
Defective nickel sulfide hierarchical structures for efficient electrochemical conversion of plastic waste to value-added chemicals and hydrogen fuel†
Zhijie
Chen
 a,
Wei
Wei
a,
Yansong
Shen
b and
Bing-Jie
Ni
*a
a,
Wei
Wei
a,
Yansong
Shen
b and
Bing-Jie
Ni
*a
aCentre for Technology in Water and Wastewater, School of Civil and Environmental Engineering, University of Technology Sydney, Sydney, NSW 2007, Australia. E-mail: bingjieni@gmail.com
bSchool of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia
First published on 4th July 2023
Abstract
Valorizing plastic waste into value-added chemicals/fuels in a green manner (e.g., electrochemical reforming) is a sustainable way to address the global pressing problem of plastic pollution. For the electro-reforming of plastic waste (e.g., polyethylene terephthalate (PET)), the development of highly active and selective catalysts remains a challenge. Herein, we have designed a low-cost and highly defective nickel sulfide to selectively convert ethylene glycol (PET monomer) into formate, with the co-production of hydrogen fuel. The optimal B and Co co-doped Ni3S2 (B,Co-NiS) only takes 1.341 V compared to the reversible hydrogen electrode (RHE) at 100 mA cm−2 for ethylene glycol oxidation, and it can realize high faradaic efficiency (>93%) and selectivity (>92%) for EG-to-formate conversion over a wide potential range. Furthermore, the bifunctional B,Co-NiS assisted real PET waste hydrolysate electrolysis in a membrane-electrode assembly (MEA) can generate 15.24 mmol formate per hour and attain H2 production efficiency over 70 times compared to conventional water electrolysis. Mechanism analyses indicate that the excellent catalytic performance of B,Co-NiS stems from the hierarchical structure, rich S defective sites, regulated electronic properties, and the dopant-facilitated in situ formation of an active sulfide@oxyhydroxide phase. This work provides an integrated nanostructure controlled-composition regulation strategy to design cost-effective catalysts for plastic electro-upcycling and paves the way for low-carbon plastic waste management.
1. Introduction
Plastic waste has become a global issue and poses a severe threat to the ecosystem. To date, about 80% of produced plastics turn into waste,1 leading to a large amount of organic and persistent pollutants.2,3 To address the troublesome issue of plastic waste, diverse techniques have been developed, including incineration, landfilling, gasification, pyrolysis, mechanical recycling, catalytic conversion, etc.4–6 Most of these methods require extreme conditions and generate multiple pollutants, and the repurposing of plastic waste under mild conditions remains a great challenge. Under these circumstances, electrochemical upcycling (electro-upcycling) of plastic waste (e.g., polyethylene terephthalate (PET)) has gained growing attention due to its eco-friendliness, full recovery feature, and value-added products (Scheme 1).7 In the electro-upcycling of PET, the typical monomer ethylene glycol (EG) can be selectively oxidized into high value formate/formic acid with the co-production of hydrogen fuel at the cathode. Moreover, the coupling of the EG oxidation reaction (EGOR) with the hydrogen evolution reaction (HER) can accomplish energy-saving H2 generation in comparison with the conventional water electrolyzers.7,8 Accordingly, it is highly suggested to convert PET waste into value-added hydrogen fuels and chemicals, which can not only turn waste into value-added products and achieve the circular economy goal, but also help resolve the pressing issue of plastic pollution around the world.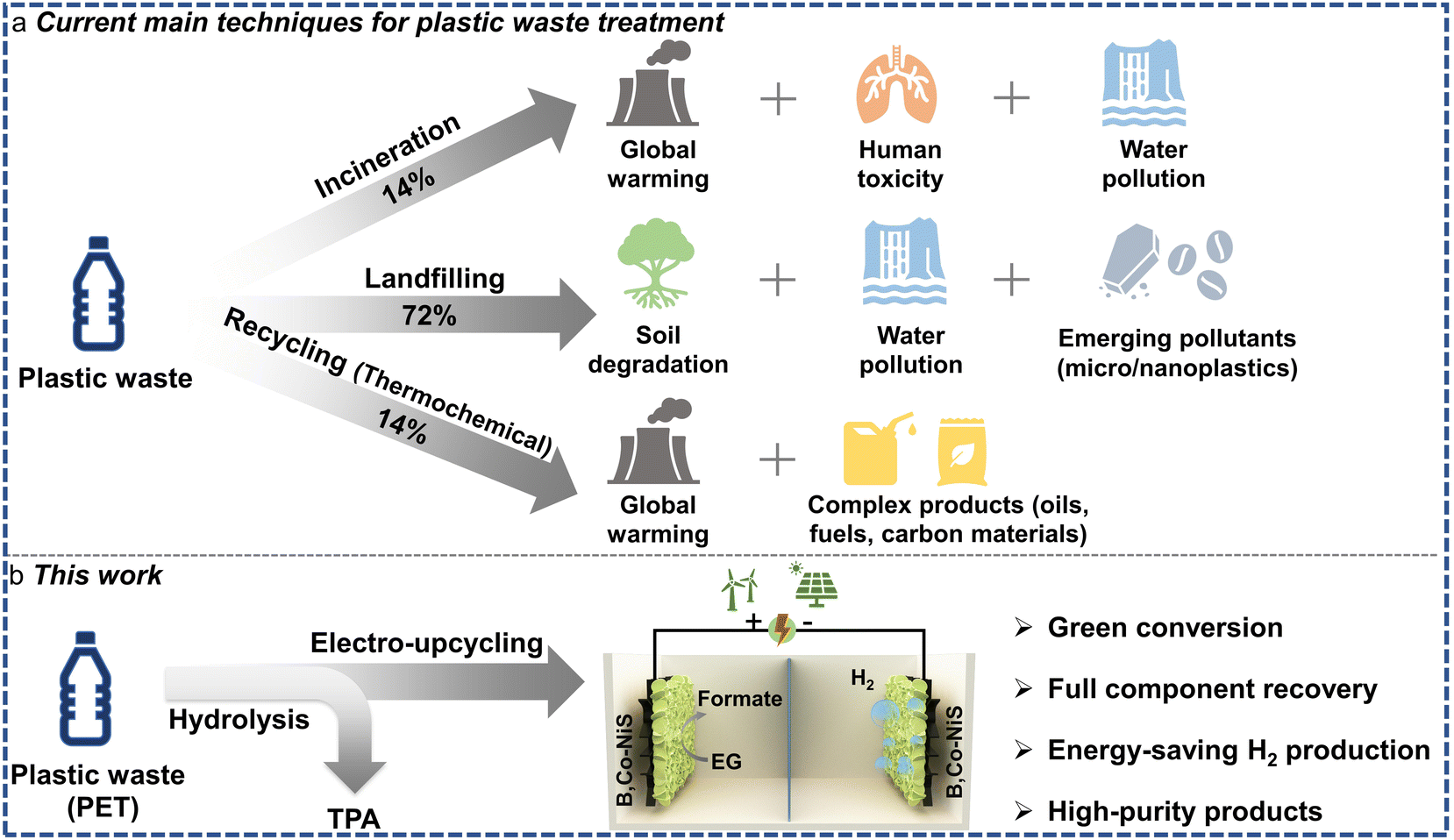 | ||
| Scheme 1 (a) Current techniques for plastic waste treatment and related environmental impacts. (b) Proposed electro-upcycling route for plastic waste (PET) valorization (TPA: terephthalic acid). | ||
To enhance electrolysis efficiency, developing cost-effective electrocatalysts for EG-involving electrolysis is an urgent mission. Although the development of HER electrocatalysts has achieved great progress,9–11 low-cost electrocatalysts for the EGOR are still limited and the catalytic performance needs further improvement. Several transition metal (TM)-based catalysts (e.g., oxides,8 nitrides,12 and phosphides7) have been designed for the formate product-targeted EGOR. However, the development of highly active, selective, and stable EGOR electrocatalysts remains a big challenge. TM sulfides, with good electronic/chemical properties, high Earth abundance, and eco-friendliness, display good electrocatalytic performance for diverse reactions, such as the HER, urea oxidation reaction, and biomass oxidation.13–15 In spite of this, the application of TM sulfides for the efficient EGOR is highly unexplored. Interestingly, previous studies found that TM-based electrocatalysts would undergo structural reconstruction during the EGOR process, and the in situ generated metal (oxy)hydroxides played a critical role in determining the catalytic activity.7,16,17 Hence, developing TM sulfides with favourable structural reconstruction by advanced strategies (e.g., doping, defect engineering, and nanostructure control) would significantly promote the EGOR process and improve the electro-reforming of plastic waste. Furthermore, unveiling the EG conversion mechanism over TM sulfides is of great interest for constructing novel electrocatalysts for plastic utilization, which remains unexplored.
