
Exploring the electrochemical ring hydrogenation of furanic compounds†
Thorben
Lenk
 ac,
Valentin
Rueß
a,
Janko
Gresch
a and
Uwe
Schröder
*bc
ac,
Valentin
Rueß
a,
Janko
Gresch
a and
Uwe
Schröder
*bc
aInstitute of Environmental and Sustainable Chemistry, Technische Universität Braunschweig, Braunschweig, Germany
bInstitute of Biochemistry, Universität Greifswald, Greifswald, Germany. E-mail: uwe.schroeder@uni-greifswald.de
cCluster of Excellence SE2A-Sustainable and Energy-Efficient Aviation, Technische Universität Braunschweig, Braunschweig, Germany
First published on 28th March 2023
Abstract
The electrochemical ring hydrogenation of aromatic compounds is an important synthetic tool, though scarcely described in the literature. This research paper investigates the utilization of different Pd- and Pt-containing electrocatalyst materials for an electrosynthesis of tetrahydrofuran derivatives from furfural and related furans. It focuses on the selectivity of the ring hydrogenation as a function of the electrocatalyst composition, as well as on a preliminary elucidation of the electrocatalytic activity during repetitive electrode use. Furthermore, different furanic derivatives are employed as starting material for mechanistic considerations. While furfural, furfuryl alcohol and furan are found to be highly reactive in ring hydrogenation reactions, furoic acid and 2-methyl furan do not show this behavior. Selectivities for tetrahydrofurfuryl alcohol of up to 15.3% (from furfural) and 33% (from furfuryl alcohol) are observed in this context.
Introduction
Replacing fossil resources with sustainable alternatives has become more necessary than ever.1 This applies for combustion fuels as well as for chemicals, for which solutions have to be found. One option for an environmentally more friendly production of chemicals and fuels is the utilization of waste biomass constituents, valorized by reactions powered by regenerative electric energy – i.e., by electrosynthesis.2 The majority of studies using electrosynthesis for the valorization of biomass components deal with the removal of oxygen functionalities. Hereby, besides the decarboxylation (Kolbe and Non-Kolbe reaction),3 especially hydrogenation (and hydrodeoxygenation) reactions of carbonyl and hydroxyl functional groups have been intensively studied.4–9 In contrast, reactions employing the hydrogenation of aromatic compounds are scarce due to the high stability of aromatic systems.7,10Since the discovery of benzene – its remarkable stability, resonance structures, and aromaticity – aromatic compounds have been an essential class of chemicals.11 The hydrogenation of the aromatic ring has been of particular interest as it opens an entire field of new synthetic pathways to saturated cyclic compounds. As the reactivity is relatively low due to the aromaticity, the reactions require relatively harsh conditions in, e.g., the Birch reduction with metals as reducing agents.12 Other methods employ precious metals as heterogeneous13 or homogeneous catalysts, hydrogen atmospheres with elevated pressures (0.1–10 MPa) and temperatures (150–380 °C).13–15
By providing the necessary reduction equivalents in situ at the electrode surface, electrosynthesis avoids chemical wastes that are unavoidable when using chemical reducing agents. In combination with mild reaction conditions, the use of water as the reaction solvent, electrosynthesis obeys the major rules of Green Chemistry.16,17 Hence, an electroorganic hydrogenation of aromatic compounds is desirable. In electroorganic reactions, the reduction of aromatic systems remains challenging,18 although some research groups were successful with the electrochemical hydrogenation of aromatic rings.7,19–22 These findings show possibilities ranging from electrolytic Birge-type reductions23,24 to the use of electrocatalysts such as RANEY®-Ni, Pt- or Ru-electrodes.7,19,20
As they can easily be derived from carbohydrates via dehydration, the two aromatic compounds furfural (FF) and 5-hydroxymethylfurfural (HMF) have repeatedly been the subject of electrosynthesis studies.25,26 These studies usually focused on the aldehyde hydrogenation to yield the respective methyl (MF) or alcohol (FA) derivative (see Fig. 1) and typically employ Cu working electrodes in acidic media.27–30 The ring hydration was less often investigated. In this regard, Green et al. demonstrated ring hydrogenation of FF towards the saturated products 2-methyl tetrahydrofuran (MTHF) and tetrahydrofurfuryl alcohol (THFA) in membrane electrode assemblies using Pt/C and Pd/C cathodes (see Fig. 1).31 In these experiments, Pd/C was a more suitable catalyst material than Pt/C, producing primarily FA, with THFA as a secondary product with up to 25% selectivity on Pd/C. MF and MTHF were also formed as minor products.31 Another approach for ring hydrogenation using membrane reactors was presented by Delima et al., comparing different solvent and catalyst conditions using Pd/Pd membranes as the foundation for their catalyst material.32 All the presented hydrogenation reactions have in common that they primarily obtain aldehyde hydrogenation products, while ring hydrogenation products show lower selectivities. These observations are expectable due to the higher energy barrier of the ring hydrogenation.31 Only Delima et al. could show reaction conditions with THFA as the primary product.32 Besides FF, also HMF was investigated for its ability to undergo a ring hydrogenation. In this regard, Kwon et al. presented a variety of electrode materials that can serve as electrocatalysts towards the ring-hydrogenated bis(hydroxymethyl)tetrahydrofuran as a minor product with Bi, Sb, and Ag showing the highest rate of ring hydrogenation.33,34 A mechanistic study on the hydrogenation of furan (FUR) was published by Wang et al.35
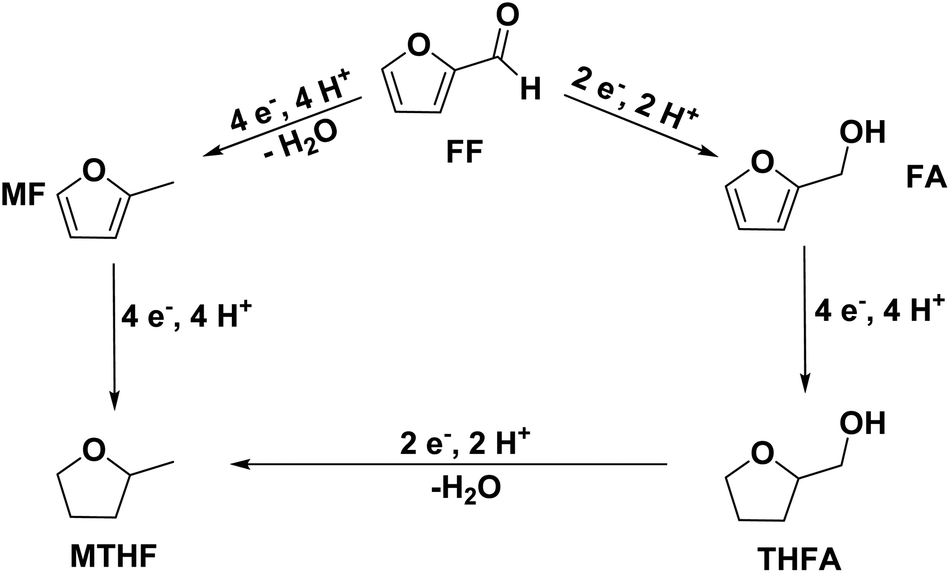 | ||
| Fig. 1 Schematic overview of the possible pathways of the electrochemical hydrogenation of furfural. (FF: furfural; MF: 2-methyl furan; FA: furfuryl alcohol; THFA: tetrahydrofurfuryl alcohol; MTHF: 2-methyl tetrahydrofuran. | ||
An exploration of the electrosynthesis route of furfural towards THFA and MTHF is desirable as it allows the production of both components from the same regenerative precursor. THFA is typically used as a solvent for various applications (including dyes, inks, pesticides, and herbicides), polymer production, and the preparation of pharmaceuticals.36 Especially its high boiling point and biodegradability make it an interesting molecule.36 MTHF is also used as a solvent and as a fuel component.2,36 Compared to the corresponding furans, the ring-hydrogenated compounds have a reduced oxidation state, providing a potential route for the storage of excess electrical energy – and for a potential combustion fuel for, e.g., regenerative mobility applications.
