
Renewable lignin and its macromolecule derivatives: an emerging platform toward sustainable electrochemical energy storage
Xueru
Yang
,
Yufei
Zhang
,
Minghui
Ye
,
Yongchao
Tang
,
Zhipeng
Wen
,
Xiaoqing
Liu
* and
Cheng Chao
Li
 *
*
School of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China. E-mail: licc@gdut.edu.cn; liu@gdut.edu.cn
First published on 11th May 2023
Abstract
The conversion of natural renewable resources to high-value chemical products for electrochemical energy storage is becoming an effective measure to alleviate the energy crisis caused by the fossil shortage. As the second largest renewable biomass material in the world, lignin has been successfully utilized to construct sustainable energy storage devices (ESDs), both aqueous and organic ones. In this review, we provide a comprehensive overview of recent advances in the application of lignin-based/-derived macromolecules as key ESD components. A brief introduction to the origin and classification of lignin and its basic chemistry for electrochemical energy storage is first presented. The latest progress in lignin-based ESDs is then specifically elaborated on and discussed in detail from the perspective of organic electrodes, binders, electrolytes, and separators. Critical challenges and prospects in this area are put forward at the end, which is anticipated to shed light on the rational design of advanced lignin-based economic materials for sustainable development.
1. Introduction
Energy is a fundamental need for modern life and has become a key well-being issue in terms of economy, security, and the environment.1 Facing the environmental pollution from and cost escalation of conventional fossil fuels, sustainable alternative energies are needed, which thereby stimulates the exploration of renewable feedstocks such as wind and solar energy.2 Energy storage devices (ESDs), mainly including batteries and supercapacitors, play a critical role in the transition toward sustainable energy utilization.3 In rechargeable batteries, chemical transformations of the anode and cathode materials repeatedly take place. In the discharging course, electrons are transported from anode to cathode through the external circuit, and vice versa in the charging course.4 In capacitors, no chemical and phase changes are involved during charging and discharging. Typically, batteries have high storage capacity but low charging rates, and supercapacitors are the opposite. The trade-off between energy and power density originates from diffusion limitations in the electrolyte.5 Recent years have witnessed the research surge of advanced ESDs and considerable attention has been paid to the development of new energy storage materials.6 Biomass, biomaterials that integrate the advantageous properties of a low carbon footprint, natural abundance, low cost, and recyclability, is a very promising choice. Next-generation ESDs based on biomass, either for the fabrication or modification of the key components, are anticipated to improve energy independence and alleviate the pressures of climate change.7 To date, representative biomass materials, including cellulose, chitosan, lignin, soybean protein, gum, alginate, gelatin, etc., have been demonstrated to be capable of modifying the electrochemical performance of ESDs.8,91.1 Structure and properties of lignin
Lignin is the most abundant aromatic natural polymer on Earth, accounting for 15–30 wt% of biomass. In nature plants, lignin fills the gap between cellulose and hemicellulose, acting as a binder (Fig. 1a).10,11 It is a complex racemic aromatic hetero-polymer, mainly derived from three monolignols with different degrees of methoxylation: p-coumaryl, coniferyl, and sinapyl. These monomers produce p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units respectively (Fig. 1b). When bound to a lignin polymer, these three units are linked by β-O-4 (β-aryl ether), β–β, β-1, 5–5, 4-O-5 and β-5 to form a complex molecular structure (Fig. 1c). β-O-4, accounting for about 50%, is the most easily broken chemical bond.12 The contents of these three units vary in different plants, and exceptions to these bonding bonds can also be found in some specific cases.2,13 Therefore, the exact molecular weight of lignin is not a fixed value.14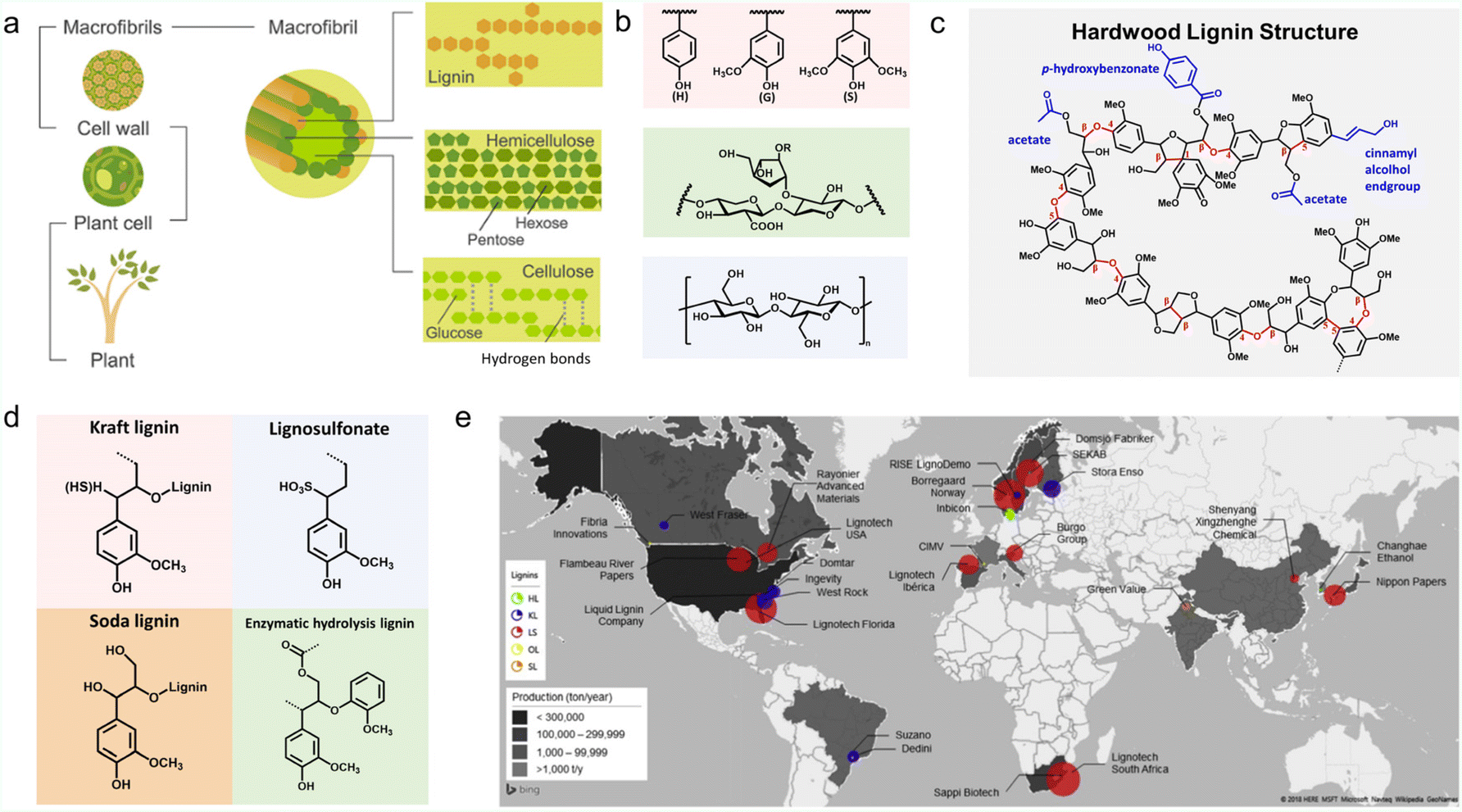 | ||
| Fig. 1 Properties and production of lignin. (a) Schematic structure of lignocellulose. Reproduced with permission.138 Copyright 2016, Wiley-VCH. (b) Schematic of the molecular structure of the lignin monomer units (pink box), hemicellulose (green box) and cellulose (blue box). Reproduced with permission.47 Copyright 2022, The Royal Society of Chemistry. (c) A proposed structural model for a hardwood lignin polymer. Reproduced with permission.62 Copyright 2020, The Royal Society of Chemistry. (d) Molecular structures of four most common industrial lignin derivatives. Reproduced with permission.47 Copyright 2022, The Royal Society of Chemistry. (e) Schematic of lignin production throughout the world. Reproduced with permission.34 Copyright 2022, The Royal Society of Chemistry. | ||
Lignin is the only phenolic polymer in plant resources, and displays hydrophobicity and thermal stability.15 The possession of massive oxygen-containing functional groups like methoxyl, hydroxyl, carboxyl, and ether endow it with many attractive features for energy storage.16 For example, the phenolic and alcoholic hydroxyl groups affect the hydrogen-bonding effect of the electrolyte, thus altering the energy storage behaviors of ESDs. The –SO3− groups can prevent the dissolution or induce the chemical bonding of polysulfides, holding great potential in addressing the notorious shuttle effect issue of lithium–sulfur batteries. Most importantly, for those containing phenolic groups (Ar-OH), two-electron-transfer-involved redox reactions (the conversion of the quinone/hydroquinone redox couple, denoted as Q/QH2 groups, Q + 2e− + 2H+ ⇌ QH2) can take place reversibly in the repeated charge/discharge process, and are capable of transporting charge carriers like Li+, K+, and Zn2+ with high efficiency.17
It has been proved that Q/QH2 are the major active sites for electrochemical energy storage for most lignin-based/-derived materials and their fraction strongly depends on the origin and post-processing courses of lignin.18 Namely, the electrochemical characteristic of lignin are highly correlated to its chemical structure, i.e. the G/H/S unit distribution. Generally, softwoods are G-lignins (with low H levels), and hardwoods are S/G-lignins (with very low H levels).19 Grass lignins are considered to have G/S/H units (with H units still comparatively minor) besides p-hydroxycinnamates. The p-coumarate of the latter acylated ∼10% of the γ-OH of the lignin monolignols.20 Note that, in G or S units, as the phenyl ring is 4-hydroxy-3-methoxy-(guaiacyl-) or 4-hydroxy-3,5-dimethoxy-(syringil-) substituted,21 they can function as active sites for charge storage once demethylated.18 Yet the phenol groups in H units and p-coumarate are methoxy-free and cannot be converted to redox-active quinones.22 Given the redox inactive feature of H units and p-coumarate, lignins possessing more S and G units have more redox-active Q/QH2 sites and are more appropriate for energy storage. Notably, any catechol, pyrogallol or 3-methoxycatechol is considered as an active redox group when referring to Q/QH2-type sites in lignin-based/derived materials.
Because the redox activity is dependent on the phenolic moieties of different degrees of substitution, electrochemical activation, or additional pre-treatment (electro-oxidative depolymerization or hydrothermal liquefaction),19,21–27 for the purpose of enriching the phenolic sites for charge storage capacity, is essential to realize the demethylation and cleavage of the aryl ether bonds of lignins.19,23 As a proof of concept, the Ar-OH content of alkali lignin (AL) could be increased by 1.9 times after 4 h stirring in LiBr solution at 80 °C, which is produced by the effective breakage of aryl ether bonds, especially methyl aryl ether bonds.27 A similar effect can also be achieved by applying an appropriate voltage or adding reactive additives.
1.2 Classification of lignin
At the industrial level, a series of technical lignins have been successfully derived from papermaking waste liquors or biorefinery by-products. They can be roughly classified into sulfur-containing types (kraft lignin (KL) and lignosulfonate (LS)) and sulfur-free types (soda lignin (SL) and enzymatic hydrolyzed lignin (EHL)) etc.6 Their molecular formulas can be found in Fig. 1d.LS is extracted from the liquid waste of sulfite pulping (Na2SO3, MgSO3, CaSO3) and is soluble in aqueous solutions due to its rich sulfonic acid groups on the side chain.16 Using nickel compound catalysts at 200 °C, LS could be transformed into G units with more than 60% conversion and 75–90% selectivity.28 KL and SL are two subtypes of AL extracted from the black liquor produced by the alkaline pulping process,29 in which an alkaline catalyst is utilized to deconstruct the lignin's β-O-4 bond to enrich the phenolic groups.28 For instance, KL can be obtained from the waste liquid of sulphate pulping by treating wood with NaOH and NaHS solutions in the temperature range of 150–170 °C.30 Unlike LS, KL is insoluble in water, but it can also be wetted by water due to the presence of hydrophilic functional groups such as hydroxyl, phenol, and carboxylic acids.31 Notably, hardwood species are easier to fractionate than softwood species. KL derived from hardwood is more condensed with β-O-4 bonds than is softwood, which promises a higher concentration of S-type mono-phenolic group than native lignin by depolymerization.28 SL is obtained from the soda pulping process, which has a lower environmental impact than other lignin extraction methods, such as kraft and organosolv. The main source of SL is non-wood, compared with wood-derived lignin, which has a higher content of mono-phenolic groups.32 The sulfur-free characteristic makes SL poorly soluble in water.12,28,33 EHL is mainly produced by the biorefinery industry, deriving from enzymatic pretreatment concepts of lignin hydrolysis.32,34 The resource of EHL could be woody or non-woody.35,36 For example, the transformation of cellulose and hemicellulose from agricultural straw into bioethanol would leave behind a large number of EHL by-products. EHL is closer to primary lignin in terms of chemical composition. It cannot be dissolved in water and alkali but possesses high solubility and good compatibility in many organic solvents with strong hydrogen-donating characteristics, such as methanol, ethanol, tetralin, and formic acid.28,35,37 In comparison with other lignins, N and O atoms could be doped into the carbon skeleton of EHL during biological fermentation, which is favorable for electrochemical energy storage.38
The worldwide production distribution of different kinds of lignin is presented in Fig. 1e. The circled areas are proportional to the amount of lignin produced by the company and the colors correspond to the types of lignin. It can be seen that LS dominates the market, seeing a high production of ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 tons per year, with a market value of 180–500 USD per ton.16 The worldwide production capacity is followed by KL at around 265000 tons per year,39 with a price of 260–500 USD market value.34,39 SL has around 5000–10000 tons of production per year, with a price of 200–300 USD per ton.32 EHL has a production capacity of 100 tons per year.39 Produced from forest/agriculture waste, the very high purity EHL from TMP-BIO™ technology is predicted to have a market value of 1.12 billion by the year 2027.32,34 It is suitable for laboratory depolymerization research and carbon nanofiber synthesis.40 Using low-purity lignin as raw materials, other high-value technical lignin-derived products like benzene (1100–1300 USD per ton), toluene (1150–1300 USD per ton), xylene (865–1100 USD per ton), aromatic monomers (∼2160 USD per ton), and vanillin (∼16200 USD per ton) have great potential in valorization.40,41 For practical application, the cost budget from key technologies such as electro-oxidative depolymerization or hydrothermal liquefaction has to be taken into consideration.42,43 Currently, the application of lignin in ESDs represents an emerging platform toward sustainable electrochemical energy storage and is still at the laboratory level.
000000 tons per year, with a market value of 180–500 USD per ton.16 The worldwide production capacity is followed by KL at around 265000 tons per year,39 with a price of 260–500 USD market value.34,39 SL has around 5000–10000 tons of production per year, with a price of 200–300 USD per ton.32 EHL has a production capacity of 100 tons per year.39 Produced from forest/agriculture waste, the very high purity EHL from TMP-BIO™ technology is predicted to have a market value of 1.12 billion by the year 2027.32,34 It is suitable for laboratory depolymerization research and carbon nanofiber synthesis.40 Using low-purity lignin as raw materials, other high-value technical lignin-derived products like benzene (1100–1300 USD per ton), toluene (1150–1300 USD per ton), xylene (865–1100 USD per ton), aromatic monomers (∼2160 USD per ton), and vanillin (∼16200 USD per ton) have great potential in valorization.40,41 For practical application, the cost budget from key technologies such as electro-oxidative depolymerization or hydrothermal liquefaction has to be taken into consideration.42,43 Currently, the application of lignin in ESDs represents an emerging platform toward sustainable electrochemical energy storage and is still at the laboratory level.
1.3 Application of lignin in ESDs
Profiting from its non-toxic and renewable nature, both for its preparation and potential applications, the strategic conversion of lignin to high-value chemical products is probably a promising strategy to alleviate environmental pollution.16,44 Encouragingly, in recent years, successive achievements have been made in ESD R&D based on lignin-involved or -derived materials, both macromolecular composite and carbonaceous matrix.6Lignin is an ideal precursor for carbonization, originating from its high carbon content, and pyrolysis treatment is a common way to achieve this goal. Generally, at temperatures below 200 °C, dehydration occurs. At the range of 200–450 °C, the unstable β-O-4 linkages are broken and heteroatoms are eliminated. At higher temperatures, functional groups, oxygen groups, and redox-active sites decompose, while carbon material emerges.45 So far, graphite-like, porous, or foreign element-doped carbons (N, S, P) have been reported by using lignin as raw materials. However, the oxygen redox-active functional groups (Q/QH2 sites) diminish when lignin is carbonized under high temperatures and an inert atmosphere.46 Therefore, unlike in lignin-based materials of which the redox-active sites are confined at the surface, the bulk structure and physiochemical properties (structure, mechanical properties, and electrical conductivity) are more important for enhancing the capacity of lignin-derived carbons. In this context, great attention has been paid to tailoring these properties by varying the pyrolysis parameters or post-treatment (chemical activation, template methods, physical activation, etc.45), for the sake of regulating the ion kinetics, electron transport, and structural stabilization of ESDs. Since some recent reviews have summarized in detail the application progress of lignin-based carbonaceous matrix in ESDs, herein we only discuss the characteristics, pros and cons, and developments of composite macromolecules, either lignin-based or lignin-derived ones.6,16,29,44,47 The former refer to the physical mixture of lignin with other reagents, which well maintains the functional groups of lignin itself without changing the overall chemical properties. The latter experience substantial chemical modification to achieve specific functionalization and their overall properties substantially differ from those of lignin. Major ways to functionalize lignin towards macromolecules include: (i) grafting copolymerization; (ii) hydrogen bonding, and (iii) electrosynthesis, as presented in Fig. 2. In this way, some specific physiochemical properties (ionic conductivity, structural stability, and chemical reaction mechanism, etc.) can be precisely adjusted or specifically introduced by grafting special functional groups onto the hydroxyl groups or tuning the hydrogen bonding, complexation or π–π stacking effects. However, some inherent deficiencies of the lignin-based macromolecular composite are inevitable, like the difficulty in controlling their molecular structure and poor electrical conductivity, leaving a large space for their performance optimization.2
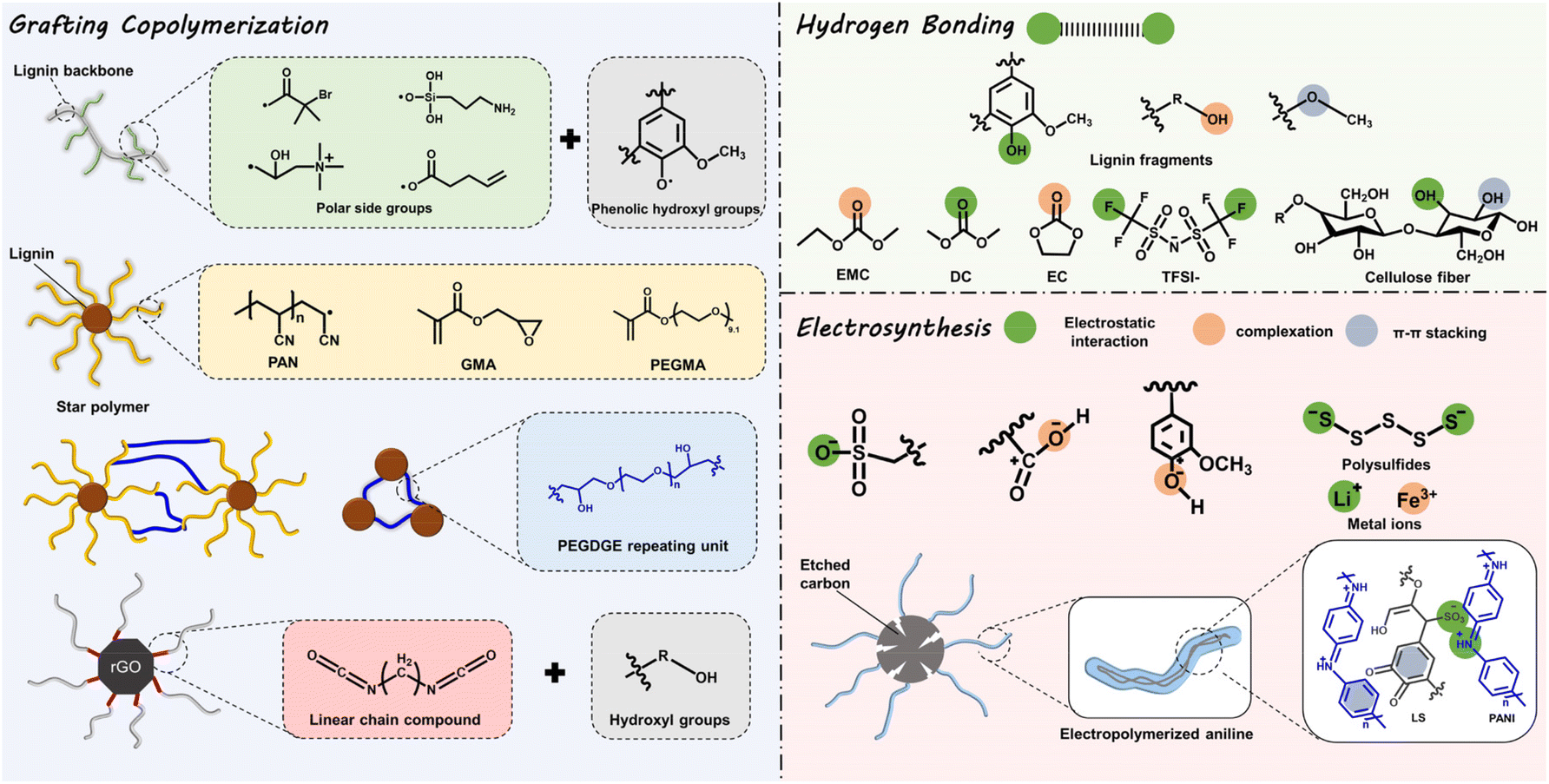 | ||
| Fig. 2 Schematic structure diagram of the functionalization of lignin polymer. | ||
Overall, lignin and its derived macromolecules see a bright future for large-scale application in constructing advanced ESDs in view of their remarkable unique advantages like low cost, ease of synthesis, and potential for mass production. In the past decades, comprehensive research work has been carried out and concentrated on rechargeable batteries and supercapacitors. In this review, we summarize the recent progress of lignin-based and -derived materials functioning as fundamental elements of these two systems including electrodes, binders, electrolytes, and separators (Fig. 3). Some key contributions in this field are discussed in detail and the structure–performance relationship is highlighted. Insights into the current challenges and probable development directions of lignin-based/-derived energy storage materials are proposed in the end.