Herein, we integrate defect engineering and nanostructure control with anion–cation co-doping to upgrade the electrocatalytic performance of nickel sulfide for selectively converting real plastic (PET) waste into hydrogen and formate. The B and Co co-doped defective nickel sulfide hierarchical structure synthesized by a two-step hydrothermal treatment–reduction method shows better performance toward PET electrolysis than its single-doped and undoped analogues. B,Co-NiS only needs 1.346 V compared to RHE to achieve 100 mA cm−2 for the EGOR, and it can realize high efficiency and selectivity (>90%) for EG-to-formate conversion at high current densities. The bifunctional B,Co-NiS driven real PET waste hydrolysate electrolysis in a membrane electrode assembly (MEA) can generate 15.24 mmol formate per hour and attain H2 production efficiency over 70 times compared to conventional water electrolysis. Further analyses indicate that the excellent catalytic properties of B,Co-NiS result from the defective hierarchical structure and the dopant facilitated formation of the sulfide@oxyhydroxide phase via in situ structure reconstruction. The noble metal-free nickel sulfide-based catalyst-mediated plastic upcycling technique demonstrated here would help to innovate further high-performance catalyst design and address the current pressing issue of plastic pollution.
2. Experimental
2.1. Fabrication of catalysts
2.2. Electrolysis of real plastics
Details of chemicals, material characterization, and electrochemical tests are given in the ESI.†
3. Results and discussion
3.1. Characterization of catalysts
The boron and cobalt dual-decorated defective nickel sulfide catalyst (B,Co-NiS) was synthesized by a two-step hydrothermal treatment–NaBH4 reduction process, as illustrated in Fig. S1 (ESI†). Using nickel foam as the substrate and nickel source, the hydrothermal process leads to the formation of nickel sulfides. In the NaBH4 reduction treatment, NaBH4 acts as the reductant and boron source. For comparison, undoped and single B or Co doped nickel sulfide catalysts were also synthesized. As depicted in Fig. 1a, all characteristic diffraction peaks in NiS catalyst's X-ray diffraction (XRD) pattern are well matched with the Ni3S2 phase (JCPDS no. 44-1418), except for the two strong peaks of the NF substrate (JCPDS no. 04-0850). Compared to bare NiS, the XRD patterns of B- or Co-doped NiS and the B, Co co-doped NiS catalysts exhibit no new peaks, implying that the doping of B and/or Co does not alter the main Ni3S2 phase. The morphological structure of the as-obtained B,Co-NiS was imaged by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). In Fig. 1b, c, and Fig. S2,† it can be seen that B,Co-NiS contains abundant interconnected ultrathin nanosheets (with a thickness of ∼20 nm) which self-assemble into ball-like microstructures (with an average diameters of 1–2 μm). The as-formed hierarchical B,Co-NiS catalyst with a large surface area and a highly open structure benefits (i) the enlargement of the electrochemically active surface area of the catalyst and (ii) the reactant migration and electron transfer processes,18–20 leading to improved electrocatalytic performance. The SEM images of NiS, Co-NiS, and B-NiS also suggest a similar hierarchical structure of these NiS-based catalysts (Fig. S3†), since the NiS, Co-NiS, and the NiS precursors of B-NiS/B,Co-NiS were hydrothermally synthesized under the same conditions (temperature and time). They also suggest that the B and/or Co doping processes have limited effects on the morphology of NiS. The images of B,Co-NiS in Fig. 1d and e further confirm the formation of a nanosheet aggregate-like structure with many distinctive nano-holes (with a size of about 1–3 nm) (Fig. S4a†) in nickel sulfide nanosheets. Compared with the TEM images of Co-NiS (Fig. S4b†), it can be found that these holes in B,Co-NiS should be generated during the NaBH4 reduction process. The abundant holes can help mass/charge transfer during electrocatalytic processes and accelerate the reaction kinetics. In Fig. 1f, the clear lattice fringes with interplanar distances of 0.237 and 0.203 nm are ascribed to the (003) and (202) planes of Ni3S2, respectively. Moreover, it is clear that many amorphous regions and defects co-exist with discontinuous lattice fringes, which leads to a high density of structural defects/grain boundaries and crystalline–amorphous interfaces, which would play a critical role in governing the ultimate electrocatalytic performance of B,Co-NiS. The formation of such rich defects and amorphous regions should be ascribed to the great reducing ability of NaBH4, which has shown strong potential in regulating the crystallinity and defect creating ability of the catalyst.21,22 To further identify NaBH4-induced structure evolution, electron paramagnetic resonance (EPR) spectra of Co-NiS and B,Co-NiS were collected. As shown in Fig. 1g, the much stronger signal of B,Co-NiS at g = 2.004 can be attributed to the capture of unpaired metal–S electrons in the presence of a higher concentration of sulfur vacancies in the B,Co-NiS lattice than that in its Co-NiS counterpart.23,24 The elemental mapping images in Fig. S5† show an even distribution of S (yellow), B (red), Ni (blue), and Co (green) elements in B,Co-NiS, indicating the successful introduction of B and Co elements.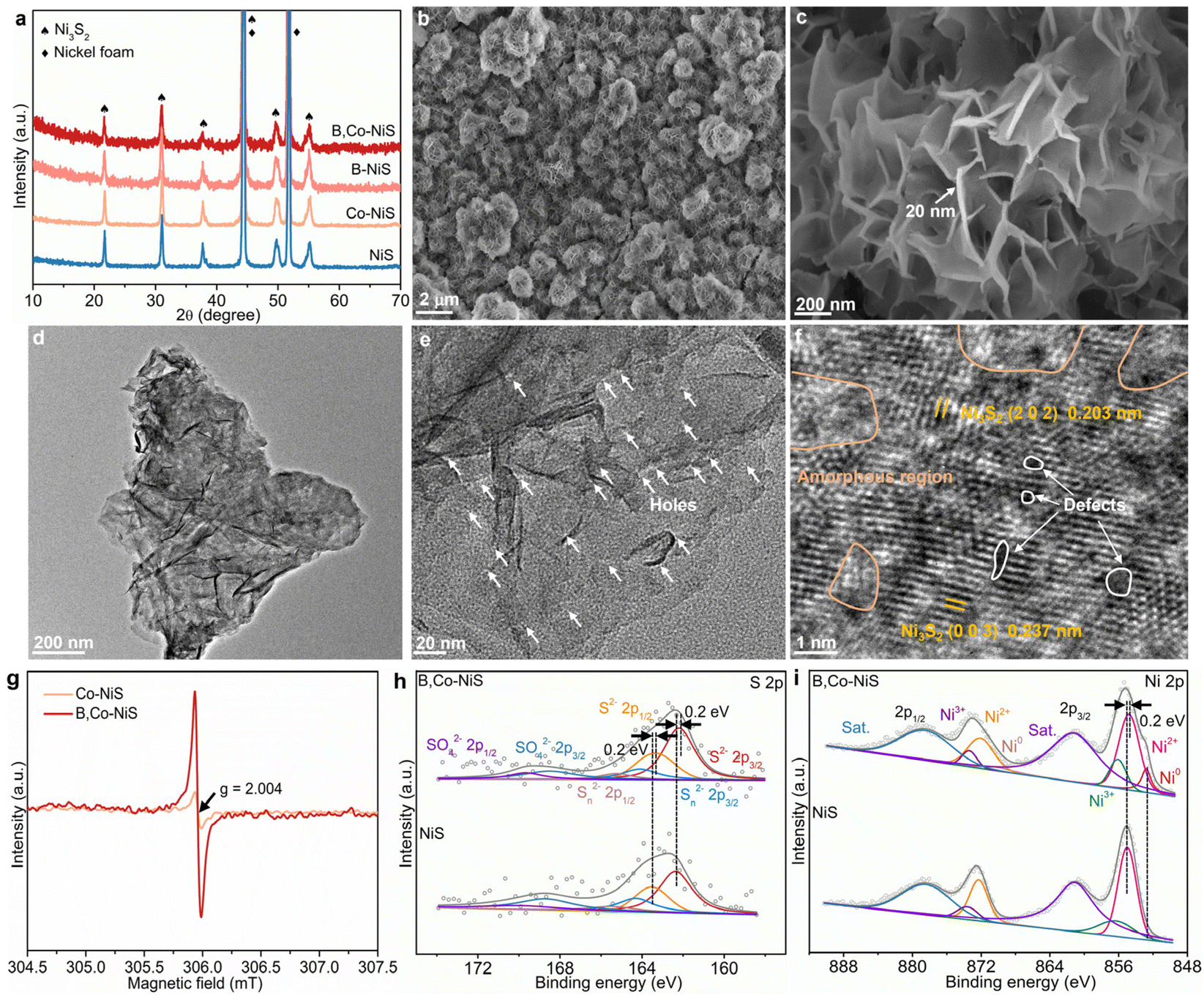 | ||