This research paper explores the ring hydrogenation reaction of FF under consideration of different electrode material compositions and their behavior over time. Mechanistic considerations are made evaluating the influence of the starting material on the ring hydrogenation. Different studies suggest that the electrochemical hydrogenation of aldehyde groups30 and aromatic groups37 proceeds via adsorbed hydrogen Hads acting as reducing agent. This reaction mechanism was also the rationale for the selection of Pd and Pt – both efficient electrocatalysts for the reduction of protons and for the dissociative hydrogen adsorption – for this study.
Results and discussion
Time dependence of the electrochemical hydrogenation
In this study, we used a platinized Pt electrode (Pt/Pt) as reference material for an estimation of the optimal amount of electric charge for the electrochemical reaction. Fig. 2 shows (a) the concentration profile and (b) the reaction performance parameters conversion (X), mole balance (MB), Coulomb efficiency (CE), and selectivity (S) for the hydrogenation reaction of FF on Pt/Pt. The results of this experiment show increasing concentrations of FA, MF, and THFA over time, with FA being the most favored product. Furthermore, Fig. 2b shows decreasing selectivities for all products along with a decreasing mole balance the more charge was transferred respectively time passed. This corresponds to the high susceptibility of furanic compounds to react in oligomerization reactions.38 An occurring yellow color of the solution confirms this. Such oligomeric products may potentially inhibit the electrocatalyst by blocking the active sites. In order to avoid considerable substance loss due to these side reactions, while still enabling sufficient conversion, this study considers the performance after 6 Faraday Equivalents (FEq) (80.4 min) for FF hydrogenation, although theoretically, 8 FEq (107.2 min) for a complete conversion of furfural towards MTHF are necessary. Longer reaction times and more transferred charge would certainly increase the overall yield, but it also comes – in this kind of batch experiment – with decreased S and CE – especially due to the increasing contribution of the hydrogen evolution reaction to the overall process.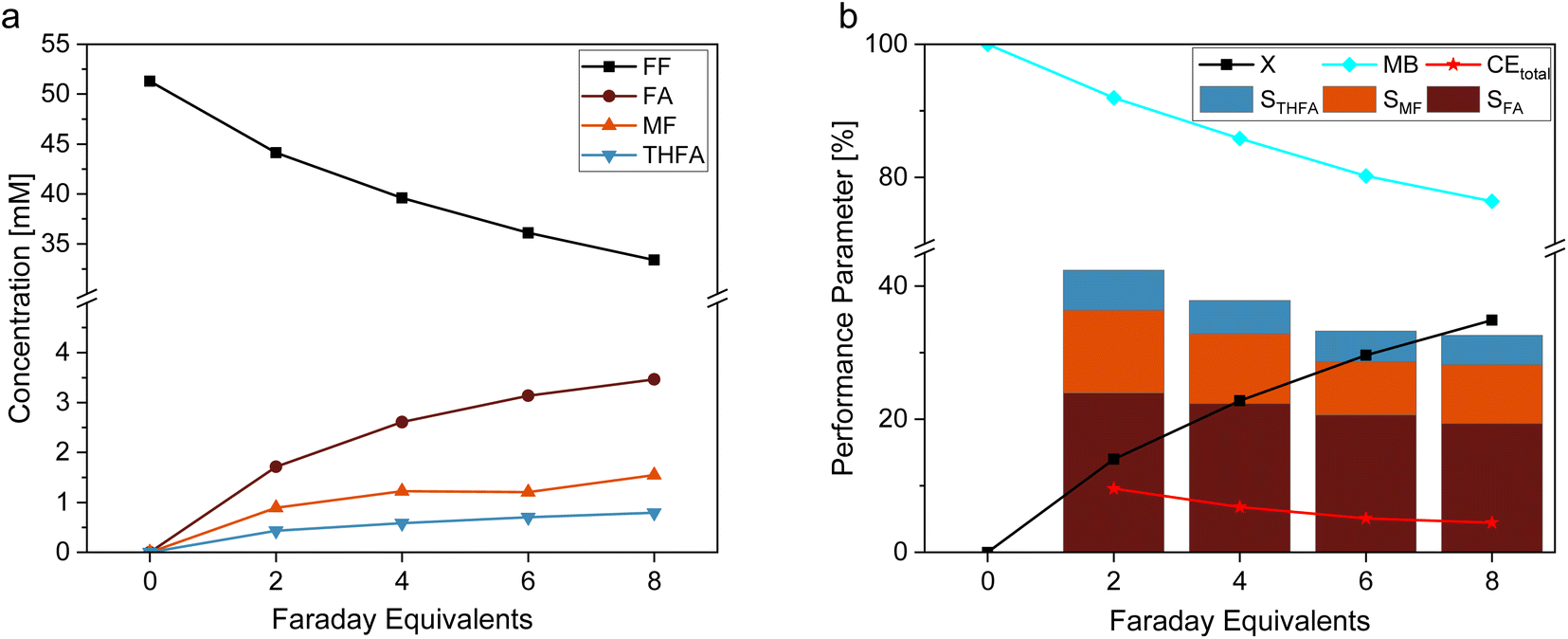 | ||
| Fig. 2 (a) Concentration profile and (b) reaction performance parameters of the hydrogenation reaction of FF on platinized platinum (Pt/Pt) electrodes under variation of the total amount of transferred charge with 0.05 M FF solution in 0.5 M H2SO4 electrolyte with 15% acetonitrile under galvanostatic conditions with −300 mA. | ||
Dependence of the furfural hydrogenation on the electrocatalyst composition
Fig. 3 shows a comparison of the FF hydrogenation at different electrode materials. Electrodes made of unmodified Pd-sheets yielded only small amounts of ring-hydrogenated THFA, however, showing the highest selectivity towards FA formation. Unmodified Pt sheets did not produce any measurable THFA, and the overall FF conversion and Coulomb efficiency were the lowest of all tested electrode materials. Interestingly, platinized platinum (Pt/Pt) was the only material with Pt as the sole electrocatalytic material that produced a ring-hydrogenated species. In comparison, the Pt–Pd ink-coated GC electrodes possessed a generally higher selectivity towards THFA. With a selectivity of STHFA = 15.3%, the coated GC electrodes with a Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratio of 5:1 show the highest selectivity towards THFA. A high Pd content seems necessary for a high ring hydrogenation selectivity, while adding a minor Pt content enhances STHFA (Fig. 3a). The enhancement of STHFA through a combination of Pd and Pt is comparable to reports in the literature for membrane reactors.32 Additionally, the Pd and Pt electrodes with a high Pd:Pt ratio show a comparingly high Coulomb efficiency