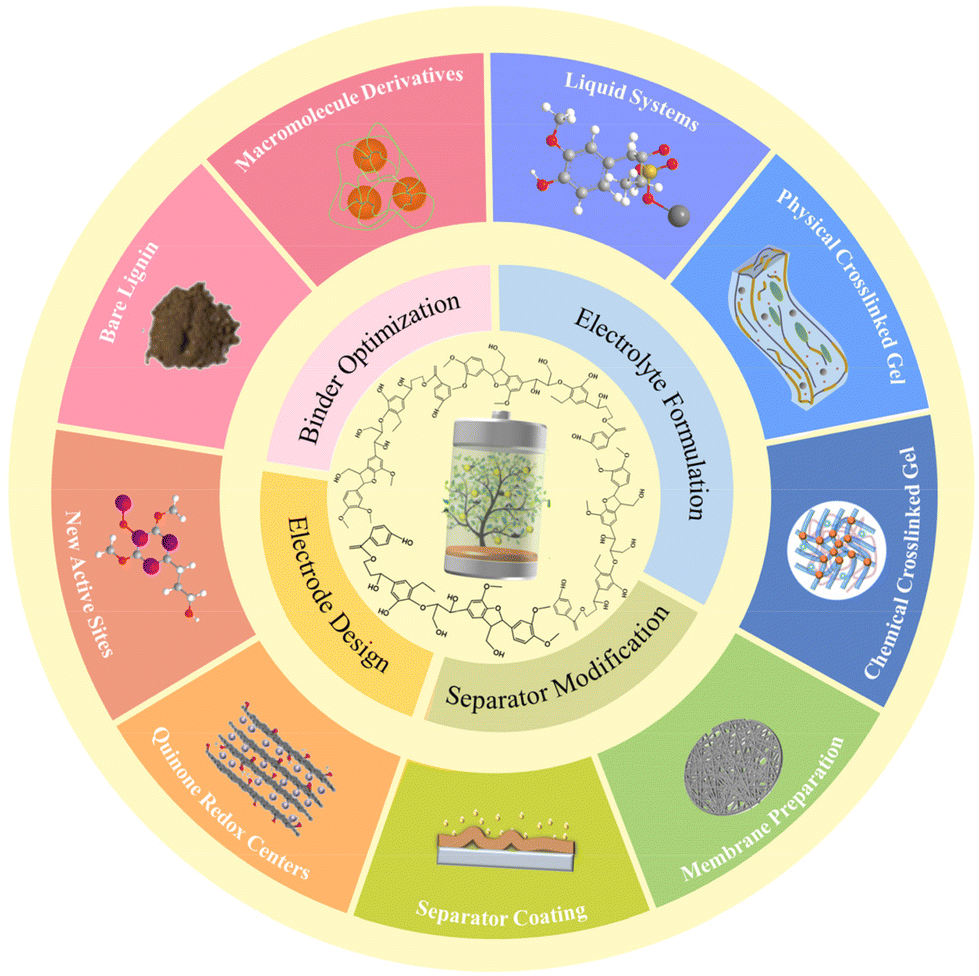 | ||
| Fig. 3 Overview of representative lignin-based/-derived macromolecules for energy storage device components. | ||
2. Lignin-based/-derived organic electrodes
Lignin could be used as an electrode material for both supercapacitors and batteries.10,48 It belongs to n-type organic materials of which the electrochemical transformation takes place between its neutral state and negatively charged state. As technical lignin is more stable in an acidic environment, most reported devices are mainly built under acidic conditions. The main active sites of lignin-derived organic electrodes are Q/QH2 functional groups that can afford two-electron-mediated reversible redox processes during the charge/discharge courses.4,49,502.1. Organic electrodes for supercapacitors
Considering their great potential to achieve a high power density and high redox potential, lignin-based materials had been widely used as organic electrodes for supercapacitors.51 The electrochemical performance of various recently-reported lignin-involved electrodes with different characteristics for supercapacitors were summarized in Table 1. In general, the charge storage form and relevant redox reaction of the electrode materials determine the energy storage mechanism of supercapacitors. Depending on the electric double-layer capacitance and pseudocapacitance behaviors, the corresponding devices can be classified into electric double-layer capacitors (EDLCs) and pseudocapacitors, respectively. As this paper does not refer to the progress of lignin-derived carbon materials, the subsequent devices based on Q/QH2 conversion are most pseudocapacitors, of which the charge/discharge profiles show both non-faradaic and faradaic contributions.| Cathode | Anode | Electrolyte components | Potential (V) | Specific capacitance (F g−1) | Capacity retention | Energy density (W h kg−1) | Power density (kW kg−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| PANI-LS | PANI-LS | PVA-H2SO4 | −0.2–0.8 | 1200 (1 A g−1) | 87% (15000 cycles) |
21.2 | 26 | 52 |
| LS/SWCNTHNO3 | LS/SWCNTHNO3 | Cellulose-Li2SO4 hydrogel | 0–1.0 | 156 (20 A g−1) | 80.1% (10000 cycles) |
17.1 | 0.32 | 54 |
| Ti3C2Tx/HCNF@Lig (3@1)97/3 | Pt | 1 M H2SO4 | 0–0.75 | 273 (0.5 A g−1) | 87.6% (10000 cycles) |
633.1 W L−1 | 16.2 W h L−1 | 55 |
| PAAm-Ag-lignin NPs-Tar-CNT | PAAm-Ag-lignin NPs-Tar-CNT | PVA/H3PO4 gel | 0–0.8 | 298.6 (10 mV s−1) | 89.9% (10000 cycles) |
13.7 | 0.2 | 58 |
| OKL/TAC | Pt | 1 M H2SO4 | 0–0.8 | 390 (0.5 A g−1) | 99.7% (1000 cycles) | N/A | N/A | 61 |
| rGO/SP | Pt | 0.1 M HClO4 | 0.2–0.8 | 365 (10 A g−1) | 88% (200 cycles) | N/A | N/A | 62 |
| rGO/SP-AOF | Pt | 0.1 M HClO4 | −2.0–2.0 | 210 (10 A g−1) | N/A | N/A | N/A | 50 |
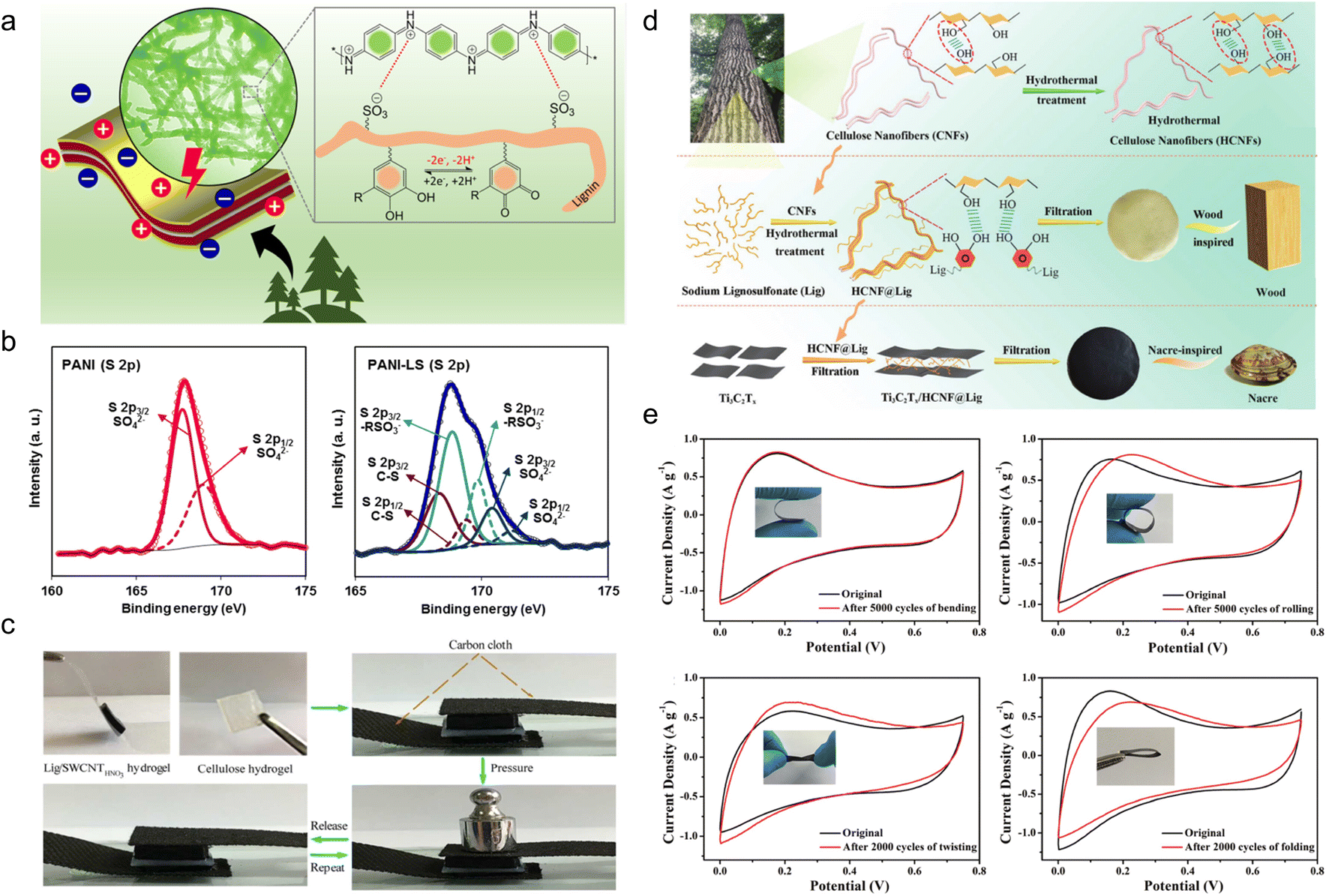 | ||
| Fig. 4 Lignin-based organic electrodes for supercapacitors. (a) Schematic illustration of the electrochemical activity of LS moieties as a dopant and a structure-directing agent for the PANI film and the redox activities of LS by forming catechol moieties in the PANI-LS nanocomposite. (b) Deconvoluted core level XPS spectra of the PANI electrode (S 2p) and PANI-LS electrode (S 2p). Reproduced with permission.52 Copyright 2021, The Royal Society of Chemistry. (c) Real-time optical images of the compressible supercapacitor showing a compressing and recovering process. Reproduced with permission.54 Copyright 2018, American Chemical Society. (d) Schematic illustration of the synthesis of HCNFs (top), wood-inspired HCNF@Lig nanofibers and composite films (middle), and nacre-inspired Ti3C2Tx/HCNF@Lig composite films (bottom). Reproduced with permission.55 Copyright 2021, The Royal Society of Chemistry. (e) CV curves of Ti3C2Tx/HCNF@Lig (3@1)97/3 FSC before and after bending (upper left), rolling (upper right), twisting (lower left) and folding (lower right) at a scan rate of 10 mV s−1. (Ti3C2Tx/HCNF@Lig (3@1)97/3: HCNF@Lig composite in a 3/1 mass ratio and Ti3C2Tx/HCNF@Lig (3@1) composite in a 97/3 mass ratio). Reproduced with permission.31 Copyright 2021, The Royal Society of Chemistry. | ||
In spite of electrosynthesis, conventional hydrothermal methods are also wise choices for the preparation of conductive lignin-based organic electrodes as they can introduce carbon materials to lignin via hydrogen bonding or π–π stacking interactions. For example, Peng et al.54 synthesized LS/SWCNTsHNO3 pressure-sensitive hydrogel electrodes by hydrothermal treatment at 180 °C with an HNO3 additive. Acid pretreatment endowed single-wall carbon nanotubes (SWCNT) with abundant hydroxyl and carboxyl groups tidily incorporated with nitrated LS, which constructed a three-dimensional (3D) porous complex structure with better conductivity and improved mechanical properties. In this case, the lignin content is 16 times higher than for SWCNTs, implying its great contribution to energy storage. The symmetric flexible all-solid-state supercapacitor (FSC) device delivered excellent compressible ability (Fig. 4c), durability of only 2% capacity loss after 1000 times of 90° bending cycles, high capacity of 131 F g−1 at 50 A g−1, power density of 324 W kg−1 and energy density of 17.1 W h kg−1. Similarly, Chang and coworkers synthesized a nacre-like Ti3C2Tx/HCNF@Lig composite membrane consisting of Ti3C2Tx, cellulose nanofiber (HCNF), and sodium lignosulfonate (LSNa) by a simple hydrothermal process (Fig. 4d).55 The LSNa not only provided pseudo-capacitance but also promoted electron and/or ion transportation through the hydrogen bonding effect with both Ti3C2Tx and HCNFs. Benefiting from the abundant hydrogen bonds, LSNa/HCNFs provided enough strength in ultrathin 3–5 μm film.56 The composite membrane exhibited high mechanical properties and arbitrary deformability.57 The FSC device assembled with the as-prepared film electrode saw near 100% capacity retention after 5000 times bending, rolling cycles and 2000 times twisting, and folding cycles (Fig. 4e), reaching a high specific capacitance of 248 F g−1 (748.96 F cm−3) at 0.5 A g−1. In the above discussion, the lignin provided pseudocapacitance, as affirmed by the slight redox peaks in the CV curves. In comparison, Wang et al.58 synthesized PAAm-Ag-lignin NPs-Tar-CNT hydrogel electrodes that are composed of a synthetic hydrogel matrix and conductive carbon nanotubes. The hydrogel matrix was integrated by a polyacrylamide network, tartaric acid, Ag-lignin composite nanoparticles, and non-covalent interactions formed by the lignin's phenol groups (Fig. 5a). The hydrogen bond endowed the hydrogel with a self-healing behavior,59 and the supercapacitor exhibited a slight capacity retention decline after 5000 cycles. This electrode material realized energy storage via an electrical double-layer behavior (Fig. 5b), which might be caused by its extremely low quinone content. In contrast to the pseudocapacitive ones, the energy density of the EDLC was lower, about 13.7 W h kg−1, indicating that the pseudocapacitive contribution of Q/QH2 groups was the key to elevating the energy density of lignin-involved supercapacitors. Along with the mechanical properties’ improvement, the all-polymer supercapacitor does not need extra conductive patches, binders, or separators, further increasing the efficiency and reducing the use of synthetic materials.60
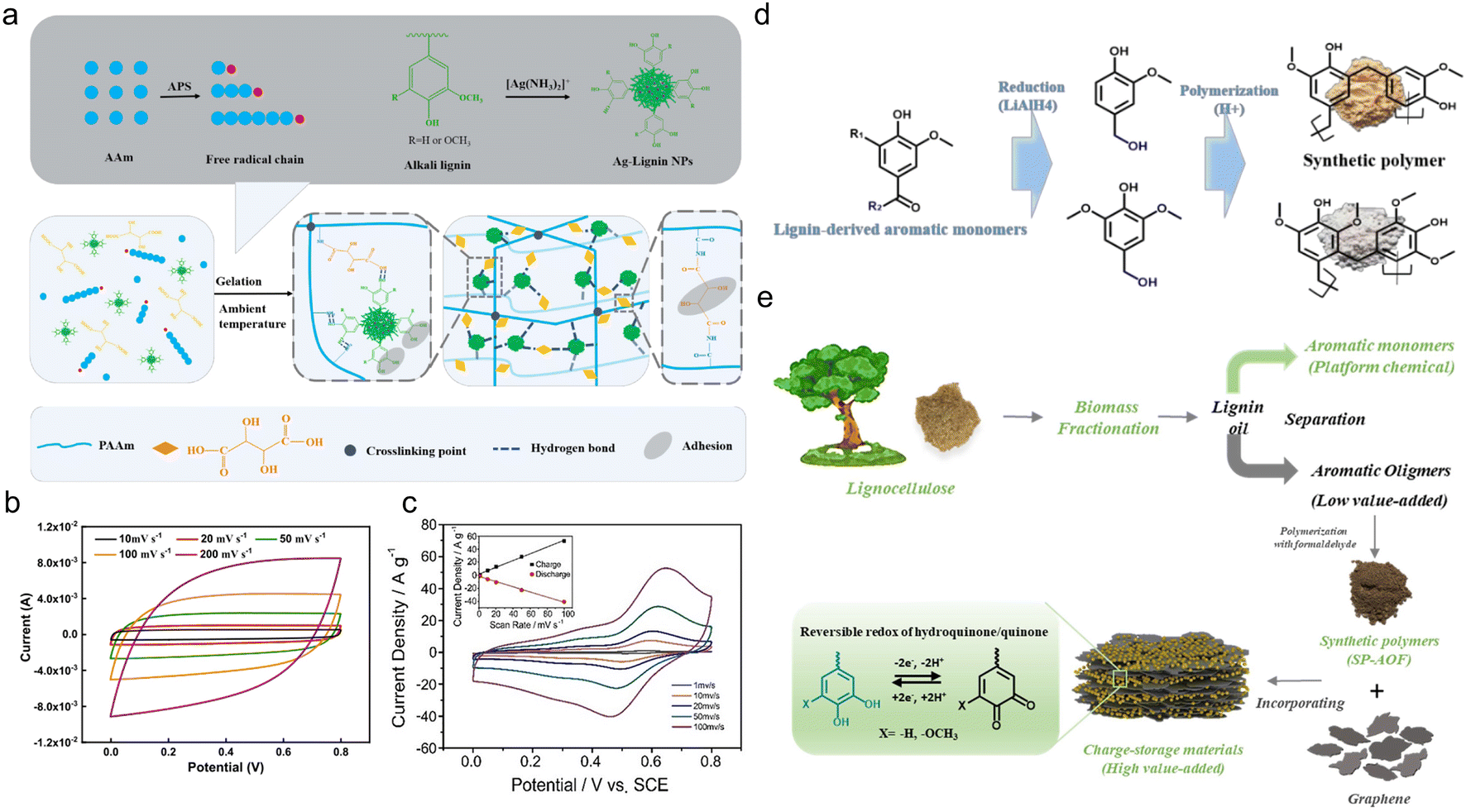 | ||
| Fig. 5 Lignin-derived organic electrodes for supercapacitors. (a) Schematic illustration of the gelation process of PAAm-Ag-lignin NPs-Tar hydrogel matrix at ambient temperature. (b) CV curves of the all-hydrogel supercapacitor at different scanning rates. Reproduced with permission.58 Copyright 2022, Elsevier B.V. (c) CV curves of an OKL/TAC sample in 1 M H2SO4 solution at scan rates from 1 mV s−1 to 100 mV s−1 (inset: plots of peak currents vs. scan rates for charge (R2 = 0.9969) and discharge (R2 = 0.9961) curves). Reproduced with permission.61 Copyright 2019, Wiley-VCH. (d) Schematic illustration of SP preparation based on LDAMs. Reproduced with permission.62 Copyright 2020, The Royal Society of Chemistry. (e) Scheme illustrating the preparation of an rGO/SP-AOF composite electrode material and the proposed energy storage mechanism. Reproduced with permission.50 Copyright 2021, Elsevier B.V. | ||
The first strategy to create active centers is to oxidize lignin with specific chemicals. By using hydrogen peroxide as an oxidant, Zhou and his team selectively converted the methoxyl of KL to phenolic hydroxyls.50,61 The hybridization of the oxidized KL (OKL) with HNO3-treated active carbon (TAC) for conductivity improvement was realized by simple ultrasonic treatment. The as-prepared composite electrode, of which the OKL content accounted for 17 wt%, saw a 25.6% improvement of the phenolic hydroxyl content in contrast to the pristine KL/TAC counterpart. Owing to the enhanced pseudocapacitive behavior of the OKL (Fig. 5c), the capacitance of the assembled supercapacitor rose from 322 F g−1 to 390 F g−1 at 0.5 A g−1. Although the degree of modification was slight, as revealed by the insufficient capacitance improvement, this work did provide a new train of thought to modify bulk industrial lignin for importing extra active redox centers.
