
Reprogramming the metabolism of oleaginous yeast for sustainably biosynthesizing the anticarcinogen precursor germacrene A†
Qi
Liu
abc,
Ge
Zhang
bc,
Liqiu
Su
bc,
Pi
Liu
bc,
Shiru
Jia
a,
Qinhong
Wang
bc and
Zongjie
Dai
 *bc
*bc
aCollege of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China
bKey Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China. E-mail: daizj@tib.cas.cn
cNational Technology Innovation Center of Synthetic Biology, Tianjin 300308, China
First published on 4th August 2023
Abstract
Due to the diverse structures and broad functions of terpenes, microbial biosynthesis of these compounds has been favored as a sustainable alternative to phytoextraction and chemosynthesis. Here, systematic metabolic engineering strategies were explored in the oleaginous yeast Yarrowia lipolytica for hyperproducing sesquiterpene germacrene A which serves as an important intermediate of numerous anticarcinogens. By identifying the most efficient germacrene A synthase to date, reconstructing the endogenous mevalonate pathway and extending the cytosolic acetyl-CoA pool by regulating lipid metabolism, the resulting strain overproduced 2.794 g L−1 germacrene A in shake flasks, which represented a 38-fold improvement over the initial strain. The engineered strain was subsequently capable of producing 39 g L−1 germacrene A at a yield of 0.181 g g−1 glucose during optimized bioreactor fermentation, with this being the highest sesquiterpene production level reported to date for Y. lipolytica. These results demonstrate that reprogramming the metabolism of the host cell by systematic metabolic engineering plays an essential role in diverting its inherent metabolic fluxes for sesquiterpene biosynthesis and these approaches can be extensively applied for synthesizing natural terpenes.
1. Introduction
Plant-derived natural terpenes are considered to be an interesting group of compounds with remarkable bioactivities and are widely applied as medicines, flavors, biofuels or food additives.1,2 In particular, the sesquiterpene germacrene A is considered a critical intermediate of the germacrane class compounds, with its spontaneous Cope rearrangement to form commercialized anticarcinogen β-elemene.3,4 Furthermore, germacrene A can be oxidized into germacrene A acid for producing parthenolide,5 guaianolide and germacranolide,6 all of which are natural products of therapeutic value against physiological diseases (ESI Fig. 1†).3,7The traditional plant-based manufacturing or chemical synthesis of germacrene A is usually inefficient and difficult due to its naturally low abundance and structural complexity. In recent years, with the development of synthetic biology, microorganisms have been considered as alternatives to conventional methods for germacrene A production.5,8–10 In these cases, by overexpressing critical endogenous genes of the mevalonate (MVA) pathway and introducing a heterogeneous germacrene A synthase (GAS), including LTC2 from the plant Lactuca sativa or AvGAS from the bacteria Anabaena variabilis, previous studies have successfully constructed Saccharomyces cerevisiae cell factories to produce germacrene A.9,10 However, due to the inefficient GAS, insufficient flux of the MVA pathway and deficient fine metabolic control, the production of germacrene A was only 309.8 mg L−1. Recently, Ye et al. selected an industrial yeast Ogataea polymorpha as a host for germacrene A biosynthesis; by optimizing the MVA pathway, enhancing the supply of NADPH and acetyl-CoA, and downregulating competitive pathways, the final titer of germacrene A reached 4.7 g L−1 with a yield of 0.028 g g−1 glucose.11 Although this represents a notable improvement compared with previous reports, the concentration and yield are still not competitive enough for industrial production. The key challenges for achieving efficient biomanufacturing of terpenes include but are not limited to suitable chassis cells, conspicuous terpene synthases and fine-tuning the biosynthetic route.
Due to its endogenous MVA pathway and sufficient supply of acetyl-CoA and co-factors, the unconventional oleaginous yeast Yarrowia lipolytica has been highlighted as an attractive chassis cell for the biosynthesis of terpenes,12–14 such as limonene,15 α-farnesene,16 gibberellin17 and squalene.18 In particular, oleaginous Y. lipolytica possesses inherent advantages for the production of liposoluble terpenes such as β-carotene and lycopene.19–21 But its production level is still far from that of the model strains S. cerevisiae22 and Escherichia coli,23 with this disparity being mainly caused by the lack of diverse and multilayered systematic metabolic engineering in this species.
The current study aimed to explore the potential of terpene biosynthesis in Y. lipolytica; for this purpose, multi-modular metabolic engineering strategies were used to construct a suitable strain for overproducing germacrene A (Fig. 1). This involved identifying the most efficient germacrene A synthases, optimizing the germacrene A biosynthetic route and evaluating novel means of supplying acetyl-CoA towards the MVA pathway. Ultimately, through this approach, a strain capable of producing 39 g L−1 germacrene A with a glucose yield of 0.181 g g−1 in bioreactor fermentations was established. This capacity for biomanufacturing sesquiterpene was the highest titer in Y. lipolytica and the highest yield reported in yeast so far. The findings of this study can therefore serve as a paradigm to sustainably produce other value-added terpenes at industrial levels.
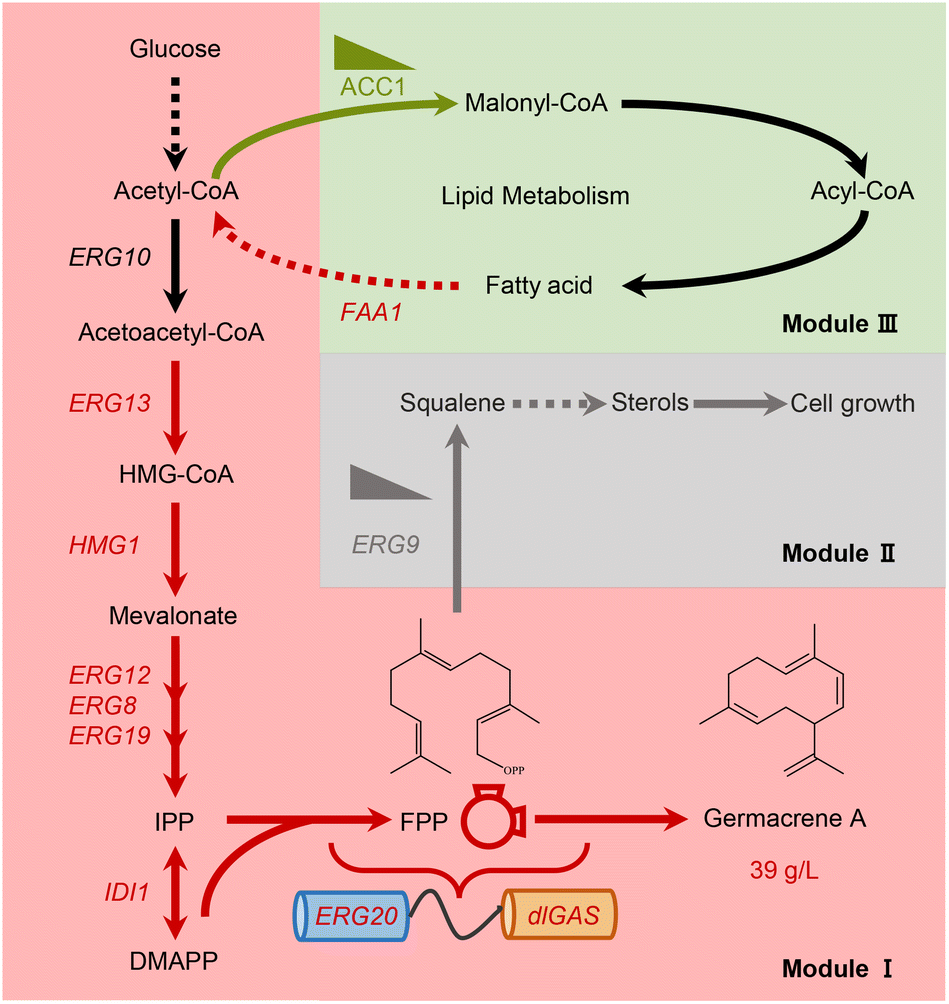 | ||
| Fig. 1 Engineering the heterologous biosynthesis of germacrene A in Y. lipolytica. A multi-module systematic metabolic engineering strategy was implemented to enable the production of germacrene A by following the components that enhanced the synthetic route (pink box, module I), eliminating competition (grayish box, module II) and supplying precursors (reseda box, module III). Intermediates: HMG-CoA, 3-hydroxy-3-methyl-glutaryl coenzyme A; IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; FPP, farnesyl diphosphate; enzymes: ERG10, acetoacetyl-CoA thiolase; ERG13, HMG-CoA synthase; HMG1, HMG-CoA reductase; ERG12, mevalonate kinase; ERG8, phosphomevalonate kinase; ERG19, mevalonate diphosphate decarboxylase; IDI1, isopentenyl diphosphate isomerase 1; ERG20, FPP synthase; ERG9, squalene synthase; ACC1, acetyl-CoA carboxylase 1; and FAA1, fatty acyl-CoA synthetase. | ||
2. Experimental
2.1 Strains and culture conditions
E. coli DH5α was used for the construction and propagation of plasmids. The strains were cultured at 37 °C and 250 rpm in Luria–Bertani (LB) liquid medium or at 37 °C on LB agar plates. Ampicillin was added to the medium at a final concentration of 100 mg L−1 when necessary. Wild type Y. lipolytica W29, obtained from the ARS Culture Collection (NRRL), was chosen as the basic strain for construction. Y. lipolytica strains were cultured in yeast extract peptone dextrose (YPD) liquid medium or on YPD plates, supplemented with 20 g L−1 of agar, at 30 °C. In addition, 250 mg L−1 of nourseothricin was added to YPD for screening of recombinant strains containing nat resistance markers. A description of all recombinant plasmids and strains used in this study is provided in ESI Tables 1 and 2.†2.2 gRNA plasmid construction and DNA manipulation
The optimal target-specific gRNA sequences (20 bp) for a selected gene or genomic locus were identified and ranked with W29 genetic background using CHOPCHOP (https://chopchop.cbu.uib.no/).24 Plasmid pCfB3405![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 was then employed to construct vectors harboring a single gRNA expression cassette by standard restriction enzyme digestion and ligation or Gibson assembly.