| Fig. 1 (a) XRD patterns of catalysts. (b and c) SEM images of B,Co-NiS. (d and e) TEM images of B,Co-NiS. (f) High-resolution TEM (HRTEM) image of B,Co-NiS. (g) EPR spectra of Co-NiS and B,Co-NiS. XPS spectra for (h) S 2p and (i) Ni 2p of B,Co-NiS and NiS. | ||
The surface compositions and chemical states of undoped NiS and B,Co-NiS were examined by X-ray photoelectron spectroscopy (XPS). In Fig. S6a.† the XPS survey spectra indicate the co-existence of Ni, S, B, and Co elements in B,Co-NiS and the co-existence of S and Ni elements in NiS. As shown in Fig. 1h, the S 2p spectrum of NiS can be fitted with six peaks. Peaks at 162.36 and 163.52 eV correspond to S 2p3/2 and S 2p1/2 of S2−, and two peaks located at 164.30 and 165.46 eV are assigned to S 2p3/2 and S 2p1/2 of Sn2−, respectively. Two other peaks in the higher energy region are indexed to SO42−.25 Compare with bare NiS, all peaks of S2−, Sn2−, and SO42− in the S 2p spectrum of B,Co-NiS show negative shifts (∼0.2 eV), suggesting increased electron density around S atoms due to the presence of B and Co dopants as well as the formation of S vacancies.26 In the Ni 2p spectrum of NiS, the distinct peaks at 855.0 and 872.3 eV can be indexed to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively (Fig. 1i). Two peaks at 856.3 and 873.6 eV are ascribed to Ni3+ 2p3/2 and Ni3+ 2p1/2, respectively, with two satellite peaks at 861.1 and 878.4 eV.27,28 Compared to NiS, the binding energies of Ni2+ 2p3/2 and Ni3+ 2p3/2 in B,Co-NiS show a negative shift, with the formation of a small ratio of Ni0, indicating an increased electron density around Ni atoms. The reason should be the formation of S vacancies,26 the electron transfer from B dopants to Ni atoms,29,30 and the coupling between Ni and Co atoms.31 The Co 2p spectrum of B,Co-NiS in Fig. S6b† contains six peaks, including Co 2p3/2 (777.60 eV) and Co 2p1/2 (792.59 eV) of Co3+, Co 2p3/2 (780.60 eV) and Co 2p1/2 (795.59 eV) of Co2+, and two satellite peaks at 785.50 and 800.49 eV.32,33 Moreover, in the B 1s spectrum of B,Co-NiS (Fig. S6c†), two characteristic peaks at 187.51 and 191.55 eV can be found, revealing the co-existence of both elemental and oxidized states of boron, respectively.34 The spectra of Co and B validate that both Co and B elements have been incorporated into Ni3S2 successfully, and Co and B dopants together with S defects can significantly regulate the electronic structure of Ni3S2.
3.2. Electrochemical EGOR performance of catalysts
Previous reports indicate that the EGOR is a thermodynamically more favourable reaction than the oxygen evolution reaction (OER) and the anode reaction of conventional water electrolysis. Thus, the coupling of the EGOR with the HER might lead to energy-saving hydrogen production at the cathode part of the electrolyzer. In this study, the catalytic performance of nickel sulfides for the OER and the EGOR are examined with a H-type cell. Aside from the prepared nickel sulfide catalysts, the purchased RuO2 catalyst and NF were also studied. The OER activities were tested in 1 M KOH. As illustrated in Fig. 2a, the nickel sulfide-based electrocatalysts exhibit better OER performance than the RuO2 catalyst and NF, and the B and Co dual-decorated NiS outperforms B or Co single-doped and pure NiS. At a fixed current density of 100 mA cm−2, B,Co-NiS takes a significantly smaller overpotential (η, 297 mV) than B-NiS (338 mV), Co-NiS (343 mV), NiS (367 mV), the RuO2 catalyst (410 mV), and bare NF (470 mV). The current density of the NF support is quite low, indicating that the electrocatalytic activity of as-prepared electrodes mainly comes from the nickel sulfide phase. Of note, B,Co-NiS shows a better OER activity (η10 = 239 mV) than the state-of-the-art TM electrocatalysts, such as CeO2–CoS1.97 (η10 = 264 mV),35 NH4CoPO4·H2O (η10 = 252 mV),36 and CoFeZr oxides (η10 = 248 mV),37 and more comparisons are shown in Table S1.†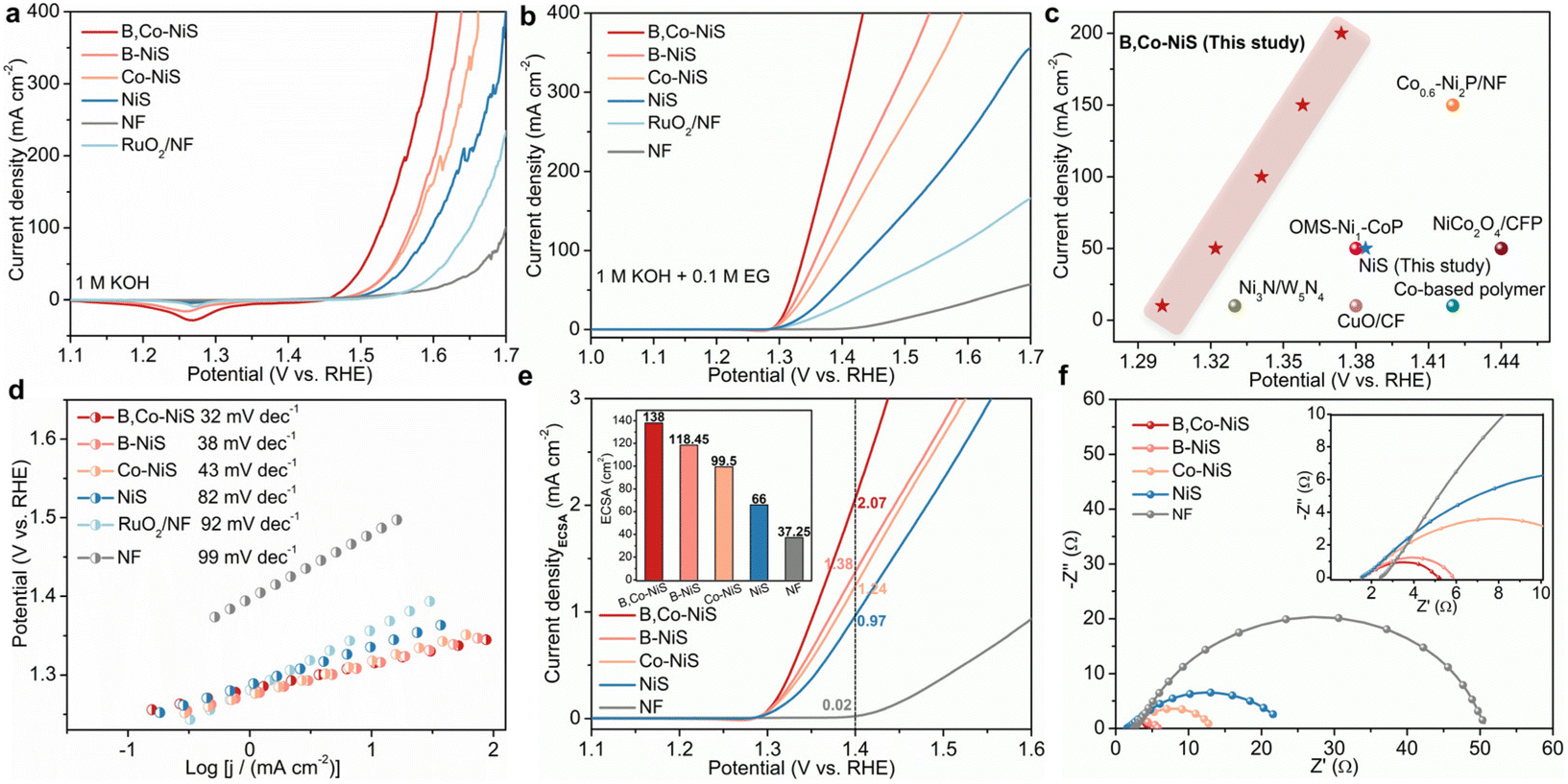 | ||