The employment of depolymerization products of industrial lignin as raw material is also an effective way to achieve a high quinone content. Yang et al.62 selected lignin-derived aromatic monomer (LDAM) to prepare an rGO/synthetic polymer (SP) electrode, where rGO and SP were composited through ultrasonic-assisted self-assembly (SP was 1.5 times more than rGO). The phenolic condensation polymerization of LDAM raised the quinone content of commercial KL by 6–7 times (Fig. 5d), which ensured its high redox activity for energy storage. The supercapacitor assembled with an rGO/SP electrode acquired a high specific capacitance of 465 F g−1, about 1.9 times higher than that of rGO/KL. This device also presented a favorable rate performance (365 F g−1 at 10 A g−1), demonstrating the high reversibility of the redox reactions for energy storage. Another residue of lignin depolymerization holding great potential in this field is lignin-derived aromatic oligomers (LDAOs).50 Using phenol-formaldehyde condensation, synthetic polymer (SP-AOF) could be obtained from LDAOs. When combined with rGO, LDAO behaved in a capacitive-controlled rather than diffusion-controlled mechanism (Fig. 5e). This was conducive to fast ion transport, leading to an admirable rate performance.63 The RSP-60 electrode where the weight ratio of rGO/SP-AOF was 40/60 had a capacity of 250 F g−1 at 1 A g−1, 1.67 and 1.23 times that of OL and AL.
2.2. Organic electrodes for rechargeable batteries
Similar to pseudo-capacitors, the application of lignin as organic electrodes for batteries contributes capacity primarily through Faraday reactions. Yet, the electrodes adapted to batteries experience a deeper reaction depth and larger volume change because of their higher requirements for specific energies.64 Therefore, the structural design for these electrodes should meet the needs of reducing volume effects which would lead to an irreversible loss of capacity during the charge and discharge process in actual work.29 Until now, lignin-based/-derived biomacromolecule composite electrodes have been reported in both aqueous and organic rechargeable batteries, including zinc ion batteries (ZIBs), lithium ion batteries (LIBs), sodium ion batteries (SIBs), potassium ion batteries (PIBs) and proton batteries.65 In these applications, the most-commonly used types of lignin are commercial LS materials with Q/QH2 redox pairs as they are the main by-product of the papermaking industry. A typical chemical structure of LS is shown in Fig. 6a left. Lignin itself was not conductive, so it had to be combined with conductive polymers or carbon to become electro-chemically conductive. In LS/conducting polymer composite, electrons passed through the composite by hopping between adjacent conducting polymer chains (dark blue rectangles) and reached the redox quinone site on the LS (red color lines) (Fig. 6a middle). In the lignin/carbon nanoparticle composite, the electrochemical redox process of lignin was displayed on the interfacial volume between the carbon and lignin (Fig. 6a right). Recently, some attempts on other chemically modified lignins with new active sites have also been reported in this area.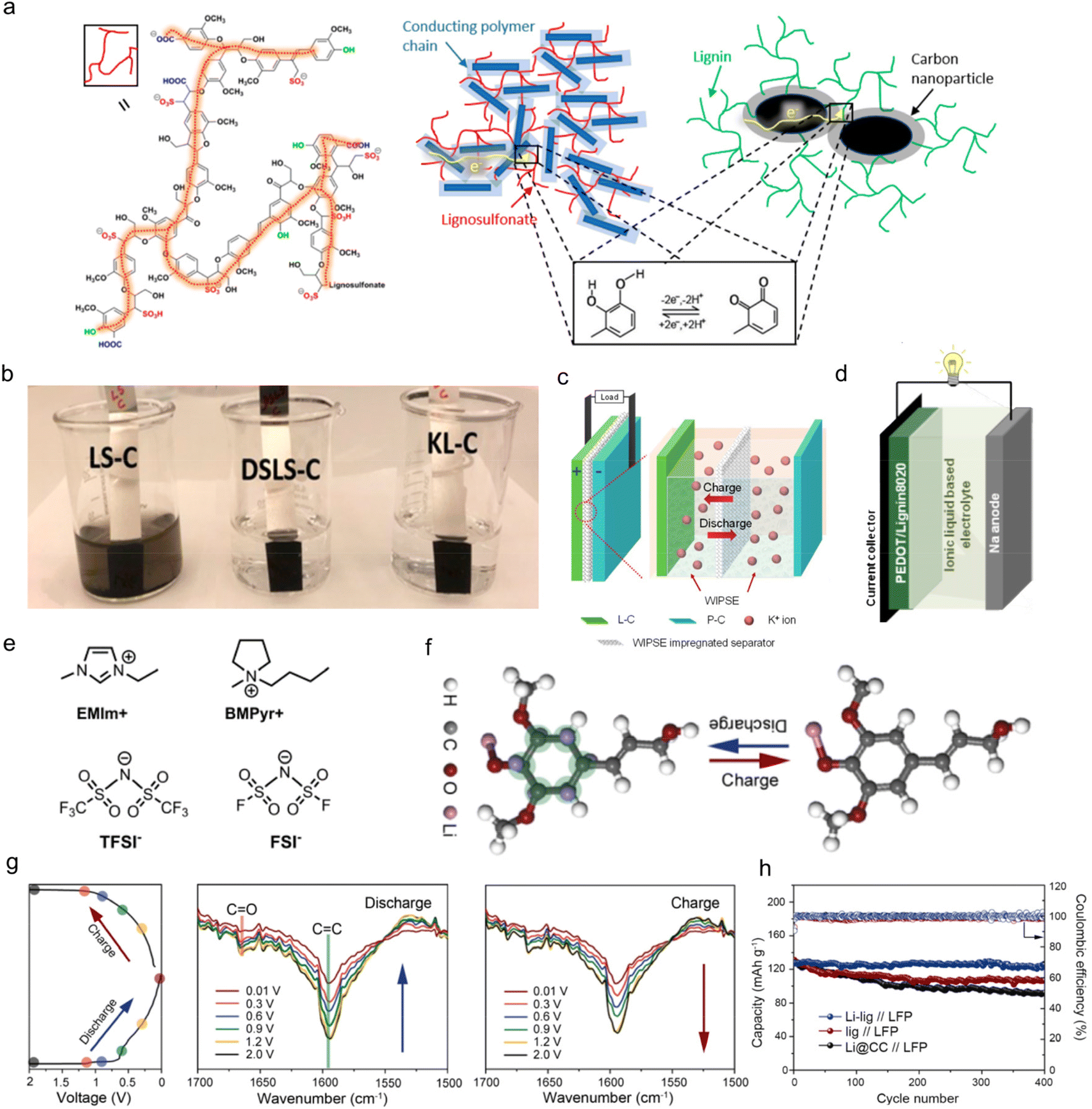 | ||
| Fig. 6 Lignin-based/-derived organic electrodes for rechargeable batteries. (a) Sketch of the chemical structure of LS (left), the LS/conducting polymer composite (middle), and the KL/carbon nanoparticle composite (right) with the bottom inset illustrating the redox reaction of lignin. (b) Optical image of composite electrodes dipped in water for 2 h. Reproduced with permission.31 Copyright 2020, American Chemical Society. (c) Sketch of the device concept with a mechanistic illustration of a cation rocking chair battery. (P–C: polyimide/carbon composite electrode). Reproduced with permission.71 Copyright 2022, Elsevier B.V. (d) Schematic diagram of a PEDOTLignin8020/sodium battery device. (PEDOTLignin80/20: PEDOT/lignin composites in a 80/20 weight ratio). (e) Sketch of the chemical structure of the cationic and anionic species of the ionic liquids used in this study. Reproduced with permission.65 Copyright 2017, WILEY-VCH. (f) Schematic illustration of the structural evolution during the lithium intercalation/deintercalation process, where the binding sites between the Li-ions and the Li-lig are highlighted in green. (g) The discharge/charge cycle profile at 0.1 A g−1 with the corresponding stepwise stages where the FTIR data were obtained (left), and the in situ FTIR spectra of the Li-lig for the first discharge (middle) and charge cycle (right). (h) Cycling performances of the full cells with different anodes at 1 C. (LFP: LiFePO4). Reproduced with permission.17 Copyright 2020, Elsevier B.V. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the benzene ring with abundant CC bonds were activated to be appropriate accommodation sites for lithium intercalation/deintercalation through the “superlithiation” transport mechanism (Fig. 6f). In situ FTIR measurement confirmed the mechanism. As illustrated in Fig. 6g, the CC bands at 1590 cm−1 and CO bands at 1670 cm−1 became weaker when discharged to 0.01 V, in contrast to when fully charged to 2.0 V. The effective use of multiple energy storage sites was helpful to improve the conductive activity of lignin itself, thus improving the electrode performance. As a result, the Li-lig//LiFePO4 full battery with organic electrolyte maintained a capacity of 119 mA h g−1 at 1 C, and 94.4% capacity retention after 400 cycles, and the cycling performance exceeded that of the Lig anode and carbon cloth anode (Li@CC) (Fig. 6h).
O group and the benzene ring with abundant CC bonds were activated to be appropriate accommodation sites for lithium intercalation/deintercalation through the “superlithiation” transport mechanism (Fig. 6f). In situ FTIR measurement confirmed the mechanism. As illustrated in Fig. 6g, the CC bands at 1590 cm−1 and CO bands at 1670 cm−1 became weaker when discharged to 0.01 V, in contrast to when fully charged to 2.0 V. The effective use of multiple energy storage sites was helpful to improve the conductive activity of lignin itself, thus improving the electrode performance. As a result, the Li-lig//LiFePO4 full battery with organic electrolyte maintained a capacity of 119 mA h g−1 at 1 C, and 94.4% capacity retention after 400 cycles, and the cycling performance exceeded that of the Lig anode and carbon cloth anode (Li@CC) (Fig. 6h).
Lignin-derived organic compound electrodes have been successfully applied in the most widely used ESDs. The comparison of various electrodes on compositions, potentials, specific capacities and cycling stabilities were listed in Table 2. However, how to simultaneously achieve a high energy density and rate performance remains a critical challenge, mainly restricted by the ineffective electrical and ion-transporting structure.63 In the future, the depolarization of ions and charge transfer on the electrode material surface should be given priority.73
| Cathode | Anode | Electrolyte components | Potential (V) | Specific capacity (mA h g−1) | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| FEC = fluoroethylene carbonate. | ||||||
| PPy/LS | Zn | ZnAc2 in (50 wt% choline acetate + 50 wt% water) | −0.4–1.1 | 130 (0.2 A g−1) | ∼16.7% (200 cycles) | 67 |
| DSLS/C | Pt | 0.1 M HClO4 | −0.2–0.75 | 38 (0.25 A g−1) | 69% (2200 cycles) | 31 |
| L–C | P–C | PAAK water-in-polymer salt electrolyte | −1.6–1.5 | 35 (0.1 A g−1) | 51% (2400 cycles) | 71 |
| PEDOTLignin80/20 | Na | EMImFSI: NaFSI (20 mol%) | 1.5–3.9 | 70 (0.02 A g−1) | 94% (100 cycles) | 65 |
| Li-lig | LiFePO4 | 1 M LiPF6 in EC/DEC/EMC (1:1:1, v/v/v) + 1 wt% FEC |
2.5–3.8 | 135 (0.5 C) | 81.8% (180 cycles) | 17 |
| LNMO + lignin powder | Graphite | 1 M LiPF6 in EC/DMC (1:1, v/v) |
3.5–5.0 | 110.8 (1 C) | 94.1% (1000 cycles) | 78 |
| Li | Sulfur + LA/LSS | 1 M LiTFSI + 0.1 M LiNO3 in DME/DOL (v/v = 1:1) |
1.7–2.7 | 661 (1.68 A g−1) | 60% (100 cycles) | 80 |
| Li | Sulfur + LSCa | 1 M LiTFSI in DOL/DME (1:1, v/v) + 1 wt% LiNO3 |
1.7–2.8 | 571 (5 C) | 33.3% (500 cycles) | 81 |
| Li | Si + PAL-NaPAA | 1 M LiPF6 in EC/DMC/EMC (1:1:1, v/v/v) |
0.01–1.0 | 1914 (0.84 A g−1) | N/A | 82 |
| Li | LiFePO4 + LPG | LPGP10 | 0–5.0 | 140 (0.1 C) | 99.2% (100 cycles) | 83 |
3. Binders
In the above section, we have fully discussed the feasibility of lignin as an active electrode material. Another important component that determines the electrochemical properties of the electrode is the binder. The binder is utilized to adhere to all the active materials in the electrode, as well as the electrode and the current collector, thus profoundly affecting the structure and mechanical properties of the entire electrode.8 On top of that, appropriate lignin-based/-derived binders should also have several additional functions, such as the ability to withstand volume changes during the charge–discharge cycle and good chemical stability while maintaining the energy density of the battery.74,75 At present, lignin-based/-derived binders are mainly reported in lithium batteries.3.1. Lignin-based macromolecular binders
The commonly-used binders in lithium batteries include polyvinylidene difluoride (PVDF) and water-soluble ones such as carboxymethyl cellulose (CMC), sodium alginate (SA), etc. PVDF is an effective binder choice for most intercalation-based electrodes. Despite its weak flexibility, it embeds the ability to interact with active materials to stabilize their interfaces with the liquid electrolyte.76 As for water-soluble binders, the caboxylic-group-containing CMC and SA could effectively form a stable solid electrolyte interphase (SEI) layer, while the single type of functional group makes them lack diversified functions.77 Lignin with abundant phenolic groups and sulfonate groups can regulate ion-deposition behavior, which is anticipated to govern the interface formation and control the side effects during battery operation.In terms of the cathode, technical lignin could be directly used as a water-soluble binder.47 Ma and his co-workers firstly used AL aqueous solutions as a binder for a LiNi0.5Mn1.5O4 (LNMO) cathode to achieve high-voltage 5.0 V LIBs.78 First-principle simulations by density functional theory revealed that due to the existence of rich phenol groups, the capture energy of free radicals for lignin (3.37 eV) was stronger than that of PVDF (0.29 eV). So, AL could effectively scavenge the free radicals, which was the decomposition product of the organic electrolyte, while PVDF didn't have that feature (Fig. 7a). This was helpful to construct a smooth, regular and compact multi-dimensional cathode electrolyte interphase (CEI) and SEI (Fig. 7b). Consequently, the interphase impedance and electron transfer impedance of the lignin/LNMO electrode was much smaller than that of PVDF counterparts even after 1000 cycles (Fig. 7c left). The electrochemical properties of the as-assembled half-cell far exceeded those using commercial PVDF binders, as testified by its 99.5% coulombic efficiency and 110.8 mA h g−1 specific capacity after 1000 cycles at 1 C.