towards THFA (Fig. 3b), indicating a lower portion of side reactions, like, e.g., the hydrogen evolution reaction (HER). The latter process is the major side reaction in organic hydrogenation reactions and thus main limiter of the CEs. Again, a Pd:Pt ratio of 5:1 shows the highest overall CE (14.65%) and CETHFA (8.00%). Pd and Pt are both highly active for HER, though Pt is slightly more efficient.39 Balancing Pd and Pt content on a GC electrode, the reaction can thus be optimized towards high STHFA with a sufficient hydrogen supply without unnecessary gaseous hydrogen formation lowering the CE. The comparably high conversion for pure GC electrodes can be attributed to various oligomerization reactions occurring on carbon electrodes (compare also mole balance (MB) in ESI Table S5†).38,40 The comparably low overall selectivities can be attributed to the oligomerization reactions described for furanic compounds in the literature38 and losses due to evaporation of products with low vapor pressure as e.g. MF. Contrary to thermal catalytic reactions for FF on Pd/C, or electrochemical conversion on Zn, no decarbonylation or ring opening was found.41–43
:Pt ratio of 5:1 show the highest selectivity towards THFA. A high Pd content seems necessary for a high ring hydrogenation selectivity, while adding a minor Pt content enhances STHFA (Fig. 3a). The enhancement of STHFA through a combination of Pd and Pt is comparable to reports in the literature for membrane reactors.32 Additionally, the Pd and Pt electrodes with a high Pd:Pt ratio show a comparingly high Coulomb efficiency towards THFA (Fig. 3b), indicating a lower portion of side reactions, like, e.g., the hydrogen evolution reaction (HER). The latter process is the major side reaction in organic hydrogenation reactions and thus main limiter of the CEs. Again, a Pd:Pt ratio of 5:1 shows the highest overall CE (14.65%) and CETHFA (8.00%). Pd and Pt are both highly active for HER, though Pt is slightly more efficient.39 Balancing Pd and Pt content on a GC electrode, the reaction can thus be optimized towards high STHFA with a sufficient hydrogen supply without unnecessary gaseous hydrogen formation lowering the CE. The comparably high conversion for pure GC electrodes can be attributed to various oligomerization reactions occurring on carbon electrodes (compare also mole balance (MB) in ESI Table S5†).38,40 The comparably low overall selectivities can be attributed to the oligomerization reactions described for furanic compounds in the literature38 and losses due to evaporation of products with low vapor pressure as e.g. MF. Contrary to thermal catalytic reactions for FF on Pd/C, or electrochemical conversion on Zn, no decarbonylation or ring opening was found.41–43
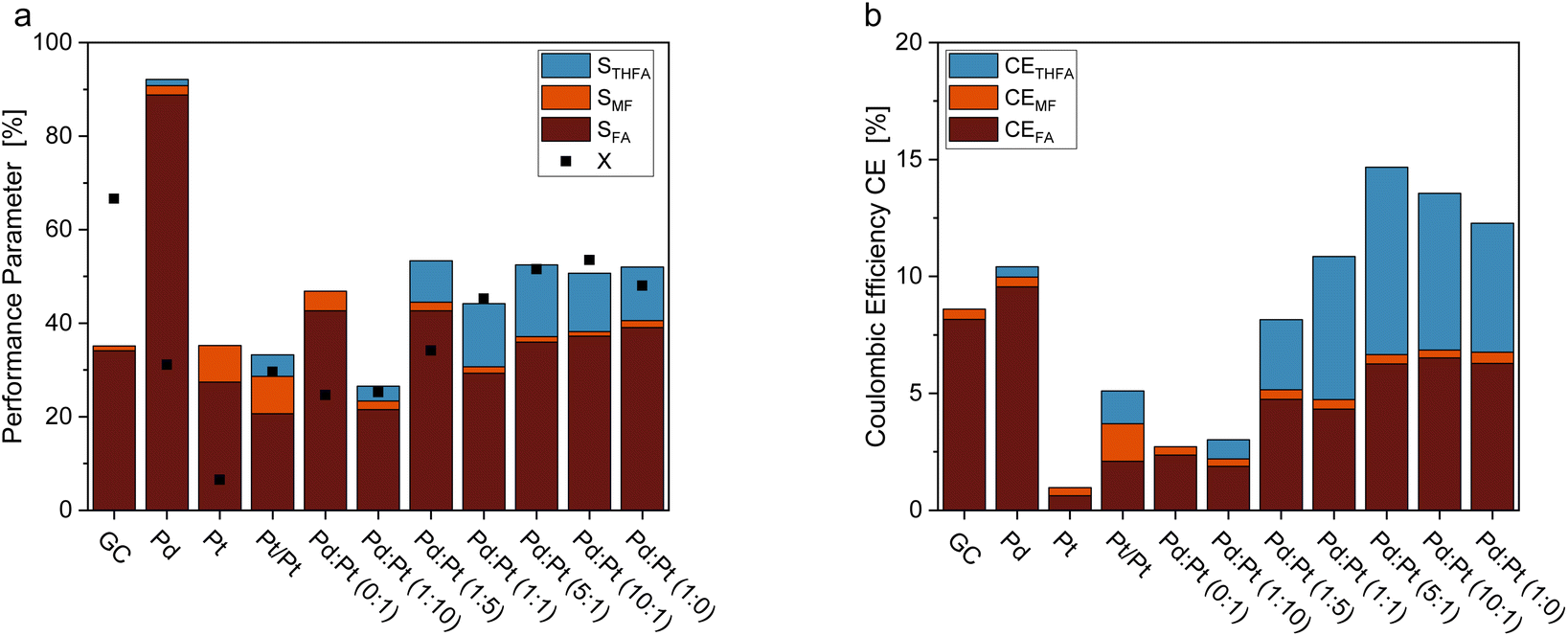 | ||
| Fig. 3 Comparison of different working electrodes for their reaction performance indicators ((a) X and S, (b) CEs), including metal sheet electrodes (Pd, Pt), platinized platinum sheet (Pt/Pt), and Glassy Carbon (GC) electrodes coated with catalytic inks containing varying Pd:Pt ratios. With 0.05 M FF solution in 0.5 M H2SO4 electrolyte with 15% acetonitrile under galvanostatic conditions with −300 mA, and 6 FEq transferred electrons. | ||
A formation of MTHF was not observed in any of the described cases. This phenomenon potentially can be ascribed to the slow kinetics of the THFA hydrodeoxygenation.32 From the thermodynamic perspective, this finding is also supported by previous findings that the reduction step from THFA towards MTHF was the most/second most energy-consuming step in the reaction network from FF – indicating that the thermodynamic conditions are contrary to the formation of MTHF.31 A further reason for the absence of this compound in our analysis may be its low impact on the refractive index (and hence on the RI detector of the used HPLC system), impeding the detection of small amounts (<1 mM) of MTHF.
