformly deposited Ni0 on the electrode surface for the reductive, catalytic cleavage to yield phenol and toluene. To better understand the reaction mechanism, the surface morphology and composition of the Ni/CP electrode were investigated by SEM, XRD, and XPS. Additionally, the role of methanol as a proton donor was established, and electrochemical hydrogenation/hydrogenolysis (ECH) experiments of BPE with sterically hindered substituents revealed that the reaction mechanism shares similarities with Pd/C hydrogenation/hydrogenolysis chemistry. DFT calculations further supported this mechanistic route and were consistent with the experimental observations. Based on both experimental and calculation results, a mechanism including (1) the interaction of the benzylic side of BPE with the catalyst surface, (2) adsorbed hydrogen interacting with the benzylic carbon to induce C–O bond scission, and (3) proton transfer to the phenoxy anion from the methanol was proposed.
1. Introduction
Pressing concerns over the depletion of fossil fuels and the need for climate change mitigation has prompted research on sustainable solutions that include biomass conversion into fuel and chemicals.1 One promising approach is the valorization of lignin, which is currently simply used for its energy value.2 However, the ability to selectively cleave C–O bonds present in lignin, such as the α-O-4 bond, would help to unlock lignin's potential as a renewable source of aromatic molecules to produce fuels and commodity chemical precursors.3,4 The catalytic cleavage of the C–O bond is a challenging chemical transformation due to the relatively high bond strength of these linkages and the general recalcitrant nature of lignin. In addition to lignin valorization, the C–O cleavage is also essential to organic synthesis, for instance, benzyl ethers deprotection which is a common protection strategy for hydroxy and amino functionalities. In this regard, removal of benzyl protecting group typically requires transition metal catalysts, such as Raney® nickel (Ni) or palladium on carbon (Pd/C) in combination with various reductants such as NaBH4 and H2.5,6With a growing interest in C–O bond cleavage, researchers have focused on exploring inexpensive alternatives to replace traditional, expensive, hetero- and homogeneous Pd-based catalysts for C–O cleavage.7,8 To date, progress has been made in using nickel (Ni) for the reductive and selective cleavage of C–O bonds due to Ni's natural abundance and robust nature. For example, Hartwig's group introduced the use of homogenous Ni complexes that can be operated at relatively mild conditions for selective hydrogenolysis of aryl ethers.9 Since then, efforts have been extended to the development of heterogenous Ni-based catalysts. Significant contributions to this field were reported by the groups of Hartwig,10,11 Lercher,12–15 Esposito,16,17 and others.18–21 Moreover, Jackson's group intensively studied Raney®-Ni's applications in electrocatalytic hydrogenation (ECH) of aryl ethers.22–24 The employed ECH is a promising method for the generation of active species at room temperature and atmospheric pressure. The so-called Volmer reduction, (H+ + e− + surface → Hads), supplies the reducing equivalents via electrochemical proton reduction on the electrode surface, thus offering a safer alternative to flammable hydrogen gas. However, the preparation of any Raney®-type catalyst is a chemical and energy-intensive process, and its storage requires special caution due to its air-sensitive and pyrophoric nature. Recently, we reported a Ni-promoted electro-reductive cleavage that is selective for C–O bond scission in various benzyl phenyl ethers as model compounds for the α-O-4 bond in lignin under mild conditions in methanol. The optimized protocol was selective to α-O-4 linkages only, but was inert to other common lignin C–O linkages, such as in lignin β-O-4 or 4-O-5 model compounds (i.e., 2-phenylethyl ether and diphenyl ether, respectively).25 We observed that Ni ions were uniquely effective for benzylic C–O bond cleavage compared to other divalent metal ions such as Cu, Co, and Fe. The selective and effective electrochemical cleavage of C–O warrants a deeper investigation into the mechanistic details, specifically the role of Ni as well as how the substrate interacts with the growing Ni surface. Mechanistic investigations of electrochemical reactions can be challenging because the reaction occurs at the interface of the active electrode surface and the solution, thereby potentially complicating mechanistic observations. Consequently, this study aimed to examine and propose a mechanism for the observed benzylic C–O bond cleavage through a dual empirical and computational approach.
2. Experimental section
2.1. Materials and reagents
Solvents and reagents were purchased from commercial suppliers and used as received: benzyl phenyl ether (>98% purity, TCI, Portland, OR), nickel chloride hexahydrate (>98.5%,), 1-isopropyl-4-(phenoxymethyl)benzene (>99%; Scheme 4d), benzyl benzoate (>99%; j), benzyl acetate (>99%; k; all from Sigma-Aldrich, St Louis, MO). The benzyl phenyl ether derivative model compounds (Scheme 4, compounds c, e–i) were synthesized using the Williamson ether synthesis according to literature26 with small modifications (see ESI† for details). 1H NMR and 13C NMR spectra were recorded at 600 MHz (Agilent DD2, Santa Clara, CA). Cyclic voltammetry was conducted using a potentiostat (CHI660C, CH Instruments, Austin, TX) using a 3 mm diameter glassy carbon disk as working electrode, a Pt wire counter electrode, and a Ag/AgCl reference electrode.2.2. Preparation of pre-deposited Ni carbon paper electrodes (Ni/CP)
The cathode materials were prepared via in situ plating of NiCl2·6H2O (2 mM to 40 mM) The electrolyte solution was prepared by dissolving 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6; >99%; Sigma-Aldrich) as supporting electrolyte in methanol (HPLC grade, Fisher Scientific, Waltham, MA) onto carbon paper (CP) with a 2 cm2 surface area using a current range from 2 to 10 mA under air or Ar atmosphere. Pt wire (Alfa Aesar, Ward Hill, MA) was used as anode material. The standard plating conditions were 40 mM NiCl2·6H2O in 0.1 M TBAPF6, and 10 mA constant current unless specified.2.3. General procedures for electrolysis
A two-chambered glass H-cell (manufactured by the Yale scientific glassblowing laboratory) separated by a Nafion 117 membrane (DuPont, Wilmington, DE) was used. The specified amounts of reactants were dissolved in the electrolyte and the mixture was added to the cathode chamber in the presence or absence of NiCl2·6H2O as required. Pre-deposited Ni on carbon paper (Ni/CP) (see section 2.2) was used as the cathode, and Pt wire (Alfa Aesar) was used as the anode material. A DC power supply (Kaito Electronics Tekpower TP3005T, Montclair, CA) was used to provide a constant current at room temperature.2.4. Sample analysis
Aliquots of the reaction mixture (0.2 ml) were taken from the electrolysis cell at the desired time point, and a 0.2 mL of diphenyl (1 mg mL−1) was added to each sample as an internal standard. After filtration (0.2 μm filter; Fisher Scientific), 1 μL was injected into a GC-MS (PerkinElmer Clarus 580 with connected SQ8S mass spectrometer; Waltham, MA), fitted with an Elite-5-MS (60 m × 0.25 mm i.d. × 0.25 μm) capillary column (PerkinElmer) using the following program: 50 °C hold 2 min, ramp 15 °C min−1 to 150 °C, ramp 20 °C min−1 to 290 °C, hold 12 min. Quantitative analyses were performed after obtaining calibration curves using commercially available standards with diphenyl ether as internal standard.2.5. Electrochemical active surface area (ECSA) measurement
To estimate the active surface area of the pre-deposited Ni/CP electrodes (2 cm2 geometric area), the double-layer capacitance (Cdl) method was utilized. It was carried out by running a series of cyclic voltammetry (CV) experiments at different scan rates (35, 50, 60, 75 and 100 mV s−1) ± 50 mV around the open circuit potential (OCP). This method was performed in 0.1 M KOH using a saturated calomel electrode (SCE) as reference electrode (CH Instruments) and Pt mesh (2 cm × 1 cm, 0.1 mm wire diameter, 99.9%) (Alfa Aesar) as counter electrode. Then, plotting the charging current at OCP vs. scan rate yields a linear slope, which is equivalent to the value of Cdl. The ECSA can then be obtained by dividing Cdl by the specific capacitance (Cs) of the electrode material, which is assumed to be 0.04 mF cm−2.27,28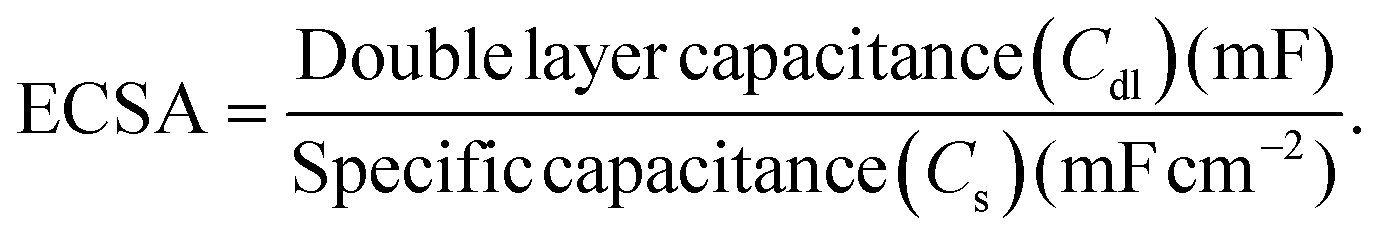