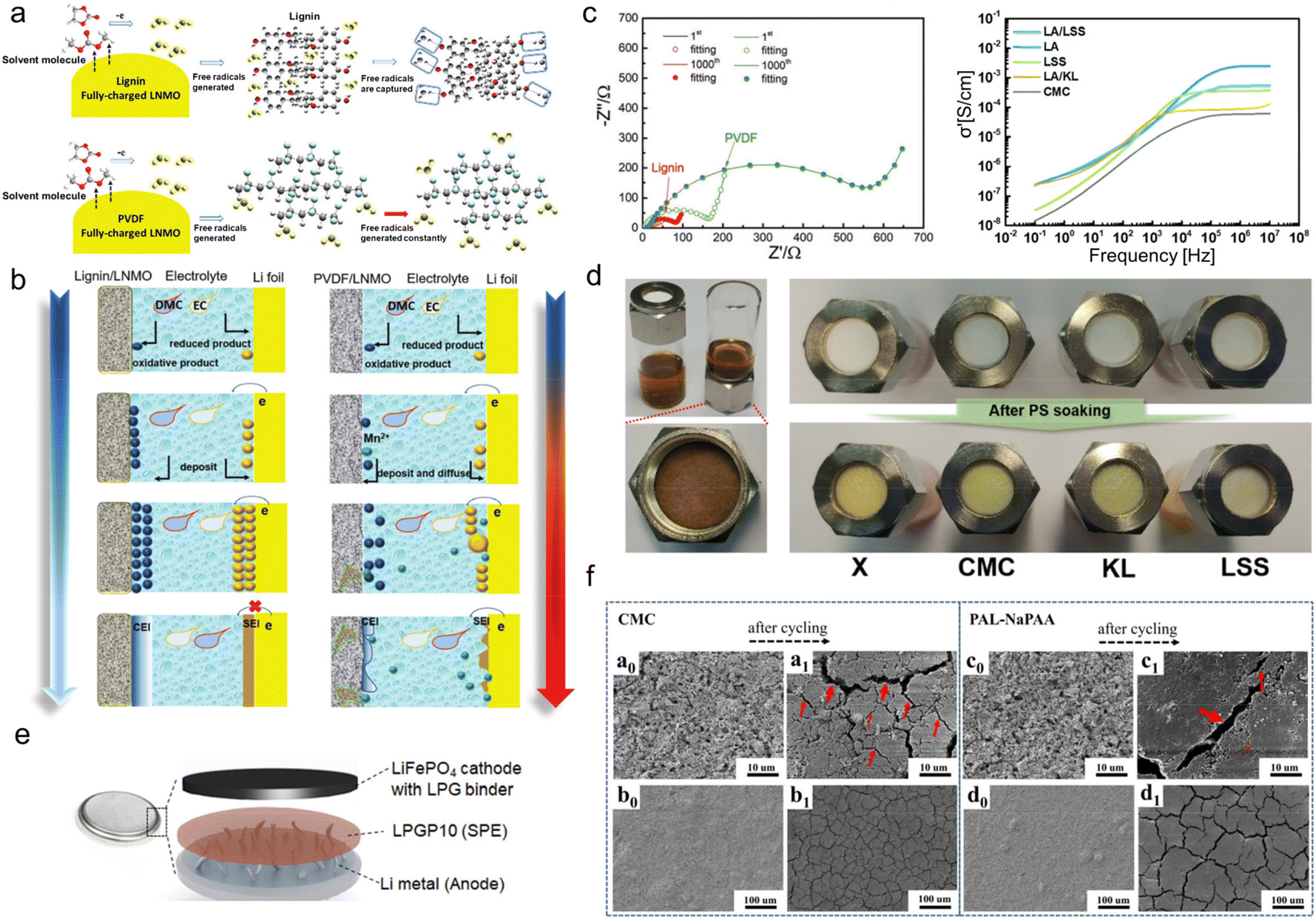 | ||
| Fig. 7 Lignin-based/-derived binders for lithium-based batteries. (a) Diagrammatic illustration of the free radical scavenging ability of lignin (top) and PVDF (bottom). (b) Schematic illustrations of CEI film formation processes based on a lignin binder and PVDF binder. Reproduced with permission.78 Copyright 2019, The Royal Society of Chemistry. (c) Impedance spectra of fully-discharged lignin- and PVDF-based electrodes after long-term cycling (left). Reproduced with permission.78 Copyright 2019, The Royal Society of Chemistry. Plots of the lithium ionic conductivity of each polymer when mixed with lithium salt at 25 °C (right) (LA: LA133 binder). Reproduced with permission.80 Copyright 2019, American Chemical Society. (d) Photograph of a polysulfide repulsion test using pristine glass fiber filter (denoted as X) and polymer-soaked glass fiber filters with CMC, KL, and LSS solution, respectively. (PS: polysulfide). Reproduced with permission.80 Copyright 2019, American Chemical Society. (e) A schematic configuration of all-solid-state lithium metal battery in this study (LPGP10: the solid electrolyte contains 10 wt% PEGDE). Reproduced with permission.83 Copyright 2020, WILEY-VCH. (f) SEM images of CMC and PAL-NaPAA electrodes at different magnifications, respectively. Reproduced with permission.82 Copyright 2018, American Chemical Society. | ||
Apart from hydrophobic phenolic groups, sulfonate groups were also demonstrated to be beneficial to binder performance. As an anionic surfactant, sulfonate-containing LS could be dispersed homogeneously in water and was used as water-based binders for lithium–sulfur Li–S batteries composed of a lithium metal anode and sulfur composite cathode. When discharged, lithium anodes were oxidized into Li+, transferred into the electrolyte and S cathode, and reduced to lithium sulfite. When charged, lithium sulfite could be oxidized to S and release Li+ back to the anode. In this process, a complex reaction of S could result in the production of polysulfide. The crossover of polysulfide from cathode to anode, which is the notorious shuttle effect, is one of the main culprits causing the capacity decline of Li–S batteries.79 Jeon et al.80 discovered that lignosulfonate salt (LSS) solution could function as a decent binder to alleviate the shuttle effect faced by the sulfur electrodes of Li–S batteries. To highlight the important role of sulfonate groups, they compared the electrochemical performance of LSS with KL (rich in –SH groups) and CMC (abundant negatively charged –COO−). It was demonstrated that the large amount of negatively charged sulfonate groups in the LSS could attract Li+ electrostatically to increase the rate of lithium-ion conduction and repel polar lithium polysulfides. Therefore, LSS-containing glass fiber filter paper was harder for polysulfide solution to infiltrate compared with CMC- and KL-containing ones (Fig. 7d), signifying its great potential for shuttle effect suppression. When using LSS as a binder, the commercial LA133 (a kind of aqueous dispersion of acrylonitrile multipolymer) electrode performed better lithium ionic conductivity (Fig. 7c right) and cycling lifespan than LA/KL and LA/CMC. With a high mass loading of 3.2 mg cm−2, it still provided an areal capacity of 1.55 mA h cm−2 at a current density of 0.2 C after 100 cycles. Wu's group further uncovered the working mechanism of LSS for shuttle effect inhibition.81 It was ascribed to the better adsorption ability of sulfonate groups with electron-rich oxygen with lone pairs, which confined polysulfide by forming Li–O bonds. Among various LSS, calcium lignosulfonates (LSCa) had the best adhesion property, and the assembled electrode maintained an areal capacity of 4.16 mA h cm−2 for 100 cycles.
3.2. Lignin-derived graft macromolecular binders
It should be noted that although lignins are electrochemically stable as binders, their molecular weights (500–50000) are much lower than conventional binders and thus cannot provide sufficient viscosity.47 Therefore, some researchers explored the grafting copolymerization of lignin to manufacture higher molecular weight binders. Because of the complex structure of lignin, when grafted with a linear polymer chain, the product will turn into a non-linear structure. Such a structural evolution makes the binder able to endure a certain volume expansion of the electrode. By using a graft polymerization method, Luo et al.82 developed a novel, non-linear, high-performance lignin-sodium polyacrylate (PAL-NaPAA) binder which was adapted to the fume microsilica (SiMP) anode of LIBs. They first grafted polyacrylonitrile (PAN) chains onto lignin fragments through free radical copolymerization. Then, by virtue of an alkaline hydrolysis step, –CN groups were transformed to –COO−, which greatly improved the compatibility of the PAL-NaPAA with the SiMP electrodes. The PAL-NaPAA-bonded SiMP anode could effectively grab free crushed silicon units during cycling and alleviate the volume-change-induced peeling of active materials. That is, the employment of PAL-NaPAA as the binder could completely solve the limited cycling durability issue encountered by the anode using traditional CMC binders. The scanning electron microscope (SEM) image comparison of the anodes before and after cycling using the two binders showed that there were fewer cracks on the anode surface using PAL-NaPAA (Fig. 7f). The maintenance of the continuous and intimate contact was ascribed to the unique properties of the as-developed new binder. The PAL-NaPAA/SiMP (silicon loading ∼0.8 mg) electrode assembled LIBs could be stably cycled for 940 cycles and maintained 800 mA h g−1 specific capacity at 840 mA g−1, far exceeding the CMC electrode that underwent apparent capacity decay after only 250 cycles.
Jeong and his research group synthesized a star-shaped lignin-derived multifunctional macromolecular binder by grafting ion-conducting and cross-linkable moieties to the lignin center.83 This was achieved via a well-defined synthetic route with simple chemical modification and atom transfer radical polymerization (ATRP). The chemical modification brought lignin brominated functional groups, leading to a multi-arm macroinitiator for the subsequent ATRP. FTIR and THF solubility tests demonstrated that flexible poly(ethylene glycol) methyl ether (PEGMA) and glycidyl methacrylate (GMA) chains were successively anchored onto bulk lignin core upon ATRP under ultraviolet (UV) irradiation. The resultant macromolecular network with cross-linking displayed high mechanical stability even at high temperatures whereas the chain mobility was maintained. Such specific architecture of the as-prepared star polymer LPG was beneficial for ionic conduction, enabling it to serve as an excellent binder. It was assembled with poly(ethylene glycol) dioxalyl ether (PEGDE), a solid electrolyte of which the flexible ion-conducting polyethylene glycol (PEG) chain could promote ionic conductivity and mechanical integrity. Compared with the conventional liquid electrolyte-Celgard system, LPG provided a facile ion conduction pathway and efficiently suppressed lithium dendrite growth during cycling. On this account, all-solid-state lithium metal batteries (Fig. 7e) with a satisfactory cycling lifespan at high temperatures were obtained (150 mA h g−1 after 50 cycles at 60 °C).
In brief, lignin-involved binders are mainly used in LIBs, which could be divided into lignin-based binders and lignin-derived graft macromolecular binders. The former mainly utilized the functional groups of lignin itself to achieve goals such as free radical scavenging, repulsion of polysulfides, etc. They have the advantages of a convenient and environmentally friendly manufacturing process. However, the molecular weight of lignin is greatly affected by factors such as manufacturing methods and raw material sources, bringing insufficient viscosity. In contrast, lignin-derived binders present a larger, homogenous and nonlinear structure by grafting functional polymers, which endows the electrode with structural and thermal stability. Besides, pollution can be avoided by applying UV irradiation to initiate polymerization. In addition, it is a promising development direction for lignin-derived binders to be used in aqueous batteries.
4. Electrolytes
Electrolytes are one of the most influential components in the performance of ESDs, since the synergistic effect between the electrodes and electrolytes would profoundly affect the performance of the electrode and even the whole device, both for organic and aqueous systems.84 Electrolyte modulation can regulate a series of important factors involved in the energy storage courses, like the electrochemical stability window, the redox routes, the rate ability and the formation of an SEI or CEI (a decisive factor of the cycling stability of ESDs).85–87 In recent years, electrolyte formulation has been identified as a common, effective way to optimize the performance of various ESDs, either by altering the main electrolyte component or adding some functional additives. Taking advantage of its rich polar functional groups which could compound with commercial synthetic polymers and metal salts effectively, lignin has been also utilized to tailor the electrolyte properties of various ESDs.4.1. Liquid electrolyte formulation
Admiring the environmental friendliness feature of lignin, Mukhopadhyay et al.48 directly used 0.1 M LS aqueous solution as an anolyte to build flow batteries (Fig. 8a). The anolyte realized energy storage based on the redox reaction of Q/QH2, similar to the application of lignin in organic electrodes. There were various advantages for lignin to serve as the anolyte. The high content of phenol groups ensured a sufficient concentration of the active substance, contributing to energy density improvement. The redox reaction stably went through multiple cycles without any activity decline in acidic conditions. The low molecular weight of lignin led to a low viscosity that reduced the electrolytic crossover, ensuring high-capacity retention. Based on above merits, the assembled full cell composed of an LS anolyte and Br2/Br− catholyte could operate at an ultra-high current density of 20 mA cm−2 (Fig. 8b), with polarization resistance as low as 15.1 Ohm cm−2 (Fig. 8c) and voltage efficiency of up to 85% at low current densities.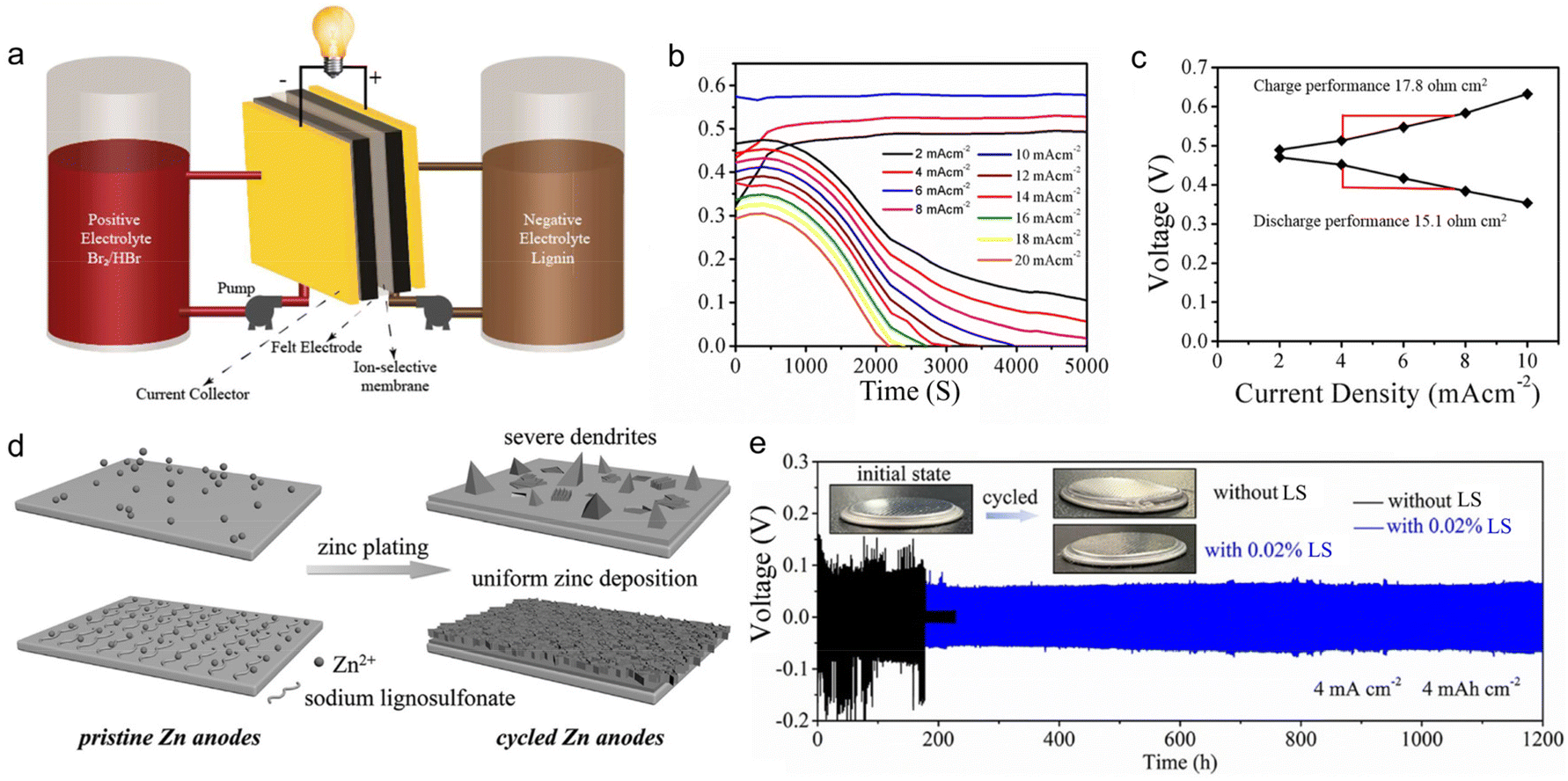 | ||
| Fig. 8 Lignin-based liquid electrolyte formulation for rechargeable batteries. (a) Schematic of the lignin flow battery system assembly. (b) The plot of cell voltage vs. time at various current densities of the lignin flow battery. (c) Polarization curve of the lignin flow battery during charge and discharge at various current densities. Reproduced with permission.48 Copyright 2018, American Chemical Society. (d) Schematic illustration of the zinc deposition with and without LS electrolyte additive. (e) Galvanostatic charge/discharge curves of Zn//Zn cells with and without 0.02% LS at 4 mA cm−2 and 4 mA h cm−2 where the insets show the photographs of the cells before and after cycling. Reproduced with permission.89 Copyright 2020, WILEY-VCH. | ||
Lignin could also work as a functional additive to tailor the physiochemical properties of regular electrolytes, thereby modulating the energy storage behaviors of ESDs. Lota et al.88 found that the presence of sulfonic groups in LS enabled it to strongly adsorb on carbon electrodes and to behave as redox active films after initial oxidation (assignable to quinone-type moieties). When 5 g L−1 LS was added to 1 M H2SO4 electrolyte, the overall capacitance of the supercapacitors underwent a sharp rise owing to LS-induced pseudo-capacitive effects. They selected two kinds of LS material with different molecular masses (8000 and 42700 g mol−1, denoted as LS1 and LS2 respectively) for further investigation. The results showed that LS2 gave better specific capacities than LS1 when employing a less porous carbon as an electrode. The relative capacitance increased by 4.5% after 5000 charge/discharge cycles at 1 A g−1. In addition to its role in ion storage, lignin could also work as an effective corrosion inhibitor for the Zn metal, benefitting from its hydrophobic main chain and abundant hydrophilic groups (i.e., sulfonic group and phenolic hydroxyl). Zhou et al.89 reported that the addition of 0.02% LS to 2 M ZnSO4 + 0.2 M MnSO4 aqueous solution could improve the ionic conductivity of the electrolyte, confine the 2D planar diffusion of Zn2+ ions at the interface, lower the nucleation overpotential of Zn2+ ions, inhibit side reactions and suppress the corrosion of the Zn surface (Fig. 8d). In this way, zinc dendrites and side reactions were effectively prohibited. The Zn//Zn symmetric cell with LS-modified electrolytes delivered a long lifespan of 1200 h under a large areal specific capacity of 4 mA h cm−2 (Fig. 8e), while the cell assembled without LS was broken during cycling due to the harsh corrosion of Zn metal. Consequently, the Zn//α-MnO2 full battery with 0.02% LS electrolyte additive exhibited a capacity of 146 mA h g−1 at 1 A g−1 after 1000 cycles.
4.2. Gel electrolyte development
As a natural biopolymer with multiple hydrophilic or hydrophobic functional groups, lignin is a good candidate for gel fabrication. There are two major ways to transform lignin-based materials into gels, one of which is physical crosslinking. The steps of this approach are as follows. First, select an appropriate solvent for different types of technical lignin species which are generally dissolved in water or organic solvents. Second, use a convenient solution casting method to provide physically crosslinked gel electrolytes where the main chains interact by physical entanglement and intertwining, like hydrogen bonds or polar bonds.90 The key factor of this method is to formulate suitable solutions that could help overcome the difficulty of lignin gelation. The other method is chemical crosslinking. Through a chemical grafting method, a chemically crosslinked gel electrolyte could be provided, where different types of main chain are chemically covalently bonded to each other. The key factor of this method is to select an appropriate functional graft that enriches the product properties such as gel robustness, ionic conductivity, etc.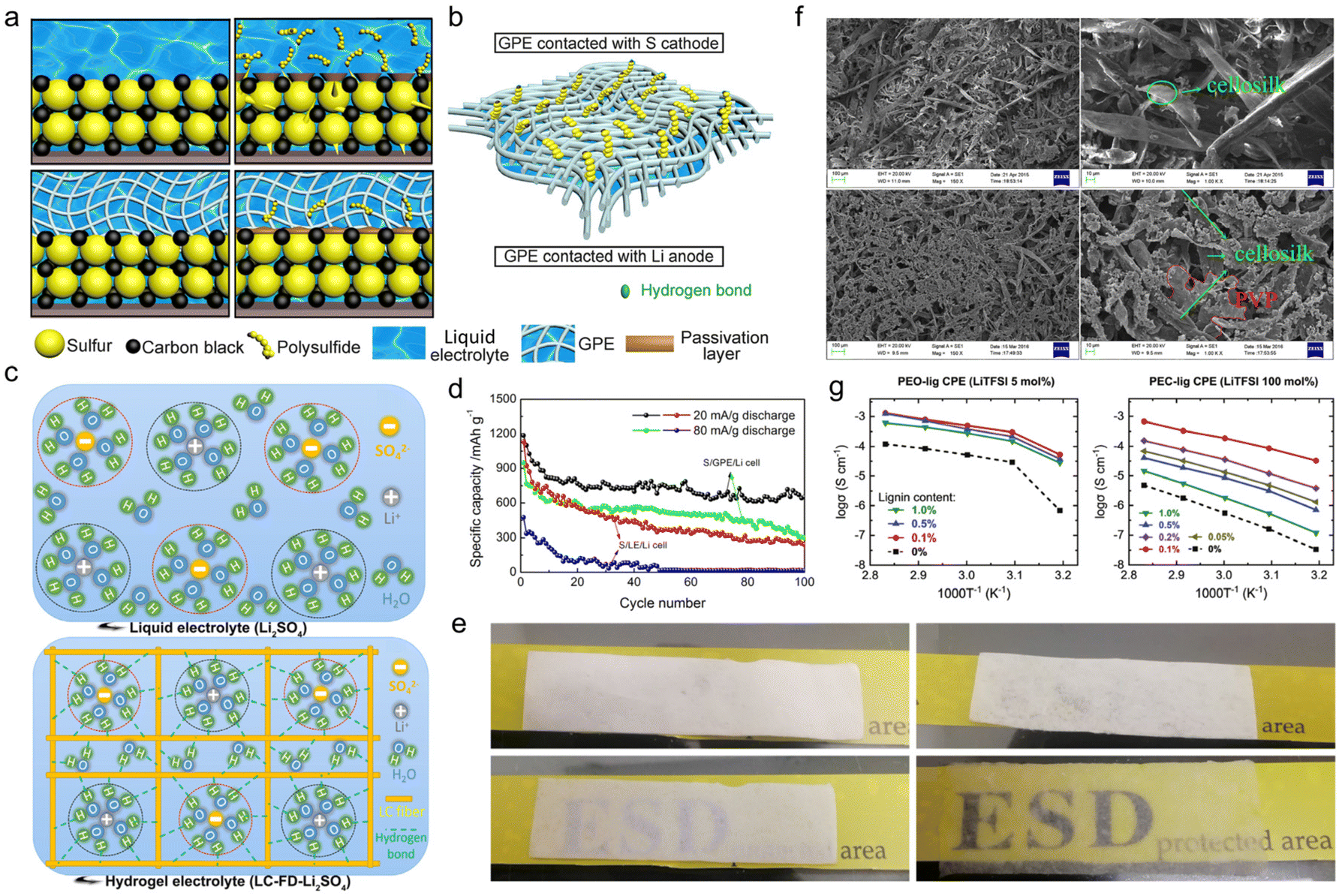 | ||
| Fig. 9 Lignin-based physically cross-linked gel electrolyte. Schematic illustration of (a) the immobilization mechanism for polysulfides within a Li/liquid electrolyte/S (upper left, upper right) and Li/G2/S (lower left, lower right) cell at the initial state (upper left, lower left) and after charge–discharge cycles (upper right, lower right), and (b) the hydrogen bonding between H–O and part of the polysulfides (–SO4, –SO3 and –SO2) on the surface of GPE contact with sulfur cathode. Reproduced with permission.92 Copyright 2018, Elsevier B.V. (c) Schematic diagram of the liquid electrolyte (upper) and hydrogel electrolyte (bottom). Reproduced with permission.93 Copyright 2020, Elsevier Ltd. (d) Cycling performances of a Li/LE/S cell and Li/G2/S cell. Reproduced with permission.92 Copyright 2018, Elsevier B.V. (e) Optical image of an LC membrane (upper left), LC-FD membrane (upper right), LC membrane-based hydrogel electrolyte LC-Li2SO4 (lower left) and LC-FD membrane-based hydrogel electrolyte LC-FD-Li2SO4 (lower right). Reproduced with permission.93 Copyright 2020, Elsevier Ltd. (f) SEM micrographs of lignin (top) and lignin-PVP (bottom) membranes at different magnifications, respectively. Reproduced with permission.94 Copyright 2018, Springer. (g) Plots of the temperature dependence of ionic conductivity for PEO-lig CE (left) and PC-lig CE (right). Reproduced with permission.96 Copyright 2022, The Royal Society of Chemistry. | ||
Apart from fiber length, the porosity of the gel electrolyte far-reachingly impacted the liquid uptake ability. Qiu et al.93 compared the electrochemical performance of lignocellulosic neutral hydrogel (LC-FD) as a supercapacitor electrolyte prepared by solution casting and freeze-drying methods. In contrast to the one obtained by solution casting alone, the gel sample treated by freeze-drying embedded more porosity and a larger specific surface area, integrating self-supporting capability, lifted electrolyte uptake and improved ionic conductivity. It could be seen from optical images that the LC-FD exhibited a much higher liquid adsorption ability than the LC membrane (Fig. 9e). More importantly, in this case, water molecules were stable in hydrogel electrolyte as oxygen-containing functional groups in the LC-FD film could form hydrogen bonds with water molecules in the solvated shell (Fig. 9c). The confined reactivity of water extended the electrochemical stability window of this gel electrolyte, presenting a decent performance even at high voltages. The supercapacitor assembled with LC-FD-Li2SO4 gel electrolyte operated correctly at 1.8 V, delivering an energy density of 16.08 W h kg−1 and a power density of 225.25 W kg−1, much higher than the maximum energy density of the liquid electrolyte-based supercapacitor. The gel electrolytes fabricated by solution casting methods generally suffered from insufficient mechanical properties. The addition of film-forming agents, such as polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP), poly(ethyleneoxide) (PEO), poly(ethylene carbonate) (PEC), etc. in the manufacturing process was an effective way to circumvent this problem. For instance, Liu et al.94 selected silanization-modified AL and PVP as raw materials to prepare a final gel electrolyte product through a solution casting method. After graft modification of the AL, the PVP and modified lignin were strongly combined by hydrogen bonds. It could be clearly seen from SEM images that PVP filled the gaps of the lignin cellosilk (Fig. 9f). When the PVP content was 22 wt%, the dense structure elevated the maximum stress of the membrane to 7.27 MPa, which increased by 670% more than that of bare lignin. However, the stress was weakened when the PVP content exceeded 22 wt%. Under this circumstance, the as-prepared gel electrolyte led to a full battery with a capacity of 117 mA h g−1 at 1 C.