In order to study a potential impact of the electrochemical utilization of the electrocatalyst coatings on their long term performance, consecutive batch syntheses were performed and the change in the electrode performance were examined. For comparison, Fig. 4 shows selected reaction performance parameters (X, STHFA, CEtotal) for a first and a second utilization (batch conversion of FF) of different electrode compositions. It reveals a decline in electrode performance, most prominent for the total FF conversion, where decreases of up to 10% can be observed. Additionally, STHFA and CEtotal are lower in a second utilization. Reasons for this performance decline could be either losses in electrocatalyst particles or the blocking of electrocatalytic sites by formed polymers/oligomers of a furfural decomposition.
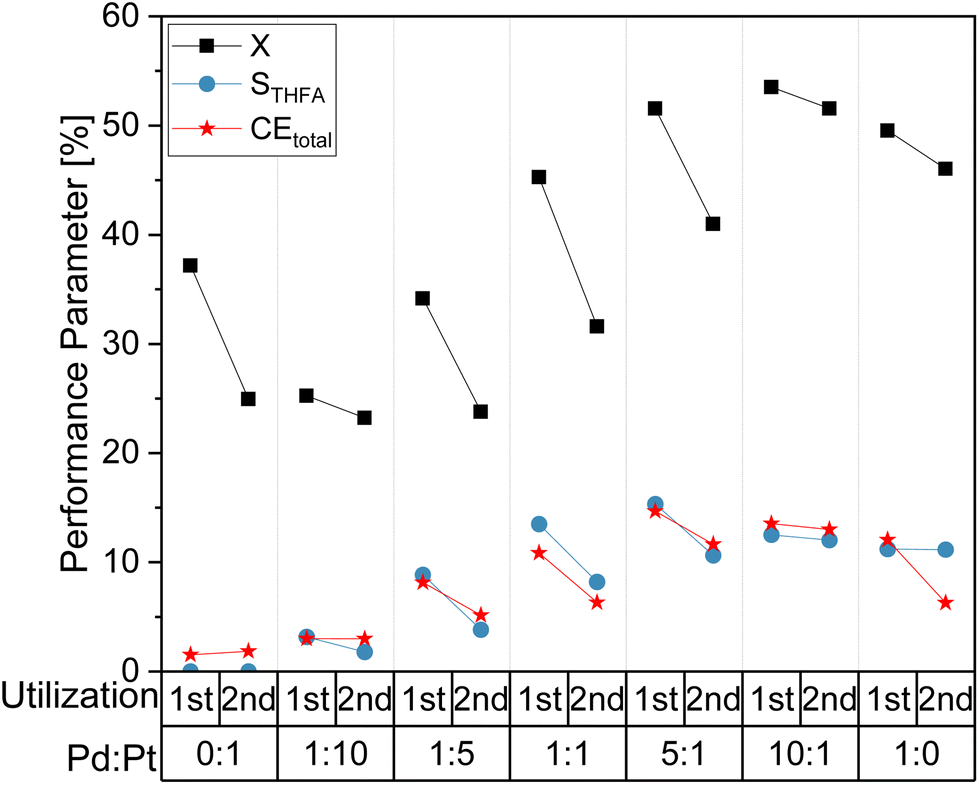 | ||
| Fig. 4 Comparison of selected reaction performance parameters for the first and second utilization of different electrode material compositions. With 0.05 M FF solution in 0.5 M H2SO4 electrolyte with 15% acetonitrile under galvanostatic conditions with −300 mA and 6 FEq transferred electrons. | ||
A comparison of scanning electron microscopic (SEM) images did not show any apparent differences in surface morphology (see ESI, Fig. S1†). However, energy dispersive X-ray spectroscopy (EDX) measurements revealed a decrease in catalyst content between freshly prepared electrode surfaces and utilized surfaces (ESI, Tables S1 and S2†), which may explain the decreased electrode performance.
Dependence of the furfural hydrogenation on the electrolyte composition
As an alternative to the beforehand described experiments using 0.5 M sulfuric acid as an electrolyte solution, a hydrochloric acid solution of the same concentration was employed to compare the impact of the electrolyte composition on the reaction performance. This experiment is based on previous findings that changing between sulfate and chloride ions can significantly alter the performance of electrochemical hydrogenation.44 With a pH of 0.39 (H2SO4) and 0.42 (HCl) of the respective 0.5 M acid solutions, comparable pH conditions are assured. Fig. 5a shows the differences in STHFA for both electrolyte types for FF as starting material. While STHFA is higher for the sulfuric acid solution, X, SMF, SFA, and CEtotal show higher values for the hydrochloric acid solution. A comparable but more distinct trend can also be observed in the case of FA as starting material (Fig. 5b). Here again, STHFA is lowest for the hydrochloric acid electrolyte, while SMF is higher for the hydrochloric acid solution as an electrolyte. A 0.5 M sulfuric acid solution seems to be beneficial for a high selectivity toward ring hydrogenation products. A likely explanation for this behavior is a supporting anion effect of the sulfate anion, which is more pronounced than for chloride. Since both acids possess very similar pH values, a pH effect can be excluded.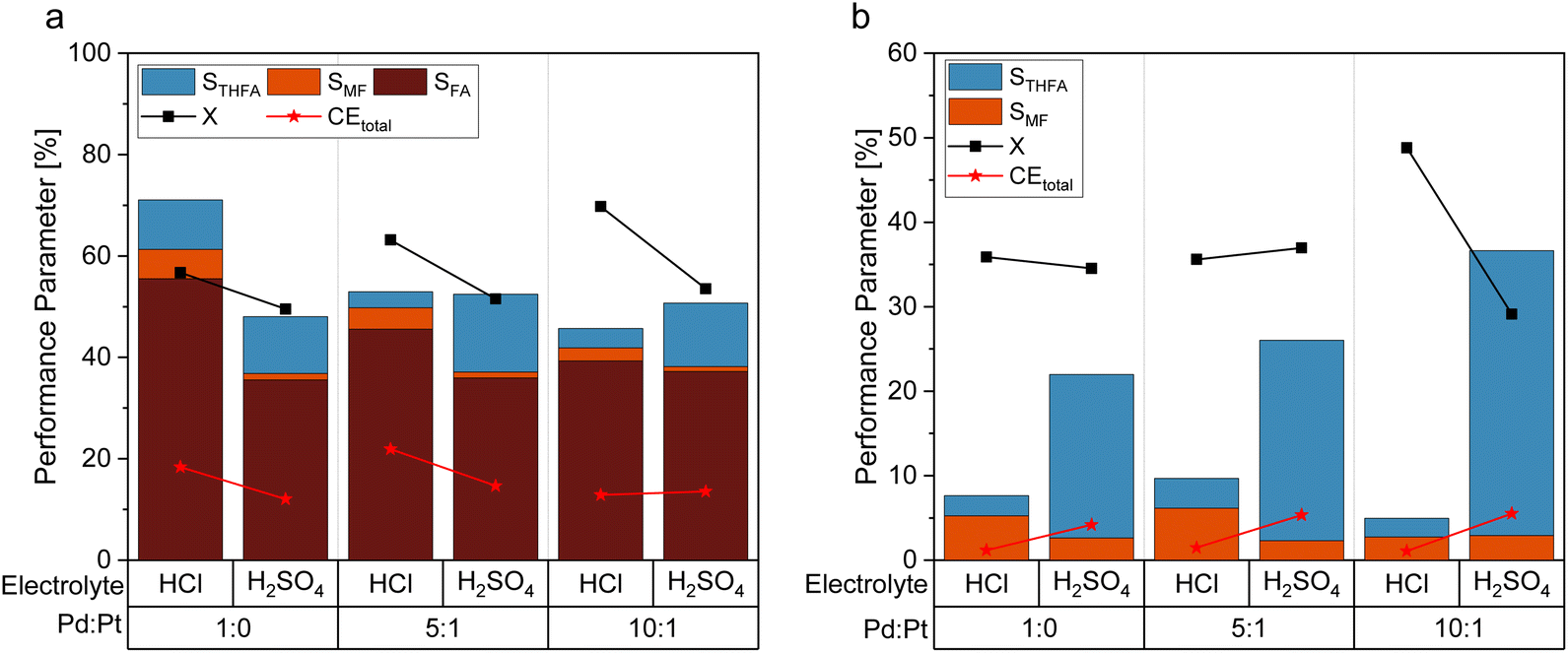 | ||
| Fig. 5 Comparison of selected reaction performance parameters for the utilization of 0.5 M HCl solution and 0.5 M H2SO4 solution with 15% acetonitrile for the utilization of (a) furfural and (b) furfuryl alcohol as starting material with 0.05 M FF solution under galvanostatic conditions with −300 mA and 6 FEq transferred electrons. | ||
Mechanistic considerations towards the ring hydrogenation
Theoretically, furfural and its derivatives can react at both – the aromatic ring and the side chain (substituent). By the reaction of the substituent alone, the FF hydrogenation yield products such as FA and MF – which can again serve as intermediates for subsequent ring hydrogenation. The nature of the respective substituents (the aldehyde group of the FF, the hydroxyl group of the FA, and the methyl group of the MF) may have an impact on a potential ring hydrogenation of these compounds. Following literature, the electrochemical hydrogenation of aromats should be most efficient for derivatives with electron-pulling substituents, i.e., with electron-poor aromatic systems.18 In order to compare the impact of substituents with varying electron pulling or pushing properties, we studied a number of furanic compounds (see Table 1) for their ring hydrogenation behavior.| MF | +I |
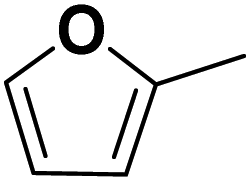
|
Small amount MTHF formed |
| FA | +I |
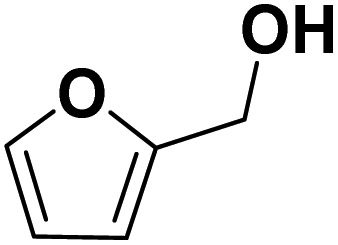
|
THFA formed |
| FUR | +0 |
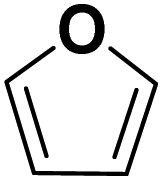
|
THF formed |
| FCA | −I, −M |
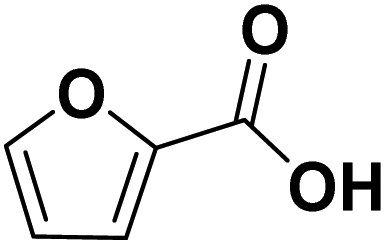
|
No conversion |
| FF | −I, −M |
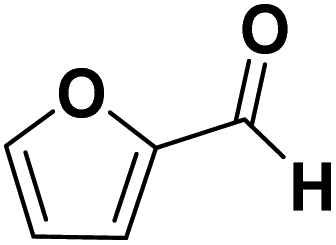
|
THFA formed |
For a reactivity comparison of the different furanic compounds, electrodes with Pd:Pt ratios of 5:1, 10:1, and 1:0 were chosen. These three electrocatalysts were further utilized in electrochemical reactions comparing the hydrogenation behavior of FF, FA, MF, furan carbonic acid (FCA), and furan (FUR).
Using FCA as reaction educt, no conversion at all was observed for the three tested electrode materials, indicating a remarkable stability of the carboxylic acid derivative. Concerning the hydrogenation of the carboxylic function, this stability is evidenced by only few literate examples for a successful hydrogenation (hydrodeoxygenation) of carboxylic acids.19,25 Hereby, the stability can be explained by the formation of hydrogen bonding based dimers stabilizing carboxylic acids in aqueous media.45 Yet, for the FCA, also the ring hydrogenation is inhibited – which seems contrary to the expectation based on the −M and −I of the carboxylic group.
The aldehyde group of FF also possesses a −M and −I effect, and one may argue this is the reason for reactivity towards THFA formation. However, the FF ring hydrogenation does not appear to proceed directly from FF but appears to require a prior reduction of the aldehyde function. This is supported by the finding that neither in our study nor previous studies, the generation of tetrahydrofurfuryl aldehyde was detected.31,32
The functional groups of the products of the FF aldehyde group hydrogenation – the respective hydroxyl and the methyl group of FA and MF, respectively, possess +I effects. As Fig. 6a shows by means of a comparative evaluation of the hydrogenation of FA and FF at different electrocatayst compositions, a considerably higher STHFA was found for FA as starting compound than for FF. As this result is biases through the different numbers of (consecutive) products, product formation rates (r) were computed (Fig. 6b). They reveal comparable product formation rates (rTHFA) for FA and FF as starting material. In contrast to findings in the literature, suggesting FF aldehyde hydrogenation being the rate determinant reaction step,32 we conclude from our results that the ring hydrogenation represents the actual rate determination step. A direct utilization of FF as starting material seems to be convenient for a higher productivity at the electrode surface in contrast to FA as reaction educt. Surprisingly, the FA reduction at Pd/Pt also leads to the formation of MF as a side product. In contrast, copper electrodes, often used for similar reactions, are incapable of activating the alcohol group.29,38
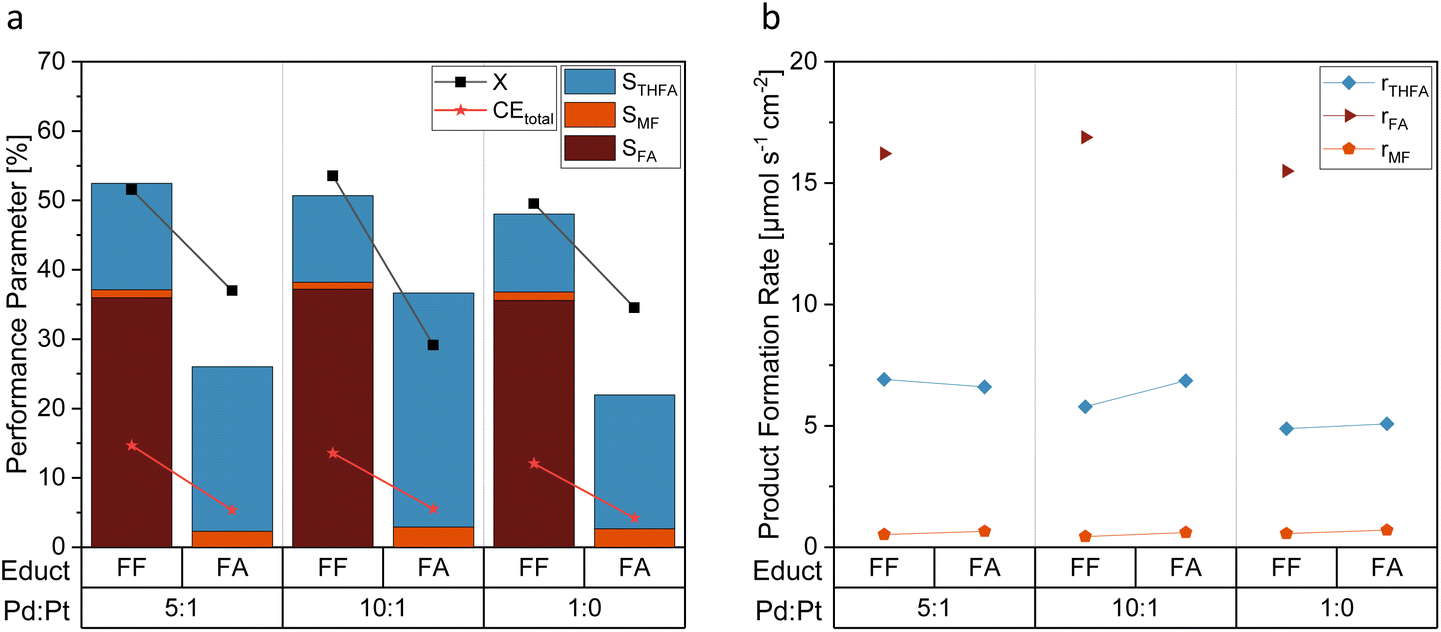 | ||
| Fig. 6 Comparison of FF and FA as starting material with selected Pd:Pt on GC electrodes. (a) Reaction performance parameters and (b) product formation rates. With 0.05 M FF respectively FA solution in 0.5 M H2SO4 electrolyte with 15% acetonitrile under galvanostatic conditions with −300 mA and 6 FEq transferred electrons. | ||
The MF hydrogenation was affected by MF losses, most likely by evaporation (the MF boiling point is only 63 °C).36 Nevertheless, the generation of methyl tetrahydrofuran (MTHF) was qualitatively validated for all three tested electrode materials, although not quantitatively determined due to the low content and weak activity in refractive index detectors during HPLC analysis. Here, the high energy barrier for the transformation leads to low conversion respectively MTHF formation.
Performing the reaction with FUR proved to be a challenge due to the high vapor pressure and low boiling temperature of FUR and its hydrogenation product, tetrahydrofuran (THF).36 Selected reaction performance parameters for this reaction on the three electrode materials can be found in the ESI, Fig. S2.† They show a high X, SFUR, and CEFUR, making the electrochemical ring hydrogenation of FUR a promising reaction. The absence of reactive substituents that compete in the hydrogenation process, aids the hydrogenation process of this compound. The data presented in the ESI† have to be handled with care as the high vapor pressure of the investigated substances led to a high variability in starting material concentration throughout all performed experiments. Furthermore, high vapor pressure of molecules could also lead to impaired and falsified HPLC calibration.
The above findings seem contrary to previous literature, which states a beneficial effect of −M and −I substituents and an inhibitory effect of +M and +I substituents for the hydrogenation of aromatic systems. This contradiction cannot be solved in this communication. Possibly, in addition to the electron-withdrawing or pushing properties of the substituents, further mechanistic or steric effects have to be considered.
Conclusion
This research paper explored the electrochemical hydrogenation of furfural and its derivatives, focusing on the formation of the respective tetrahydrofuran species. The results of the time dependency, respectively transferred charge of the performed batch reactions show that for an efficient tetrahydrofuran derivative formation, an optimal reaction time is crucial for selectivity and complete conversion on one hand and low side reactions toward oligomerization products on the other hand. Performing the reactions in continuous electrochemical flow cells may further improve the reactions and reduce losses.This study evaluated the performance of different Pd- and Pt-containing electrodes. While for electrodes containing only Pt as electrocatalytic material, only platinized Pt electrodes were active in ring hydrogenation, all the investigated Pd-containing electrodes yielded ring hydrogenated products. The highest selectivity showed glassy carbon electrodes with catalytic ink containing a high Pd:Pt ratio. Accordingly, a high Pd content seems to be beneficial for a high selectivity towards ring hydrogenation. Furthermore, an additional minor Pt content increased STHFA and CETHFA. A variation of the total deposited amount of catalyst material and a more detailed analysis of Pd:Pt ratio seems to be promising for future experiments. Decreases in electrode performances for consecutive utilizations should be encountered by, e.g., investigating oxidative or reductive cleaning procedures to remove deposited humins. Changing the electrolyte from 0.5 M H2SO4 to 0.5 M HCl showed higher STHFA for the sulfuric acid electrolyte revealing a preference for sulfate anions.
The presented preliminary results open a path to a variety of follow-up experiments as, e.g., studies of the impact of the electrode potential, experiments involving quantitative gas-phase analysis employing, e.g., cooling traps or online GC, the study of different catalyst morphologies, the utilization of different kinds of noble metals as Ru, Rh, Ir or the application of co-alloying for the preparation of binary electrocatalysts. Especially when using platinum group metals, the HER will always play an important role as the major side reaction of the electroorganic hydrogenation. In particular, the balance between the necessary Hads formation and the Hads combination to molecular hydrogen is the focus of the resulting optimization strategies.
A comparison of the ring hydrogenation of different furan derivatives was expected to reveal a dependency of the reaction on the electron density within the aromatic ring of the furan derivatives. As electron-withdrawing functional groups (−I, −M) lead to a lower electron density in the ring, the electrochemical reduction was expected to be favored for the respective derivatives. Yet, we could not confirm this hypothesis, as the most electron-pulling substituents, carbonic acid (FCA) and aldehyde (FF), did not show direct ring hydrogenation. For FCA, no conversion at all was observed, while FF underwent aldehyde hydrogenation to yield FA prior to the consecutive ring hydrogenation. For the reaction of FF toward THFA, the ring hydrogenation was observed to be the rate-determining step, while the aldehyde hydrogenation happened at a higher reaction rate. MF showed a low reactivity toward hydrogenation, while furan showed a high conversion and selectivity toward THF, although the high vapor pressures of furan showed to be an obstacle that should be circumvented in future experiments. All in all, more detailed studies are necessary for a more thorough understanding of the different reaction mechanisms of different furan derivatives. These studies can additionally include the utilization of a wider variety of furan derivatives for more consolidated conclusions.
Experimental section
Electrode preparation
Pd (99.9%, ChemPUR, Germany) and Pt (99.99%, ChemPUR, Germany) sheets employed as working electrodes were rinsed with acetone and deionized (DI) water prior to use. Pt sheets were additionally flame annealed if contaminations were observed. Glassy carbon (GC) (HTW Germany, Germany), electrodes were washed with acetone and water and ground with sandpaper before being used as an electrode material.For the preparation of GC electrodes coated on the front and backside with catalyst material, a suspension of 4 mg precious metal on Carbon in 187 μL 2-propanol (99.96%, Fisher Scientific, USA) and 50 μl Nafion 117 solution (5%, Merck, Germany) was prepared. The Pd:Pt ratio was adjusted by adding Pt/C (10wt%, Sigma Aldrich, Germany) and Pd/C (10wt%, Sigma Aldrich, Germany) in the specified mass ratio. The suspension was treated for several minutes in an ultrasonic bath before the catalytic ink was deposited onto the surface of a glassy carbon electrode in an area of 2 × 2 × 1.5 cm. The ink was dried in an oven at 50 °C for 20 min for the frontside and overnight for the backside. If not stated otherwise, the coating was renewed for each reaction.
Platinized Pt electrodes were prepared according to an adapted literature procedure.46 The respective Pt sheet (99.99%, ChemPUR, Germany) was cleaned for 4–5 min in warm (50 °C–60 °C) aqua regia before it was platinized in an electrochemical procedure with an H2PtCl6 hydrate (99.9%, Alfa Aesar, USA) and Pb(II)acetate trihydrate (99%, Alfa Aesar, USA) solution containing 3.5 wt% H2PtCl6 and 0.005 wt% Pb(II)acetate in DI water under constant stirring in an undivided cell. The working electrode was platinized with −30 mA cm−2 (total immersed area 8 cm2 considering front and backside) for a total amount of 10 min. After 5 min, the platinum electrode was turned to have the opposite side facing the counter electrode. A Pt electrode (8 cm2 immersed in electrolyte) served as the counter electrode. The Pb content in the solution enhances the electrochemical Pt precipitation, however, Pb itself is not incorporated in the formed metal deposit. This is supported by the described literature46 and performed EDX measurements, where 90% Pt and 10% C were found.
Chemicals and electrolytes
Furfural (99%, Sigma-Aldrich, Germany), furfuryl alcohol (98%, Sigma-Aldrich, Germany), 2-methylfuran (99%, Sigma-Aldrich, Germany), 2-methyl tetrahydrofuran (99%, Alfa Aesar, USA), tetrahydro furfuryl alcohol (98%, TCI, Japan), furoic acid (98%, Sigma-Aldrich, Germany), tetrahydrofuran (99.5%, Carl Roth, Germany), and furan (99%, TCI, Japan) were used as purchased and employed as starting material and for qualitative and quantitative analysis.Electrolyte solutions were prepared from an 85:15 mixture of deionized water and acetonitrile (99.9% HPLC grade, Sigma-Aldrich, Germany). Acetonitrile was utilized as a co-solvent to minimize the evaporation of more volatile products from the solution. The experiments employed sulfuric acid (95.0–97.0%, Sigma-Aldrich, Germany) and hydrochloric acid (37%, Carl Roth, Germany) in 0.5 M concentration in the solvent mixture. If not stated otherwise, 0.5 M sulfuric acid solution was utilized for the experiments.
Electrochemical reactions
For the amount of transferred charge, Faraday Equivalents (FEq) were considered, with 1 FEq being 1 transferred electron to the amount of starting material in the solution. This way, FEq corresponds to the transferred charge per molecule considering 0.05 M starting material concentration (⇔0.0025 mol⇔ 241.213 C) according to Faraday's law. All electrochemical reactions were carried out under galvanostatic conditions until 6 FEq were transferred for the hydrogenation of FF and FA, while 2 FEq were transferred for the hydrogenation of FUR. For time/conversion-dependent experiments, 2, 4, 6, and 8 FEq were investigated. A reductive current of −300 mA, respectively −50 mA cm−2, was applied until the stated FEqs were transferred (13.4 min for 1 FEq). The WE area was 6 cm2, including front and backside. Galvanostatic operation was employed as it facilitates the comparability of the reaction outcome of different catalysts and starting materials. The experiments were carried out in divided H-type cells using a three-electrode configuration with an Ag/AgCl sat. KCl (SE11, Sensortechnik Meinsberg, Germany, 0.197 V vs. standard hydrogen electrode (SHE)) as the reference electrode, a Pt-sheet (99.9%, ChemPUR, Germany) as the counter electrode (8 cm2 immersed in the electrolyte), and the respective prepared electrode as the working electrode. The half cells were filled with 50 mL of solution. While the working electrode chamber contained 0.05 M starting material dissolved in the electrolyte, the counter electrode chamber only contained the electrolyte. The two chambers were separated by a cation exchange membrane (fumasep FKE-50, Fumatech, Germany). The potentials and currents were measured and applied using an SP-150 Potentiostat (BioLogic SAS, France). Except for the reactions using furan as precursor, all experiments were performed as duplicate or triplicate. Variances for the presented measured concentrations can be found in the ESI (Tables S4–S9†). An exemplary plot of potential over time for Pd:Pt 10:1 on GC can be found in ESI Fig. S3.†
Quantitative analysis
Reaction performance parameters were obtained from quantitative analysis using an HPLC (Agilent 1260 Infinity II LC, USA) with a diode array detector (DAD) and a refractive index (RI) detector (40 °C). The utilized column was a Phenomenex Hydro-RP 80 Å (4 μm, 250 × 4.6 mm). The HPLC was operated with an eluent flow rate of 1 mL min−1 at 25 °C using a 1:1 mixture of deionized water and acetonitrile (99.9% HPLC grade, Sigma-Aldrich, Germany). Aromatic compounds such as FF, FA, FCA, FUR, or MF were quantified using the DAD with 1 μL injection volume, while saturated compounds such as MTHF, THFA, or THF were qualified and quantified with the RI detector and 10 μL injection volume. Retention times are given in the ESI (Table S1†). Oligomerization and polymerization products of the furanic compounds were not closer determined or quantified. Using the product and educt concentrations determined with HPLC, different reaction performance parameters were computed. For measured concentrations, variances s2, and calculated reaction performance parameters see ESI Tables S4–S9. The conversion X, Selectivity S, and Coulomb efficiency CE follow their standard definition. For clarity they are provided in the ESI (eqn (S1)–(S3)). The mole balance MB is the ratio of the amount n of the educt and all products at the end of the reaction to the amount of starting material used at the beginning (eqn (1)). Stoichiometric coefficients are not introduced as 1 mol of educt reacts to 1 mol of product.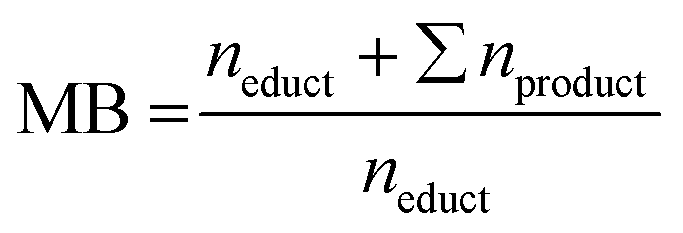 | (1) |
The product formation rate r (eqn (2)) was calculated using the amount of produced product n, the reaction time t, and the electrode area A.
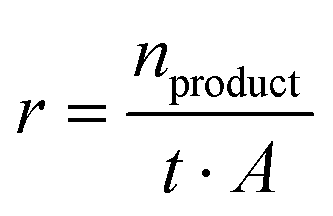 | (2) |
Surface morphologies and constitutions were examined using a scanning electron microscope (SEM, Zeiss EVO LS 10, Germany) and energy-dispersive X-ray spectroscopy (EDX, Ametek-EDAX Z2e Analyzer, USA). Exemplary data for the SEM and EDX measurements are shown in the ESI (Fig. S1 and Tables S1, S2†).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
T. L. and U. S. want to acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2163-1 – Sustainable and Energy Efficient Aviation – Project ID 390881007. U. S. also acknowledges DFG grant SCHR 753/12-1. Additionally, the authors would like to thank Dipl.-Min. Christiane Schmidt for providing SEM/EDX measurements.References
- S. A. Matlin, S. E. Cornell, A. Krief, H. Hopf and G. Mehta, Chem. Sci., 2022, 13, 11710–11720 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- V. Ramadoss, Y. Zheng, X. Shao, L. Tian and Y. Wang, Chem. – Eur. J., 2021, 27, 3213–3228 CrossRef CAS PubMed.
- T. R. Dos Santos, F. Harnisch, P. Nilges and U. Schröder, ChemSusChem, 2015, 8, 886–893 CrossRef CAS PubMed.
- M. Guschakowski and U. Schröder, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- C. H. Lam, S. Das, N. C. Erickson, C. D. Hyzer, M. Garedew, J. E. Anderson, T. J. Wallington, M. A. Tamor, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2017, 1, 258–266 RSC.
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- Y. Kwon, K. J. P. Schouten, J. C. Van Der Waal, E. De Jong and M. T. M. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- G. Li, N. Li, J. Yang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 594–599 RSC.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17, 601–609 RSC.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1419 CrossRef CAS PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn., 2012 Search PubMed.
- Y. Wan and J.-M. Lee, ChemSusChem, 2022, 15(13), e202102041 CrossRef CAS PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- A. Stanislaus and H. C. Barry, Catal. Rev., 1994, 36, 75–123 CrossRef CAS.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370 CrossRef CAS PubMed.
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, J. Am. Chem. Soc., 2020, 142, 4037–4050 CrossRef CAS PubMed.
- D. Robin, M. Comtois, A. Martel, R. Lemieux, A. K. Cheong, G. Belot and J. Lessard, Can. J. Chem., 1990, 68, 1218–1227 CrossRef CAS.
- N. Singh, Y. Song, O. Y. Gutiérrez, D. M. Camaioni, C. T. Campbell and J. A. Lercher, ACS Catal., 2016, 6, 7466–7470 CrossRef CAS.
- R. A. Benkeser and E. M. Kaiser, J. Am. Chem. Soc., 1963, 85, 2858–2859 CrossRef CAS.
- J. Chaussard, C. Combellas and A. Thiebault, Tetrahedron Lett., 1987, 28, 1173–1174 CrossRef CAS.
- C. H. Lam, W. Deng, L. Lang, X. Jin, X. Hu and Y. Wang, Energy Fuels, 2020, 34, 7915–7928 CrossRef CAS.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- P. Nilges, T. R. Dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- S. Jung and E. J. Biddinger, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508 CrossRef CAS.
- A. S. May and E. J. Biddinger, ACS Catal., 2020, 10, 3212–3221 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- S. K. Green, J. Lee, H. J. Kim, G. A. Tompsett, W. B. Kim and G. W. Huber, Green Chem., 2013, 15, 1869–1879 RSC.
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215–224 RSC.
- Y. Kwon, E. De Jong, S. Raoufmoghaddam and M. T. M. Koper, ChemSusChem, 2013, 6, 1659–1667 CrossRef CAS PubMed.
- Y. Kwon, Y. Y. Birdja, S. Raoufmoghaddam and M. T. M. Koper, ChemSusChem, 2015, 8, 1745–1751 CrossRef CAS PubMed.
- S. Wang, V. Vorotnikov and D. G. Vlachos, Green Chem., 2014, 16, 736–747 RSC.
- H. E. Hoydonckx, W. M. Van Rhijn, W. M. Van Rhijn, D. E. De Vos and P. A. Jacobs, Ullmann’s Encycl. Ind. Chem., 2007, d, 285 Search PubMed.
- N. Singh, U. Sanyal, G. Ruehl, K. A. Stoerzinger, O. Y. Gutiérrez, D. M. Camaioni, J. L. Fulton, J. A. Lercher and C. T. Campbell, J. Catal., 2020, 382, 372–384 CrossRef CAS.
- S. Jung and E. J. Biddinger, Energy Technol., 2018, 6, 1370–1379 CrossRef CAS.
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- S. Wang, V. Vorotnikov and D. G. Vlachos, ACS Catal., 2015, 5, 104–112 CrossRef CAS.
- T. Ishida, K. Kume, K. Kinjo, T. Honma, K. Nakada, H. Ohashi, T. Yokoyama, A. Hamasaki, H. Murayama, Y. Izawa, M. Utsunomiya and M. Tokunaga, ChemSusChem, 2016, 9, 3441–3447 CrossRef CAS PubMed.
- J. J. Roylance and K. S. Choi, Green Chem., 2016, 18, 2956–2960 RSC.
- W. Sauter, O. L. Bergmann and U. Schröder, ChemSusChem, 2017, 10, 3105–3110 CrossRef CAS PubMed.
- J. Chen, C. L. Brooks and H. A. Scheraga, J. Phys. Chem. B, 2008, 112, 242–249 CrossRef CAS PubMed.
- A. M. Feltham and M. Spiro, Chem. Rev., 1971, 71, 177–193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00515a |
| This journal is © The Royal Society of Chemistry 2023 |