| Fig. 2 (a) OER linear sweep voltammetry (LSV) curves of catalysts. (b) EGOR LSV curves of catalysts. (c) Comparison of B,Co-NiS with state-of-the-art TM-based EGOR catalysts. (d) Tafel plots of electrocatalysts for the EGOR. (e) ECSA normalized EGOR LSV curves, and inset shows the calculated ECSA value of electrocatalysts. (f) EIS Nyquist plots of electrocatalysts, and inset shows an enlarged part of Nyquist plots. | ||
The EGOR performance of electrocatalysts was studied in 1 M KOH upon the addition of 0.1 M EG. LSV curves of catalysts are shown in Fig. 2b, and the tendency of EGOR activity of investigated samples is in line with the OER scheme. Compared with single B or Co doped and undoped nickel sulfide catalysts, B,Co-NiS exhibits better EGOR activities. At 100 mA cm−2, the needed potentials for B,Co-NiS, B-NiS, Co-NiS, NiS and the RuO2 catalyst are 1.341, 1.364, 1.384, 1.444, and 1.572 V compared to RHE respectively. Compared to the OER, all tested samples need a lower input energy at each current density for the EGOR. As for B,Co-NiS, necessitated potentials at 100, 200, and 300 mA cm−2 for the EGOR decreased by 187, 191, and 184 mV compared to those for the OER (Fig. S7†). In this context, the B,Co-NiS-mediated EGOR process is a favourable electrochemical oxidation reaction to replace the OER, in terms of energy saving for hydrogen generation. In addition, compared with recently reported EGOR catalysts, B,Co-NiS exhibits a better catalytic performance (Fig. 2c and Table S2†), indicating that sulfides are favourable candidates for the efficient EGOR. The electrochemical kinetics for the EGOR of catalysts were investigated through the LSV curve-derived Tafel plots (Fig. 2d). B,Co-NiS shows a lower Tafel slope (32 mV dec−1) than B-NiS (38 mV dec−1), Co-NiS (43 mV dec−1), NiS (82 mV dec−1), the RuO2 catalyst (92 mV dec−1), and NF (99 mV dec−1), suggesting efficient reaction kinetics over B,Co-NiS for the EGOR. Therefore, B,Co-NiS exhibits excellent thermodynamic and kinetic properties for the EGOR.
Electrochemically active surface area (ECSA) is an important parameter to quantify electroactive sites on catalysts. By conducting cyclic voltammetry (CV) scans (Fig. S8a–e†), electrochemical double-layer capacitances (Cdl) of nickel sulfide catalysts and NF were first obtained. As depicted in Fig. S8f,† the Cdl value of B,Co-NiS (5.52 mF cm−2) is higher than those of B-NiS (4.74 mF cm−2), Co-NiS (3.98 mF cm−2), NiS (2.64 mF cm−2), and NF (1.49 mF cm−2). Therefore, B,Co-NiS which has the largest ECSA (138 cm2) will ensure more active area/sites for the EGOR. Furthermore, the EGOR LSV curves were normalized with ECSA to exclude the impact of ECSA on the intrinsic catalytic activity of catalysts. As shown in Fig. 2e, B,Co-NiS can attain a high ECSA-normalized current density (jECSA, 2.07 mA cm−2) at 1.4 V compared to RHE, which is obviously larger than those of B-NiS (1.38 mA cm−2), Co-NiS (1.24 mA cm−2), NiS (0.97 mA cm−2), and NF (0.02 mA cm−2). Such results hint the large active surface area (rich active sites) and high intrinsic catalytic properties of B,Co-NiS collectively contributing to its excellent EGOR performance. Furthermore, B or Co single doping can upgrade the ECSA and intrinsic activity of bare nickel sulfide, which is further enhanced by the co-presence of B and Co dopants.
The charge transfer characteristics (charge-transfer resistance (Rct)) of electrocatalysts during the EGOR process were studied by electrochemical impedance spectroscopy (EIS). The Nyquist plots displayed in Fig. 2f were analyzed with an equivalent circuit model (Fig. S9†), and the results are shown in Table S3.† B,Co-NiS has a smaller Rct (3.72 Ω) than NiS (23.26 Ω), Co-NiS (12.47 Ω), B-NiS (4.53 Ω), and NF (50.43 Ω). Accordingly, when B,Co-NiS is used for the EGOR, efficient charge transfer can be achieved at the interface of the electrolyte and the catalyst. Both B and Co doping can enhance the charge transfer kinetics of nickel sulfide, which is further improved by B and Co co-doping.
Besides catalytic activities, the stability of B,Co-NiS for the EGOR is also measured. As shown in Fig. S10,† the catalytic activity can maintain at about 39% in the 12 h chronoamperometry (CA) test, with an initial current density of about 100 mA cm−2. Additionally, the LSV curve of B,Co-NiS after the CA test in the pristine 1 M KOH + 0.1 M EG solution is almost the same as the one before the CA test, implying that the catalytic activity of B,Co-NiS is well preserved in the CA test. Hence, the main reason for the notable decreased current density during the long-term CA test should be the reduced concentration of EG during the electrooxidation process.16 In general, B,Co-NiS is a highly promising EGOR electrocatalyst for its high ECSA, excellent intrinsic catalytic activity, low Rct, and good durability.