Even though the film-forming agent reinforced the mechanical properties, the ionic conductivity of the resultant gel electrolytes was usually unsatisfactory, demanding further improvement of the solution casting process. In this context, Shabanov and his research team combined sulfonated- and subsequent chlorinated-lignin with PVA to prepare a mono-ionic conductive gel electrolyte (GE-LS-Cl) for lithium batteries.95 Chloro-lignosulfonic acid (LS-Cl), the product after two-step modification of the original lignin, was neutralized by 0.1 M LiOH solution to induce the pre-absorption of lithium ions on lignin. The product was mixed with PVA, dissolved in DMSO, and then used as the solution for the GE-LS-Cl preparation. A semi-empirical PM3 method investigation uncovered that, compared with unmodified and single-sulfonated lignin, LS-Cl after two-step modification had greater molecular polarity and more active sites for lithium-ion transport. These properties provided the GE-LS-Cl electrolyte with the ability for predominant cationic conductivity. Thus, the lithium-ion transport number tLi+ of the LS-Cl GE was 0.89, far exceeding commercial Celgard 2730 membrane (0.27). In addition to chemical modification, the chemical structure of the lignin could also be altered by changing the lignin extraction process. Liu et al.96 proposed a novel lignin extraction process called simultaneous enzymatic saccharification and comminution (SESC). The electrolyte sample was prepared firstly by a simple solution casting method using SESC lignin powder and PEC mixed acetonitrile solution to get a pre-mixed solid. Secondly, the pre-mixed solid underwent hot drying, mixing, hot-pressing and cold-pressing to obtain the final composite electrolytes (CE) film. The as-prepared lignin had free radical scavenging activity originating from the guaiacol groups of the SESC lignin, which simultaneously weakened the Li+ coordination with the polymer chains and facilitated fast ion migration. As a result, the content of 0.1% SESC lignin increased the conductivity of the lignin-containing CE (namely PC-lig CE) by three orders of magnitude, which improved two orders of magnitude in the PEO-based CE fabricated by the same methods (Fig. 9g).
With the deepening of the research, the solution casting method had been further simplified. By adjusting the solution formula, the casting step could be completed at room temperature to construct polyelectrolyte gel electrolyte products, which enabled high-capacity and long-life ESDs. Lignin-based polyelectrolyte gels were synthesized by the action of various electric charges. In the case of chitosan and cellulose-derived biomaterials, the hydroxyl groups would only complex with metal ions like Cu2+,97,98 while in the case of lignin, its catechol groups could serve as redox active sites for metal ions.10,99 So, using a metal-based polymerization catalyst, the lignin macromolecular self-catalytic system would provide a fast gelation process, thus forming a robust gel under ambient temperature.100 As a proof of concept, Wang et al.101 fabricated lignin-derived ionic gel (DGel-Fe1L0.5) to serve as a supercapacitor electrolyte. They utilized the dynamic redox reaction between the lignin catechol group and Fe2(SO4)3 to catalyze the polymerization of acrylic acid within a deep eutectic solvent (DES) which is composed of betaine and ethylene glycol. The instant cross-linking process provided a large number of ions and hydrogen bonds between DES, polyacrylic acid (PAA) and lignin, ensuring the ionic gel's conductivity and stretchability (Fig. 10a). On this account, the as-prepared gel DGel-Fe1L0.5 with a decent mass ratio of Fe3+ and lignin was flexible at −50 °C (Fig. 10b). When it was employed as an electrolyte, the assembled flexible quasi-solid supercapacitor saw 89.2% capacity retention after 3000 cycles (51.4 F g−1 capacity at 0.5 A g−1).
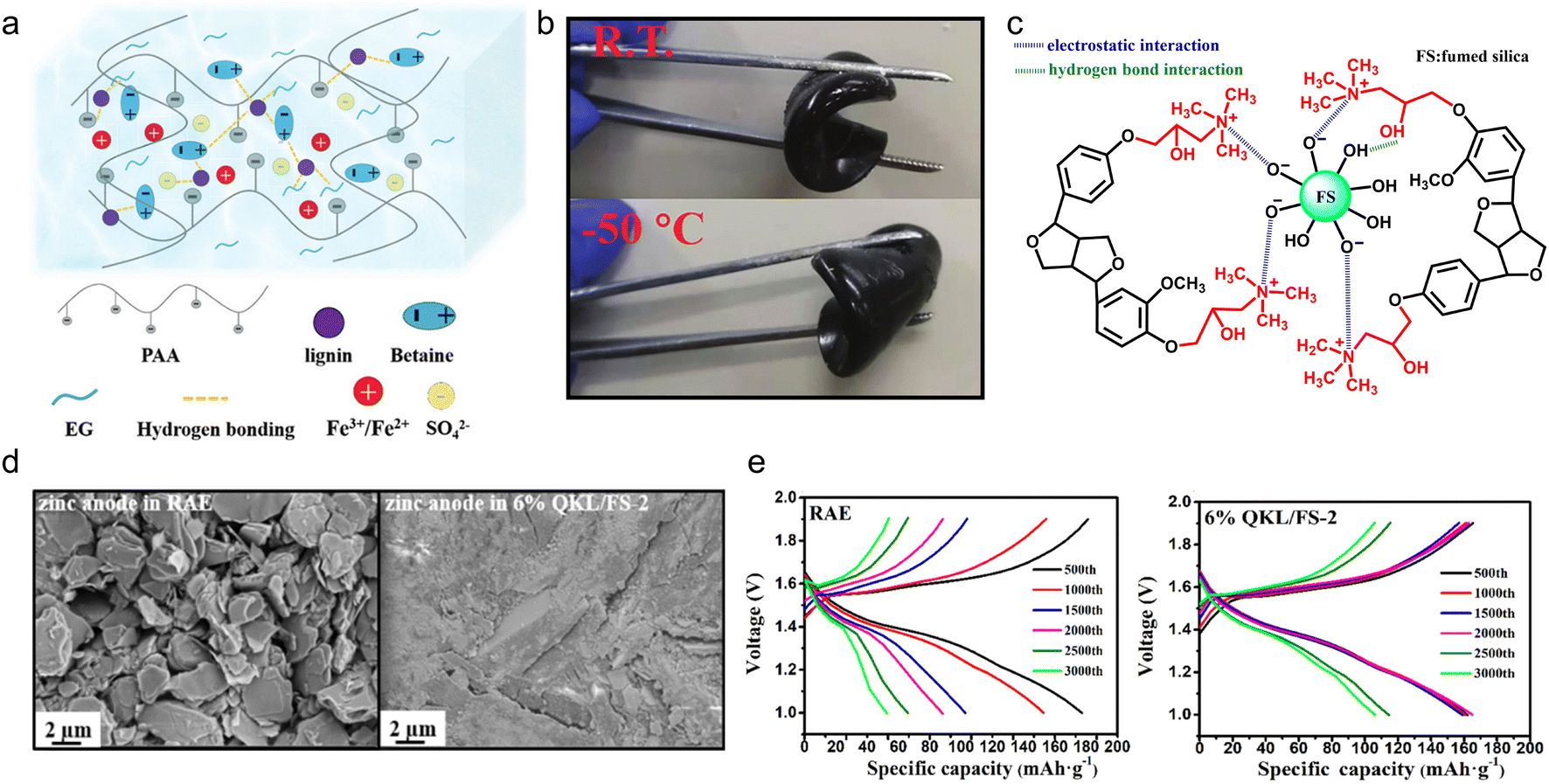 | ||
| Fig. 10 Lignin-based/-derived physically cross-linked gel electrolyte. (a) Structural characteristics of the DES gel. (b) Photos of the bending of the gel at room temperature and −50 °C. Reproduced with permission.101 Copyright 2021, The Royal Society of Chemistry. (c) Chemical structure of the as-prepared QKL/FS. (d) SEM images of the corresponding zinc anodes. (e) The charge/discharge curves of the batteries with the RAE and 6% QKL/FS-2 at a current density of 1.5 A g−1 in 3000 cycles. Reproduced with permission.102 Copyright 2022, American Chemical Society. | ||
In the above polyelectrolyte synthesis process, the charge was supplied through the DES and catalyst. Another way to provide charge was to directly charge the lignin, which facilitated the construction of the polyelectrolytes. Xu et al.102 chemically modified the phenolic hydroxyl of KL by graft copolymerization and the product was denoted as QKL. The QKL/FS mixture was obtained by mixing FS solution with QKL solution and filtering, where QKL and FS interacted by an electrostatic and hydrogen-bonding effect (Fig. 10c). Then, the mixture was utilized as an additive to a reference aqueous electrolyte (RAE) (2 M ZnSO4 + 0.2 M MnSO4) for the preparation of the gel electrolyte. The zinc dendrite formation issue confronted by the RAE no longer occurred in the case of the gel electrolyte (Fig. 10d), which slowed down the deterioration rate of the Zn anode. The Zn//MnO2 rechargeable battery containing 6% QKL/FS-2 gel electrolyte (the mass proportion of QKL to FS was 2) retained 106.2 mA h g−1 after 3000 cycles at 1.5 A g−1, more than 2 times higher than that in unmodified RAE (Fig. 10e).
How to connect bulk lignin with long chain polymers was a vital problem in the synthesis of chemically crosslinked gel electrolytes. Liu and his team functionalized lignin and PEG respectively, aiming at accessing the chemically active sites that trigger the photo-redox thiolene reaction to link the two polymers.103 The pre-functionalization process included the conversion of the hydroxyl groups of lignin to alkene and the addition of thiol groups to PEG through esterification. The final product, lignin-graft-PEG 2000, derived from the chemical reaction of the two pre-functionalized materials, presented as a dark brown slurry solid containing 34% lignin. When mixed with lithium bis-trifluoromethanesulfonimide (LiTFSI), it demonstrated a decent ionic conductivity that could be applied in a solid electrolyte field. Although the study did not employ the electrolyte to the assembly of a full battery, it did verify the possibility of building novel polymer electrolytes by grafting and copolymerization of lignin through chemical reactions.
In the above study, the modification reaction of PEG itself increased the complexity of the whole synthesis process. To simplify the procedure, researchers chose chemically reactive PEGDGE as an alternative to PEG. Park and his research team firstly synthesized AL-graft-PEGDGE hydrogel electrolyte by one-step alkali-catalyzed ring-opening polymerization.104 At both ends of the PEGDGE, a phenoxy nucleophile formed a crosslinking structure from the attack of the primary terminal of the less blocked epoxide. The cross-linked networks of the hydrogel ensured high ionic conductivity (10.35 mS cm−1) and mechanical robustness (523% of swelling ratio). The supercapacitor composed of a lignin-derived hydrogel and lignin/PAN electrospun free-standing electrodes (Fig. 11a) showed excellent cycling ability, maintaining 95% of its initial capacitance over 10000 cycles. The mechanical properties and conductivity are currently the main concerns of lignin-derived gel electrolytes. Based on the concept of stiffening to provide better mechanical properties, Liu et al.105 took advantage of the pH responsiveness characteristic of this gel, and formulated double-crosslinked hydrogel (DC) electrolytes for supercapacitors.106 Using the same chemical crosslinking method developed by Park et al., a further step of physical crosslinking was applied by introducing H2SO4 solution treatment. The DC had a denser network structure compared with single-crosslinked hydrogel (SC) (Fig. 11b). A smaller pore size in DC seen from SEM was believed to be beneficial for avoiding stress concentration and crack propagation, which resulted in a significantly improved compression stress of 4.74 MPa. Composed of the hybrid DC hydrogel electrolyte and PANI electrode, the supercapacitor was able to maintain high capacitance under a severe compression strain and after various bending angles owing to the flexibility of the electrolytes. This supercapacitor was suitable for flexible device scenarios. In order to increase the energy density of flexible SCs, Mondal et al.107 provided a highly conductive gel LSNa-PEGDGE hydrogel using the same chemical crosslinking reaction.108 By further immersing the hydrogel in 1.25 M FeCl3 solution for 10 min, Fe3+ chelated with sulfonic, catechol, phenolic hydroxyl and carboxyl groups in the lignin, improving the ionic conductivity up to 66.9 mS cm−1 (Fig. 11c). Thus, the SC assembled with such gel electrolyte showed both excellent cycling stability (94.1% capacitance retention rate after 10000 continuous charge–discharge cycles) and energy density (26.73 W h kg−1).
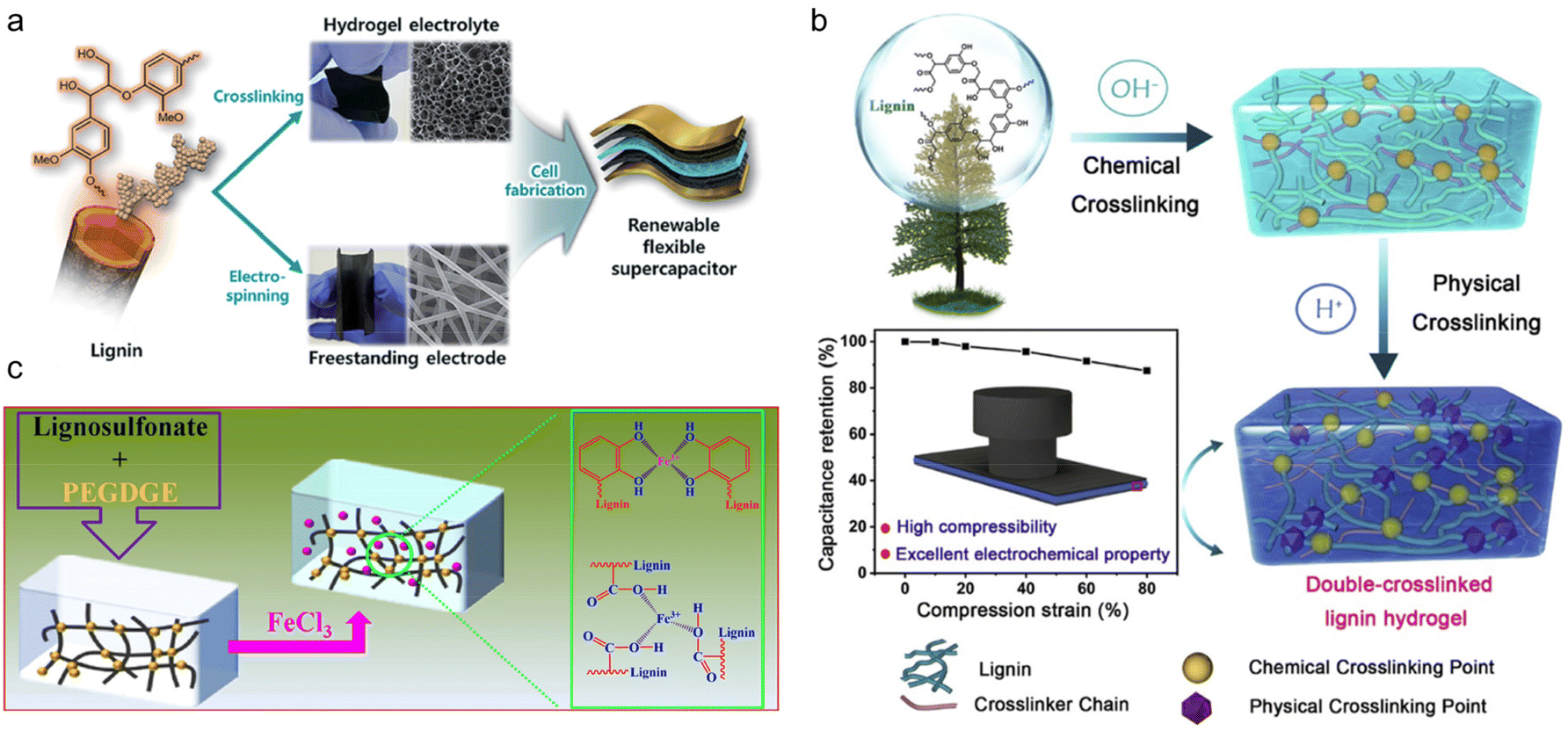 | ||
| Fig. 11 Lignin-derived chemically cross-linked gel electrolyte. (a) Experimental scheme of the fabrication of an all-solid-state flexible supercapacitor based on a lignin hydrogel electrolyte and lignin/PAN carbon nanofiber electrode. Reproduced with permission.104 Copyright 2019, The Royal Society of Chemistry. (b) Schematic illustration of a renewable double-crosslinked lignin hydrogel formed via a sequential chemical crosslinking and physical crosslinking strategy, which could be an excellent hydrogel electrolyte to be used in highly compressible and foldable supercapacitors. Reproduced with permission.105 Copyright 2019, Elsevier Ltd. (c) Schematic illustration of the preparation of the LSNa-PEGDGE hydrogel along with impregnation of Fe3+. Reproduced with permission.107 Copyright 2022, American Chemical Society. | ||
Apart from supercapacitors, lignin/PEGDGE electrolyte had a variety of application scenarios in ESDs. de Haro et al.109 explored its application in photovoltaic device aqueous dye-sensitized solar cells (DSSCs). They firstly oxidized KL using the Fenton reaction, with the purpose of increasing the content of phenolic hydroxyl that favors the crosslinking reaction. The higher the amount of PEGDGE in the formulation, the more flexible segments there were in the three-dimensional polymeric network and the higher the ionic transportation ability. Lignin had a UV-protection behavior in DSSCs, which protected photosensitive molecular dyes from photoinduced degradation by absorbing high-energy photons. Based on the structural stability of PEGDGE and the light resistance of lignin, the quasi solid-state lignin/PEGDGE electrolyte led to a DSSC device with decent cycling stability under UV-Vis light, especially compared with the carboxymethylcellulose electrolyte assembled one. The chemically crosslinked gel electrolytes also had the prospect of being used in rocking-chair batteries. Trano et al.110 used pre-oxidized kraft lignin (OxL)/PEGDGE crosslinking film soaked with an ethylene carbonate (EC)/diethyl carbonate (DEC) composite electrolyte to serve as a GPE in potassium batteries. The pre-oxidized lignin matrix with an ethoxylated difunctional oligomer promoted a good adaptation to solvated potassium salts. Under this circumstance, the potassium metal cell had a wide voltage window (4 V) and delivered a capacity of 168 mA h g−1 at 0.05 A g−1.