25 was then employed to construct vectors harboring a single gRNA expression cassette by standard restriction enzyme digestion and ligation or Gibson assembly.
The genomic DNA of W29 was used as a template for amplifying native DNA fragments including promoters, endogenous genes, terminators and homologous arms. All heterologous GAS genes were codon-optimized and synthesized by Genewiz. The primers used are listed in ESI Table 3,† while the GAS genes and mentioned endogenous fragments are provided in ESI Table 5.† All seamlessly integrated fragments were generated by overlapping extension PCR. The typical flexible polypeptide (GGGGS) was used for fusing the proteins dlGAS and ERG20. To obtain a recombinant strain, the integration fragment and specific gRNA plasmid were mixed and transformed into the Y. lipolytica host strain through the lithium acetate-mediated yeast transformation method as described in a previous report.25
2.3 RNA extraction and quantitative real-time PCR
Yeast cells were harvested during the early log phase, employing the same conditions as those in germacrene A fermentation. A total of 2 × 107 cells were collected and subjected to RNA extraction using the R6870 Yeast RNA Kit (OMEGA, USA). Subsequently, approximately 500 ng of RNA was reverse transcribed into cDNA using the Evo M-MLV RT Premix (Accurate Biotechnology, China). The primers of quantitative real-time PCR (qPCR) were designed using the online tool available at https://www.ncbi.nlm.nih.gov/tools/primer-blast/ and are listed in ESI Table 4.† Quantitative PCR analysis was performed on an Applied Biosystems 7500 Real Time PCR System using SYBR® Green Pro Taq HS (Accurate Biotechnology, China), following standard instructions. The ACT1 gene was selected as the reference housekeeping gene.2.4 Fermentation
The engineered Y. lipolytica strains were assessed for the production of germacrene A in minimal medium,26 which contained 7.5 g L−1 (NH4)2SO4, 14.4 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 20 g L−1 glucose, 2 mL L−1 trace metal and 1 mL L−1 vitamin solutions supplemented with 40 mg L−1 uracil if needed. All strains were pre-cultured in 15 mL culture tubes containing 2 mL of minimal medium, with incubation performed at 30 °C and 250 rpm until an OD600 of about 4.0 was reached. After pre-culture, the cell broth was inoculated into 20 ml of fresh medium in a 100 mL flask at an initial OD600 of 0.1. After cultivating the main cultures for 24 h, 2 mL of isopropyl myristate (IPM) was added for the organic extraction phase. All shake-flask cultivation strains were grown at 30 °C and 250 rpm for 96 h.To scale up germacrene A production, fed-batch fermentations were then performed in a 5 L bioreactor system (BIOTECH-5BG, Bxbio, China). In this case, 2 L of complete synthetic medium containing 5 g L−1 (NH4)2SO4, 3 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 40 mg L−1 uracil, 60 g L−1 glucose, 2 mL L−1 trace metal and 2 mL L−1 vitamin solutions was prepared for the initial batch stage. During the fermentation process, the fermenters were aerated at 1.5 volume per volume per minute (VVM) while maintaining 30% dissolved oxygen by adjusting the agitation speed from 200 to 700 rpm. The temperature was maintained at 30 °C, and the pH was set at 5.0 by automatically dropping 4 M KOH. At the fed-batch culture stage, the fermentation system was fed with supplementary components containing 25 g L−1 (NH4)2SO4, 15 g L−1 KH2PO4, 2.5 g L−1 MgSO4·7H2O, 200 mg L−1 uracil, 10 mL L−1 trace metal, 10 mL L−1 vitamin solutions and 600 g L−1 glucose. The feeding stage was started once the detected glucose titer was less than 10 g L−1. At this point, two different feeding approaches were performed, and these involved either maintaining the glucose concentration at around 10 g L−1 or pulse-feeding between 10 and 50 g L−1 of glucose. The organic layer of 200 mL of IPM was aseptically added after 24 h of cultivation, and another 200 mL of IPM was added after 96 h. Sampling was regularly carried out during fermentation to monitor OD600, glucose, germacrene A and other metabolites. Furthermore, in order to determine the ratio between the organic layer and the medium, we measured the volumes of IPM and the aqueous phase in timed samples, in conjunction with quantifying the amount of the feeding medium.
2.5 Quantification of products and metabolites
For extracting and analyzing germacrene A production, IPM was added during shake-flask cultivation and fed-batch fermentation as described above. For germacrene A detection, the organic phase was quantified by gas chromatography-mass spectrometry (GC-MS; Agilent 7890A-5975C) with an HP-5 column (30 m × 0.25 mm × 0.25 μm) and an ESI mass spectrometer. The initial temperature of the column was set at 60 °C and held for 3 min; followed by a ramp to 90 °C at a rate of 3 °C min−1. Then, there was a second ramp to 260 °C at a rate of 20 °C min−1 and held for 5 min. At a temperature of approximately 250 °C, germacrene A undergoes a spontaneous Cope rearrangement, resulting in the formation of β-elemene, which is commonly regarded as the standard compound for quantifying germacrene A.4,9–11 Germacrene A production is presented as the β-elemene equivalent.Samples from the aqueous layer were analysed by diluting them to measure OD600 and mevalonate. For mevalonate analysis, 1 mL of broth was centrifuged at 14000 rpm for 5 min, and the resulting supernatant was filtered through a 0.22 μm syringe filter prior to analysis. An HPLC system (Agilent Infinity 1200) equipped with an Aminex HPX-87H column (BioRad) and a refractive index detector was used for analysis. Integration of the peak areas was used to quantify MVA using standard curves. In addition, squalene was extracted and quantified as described in a previous report.27
For the acetyl-CoA assay, the engineered strains were cultivated under identical conditions as those employed in shake-flask fermentation. After 24 h of cultivation, yeast cells were collected for acetyl-CoA extraction and quantification using the Acetyl CoA Assay Kit (Abcam, UK) according to the provided standard protocols. The specific fluorescence was measured at excitation and emission wavelengths of 535 nm and 587 nm, respectively.
2.6 Sequence analysis and phylogenetic tree construction
The amino acid sequences annotated as GAS or terpene synthase were acquired from NCBI or UniProt databases and aligned by ClustalW. The maximum likelihood phylogenetic trees were then generated using MEGA-1128 software based on the LG model, with the strength of the nodes determined with 100 bootstrap replicates.