3.3. Investigation of the EG electrochemical conversion process
To figure out the conversion mechanism of EG, 1H and 13C NMR spectroscopy analyses were employed to analyze the products of EG electrooxidation qualitatively and quantitatively. As presented in Fig. 3a, formate is the main product with high faradaic efficiencies (FE, over 93%) ranging from 1.3 to 1.7 V compared to RHE, suggesting the high EG-to-formate conversion efficiency over B,Co-NiS. The highest FE (95.7%) and selectivity (94.3%, Fig. S11†) are obtained at 1.5 V compared to RHE, and further higher potentials result in slightly reduced FE and selectivity. This should be ascribed to the deep oxidation of formate or growing water oxidation at high potentials.38 The formate yields were also calculated, which roughly shows a positive correlation with the applied potential. The formate yield reaches 4.25 mmol cm−2 h−1 at 1.7 V compared to RHE. To gain insights into the dynamic conversion of EG during the oxidation process, input charge-dependent NMR spectra were recorded. As the electro-oxidation proceeds, the concentration of EG decreases gradually and the concentration of formate shows a continuous increase (Fig. 3b). When all EG has been oxidized, just a trace amount of glycolate (GA) can be identified during the oxidation process, which is further evidenced by the 1H and 13C NMR spectra (Fig. 3c and d). In the 1H NMR spectra, the peak of EG gradually weakens until it disappears, and the peak of formate becomes stronger, with the continuous electro-oxidation process. An enlarged part of the 1H NMR spectra in Fig. S12† shows the evolution of GA. It can be seen that a small peak in the chemical shift region of 3.80–3.85 ppm which can be indexed to GA generated first and then disappears. Therefore, a little amount of GA was generated by EG oxidation, and it was further converted to other chemicals as the oxidation process proceeded. The 13C NMR spectra of the post-EGOR electrolyte also suggest the production of formate and a small amount of carbonate. Combining the 1H and 13C NMR spectra, it can be concluded that the GA intermediate converts into formate and carbonate, and thus a possible pathway of the B,Co-NiS-mediated EGOR is recommended as shown in Fig. S13.† EG is first converted into glycolic aldehyde which is then oxidized into intermediates, i.e., GA and glyoxal. Second, the cleavage of C–C bonds in glyoxal generates formate, and the C–C bond cleavage in GA leads to formate and carbonate. Notably, only a little amount of GA and carbonate can be noticed in the whole EGOR, implying that GA is a minor intermediate in EG-to-formate conversion.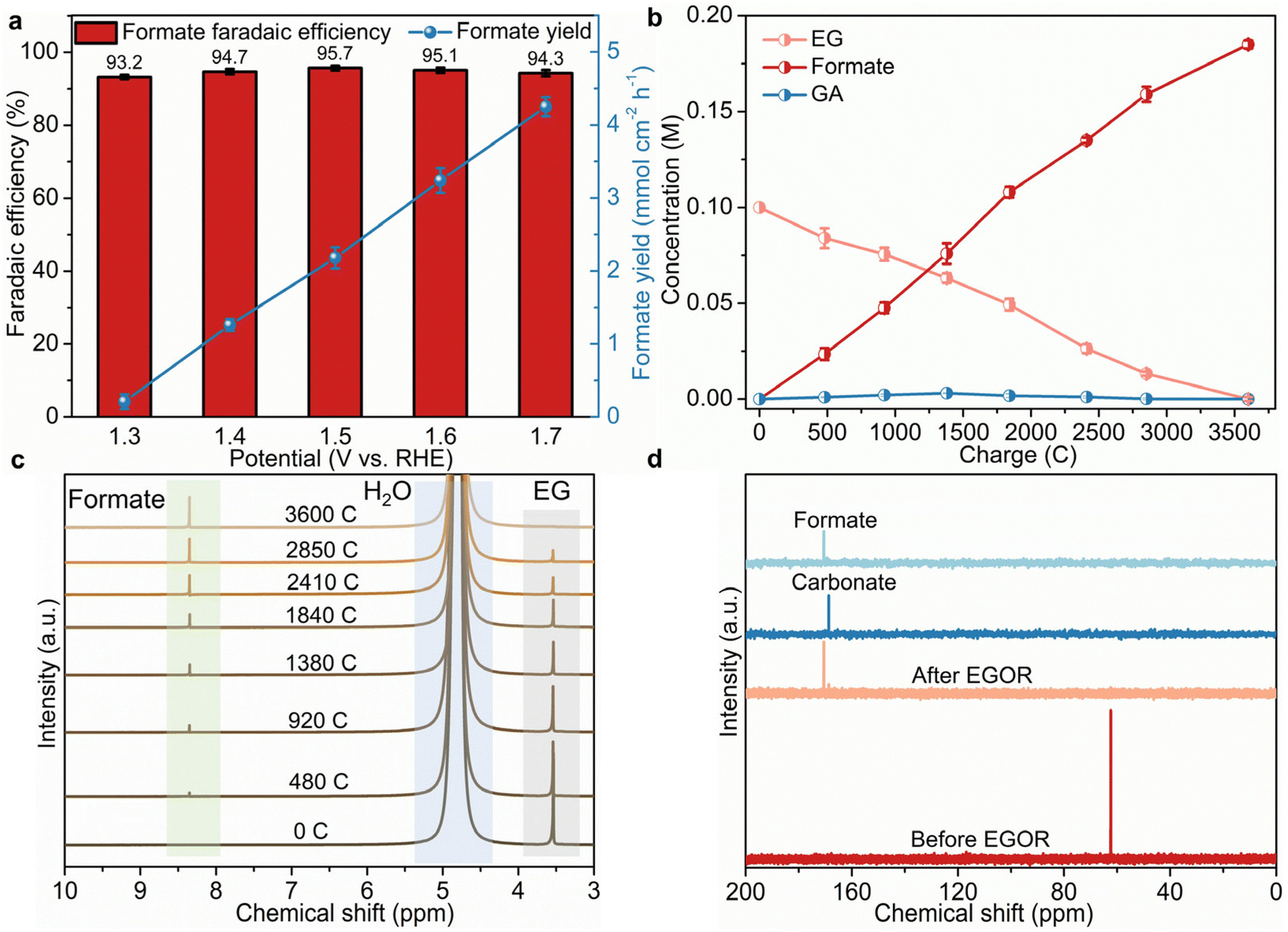 | ||
| Fig. 3 (a) faradaic efficiency and yield for formate production of B,Co-NiS from 1.3 to 1.7 V vs. RHE. (b) Charge-dependent concentration of reactants, intermediates, and products. (c) 1H NMR spectra of products during the EGOR process. (d) 13C NMR spectra of initial electrolytes and electrolytes after the EGOR. | ||
3.4. Origin of excellent EGOR performance of B,Co-NiS
To explore the origin of the high EGOR performance of B,Co-NiS, the post-reaction B,Co-NiS was thoroughly characterized using spectroscopic, microscopic, and analytical tools. The Raman spectra of the catalyst were first recorded. For the pristine B,Co-NiS, the Raman shifts at 184, 198, 223, 306, 325 and 351 cm−1 are in line with the typical vibrations of Ni3S2 (Fig. 4a).39,40 Maintaining the characteristic peak of Ni3S2, post-EGOR B,Co-NiS shows two new strong peaks at ∼478 and ∼556 cm−1 which can be indexed to the Ni–O bending and stretching vibrations of the NiOOH phase.41,42 Moreover, a broad peak at ∼1050 cm−1 is also generated after the EGOR process, which can be ascribed to the formation of NiOO−.43 Hence, B,Co-NiS undergoes structure reconstruction during the EGOR process, and the co-existing Ni3S2 and NiOOH phases contribute to the high EGOR performance. The structure evolution of