For lignin-based/-derived electrolytes, the basic physicochemical properties such as ionic conductivity and mechanical strength, compression stress et al. have surpassed those of some commercial materials. In future directions, from the perspective of liquid additives, the mass transfer behavior of lignin in large and small ESDs should be considered, since it affects the mass transfer efficiency in liquid electrolytes. In terms of the physically crosslinked hydrogel electrolytes, a stable ultra-thin electrolyte is expected to be constructed to meet the need for high energy density and flexible devices. For chemically crosslinked hydrogel electrolytes, more environmentally friendly grafted materials could be considered so as to achieve both distinguished properties and the purpose of waste recycling.
5. Separator modifications
The separator, a significant component that electrically isolates the cathode and anode to avoid short-circuits, plays a key role in assuring the safe operation of ESDs. As the separator also provides the necessary channels for the transport of ionic charge carriers between the two electrodes, the efficiency and cycling life of the ESDs depends on its intrinsic properties. Besides, it determines the cost of the cell to a great extent.111 In the case of aqueous batteries, a separator should be electrochemically stable under various pH conditions while beneficial for the interface chemistry.112 For lithium batteries, a separator should avoid the electrochemical abuse, thermal abuse and mechanical abuse that may trigger safety concerns by internal heat generation.113 In Li–S batteries, particularly, the inhibition of the shuttle effect, the main problem that leads to the irreversible loss of active materials and rapid capacity degradation, should be addressed.114 Lignin-based/-derived materials have been applied for separator modification in the form of membranes and membrane coatings in ESDs, including both aqueous and organic devices.5.1. Aqueous device membranes
As the first and most mature separators for hydrous alkaline batteries, ion exchange membranes are frequently used in aqueous ESDs.115 Among numerous candidates, Nafion represents the most widely applied proton exchange membranes but the high price severely limited its further practical application. To tackle this problem, there are two main solutions. First, introduce some functional materials with a lower price for composition membrane fabrication. Second, replace the Nafion with other cheap proton-exchange membranes.It has been reported that the hybridization of cheap lignin with Nafion could reduce the cost but simultaneously maintain or even optimize the electrochemical performance of ESDs. Inspired by the fact that the protection of the Zn anode surface by LS was able to induce homogeneous Zn deposition in ZIBs, Yuan et al.116,117 synthesized an LS/Nafion composite separator by a re-casting method using N,N-dimethylformamide as solvent, whose structure could be seen in Fig. 12a. The lignin-Zn2+ interface as SEI effectively protected the Zn surface. It could be seen from the SEM images that a lateral zinc hydroxide layer was present on the Zn surface after 1000 cycles, while perpendicular zinc deposition was found on the Zn surface when using glassy fiber (Fig. 12b). It had been confirmed that a horizontal orientation had a relatively dense stacking structure.118,119 For the purpose of enriching the water channels and conductivity, 10 wt% lignin was incorporated into the Nafion substrate, resulting in the lateral ZHS layer favoring a grown Zn(100) direction and 410 h Zn||Zn symmetric cell cycling life better than the pure Nafion one (345 h) (Fig. 12c).117,120,121 Besides, the LS/Nafion with enriched water channels reduced the interfacial resistance (Rct) compared with pure Nafion, resulting in the formation of a robust SEI. Based on a well-regulated Zn anode surface, the Zn||ZnSO4||β-MnO2 full battery remained a specific capacity of 236 mA h g−1 under a current density of 0.1 C.
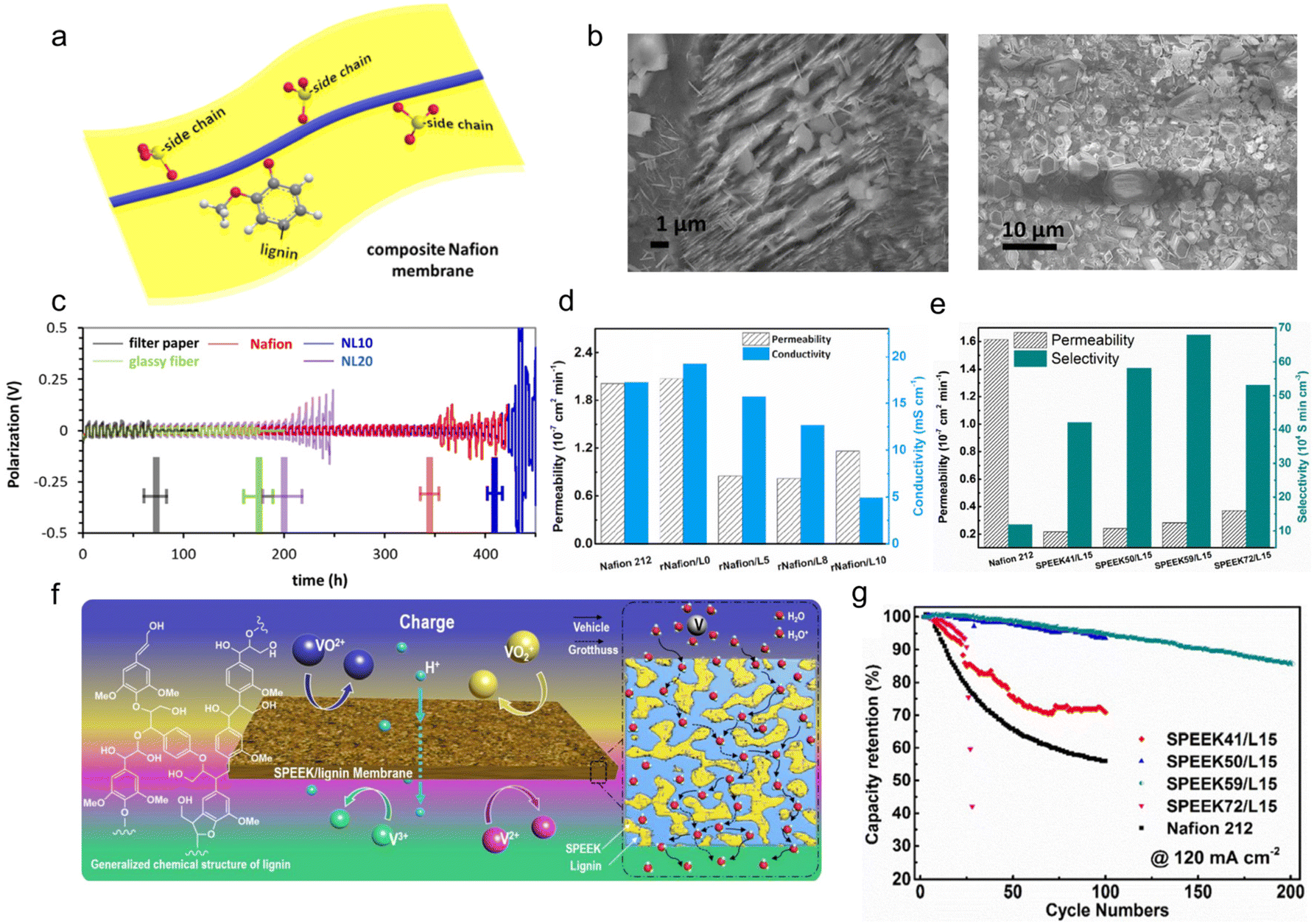 | ||
| Fig. 12 Lignin's application on membrane modification. (a) Scheme illustrating the design of composite LS/Nafion membranes. (b) SEM images of the zinc surface using a glassy fiber membrane (left) and the one using an LS/Nafion membrane (right) after the 1000th cycle. (c) Stripping/plating of symmetric cells with different membranes until a constant capacity of 0.6 mA h cm−2. Reproduced with permission.117 Copyright 2019, Wiley-VCH. (d) The permeability values and the proton conductivities of the membranes. Reproduced with permission.123 Copyright 2020, Elsevier B.V. (e) Comparison between Nafion 212 and SPEEK/lignin membranes in aspects of permeability and ion selectivity. Reproduced with permission.124 Copyright 2018, Frontiers. (f) Schematic illustration of a vanadium redox flow battery employing the SPEEK/lignin composite membrane. Reproduced with permission.125 Copyright 2018, Elsevier Ltd. (g) The discharge capacity retention of VRFB cells using different membranes under a current density of 120 mA cm−2. Reproduced with permission.124 Copyright 2018, Frontiers. | ||
In addition to metal batteries, lignin was also utilized as separator modulators to prolong the lifespan of a vanadium redox flow battery (VRFB), a device holding great potential for large-scale energy storage. In this case, the redox activity of vanadium ions is particularly vital for the energy efficiency of the whole battery.122 Herein, a desirable separator should possess high proton conductivity, low vanadium permeability, excellent chemical and mechanical stability, and low cost. Despite its high cost (almost 30–40% of the VRFB stack), Nafion is still widely used in the VRFB system but it suffers from a high permeability of vanadium ions due to its relatively larger proton conduction channel size (∼1.5–5 nm) than the hydrated multivalent vanadium ions (∼0.6 nm). Such an inherent deficiency of Nafion causes a severe self-discharge and poor cycling stability of the VRFB. To extend the cycle life of the VRFB, by simple solution blending and casting, Ye et al.123 prepared a cost-competitive and eco-friendly Nafion/lignin composite membrane to serve as the separator. The rational incorporation of lignin with abundant hydroxyl groups into the Nafion matrix not only enlarged the size of the proton conduction channels, but also lowered the vanadium ion permeation. Among the numerous samples in Fig. 12d, the rNafion/L5 membrane (mass content of lignin was 5%) experienced the smallest swelling ratio, which indicated a dimension stability, and possessed the lowest VO2+ permeability, accompanied by decent ionic conductivity. It performed the best to tackle the aforementioned self-discharge and poor cycling stability issues, highlighting the shining characteristic of lignin for modulating the physiochemical features of conventional polymer separators. The replacement of commercial Nafion 212 membrane by rNafion/L5 extended the capacity retention of the VRFB from ∼34.8% (after 150 cycles) to 52.8% (after 1000 cycles).
From the perspective of cost, the Nafion membrane accounts for almost 30–40% of the cost of the whole VRFB. Even though the addition of lignin cut down the cost of the membrane by 28% (in contrast to commercial Nafion 212), the cost of rNafion/L5 remained quite high, mainly attributed to the high mass content of Nafion in the composite. Therefore, it is appealing to explore low-cost alternative systems without the usage of Nafion toward practical application. Ye et al.124 proposed the utilization of sulfonated poly(ether ether ketone) (SPEEK) as the substitutional membranes. This material integrates the advantages of low vanadium ion permeability, simple preparation, and high chemical and mechanical stability. More importantly, the price of SPEEK membrane merely accounts for about several tenths of that of commercial Nafion. They prepared a series of lignin/SPEEK composite membranes by solution casting and systematically investigated how the sulfonation degree of the SPEEK matrix affected the ion selectivity and stability. Encouragingly, the incorporation of lignin into the SPEEK substrate substantially suppressed the permeability of VO2+ ions, benefitting from its rich hydroxyl groups. The ion selectivity of the as-designed lignin/SPEEK composite membranes, namely the ratio of proton conductivity to ion permeability, was much higher than that of the Nafion 212, regardless of the degree of sulfonation in SPEEK. In particular, the SPEEK59/L15 membrane (the mass ratio of SPEEK to lignin was 59/15) presented the best performance (61.96 × 104 S min cm−3), far outstripping commercial Nafion 212 (12.78 × 104 S min cm−3) (Fig. 12e). Meanwhile, its vanadium ion permeability was lower than the rNafion/L5 sample illustrated above. Based on the synergistic effect of the lignin additives and SPEEK matrix, proton conductivity was improved and crossover of the vanadium ions was suppressed (Fig. 12f).125 As a consequence, the VRFB cell based on the SPEEK59/L15 membrane exhibited 95.95% capacity retention after 100 cycles at 120 mA cm−2 (Fig. 12g).
Up to now, lignin-derived ion exchange membranes with low cost and excellent performance have been developed in aqueous batteries. But it still has room for improvement. The manufacturing process is limited to the casting method, which consumes a large amount of organic solvents. A novel, facile and environmentally friendly method needs to be developed.
5.2. Organic device membranes
In organic devices like LIBs and Li–S batteries, polyolefin-based microporous materials such as polyethylene (PE) and polypropylene (PP) membranes are widely used as separators. Pitifully, they have some intrinsic limitations for future applications, like poor thermal stability, sluggish ionic transport and environmental pollution. Such deficiencies stimulate the exploration of new-type separators with competitive performance (mechanical strength, electrochemical stability, thermal stability and ion transport efficiency), as well as environmental benignity. With the rapid development of the battery market, tremendous efforts have been made to develop novel separators based on biodegradable materials.Cellulose separators have gained their great reputation in this field due to their cheap price, green sustainability and superb wettability. Restricted by its unique hygroexpansion feature, this material faces unsatisfactory mechanical strength and suffered from inevitable swelling in organic electrolytes. Nevertheless, appropriate mechanical properties are of essential importance to maintain the structural integrity and battery safety in behaving as LIBs separators. Aiming to develop a high-strength separator based on cellulose membranes, Xie et al.126 resorted to the hydrophobicity peculiarity of lignin to limit the swelling-induced volume expansion. Lignin was introduced to the cellulose pulp by a conventional solution casting method. The experimental and molecular study indicated that, in the composite membrane containing 2.5% and 5% lignin, the deformation of the amorphous region of cellulose under electrolyte immersion, the culprit that weakened the separator strength, did not take place any more. Yet, overdosing of lignin addition (7.5% and 10%) impaired the stability of the well-formed cellulose fibrous network, hence bringing about severe weakening of the Young's modulus (Fig. 13a top). This was because an appropriate amount of intermolecular hydrogen bonds could be formed between the cellulose molecules and lignin, creating a stronger and larger fibrous network with better mechanical strength in the electrolyte. Plus, the chemical stable voltage window of the as-designed lignin/cellulose composite membrane (4.2–4.7 V) was comparable to that of the commercial polypropylene separator, further attesting to its qualification as an LIB separator.
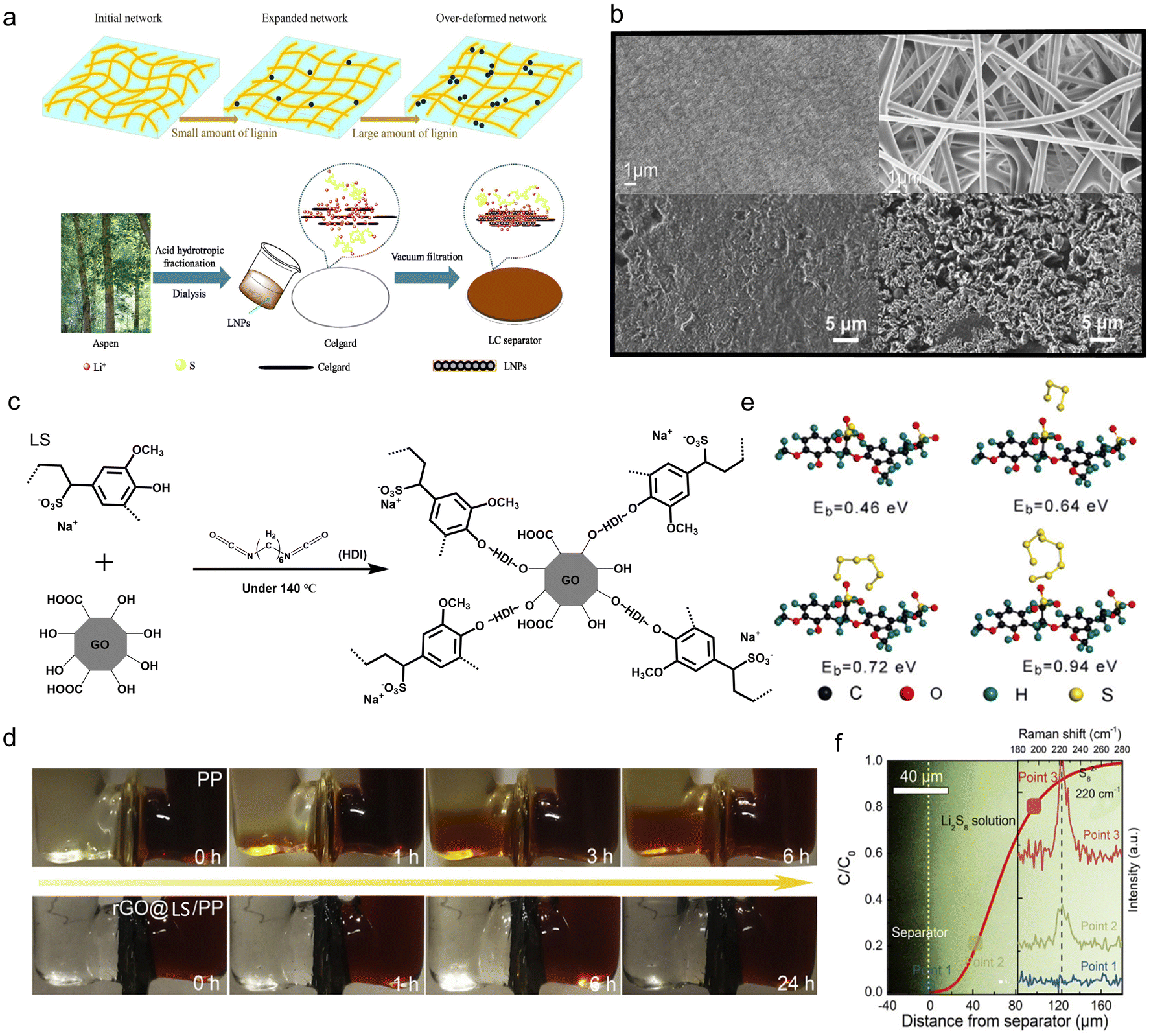 | ||
| Fig. 13 Lignin's application for membrane modification. (a) Effect mechanism of lignin particles(top). Reproduced with permission.126 Copyright 2020, Elsevier Ltd. Schematic of the fabrication process to produce the lignin nanoparticle (LNP)-coated Celgard (LC) separator (bottom). Reproduced with permission.131 Copyright 2019, MDPI. (b) SEM images of the Celgard separator at 20,000 times magnification (upper left) and lignin/PVA membrane at 10k× magnification (upper right). Top-view of deposited Li (1.0 mA h cm−2) in Li||Cu cells with L-SEI (lower left) and E-SEI (lower right). Reproduced with permission.127,128 Copyright 2017 and 2020, Elsevier B.V. (c) Molecular structure and synthetic steps to produce rGO@LS. (d) Permeation experiment with the double-L permeation device for the PP (top) and rGO@LS/PP separator (bottom). (e) Schematics and theoretical calculation of various PS conformations on LS. (f) The concentration of PS at different potentials where the inset image shows the PS concentration evolution and the corresponding Raman spectra at points 1, 2, and 3. Reproduced with permission.129 Copyright 2018, Elsevier Inc. | ||
Apart from mechanical stability, high ionic conductivity is also a vital requirement for separators in LIBs. Non-woven membranes have gained popularity in ion transport enhancement considering their high porosity. By virtue of an electrospinning method, Uddin et al.127 obtained lignin-based non-woven membranes with a fully interconnected pore structure and high porosity. The precursor was a water-based solution consisting of lignin and PVA with the mass ratio of 1:1, a biocompatible polymer with good chemical and thermal stability. In this case, the PVA polymer provided the necessary skeleton for electrospinning while lignin was employed to improve the porosity, wettability, and thermal stability. In terms of thermal endurance, the as-fabricated lignin/PVA membrane underwent only ∼15% shrinkage under 150 °C heat treatment for 1 h (45% for a commercial Celgard separator), exhibiting excellent flame-retardant properties. Meanwhile, unlike the flat and poreless morphology of the Celgard membrane (Fig. 13b upper left), the composite membrane had a porous interpenetrating network structure (Fig. 13b upper right). Such specific architecture endowed the separator with superior wettability (533% electrolyte uptake) and improved ionic conductivity. Despite its higher thickness than the Celgard separator (90 μm vs. 25 μm), the lignin/PVA membrane delivered an enhanced electrochemical performance due to the high ion transport efficacy. The Li//Li(Ni0.33Mn0.33Co0.33)O2 half-cell with a lignin/PVA membrane showed a better rate performance (133.3, 117.9, 91.2 and 33.4 mA h g−1 at 0.5 C, 1 C, 2 C, and 5 C) than those with the Celgard separator (122.6, 88.8, 47.8, and 7.6 mA h g−1 at the same respective C-rates).