2.7 Computational GAS tunnel
The protein structures of dlGAS, deGAS and lsGAS were predicted using AlphaFold2.29 To predict the binding pocket, the substrate FPP was docked into GAS models using AutoDock.30 The CAVER Analyst software31 was then used to visualize the substrate transport tunnel and calculate access pathways to buried active sites within the proteins. The statistical parameters, involving residue hydrophobicity around the tunnel and volume of the cavity, were eventually determined using PyMOL and CAVER Analyst.3. Results and discussion
3.1 Identifying efficient enzymes for germacrene A biosynthesis
The structural complexity and functional diversity of terpenes are the result of evolutionary terpene synthases among widespread species to tackle their changing environment.32 Hence, mining and characterizing specific terpene synthases are considered to be the rate-limiting factors in establishing terpene cell factories. To identify efficient GASs for converting farnesyl pyrophosphate (FPP) to germacrene A, preliminary screening was performed using a phylogenetic tree based on hypothetical GASs or terpene synthases available in NCBI and UniPort databases (ESI Fig. 2†). Synthases from different evolutionary branches (Fig. 2A), especially those originating from fungi and exhibiting unique evolutionary routes, were subsequently selected to evaluate their functions and efficiency by chromosomally integrating them into CRISPR–Cas9 systems embedded in Y. lipolytica strain YLZG03 (ESI Table 2†). During shake flask fermentations, most screened GASs showed no germacrene A biosynthesis. However, deGAS from Daldinia eschscholtzii and dlGAS from Daldinia loculata generated germacrene A (ESI Fig. 3†) and showed a 2.2-fold and 4.3-fold increase over previously reported lsGAS, respectively10 (Fig. 2B).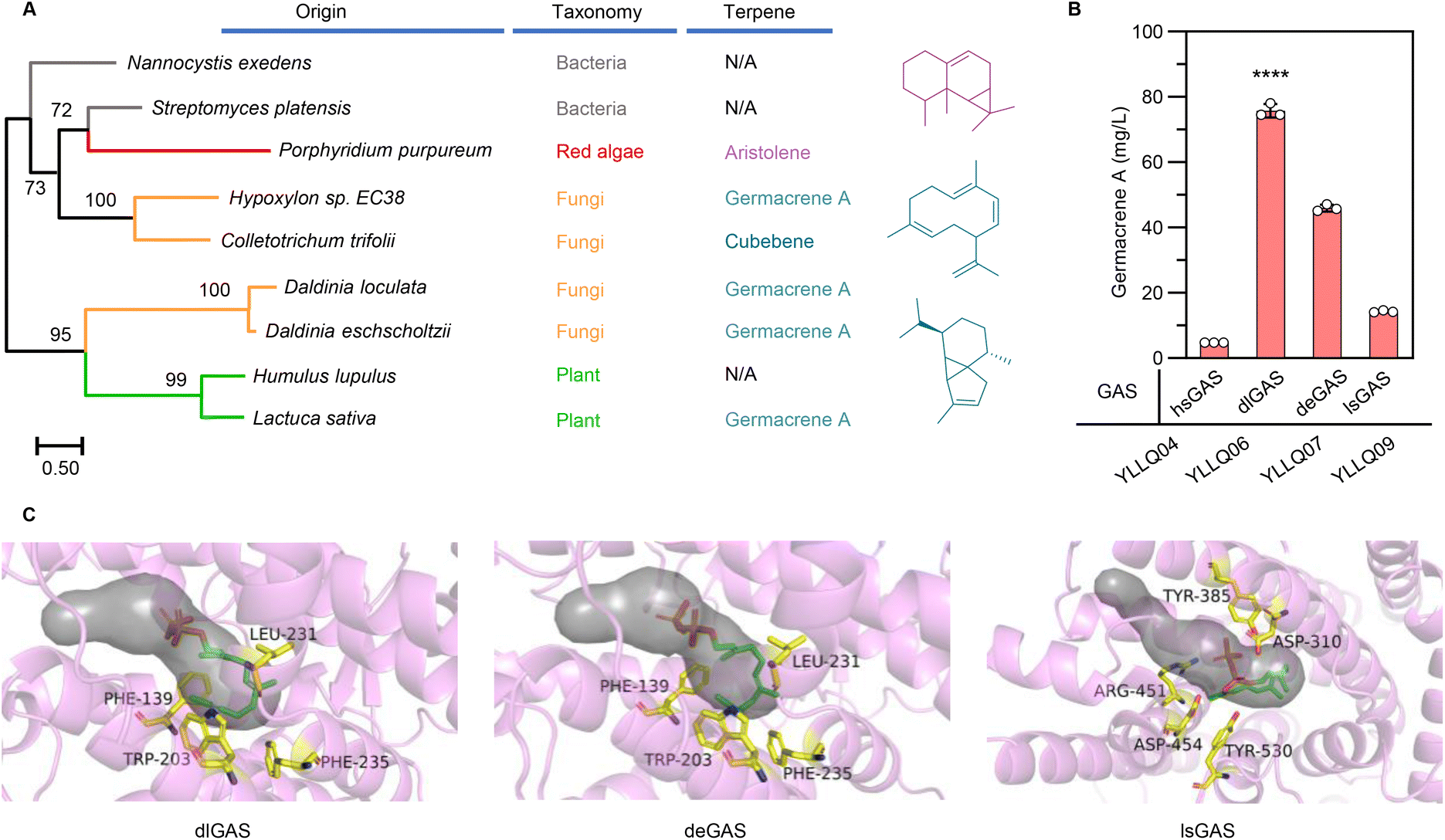 | ||
| Fig. 2 Bioinformatics-guided screening of efficient GASs. (A) Phylogenetic analysis and the functions of primary GASs. The main functional products of the selected GASs are listed and represented in distinct colors: aristolene (in violet), germacrene A (in mazarine) and cubebene (in dark cyan). (B) Germacrene A production by strains expressing identified GASs. Error bars represent the SD of triplicate samples. (C) Computational analysis of the active cavity in dlGAS, deGAS and lsGAS. The tunnels are shown in grayish. The surrounding residues are shown in yellow. | ||
To elucidate the differences between the activities of these GASs, the three-dimensional structures of deGAS, dlGAS and lsGAS were predicted using AlphaFold229 (ESI Fig. 4A†). As shown in Fig. 2C, the active sites of both deGAS and dlGAS are surrounded by Thr138, Phe139, Trp203, Leu231 and Phe235, which exhibit significant hydrophobicity compared with those of lsGAS. This difference in the hydrophobicity of residues surrounding active sites revealed that a hydrophobic environment facilitates the biosynthesis of lipophilic terpenes.33 In addition, lsGAS exhibited a larger cavity (1617.8 Å3) than deGAS (757.1 Å3) and dlGAS (624.9 Å3) (ESI Fig. 4B†), hence indicating that a cavity of an appropriate size might prevent the escape of the ligand molecule. Taken together, the above results demonstrated that the high bioactivity of GAS was the result of a modest cavity volume and a hydrophobic environment, both of which were beneficial for FPP transportation and cyclization. Undoubtedly, the newfound efficiency of GASs raised biomanufacturing germacrene A production and provided the basis for further improvement by protein engineering. Therefore, the strain YLLQ06 and dlGAS were selected for subsequent experiments.
3.2 Fusing critical enzymes to facilitate substrate trafficking
Narrowing the physical distance between substrates and catalytic enzymes within spatial organization through protein fusion has been recognized as being critical for increasing the local concentration of intermediates and improving reaction flux.34 FPP originating from the MVA pathway is catalyzed by FPP synthase (ERG20) and is also the direct precursor to synthesizing germacrene A. Therefore, to speed up substrate trafficking, dlGAS was fused with ERG20 in different orientations using a flexible peptide linker GGGGS to create strains YLLQ16 and YLLQ17 (Fig. 3B). In addition, a strain YLLQ18 which individually expressed ERG20 and dlGAS was also obtained. The most significantly enhanced germacrene A production was observed in strain YLLQ16, whereby the fused enzyme in the dlGAS–ERG20 orientation provided the highest germacrene A titer of 95.9 mg L−1, which represented a 26.7% and 10.6% improvement compared with strains YLLQ06 and YLLQ18, respectively (Fig. 3B). In contrast, only half of germacrene A was detected in strain YLLQ17, presumably due to the ERG20–dlGAS orientation blocking structural configurations and enzyme activities. These results suggested that physically fusing enzymes for the formation and consumption of FPP was beneficial for driving the metabolic flux towards germacrene A biosynthesis.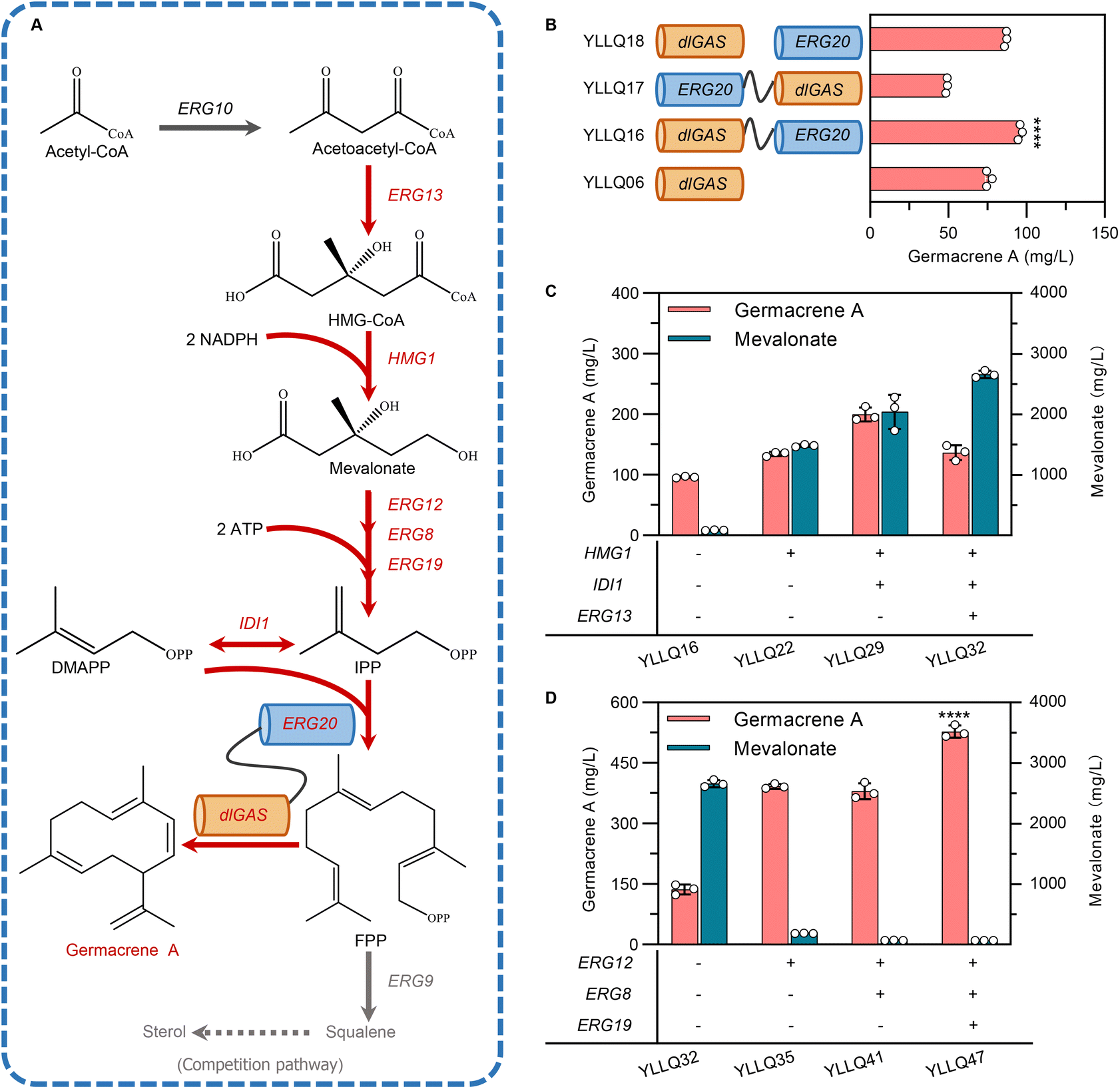 | ||
| Fig. 3 Reconstructing the biosynthetic route of germacrene A in Y. lipolytica. (A) Schematic illustration of the biosynthetic pathways leading to the production of germacrene A and related byproducts. Red arrows: increased germacrene A biosynthesis; gray arrows: byproduct pathway. (B) Quantification of the resulting product synthesized by strains carrying a fused enzyme of dlGAS and ERG20. (C) Production profiles of resulting germacrene A and intermediate mevalonate after overexpressing key enzymes of the MVA pathway. (D) Changes in the production profiles of germacrene A and mevalonate after increasing the flux that converts mevalonate to IPP. Error bars represent the SD of triplicate samples. | ||
3.3 Enhancing germacrene A production by strengthening the expression of the MVA pathway
Currently, most traditional cases of metabolic engineering employ model organisms as host systems due to their genetic tractability and unambiguous metabolism.35 However, the non-conventional yeast Y. lipolytica is considered a promising host for biomanufacturing isoprenoid products, especially by taking advantage of its naturally abundant acetyl-CoA, cofactor synthesis and endogenous MVA pathway. The established Y. lipolytica platform focused on overexpressing critical enzymes of the MVA pathway while ignoring the manipulation of gene expression across the entire continuum.36–38 In this study, promoter engineering was implemented to enhance expression intensity and the synergistic effect of the MVA pathway.Here, the strong promoter PTEFin was selected to replace the endogenous ones controlling critical genes in the MVA pathway to avoid background interference. As expected, strain YLLQ22 overexpressing HMG1 produced 40% more germacrene A compared with YLLQ16, reaching a titer of 134.5 mg L−1 (Fig. 3C). To improve the flow of the MVA pathway, key genes including IDI1 and ERG13 were further overexpressed. In this case, strain YLLQ29, with IDI1 overexpression, accumulated 199.7 mg L−1 of germacrene A which was 48.5% higher than that of YLLQ22. However, the ERG13-overexpressing strain YLLQ32 did not produce higher germacrene A (Fig. 3C). At the same time, it was found that the concentration of MVA unexpectedly increased (Fig. 3C). These results indicated that insufficient flux of the downstream MVA pathway leads to the accumulation of MVA, which is not conducive to the production of germacrene A.
To overcome this issue, three genes, namely ERG12, ERG8 and ERG19 that convert MVA to IPP, were overexpressed in sequence. As shown in Fig. 3D, the byproduct MVA was successfully reduced, while the titer of germacrene A was significantly enhanced. More specifically, by overexpressing ERG12, the titer of germacrene A for strain YLLQ35 reached 392.1 mg L−1, representing approximately a 3-fold increase compared with strain YLLQ32. Interestingly, the titer of MVA was simultaneously reduced by 93%. Another strain, YLLQ41, was obtained by overexpressing ERG8 in YLLQ35; although MVA levels decreased further, its OD600 was only half that of strain YLLQ35, which can be attributed to the imbalanced provision and consumption of 5-diphosphomevalonate, a cytotoxic intermediate in the MVA pathway (ESI Fig. 5A†).39 In order to restore balance to MVA pathway flux, we subsequently increased the expression level of ERG19 and obtained strain YLLQ47 for which the germacrene A titer reached 527.8 mg L−1 with recuperative cell growth as YLLQ35. In comparison with the initial strain YLLQ16, the substitution of the native promoter of the MVA pathway with strong promoters PTEFin or PGPD resulted in an increased level of this gene expression in strain YLLQ47, particularly for the pivotal genes HMG1, ERG12, and IDI1 (ESI Fig. 5C†). With the successful generation of efficient germacrene A-producing strains, these results demonstrated that the synergistic expression of genes within the MVA pathway is beneficial for cell growth as well as the production of terpenes.
3.4 Fine tuning of the germacrene A synthetic route by a ‘restrain–pull’ strategy
FPP is the direct precursor of sesquiterpene biosynthesis, as well as of squalene, ubiquinone, ergosterol, and other essential substances for cell growth.40,41 As shown in Fig. 4A, squalene synthase (ERG9) is a key node for balancing cell growth and germacrene A production. Truncating the native promoter of ERG9 (PERG9) was successfully used to overproduce β-carotene and α-bisabolol in Y. lipolytica.27,42 In the present study, PERG9 was truncated to 50 base pairs to create strain YLLQ52 which produced 787.5 mg L−1 of germacrene A, with this titer being 49% higher than strain YLLQ47 harboring endogenous PERG9 (Fig. 4B). As expected, the level of competitive product squalene was not detectable (Fig. 4B). Additionally, the OD600 of YLLQ52 was notably reduced, leading to a higher specific titer of germacrene A (77.7 mg per L per OD) compared with that of YLLQ47 (36.2 mg per L per OD). These results indicated that restraining the downstream squalene synthesis pathway redirected more metabolic fluxes of FPP towards germacrene A production rather than cell growth.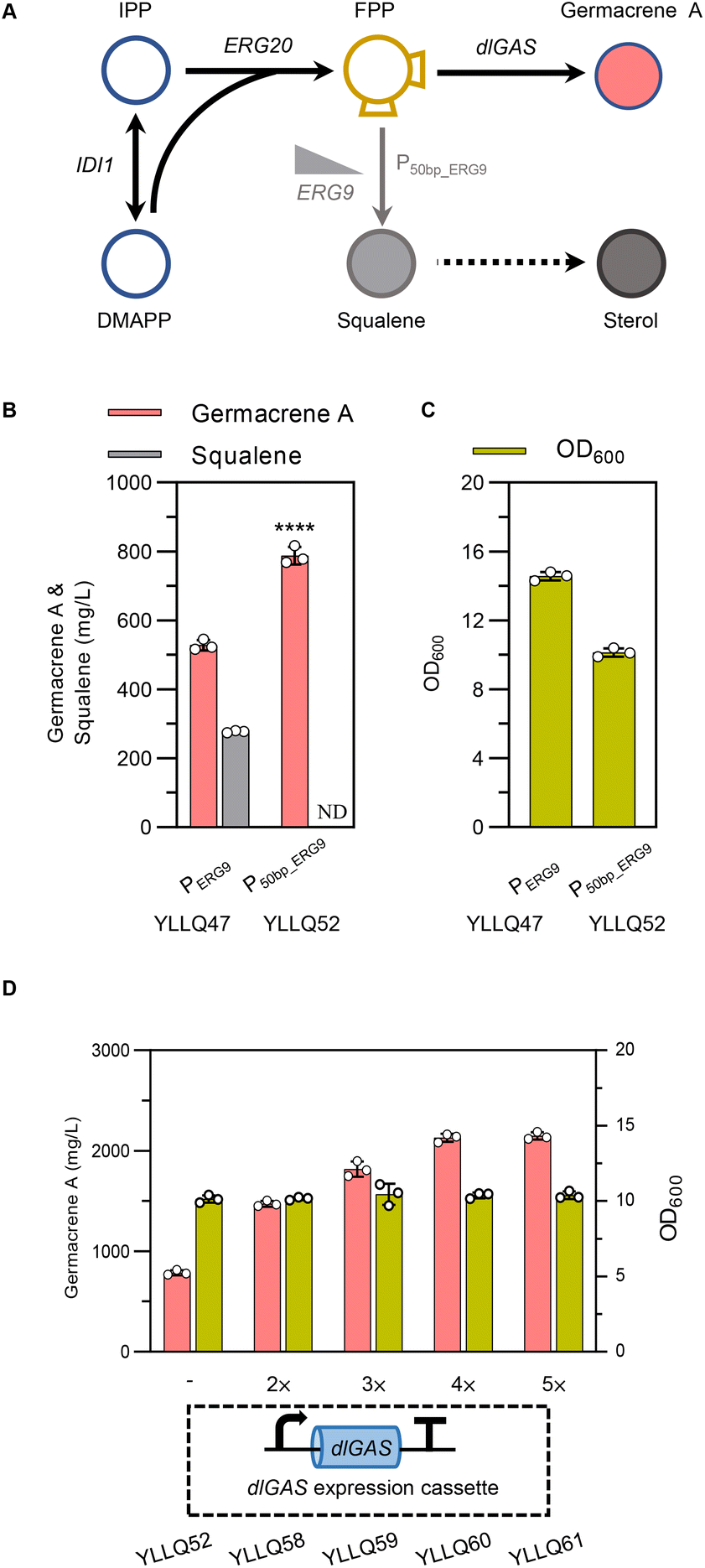 | ||
| Fig. 4 Regulating the flux of the germacrene A biosynthetic pathway. (A) Schematic illustration of the downregulation of the byproduct pathway with an FPP-mediated flux flow restrictor. The repressed reaction is shown with a grayish arrow. (B and C) Effects of downregulating squalene synthesis on the production profiles of germacrene A, squalene and biomass, respectively. (D) Effects of optimizing dlGAS copy numbers on the production and biomass. Error bars represent the SD of triplicate samples. | ||
It was then speculated that increasing the copy number of dlGAS could pull more FPP towards germacrene A biosynthesis. Hence, strains YLLQ58–YLLQ61 were generated through chromosomal integration of dlGAS. In doing so, germacrene A production of the resulting strains was further improved. Specifically, strains YLLQ60 and YLLQ61, holding three and four additional dlGAS copies, respectively, produced 2129.4 mg L−1 and 2147.1 mg L−1 of germacrene A, representing a 3-fold increase over YLLQ52 (Fig. 4D). Moreover, the minimal alteration in biomass suggested that the augmented copy number of dlGAS did not impose an additional metabolic load on the strains (Fig. 4D). On the other hand, overexpressing acetyl-CoA acetyltransferase ERG10 (the initial gene of the MVA pathway) did not promote germacrene A production (ESI Fig. 6†). These results strongly suggested that ‘restrain–pull’ strategies are effective for germacrene A formation, and further boosting target compound production requires an increased supply of acetyl-CoA.
3.5 Rewiring cytosolic acetyl-CoA towards the MVA pathway
In Y. lipolytica, acetyl-CoA flux is mainly used for lipid biosynthesis, which weakens the potential of terpene formation.14 To increase cytosolic acetyl-CoA towards the MVA pathway, heterogeneous pathways like the non-oxidative pentose-phosphate pathway (PK-PTA) or the native pyruvate bypass pathway have been applied. Despite recognizing these pathways for their capacity to overproduce acetyl-CoA derivatives, their impact on terpene synthesis remains inconspicuous.43–45 Therefore, shutting down carbon fluxes from lipid metabolism could be a key target for increasing terpene production in Y. lipolytica. To reduce lipid synthesis, the genes DGA1 and DGA2, which are essential for TAG synthesis, were generally knocked out, while simultaneously breaking them has been shown to slow down growth and yield a smaller amount of biomass.46 Thus, it is crucial to design effective compensatory strategies to regulate metabolism involved in lipid biosynthesis and degradation (Fig. 5A).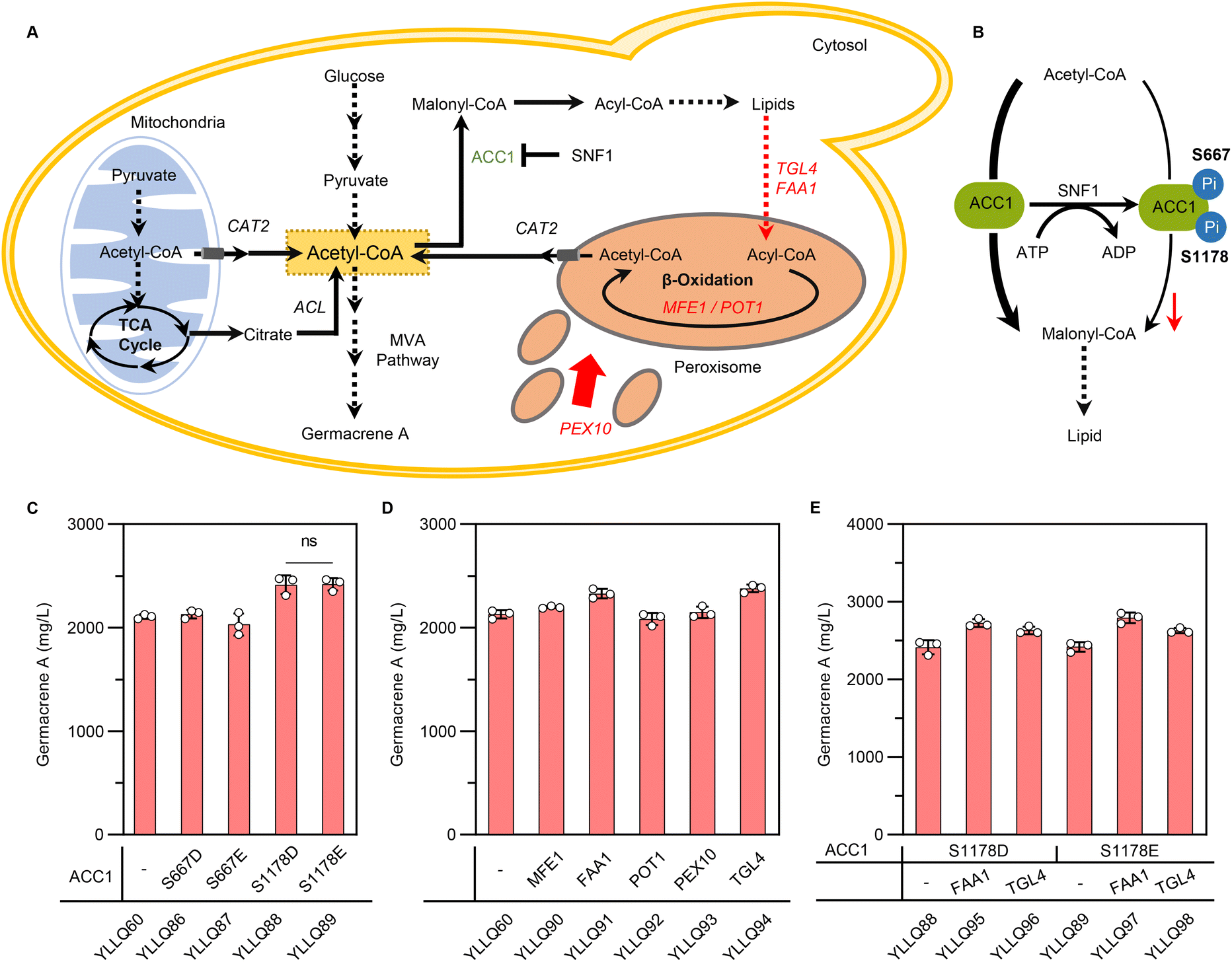 | ||
| Fig. 5 Effects of rewiring lipid metabolism on germacrene A biosynthesis. (A) Schematic illustration of alternative cytosolic acetyl-CoA pathways engineered in Y. lipolytica. Overexpressed genes are shown by red arrows, with the repressed enzyme shown in reseda font. Intermediates: ACC1, acetyl-CoA carboxylase 1; TGL4, triacylglycerol lipase; FAA1, fatty acyl-CoA synthetase; MFE1, multifunctional β-oxidation enzyme; POT1, 3-ketoacyl-CoA thiolase; PEX10, peroxisomal matrix protein; CAT2, carnitine acetyltransferase; and ACL, ATP dependent citrate lyases. (B) Phosphorylation approach to reduce the activity of ACC1. (C) Effects of simulating phosphorylation of ACC1 on germacrene A titer. (D) Germacrene A production from strains with MFE1, FAA1, POT1, PEX10 and TGL4 overexpression in alternative cytosolic acetyl-CoA pathways improves germacrene A content. (E) Production profile of germacrene A by simultaneously performing the processes mentioned in (C) and (D). Error bars represent the SD of triplicate samples. | ||