B,Co-NiS is further confirmed by the TEM images. As shown in Fig. 4b, several crystalline regions closely covered by amorphous NiOOH layers are observed, with the formed abundant crystalline–amorphous phase boundary further boosting the catalytic performance.44,45 The HRTEM images shown in Fig. 4c indicate that the lattice fringes are still ascribed to Ni3S2, in line with the Raman spectra. In addition, defects in the discontinuous lattice fringes of Ni3S2 are well preserved (Fig. S14†), which might contribute to the high electrocatalytic performance of B,Co-NiS during the EGOR process. Therefore, the original B,Co-NiS is converted to the sulfide@oxyhydroxide structure under EGOR conditions.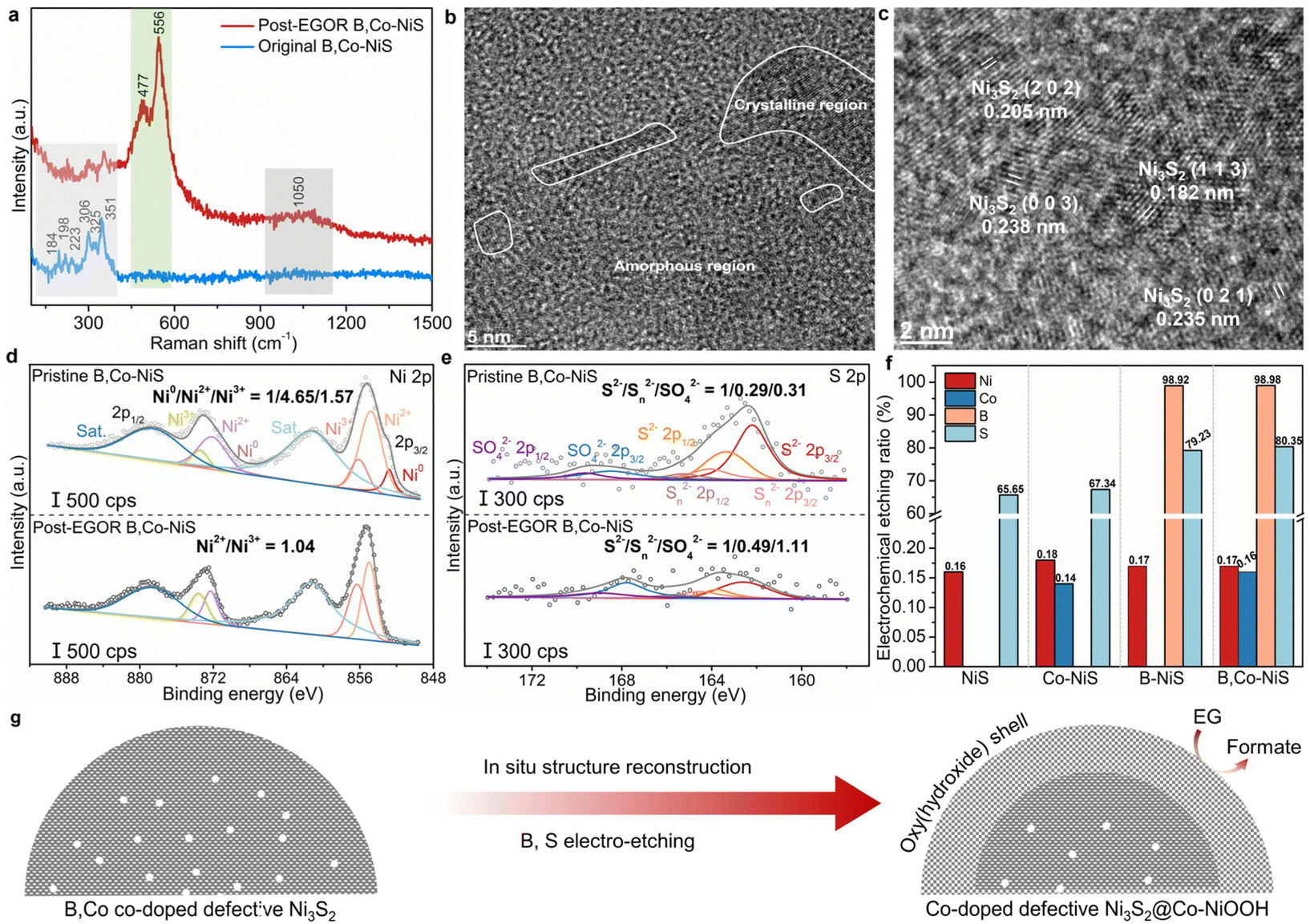 | ||
| Fig. 4 (a) Raman spectra of B,Co-NiS before and after the EGOR process. (b) TEM and (c) HRTEM images of B,Co-NiS after the EGOR process. XPS spectra for (d) Ni 2p and (e) S 2p of B,Co-NiS before and after the EGOR process. (f) Ion concentrations in electrolytes of different catalysts after the EGOR process. (g) Scheme of the structural reconstruction of B,Co-NiS during the EGOR. | ||
XPS spectra of post-EGOR B,Co-NiS were also collected to study the evolution of surface chemical properties. In the high-resolution Ni 2p spectra, the Ni0 species in the pristine catalyst have been totally converted into Ni2+/3+, and the Ni2+/Ni3+ ratio decreases from 2.96 (pristine B,Co-NiS) to 1.04 (post-EGOR B,Co-NiS) (Fig. 4d), suggesting that Ni has been significantly oxidized during the EGOR process and the main species in the post-EGOR B,Co-NiS are Ni2+/3+. Similarly, the Co 2p spectrum of the post-EGOR B,Co-NiS shown in Fig. S15a† also exhibits a higher proportion of high-valence Co3+, with the Co2+/Co3+ ratio reduced from 2.18 (pristine B,Co-NiS) to 0.78 (post-EGOR B,Co-NiS). The oxidation of metal species is accompanied by the oxidation of sulfur. In Fig. 4e, the S2−/Sn2−/SO42− ratio of pristine B,Co-NiS is 1/0.29/0.31, which changes to 1/0.49/1.11 in the post-EGOR sample. Aside from the growth in the high-valence S species, it is obvious that the content of the S element is significantly reduced. Also, the XPS spectra of B show that the signal of the B element is almost not detected in the post-EGOR B,Co-NiS (Fig. S15b†). The significant reduction of S and B elements in the post-EGOR B,Co-NiS is due to in situ electrochemical etching during the electro-oxidation process. In the O 1s spectra (Fig. S15c†), the peak belonging to lattice oxygen species of post-EGOR B,Co-NiS (530.4 eV) increases considerably compared to the pristine catalyst, further verifying the evolution of NiOOH on the catalyst surface.
The chemical composition of the catalyst also undergoes alteration during the EGOR. In Fig. 4f, the electrochemical etching ratios of metals (i.e., Co and Ni) are quite low (∼0.17%), suggesting the high stability of the catalyst framework. However, both S and B show high etching ratios, especially B. B,Co-NiS shows a higher S etching ratio of 80.35% than B-NiS (79.23%), Co-NiS (67.34%), and NiS (65.65%). The presence of the B dopant has a prominent effect on facilitating the dissolution of S during the EGOR process, which may be because the NaBH4 treatment induced more defects in B,Co-NiS. In addition, the dissolved B amounts of B-NiS and B,Co-NiS are 98.92% and 98.98% respectively, which implies the complete etching of B elements. The high electrochemical dissolution of B has also been reported in previous studies on the OER.46,47 The prominent co-etching of anions (S and B) contributes to more vacancies in the catalyst (Fig. S16†) and facilitates the in situ structure reconstruction for the formation of the electroactive Co-doped NiOOH phase. Overall, the defective B,Co-NiS undergoes structure evolution during the EGOR process and forms the stable and electroactive Co-doped defective Ni3S2@Co-doped NiOOH structure which accounts for the high EGOR performance of B,Co-NiS (Fig. 4g).