2.6. XPS measurements
The prepared Ni/CP electrodes (see section 2.2) were transferred into an X-ray photoelectron spectrometer (XPS; PHI VersaProbe II Scanning XPS Microprobe; ULVAC-PHI, Chigasaki, Japan) using a protective transfer method as outlined in the literature to prevent the catalyst surface from being oxidized by air during the transfer.29 Briefly, the working electrode was taken out of the electrochemical cell after plating or electrolysis of reactants, immediately immersed in Ar-purged methanol, and placed inside an N2-filled glove box for XPS sample preparation. The samples were then transferred into the XPS spectrometer with a vacuum sample transfer vessel. XPS spectra were recorded with 23.5 eV pass energy, 20 ms integration time and 100 meV step size using monochromatic 1486.7 eV Al Kα X-ray source on the XPS with a 0.47 eV system resolution. All spectra were referenced to the C peak at 284.8 eV.2.7. Materials characterization
SEM images and EDX spectra of the electrodes before and after the reactions were collected with a Hitachi SU8230 UHR Cold Field Emission Scanning Electron Microscope (Santa Clara CA) equipped with a BRUKER XFlash 5060FQ Annular EDX detector (Billerica, MA). XRD was measured under the Bragg–Brentano focusing mode with a Rigaku Smartlab X-Ray Diffractometer (The Woodlands, TX) with Cu Kα radiation.2.8. DFT calculations
The Density Functional Theory (DFT) approach was used to explore the mechanism of C–O bond cleavage. DFT methods, as implemented in the Gaussian 16 suite of programs,30 combine relatively low computational cost and high accuracy and have been successfully used for several decades to predict a range of molecular properties, interaction energies, reaction mechanisms, and thermochemical parameters.31,32 Ground-state geometries of all molecules were optimized using ωB97XD33 density functional and f2SVP34 basis sets in a vacuum as implemented in Gaussian 16. A pruned integration grid with 99 radial shells and 590 angular points (ultrafine grid, default in Gaussian 16) was used for all calculations.3. Results and discussion
3.1 Comparison of ECH of BPE under air and inert conditions using NiCl2·6H2O
The addition of NiCl2·6H2O in the catholyte was essential to enable the selective electrochemical reductive cleavage of C–O bond in BPE in mild conditions. The optimal product yields with 90% toluene and 84% phenol were achieved using a mole ratio of NiCl2·6H2O/BPE = 4 under an atmospheric environment (Table 1, entry 1). However, decreasing the amount of NiCl2·6H2O resulted in lower performance. For example, a mole ratio of NiCl2·6H2O/BPE = 0.5 (entry 2) decreased the yields to 56% toluene and 37% phenol. When the electrolysis was performed in an Ar atmosphere, the cleavage yield was greatly promoted, and the yield was still high even at a lower sub-stoichiometric amount of NiCl2·6H2O. From Table 1, the comparable conversion and product yields could be achieved by only using sub-stoichiometric amount NiCl2·6H2O (entries 3–5) when the reactions were performed in an Ar atmosphere. Even though the mole ratio of NiCl2·6H2O/BPE was down to 0.125 (entry 5), the BPE was cleaved to toluene and phenol with 84% and 72% yields, respectively, which was comparable to entry 1 when amount of NiCl2·6H2O was 8 times greater.| Entry | NiCl2·6H2O (mM) | Mole ratio of NiCl2·6H2O/BPE | Conversion (%) | Product yielda (%) | |
|---|---|---|---|---|---|
|
|
|
||||
| Reaction conditions: divided cell, 10 mM BPE, 15 mL MeOH, 0.1 M TBAPF6, constant current at 10 mA, 2 cm2 carbon paper electrode, 5 h, under air.a Based on GC analysis with diphenyl as an internal standard.b The reactions were run under Ar atmosphere. Three replicates of the measurements were produced, and the reported value is the average within = 5% of error. | |||||
| 1 | 40 | 4 | 90 | 90 | 84 |
| 2 | 5 | 0.5 | 64 | 56 | 37 |
| 3b | 5 | 0.5 | 93 | 97 | 74 |
| 4b | 2.5 | 0.25 | 94 | 96 | 75 |
| 5b | 1.25 | 0.125 | 85 | 84 | 72 |
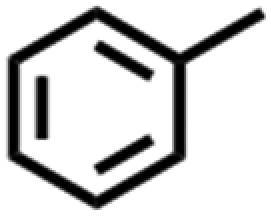
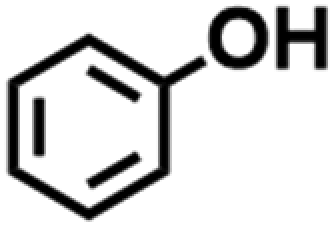
Based on these results, we explored how the inert atmosphere provided by Ar could result in the observed improvements in required NiCl2·6H2O. Ni2+ ions were reduced to Ni0 in the present reductive environment due to its low reduction potential (−0.257 V vs. SHE in standard conditions), and that the inert atmosphere can prevent the newly formed Ni0 from oxidation. These features indicated freshly deposited Ni0 was essential to the catalytic cleavage of BPE. To validate our hypothesis, the ECH experiments were conducted in the absence of NiCl2·6H2O but using carbon paper electrodes on which Ni0 was pre-deposited in Ar and normal atmosphere.
3.2 Characterization and application of pre-deposited Ni (Ni/CP) electrodes for the ECH of BPE
Two working electrodes were prepared by electrochemical deposition of Ni on carbon paper either under air (Ni/CP-Air) or under Ar (Ni/CP-Ar). The mass balance of Ni2+ ions in the plating solution before and after the electroplating was measured by ICP-MS to quantify the amount of nickel deposition. For example, it was found 0.425 mmol Ni was plated onto carbon paper after 5 h electroplating for Ni/CP-Air electrode (see Table S1†). The catalytic activities of the plated Ni/CP-Air/-Ar electrodes were then investigated in the absence of NiCl2·6H2O salt. Structural analysis of the Ni/CP-Air electrode was conducted using high magnification scanning electron microscope (SEM), energy dispersive X-ray spectroscopy (EDS), and X-ray diffraction (XRD). SEM of the pristine carbon paper (CP) showed that it consists of carbon fibers and a smooth surface (Fig. 1a(i) & a(ii)). After Ni electro-deposition, spherical, metallic particles were observed (Fig. 1a(iii) & a(iv)). The surface of the particles was composed of Ni as determined by EDS elemental mapping (Fig. 1b(i) & b(ii)). To further confirm this, XRD patterns showed three diffraction peaks at 2θ = 44.6°, 51.8°, and 76.3°, which can be assigned to (111), (200), and (220) reflections of metallic Ni with a face-centered cubic structure (Fig. 1c). | ||
| Fig. 1 (a) (i &ii) SEM images of pristine carbon paper (CP); (iii & iv) CP after electroplating from a 40 mM solution of NiCl2·6H2O for 5 h under air (Ni/CP-Air); (b) (i &ii) SEM &EDX elemental mapping of Ni/CP-Air; (c) XRD spectrum of pristine CP & Ni/CP-Air. | ||
The Ni/CP-Air electrode was then utilized immediately in an ECH experiment in the absence of NiCl2·6H2O to cleave BPE after gently rinsing with methanol to remove any NiCl2·6H2O residue. The results indicate that the Ni/CP-Air electrode was indeed able to cleave BPE in these conditions. However, the reaction only showed a low conversion (29%) and low product yields (22% toluene; 10% phenol; Scheme 1, condition A). Similar to the findings in section 3.1, the inferior catalytic performance was suspected of being a result of an oxidation process of the freshly electroplated Ni0 surface, as Ni0 is very sensitive to air.35 X-ray Photoelectron Spectroscopy (XPS) was performed to investigate the surface composition of as prepared Ni/CP-Air electrode, and the results were compared to a Ni/CP-Ar electrode (Ni/CP-Ar). Fig. 2 shows the Ni 2p spectra of both electrodes ranging from 850 eV to 884 eV. For the Ni/CP-Air electrode, Ni 2p peaks located at 855.6 eV and 873.8 eV are attributed to the characteristic peaks of nickel oxide (NiO) (Fig. 2, black line).36,37 In addition, the peaks located at 861.4 eV and 879.1 eV are the corresponding satellite peaks of NiO of Ni 2p3/2 and Ni 2p1/2 respectively.37 In contrast, the XPS spectrum of nickel-plated electrode that was prepared under argon (Ni/CP-Ar) shows the major metallic Ni0 peaks at 852.4 eV (2p3/2) and 869.5 eV (2p1/2) with only insignificant fractions of NiO present at 855.6 eV and 873.8 eV.(Fig. 2, red line)38 The obvious shift of binding energy to a higher value from Ni/CP-Ar to Ni/CP-Air confirmed that the freshly deposited Ni surface was indeed oxidized to NiO when electroplating was performed under air.
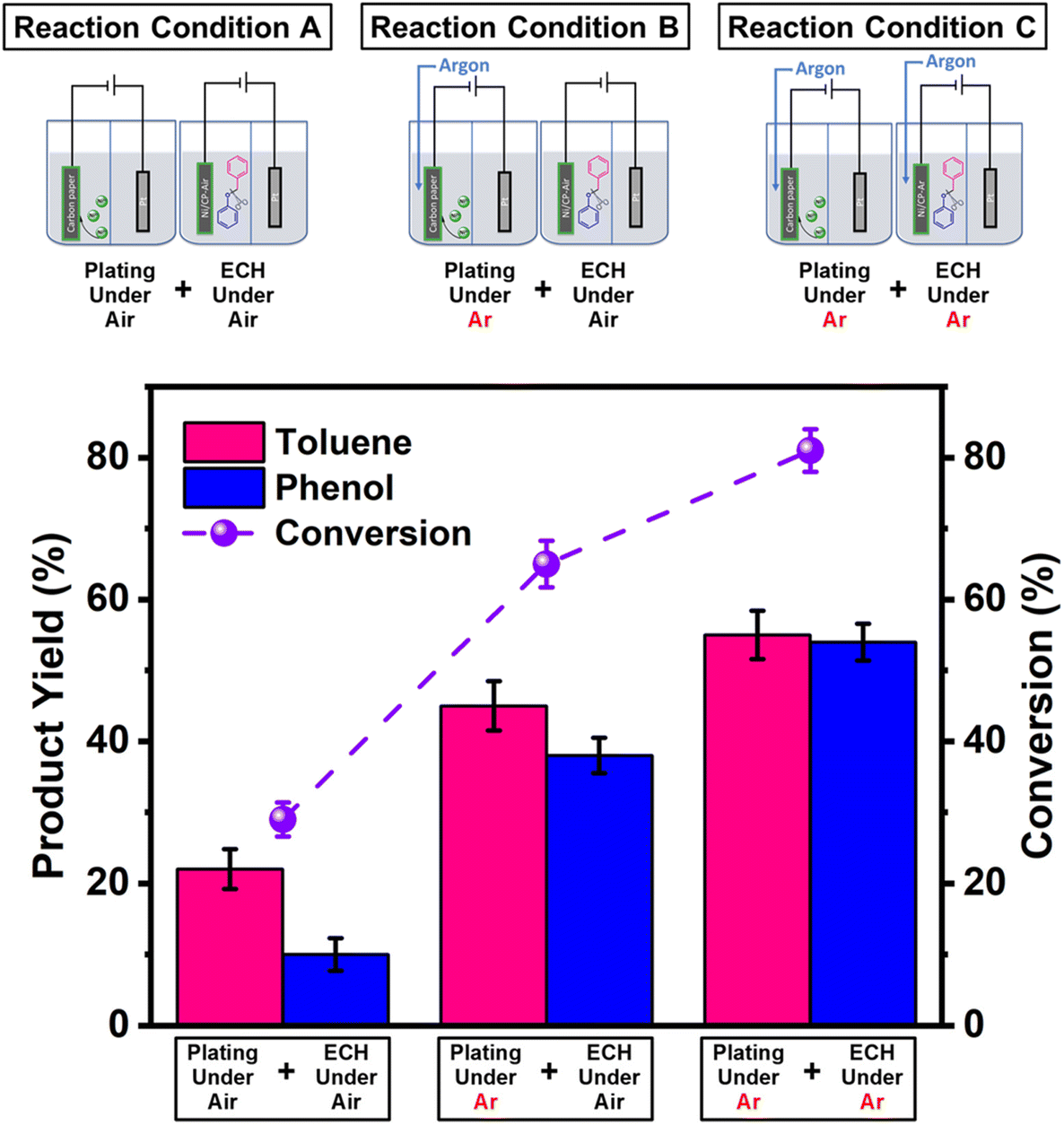 | ||
| Scheme 1 Top: schematic representation of the plating atmosphere (air or argon; left side) and ECH atmosphere (air or argon; right side) combinations; bottom: product yields and conversion for ECH carried out under the three outlined conditions above. | ||
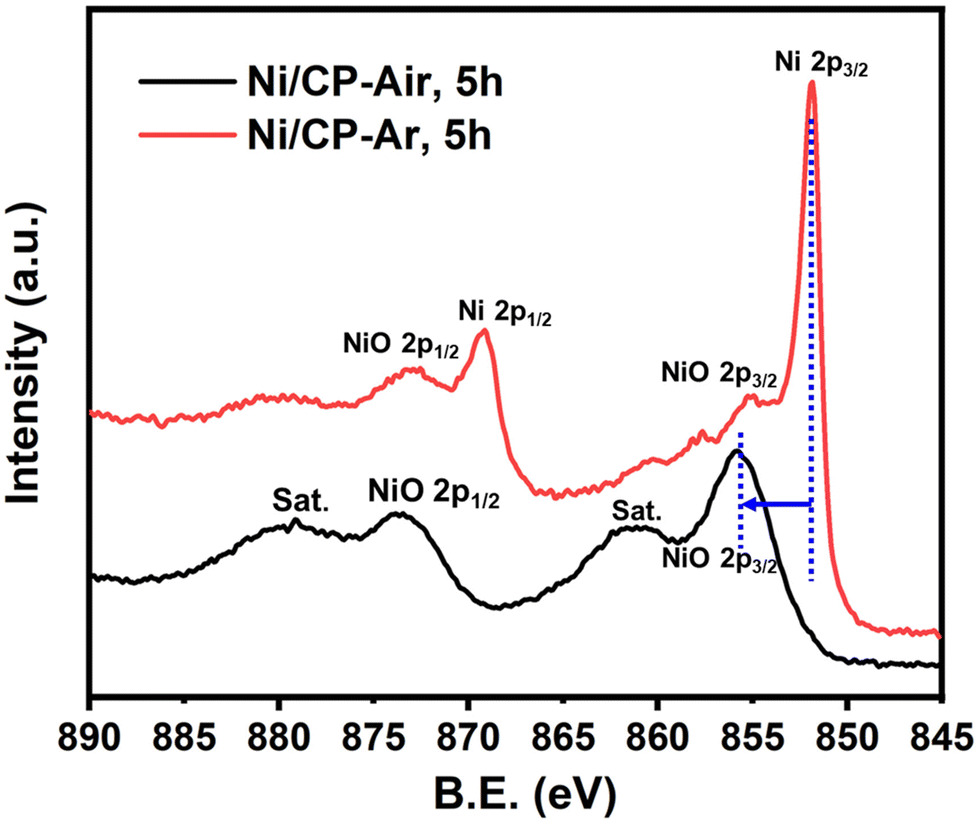 | ||
| Fig. 2 XPS comparison between Ni/CP-Air (black line) & Ni/CP-Ar (red line). The Ni/CP-Air electrode was prepared under air atmosphere & the CP was undergone 5 h electroplating in 40 mM of NiCl2·6H2O in methanol at 10 mA. The Ni/CP-Ar electrode was prepared under Ar atmosphere & the CP was undergone 5 h electroplating in 40 mM of NiCl2·6H2O in methanol at 10 mA. | ||
The Ni/CP-Ar electrodes were then employed as cathodes to perform the ECH of BPE under identical conditions to Ni/CP-Air; one in air and the other in argon (Scheme 1). While the reaction using Ni/CP-Ar yielded 45% toluene and 38% phenol with a 65% conversion under air (Scheme 1, condition B), performing both the electroplating (Ni/CP-Ar) and the ECH under Ar yielded 55% toluene and 54% phenol with an 81% conversion (Scheme 1, condition C). These results demonstrated that maintaining an intact Ni0 surface under argon was beneficial to the catalytic activity.
3.3 Optimization of ECH of BPE in Ni/CP systems
To further improve both conversion and product yields of the pre-deposited Ni system, several deposition conditions including plating time, current flow, and Ni2+ concentration in the electroplating bath were varied, and the so-produced electrodes were employed in ECH experiments of BPE. While current flow (2 or 10 mA) and Ni2+ concentration (2 or 40 mM NiCl2·6H2O) did not significantly change conversion and product yields using Ni/CP-Ar electrodes for the ECH of BPE (see Fig. S3†), shortening the plating time from 5 h to 1 h resulted in significant conversion changes. Ni/CP-Ar electrodes plated for 1 h increased conversion by 14% (conversion 94%) and product yields by 45% (toluene yield 80%) and 33% (phenol yield 72%) when compared to a plating time of 5 h (both Ni/CP-Ar; see Fig. 3). This observation was also found to be true for the Ni/CP-Air conditions.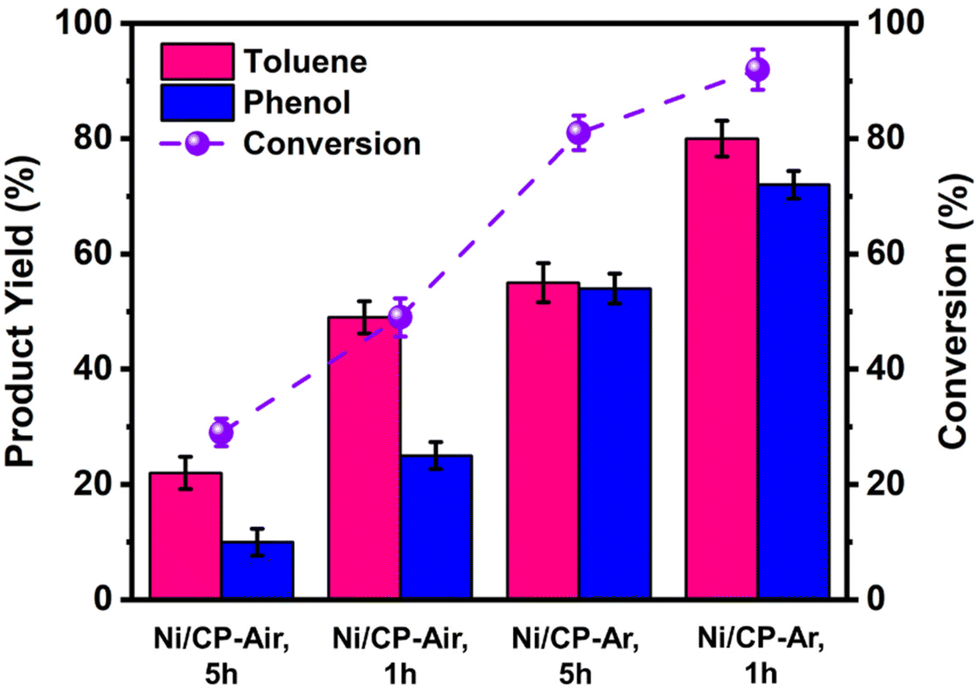 | ||
| Fig. 3 Product yields (left Y-axis) and conversion (right Y-axis) results of ECH experiments of benzyl-phenyl ether (BPE) carried out with different cathode materials listed on the X-axis. All cathode materials were prepared by electroplating NiCl2·6H2O onto carbon paper (CP), under air or argon, and for 1 h or 5 h. ECH of entries 1 & 2 were performed under air, 3 & 4 were performed under argon. | ||
To understand the observed differences resulting from differential plating times, the morphology of Ni/CP-Ar was investigated by SEM. The results show that the shorter plating time (1 h) resulted in smaller and more defined particles, such as elongated needle-shapes, and high uniformity and dispersity for Ni/CP-Ar-1 h (Fig. 4a(i) & (ii)) while the longer plating time (5 h) (Fig. 4b (i) & (ii)) resulted in nickel particle aggregation and less uniform particles dispersion. This observation was also found to be true for the Ni/CP-Air conditions (Fig. 4c (i) & (ii) vs.Fig. 1a (iii) & (iv)). A more uniform particle dispersion provides a higher surface area, which could be a key factor in heterogenous catalysis. To determine this, the electrochemically active surface area (ECSA) was estimated by measuring the double layer capacitance (Cdl) via cyclic voltammetry (CV) scans in the non-faradaic region (see ESI, Fig. S6†). The Cdl value of Ni/CP-Ar-1 h was estimated to be 6.5 mF, which is 1.7× and 3.4× higher than those of Ni/CP-Ar-5 h (3.7 mF) and Ni/CP-Air-1 h (1.9 mF), respectively (Fig. 4d). The enhanced ECSA indicates the availability of more active sites which enable fast charge-transfer kinetics and optimal electrode–electrolyte interaction,27,39 resulting in increased conversion and yield. Therefore, the presented results are consistent with a morphology-property correlation for the investigated system.
 | ||
| Fig. 4 (a) (i & ii) SEM images of the Ni/CP-Ar, 1 h; (b) (i & ii) Ni/CP-Ar, 5 h; (c) (i & ii) Ni/CP-Air, 1 h; (d) double-layer charging current vs. scan rate of Ni/CP-Ar, 1 h; Ni/CP-Ar, 5 h; Ni/CP-Air, 1 h. The linear slope is equivalent to the double-layer capacitance (Cdl). | ||
Because the morphology of the Ni/CP cathodes played an important role in BPE cleavage and product yields, as outlined above, the morphology of the cathode after ECH was also investigated by SEM and no obvious changes were observed (Fig. S2a†). In addition, post-reaction characterizations were also performed to investigate the structure change of Ni/CP-Ar after ECH of BPE. XRD was employed to detect bulk structure, while XPS was used to investigate the surface composition changes. The XRD results indicated that the Ni/CP-Ar electrode did not proceed phase shift after electrolysis (Fig. S2b†) as the fcc Ni structure was retained. The XPS results showed that the electrode surface remained in the metallic Ni0 state after ECH of BPE (Fig. S2c†). These results indicate that the electroplated Ni surface was well protected from auto-oxidation to NiO under an argon atmosphere during the electrolysis. Furthermore, it re-emphasized the essential role of Ni0 toward the ECH of BPE.
3.4 Mechanism discussion
In earlier work, we have demonstrated similar cleavage efficiency of BPE when Ni2+ salts were present in the methanol/electrolyte solution. Because other divalent ions such as Fe2+, Co2+, or Cu2+ could not catalyze the cleavage reaction of BPE, we hypothesize based on the presented results, that Ni2+ was freshly deposited continually on the cathode, leading effectively to a “quasi-constant” Ni0 surface. This would also explain why the reaction could be carried out in air when Ni2+ is present in the electrolyte, as the oxidized and thus deactivated Ni on the surface could be replenished continuously. Of note, control experiments with pre-deposited Fe/CP, Co/CP, and Cu/CP under the chosen conditions also did not result in BPE cleavage (see Table S3†), further highlighting the unique role of Ni.To further elucidate the mechanism of the cleavage reaction, we designed experiments where pre-deposited Ni/CP-Ar cathodes were utilized, but the ECH was carried out in the aprotic solvents acetonitrile (ACN), dimethylformamide (DMF), or tetrahydrofuran (THF) using the same conditions. No conversion was observed in any of the aprotic solvents (see Table S2†), suggesting that methanol played a role in the mechanism. To probe the role of methanol in the proposed reaction mechanism, experiments with deuterium labeled methanol-d4 were carried out, which confirmed that the protons incorporated into the cleavage products were sourced from methanol (Scheme 2). The relevant spectra (NMR and MS) are included in ESI.† Besides acting as a proton source, the methanol solvent was also capable of delivering the hydrogen evolution reaction (HER) when sufficiently negative reduction potential was applied.40 In our chosen reaction conditions, protons from methanol are expected to be adsorbed on the Ni surface during the Volmer step (H+ + e− → Hads), subsequently forming a hydride complex with the Ni surface.41,42
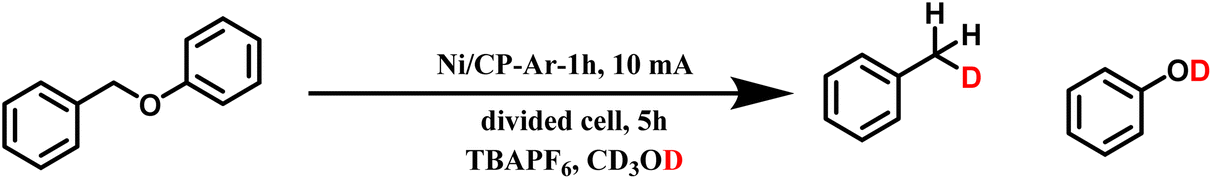 | ||
| Scheme 2 Deuterium-labeling experiments. | ||
To further explore the role that group 10 metals play for electrochemical hydrogenation and hydrogenolysis (ECH), we carried out ECH experiments of BPE in the model system conditions, but with Pd instead of Ni (Table S3†). Results demonstrate that using PdCl2 produced similar catalytic activity and product yields (Table S3,† entry 2) as observed with NiCl2, which was also true for pre-deposited Pd/CP electrodes in comparison to Ni/CP electrodes (Table S3,† entry 3). The adsorbed hydrogen (Hads) which was proposed in the hydrogenation reactions catalyzed by the group 10 metal catalysts such as Raney® Ni,43 Pd/C,44 and Pt/C45 may contribute to the selective cleavage of the benzylic C–O bond in BPE as well. To probe for this, a reaction without an applied current but while bubbling hydrogen gas (H2) around the Pd/CP cathode to introduce Hads was carried out (Table S3,† entry 7). It yielded 31% toluene and 22% phenol, which arguably demonstrated the contribution of Hads to the selective cleavage of the benzylic C–O bond in BPE.
The hydrogenolysis of the benzylic C–O bond is relatively well studied in the Raney® nickel and Pd/C systems.46–48 Hydrogenolysis reactions achieved by these two catalysts have been shown to follow two different reaction mechanisms: Raney® nickel creates strong interactions between the benzylic ring and the neighboring oxygen on the catalyst surface (Scheme 3a), while the surface of Pd/C catalysts lacks the latter and only exhibits strong interactions with the benzylic ring of the substrate (Scheme 3b).49,50 To further probe whether our reaction shares similarities with these mechanisms, we investigated how well the Ni/CP system performed with BPE derivatives that were sterically hindered on the benzylic side (Scheme 4, compounds b–f). There is a clear trend between decreasing product yield with increasing degree of steric hindrance. When the bulky t-butyl group was present to the para-position of benzylic ring (R1), low product yields were found (Scheme 4, compound e; 35% 4-tertbutyl toluene, and 23% phenol). Similarly, the reaction was completely impeded in the case of 1,3-di-tert-butyl-5-(phenoxymethyl) benzene with 2× t-butyl groups in the meta positions of R1 (Scheme 4, compound f). The variation in conversion yield due to steric hindrance suggests that interactions of the benzylic side of BPE with the Ni surface play a crucial role in the cleavage reaction, similar to both the Pd/C and the Raney® Ni mechanisms. Moreover, these results indicate the reductive C–O cleavage occurred directly on the catalyst surface rather than at a moderate distance away from the surface.51–53
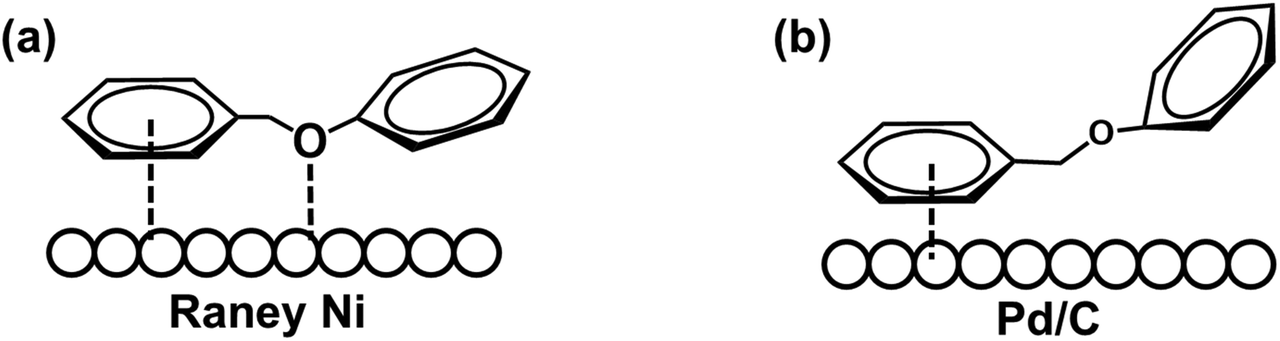 | ||
| Scheme 3 Schematic illustration of hydrogenolysis of the benzylic C–O bond over (a) Raney® Ni system; (b) Pd/C system. | ||
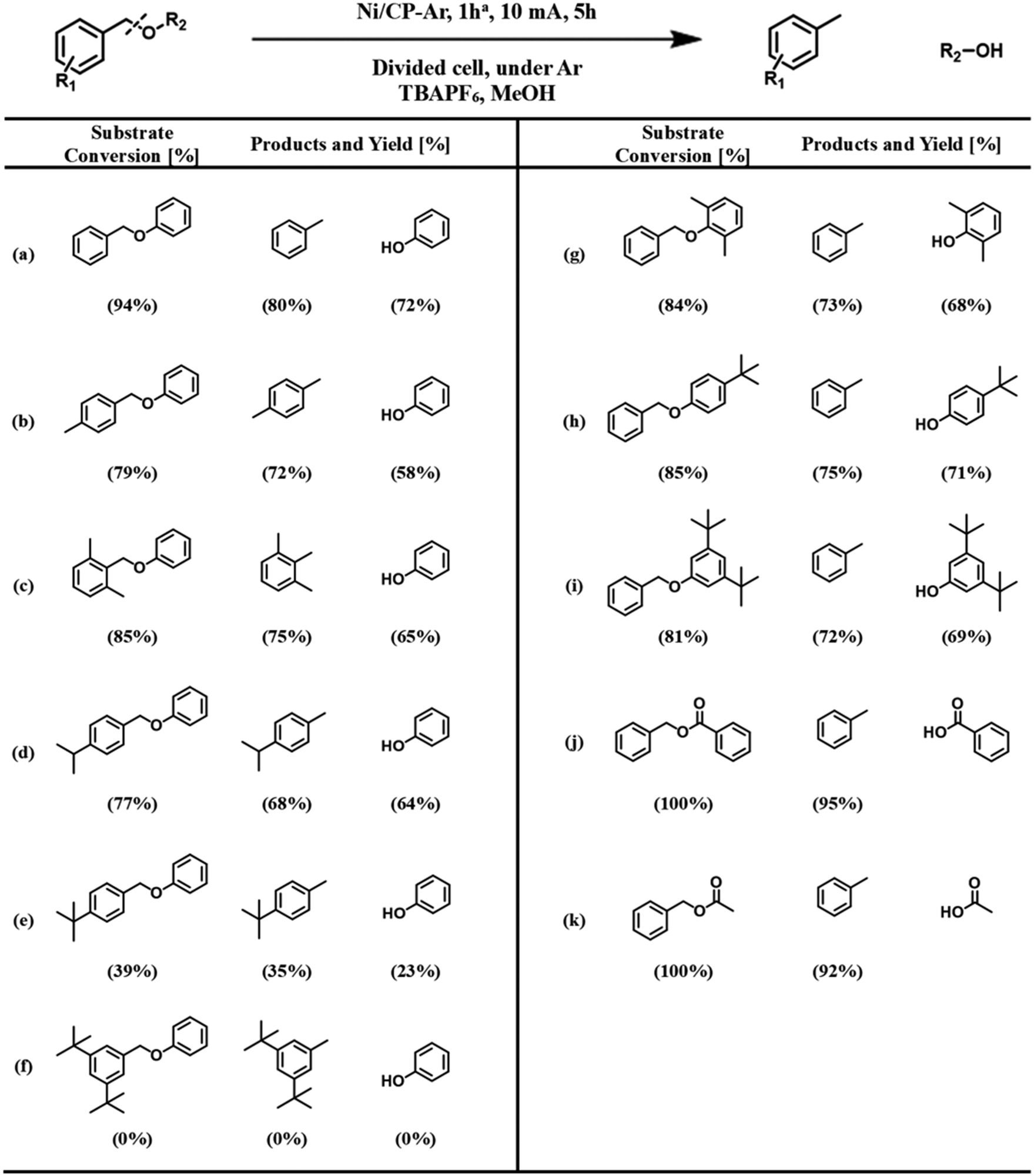 | ||
Scheme 4 Steric hindrance study: ECH performance of substituted BPE using Ni/CP-Ar, 1 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The Ni/CP-Ar, l h electrode was prepared under Ar for 1 h electroplating time in 40 mM of NiCl2·6H2O in methanol at 10 mA. The yields are based on GC analysis with diphenyl as an internal standard. Three replicates of the measurements were produced, and the reported value is the average within = 5% of error. The Ni/CP-Ar, l h electrode was prepared under Ar for 1 h electroplating time in 40 mM of NiCl2·6H2O in methanol at 10 mA. The yields are based on GC analysis with diphenyl as an internal standard. Three replicates of the measurements were produced, and the reported value is the average within = 5% of error. | ||
To investigate whether or not the interaction between the bridging oxygen and the Ni surface plays a role, such as for Raney® nickel, substituents with different levels of bulkiness at the phenyl position (R2) of BPE were subjected to ECH reactions in our system (Scheme 4, compounds g–i). Since these compounds were cleaved with comparable product yields to the un-substituted BPE (Scheme 4, compound a), these results suggest that our proposed reaction pathway shares more similarity with the reaction mechanism found in Pd/C than that of Raney® nickel.
For the hydrolysis found in the Pd/C system, the benzylic ring is strongly adsorbed on the metal surface, while the phenyl ring of the substrate acts as a leaving group. We next examined the influence of different leaving groups on the R2 side of BPE on reaction yields, namely as benzyl benzoate and benzyl acetate (Scheme 4, compounds j–k). Results indicate slightly improved yields compared to BPE for both compounds, likely because the benzoate and acetate anions are better leaving groups than the phenolate anion. Note that benzoic acid and acetic acid reacted with the TBAPF6 electrolyte to their respective butyl esters. These results demonstrate that the protocol can be applied to a wide range of benzylic ethers. However, the average faradaic efficiency (%FE) of reactions with conversion >0 (Scheme 4) was 11.5% ± 2.6% (standard deviation). The somewhat low %FE values (Table S4†) can be explained by the visually observed hydrogen evolution reaction, representing a major competitive side reaction.
In addition, the free radical mechanism has also been considered for electrochemical reactions.54 To probe our system for such mechanism, we carried out two control experiments. First, an ECH with Ni/CP electrodes was carried out with a higher substrate concentration of 100 mM BPE (vs. 10 mM at optimized conditions), with no dimerization product that would result from the recombination of 2 radicals (e.g. bibenzyl) being observed. It has been demonstrated that higher substrate concentrations will promote dimerization reactions if the reaction follows the free radical mechanism. For instance, the phenomenon is commonly observed in Kolbe electrolysis where the dimerization of alkyl radicals benefits from a high concentration of the starting materials.55–57 For the second experiment, the radical inhibitor butylated hydroxytoluene was added into the system using Ni-CP electrode at the optimized conditions, the reaction still proceeded smoothly with up to 90% conversion. Based on these observations, it is unlikely that the reaction system proceeded via a free radical mechanism.
DFT calculations were also carried out to investigate the nature of the interaction between the Ni-plated electrode surface and BPE, and the possible mechanism of C–O bond cleavage. The complexity of the system and the computational demands of modeling a larger surface of Ni0 dictated that the calculations focused on several key steps in the C–O cleavage mechanism and a Ni0 surface represented by 10 Ni0 atoms arranged in a (111) crystal cell to simulate a relatively rigid surface. A Ni (111) facet was employed for the DFT calculation since the XRD showed that the pre-deposited Ni particles adopted the fcc crystal structure. The Ni (111) facet features the lowest surface energy of the fcc crystal structures and has also been observed as the most commonly exposed facet in synthesized Ni particles.58–61 Because the experimental data suggested that the benzylic side of BPE was favored in the interaction with the Ni-plated electrode, the model systems were designed to allow for systematically varying the normal distance between the centroid (central plane) of the Ni(111) model system and either the centroid of the phenyl side or the benzyl side, arranged in a parallel orientation to the surface (Fig. 5). The normal distances between the plane of either aromatic ring (benzyl or phenyl side) of BPE and the centroid of Ni (111) were set to 2 Å, 2.5 Å, and 3 Å. The interaction energies calculated for these distances were then averaged to account for the additional influence of the steric interactions of the respective ring of BPE perpendicular to the centroid of the Ni (111). Calculated interaction energies suggest repulsion between BPE and Ni (111) when the phenyl ring is in parallel orientation (ΔG = 4.7 kcal mol−1; Fig. 6 left side), while the interaction is favorable/attractive if the benzyl side is in parallel orientation (ΔG = −2.3 kcal mol−1; Fig. 5 right side; also see Table 2). This suggests that the benzyl side of BPE would preferably approach the Ni electrode surface which is in accordance with the empirical observations.
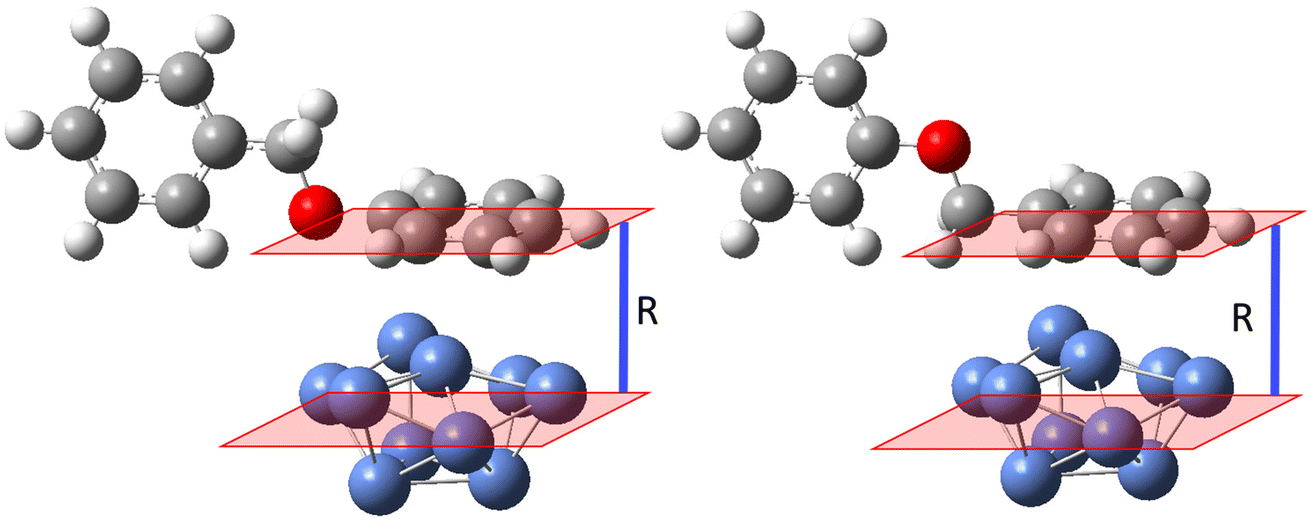 | ||
| Fig. 5 Interaction between the phenyl/benzyl side and Ni (111) at varying normal distance R. | ||
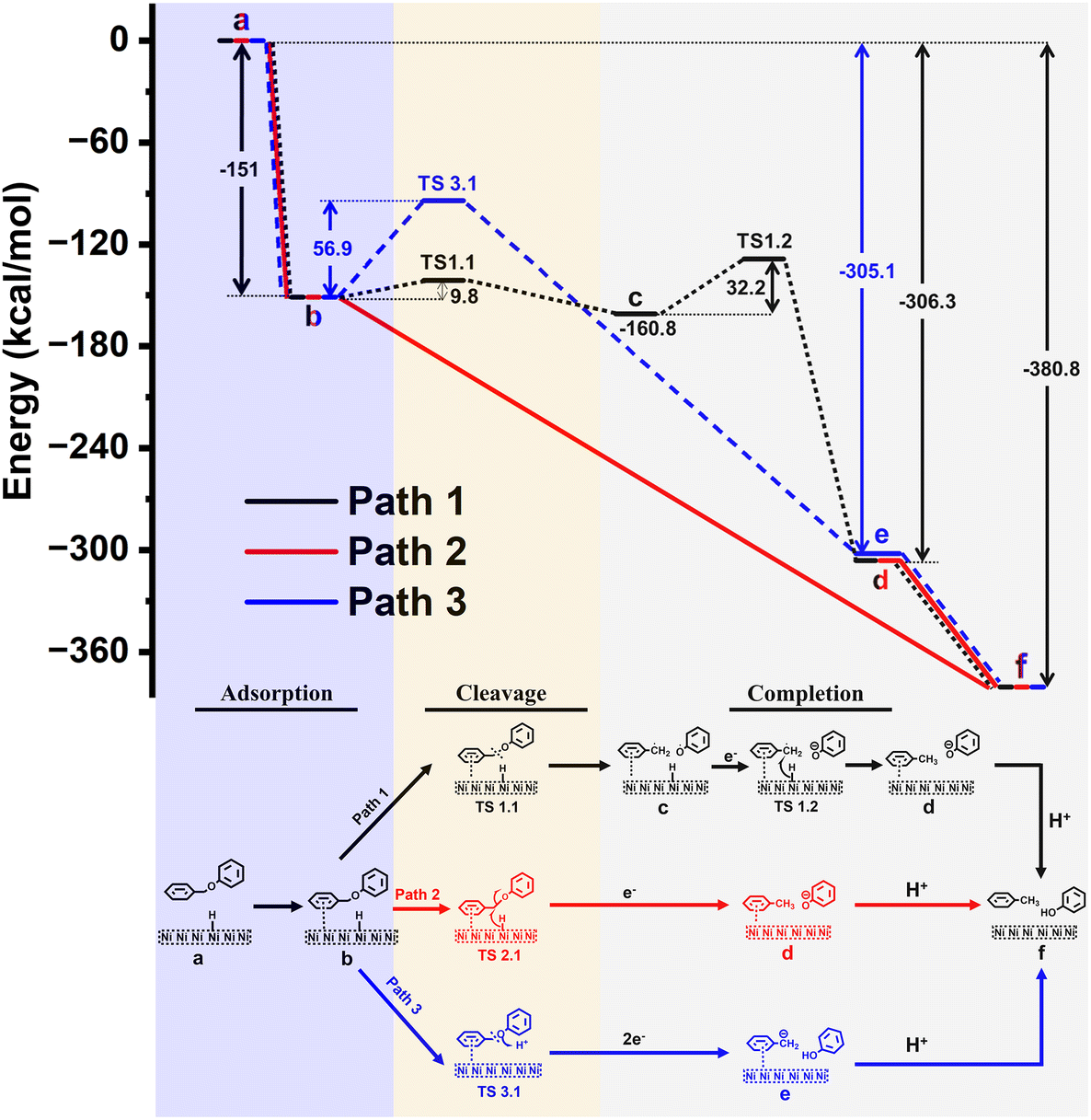 | ||
| Fig. 6 Relative free energy diagram for C–O bond cleavage in BPE by a Ni catalyst (upper sector); proposed pathways for the catalytic C–O bond cleavage (lower sector). | ||
| Benzyl side of BPE parallel to Ni surface | Phenyl side of BPE parallel to Ni surface | |
|---|---|---|
| R | ΔG (kcal mol−1) | ΔG (kcal mol−1) |
| 2 Å | 9.6 | 9.2 |
| 2.5 Å | −8.1 | 4.5 |
| 3 Å | −8.3 | 0.4 |
| Avg. | −2.3 | 4.7 |
Relative free energy diagram for the proposed mechanisms containing free Gibbs energies (and activation barriers) is presented in Fig. 6. Corresponding optimized structures of the intermediates and transition states (TS) are shown in Fig. S16.†
To investigate potential mechanisms, we have focused on the three key stages in the reaction: (1) adsorption of BPE to the surface, (2) BPE cleavage, and (3) reaction completion. In the first stage of the reaction (1), BPE (Fig. 6, state b) is adsorbed to the surface of the Ni electrode in an exergonic approach (ΔG = −151 kcal mol−1). The next step (2) describes three different potential pathways by which the C–O bond could be cleaved (i.e., black, red, or blue line).
In the first possible pathway (black line), the C–O bond undergoes a non-electrocatalytic, homolytic cleavage through the transition state TS1.1 with an energy barrier of ΔG = 9.8 kcal mol−1 to form short-lived benzyl and phenoxy radicals (state c). Although we ruled out the presence of radicals in the electrolyte experimentally, we nevertheless computationally explored the possibility of a surface-adsorbed, short-lived radical species that was ready to interact with other electrode surface species. Then, an adsorbed hydrogen is delivered from the Ni surface to benzyl radical with an energy barrier of ΔG = 32.2 kcal mol−1 through TS1.2, followed by an electron transfer from the electrode to form toluene and a phenoxy anion (state d). The reaction is finalized by a proton transfer to the phenoxy anion from the protic solvent (methanol) to form the final reaction products (state f) with a total energy gain of ΔG = −380.8 kcal mol−1.
The second possible pathway was a transfer of adsorbed hydrogen from the nickel surface onto the benzylic carbon along with an electron, to induce the C–O bond cleavage which would directly yield toluene and a phenoxy anion (state d; red line). However, it was not possible to isolate a transition state (TS2.1) to describe this process, indicating this step was barrierless with a total energy gain of ΔG = −306.3 kcal mol−1 relative to BPE. The reaction is completed as described for path 1 above, to yield a total energy gain of ΔG = −380.8 kcal mol−1.
The third possible pathway for the C–O bond cleavage is the protonation of the connecting ether oxygen of BPE (blue line) with an energy barrier of ΔG = 56.9 kcal mol−1 (TS3.1). This is followed by a two-electron transfer from the electrode to carbon anion and subsequent C–O bond cleavage to form phenol and a benzyl anion (state e) with a total energy gain of ΔG = −305.1 kcal mol−1, which is energetically less favorable than path 1. In the final stage, the reaction is completed by a proton transfer from the protic solvent to the benzyl anion to form the final products toluene and phenol (state f).
Based on this model system for calculations and the outlined limitations, the comparison of the energetics for the three proposed pathways suggests that path 2 (Fig. 6, red line) is the most likely reaction pathway, as it is an exergonic process without activation barriers, while paths 1 and 3 would require steps that would involve overcoming activation barriers (9.8 and 32.2 kcal mol−1 for path 1, and 56.9 kcal mol−1 for path 3). This computational result is in agreement with the experimental findings (see section 3.4), which suggested a mechanism similar to Pd/C systems in which the aromatic ring of the benzyl side most interacts with the Ni surface, but not the phenyl side (aligned with path 2 but not path 3 in Fig. 6). In addition, no radicals were trapped experimentally which is consistent with path 2 but not with path 1 in Fig. 6.
3.5 Green Chemistry considerations
The purpose of this mechanistic study of electrochemical cleavage of BPE is to advance the valorization of highly abundant lignin and to make full use of it as a rich source of aromatic compounds as we move toward the biobased economy. The green chemistry benefits of the transformation are numerous: 1. being carried out at room temperature, 2. using methanol – a solvent that has been evaluated as less problematic compared to many other solvents,62 3. employing electrocatalysis based on the abundant metal nickel and Ni2+ salt, and 4. being selective toward the products, thus make it highly aligned with the Principles of Green Chemistry.1,63 This mechanistic study importantly shows that these mild transformations identified can proceed using simple Ni0 and Ni2+salts in the electrocatalytic system either in air or under an inert argon atmosphere. With mechanistic understanding the further development and application of electrocatalytic lignin utilization will be advanced.4. Conclusion
In conclusion, this manuscript outlines a plausible mechanism based on experimental and computational results for the reported electrochemical cleavage of benzyl phenyl ether (BPE) as model compound for the α-O-4 bond in the lignin. We could demonstrate that an active Ni0 surface prepared by a facial electroplating method plays a central role in facilitating proton transfer to enable the C–O cleavage. As-prepared Ni0 surface was found to be prone to be oxidized. Therefore, Ar inert atmosphere was employed to enhance its catalytic activity. The mechanism was found to resemble Pd/C hydrogenation chemistry where the benzylic side approaches the catalytic surface, but an interaction of the ether oxygen, such as observed in Raney® nickel systems is not crucial. Taken together, the developed catalyst system serves as an alternative to traditional hydrogenation catalysts such as Pd/C due to the reliance on abundant nickel, as well as Raney® nickel systems due to its lower inherent hazards such as non-flammable nature, operational simplicity, and improved handling. These insights will be potentially valuable to the design of a more efficient heterogeneous earth-abundant metal catalysts, as well as the selective lignin degradation scheme that is essential to enable a “lignin first” valorization strategy.Conflicts of interest
There are no conflicts to declare.References
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Designing for a green chemistry future, Science, 2020, 367(6476), 397–400 CrossRef CAS PubMed.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, Greener routes to biomass waste valorization: Lignin transformation through electrocatalysis for renewable chemicals and fuels production, ChemSusChem, 2020, 13(17), 4214–4237 CrossRef CAS PubMed.
- F. Mauriello, E. Paone, R. Pietropaolo, A. M. Balu and R. Luque, Catalytic Transfer Hydrogenolysis of Lignin-Derived Aromatic Ethers Promoted by Bimetallic Pd/Ni Systems, ACS Sustainable Chem. Eng., 2018, 6(7), 9269–9276 CrossRef CAS.
- C. Zhang and F. Wang, Catalytic Lignin Depolymerization to Aromatic Chemicals, Acc. Chem. Res., 2020, 53(2), 470–484 CrossRef CAS PubMed.
- S. Vidal, Protecting groups: strategies and applications in carbohydrate chemistry, John Wiley & Sons, 2019 Search PubMed.
- Y. Yamamoto, E. Shimizu, K. Ban, Y. Wada, T. Mizusaki, M. Yoshimura, Y. Takagi, Y. Sawama and H. Sajiki, Facile Hydrogenative Deprotection of N-Benzyl Groups Using a Mixed Catalyst of Palladium and Niobic Acid-on-Carbon, ACS Omega, 2020, 5(6), 2699–2709 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Bright Side of Lignin Depolymerization: Toward New Platform Chemicals, Chem. Rev., 2018, 118(2), 614–678 CrossRef CAS PubMed.
- P. J. Deuss and K. Barta, From models to lignin: Transition metal catalysis for selective bond cleavage reactions, Coord. Chem. Rev., 2016, 306, 510–532 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Selective, nickel-catalyzed hydrogenolysis of aryl ethers, Science, 2011, 332(6028), 439–443 CrossRef CAS PubMed.
- F. Gao, J. D. Webb and J. F. Hartwig, Chemo–and Regioselective Hydrogenolysis of Diaryl Ether C− O Bonds by a Robust Heterogeneous Ni/C Catalyst: Applications to the Cleavage of Complex Lignin–Related Fragments, Angew. Chem., Int. Ed., 2016, 55(4), 1474–1478 CrossRef CAS PubMed.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, A heterogeneous nickel catalyst for the hydrogenolysis of aryl ethers without arene hydrogenation, J. Am. Chem. Soc., 2012, 134(50), 20226–20229 CrossRef CAS PubMed.
- J. He, C. Zhao and J. A. Lercher, Ni-Catalyzed Cleavage of Aryl Ethers in the Aqueous Phase, J. Am. Chem. Soc., 2012, 134(51), 20768–20775 CrossRef CAS PubMed.
- J. He, L. Lu, C. Zhao, D. Mei and J. A. Lercher, Mechanisms of catalytic cleavage of benzyl phenyl ether in aqueous and apolar phases, J. Catal., 2014, 311, 41–51 CrossRef CAS.
- S. Kasakov, H. Shi, D. M. Camaioni, C. Zhao, E. Baráth, A. Jentys and J. A. Lercher, Reductive deconstruction of organosolv lignin catalyzed by zeolite supported nickel nanoparticles, Green Chem., 2015, 17(11), 5079–5090 RSC.
- M. Wang, Y. Zhao, D. Mei, R. M. Bullock, O. Y. Gutiérrez, D. M. Camaioni and J. A. Lercher, The Critical Role of Reductive Steps in the Nickel–Catalyzed Hydrogenolysis and Hydrolysis of Aryl Ether C− O Bonds, Angew. Chem., Int. Ed., 2020, 59(4), 1445–1449 CrossRef CAS PubMed.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, Titanium nitride-nickel nanocomposite as heterogeneous catalyst for the hydrogenolysis of aryl ethers, J. Am. Chem. Soc., 2014, 136(5), 1758–1761 CrossRef CAS PubMed.
- V. Molinari, G. Clavel, M. Graglia, M. Antonietti and D. Esposito, Mild continuous hydrogenolysis of kraft lignin over titanium nitride–nickel catalyst, ACS Catal., 2016, 6(3), 1663–1670 CrossRef CAS.
- S. Lange, D. Formenti, H. Lund, C. Kreyenschulte, G. Agostini, S. Bartling, S. Bachmann, M. Scalone, K. Junge and M. Beller, Additive-Free Nickel-Catalyzed Debenzylation Reactions via Hydrogenative C–O and C–N Bond Cleavage, ACS Sustainable Chem. Eng., 2019, 7(20), 17107–17113 CrossRef CAS.
- M. Zaheer, J. Hermannsdörfer, W. P. Kretschmer, G. Motz and R. Kempe, Robust heterogeneous nickel catalysts with tailored porosity for the selective hydrogenolysis of aryl ethers, ChemCatChem, 2014, 6(1), 91–95 CrossRef CAS.
- C. Zhu, J. P. Cao, X. Y. Zhao, T. Xie, J. Ren and X. Y. Wei, Mechanism of Ni-catalyzed selective C-O cleavage of lignin model compound benzyl phenyl ether under mild conditions, J. Energy Inst., 2019, 92(1), 74–81 CrossRef CAS.
- X. Wang and R. Rinaldi, Exploiting H-transfer reactions with Raney® Ni for upgrade of phenolic and aromatic biorefinery feeds under unusual, low-severity conditions, Energy Environ. Sci., 2012, 5(8), 8244–8260 RSC.
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, Multiple Mechanisms Mapped in Aryl Alkyl Ether Cleavage via Aqueous Electrocatalytic Hydrogenation over Skeletal Nickel, J. Am. Chem. Soc., 2020, 142(8), 4037–4050 CrossRef CAS PubMed.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron and D. J. Miller, Electrocatalytic upgrading of model lignin monomers with earth abundant metal electrodes, Green Chem., 2015, 17(1), 601–609 RSC.
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, Skeletal Ni electrode-catalyzed CO cleavage of diaryl ethers entails direct elimination via benzyne intermediates, Nat. Commun., 2022, 13(1), 2050 CrossRef CAS PubMed.
- F. Lin, T. Ho-Yin, H. C. Erythropel, P. V. Petrović, M. Garedew, J. Chen, J. C.-H. Lam and P. T. Anastas, Development of a Ni-promoted, selective electrochemical reductive cleavage of the C–O bond in lignin model compound benzyl phenyl ether, Green Chem., 2022, 24(16), 6295–6305 RSC.
- L. Bering, K. Jeyakumar and A. P. Antonchick, Metal-Free C–O Bond Functionalization: Catalytic Intramolecular and Intermolecular Benzylation of Arenes, Org. Lett., 2018, 20(13), 3911–3914 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices, J. Am. Chem. Soc., 2015, 137(13), 4347–4357 CrossRef CAS PubMed.
- E. Cossar, M. S. Houache, Z. Zhang and E. A. Baranova, Comparison of electrochemical active surface area methods for various nickel nanostructures, J. Electroanal. Chem., 2020, 870, 114246 CrossRef CAS.
- Z. Wu, Q. Gan, X. Li, Y. Zhong and H. Wang, Elucidating surface restructuring-induced catalytic reactivity of cobalt phosphide nanoparticles under electrochemical conditions, J. Phys. Chem. C, 2018, 122(5), 2848–2853 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- L. Goerigk and N. Mehta, A Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User, Aust. J. Chem., 2019, 72(8), 563–573 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115(19), 2315–2372 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- Y. Liu, C. M. Hangarter, D. Garcia and T. P. Moffat, Self-terminating electrodeposition of ultrathin Pt films on Ni: An active, low-cost electrode for H2 production, Surf. Sci., 2015, 631, 141–154 CrossRef CAS.
- G. Ertl, R. Hierl, H. Knözinger, N. Thiele and H. Urbach, XPS study of copper aluminate catalysts, Appl. Surf. Sci., 1980, 5(1), 49–64 CrossRef CAS.
- A. Mansour, Characterization of NiO by XPS, Surf. Sci. Spectra, 1994, 3(3), 231–238 CrossRef CAS.
- N. McIntyre and M. Cook, X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper, Anal. Chem., 1975, 47(13), 2208–2213 CrossRef CAS.
- Z. Hu, L. Zhang, J. Huang, Z. Feng, Q. Xiong, Z. Ye, Z. Chen, X. Li and Z. Yu, Self-supported nickel-doped molybdenum carbide nanoflower clusters on carbon fiber paper for an efficient hydrogen evolution reaction, Nanoscale, 2021, 13(17), 8264–8274 RSC.
- J. Rivera, R. Garcia-Garcia, E. Coutino-Gonzalez and G. Orozco, Hydrogen evolution reaction on metallic rhenium in acid media with or without methanol, Int. J. Hydrogen Energy, 2019, 44(50), 27472–27482 CrossRef CAS.
- M. Bernardini, N. Comisso, G. Davolio and G. Mengoli, Formation of nickel hydrides by hydrogen evolution in alkaline media, J. Electroanal. Chem., 1998, 442(1–2), 125–135 CrossRef CAS.
- U. Sanyal, J. Lopez-Ruiz, A. B. Padmaperuma, J. Holladay and O. Y. Gutiérrez, Electrocatalytic hydrogenation of oxygenated compounds in aqueous phase, Org. Process Res. Dev., 2018, 22(12), 1590–1598 CrossRef CAS.
- P. Tundo, A. Perosa and S. Zinovyev, Modifier effects on Pt/C, Pd/C, and Raney-Ni catalysts in multiphase catalytic hydrogenation systems, J. Mol. Catal. A: Chem., 2003, 204, 747–754 CrossRef.
- U. Sanyal, S. F. Yuk, K. Koh, M. S. Lee, K. Stoerzinger, D. Zhang, L. C. Meyer, J. A. Lopez-Ruiz, A. Karkamkar and J. D. Holladay, Hydrogen bonding enhances the electrochemical hydrogenation of benzaldehyde in the aqueous phase, Angew. Chem., 2021, 133(1), 294–300 CrossRef.
- Y. Song, O. Y. Gutiérrez, J. Herranz and J. A. Lercher, Aqueous phase electrocatalysis and thermal catalysis for the hydrogenation of phenol at mild conditions, Appl. Catal., B, 2016, 182, 236–246 CrossRef CAS.
- P. G. Wuts and T. W. Greene, Greene's protective groups in organic synthesis, John Wiley & Sons, 2006 Search PubMed.
- E. W. Garbisch, L. Schreader and J. J. Frankel, Mechanism of benzylic substituent hydrogenolysis, J. Am. Chem. Soc., 1967, 89(16), 4233–4235 CrossRef CAS.
- A. Kieboom, J. De Kreuk and H. Van Bekkum, Substituent effects in the hydrogenolysis of benzyl alcohol derivatives over palladium, J. Catal., 1971, 20(1), 58–66 CrossRef CAS.
- F. M. Harvey and C. G. Bochet, Stereospecific hydrogenolysis of benzylic alcohols over Pd/C, J. Org. Chem., 2020, 85(11), 7611–7619 CrossRef CAS PubMed.
- S. Mitsui, S. Imaizumi and Y. Esashi, Stereochemistry and Mechanism of Catalytic Hydrogenation and Hydrogenolysis. III. Catalytic Hydrogenolysis of Benzyl-type Alcohols and Their Derivatives, Bull. Chem. Soc. Jpn., 1970, 43(7), 2143–2152 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, Mechanisms of Furfural Reduction on Metal Electrodes: Distinguishing Pathways for Selective Hydrogenation of Bioderived Oxygenates, J. Am. Chem. Soc., 2017, 139(40), 14120–14128 CrossRef CAS PubMed.
- S. Huang, Y. Jin, M. Zhang, K. Yan, S.-P. Feng and J. C.-H. Lam, MoS2-Catalyzed Selective Electrocatalytic Hydrogenation of Aromatic Aldehydes in an Aqueous Environment, Green Chem., 2022, 24(20), 7974–7987 RSC.
- S. Huang, B. Gong, Y. Jin, P. H. L. Sit and J. C.-H. Lam, The Structural Phase Effect of MoS2 in Controlling the Reaction Selectivity between Electrocatalytic Hydrogenation and Dimerization of Furfural, ACS Catal., 2022, 12(18), 11340–11354 CrossRef CAS.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Modern strategies in electroorganic synthesis, Chem. Rev., 2008, 108(7), 2265–2299 CrossRef CAS PubMed.
- Y. Qiu, J. A. Lopez-Ruiz, U. Sanyal, E. Andrews, O. Y. Gutiérrez and J. D. Holladay, Anodic electrocatalytic conversion of carboxylic acids on thin films of RuO2, IrO2, and Pt, Appl. Catal., B, 2020, 277, 119277 CrossRef CAS.
- L. Lang, Y. Li, J. C.-H. Lam, Y. Ding, X. Yin and C. Wu, Enhancing the selectivity of hydrocarbons during the Kolbe electrolysis of biomass-derived short-chain carboxylic acids by anionic surfactants, Sustainable Energy Fuels, 2022, 6(11), 2797–2804 RSC.
- C. Wang, K. Liu, Y. Jin, S. Huang and J. C.-H. Lam, Amorphous, RuO2 Catalyst for Medium Size Carboxylic Acid to Alkane Dimer Selective Kolbe Electrolysis in an Aqueous Environment, ChemSusChem, 2023, 16(16), e202300222 CrossRef CAS PubMed.
- Z. Jiang, J. Xie, D. Jiang, X. Wei and M. Chen, Modifiers-assisted formation of nickel nanoparticles and their catalytic application to p-nitrophenol reduction, CrystEngComm, 2013, 15(3), 560–569 RSC.
- D.-H. Chen and C.-H. Hsieh, Synthesis of nickel nanoparticles in aqueous cationic surfactant solutions, J. Mater. Chem., 2002, 12(8), 2412–2415 RSC.
- Z.-X. Zhang, Y. Liu, W.-J. Meng, J. Wang, W. Li, H. Wang, D. Zhao and J.-X. Lu, One-pot synthesis of Ni nanoparticle/ordered mesoporous carbon composite electrode materials for electrocatalytic reduction of aromatic ketones, Nanoscale, 2017, 9(45), 17807–17813 RSC.
- A. Mahata, K. S. Rawat, I. Choudhuri and B. Pathak, Octahedral Ni-nanocluster (Ni85) for efficient and selective reduction of nitric oxide (NO) to nitrogen (N2), Sci. Rep., 2016, 6(1), 25590 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, CHEM21 selection guide of classical-and less classical-solvents, Green Chem., 2016, 18(1), 288–296 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković and Q. Tu, The Green ChemisTREE: 20 years after taking root with the 12 principles, Green Chem., 2018, 20(9), 1929–1961 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01814h |
| This journal is © The Royal Society of Chemistry 2023 |