Interestingly, besides tailoring the physical parameters, separator modification by lignin in organic devices is also capable of modulating the interfacial chemical reactions. In lithium metal batteries, the properties of the solid electrolyte interphase (SEI) are decisive for the cycling life of the Li anode as it is closely related to the lithium dendrite growth. The SEI resulting from spontaneous electrolyte decomposition (E-SEI) is usually heterogeneous and fragile, which is not firm enough to immune an even Li deposition and dendrite growth. LS contains rich cross-linkable aromatic skeletons and negatively charged sulfonic groups, displaying high reactivity with Li metal. When an ultra-thin (∼1.7 μm) LS was coated on PP via a doctor-blade method, the composite separator could in situ react with the Li metal to form a robust SEI film at the anode surface.128 The chemical composition comparison of E-SEI and LS-induced SEI (L-SEI) revealed that they were both composed of inorganic species (such as Li2CO3, LiF and Li2S) and organic components (C–C/C–H, ROLi and ROCO2Li). However, the content of inorganic and aromatic species dramatically increased in L-SEI, ensuring its high ionic conductivity (produced by abundant inorganic species) and good mechanical integrity (inherited from the aromatic groups). As depicted in Fig. 13b lower left, L-SEI produced a highly reversible, dendrite-free Li anode with long-term durability, while the one with E-SEI (Fig. 13b lower right) saw apparent dead Li accumulation. Benefitting from the efficient plating/stripping manner of the Li anode, the Li//LiNi0.5Co0.2Mn0.3O2 full cell containing L-SEI had the discharge capacity of 148.5 mA h g−1 and 73.5% capacity retention after 100 cycles under 0.4 C.
Similar to the strategy for binders, negatively charged –SO3− groups in LS are beneficial for suppressing the shuttle effect caused by soluble polysulfides in Li–S batteries, an insurmountable challenge that significantly damages the battery lifecycle and sulfur utilization. Lei et al.129 coated a thin layer of rGO and sodium LS composite on a PP membrane by a simple filtration method and the resulting product was called rGO@LS/PP. The rGO@LS was produced by connecting the hydroxyl groups of lignin with the rGO through linear chain compound hexamethylene diisocyanate by a chemical reaction (Fig. 13c). It could be clearly seen from permeation experiment that the rGO@LS/PP membrane effectively blocked the polysulfides (Fig. 13d), which was contributed by the dendrite structure and negatively charged –SO3− groups in the LS. Raman spectra also confirmed that the closer the polysulfides were to the rGO@LS/PP membrane, the weaker the polysulfide signals were (Fig. 13f). They reasoned from this that LS displayed an electrostatic repulsion effect on all polysulfide species, especially with a larger repulsive force for long-chain polysulfides (such as Li2S8 and Li2S6), as confirmed by theoretical calculation in Fig. 13e. As a result, the Li–S battery achieved a capacity of 816 mA h g−1 at 1 C after 500 cycles, and could run for 1000 cycles at 2 C. That cycling performance stayed comfortably ahead in recent Li–S battery separator coatings studies.130 In addition to sulfonate groups, other functional groups with the electron-donating features of lignin could also suppress polysulfide dissolution. The strategic loading of lignin nanoparticles (LNPs) on commercial Celgard membranes by a vacuum filtration method (Fig.13a bottom) raised the capacity retention ratio from 40.1% to 72.9% after 400 cycles.131 There were two ways for the sulfur-free LNPs to inhibit the shuttle effect. First, LNPs served as a temporary electrolyte reservoir to restrain polysulfides from directly diffusing into the bulk electrolyte. Second, the electron-donating groups (CC benzene ring and –CH3) induced chemical binding with the polysulfides. Besides, the overwhelming cycling stability of the LC separator over the Celgard separator was maintained at a high rate. The details of various electrolytes and membranes in terms of compositions, potentials, specific capacities, cycling stabilities and ionic conductivities were listed in Table 3.
| Cathode | Anode | Electrolyte components | Potential (V) | Specific capacity (mA h g−1) | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| AC = activated carbon; SS = stainless steel; DOL = 1,3-dioxolane; DME = dimethyl ether; ACET = acetylene black. | ||||||
| N/A | N/A | LS/HClO4 (anolyte) Br/HBr (catholyte) | 0.1–0.7 | 7 W h L−1 | N/A | 48 |
| Carbon | Carbon | 5 g L−1 LS2 in 1 M H2SO4 | 0–0.8 | 107 F g−1 (1 mV s−1) | 104.5% (5000 cycles) | 88 |
| MnO2 | Zn | 0.02 wt% LS in 2 M ZnSO4 + 0.2 M MnSO4 | 1.0–1.85 | 146 (1 A g−1) | 83.5% (1000 cycles) | 89 |
| Li | LiFePO4 | GE | 1.0–7.5 | 129 (1 C) | N/A | 91 |
| Li | Sulfur | G2 | −1.0–7.0 | 1186.3 (0.02 A g−1) | 55.1% (100 cycles) | 92 |
| AC | AC | LC-FD-Li2SO4 gel electrolyte | 0–2.1 | 138.4 F g−1 (1 A g−1) | 88% (10000 cycles) |
93 |
| Li | LiFePO4 | LP-GE | 0–8.0 | 117 (1 C) | 95.3% (100 cycles) | 94 |
| Li | Li | GE-LS-Cl | N/A | N/A | N/A | 95 |
| Graphene | Graphene | DGel-Fe1L0.5 | 0–2.0 | 51.4 F g−1 (0.5 A g−1) | 89.2% (5000 cycles) | 101 |
| α-MnO2 | Zn | QKL/FS-2 | 1.0–1.9 | 106.2 (1.5 A g−1) | N/A | 102 |
| Lignin/PAN | Lignin/PAN | AL-graft-PEGDGE | 0–1.0 | 129.23 (0.5 A g−1) | 95% (10000 cycles) |
104 |
| PANI@CC | PANI@CC | DC | 0–0.8 | 190 (0.25 A g−1) | 91% (10000 cycles) |
105 |
| PANI@CC | PANI@CC | LSNa-PEGDGE | 0–0.8 | 301.8 (0.5 A g−1) | 94.1% (10000 cycles) |
107 |
| SS | K | OxL/PEGDGE | 0–3.0 | 168 (0.05 A g−1) | 64.18% (200 cycles) | 110 |
| β-MnO2 | Zn | 2 M ZnSO4 | 0.9–1.9 | 236 (0.1 C) | N/A | 117 |
| N/A | N/A | 1.5 M V3+ in 3 M H2SO4 (anolyte) | 0.7–1.75 | ∼560 (120 mA cm−2) | 52.8% (1000 cycles) | 123 |
| 1.5 M VO2+ in 3 M H2SO4 (catholyte) | ||||||
| N/A | N/A | 1.5 M V3+ in 3 M H2SO4 (anolyte) | 0.7–1.75 | ∼550 (50 mA cm−2) | 85% (200 cycles) | 124 |
| 1.5 M VO2+ in 3 M H2SO4 (catholyte) | ||||||
| Li | Li(Ni0.33Mn0.33Co0.33)O2 | 1 M LiPF6 in EC/EMC (1:1, v/v) |
3.0–4.3 | 153.1 (0.1 C) | N/A | 127 |
| Li | LiNi0.5Co0.2Mn0.3O2 | 1 M LiTFSI in DOL/DME (1:1, v/v) + 2 wt% LiNO3 |
1.7–2.8 | 148.5 (0.4 C) | 73.5% (100 cycles) | 128 |
| Li | Sulfur | 1 M LiTFSI in DOL/DME (1:1, v/v) + 1 wt% LiNO3 |
1.7–2.7 | 816 (1 C) | 74% (1000 cycles) | 129 |
| Li | ACET/sulfur | 1 M LiTFSI in DOL/DME (1:1, v/v) + 1 wt% LiNO3 |
1.7–2.8 | 272 (1 C) | 72.9% (400 cycles) | 131 |
6. Conclusions and prospect
As the most abundant aromatic polymer in nature and a major by-product of the pulping industry, renewable lignin provides an sustainable platform for the construction of green and economical electrochemical energy storage systems including rechargeable batteries and supercapacitors. This should be attributed to its shining features including its low cost, huge reserves and abundant functional groups. In turn, the fabrication of ESD components by using lignin also provides a promising value-added pathway for lignin utilization, as demonstrated by the considerable inspiring achievements made in this field. This review does not include ESDs based on lignin-derived carbonaceous materials, to avoid possible duplication with some recent reviews. It begins with a detailed description of the sources, species and structural characteristics of lignin. Afterwards, the recent progress of lignin-based/-derived macromolecules as different ESD components is summarized in terms of organic electrode, binders, electrolyte and separators. It can be concluded that the utilization of lignin for electrochemical energy storage is a promising, fast-growing area but still at an early stage. Even though in some reported work, there are still some problems, like the low content or unclear role of lignin, they do demonstrate the application potential of lignin for ESDs and are enlightening for further performance optimization in the future. In view of the current status of lignin-involved ESDs, continued efforts should be directed towards electrode structure design, aiming at promoting efficient ion and electron transportation.132 In terms of binder optimization, their adaptability in aqueous environments needs urgently addressing. In addition, the application of lignin-based/-derived separators in some newly emerged battery systems (such as Zn–S or Zn–I2 batteries) is worth trying, focusing on solving the shuttle effect.133 For the electrolyte formulation, co-solvent electrolyte and organohydrogel electrolyte should be considered.134 The combination of lignin with ESDs is also a promising way to widen its application scenarios.To give some basic guidance in this area, some critical obstacles and challenges for the further practical application of lignin in ESDs are summarized as follows:
(1) Intricate structural randomness of macromolecular lignin. Apart from the similarity of the main structure, the exact molecular weight of different lignin molecules varies significantly from one to another. Depending on the origin, fabrication and derivation processes, some critical physicochemical properties of different lignins, parameters closely related to electrical energy storage, show vast discrepancies, like purity, solubility, mechanical strength, thermal stability, and the type and content of heteroatoms. Such intricate structural heterogeneity is currently the most pivotal bottleneck in the further fabrication of lignin-based materials because a slight variation of these parameters can substantially alter the electrochemical properties of the electrode. Plus, the chemical consistency of lignin is a basic requirement for the further development and optimization of the state-of-the-art materials. Therefore, efforts should also be dedicated to the development of advanced methods for lignin extraction and purification. The research direction includes upstream (depolymerizing lignin into molecule fragments or funtionalizing lignin into polymers) and downstream (upgrading lignin to the desired pure product via chemo- and bio-pathways) processes. For upstream-depolymerization processes, to obtain a monomeric product, the rational design of the catalytic system avoiding repolymerization is crucial. The ‘flow-through’ concept and ‘lignin-first’ strategies are examples of solutions in this regard.135 For downstream processes, a ‘funneling process’ is necessary to convert a mixture of depolymerised lignin into a single stream product rather than another mixture of value-added chemicals. In parallel, a one-pot conversion including both upstream and downstream processes is also a delightful future direction, for the sake of reducing product loss and energy consumption.136
(2) Limited understanding of the working mechanism of lignin-based/-derived electrodes. Restricted by the complex fabrication course and highly entangled structure of lignin-derived organic electrodes, the mechanism investigation is extremely difficult, especially referring to the interfacial chemistry. Currently, Q/QH2 redox pairs are recognized as the redox centers of the overwhelming majority of reported lignin-involved electrodes, both for supercapacitors and batteries. However, the present mechanism study is generally confined to the identification of reversible Q/QH2 transformation whereas the deep investigation of ion/electron conduction dynamics at the electrode/electrolyte interface is quite limited. The specific active sites of the electrode for energy storage remain mysterious, as well as its impact factors. New in situ characterization techniques, such as cryogenic electron microscopy that offers the possibility of detecting chemical reactions and beam-sensitive samples at the atomic scale without interfering with their natural state, may contribute to studying the complex chemical processes. In addition, it has been recently demonstrated that the CO and CC bonds can be activated for Li+ accommodation by suitable pre-treatment. The extension of activation methods and relevant mechanism study seem to be an untapped yet interesting area to explore.
(3) Narrow application scenarios in aqueous ESDs. The lignin-based/-derived macromolecules are mainly applied in organic energy storage systems like LIBs and Li–S batteries. In view of the demanding requirements for grid-scale energy storage, the research interest on economical, safe, and efficient aqueous batteries has boomed in recent years, especially stimulated by the continuously increased usage of renewable resources like solar and wind power. Encouragingly, some initial attempts have been reported in aqueous systems including supercapacitors, redox flow batteries as well as ZIBs, mainly for electrolyte formulation. In future, more attention should be paid to discovering the potential of lignin-based/-derived macromolecules to function as electrodes, binders and separators. However, the selected polymer should meet the requirements for water-dissolving resistance. Taking account of the solubility of some polar groups in aqueous electrolytes, tailoring the physiochemical properties of lignin should be considered, including chemical modification of the existing groups and grafting polymerization.
(4) Limited lignin resources usage. Most current studies focus on the application of LS and AL as the raw materials for investigation. Other lignin products of various plant types in different biorefinery processes such as organosolv lignin and EHL have been seldom discussed, and could be further developed and applied in ESD components considering their high-purity feature.34,137 Besides, compared with LS and AL, they possess higher homogeneity and narrower molecular distributions, displaying a distinct priority in post-processing. The deficiency of active sites is the main drawback of the sulfur-free lignins in ESDs. Converting these lignins into carbon materials is a straightforward way for their further application in supercapacitors or rechargeable batteries. The strategic introduction of new groups as the active redox sites in aromatic rings is also crucial to extend their energy storage applications.
(5) Lack of cost evaluation. The lignin-based/-derived ESDs reported in the literature are now restricted at the laboratory level. Although there is still great room for performance improvement for their commercial application, some of their optimal performances can be comparable to or even exceed that of conventional ESD components, as we previously discussed in the main text. Considering their high potential for practical application, the cost and sustainability is necessary to consider. Currently, lignin is considered as an industrial waste and commonly accepted as a cheap raw material for manufacturing various high-value chemical products. However, the commercial success of lignin application in ESDs is driven by the combined cost involved in the entire manufacturing process. Detailed understanding of the total cost of purification and chemical post-treatment like chemical, physical or thermal modification, which is especially important in the expansion from laboratory processing to commercial scale utilization, is still lacking. This area requires immediate attention in view of the commercial viability of lignin as an ESD component.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 51771058, 52271204, 22075330), Pearl River Talent Program of Guangdong Province (2017GC010030), Guangdong Basic and Applied Basic Research Foundation (2021A0505030067), and Guangdong Provincial Key Laboratory of Plant Resources Biorefinery (2021B1212010011).References
- P. Yan, C. Xiao, L. Xu, G. Yu, A. Li, S. Piao and N. He, Renewable Sustainable Energy Rev., 2020, 127, 109857 CrossRef.
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- D. Columbus, V. Arunachalam, F. Glang, L. Avram, S. Haber, A. Zohar, M. Zaiss and M. Leskes, J. Am. Chem. Soc., 2022, 144, 9836–9844 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- C. Heubner, M. Schneider and A. Michaelis, Adv. Energy Mater., 2020, 10, 1902523 CrossRef CAS.
- J. L. Espinoza-Acosta, P. I. Torres-Chávez, J. L. Olmedo-Martínez, A. Vega-Rios, S. Flores-Gallardo and E. A. Zaragoza-Contreras, J. Energy Chem., 2018, 27, 1422–1438 CrossRef.
- K. Yang, Q. Gao, Y. Tan, W. Tian, W. Qian, L. Zhu and C. Yang, Chemistry, 2016, 22, 3239–3244 CrossRef CAS PubMed.
- X. Fu and W. H. Zhong, Adv. Energy Mater., 2019, 9, 1901774 CrossRef CAS.
- K. Wu, J. Huang, J. Yi, X. Liu, Y. Liu, Y. Wang, J. Zhang and Y. Xia, Adv. Energy Mater., 2020, 10, 1903977 CrossRef CAS.
- H. Wang, Y. Yang and L. Guo, Adv. Energy Mater., 2017, 7, 1700663 CrossRef.
- P. G, S. As, J. S. Jayan, A. Raman and A. Saritha, Process Saf. Environ., 2021, 145, 395–410 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- H. Zhu, W. Luo, P. N. Ciesielski, Z. Fang, J. Y. Zhu, G. Henriksson, M. E. Himmel and L. Hu, Chem. Rev., 2016, 116, 9305–9374 CrossRef CAS PubMed.
- J. K. Weng and C. Chapple, New Phytol., 2010, 187, 273–285 CrossRef CAS PubMed.
- W. Zhang, J. Yin, C. Wang, L. Zhao, W. Jian, K. Lu, H. Lin, X. Qiu and H. N. Alshareef, Small Methods, 2021, 5, 2100896 CrossRef CAS PubMed.
- G. Lu, J. Zheng, C. Jin, T. Yan, L. Zhang, J. Nai, Y. Wang, Y. Liu, T. Liu and X. Tao, Chem. Eng. J., 2021, 409, 127454 CrossRef CAS.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- J. C. del Rio, J. Rencoret, P. Prinsen, A. T. Martinez, J. Ralph and A. Gutierrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef CAS PubMed.
- G. Milczarek, Langmuir, 2009, 25, 10345–10353 CrossRef CAS PubMed.
- G. Milczarek, Electroanalysis, 2008, 20, 211–214 CrossRef CAS.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- G. Milczarek, Electrochim. Acta, 2009, 54, 3199–3205 CrossRef CAS.
- F. N. Ajjan, M. Ambrogi, G. A. Tiruye, D. Cordella, A. M. Fernandes, K. Grygiel, M. Isik, N. Patil, L. Porcarelli, G. Rocasalbas, G. Vendramientto, E. Zeglio, M. Antonietti, C. Detrembleur, O. Inganäs, C. Jérôme, R. Marcilla, D. Mecerreyes, M. Moreno, D. Taton, N. Solin and J. Yuan, Polym. Int., 2017, 66, 1119–1128 CrossRef CAS.
- M. Gandhi, D. Rajagopal and A. Senthil Kumar, ACS Omega, 2020, 5, 16208–16219 CrossRef CAS PubMed.