3.5. B,Co-NiS driven real plastic electro-upcycling
Considering the good EGOR performance of B,Co-NiS, a further attempt was made to demonstrate the energy-saving hydrogen production by real plastic electro-upcycling. First, the HER performance of catalysts was tested, and the LSV curves are depicted in Fig. S17a.† Surprisingly, B,Co-NiS shows good HER activity, with η10 = 86 mV. Although inferior to the commercial Pt/C catalyst, B,Co-NiS is better than other nickel sulfide-based analogues (Fig. S18†), as well as state-of-the-art TM-based catalysts (Table S4†). Additionally, HER kinetics is further analyzed by Tafel plots. B,Co-NiS shows a small Tafel slope (81 mV dec−1) (Fig. S17b†), suggesting favourable reaction kinetics of B,Co-NiS for the HER. The stability of B,Co-NiS for the HER was also tested via recording the chronoamperometric curve for 24 h. B,Co-NiS can maintain the current density for the 24 h test at ∼50 mA cm−2, with only minor (1.1%) activity loss (Fig. S19†). Overall, B,Co-NiS is a high-performance bifunctional catalyst for the EGOR and the HER, which holds great promise for real plastic electrolysis.The real PET waste (Fig. S20†) was first cleaned with water and hydrolyzed in KOH solution at 80 °C, and the PET hydrolysate was then used as the electrolyte. As suggested by previous studies, the EG monomer of PET can be selectively oxidized into formate while the TPA monomer will remain in the solution.7 The PET hydrolysate electrolysis was conducted in a self-made MEA flow electrolyzer, as illustrated in Fig. 5a and Fig. S21.† The bifunctional B,Co-NiS catalyzed PET hydrolysate electrolysis only takes low voltages of 1.491 and 1.538 V to achieve 100 and 200 mA cm−2, respectively, which are 0.258 and 0.346 V lower than those of water electrolysis (Fig. 5b). The stability of the PET hydrolysate electrolyzer was evaluated by recording the CA curve, with a starting current density of ∼300 mA cm−2. The current density shows negligible attenuation in the 48 h test, indicating highly stable operation of the system (Fig. S22†). The hydrogen production rate was further compared, and the PET hydrolysate electrolyzer can produce hydrogen gas at an average rate of 45.38 mmol h−1, which is over 70 times higher than the conventional water electrolyzer (Fig. 5c). In addition, the anode product yields were also calculated, and the PET hydrolysate electrolyzer can generate 15.24 mmol formate per hour while the water electrolyzer can generate 0.32 mmol oxygen gas in the same period. Moreover, the PET electrolyte was treated with H2SO4 solution after electrolysis to regenerate white-coloured pure TPA (Fig. S23†), and formate was recovered in the form of formic acid (FA) by a vacuum distillation process. For 100 g of PET waste, the low-cost nickel sulfide-assisted electro-reforming process can produce 39.1 g FA, 3.36 g H2, and 84.8 g TPA (Fig. 5d). Overall, the PET hydrolysate electrolyzer can not only realize energy-saving hydrogen production compared to conventional water electrolyzer but also produce value-added chemicals (formate and TPA) and contribute to the closed-loop utilization of plastic wastes.
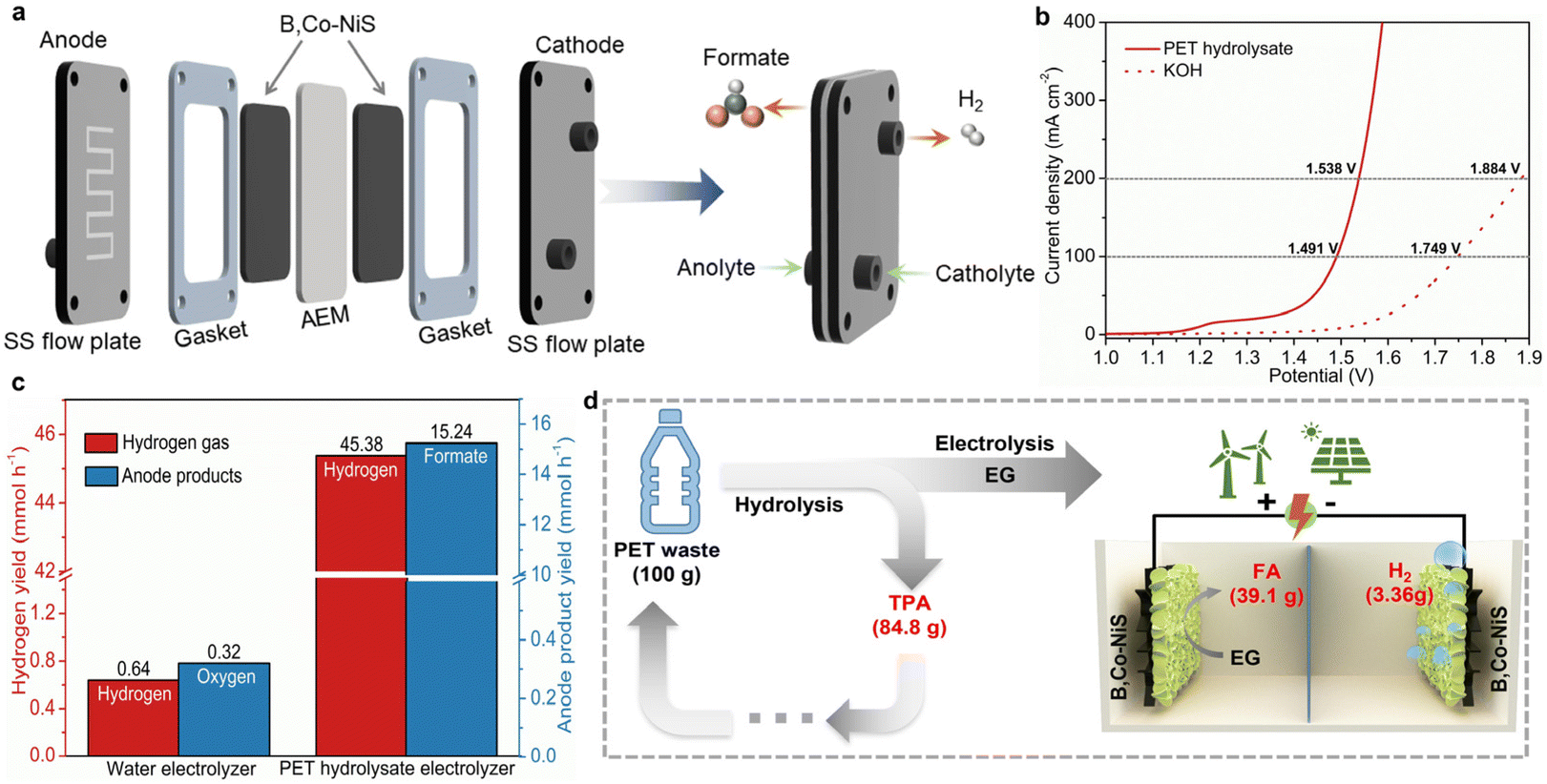 | ||
| Fig. 5 (a) The MEA setup for PET hydrolysate and water electrolysis. (b) LSV curves of the PET hydrolysate and water electrolysis. (c) Comparisons of hydrogen gas and anode product yields for PET hydrolysate and water electrolysis systems. (d) Schematic illustration of the PET electro-reforming process and mass flow of products. | ||
4. Conclusions
Developing cost-effective electrocatalysts for the selective and efficient conversion of plastic waste is the main mission to tackle the global plastic pollution problem. In this work, we demonstrate a high-performance and low-cost B,Co-NiS electrocatalyst for PET upcycling. Benefiting from the self-supported hierarchical structure and rich S defective sites, B,Co-NiS possesses abundant catalytically reactive sites and improves the mass/charge transfer process for electrochemical reactions. Furthermore, the presence of B and Co dopants regulates the electronic structure of the catalyst and accelerates the in situ structure reconstruction of B,Co-NiS during the EGOR process, which leads to the highly active and stable sulfide@oxyhydroxide phase. Taking advantage of the defective hierarchical structure and dopant regulation-induced high intrinsic activity, B,Co-NiS only needs 1.341 V compared to RHE to achieve 100 mA cm−2 for the EGOR, and it can realize high FE (>93%) and selectivity (>92%) for EG-to-formate conversion over a wide potential range. Moreover, bifunctional B,Co-NiS involved real PET waste hydrolysate electrolysis in the MEA reactor can generate formate at a high rate of 15.24 mmol h−1. Importantly, PET waste hydrolysate electrolysis can realize energy-saving hydrogen production via attaining an H2 production efficiency over 70 times compared to conventional water electrolysis. By converting PET plastic waste into value-added formate, TPA, and hydrogen fuel in a mild, selective, and low-carbon manner, the cost-effective catalyst-assisted plastic electro-upcycling practice shows great potential for the circular economy of plastics and holds promise for accelerating global hydrogen energy development.Author contributions
Zhijie Chen: experiments, conceptualization, discussion, and writing – original draft; Wei Wei: data analysis and discussion; Yansong Shen: data analysis and discussion; Bing-Jie Ni: discussion, writing – review & editing, and project acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the Australian Research Council (ARC) Discovery Project (DP220101139). Dr Wei Wei acknowledges the support from the Australian Research Council (ARC) through Project DE220100530.References
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- X. Shi, Z. Chen, X. Liu, W. Wei and B.-J. Ni, Sci. Total Environ., 2022, 157498 CrossRef CAS PubMed.
- T. Luo, X. Dai, Z. Chen, L. Wu, W. Wei, Q. Xu and B.-J. Ni, Water Res., 2023, 228, 119356 CrossRef CAS PubMed.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri and J. Dilworth, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- Z. Chen, W. Wei, B.-J. Ni and H. Chen, Environ. Funct. Mater., 2022, 1, 34–48 Search PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- F. Liu, X. Gao, R. Shi, C. Edmund and Y. Chen, Green Chem., 2022, 24, 6571–6577 RSC.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, Appl. Catal., B, 2021, 297, 120396 CrossRef CAS.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2023, 16, 4625–4633 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- B. Zhu, Z. Liang and R. Zou, Small, 2020, 16, e1906133 CrossRef PubMed.
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Appl. Catal., B, 2022, 307, 121209 CrossRef CAS.
- N. Wang, X. Li, M.-K. Hu, W. Wei, S.-H. Zhou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2022, 316, 121667 CrossRef CAS.
- Z. Chen, N. Han, R. Zheng, Z. Ren, W. Wei and B.-J. Ni, SusMat, 2023, 3(3), 290–319 CrossRef CAS.
- P. Kuang, M. Sayed, J. Fan, B. Cheng and J. Yu, Adv. Energy Mater., 2020, 10, 1903802 CrossRef CAS.
- C. C. Yang, S. F. Zai, Y. T. Zhou, L. Du and Q. Jiang, Adv. Funct. Mater., 2019, 29, 1901949 CrossRef.
- J. M. Kim, J.-H. Kim, J. Kim, Y. Lim, Y. Kim, A. Alam, J. Lee, H. Ju, H. C. Ham and J. Y. Kim, Adv. Mater., 2020, 32, 2002210 CrossRef CAS PubMed.
- Y. Wang, D. Jia, W. Zhang, G. Jia, H. Xie, W. Ye, G. Zhu and P. Gao, Chem. Commun., 2022, 58, 6132–6135 RSC.
- W. Lu, M. Yuan, J. Chen, J. Zhang, L. Kong, Z. Feng, X. Ma, J. Su and J. Zhan, Nano Res., 2021, 14, 3514–3522 CrossRef CAS.
- Y. Xu, M. Wang, Q. Xie, Y. Wang, X. Cui and L. Jiang, ACS Sustainable Chem. Eng., 2022, 10, 2938–2946 CrossRef CAS.
- H. Su, S. Song, Y. Gao, N. Li, Y. Fu, L. Ge, W. Song, J. Liu and T. Ma, Adv. Funct. Mater., 2022, 32, 2109731 CrossRef CAS.
- M. Liu, Z. Sun, C. Zhang, S. Li, C. He, Y. Liu and Z. Zhao, J. Mater. Chem. A, 2022, 10, 13410–13417 RSC.
- D. Li, L. Zhao, Q. Xia, J. Wang, X. Liu, H. Xu and S. Chou, Adv. Funct. Mater., 2022, 32, 2108153 CrossRef CAS.
- X. Yin, X. Dai, F. Nie, Z. Ren, Z. Yang, Y. Gan, B. Wu, Y. Cao and X. Zhang, Nanoscale, 2021, 13, 14156–14165 RSC.
- Z. Chen, W. Wei, W. Zou, J. Li, R. Zheng, W. Wei, B.-J. Ni and H. Chen, Green Chem., 2022, 44, 3208–3217 RSC.
- H. Yuan, S. Wei, B. Tang, Z. Ma, J. Li, M. Kundu and X. Wang, ChemSusChem, 2020, 13, 3662–3670 CrossRef CAS PubMed.
- J. M. V. Nsanzimana, R. Dangol, V. Reddu, S. Duo, Y. Peng, K. N. Dinh, Z. Huang, Q. Yan and X. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 846–855 CrossRef CAS PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. Von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- K. Chhetri, A. Muthurasu, B. Dahal, T. Kim, T. Mukhiya, S. H. Chae, T. H. Ko, Y. C. Choi and H. Y. Kim, Mater. Today Nano, 2022, 17, 100146 CrossRef CAS.
- Z. Chen, W. Zou, R. Zheng, W. Wei, W. Wei, B.-J. Ni and H. Chen, Green Chem., 2021, 23, 6538–6547 RSC.
- W. Liu, Y. Wang, K. Qi, Y. Wang, F. Wen and J. Wang, J. Alloys Compd., 2023, 933, 167789 CrossRef CAS.
- T. Dai, X. Zhang, M. Sun, B. Huang, N. Zhang, P. Da, R. Yang, Z. He, W. Wang and P. Xi, Adv. Mater., 2021, 33, 2102593 CrossRef CAS PubMed.
- Z. Song, K. Wang, Q. Sun, L. Zhang, J. Li, D. Li, P. W. Sze, Y. Liang, X. Sun and X. Z. Fu, Adv. Sci., 2021, 8, 2100498 CrossRef CAS PubMed.
- L. Huang, D. Chen, G. Luo, Y.-R. Lu, C. Chen, Y. Zou, C.-L. Dong, Y. Li and S. Wang, Adv. Mater., 2019, 31, 1901439 CrossRef PubMed.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622–627 CrossRef CAS PubMed.
- Z. Cheng, H. Abernathy and M. Liu, J. Phys. Chem. C, 2007, 111, 17997–18000 CrossRef CAS.
- Z. Wang, S. Shen, Z. Lin, W. Tao, Q. Zhang, F. Meng, L. Gu and W. Zhong, Adv. Funct. Mater., 2022, 32, 2112832 CrossRef CAS.
- Y. Jin, S. Huang, X. Yue, H. Du and P. K. Shen, ACS Catal., 2018, 8, 2359–2363 CrossRef CAS.
- Z. Chen, R. Zheng, H. Zou, R. Wang, C. Huang, W. Dai, W. Wei, L. Duan, B.-J. Ni and H. Chen, Chem. Eng. J., 2023, 465, 142684 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- H. Han, H. Choi, S. Mhin, Y.-R. Hong, K. M. Kim, J. Kwon, G. Ali, K. Y. Chung, M. Je, H. N. Umh, D.-H. Lim, K. Davey, S.-Z. Qiao, U. Paik and T. Song, Energy Environ. Sci., 2019, 12, 2443–2454 RSC.
- H. Xu, B. Fei, G. Cai, Y. Ha, J. Liu, H. Jia, J. Zhang, M. Liu and R. Wu, Adv. Energy Mater., 2020, 10, 1902714 CrossRef CAS.
- X. Wang, Y. Zuo, S. Horta, R. He, L. Yang, A. Ostovari Moghaddam, M. Ibáñez, X. Qi and A. Cabot, ACS Appl. Mater. Interfaces, 2022, 14, 48212–48219 CrossRef CAS PubMed.
- D. Wang, X. Feng, H. He, Z. Wang, R. Zheng, H. Sun, Y. Liu and C. Liu, New J. Chem., 2022, 46, 22989–22993 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01499a |
| This journal is © The Royal Society of Chemistry 2023 |