Acetyl-CoA carboxylase (ACC1) is responsible for converting acetyl-CoA to malonyl-CoA, a rate-limited step in fatty acyl-CoA metabolism and the main route to compete with terpene synthesis for the precursor acetyl-CoA.47 Phosphorylation modification induces conformational changes in the molecular structure of ACC1, resulting in hypoactivity through allosteric regulation and modulation of protein–protein interactions.48,49 Due to the absence of research about the phosphorylation sites of ACC1 in Y. lipolytica, we speculated on two possible sites based on aligning the amino acid sequences of ACC1 in Y. lipolytica (YALI1C15991p) with those of S. cerevisiae (YNR016C). As shown in ESI Fig. 7,† the residue corresponding to ylACC1S667 aligns with scACC1S659, a putative site conforming to the phosphorylation recognition motif for SNF1, while the residue corresponding to ylACC1S1178 is scACC1S1157, a positive residue identified by phosphoproteome50–54 (Fig. 5B). A highly active ACC1 mutant, denoted as ACC1S659A,S1157A, was generated through simulated dephosphorylation, resulting in an increased pool of malonyl-CoA for the biosynthesis of polyketides or flavonoids in S. cerevisiae.54 Therefore, we proposed to evaluate whether simulated phosphorylation of ACC1 could reduce its activity to minimize lipid synthesis and increase the metabolic flow of acetyl-CoA towards the MVA pathway. Specifically, site-directed mutagenesis was undertaken to mutate the serine at the phosphorylation site into glutamate or aspartate to generate four versions of ACC1: ACC1S667D, ACC1S667E, ACC1S1178D and ACC1S1178E. The results showed that germacrene A production for YLLQ88 and YLLQ89, with ACC1S1178D and ACC1S1178E mutations, respectively, increased by 13.4% and 13.6% compared with the control strain to reach 2415.2 mg L−1 and 2419.6 mg L−1 (Fig. 5C). In contrast, there was no notable increase observed in germacrene A content when comparing the mutated ACC1S667 strains with the control strain YLLQ60, suggesting that Ser1178 may indeed be the authentic phosphorylation site of ylACC1, but not Ser667.
The intracellular accumulation of TAG, which is synthesized as surplus energy and disperses carbon resources away from the desired product, was then targeted. In this case, the effects of overexpressing critical genes for supplementing extra cytosolic acetyl-CoA were assessed, especially those involved in lipid degradation and β-oxidation pathways, including the multifunctional β-oxidation enzyme MFE1 (encoded by YALI1E18441),55 fatty acid-CoA synthetase FAA1 (encoded by YALI1D22124),56 3-ketoacyl-CoA thiolase POT1 (encoded by YALI1E22238),57 peroxisomal matrix protein PEX10 (encoded by YALI1C01416)58 and triacylglycerol lipase TGL4 (encoded by YALI1F13550).59 Among these, germacrene A production for YLLQ90, the FAA1-overexpressing strain, and YLLQ94, the TGL4-overexpressing one, was significantly enhanced to a titer of 2329.3 and 2379.1 mg L−1, respectively, with both accounting for a 9.4% and 11.7% increase compared with that of strain YLLQ60 (Fig. 5D). Based on these results, the above-mentioned strategies were combined by overexpressing both FAA1 and TGL4 in background strains containing mutated ACC1S1178 to obtain strains YLLQ95–YLLQ98. All of the generated constructs resulted in improved germacrene A production, with the highest titer of 2789.4 mg L−1 and a glucose yield of 0.139 g g−1 observed for YLLQ97 (Fig. 5E). Quantification of acetyl-CoA levels directly demonstrated that both reducing ACC1 activity and enhancing lipid degradation resulted in elevated intracellular acetyl-CoA concentrations, thereby improving the synthesis of germacrene A (ESI Fig. 8†). These results suggested that our subtle approaches to balancing lipid metabolism and cell growth could play an important role in promoting the synthetic efficiency of acetyl-CoA derivatives.
3.6 Germacrene A fermentation in scale-up bioreactors
Finally, fed-batch fermentations were performed to assess the potential of the engineered strain YLLQ97 as a germacrene A cell factory. The cultivation was carried out in a 5 L bioreactor supplemented with glucose based on two different models. Superior plateaued OD600 values and germacrene A titers were observed when maintaining glucose concentrations of around 10 g L−1 (Fig. 6B), with the values being 16.7% and 54.8% higher, respectively, than those achieved by pulse-feeding between 10 and 50 g L−1 of glucose (Fig. 6A). Under both conditions, the highest titers of germacrene A reached 25.2 g L−1 (Fig. 6A) and 39 g L−1 (Fig. 6B) after 240 h of cultivation, with the corresponding glucose yields being 0.148 and 0.181 g g−1, respectively. These values highlighted significant differences between the two feeding conditions. It was therefore speculated that controlling the concentration of glucose over a stable range provided a suitable environment for strain metabolism, thereby contributing to a relatively faster productivity of germacrene A compared with pulsed-feeding of glucose over a wider range.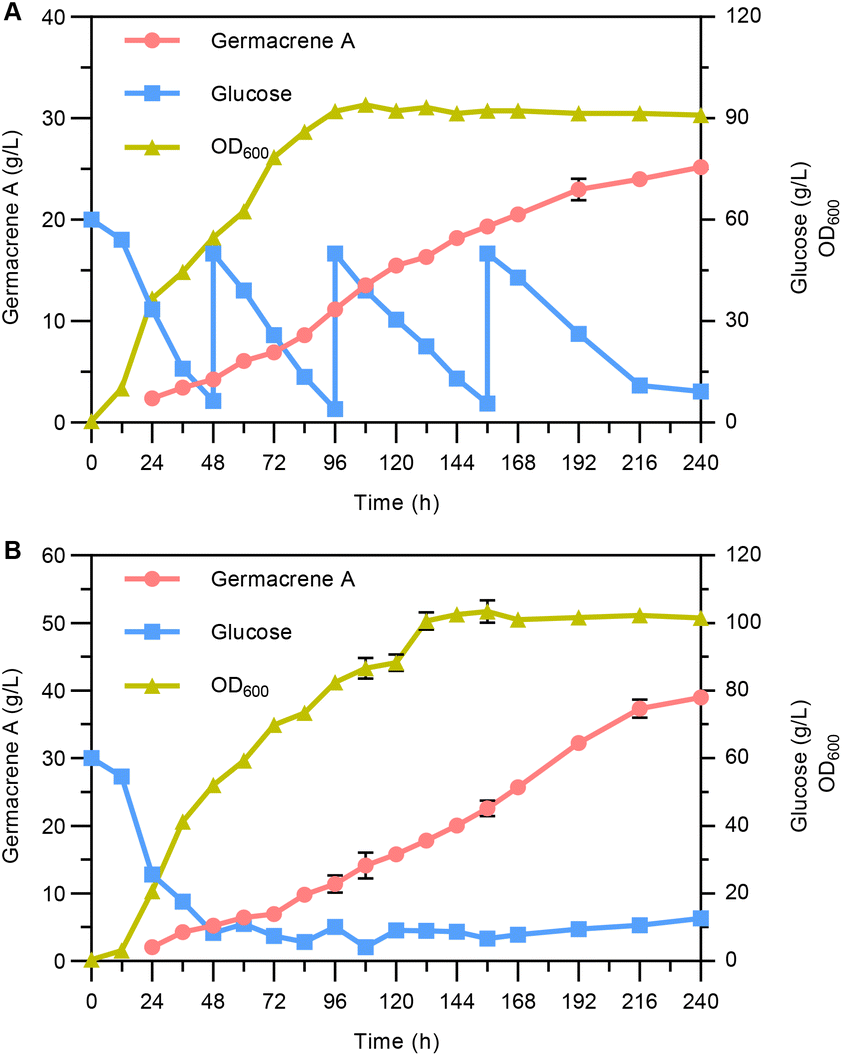 | ||
| Fig. 6 Bioreactor fed-batch fermentations for germacrene A production. Quantification of germacrene A, glucose and biomass during the cultivation of strain YLLQ97 under pulsed (A) and stabilized (B) feeding conditions. n = 3 for bioreactor fed-batch cultures, corresponding to triplicate samples. | ||
4. Conclusions
In the present study, we established a Y. lipolytica platform for sustainable production of the anticarcinogen precursor germacrene A by identifying efficient GASs, reconstructing the endogenous MVA pathway to increase the supply of precursors, and rewiring acetyl-CoA flux away from lipid metabolism toward terpene production by simulating phosphorylation of ACC1 to reduce inherent lipid biosynthesis and overexpressing FAA1 to strengthen lipid degradation. The engineered strain resulted in 2.79 g L−1 germacrene A in shake flasks and unprecedented 39 g L−1 under fed-batch fermentation, with this being the highest titer of sesquiterpenes reported for Y. lipolytica so far. In addition, the yield of germacrene A reached 0.181 g g−1 glucose, representing 71.8% of the theoretical value, the highest level reported in yeast. These engineering biological strategies for the production of germacrene A not only provide a basis for the biosynthesis of value-added terpenes but also prove that microbial synthesis possesses great potential as an alternative route to generate chemicals with complex structures.Author contributions
Q. L. and Z. D. designed research; Q. L., G. Z., L. S. and P. L. performed research; Q. L., S. J., Q. W. and Z. D. analyzed data; Q. L. and Z. D. wrote the paper; and all authors reviewed the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-002), the National Natural Science Foundation of China (32071423 and 32161133019) and the Hundreds of Talents Program of the Chinese Academy of Sciences (Y0J51009). The authors thank Professor Irina Borodina at the Technical University of Denmark for providing plasmid pCfB3405.References
- C. Liu, K. Xue, Y. Yang, X. Liu, Y. Li, T. S. Lee, Z. Bai and T. Tan, Crit. Rev. Biotechnol., 2022, 42, 73–92 CrossRef CAS PubMed.
- G. Zhang, H. Wang, Z. Zhang, K. J. Verstrepen, Q. Wang and Z. Dai, Crit. Rev. Biotechnol., 2022, 42, 618–633 CAS.
- H. Xu and J. S. Dickschat, Chemistry, 2020, 26, 17318–17341 CrossRef CAS PubMed.
- J. W. de Kraker, M. C. R. Franssen, A. de Groot, W. A. Konig and H. J. Bouwmeester, Plant Physiol., 1998, 117, 1381–1392 CrossRef CAS PubMed.
- Y. Shi, T. Dong, B. Zeng, M. Yao, Y. Wang, Z. Xie, W. Xiao and Y. Yuan, ACS Synth. Biol., 2022, 11, 2473–2483 CrossRef CAS PubMed.
- H. J. Bouwmeester, J. Kodde, F. W. A. Verstappen, I. G. Altug, J. W. de Kraker and T. E. Wallaart, Plant Physiol., 2002, 129, 134–144 CrossRef CAS PubMed.
- J. Rinkel and J. S. Dickschat, Org. Lett., 2019, 21, 2426–2429 CrossRef CAS PubMed.
- R. Chen, Y. Liu, S. Chen, M. Wang, Y. Zhu, T. Hu, Q. Wei, X. Yin and T. Xie, Front. Plant Sci., 2022, 13, 932966 CrossRef PubMed.
- W. Zhang, J. Guo, Z. Wang, Y. Li, X. Meng, Y. Shen and W. Liu, Microb. Cell Fact., 2021, 20, 7 CrossRef CAS PubMed.
- Y. Hu, Y. J. Zhou, J. Bao, L. Huang, J. Nielsen and A. Krivoruchko, J. Ind. Microbiol. Biotechnol., 2017, 44, 1065–1072 CrossRef CAS PubMed.
- M. Ye, J. Gao and Y. J. Zhou, Metab. Eng., 2023, 76, 225–231 CrossRef CAS PubMed.
- K. K. Miller and H. S. Alper, Appl. Microbiol. Biotechnol., 2019, 103, 9251–9262 CrossRef CAS PubMed.
- A. Muhammad, X. Feng, A. Rasool, W. Sun and C. Li, Biotechnol. Adv., 2020, 43, 107555 CrossRef CAS PubMed.
- A. M. Worland, J. J. Czajka, Y. Li, Y. Wang, Y. J. Tang and W. W. Su, Curr. Opin. Biotechnol., 2020, 64, 134–140 CrossRef CAS PubMed.
- J. Li, K. Zhu, L. Miao, L. Rong, Y. Zhao, S. Li, L. Ma, J. Li, C. Zhang and D. Xiao, ACS Synth. Biol., 2021, 10, 884–896 CrossRef CAS PubMed.
- Y. Liu, X. Jiang, Z. Cui, Z. Wang, Q. Qi and J. Hou, Biotechnol. Biofuels, 2019, 12, 296 CrossRef CAS PubMed.
- K. R. Kildegaard, J. A. Arnesen, B. Adiego-Perez, D. Rago, M. Kristensen, A. K. Klitgaard, E. H. Hansen, J. Hansen and I. Borodina, Metab. Eng., 2021, 66, 1–11 CrossRef CAS PubMed.
- W. Y. Tang, D. P. Wang, Y. Tian, X. Fan, C. Wang, X. Y. Lu, P. W. Li, X. J. Ji and H. H. Liu, Bioresour. Technol., 2021, 323, 124652 CrossRef CAS PubMed.
- M. Larroude, E. Celinska, A. Back, S. Thomas, J.-M. Nicaud and R. Ledesma-Amaro, Biotechnol. Bioeng., 2018, 115, 464–472 CrossRef CAS PubMed.
- Z. Luo, N. Liu, Z. Lazar, A. Chatzivasileiou, V. Ward, J. Chen, J. Zhou and G. Stephanopoulos, Metab. Eng., 2020, 61, 344–351 CrossRef CAS PubMed.
- Y. Ma, N. Liu, P. Greisen, J. Li, K. Qiao, S. Huang and G. Stephanopoulos, Nat. Commun., 2022, 13, 572 CrossRef CAS PubMed.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov, Y. Kim, L. Raetz, R. H. Dahl, A. Tai, T. Mahatdejkul-Meadows, L. Xu, L. Zhao, M. S. Dasika, A. Murarka, J. Lenihan, D. Eng, J. S. Leng, C.-L. Liu, J. W. Wenger, H. Jiang, L. Chao, P. Westfall, J. Lai, S. Ganesan, P. Jackson, R. Mans, D. Platt, C. D. Reeves, P. R. Saija, G. Wichmann, V. F. Holmes, K. Benjamin, P. W. Hill, T. S. Gardner and A. E. Tsong, Nature, 2016, 537, 694–697 CrossRef CAS PubMed.
- M. A. Rinaldi, C. A. Ferraz and N. S. Scrutton, Nat. Prod. Rep., 2022, 39, 90–118 RSC.
- K. Labun, T. G. Montague, M. Krause, Y. N. T. Cleuren, H. Tjeldnes and E. Valen, Nucleic Acids Res., 2019, 47, W171–W174 CrossRef CAS PubMed.
- C. Holkenbrink, M. I. Dam, K. R. Kildegaard, J. Beder, J. Dahlin, D. D. Belda and I. Borodina, Biotechnol. J., 2018, 13, e1700543 CrossRef PubMed.
- C. Verduyn, E. Postma, W. A. Scheffers and J. P. Vandijken, Yeast, 1992, 8, 501–517 CrossRef CAS PubMed.
- K. R. Kildegaard, B. Adiego-Perez, D. D. Belda, J. K. Khangura, C. Holkenbrink and I. Borodina, Synth. Syst. Biotechnol., 2017, 2, 287–294 CrossRef PubMed.
- K. Tamura, G. Stecher and S. Kumar, Mol. Biol. Evol., 2021, 38, 3022–3027 CrossRef CAS PubMed.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski, A. Zidek, A. Bridgland, A. Cowie, C. Meyer, A. Laydon, S. Velankar, G. J. Kleywegt, A. Bateman, R. Evans, A. Pritzel, M. Figurnov, O. Ronneberger, R. Bates, S. A. A. Kohl, A. Potapenko, A. J. Ballard, B. Romera-Paredes, S. Nikolov, R. Jain, E. Clancy, D. Reiman, S. Petersen, A. W. Senior, K. Kavukcuoglu, E. Birney, P. Kohli, J. Jumper and D. Hassabis, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. Jurcik, D. Bednar, J. Byska, S. M. Marques, K. Furmanova, L. Daniel, P. Kokkonen, J. Brezovsky, O. Strnad, J. Stourac, A. Pavelka, M. Manak, J. Damborsky and B. Kozlikova, Bioinformatics, 2018, 34, 3586–3588 CrossRef CAS PubMed.
- D. Tholl, Curr. Opin. Plant Biol., 2006, 9, 297–304 CrossRef CAS PubMed.
- N. G. H. Leferink and N. S. Scrutton, ChemBioChem, 2022, 23, e202100484 CAS.
- X. Chen, J. L. Zaro and W. C. Shen, Adv. Drug Delivery Rev., 2013, 65, 1357–1369 CrossRef CAS PubMed.
- M. Li, F. Hou, T. Wu, X. Jiang, F. Li, H. Liu, M. Xian and H. Zhang, Nat. Prod. Rep., 2020, 37, 80–99 RSC.
- X. Yang, K. Nambou, L. Wei and Q. Hua, Bioresour. Technol., 2016, 216, 1040–1048 CrossRef CAS PubMed.
- X. Cao, L.-J. Wei, J.-Y. Lin and Q. Hua, Bioresour. Technol., 2017, 245, 1641–1644 CrossRef CAS PubMed.
- L. R. R. Tramontin, K. R. Kildegaard, S. Sudarsan and I. Borodina, Microorganisms, 2019, 7, 472 CrossRef CAS PubMed.
- Z.-T. Zhu, M.-M. Du, B. Gao, X.-Y. Tao, M. Zhao, Y.-H. Ren, F.-Q. Wang and D.-Z. Wei, Metab. Eng., 2021, 68, 232–245 CrossRef CAS PubMed.
- K. Kuranda, J. Francois and G. Palamarczyk, FEMS Yeast Res., 2010, 10, 14–27 CrossRef CAS PubMed.
- Z.-J. Li, Y.-Z. Wang, L.-R. Wang, T.-Q. Shi, X.-M. Sun and H. Huang, J. Agric. Food Chem., 2021, 69, 2367–2381 CrossRef CAS PubMed.
- Y. Ma, W. Li, J. Mai, J. Wang, Y. Wei, R. Ledesma-Amaro and X.-J. Ji, Green Chem., 2021, 23, 780–787 RSC.
- P. Xu, K. Qiao, W. S. Ahn and G. Stephanopoulos, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10848–10853 CrossRef CAS PubMed.
- K. A. Markham, C. M. Palmer, M. Chwatko, J. M. Wagner, C. Murray, S. Vazquez, A. Swaminathan, I. Chakravarty, N. A. Lynd and H. S. Alper, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2096–2101 CrossRef CAS PubMed.
- Q. Guo, Y.-W. Li, F. Yan, K. Li, Y.-T. Wang, C. Ye, T.-Q. Shi and H. Huang, Biotechnol. Bioeng., 2022, 119, 2819–2830 CrossRef CAS PubMed.
- T. Shi, Y. Li, L. Zhu, Y. Tong, J. Yang, Y. Fang, M. Wang, J. Zhang, Y. Jiang and S. Yang, Biotechnol. J., 2021, 16, e2100097 CrossRef PubMed.
- M. Tai and G. Stephanopoulos, Metab. Eng., 2013, 15, 1–9 CrossRef CAS PubMed.
- A. Woods, M. R. Munday, J. Scott, X. L. Yang, M. Carlson and D. Carling, J. Biol. Chem., 1994, 269, 19509–19515 CrossRef CAS PubMed.
- M. Hunkeler, A. Hagmann, E. Stuttfeld, M. Chami, Y. Guri, H. Stahlberg and T. Maier, Nature, 2018, 558, 470–474 CrossRef CAS PubMed.
- E. J. Kerkhoven, K. R. Pomraning, S. E. Baker and J. Nielsen, npj Syst. Biol. Appl., 2016, 2, 16005 CrossRef CAS PubMed.
- J. Seip, R. Jackson, H. He, Q. Zhu and S.-P. Hong, Appl. Environ. Microbiol., 2013, 79, 7360–7370 CrossRef CAS PubMed.
- S. B. Ficarro, M. L. McCleland, P. T. Stukenberg, D. J. Burke, M. M. Ross, J. Shabanowitz, D. F. Hunt and F. M. White, Nat. Biotechnol., 2002, 20, 301–305 CrossRef CAS PubMed.
- S. Dale, W. A. Wilson, A. M. Edelman and D. G. Hardie, FEBS Lett., 1995, 361, 191–195 CrossRef CAS PubMed.
- S. Shi, Y. Chen, V. Siewers and J. Nielsen, mBio, 2014, 5, e01130–14 Search PubMed.
- T. Dulermo and J. M. Nicaud, Metab. Eng., 2011, 13, 482–491 CrossRef CAS PubMed.
- Tenagy, J. S. Park, R. Iwama, S. Kobayashi, A. Ohta, H. Horiuchi and R. Fukuda, FEMS Yeast Res., 2015, 15, fov031 CrossRef CAS PubMed.
- E. K. R. Hanko, C. M. Denby, V. Sanchez i Nogue, W. Lin, K. J. Ramirez, C. A. Singer, G. T. Beckham and J. D. Keasling, Metab. Eng., 2018, 48, 52–62 CrossRef CAS PubMed.
- L.-J. Wei, X. Cao, J.-J. Liu, S. Kwak, Y.-S. Jin, W. Wang and Q. Huaa, Appl. Environ. Microbiol., 2021, 87, e0048121 CrossRef PubMed.
- T. Dulermo, B. Treton, A. Beopoulos, A. P. K. Gnankon, R. Haddouche and J.-M. Nicaud, Biochim. Biophys. Acta, 2013, 1831, 1486–1495 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01661g |
| This journal is © The Royal Society of Chemistry 2023 |