- M. Sun, X. Wang, S. Ni, L. Jiao, H. Bian and H. Dai, Ind. Crops Prod., 2022, 187, 115441 CrossRef CAS.
- V. Patil, S. Adhikari, P. Cross and H. Jahromi, Renewable Sustainable Energy Rev., 2020, 133, 110359 CrossRef CAS.
- H. Liu, T. Xu, K. Liu, M. Zhang, W. Liu, H. Li, H. Du and C. Si, Ind. Crops Prod., 2021, 165, 113425 CrossRef CAS.
- C. Chio, M. Sain and W. Qin, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- U. Ail, J. Phopase, J. Nilsson, Z. U. Khan, O. Inganäs, M. Berggren and X. Crispin, ACS Sustainable Chem. Eng., 2020, 8, 17933–17944 CrossRef CAS.
- A. Eraghi Kazzaz and P. Fatehi, Ind. Crops Prod., 2020, 154, 112732 CrossRef CAS.
- G. Yan, S. He, G. Chen, S. Ma, A. Zeng, B. Chen, S. Yang, X. Tang, Y. Sun, F. Xu, L. Lin and X. Zeng, Nano-Micro Lett., 2022, 14, 84 CrossRef CAS PubMed.
- L. A. Zevallos Torres, A. Lorenci Woiciechowski, V. O. de Andrade Tanobe, S. G. Karp, L. C. Guimarães Lorenci, C. Faulds and C. R. Soccol, J. Cleaner Prod., 2020, 263, 121499 CrossRef CAS.
- Y. Jin, X. Cheng and Z. Zheng, Bioresour. Technol., 2010, 101, 2046–2048 CrossRef CAS PubMed.
- C. Alvarez, F. M. Reyes-Sosa and B. Diez, Microb. Biotechnol., 2016, 9, 149–156 CrossRef CAS PubMed.
- B. Zhang, D. Yang, X. Qiu, Y. Qian, H. Wang, C. Yi and D. Zhang, Carbon, 2020, 162, 256–266 CrossRef CAS.
- N. Guo, M. Li, X. Sun, F. Wang and R. Yang, Green Chem., 2017, 19, 2595–2602 RSC.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- R. Nadányi, A. Ház, A. Lisý, M. Jablonský, I. Šurina, V. Majová and A. Baco, Energies, 2022, 15, 6520 CrossRef.
- A. J. Robinson, A. Giuliano, O. Y. Abdelaziz, C. P. Hulteberg, A. Koutinas, K. S. Triantafyllidis, D. Barletta and I. De Bari, Bioresour. Technol., 2022, 364, 128004 CrossRef CAS PubMed.
- J. Wenger, V. Haas and T. Stern, Curr. For. Rep., 2020, 6, 294–308 Search PubMed.
- P. S. Chauhan, R. Agrawal, A. Satlewal, R. Kumar, R. P. Gupta and S. S. V. Ramakumar, Int. J. Biol. Macromol., 2022, 197, 179–200 CrossRef CAS PubMed.
- X. Wu, J. Jiang, C. Wang, J. Liu, Y. Pu, A. Ragauskas, S. Li and B. Yang, Biofuels, Bioprod. Biorefin., 2020, 14, 650–672 CrossRef CAS.
- W. Zhang, X. Qiu, C. Wang, L. Zhong, F. Fu, J. Zhu, Z. Zhang, Y. Qin, D. Yang and C. C. Xu, Carbon Res., 2022, 1, 14 CrossRef.
- H. Y. Jung, J. S. Lee, H. T. Han, J. Jung, K. Eom and J. T. Lee, Polymers, 2022, 14, 673 CrossRef CAS PubMed.
- W. J. Chen, C. X. Zhao, B. Q. Li, T. Q. Yuan and Q. Zhang, Green Chem., 2022, 24, 565–584 RSC.
- A. Mukhopadhyay, J. Hamel, R. Katahira and H. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 5394–5400 CrossRef CAS.
- N. R. Tanguy, H. Wu, S. S. Nair, K. Lian and N. Yan, ChemSusChem, 2021, 14, 1057–1067 CrossRef CAS PubMed.
- W. Yang, Y. Qu, B. Zhou, C. Li, L. Jiao and H. Dai, Ind. Crops Prod., 2021, 171, 113848 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- N. Dianat, M. S. Rahmanifar, A. Noori, M. F. El-Kady, X. Chang, R. B. Kaner and M. F. Mousavi, Nano Lett., 2021, 21, 9485–9493 CrossRef CAS PubMed.
- C. Han, Y. Ye, G. Wang, W. Hong and C. Feng, Chem. Eng. J., 2018, 347, 648–659 CrossRef CAS.
- Z. Peng, Y. Zou, S. Xu, W. Zhong and W. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 22190–22200 CrossRef CAS PubMed.
- L. Chang, Z. Peng, T. Zhang, C. Yu and W. Zhong, Nanoscale, 2021, 13, 3079–3091 RSC.
- C. Liu, Z. Li, X. Zhang, W. Xu, W. Chen, K. Zhao, Y. Wang, S. Hong, Q. Wu, M. C. Li and C. Mei, Adv. Sci., 2022, 9, 2202380 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- D. Wang, F. Yang, L. Cong, W. Feng, C. Wang, F. Chu, J. Nan and R. Chen, Chem. Eng. J., 2022, 450, 138025 CrossRef CAS.
- B. Li, P. F. Cao, T. Saito and A. P. Sokolov, Chem. Rev., 2023, 123, 701–735 CrossRef CAS PubMed.
- F. Mo, Q. Li, G. Liang, Y. Zhao, D. Wang, Y. Huang, J. Wei and C. Zhi, Adv. Sci., 2021, 8, 2100072 CrossRef CAS PubMed.
- B. Zhou, J. Li, W. Liu, H. Jiang, S. Li, L. Tan, L. Dong, L. She and Z. Wei, ChemSusChem, 2020, 13, 2628–2633 CrossRef CAS PubMed.
- W. Yang, X. Wang, L. Jiao, H. Bian, Y. Qiao and H. Dai, J. Mater. Chem. A, 2020, 8, 24065–24074 RSC.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 CAS.
- Y. Yin, C. Liu and S. Fan, Nano Energy, 2015, 12, 486–493 CrossRef CAS.
- N. Casado, M. Hilder, C. Pozo-Gonzalo, M. Forsyth and D. Mecerreyes, ChemSusChem, 2017, 10, 1783–1791 CrossRef CAS PubMed.
- Y. Yang, C. Liu, Z. Lv, H. Yang, Y. Zhang, M. Ye, L. Chen, J. Zhao and C. C. Li, Adv. Mater., 2021, 33, 2007388 CrossRef CAS PubMed.
- A. Lahiri, L. Yang, O. Höfft and F. Endres, Mater. Adv., 2021, 2, 2676–2683 RSC.
- Z. Tie, S. Deng, H. Cao, M. Yao, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202115180 CrossRef CAS PubMed.
- L. Zhou, L. Liu, Z. Hao, Z. Yan, X. F. Yu, P. K. Chu, K. Zhang and J. Chen, Matter, 2021, 4, 1252–1273 CrossRef CAS.
- X. Zhang, T. Xiong, B. He, S. Feng, X. Wang, L. Wei and L. Mai, Energy Environ. Sci., 2022, 15, 3750–3774 RSC.
- Z. Khan, U. Ail, F. N. Ajjan, J. Phopase, N. Kim, D. Kumar, Z. U. Khan, J. Nilsson, O. Inganäs, M. Berggren and X. Crispin, J. Power Sources, 2022, 524, 231103 CrossRef CAS.
- Z. Khan, U. Ail, F. Nadia Ajjan, J. Phopase, Z. Ullah Khan, N. Kim, J. Nilsson, O. Inganäs, M. Berggren and X. Crispin, Adv. Energy Sustainability Res., 2022, 3, 2100165 CrossRef CAS.
- K. Yang, L. Yang, Z. Wang, B. Guo, Z. Song, Y. Fu, Y. Ji, M. Liu, W. Zhao, X. Liu, S. Yang and F. Pan, Adv. Energy Mater., 2021, 11, 2100601 CrossRef CAS.
- J. Li, Y. Cai, H. Wu, Z. Yu, X. Yan, Q. Zhang, T. Z. Gao, K. Liu, X. Jia and Z. Bao, Adv. Energy Mater., 2021, 11, 2003239 CrossRef CAS.
- T. C. Nirmale, B. B. Kale and A. J. Varma, Int. J. Biol. Macromol., 2017, 103, 1032–1043 CrossRef CAS PubMed.
- Y. Zhao, Z. Liang, Y. Kang, Y. Zhou, Y. Li, X. He, L. Wang, W. Mai, X. Wang, G. Zhou, J. Wang, J. Li, N. Tavajohi and B. Li, Energy Storage Mater., 2021, 35, 353–377 CrossRef.
- J. T. Li, Z. Y. Wu, Y. Q. Lu, Y. Zhou, Q. S. Huang, L. Huang and S. G. Sun, Adv. Energy Mater., 2017, 7, 1701185 CrossRef.
- Y. Ma, K. Chen, J. Ma, G. Xu, S. Dong, B. Chen, J. Li, Z. Chen, X. Zhou and G. Cui, Energy Environ. Sci., 2019, 12, 273–280 RSC.
- W. Ren, W. Ma, S. Zhang and B. Tang, Energy Storage Mater., 2019, 23, 707–732 CrossRef.
- J. Jeon, J. K. Yoo, S. Yim, K. Jeon, G. H. Lee, J. H. Yun, D. K. Kim and Y. S. Jung, ACS Sustainable Chem. Eng., 2019, 7, 17580–17586 CrossRef CAS.
- X. Wu, C. Luo, L. Du, Y. Xiao, S. Li, J. Wang, C. Wang and Y. Deng, ACS Sustainable Chem. Eng., 2019, 7, 8413–8418 CrossRef CAS.
- C. Luo, L. Du, W. Wu, H. Xu, G. Zhang, S. Li, C. Wang, Z. Lu and Y. Deng, ACS Sustainable Chem. Eng., 2018, 6, 12621–12629 CrossRef CAS.
- D. Jeong, J. Shim, H. Shin and J. C. Lee, ChemSusChem, 2020, 13, 2642–2649 CrossRef CAS PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- S. Yuan, T. Kong, Y. Zhang, P. Dong, Y. Zhang, X. Dong, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 25624–25638 CrossRef CAS PubMed.
- P. Ding, Z. Lin, X. Guo, L. Wu, Y. Wang, H. Guo, L. Li and H. Yu, Mater. Today, 2021, 51, 449–474 CrossRef CAS.
- G. Lota and G. Milczarek, Electrochem. Commun., 2011, 13, 470–473 CrossRef CAS.
- W. Zhou, M. Chen, Q. Tian, J. Chen, X. Xu, X. Han and J. Xu, J. Colloid Interface Sci., 2021, 601, 486–494 CrossRef CAS PubMed.
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS.
- S. D. Gong, Y. Huang, H. J. Cao, Y. H. Lin, Y. Li, S. H. Tang, M.-S. Wang and X. Li, J. Power Sources, 2016, 307, 624–633 CrossRef CAS.
- A. Song, Y. Huang, X. Zhong, H. Cao, B. Liu, Y. Lin, M. Wang and X. Li, J. Membr. Sci., 2018, 556, 203–213 CrossRef CAS.
- F. Qiu, Y. Huang, G. He, C. Luo, X. Li, M. Wang and Y. Wu, Electrochim. Acta, 2020, 363, 137241 CrossRef CAS.
- B. Liu, Y. Huang, H. Cao, A. Song, Y. Lin, M. Wang and X. Li, J. Solid State Electrochem., 2018, 22, 807–816 CrossRef CAS.
- N. S. Shabanov, K. S. Rabadanov, M. M. Gafurov, A. B. Isaev, D. S. Sobola, S. I. Suleimanov, A. M. Amirov and A. S. Asvarov, Polymers, 2021, 13, 2306 CrossRef CAS PubMed.
- Z. Liu, K. Shikinaka, Y. Otsuka and Y. Tominaga, Chem. Commun., 2022, 58, 4504–4507 RSC.
- M. Wu, X. Zhang, Y. Zhao, C. Yang, S. Jing, Q. Wu, A. Brozena, J. T. Miller, N. J. Libretto, T. Wu, S. Bhattacharyya, M. N. Garaga, Y. Zhang, Y. Qi, S. G. Greenbaum, R. M. Briber, Y. Yan and L. Hu, Nat. Nanotechnol., 2022, 17, 629–636 CrossRef CAS PubMed.
- C. Yang, Q. Wu, W. Xie, X. Zhang, A. Brozena, J. Zheng, M. N. Garaga, B. H. Ko, Y. Mao, S. He, Y. Gao, P. Wang, M. Tyagi, F. Jiao, R. Briber, P. Albertus, C. Wang, S. Greenbaum, Y. Y. Hu, A. Isogai, M. Winter, K. Xu, Y. Qi and L. Hu, Nature, 2021, 598, 590–596 CrossRef PubMed.
- Y. Qian, Y. Zhou, M. Lu, X. Guo, D. Yang, H. Lou, X. Qiu and C. F. Guo, Small Methods, 2021, 5, 2001311 CrossRef CAS PubMed.
- D. Sun, Y. Feng, S. Sun, J. Yu, S. Jia, C. Dang, X. Hao, J. Yang, W. Ren, R. Sun, C. Shao and F. Peng, Adv. Funct. Mater., 2022, 32, 2201335 CrossRef CAS.
- J. Wang, Y. Deng, Z. Ma, Y. Wang, S. Zhang and L. Yan, Green Chem., 2021, 23, 5120–5128 RSC.
- J. Xu, M. Wang, M. A. Alam, G. Muhammad, Y. Lv, C. Zhu, H. Zhang and W. Xiong, ACS Sustainable Chem. Eng., 2022, 10, 2063–2071 CrossRef CAS.
- H. Liu, L. Mulderrig, D. Hallinan Jr. and H. Chung, Macromol. Rapid Commun., 2021, 42, 2000428 CrossRef CAS PubMed.
- J. H. Park, H. H. Rana, J. Y. Lee and H. S. Park, J. Mater. Chem. A, 2019, 7, 16962–16968 RSC.
- T. Liu, X. Ren, J. Zhang, J. Liu, R. Ou, C. Guo, X. Yu, Q. Wang and Z. Liu, J. Power Sources, 2020, 449, 227532 CrossRef CAS.
- L. Dai, M. Ma, J. Xu, C. Si, X. Wang, Z. Liu and Y. Ni, Chem. Mater., 2020, 32, 4324–4330 CrossRef CAS.
- A. K. Mondal, D. Xu, S. Wu, Q. Zou, F. Huang and Y. Ni, Biomacromolecules, 2022, 23, 766–778 CrossRef CAS PubMed.
- A. K. Mondal, S. Wu, D. Xu, Q. Zou, L. Chen, L. Huang, F. Huang and Y. Ni, Int. J. Biol. Macromol., 2021, 187, 189–199 CrossRef CAS PubMed.
- J. C. de Haro, E. Tatsi, L. Fagiolari, M. Bonomo, C. Barolo, S. Turri, F. Bella and G. Griffini, ACS Sustainable Chem. Eng., 2021, 9, 8550–8560 CrossRef CAS PubMed.
- S. Trano, F. Corsini, G. Pascuzzi, E. Giove, L. Fagiolari, J. Amici, C. Francia, S. Turri, S. Bodoardo, G. Griffini and F. Bella, ChemSusChem, 2022, 15, e202200294 CrossRef CAS PubMed.
- H. Hao, T. Hutter, B. L. Boyce, J. Watt, P. Liu and D. Mitlin, Chem. Rev., 2022, 122, 8053–8125 CrossRef CAS PubMed.
- Q. Ni, B. Kim, C. Wu and K. Kang, Adv. Mater., 2022, 34, 2108206 CrossRef CAS PubMed.
- Z. Liu, Y. Jiang, Q. Hu, S. Guo, L. Yu, Q. Li, Q. Liu and X. Hu, Energy Environ. Mater., 2021, 4, 336–362 CrossRef CAS.
- S. Li, W. Zhang, J. Zheng, M. Lv, H. Song and L. Du, Adv. Energy Mater., 2021, 11, 2000779 CrossRef CAS.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- Y. Liu, X. Mao, H. Wu, X. Wang, B. Shi, C. Fan, Y. Kong and Z. Jiang, J. Membr. Sci., 2022, 644, 120126 CrossRef CAS.
- D. Yuan, W. Manalastas Jr., L. Zhang, J. J. Chan, S. Meng, Y. Chen and M. Srinivasan, ChemSusChem, 2019, 12, 4889–4900 CrossRef CAS PubMed.
- D. Wang, Q. Li, Y. Zhao, H. Hong, H. Li, Z. Huang, G. Liang, Q. Yang and C. Zhi, Adv. Energy Mater., 2022, 12, 2102707 CrossRef CAS.
- W. Du, J. Yan, C. Cao and C. C. Li, Energy Storage Mater., 2022, 52, 329–354 CrossRef.
- Y. Zhang, G. Yang, M. L. Lehmann, C. Wu, L. Zhao, T. Saito, Y. Liang, J. Nanda and Y. Yao, Nano Lett., 2021, 21, 10446–10452 CrossRef CAS PubMed.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- Z. He, Y. Lv, T. Zhang, Y. Zhu, L. Dai, S. Yao, W. Zhu and L. Wang, Chem. Eng. J., 2022, 427, 131680 CrossRef CAS.
- J. Ye, D. Yuan, M. Ding, Y. Long, T. Long, L. Sun and C. Jia, J. Power Sources, 2021, 482, 229023 CrossRef CAS.
- J. Ye, X. Lou, C. Wu, S. Wu, M. Ding, L. Sun and C. Jia, Front. Chem., 2018, 6, 549 CrossRef CAS PubMed.
- J. Ye, Y. Cheng, L. Sun, M. Ding, C. Wu, D. Yuan, X. Zhao, C. Xiang and C. Jia, J. Membr. Sci., 2019, 572, 110–118 CrossRef CAS.
- W. Xie, Y. Dang, L. Wu, W. Liu, A. Tang and Y. Luo, Polym. Test., 2020, 90, 106773 CrossRef CAS.
- M. J. Uddin, P. K. Alaboina, L. Zhang and S. J. Cho, Mater. Sci. Eng., B, 2017, 223, 84–90 CrossRef CAS.
- J. Liu, R. Xu, C. Yan, H. Yuan, J. F. Ding, Y. Xiao, T. Q. Yuan and J. Q. Huang, Energy Storage Mater., 2020, 30, 27–33 CrossRef.
- T. Lei, W. Chen, W. Lv, J. Huang, J. Zhu, J. Chu, C. Yan, C. Wu, Y. Yan, W. He, J. Xiong, Y. Li, C. Yan, J. B. Goodenough and X. Duan, Joule, 2018, 2, 2091–2104 CrossRef CAS.
- N. Shi, B. Xi, J. Liu, Z. Zhang, N. Song, W. Chen, J. Feng and S. Xiong, Adv. Funct. Mater., 2022, 32, 2111586 CrossRef CAS.
- Z. Zhang, S. Yi, Y. Wei, H. Bian, R. Wang and Y. Min, Polymers, 2019, 11, 1946 CrossRef CAS PubMed.
- G. Shi, X. Peng, J. Zeng, L. Zhong, Y. Sun, W. Yang, Y. L. Zhong, Y. Zhu, R. Zou, S. A. Molla, Z. Liu, C. Liu, E. I. Iwuoha and J. Lu, Adv. Mater., 2023, 2300109 CrossRef PubMed.
- Y. Zhao, D. Wang, X. Li, Q. Yang, Y. Guo, F. Mo, Q. Li, C. Peng, H. Li and C. Zhi, Adv. Mater., 2020, 32, 2003070 CrossRef CAS PubMed.
- Z. Xing, G. Xu, J. Han, G. Chen, B. Lu, S. Liang and J. Zhou, Trends Chem., 2023, 5, 380–392 CrossRef CAS.
- Z. Sun, J. Cheng, D. Wang, T. Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- A. T. Smit, M. Verges, P. Schulze, A. van Zomeren and H. Lorenz, ACS Sustainable Chem. Eng., 2022, 10, 10503–10513 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |