
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom green innovations in oligopeptide to oligonucleotide sustainable synthesis: differences and synergies in TIDES chemistry
Lucia
Ferrazzano
 *,
Dario
Corbisiero
,
Alessandra
Tolomelli
* and
Walter
Cabri
*,
Dario
Corbisiero
,
Alessandra
Tolomelli
* and
Walter
Cabri
Department of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum – University of Bologna, Via F. Selmi 2, 40126, Bologna, Italy. E-mail: lucia.ferrazzano4@unibo.it; alessandra.tolomelli@unibo.it
First published on 2nd January 2023
Abstract
The growing market for therapeutic peptides and oligonucleotides (TIDES) draws attention towards their manufacture, aiming at efficient and sustainable productive processes in view of the predicted massive application of these molecules in several therapeutic areas in the near future. A comparative assessment of the principal innovations in the synthesis of these molecules is described herein, with a major focus on solid-phase synthesis (SPS), describing particularly the less-explored field of solid-phase oligonucleotide synthesis (SPOS). A head-to-head analysis of SPS techniques applied to peptides and oligonucleotides was performed, highlighting the strengths and weaknesses of these iterative synthetic approaches. The green innovations introduced in solid-phase peptide synthesis (SPPS), namely reduction or replacement with greener alternatives of solvents and reagents, and implementations in purification techniques, were reviewed and projected to potential targets and sustainable practices in modern SPOS, including their application in P(V) chemistry for the synthesis of stereopure oligonucleotides. By a comparative analysis, the key elements for the development of overall green procedures for oligonucleotide manufacturing were emphasized. In addition, due to the intrinsically more sustainable profile of liquid-phase synthetic techniques (LPS), recent advancements in the field reported for both TIDES were analyzed to prove the industrial interest in the manufacturing of these classes of molecules, underlining the importance of investment and modernization in the development of stronger and greener synthetic pathways.
 Lucia Ferrazzano | Lucia Ferrazzano has been a research technical assistant at the Department of Chemistry “Giacomo Ciamician” of the University of Bologna since 2019, and previously a post PhD fellow in the same department. Her research interests are mainly focused on the synthesis of peptides and peptidomimetic molecules, through green synthetic approaches. She is also interested in the synthesis of new antimicrobial peptides for biomedical applications, and in metal-catalyzed cross-coupling reactions. She is an author of 27 publications and 3 patents. She works in collaboration with international industrial companies. |
 Dario Corbisiero | Dario Corbisiero is a post PhD fellow at the Department of Chemistry “Giacomo Ciamician” of the University of Bologna. His research interests are focused on the enantioselective synthesis of N-heterocycles, by means of MacMillan catalysts, and their application to the synthesis of peptidomimetic molecules as integrin receptor ligands. His competences in catalyzed reactions were applied to the development of green procedures for Heck-Cassar-Sonogashira cross-coupling. He is also interested in green peptide synthesis and, recently, in the development of biomaterials for biomedical applications. |
 Alessandra Tolomelli | Alessandra Tolomelli has been an assistant professor at the Department of Chemistry “Giacomo Ciamician” of the University of Bologna since 2017. Her main research interests are the synthesis of bioactive peptidomimetics as ligands of biological receptors, antibiotics, and anti-inflammatory agents, using asymmetric synthesis and biocatalysis. She is also interested in the conformational analysis of peptides and peptidomimetics using NMR spectroscopy. She is an author of about 90 publications. She is involved in several national and international research projects and in collaborative research projects with industrial companies. She is also programme director of a master degree in Chemical Innovation and Regulation (ChIR) at University of Bologna, jointly with Universities of Barcelona and Algarve-Faro. |
 Walter Cabri | Walter Cabri has been a full professor of organic chemistry at the Department of Chemistry “Giacomo Ciamician” of the University of Bologna since 2019, as well as SVP of Fresenius Kabi I&D. After more than 35 years in the pharmaceutical industry, with interest spanning from drug discovery to drug development, his research interests focused mainly on greening pharmaceutical chemistry, including cross-coupling reactions and peptide synthesis. He is also interested in mechanistic studies on palladium-catalysed Heck–Cassar–Sonogashira cross-coupling. He is a coinventor of more than 100 patents, and a coauthor of more than 110 papers and books. He is involved in several national and international projects, keeping active collaborations with industrial realities. |
1. Introduction
In modern pharmaceutical chemistry, diversification of therapeutic treatments represents an important tool with which to influence the dysregulation of biological processes that cause widespread pathologies and diseases. Scientific efforts to achieve a clearer understanding of vital organic functions and their associated molecular mechanisms have facilitated the development of the so-called targeted therapies, aiming at (i) regulating/dysregulating the basic biological functions of proteins (“protein-specific” therapies), (ii) causing alterations in DNA sequences to interfere in the replication and transcription phases when these activities are responsible for specific pathologies (“DNA-specific” therapies), or (iii) modifying the transcriptional or post-transcriptional phases of mRNA-mediated synthesis of proteins (“RNA-specific” therapies).1These therapeutic targets can be reached by exogenous (e.g., small molecules) or endogenous molecules (e.g., peptides and oligonucleotides). For many years, the former class of pharmaceutical entities has been explored most extensively, due to practical difficulties in the manufacturing processes associated with the analogues of the latter. However, the consolidation of solid-phase synthetic techniques for the manufacture of peptides and oligonucleotides (TIDES) has renewed attention toward these classes of molecules.2 In 2021, 24% of new drugs approved by the Food and Drug Administration (FDA) belonged to the TIDES category, with a growing trend predicted for the next few years.3 In fact, forecasts related to TIDES suggest that the peptide market will grow with a compound annual growth rate (CAGR) of 10%, increasing from 29 billion USD in 2021 to 51 billion USD in 2026.4 The market for oligonucleotides (ONs) is also expected to grow, although conflicting views on the pace of growth have been reported by different analytical agencies. According to a report by the Allied Market Research experts published in July 2021, the global ON synthesis market will increase in revenue accumulation from 5.2 billion USD in 2020 to 26.1 billion USD in 2030, with a CAGR of 17.1% from 2021 to 2030.5
From the beginning of the new century, 38 new peptides (including insulin-like peptides) and 14 ONs have been approved in the United States, Europe, and Japan, giving a total of 52 new approved TIDES.6–8 The limitations for both modalities mainly relate to high manufacturing costs, especially for ONs, and to a weak pharmaceutical profile, related to poor chemical stability, poor oral absorption, and short half-life. The design of drugs based on oligopeptides, supported by massive process chemistry initiatives and several technological efforts, has focused predominantly on these issues, resulting in a leap forward in terms of market size, as well as the number of drug approvals and therapeutic segments covered. As a consequence, peptides are now used for the treatment of diseases that require massive volumes of drugs, including cancer and related pathologies, diabetes, inflammation and autoimmune diseases, bacterial and viral infections, and neurodegenerative and cardiovascular pathologies.9 Similarly, ONs predominantly target rare diseases, cardiovascular and metabolic disorders, oncology, neurological and muscular disorders, and ophthalmology. In addition to these large clinical targets, ONs have also been introduced in the market for the treatment of dermatological problems as well as gastrointestinal and hormonal disorders.10
Large-scale manufacturing has not been required to date, but demand growth projections mean that the limitations caused by high development and production costs will become less important by virtue of a good understanding of their mechanisms of action, generally involving the regulation of gene expression. This will make these compounds valuable in addressing rare diseases with no therapeutic alternatives.11
In this context, therapeutic ONs, together with peptides, have become pivotal modalities and consequently, a target for synthetic pharmaceutical chemists. Analyzing their structure, they are sequences of nucleic acids which are able to selectively recognize and bind a complementary target RNA sequence, modulating its functions by promoting or inhibiting the degradation of the target RNA through enzyme-dependent mechanisms or simply by acting as steric blockers, hindering protein synthesis. Their mechanism strongly depends on the modifications introduced into the structure of the synthetic ON sequence to assure enhanced pharmacokinetic stability and potency and to reduce toxicity. The main structural modifications include (i) the sugar backbone, (ii) the base, (iii) the linkage between nucleotides, and (iv) the conjugation methods (Fig. 1A).12,13 A similar analysis can be performed for peptides, consisting of sequences of amino acids connected through amide bonds. When peptides exclusively contain natural amino acids, the therapeutic effect is affected by two major drawbacks relating to the ineffective membrane permeability and the poor in vivo stability. The first is strongly influenced by the peptide length and amino acid composition, while the second depends on the easy lability of the amide bond to enzymatic hydrolysis. The stabilization of their pharmacokinetic profiles through prevention of proteolytic degradation and improvement in half-life can be achieved by incorporating structural modifications related to the (i) elongation of the peptide linkages, (ii) N-replacements, (iii) Cα-modifications, (iv) carbonyl replacements, (v) amino acid modifications, and (vi) introduction of β-turn mimetics or through intramolecular cyclization (Fig. 1B).14–16
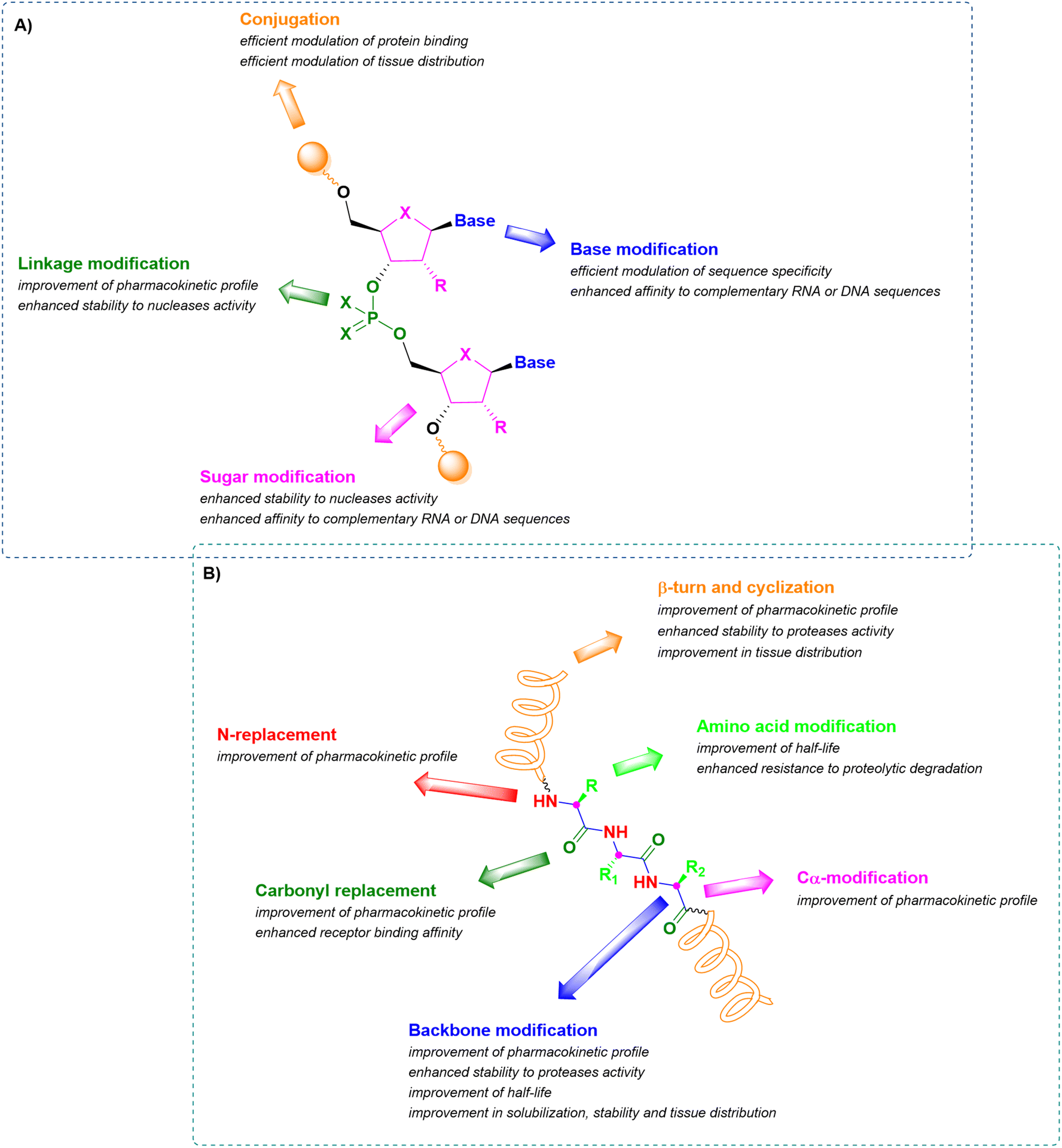 | ||
| Fig. 1 Schematic representation of common structural modifications on oligonucleotides (A) and peptides (B), with related effects on stability and bioavailability. | ||
These structural changes for both TIDES make it necessary to introduce upgrades in the synthetic processes to transform these promising new molecules into more sustainable manufacturing products. A great number of biocatalytic approaches have been developed in recent years, considerably projecting the synthesis of TIDES in a cost-effective and sustainable direction. Anyway, the biocatalytic approach requires the enzyme-catalyzed assembly of short sequences of nucleotides or amino acids, synthesized through parallel synthesis.17 In addition, the structural modifications introduced to improve the pharmacokinetic and chemical profile, discussed before, require enzyme engineering for efficient ligation of such a variety of substrates. Therefore, biotransformation will not be considered in this critical analysis, focusing on the green potential of iterative chemical TIDES synthesis. In fact, TIDES are easily produced on a multigram-scale through solid-phase synthesis (SPS), which is known respectively as solid-phase peptide synthesis (SPPS) or solid-phase oligonucleotide synthesis (SPOS). As currently applied in industry, neither process can be considered a green methodology as they require enormous use of hazardous solvents and reagents with a high carbon footprint. For peptide manufacturing by SPPS, considerable effort has been made in both academia and industry in recent years for developing green approaches to the synthetic pathways, by reducing the amount of solvents and reagents and/or choosing alternatives with a more sustainable profile.4 The same approach has not been seen for ONs. Even if the synthetic processes could potentially be improved from a sustainability point of view, it would be difficult to apply green innovations to real cases in ON synthesis, as the adopted methodologies based on the consolidated chemistry are less prone to variation compared with peptides. A description of these synthetic approaches by means of well-known green chemistry metrics should allow the quantification of their environmental performances and facilitate comparisons between the two SPS approaches. However, the simple concept of atom economy (EA) cannot be applied to these cases to guide the development of techniques. In fact, the iteration of synthetic steps and the use of on–off protections dramatically affect the overall EA, with a large mass of waste for each synthetic cycle. Analyzing the process from the point of view of process mass intensity (PMI), it is easy to see that they are far from being considered as green molecular entities (GMEs), because a mean PMI of 8400 kgwaste per kgAPI has been calculated for peptides with sequences of 50 amino acids, including the contributions of both upstream and downstream phases. Similarly, the PMI for a 20-mer ON is around 4300 kgwaste per kgAPI, not including the considerations of poor atom economy and the contribution to PMI associated with the preliminary production of reagents for SPOS.18
The evolution of SPPS in terms of greener reagents, solvents and technologies was recognized by regulatory agencies, in particular the FDA, and the result was the request for more sophisticated and precise analytical methods, and more stringent specifications for related impurities.
Peptides are obtained as single stereochemically pure chemical entities, while most ONs on the market have a phosphorothioate moiety generated via P(III) chemistry and exist in mixtures of thousands of diastereoisomers. The phosphorothioate linkage is essential to increase the stability of nuclease and protein binding; interestingly, the biological activity is thought to be independent of the configuration of the phosphorus stereocenter. Recently, technologies to produce stereopure ONs became available, and there is growing literature that supports the preference for stereopure phosphorothioate in terms of biological activity, protein binding, and development.
This critical review is intended to identify the ON process targets for the development of sustainable synthesis through P(III) and P(V) chemistry, keeping as reference the evolution of iterative modalities for oligopeptides. It is worth noting that most of the players now working on ON production originate from the peptide world, making TIDES more united than that observed previously. Therefore, the aim of this analysis is to give a clear comparison between the two solid-phase approaches for the synthesis of peptides and ONs, and to underline operational similarities and differences which could be useful for synthetic chemists in transferring and adapting the greener approaches reported for peptides to ONs. A snapshot of recent innovations in liquid-phase synthesis (LPS) will also be reported, reflecting the higher green score intrinsic to LPS techniques.
2. Historical snapshot of ON synthesis
The milestones in SPPS have been extensively described and reported in the literature over the years, as proof of concept in this chemical class of molecules. Key milestones are summarized in Fig. 2, together with the less renowned milestones in ON synthesis.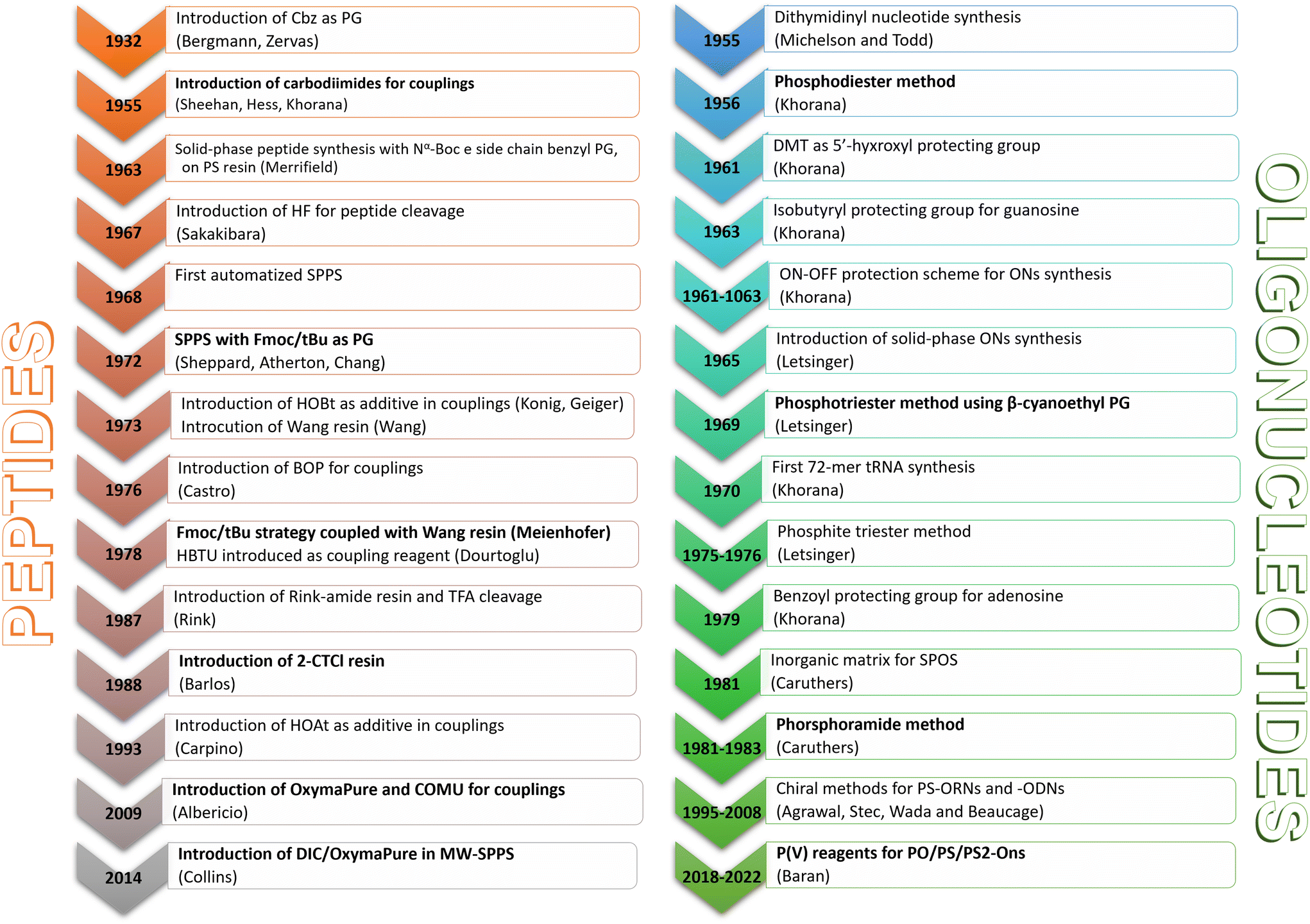 | ||
| Fig. 2 The 14 most relevant updates on the solid-phase synthesis of TIDES. | ||
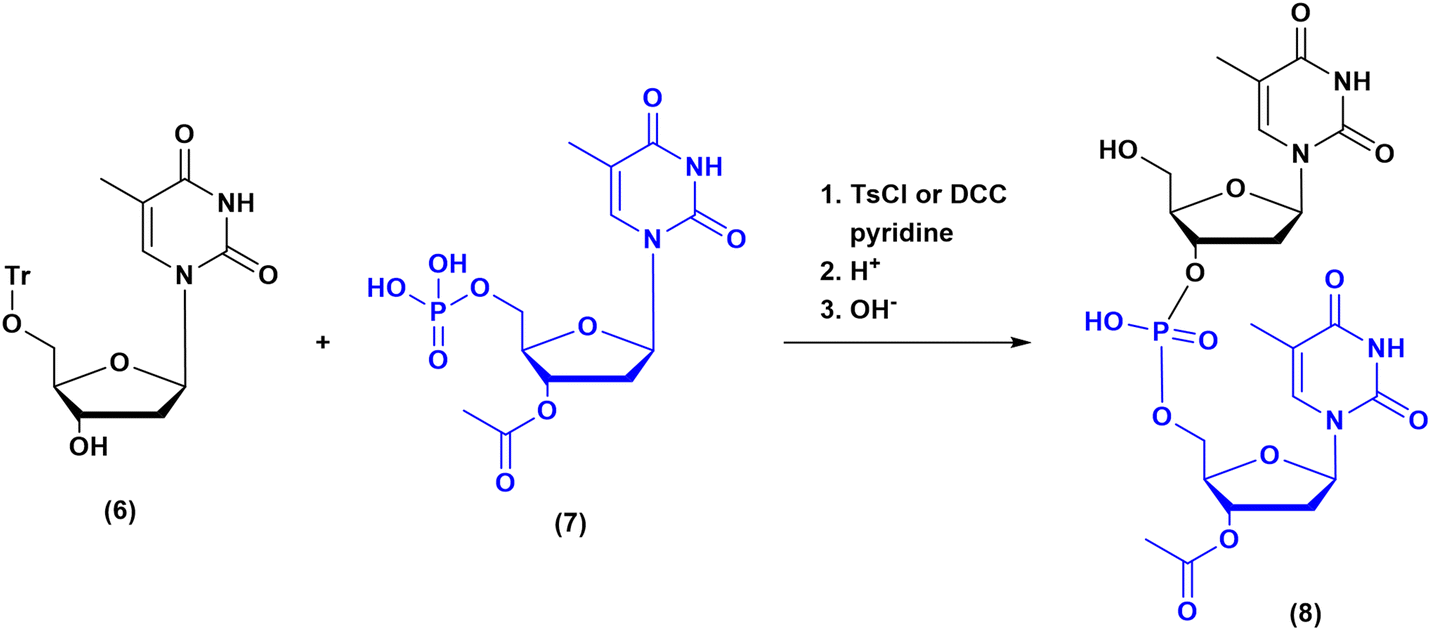
Historically, the chemistry of ONs started at the beginning of 1950 when Michelson and Todd synthesized the first fully deprotected dithymidinyl nucleotide (4) in the liquid phase, from thymidine 3′-benzylphosphoro-chloridate-5′-dibenzylphosphate (2) and 5′-hydroxyl-3′-acetylthymidine (3) (Fig. 3).19 As shown in Fig. 2, the first methodologic studies on ON synthesis emerged when the SPPS proposed by Merrifield was already an established methodology for their production. A substantial contribution to the modern synthesis of ONs emerged when Khorana and co-workers introduced two fundamental innovations: the concept of on–off protection and the phosphodiester approach. The first refers to the use of temporary protection on 5′-hydroxyl functionalities, by means of 4-monomethoxytrityl (MMTr) or 4,4′-dimethoxytrityl (DMTr) acid-sensitive protecting groups.20 The phosphodiester approach, in contrast, is based on the reactivity of a 5′-activated phosphoester nucleotide (7) with the nucleophilic 3′-alchol of the following nucleoside (6), mediated by a coupling reagent or chloride as an activating agent (Fig. 4).21–23 This methodology mimics the natural biosynthesis of nucleotides, wherein the secondary alcohol in position 3′ is the nucleophile. Khorana's contribution to the synthesis of ONs was central in achieving accurate interpretation of the genetic code and its function in protein synthesis. In recognition of these achievements, he received the Nobel prize in physiology and medicine in 1968. Later, Letsinger and co-workers introduced a reversed approach, namely the phosphotriester method, in which the nucleophilic 5′-alcohol reacts with the activated one in the 3′-position.24–26 It is worth noting that for the first time the 2-cyanoethyl group was introduced as the protecting group into P–OH and applied to SPS. While small changes were introduced into the protecting groups,27 further developments focused mainly on new methodologies for the formation of inter-nucleosides linkages, with the development in 1976 by Letsinger28 and in 1981 by Caruthers29 of phosphitetriester and phosphoramidite methodologies, respectively. According to the phosphitetriester method, it was possible to take advantage of the considerable reactivity of P(III) in the synthesis of the model dithymidinyl nucleotide by the reaction of 5′-protected phosphorochloridite thymidine (9) and 3′-protected thymidine (10). Through an in situ oxidation of P(III) to P(V) mediated by I2, the stabilized fully protected dinucleotide (12) was obtained (Fig. 5). Similarly, Caruthers and co-workers exploited the chemistry of P(III) in the reaction between 5′-O-DMTr phosphoramidite thymidine (13) and 3′-O-levolinoylthymine (14) (Fig. 6). The protecting groups on phosphorus in the phosphoramidite approach underwent changes along the years, changing from Me29 to iPr30,31 and fixing at 2-cyanoethyl,32 which is still the preferred protecting group for phosphoramidite chemistry in SPOS. According to this point, improvements have been focused on the chemically more efficient 3′–5′ sequence in which the primary alcohol is the nucleophile.
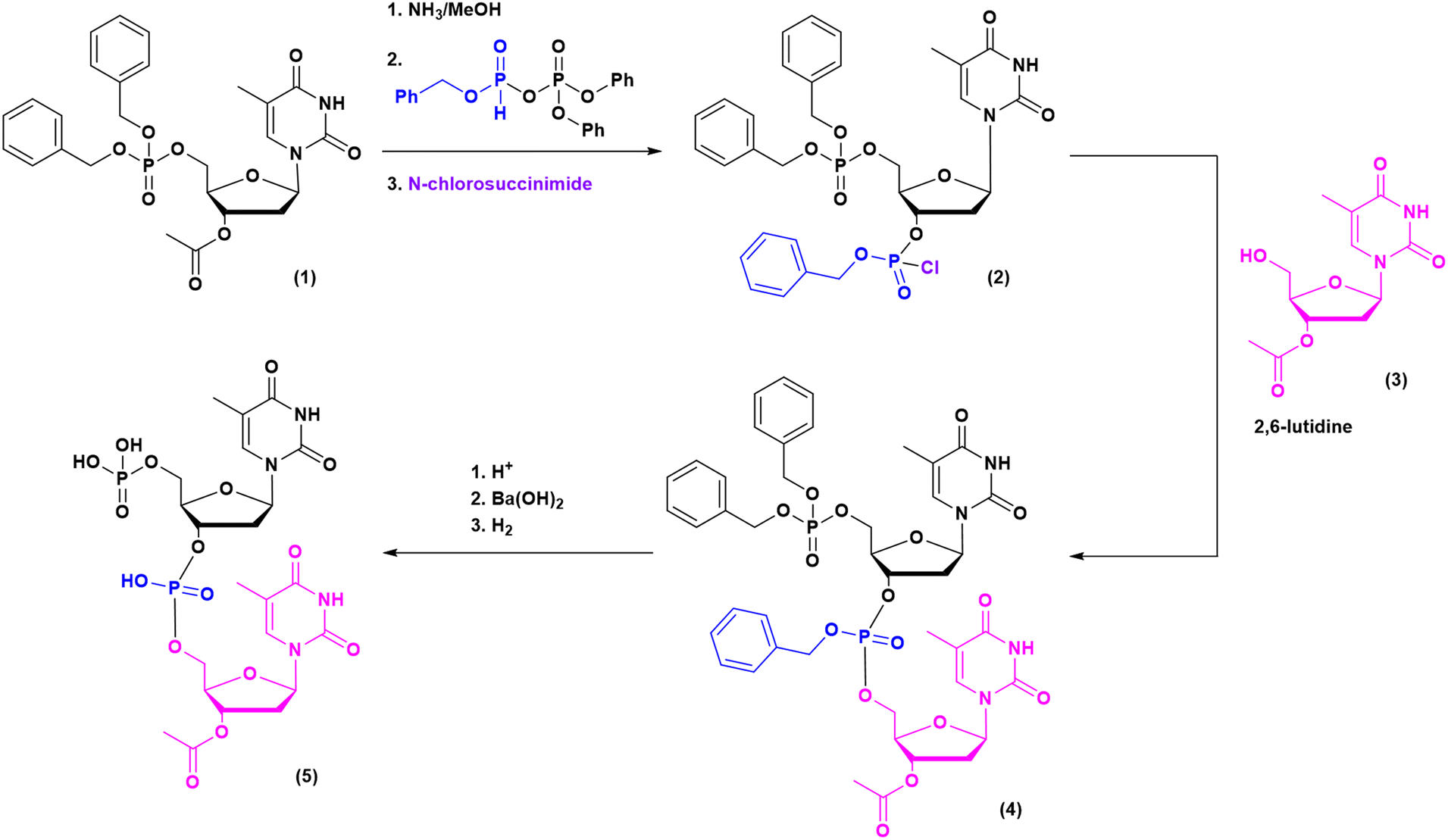 | ||
| Fig. 3 Michaelson and Todd dithymidinyl nucleotide (5) (1955). | ||
 | ||
| Fig. 4 Phosphodiester method by Khorana, applied to the synthesis of dithymidinyl nucleotide (8) (1956–1958). | ||
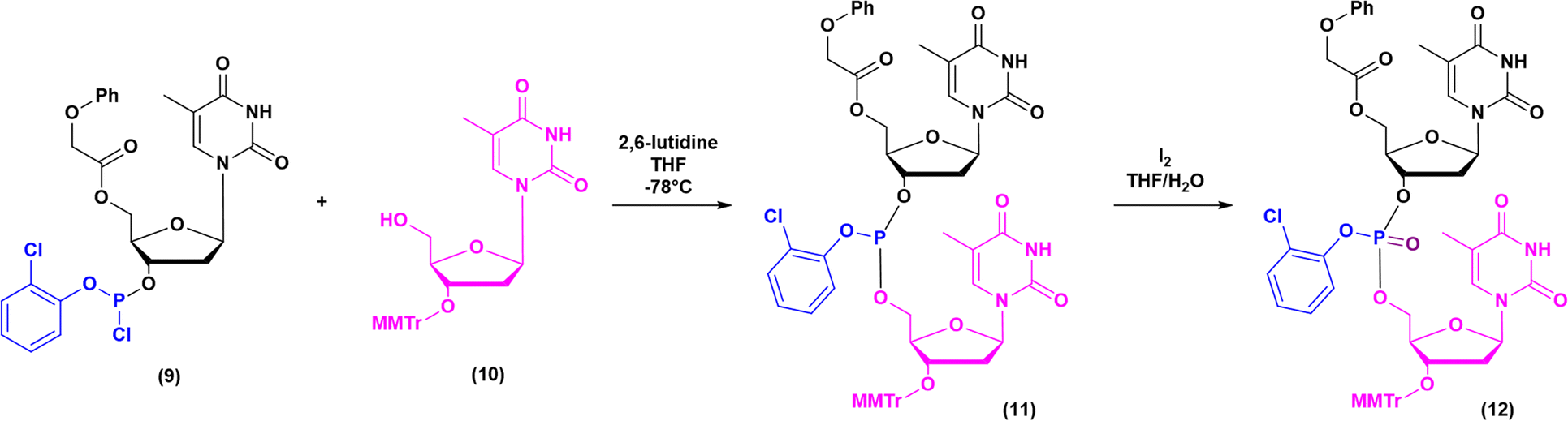 | ||
| Fig. 5 Phosphotriester method by Letsinger, applied to the synthesis of dithymidinyl nucleotide (12) (1976). | ||
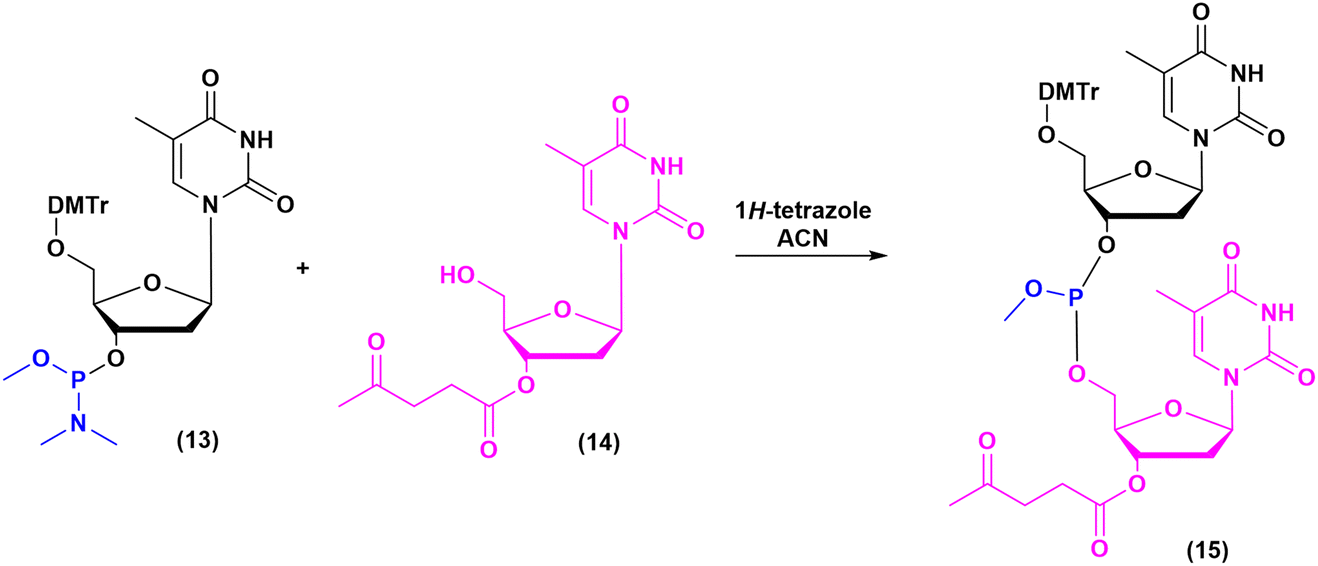 | ||
| Fig. 6 Phosphoramidite method by Caruthers, applied to the synthesis of dithymidinyl nucleotide (15) (1981). | ||
3. SPPS vs. SPOS: step-by-step analysis
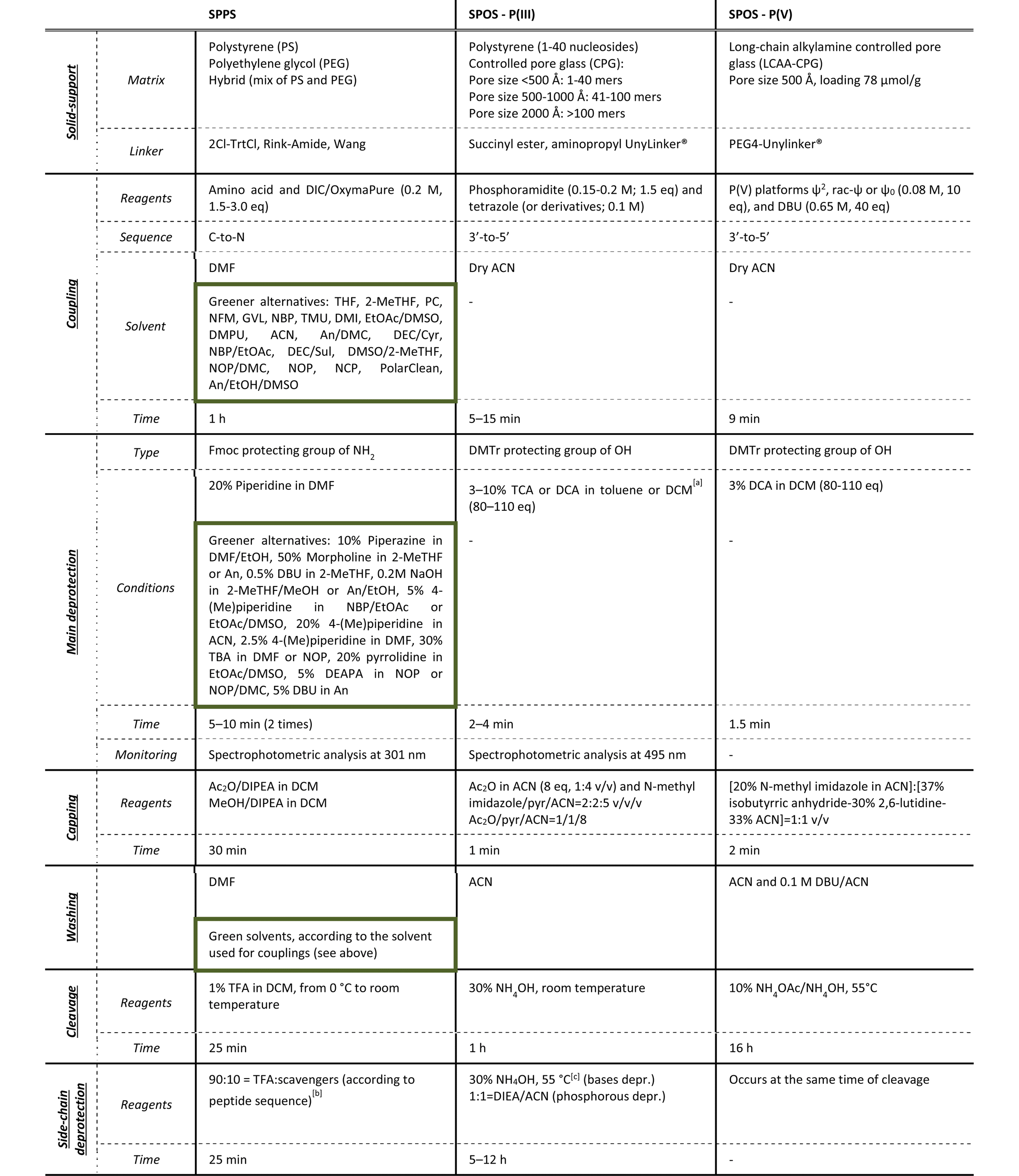
As the solid-phase approaches for TIDES synthesis are quite similar in terms of sequencing, a step-by-step analysis of reaction conditions and green improvements for both processes is described in the following paragraphs. The most common manufacturing procedures for peptides and ONs are summarized in Table 1, together with the P(V)-based methodology for SPOS that will be reviewed in detail later in the article. The phosphoramidite method represents the cornerstone in the SPS of ONs, because it allows the reaction to reach completeness in less than 1 min when performed on automatized synthesizers, and prevents the side-reactions that are usually observed in the previously reported phosphotriester-based methodology. The strength of the Caruthers’ approach stands in its possibility to be applied to the synthesis of long sequences (>50 mers or small DNA sequences).
a ACN cannot be used due to the formation of complexes with the acid (Ref. Nucleic Acids Res. 1996, 24, 15).
b Typical side-chain protections: Boc, Trt and Pbf. Deriving tBu and Trt carbocations requires scavengers (e.g., tioanisole, DDT, and DODT).
c Typical base protections: benzoyl (A, C); isobutyryl (G); dimethylformamidyl (G); phenoxyacetyl (A); isopropylphenoxyacetyl (G); acetyl (C); pivaloyloxymethyl (T); N-methyl-pyrrolidine amidine (G, C, A). Extra-steps in SPOS: oxidation by 2% I2 in pyridine/H2O (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) (2.1 eq. of I2 solution 0.5 M), 5 min; sulfurization by 0.2 M PADS solution in ACN and picoline (1:1 solution), 6–20 min. :2) (2.1 eq. of I2 solution 0.5 M), 5 min; sulfurization by 0.2 M PADS solution in ACN and picoline (1:1 solution), 6–20 min. |
|---|
|
Independently from the length of the sequence, both SPPS and SPOS require a substantial excess of reagents to achieve a complete reaction in a short time, while considerable volumes of solvents are also necessary to remove impurities and avoid intermediate purification steps. As highlighted in Table 1, green innovations were reported in the literature essentially for peptide synthesis, which has been the focus of several academic and industrial teams over the last 5 years. The more limited efforts made on ONs from a green perspective are also due to the lower volume of ON-based drugs available in the market to date.
3.1 Solid-support
In solid-phase approaches, the matrix of the solid-support has the greatest influence on the performances of the entire synthetic process, and the selection of a particular resin from all those available provides an important opportunity to predict the output of the synthesis. While the resins for SPPS are predominantly drawn from three categories (polystyrene, polyethylene glycol [PEG] or a hybrid), the solid-supports for SPOS belong mainly to two classes: polystyrene resins and controlled pore-glass (CPG) resins. The former (as for all resins adopted for SPPS) needs to be swelled, which sets limits on the choice of solvent. In contrast, the latter is a non-swellable rigid support, which makes it particularly efficient in the synthesis of long sequences of nucleotides and suitable for use with alternative solvents. The synthetic cycle in SPOS (Fig. 7) starts with a resin properly functionalized (generally with a succinyl linker or UNILINKER®), on which the first 5′-DMTr protected nucleoside is loaded.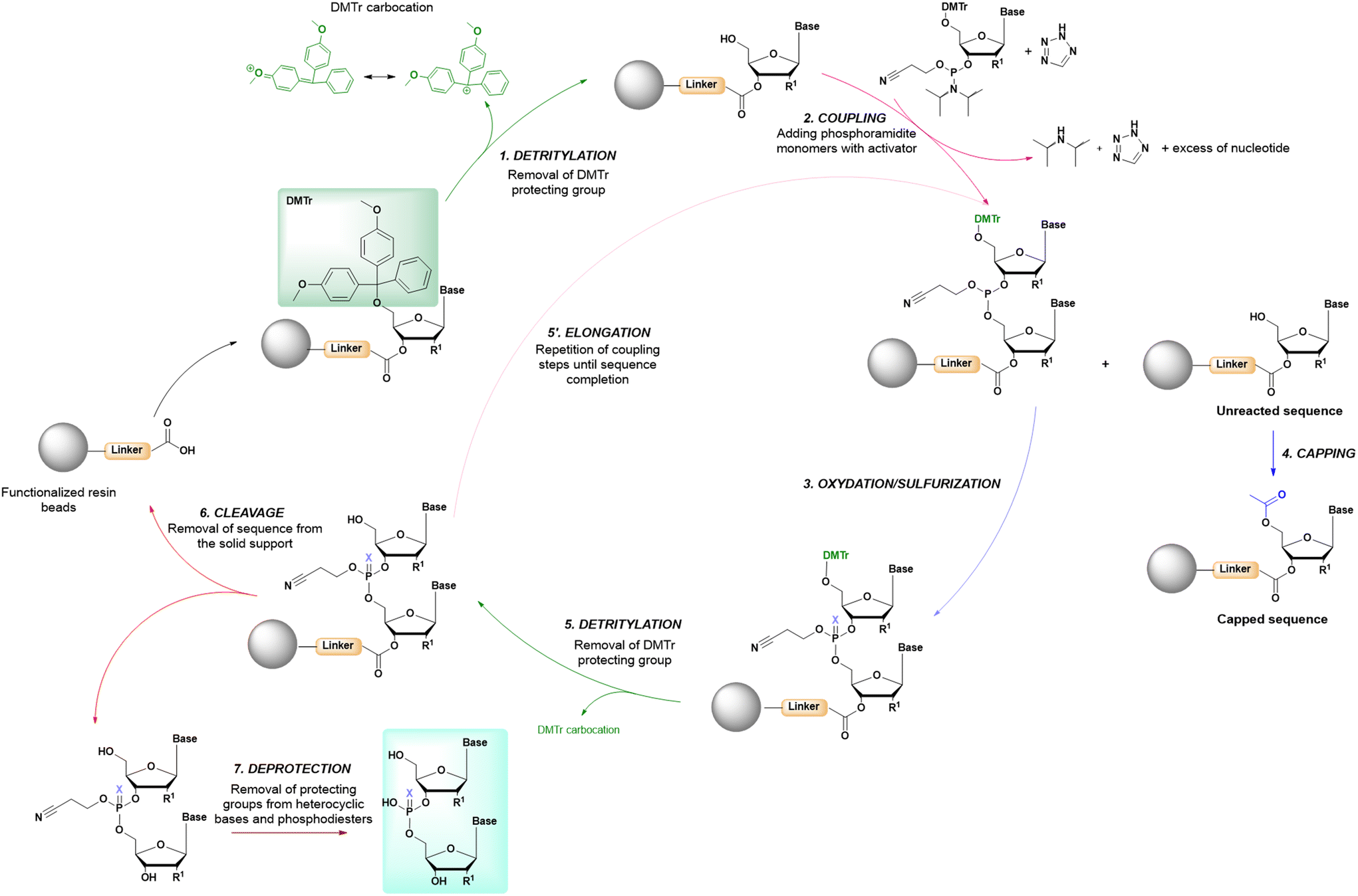 | ||
| Fig. 7 General scheme of solid-phase oligonucleotide synthesis (SPOS) steps by means of phosphoramidite chemistry (P(III)-chemistry). | ||
3.2 Synthetic method and solvents
The synthetic method for ONs is based on building the sequence in the 3′–5′ direction, using an excess of phosphoramidites in the presence of tetrazole (or derivatives) as the activating agent. Using this method, a complete conversion is achieved in acetonitrile in less than 15 min. Similarly, in SPPS, the sequences are assembled by functionalization of the N-terminal of anchored sequences, with the possibility of using different coupling reagents, among which OxymaPure/DIC is the preferred combination for reducing the racemization of the activated amino acids, ensuring good solubilization in common reaction media and fixing the reaction times close to 1 h.33In SPPS, the search for more sustainable solvents has been a target of synthetic chemists over the last 5 years. In this context, comparable results to those reported for reprotoxic dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP) were obtained by using several greener alternatives, including tetrahydrofuran (THF),34 2-methyltetrahydrofuran (2-MeTHF),35 propylcarbonate (PC),36N-formylmorpholine (NFM),35 γ-valerolactone (GVL),35,37N-butylpyrrolidone (NBP),38,39 1,3-dimethyl-2-imidazolidinone (DMI),38 1,3-dimethyl-3,4,5,6-tetrahydro-2-pyrimidinone (DMPU),38 acetonitrile (ACN),34N-octyl pyrrolidone (NOP),40N-cyclohexylpyrrolidone (NCP),40 and PolarClean.41 It has been shown that when single solvents are not sufficient to ensure good swelling of the solid-support and efficient and rapid coupling, mixtures of green solvents can also be used, including dimethyl carbonate (DMC),40,42 diethyl carbonate (DEC),42 sulfolane (Sul),42 anisole (An),42,43 cyrene (Cyr),40 NOP,40 NBP,40,44,45 dimethylsulfoxide (DMSO),43,44,46 2-MeTHF,44,47 and ethyl acetate (EtOAc).44–46
For SPOS, acetonitrile is the only solvent that has been extensively investigated regarding the coupling step, despite the drawbacks arising from its synthesis from the reduction of acrylonitrile, a petroleum-derived compound used in several industrial applications.48 In an analogy to SPPS, little research has been conducted into greener alternatives to ACN in SPOS, which represents an important area for future investigations.
3.3 Main deprotection
The 4,4′-dimethoxytrityl group (DMTr) can be removed (e.g., detritylation) by acidic treatment with trichloroacetic acid (TCA) or dichloroacetic acid (DCA) in dichloromethane (DCM), monitoring the progress of the reaction of the eliminated DMTr carbocation by spectrophotometric analysis at 495 nm. The process is similar to the SPPS monitoring of the dibenzofulvene-piperidine adduct, occurring during the removal of the fluorenylmethoxycarbonyl (Fmoc) protecting group from α-amine in peptide sequences, which is observed instead at 301 nm (Fig. 8). Again, there are no greener alternatives to the use of DCM in this synthetic process. In SPPS, the use of Fmoc chemistry for the main protection of the peptide backbone shows greater potential when looking for greener bases, as it is effective in deprotection in shorter time frames and at lower concentrations compared with the standard protocol involving 20% piperidine in DMF. In fact, due to the necessity to replace piperidine with bases displaying a better environmental, health and safety (EHS) profile, other bases are usually combined with green solvents and applied in some cases at lower concentrations to ensure efficient deprotection. In particular, 10% piperazine in DMF/EtOH,49 50% morpholine in 2-MeTHF47 or An,43 0.5% 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) in 2-MeTHF,47 0.2 M NaOH in 2-MeTHF/MeOH47 or An/EtOH,43 5% 4-(Me)piperidine in NBP/EtOAc45 or EtOAc/DMSO,46 20% 4-(Me)piperidine in ACN,50 2.5% 4-(Me)piperidine in DMF,51 30% tetrabutylamine (TBA) in DMF52 or NOP,53 20% pyrrolidine in EtOAc/DMSO,54 5% 3-diethylaminopropylamine (DEAPA) in NOP or NOP/DMC,46 and 5% DBU in An43 have been recently used for this purpose.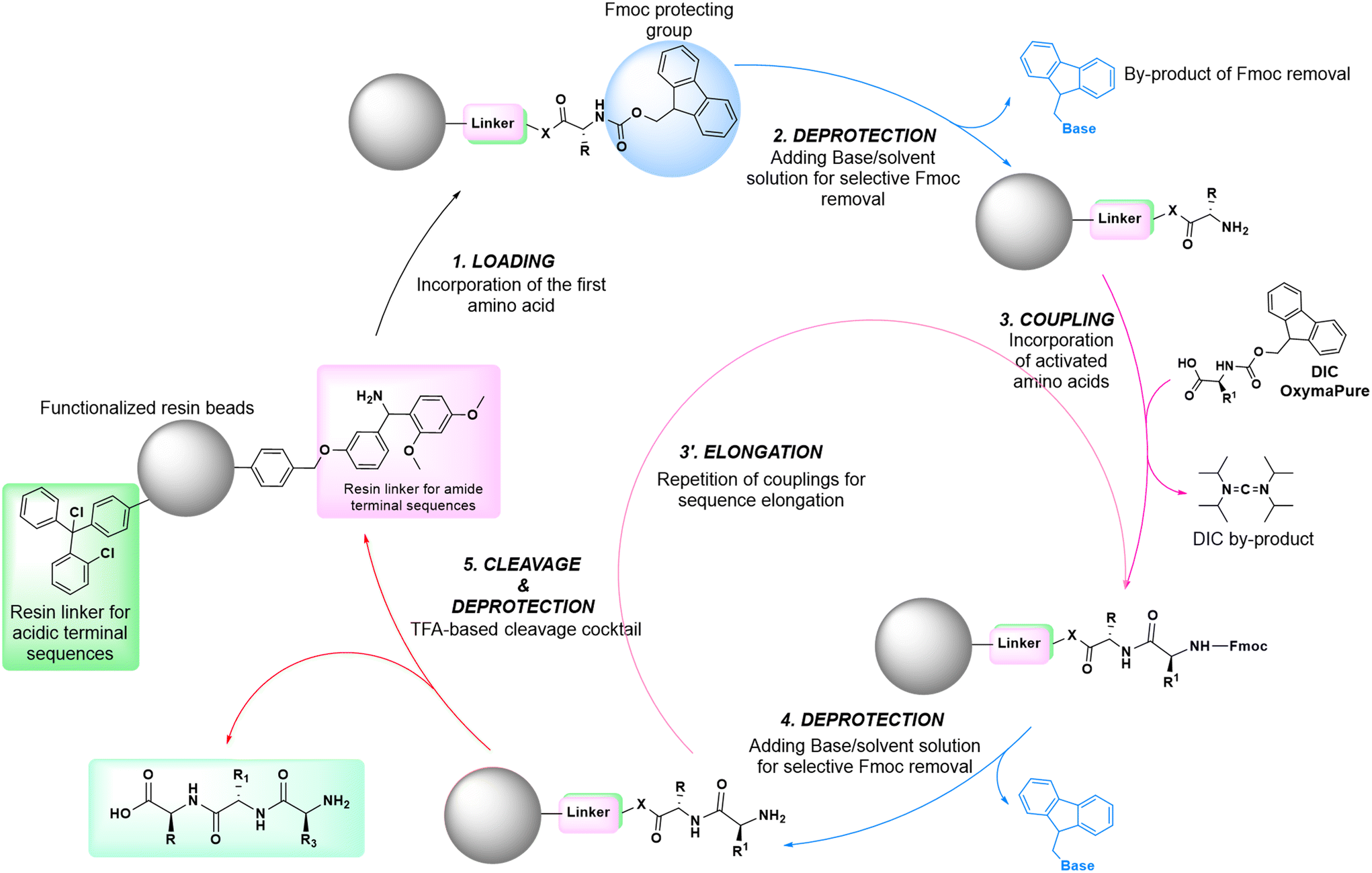 | ||
| Fig. 8 General scheme of solid-phase peptide synthesis (SPPS) steps. | ||
3.4 Capping
After detritylation, the supported deprotected nucleotide is ready for the subsequent coupling step with another phosphoramidite monomer. The 5′-hydroxyl group of the anchored nucleotide (or ON, if the process has been already iterated) reacts with the phosphoramidite of an incoming nucleotide, replacing the isopropylamino group and forming a P(III) bridge between monomers. Couplings are not complete, due to the aggregation of the growing sequences, which demonstrates the need for a capping step which plays the role of permanently blocking the 5′-hydroxyl terminals of the truncated sequences. This step is performed, as in the synthesis of peptides, with acetic anhydride in presence of a base (N-methyl imidazole for ONs and DIPEA for peptides), acetylating the terminal residue. While this step is routinely applied in SPOS, it is an option in SPPS that can be used if the chromatographic purification of the final product is not able to remove the impurities arising from des-sequences.3.5 Oxidation/sulfurization
Considering the antisense oligonucleotides (ASOs) on the drug market, five out of eight sequences contain a phosphorothioate modification at the inter-nucleotide bridge. In this case, the introduction of sulfur is achieved during the oxidation process from P(III) to P(V). Therefore, with respect to peptides, this represents an extra step required to stabilize the P(III) linkage. The oxidation is mediated by I2 in pyridine (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or a phenylacetyl disulfide (PADS)/picoline mixture (PS), obtaining a phosphotriester linkage protected with a 2-cyanoethyl group ready for the next cycle (Fig. 7).
O) or a phenylacetyl disulfide (PADS)/picoline mixture (PS), obtaining a phosphotriester linkage protected with a 2-cyanoethyl group ready for the next cycle (Fig. 7).
3.6 Cleavage and chain deprotection
When the synthetic sequence is completed, the ON can be cleaved by the solid-support exploiting the different lability of the protecting groups on both the bases and P(V), and of the growing sequence from the resin. This is similar to the approach used in peptide synthesis where the lower lability of the protecting groups on the amino acid side-chains is taken into account during the cleavage stage. In SPOS, the cleavage is performed by using the ammonium hydroxide solution at room temperature for 1 h. In fact, these conditions are mild enough to avoid the unwanted removal of the protecting group on P(V) and bases, which requires higher temperatures (55 °C) and longer reaction times (5–12 h). Similarly, in SPPS, a standard 1% TFA solution in DCM is used for simple cleavage from the solid-support, and a 90% trifluoroacetic acid (TFA) solution with scavengers is used when the removal of the contextual side-chain protecting groups is required. Recently, attempts to replace DCM in this step have been reported by Albericio55 and Rasmussen,45 who independently introduced anisole and 1,3-dimethoxybenzene, or a mixture of EtOAc/ACN as the reaction media.4. Green approaches for SPS
The lesson emerging from research teams involved in reducing the environmental impact of peptide chemistry is that every single chemical introduced into the synthesis represents a challenge from a green chemistry perspective. On analyzing and comparing the two synthetic cycles to evaluate the environmental impact of the procedures, it is clear that the main contribution to the reduction of SPPS/SPOS-associated PMI stands in (i) the possibility of reducing the volume of solvents and reagents used to drive the reaction to completion, or (ii) the recovery and reuse of solvents and reagents. Since multiple factors impact the calculation of PMIs associated with SPPS/SPOS, particularly the washing of the loaded resin after each step, any discussion would be based on rough estimations of data derived only from the upstream processes. In addition, to properly compare the PMIs of peptides and ONs, it is relevant to consider the length of the sequences and its influence on the calculations. In fact, for both TIDES with an average molecular mass between 1000 and 7000 Da, the PMI is calculated to be in the range of 2000–7000 kgmaterial per kgAPI, excluding the contribution of the synthesis of starting nucleotides to the overall PMI for ONs.4,18To numerically visualize the effect of the length of the sequences on PMI calculations, two exemplifying TIDES will be considered. The first is an octapeptide (e.g., API linear octreotide, anticancer drug mimetic of somatostatin) and the second is a 21-mer ON (e.g. Fomivirsen, applied for the treatment of cytomegalovirus [CMV] retinitis, and HIV infections). These two specific molecules were chosen on the basis of the availability of published data on the calculation of their PMIs. It is worth noting that octreotide is made up predominantly of natural amino acids (with the exception of the first amino acid, threoninol), thus requiring simple protection of the secondary amine and the side-chains of amino acids coming from fermentation. In contrast, the considered ON contains structural modifications on the ribose ring which imply a multi-step synthesis for each nucleotide. The PMIs associated with their SPS synthesis are 1726 kgmaterial per kgAPI for the octapeptide,4 with an average contribution per amino acid of 215 kgmaterial per kgAPI, and 4134 kgmaterial per kgAPI for the 21-mer ON, as recently reported by Andrews and co-workers who suggested an average contribution per nucleotide of 197 kgmaterial per kgAPI.18 Although these average PMIs are quite similar, the contribution of materials used for the synthesis of the phosphoramidite nucleotide mentioned before has not been included in the calculations. Similarly, the eventual presence of unnatural amino acids in a peptide sequence would also consistently increase its PMI. Because of all these considerations, a comparison between PMIs of TIDES turns out to be quite complex. However, overlooking the consequences of specific cases, each synthetic cycle for the peptide is always based on at least two steps, with three steps being required if capping becomes necessary. In contrast, ONs always require four synthetic steps. This means that there is no significant difference between the two standard synthetic processes; thus, the ON PMI can be considered acceptable from an environmental point of view.
Another type of analysis can be performed by taking into consideration the sustainability and greenness profile of the reagents and solvents involved. In SPPS, the use of reprotoxic and high boiling-point solvents and bases (DMF and NMP, piperidine) makes it difficult, expensive, and hazardous to secure their recovery from waste. Therefore, replacing these solvents and bases with more sustainable alternatives in the synthesis of bioactive peptides is a feasible solution, as extensively reported previously (see section 3). Attempts at minimizing solvent consumption in SPPS have mainly involved the optimization of process analytical tools (PATs) for real-time monitoring of synthetic steps, such as continuous refractive index (RI) measurements,56 or the development of protocols including the recovery and reuse of reagents and solvents, as reported by Pawlas–Rasmussen46,57 and by the authors of this paper.40,53 To a lesser extent, similar attempts were also made more than 20 years ago to recover the excess of phosphoramidite58 and the DMTr leaving group from an SPOS cycle;59 however, these routes are inconvenient when transferred to routing practice requiring multiple transformations and purification to regenerate useful reagents.
From a global analysis of synthetic processes, SPOS is greener than SPPS since greener solvents are used (ACN vs. DMF) as routine solvents for coupling, capping, and washing, despite ACN being advisable for substitution due to its extensive use in purification steps. Regarding peptides, in the last few years, several green options have been introduced to improve sustainability through an environmentally-friendly solvent choice, while for ONs, poor efforts have been made in this direction. In addition, while for peptides green alternatives are available for the base used during Fmoc removal (piperidine), which may also be combined with green solvents, in ON synthesis, to date no greener alternative to DCA or TCA has been proposed for the detritylation step. Only DCM replacement with toluene was proposed by Ravikumar in 1999,60–63 and this approach has been gradually introduced in common synthetic practice. Furthermore, in the coupling step with respect to initial tetrazole, safer 4,5-dicyanoimidazole (DCI) became the consolidated choice in the activation of nucleotides, accomplishing couplings with a 50% decrease in reaction times.64
Recently, Andrews and co-workers summarized the compliance of ON synthesis with the 12 principles of green chemistry according to a traffic-light code (Fig. 9).18
 | ||
| Fig. 9 Characteristics of oligonucleotide synthesis classified according to the 12 principles of green chemistry. | ||
When greener alternatives cannot be proposed due to intrinsic limitations of the synthetic techniques, skipping synthetic steps can be a smart trick to enhance the overall greenness of the process. This approach was applied to SPOS, for the synthesis of polystyrene (PS) ONs. Because phenylacetyl disulfide (PADS) is used during the sulfurization step, its decomposition by-product (phenylacetyl group) can act as an acetylating agent in situ by capping/scavenging the free 5′-hydroxyl group present on truncated sequences.65 Moreover, because SPOS chemistry is based on phosphoramidites, their stability is of primary importance in order to avoid the unwanted waste of starting materials. In an automatic synthesizer, the phosphoramidites remain in solution at room temperature, although the reactor should be stored at −20 °C to prevent/reduce the oxidation and hydrolysis of the substrate. This results in an easy degradation of the phosphoramidite starting materials through autocatalytic or water-catalyzed pathways, such as acrylonitrile elimination. One way to stabilize phosphoramidites has been recently proposed by Gothelf and co-workers, who proposed a protocol to immobilize the phosphorylating reagents on a solid-phase (Fig. 10).66 This approach allows the rapid phosphorylation of nucleosides, their stabilization, and the possibility of recovering the solid-support that can then be reused for >80 synthetic cycles.
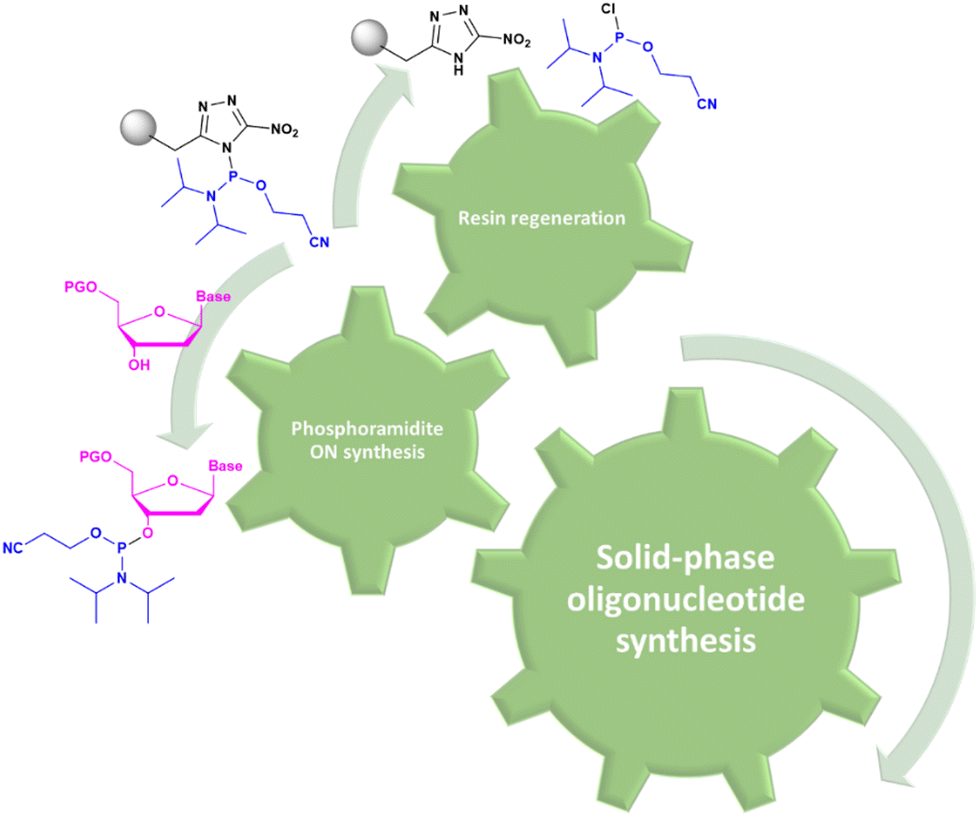 | ||
| Fig. 10 Scheme of immobilization of phosphoramidites on a solid support. | ||
An interesting demonstration of the strong interconnection between the SPS of both TIDES was reported a few months ago by Sinha and co-workers, who performed the synthesis of phosphoramidite morpholino oligonucleotides (PMOs) by using trityl/Fmoc chemistry.67 The selected solid-support to be used on an automatized synthesizer was Ramage ChemMatrix, while 5-(ethylthio)-1H-tetrazole (ETT) and N-methylpyrrolidone were selected as the activating agent and solvent, for the activation of the phosphoramidites, respectively. The innovation stands in the introduction of trityl (Trt) or Fmoc as the N-protecting group into the morpholino unit, which is removed respectively by using 3-cyanopyridine trifluoroacetic acid (CYPTFA) in DCM or 20% piperidine in DMF, similarly to SPPS. The solvent NMP was selected to guarantee the perfect solubilization of starting phosphoramidites, which are not equally solubilized by ACN. Since the linker on the resin was 6-amino capronic acid, the cleavage was accomplished with aqueous ammonium hydroxide, as in other synthetic examples. The authors reported that the Fmoc protecting group represents a better choice over Trt, since it is a base-sensitive group and thus preserves nucleobases from acid-induced depurination. Although the overall yield of the process was reported to be 20–25%, this is considered to be a more efficient, economic, and robust method compared with the similar Trt-based synthesis previously reported by Caruthers68 and Pentelute.69 In addition, since this approach is similar to that used in Fmoc-based peptide synthesis, the improvement of this SPOS can be based on the experience gained in SPPS, replacing NMP or DMF with greener alternatives, and piperidine with bases displaying a better EHS profile.
5. Stereocontrolled synthesis of ONs
As mentioned above, when implementation of green chemistry is not possible, the sustainability of SPOS becomes achievable only by reducing the number of synthetic steps in each cycle. Unlike peptide chemistry, in ON synthesis, an extra step is necessary for building the final backbone and it is represented by the oxidation/sulfurization of P(III) to P(V), as discussed before. This implies that the outcome in the case of PS linkages is a mixture of 2n diastereoisomers, where n is the number of nucleotides. None of the ONs on the market are unique stereodefined structures; this negatively affects the development of the drug and increases the complexity in identifying impurities, optimizing analytical methods, and purifying final products. In addition, most recently, considerations related to potency and safety have become the grounds to support the development of stereopure ONs. However, the available technologies for the synthesis of stereopure ASOs are still limited to the synthesis of phosphorothioate. As a result, two main approaches were proposed and applied to the synthesis of linear and cyclic dinucleotides, and longer sequences. Starting from the standard P(III)-chemistry, the use of chiral auxiliaries was initially proposed to achieve reliable stereocontrol during the synthetic process. In this sense, the work of Wada and DiRocco was pioneering. Wada proposed a chiral P(III)-stereogenic auxiliary (2-chloro-1,3,2-oxazaphospholidine (18)) for liquid- and solid-phase synthesis of dinucleotides (Fig. 11A),70,71 while DiRocco introduced a multifunctional catalyst (26, 27, 28 or 29) for assembling pronucleotides (25) from chlorophosphoramidites (24) and nucleosides via dynamic kinetic asymmetric transformations (DYKAT) (Fig. 11B).72 This last approach has been successfully transferred to the asymmetric synthesis of Remdesivir by Zhang and co-workers.73 Recently, other groups have worked on stereocontrolled P(III) or P(V) chemistry. For example, Miller and co-workers reported the stereocontrolled synthesis of cyclic dinucleotides (CDNs) using chiral phosphoric acid (CPA)-mediated couplings. They identified two different catalysts for SP or RP selectivity (34 and 35), demonstrating their efficiency in the synthesis of differently functionalized dinucleotides (33) (Fig. 11C).74,75 In 2012, researchers from Wave Life Science reported the first application claiming the use of (L)- or (D)-PSM (2-(phenylsulfonyl)-1-(pyrrolidin-2-yl)ethan-1-ol) amidites (36) for the synthesis through the Staudinger reaction of new ONs based on phosphoryl guanidine backbones (PN) (Fig. 12A).76 More recently, they extended the scope to differently substituted ribose scaffolds (2′-methoxyehtyl, 2′-H, 2′-OMe, 2′-F)77 and applied the methodology to the synthesis of chimeric ONs with stereopure PN/PS linkages. In this case, the synthesis of PS stereopure linkages uses a stereocontrolled ON synthesis with iterative capping and sulfurization (SOSICS) synthetic procedure, based on the iterative sequences of capping and sulfurization followed by the spontaneous release of the chiral auxiliary (Fig. 12B).78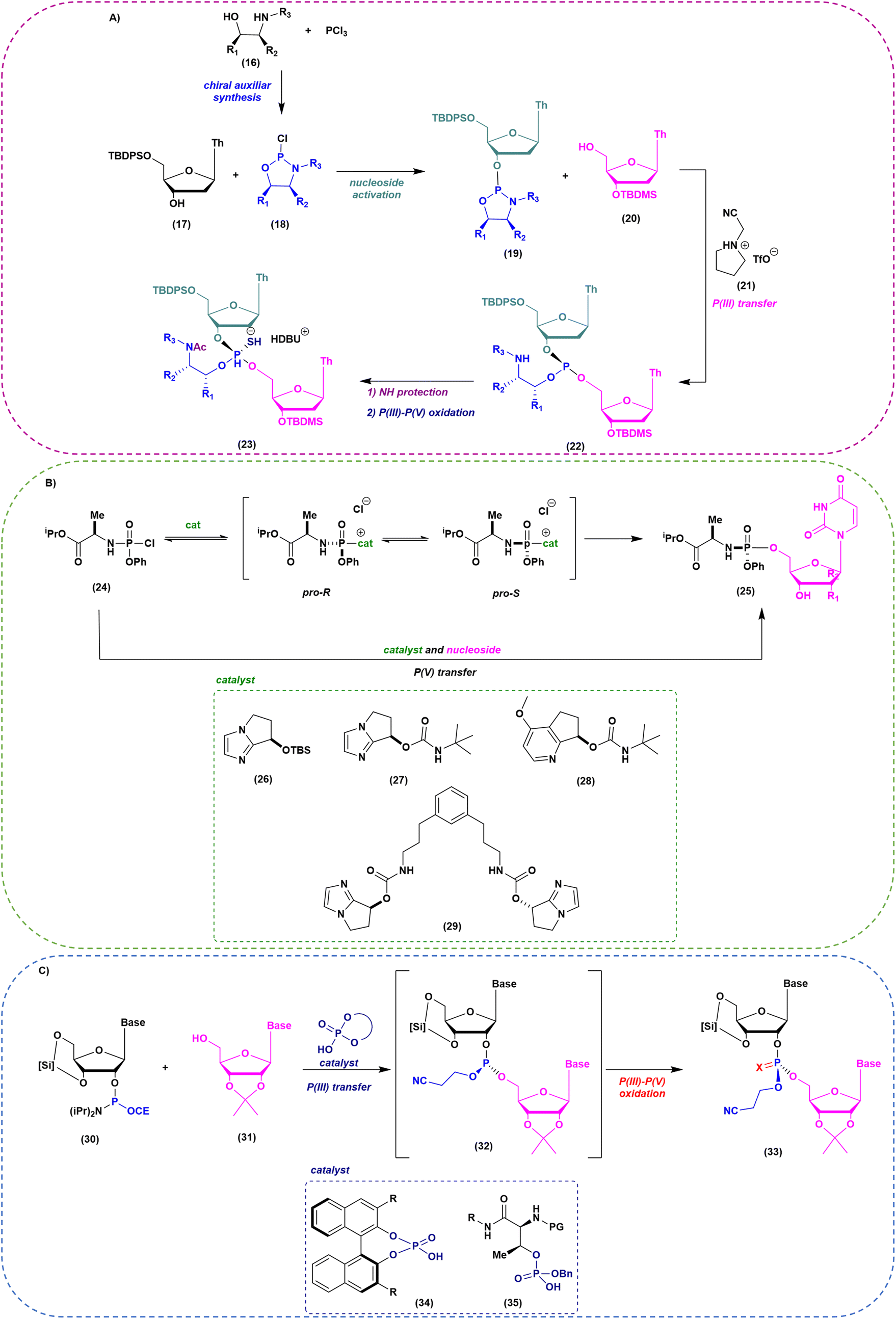 | ||
| Fig. 11 (A) Oxazaphospholidine approach for the stereocontrolled synthesis of PS-oligonucleotides. (B) Multifunctional catalyst for stereocontrolled synthesis of nucleotides via dynamic kinetic asymmetric transformations (DYKAT). (C) Catalytic stereocontrolled synthesis of oligonucleotides via chiral phosphoric acids. | ||
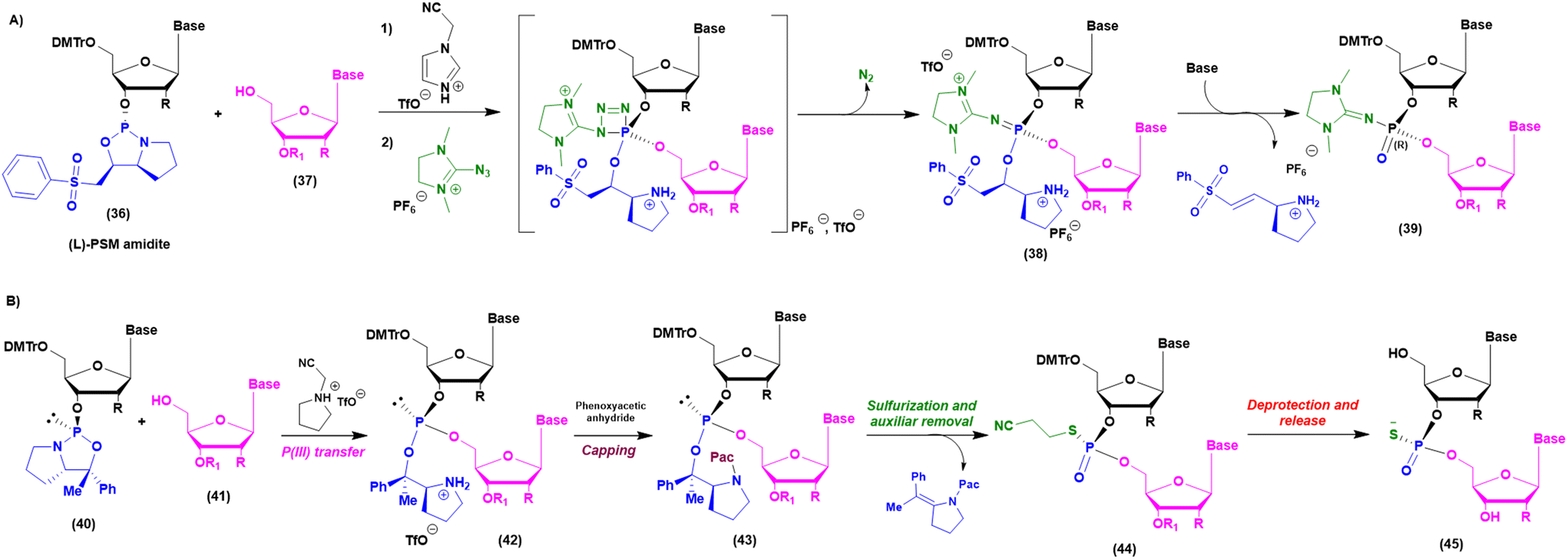 | ||
| Fig. 12 (A) Structure of stereopure PSM amidites and application to the Staudinger reaction for the chiral synthesis of PN backbone linkage. (B) Synthetic cycle of the SOSICS method for the chiral synthesis of PS backbone linkage. | ||
Evaluating the greenness, the increase in the PMI with respect to stereorandom technology is mainly related to the synthesis of the chiral auxiliaries necessary for the synthesis of PN and PS backbones, which cannot be recycled or reused. A completely different approach is the one recently developed in the context of a collaboration between Baran's group and Bristol Myers Squibb (BMS). In 2018, they patented and published, with foresight, a P(V)-based approach to stereocontrolled ON SPS based on the use of P(V)-platforms.79
The synthetic conditions of a cycle based on P(V) chemistry are summarized in the comparative analysis reported in Table 1, and reveal how this new approach maintains many common characteristics with other solid-phase synthetic cycles, being based on the alternating in sequence of loading, coupling and deprotection steps, as in previous methods (Fig. 13). However, the use of P(V)-chemistry allows to eliminate the oxidation step and the use of protecting groups like the cyanoethyl group, as used in standard SPOS.
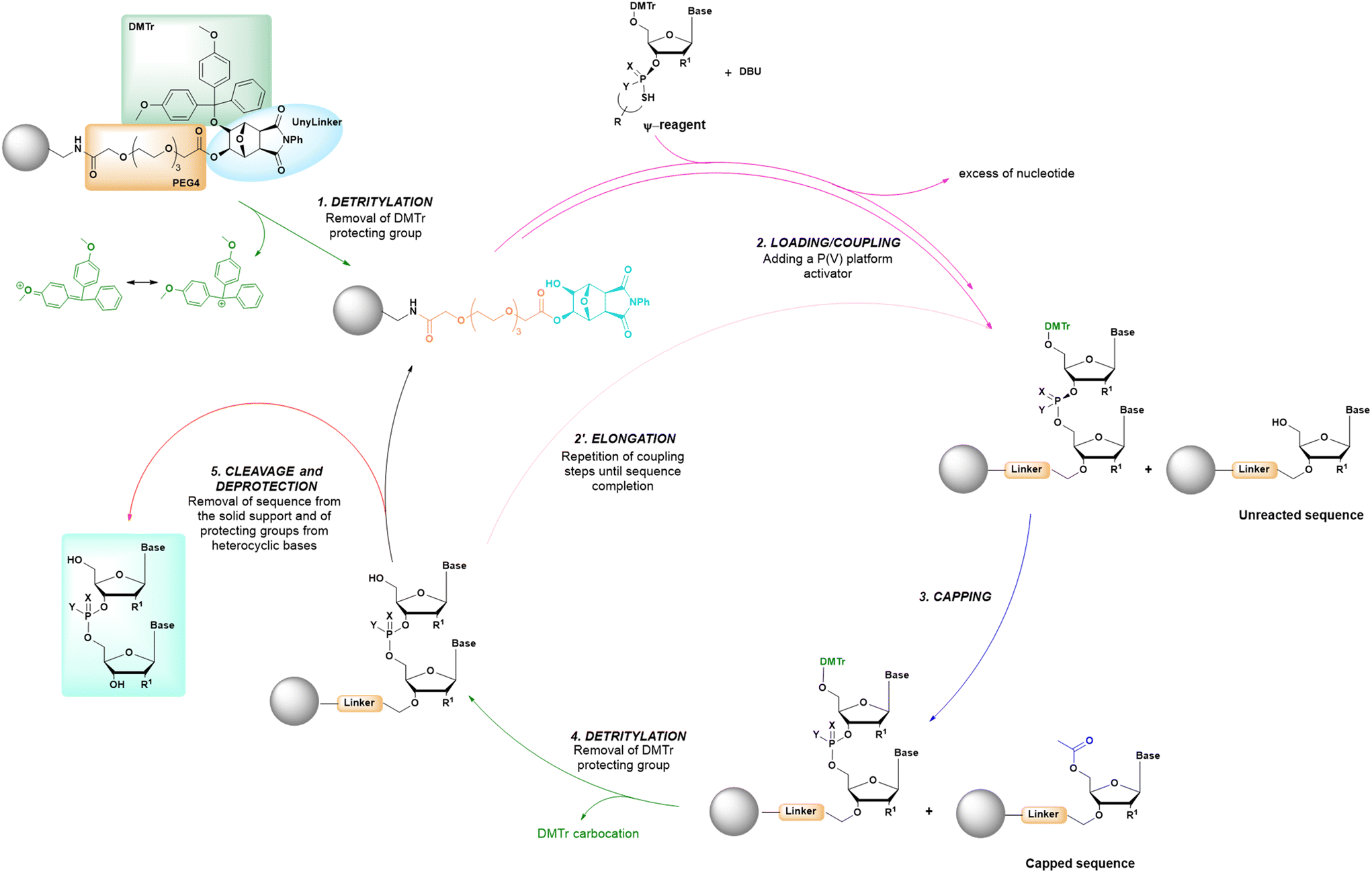 | ||
| Fig. 13 General scheme of solid-phase oligonucleotide synthesis (SPOS) steps by means of P(V) nucleotide platforms. | ||
Initially the ψ-reagent (47) was proposed for the incorporation of PS linkages (ψ = PSI, phosphorus-sulfur incorporation) by fast, scalable and stereodefined reactions, significantly reducing the length of each synthetic cycle by pre-synthesizing the ψ-reagent as a bench-stable compound.79 The reaction with the ψ-reagent was optimized in a liquid-phase to proceed at room temperature, with DBU as the activating agent and ACN as the solvent (Fig. 14), and then transferred to the solid-phase, obtaining both linear and cyclic PS-oligonucleotides (PSOs). Two years later, the attention of the same research group moved towards methylphosphonate oligonucleotides (MPOs) as interesting and stable chiral entities, proposing the first example of the stereodefined synthesis of MPOs. They introduced the П-reagent (52) (П = PI, from phosphorus incorporation), applied not only to P–O chemistry but also to P–C chemistry, to stereoselectively introduce methyl groups into phosphorus (Fig. 15).80
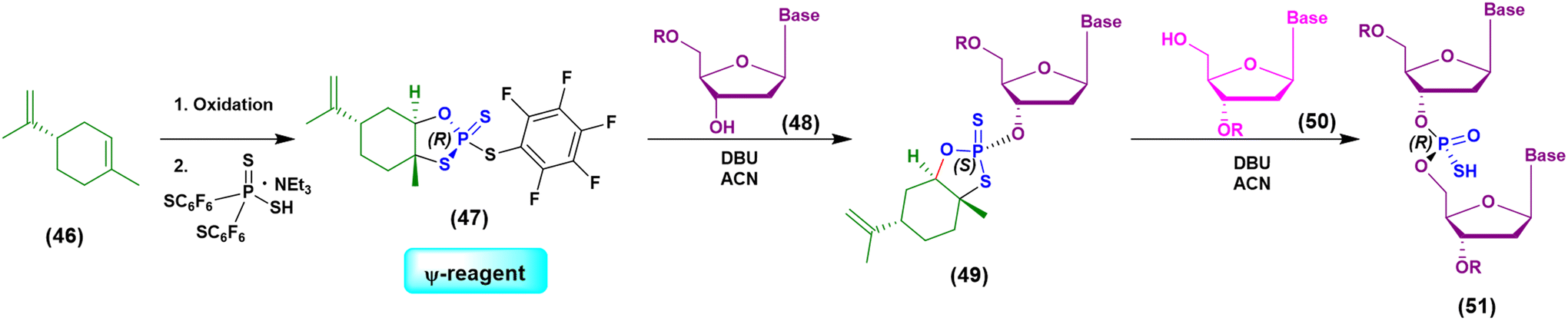 | ||
| Fig. 14 Schematization of the chemical approach based on the use of the ψ-reagent in the synthesis of a model dinucleotide. | ||
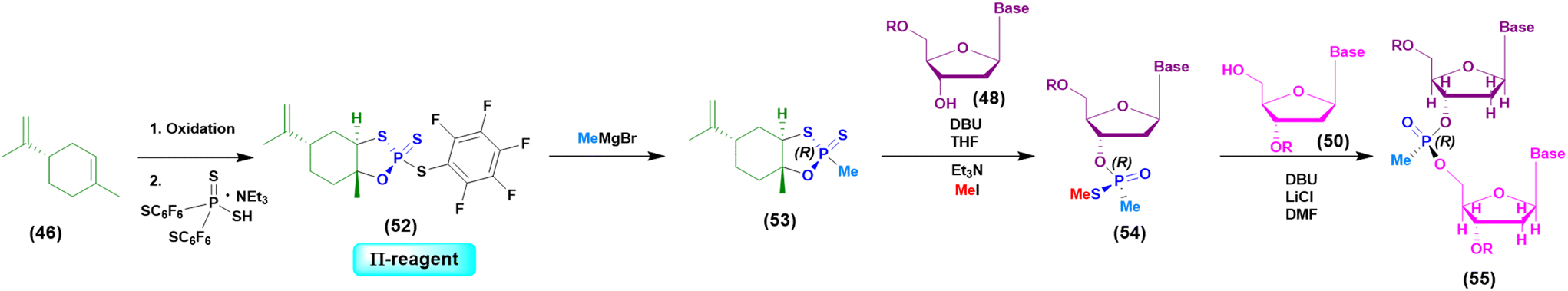 | ||
| Fig. 15 General approach based on the use of the П-reagent in the synthesis of a model dinucleotide. | ||
Then in 2021, Baran and co-workers proposed three new reagents, known as ψ2 (56), rac-ψ (57) and ψ0 reagents (58) (Fig. 16), to introduce stereodefinition into ONs containing contextual PS, PO and PS2 linkages.81,82 These reagents have been successfully applied for the synthesis of chimera ONs, but the use of a more stable solid-support (functionalized by UNILINKER®) was required to tolerate the use of DBU as an activating agent and N-methylpyrrolidine (Pya) protecting groups on the nucleobases. The synthesis of several ONs was achieved with yields ranging between 12% and 27%, which is a poorer result than the 30–60% achieved from stereorandom approaches, but it is still promising considering the upgrade to stereodefinition.
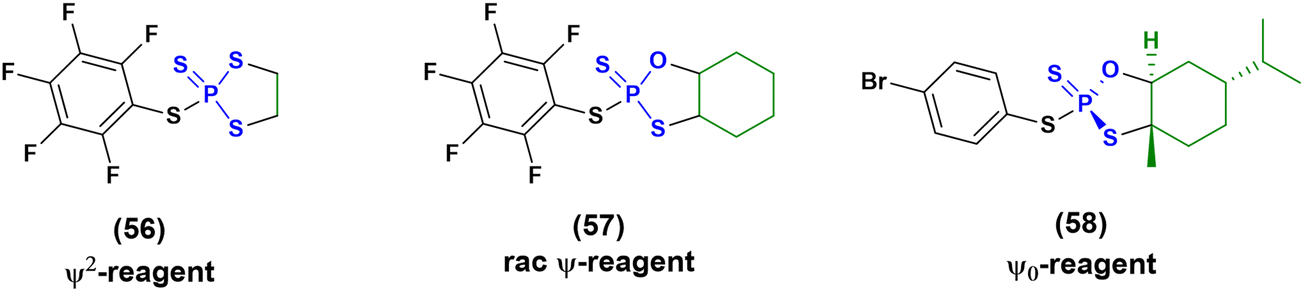 | ||
| Fig. 16 Structures of ψ2, rac- ψ and ψ0 reagents. | ||
The production of stereopure drugs is the future of ONs, and the approaches developed by Wave Life Science and Baran/BMS reveal their potential. For the time being, even if the Baran's P(V) technology allows a synthetic step to be skipped, from a greenness point of view, the complexity of the reagents and the waste of the pentafluorothiophenol moiety tip the scale in favor of Wave technology. Clearly, for both technologies, there is a large space for developments like recycling the chiral auxiliaries and other reagents, as well as replacing solvents with greener alternatives.
6. Liquid-phase TIDES synthesis (LPTS)
As for large-scale SPPS, also in SPOS the methods for monitoring the completion of reaction steps are all a posteriori, which means that it is not possible to repair errors such as incomplete couplings or deprotections (except by repeating the synthesis) without online information about the effect of gradients of concentration common in solid-phase approaches. This blind approach significantly impacts the purification stage of a full-length ON, whose impurity profile is generally more complicated than that of a peptide, including not only shortmers or longmers but also other by-products related to deamidation, depurination, formation of adducts or inefficient protecting group removal (Fig. 17).83 For this reason, LPS is now being reconsidered as a valid alternative, despite limitations from the point of view of sustainability remaining due to the necessity of purifying each intermediate. Therefore, a smart compromise for both long-chain peptides and ONs was the introduction of soluble supports, called anchors, which allow the combination of the positive characteristics of SPS and pure LPS. For peptides, these techniques were protected by patents and trademarks, such as molecular Hiving™ technology (MHT) by Jitsubo,84 Ajiphase by Ajinomoto Bio Services,85 PEPSTAR by a collaboration between Eli Lilly and Exactmer,86 and GAP by GAPPeptides,87 revealing their great potential. Similarly, for ONs, the liquid-phase synthesis (LPOS) found expression in the technique of Nanostar Sieving introduced by Exactmer with the same target (Fig. 18).86 The solid-support is a ramified macromolecule on which several chains of ONs can be built through a linear approach, removing the excess of reagents, solvents, and co-products by filtration on nanomembranes (organic solvent nanofiltration; OSN). In fact, the technology is still in the running-in phase, but it promises to become a big innovation in terms of impact on the market of ON manufacturing and, consequently, on healthcare systems.88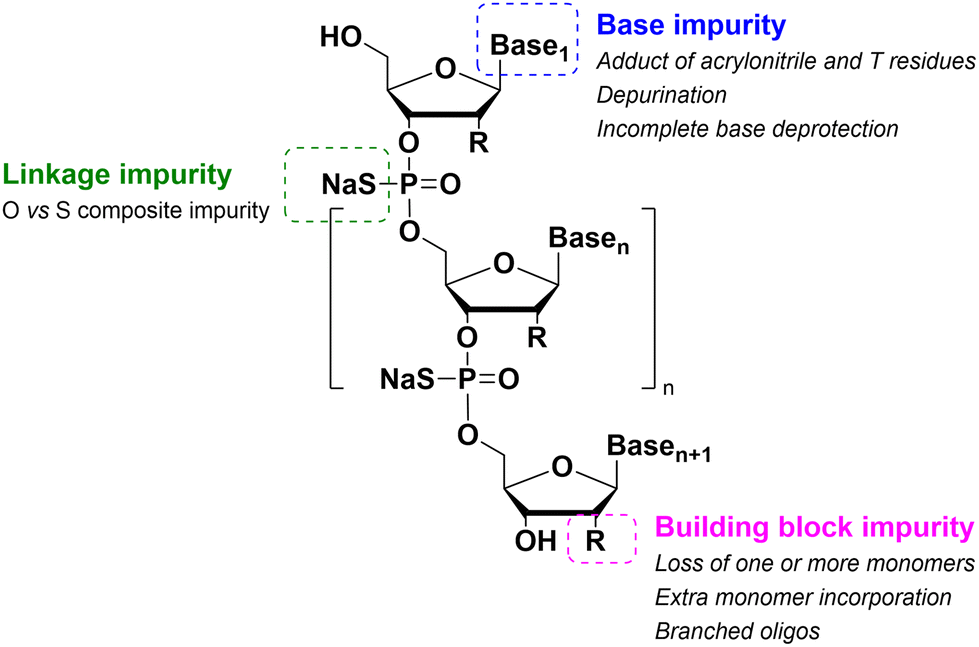 | ||
| Fig. 17 Typical impurities derived from oligonucleotide synthesis. | ||
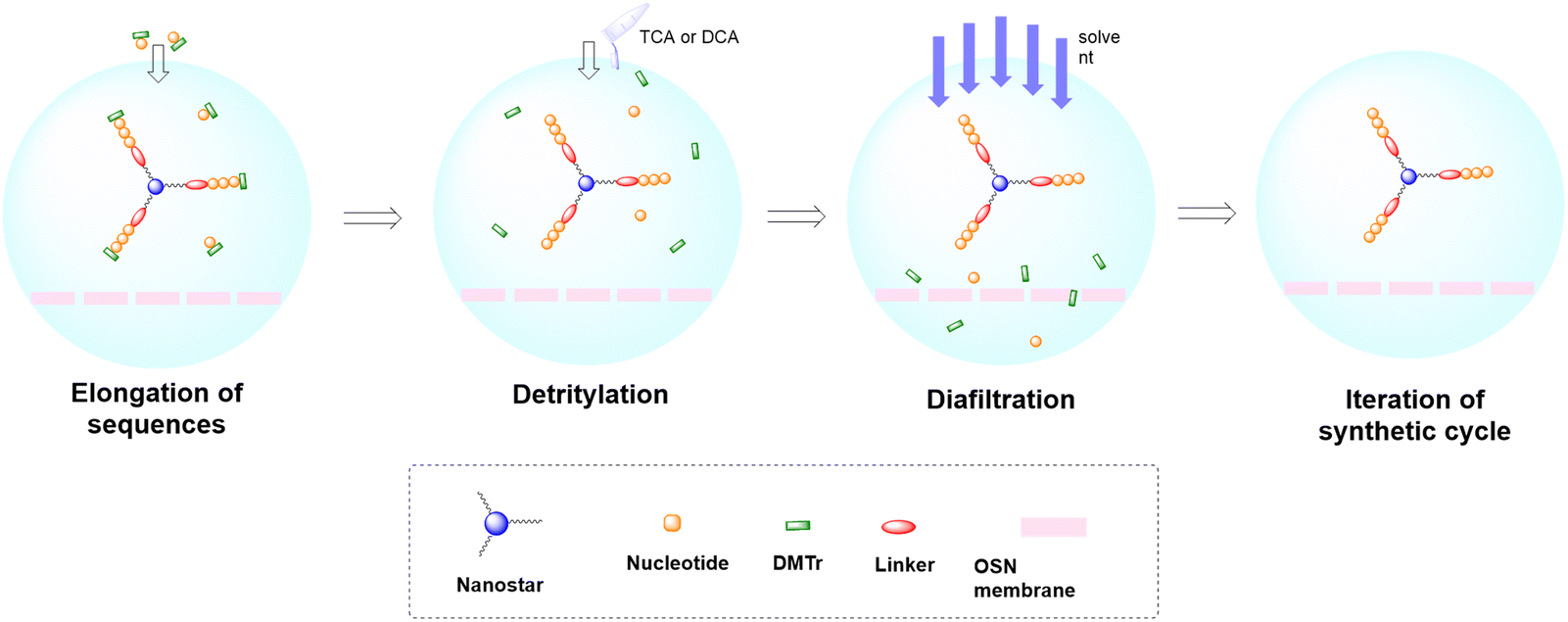 | ||
| Fig. 18 Schematic representation of Nanostar Sieving technology for oligonucleotide synthesis. | ||
To reduce the impact of purification of standard linearly-assembled ONs on their overall greenness score, an interesting example of convergent LPS of an 18-mer ON has been recently reported by Zhou and co-workers89 who demonstrated that a classical 54-reaction linear synthesis can be transformed by a synthetic approach based on 12 reactions and seven isolations (four cycles) involving solubility determining groups (SDGs) which allow the fragment impurity profile to be simply monitored by high-performance liquid chromatography (HPLC) (Fig. 19). As a scope extension, they synthesized a gram-scale 34-mer ON with complete control on the impurity profile of fragments and proved that a final full-length ON with comparable purity to that reported for the corresponding SPOS could be obtained. In addition, the authors made a comparison of the PMIs associated with LPOS and SPOS for the same 34-mer ON, reporting a lower value for the PMI associated with LPOS due to the reduced waste of phosphoramidite and solvents, with the consequent related PMIs being lowered by 48% and 27%, respectively.
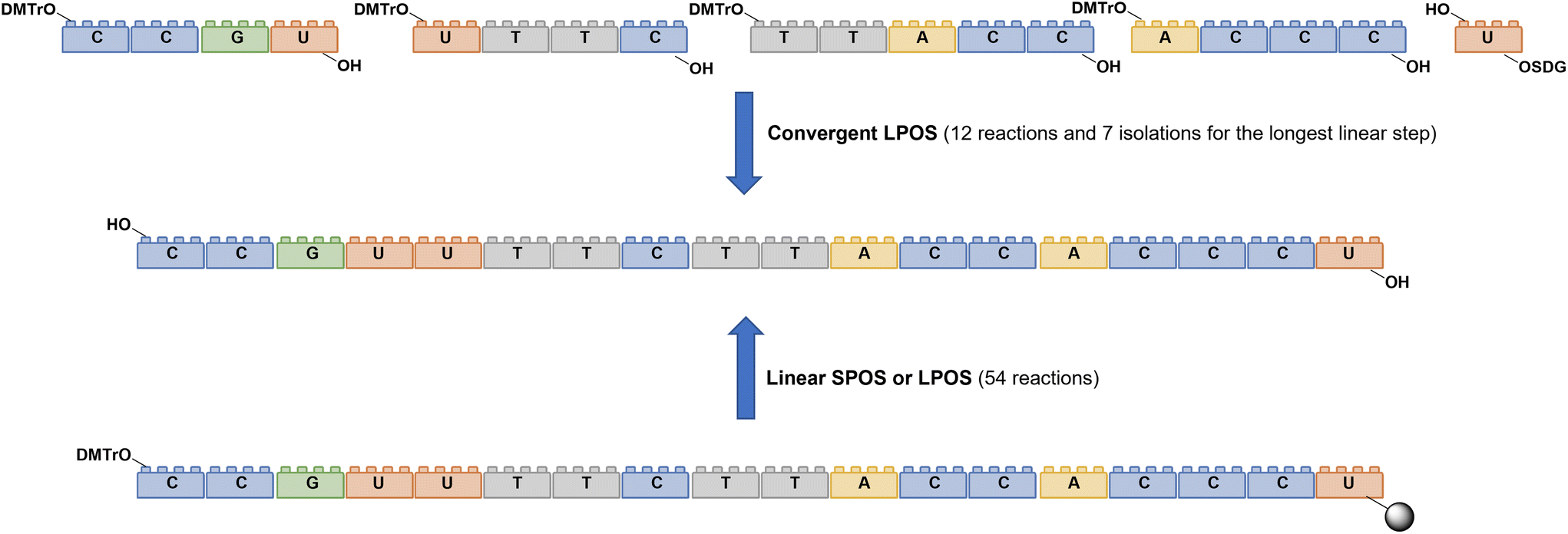 | ||
| Fig. 19 Comparison between linear and convergent syntheses of an 18-mer oligonucleotide. | ||
7. Conclusions
TIDES drug design was supported by consistent process development introduced during the last 15 years, to overcome the limitations intrinsic to their pharmaceutical profile. This has allowed the impact of peptides- and ON-based modalities to be consistently increased in the global pharmaceutical market. Among the different technologies for their manufacturing, SPPS and SPOS play a major role in the synthesis of drugs. While SPPS is ahead from a greenness perspective, with various green innovations that are now overwhelmingly being adopted in industrial practices, considerable work remains to be done for SPOS in order to reach efficiency and greenness in synthetic processes, and to fill the gap in technique development compared with peptides. Considerations on the sustainability of SPOS are particularly important when considering stereocontrolled approaches that generally imply more complex reagent synthesis, the use of auxiliaries, and the generation of toxic side-products and waste. In particular, an action space to improve the PMI in SPOS can be defined. In fact, following the golden example of peptides, it is advisable for SPOS to move towards a reduction in the amount of reagents and solvents used for the entire synthetic pathway, evaluating the possibility of optimizing the process of recovery from waste. The introduction of alternative greener solvents would then be a valuable improvement, especially considering the possibility of integrating them with the emerging chemistry of P(V), which could offer new opportunities for overcoming the limitations of previous approaches in terms of solubilization of the involved species. This is a just-born approach that needs a consistent investment in terms of innovation and development to follow a green path. Therefore, it is foreseeable that in this context a much stronger collaboration between academic and industrial disciplines would represent a powerful contribution to the consolidation of ON manufacturing processes and their scale-up for the massive production of new therapeutics.Author contributions
Conceptualization, L. F., W. C. and A. T.; resources, L. F.; data curation, L. F. and D. C.; writing—original draft, L. F. and D. C.; writing—review & editing, L. F., D. C., W. C. and A. T.; supervision, A. T. and W. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge the Alma Mater Studiorum University of Bologna, for financial support. This study was supported by EU funding within the MUR PNRR “National Center for Gene Therapy and Drugs based on RNA Technology” (Project No. CN00000041 CN3 RNA).References
- A. M. Quemener, L. Bachelot, A. Forestier, E. Donnou-Fournet, D. Gilot and M.-Q. Galibert, WIREs RNA, 2020, 11, e1594 Search PubMed.
- F. Albericio, Chem. Today, 2022, 40, 4–5 Search PubMed.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. de la Torre, Pharmaceutical, 2022, 15, 222–238 CAS.
- L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi, T. Fantoni, A. Mattellone, C. De Luca, S. Felletti, W. Cabri and A. Tolomelli, Green Chem., 2022, 24, 975–1020 RSC.
- Oligonucleotide synthesis market by product, application and end user: Global opportunity analysis and industry forecast, 2021–2030. https://www.alliedmarketresearch.com/oligonucleotide-synthesis-market-A08356 (accessed on 22nd November, 2022).
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- J. Igarashi, Y. Niwa and D. Sugiyama, Future Rare Dis., 2022, 2, FRD19 CrossRef.
- W. Cabri, P. Cantelmi, D. Corbisiero, T. Fantoni, L. Ferrazzano, G. Martelli, A. Mattellone and A. Tolomelli, Front. Mol. Biosci., 2021, 8, 697586 CrossRef CAS PubMed.
- L. Moumné, A.-C. Marie and N. Crouvezier, Pharmaceutics, 2022, 14, 260–283 CrossRef PubMed.
- J. Li, E. Decuypere, H. Aygün, N. Richardson, A. Basso, T. Lindhorst, W. Fang and K. Kobl, Chem. Today, 2022, 40, 28–38 Search PubMed.
- A. M. Quemener and M.-Q. Galibert, WIREs RNA, 2022, 13, e1703 CAS.
- W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef CAS PubMed.
- L. Gentilucci, R. De Marco and L. Cerisoli, Curr. Pharm. Des., 2010, 16, 3185–3203 CrossRef CAS PubMed.
- A. Abdildinova, M. J. Kurth and Y.-D. Gong, Asian J. Org. Chem., 2021, 10, 2300–2317 CrossRef CAS.
- A. C.-L. Lee, J. L. Harris, K. K. Khanna and J.-H. Hong, Int. J. Mol. Sci., 2019, 20, 2383–2403 CrossRef PubMed.
- T. Nuijens, A. Toplak, M. Schmidt, A. Ricci and W. Cabri, Front. Chem., 2019, 7, 829 CrossRef CAS PubMed.
- M. E. Kopach and B. I. Andrews, Curr. Opin. Green Sustainable Chem., 2022, 37, 100643 CrossRef CAS; B. I. Andrews, F. D. Antia, S. B. Brueggemeier, L. J. Diorazio, S. G. Koenig, M. E. Kopach, H. Lee, M. Olbrich and A. L. Watson, J. Org. Chem., 2021, 86, 49–61 CrossRef PubMed.
- A. M. Michelson and A. R. Todd, J. Chem. Soc., 1955, 2632–2638 RSC.
- H. Schaller, G. Weimann, B. Lerch and H. G. Khorana, J. Am. Chem. Soc., 1963, 85, 3821–3827 CrossRef CAS.
- H. G. Khorana, G. M. Tener, J. G. Moflat and E. H. Pol, Chem. Ind., 1956, 1523 CAS.
- H. G. Khorana, W. E. Razzell, P. T. Gilham, G. M. Tener and E. H. Pol, J. Am. Chem. Soc., 1957, 79, 1002 CrossRef CAS.
- P. T. Gilham and H. G. Khorana, J. Am. Chem. Soc., 1958, 80, 6212–6222 CrossRef CAS.
- R. L. Letsinger and V. Mahadevan, J. Am. Chem. Soc., 1965, 87, 3526–3527 CrossRef CAS PubMed.
- R. L. Letsinger and K. K. Ogilvie, J. Am. Chem. Soc., 1967, 89, 4801–4803 CrossRef CAS.
- R. L. Letsinger, K. K. Ogilvie and P. S. Miller, J. Am. Chem. Soc., 1969, 91, 3360–3365 CrossRef CAS.
- For review on the topic see: C. B. Reese, Org. Biomol. Chem., 2005, 3, 3851–3868 RSC.
- R. L. Letsinger and W. B. Lunsford, J. Am. Chem. Soc., 1976, 98, 3655–3661 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- S. P. Adams, K. S. Kavka, E. J. Wykes, S. B. Holder and G. R. Gallupi, J. Am. Chem. Soc., 1983, 105, 661–663 CrossRef CAS.
- L. J. McBride and M. H. Caruthers, Tetrahedron Lett., 1983, 24, 245–248 CrossRef CAS.
- N. D. Sinha, J. Biernat and H. Köster, Tetrahedron Lett., 1983, 24, 5843–5846 CrossRef CAS.
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393–2398 RSC.
- A. Kumar, Y. E. Jad, J. M. Collins, F. Albericio and B. G. de la Torre, ACS Sustainable Chem. Eng., 2018, 6, 8034–8039 CrossRef CAS.
- S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685–1691 RSC.
- A. Kumar, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2986–2988 CrossRef CAS.
- J. Lopez, S. Pletscher, A. Aemissegger, C. Bucher and F. Gallou, Org. Process Res. Dev., 2018, 22, 494–503 CrossRef CAS.
- A. Kumar, M. Alhassan, J. Lopez, F. Albericio and B. G. de la Torre, ChemSusChem, 2020, 13, 5288–5294 CrossRef CAS PubMed.
- G. Martelli, P. Cantelmi, A. Tolomelli, D. Corbisiero, A. Mattellone, A. Ricci, T. Fantoni, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and L. Ferrazzano, Green Chem., 2021, 23, 4095–4106 RSC.
- A. Kumar, A. Sharma, B. G. de la Torre and F. Albericio, Green Chem. Lett. Rev., 2021, 14, 545–550 CrossRef CAS.
- L. Ferrazzano, D. Corbisiero, G. Martelli, A. Tolomelli, A. Viola, A. Ricci and W. Cabri, ACS Sustainable Chem. Eng., 2019, 7, 12867–12877 CrossRef CAS.
- A. Přibylka, M. Pastorek, M. Grepl and E. Schütznerová, Tetrahedron, 2021, 99, 132452 CrossRef.
- V. Martin, S. Jadhav, P. H. G. Egelund, R. Liffert, H. J. Castro, T. Krüger, K. F. Haselmann, S. Thordal Le Quement, F. Albericio, F. Dettner, C. Lechner, R. Schönleber and D. S. Pedersen, Green Chem., 2021, 23, 3295–3311 RSC.
- J. Pawlas, B. Antonic, M. Lundqvist, T. Svensson, J. Finnman and J. H. Rasmussen, Green Chem., 2019, 21, 2594–2600 RSC.
- J. Pawlas and J. H. Rasmussen, Green Chem., 2019, 21, 5990–5998 RSC.
- A. Přibylka, V. Krchňák and E. Schütznerová, Green Chem., 2019, 21, 775–779 RSC.
- A. H. Tullo, Ineos to boost capacity for critical solvent, Chem. Eng. News, 2022, 100 Search PubMed.
- O. F. Luna, J. Gomez, C. Cárdenas, F. Albericio, S. H. Marshall and F. Guzmán, Molecules, 2016, 21, 1542 CrossRef PubMed.
- O. Al Musaimi, R. Wisdom, P. Talbiersky, B. G. de la Torre and F. Albericio, ChemistrySelect, 2021, 6, 2649–2657 CrossRef CAS.
- V. Rodríguez, H. Pineda, N. Ardila, D. Insuasty, K. Cárdenas, J. Román, M. Urrea, D. Ramírez, R. Fierro, Z. Rivera and J. García, Int. J. Pept. Res. Ther., 2020, 26, 585–587 CrossRef.
- I. Guryanov, A. Orlandin, A. Viola, B. Biondi, F. Formaggio, A. Ricci and W. Cabri, Org. Process Res. Dev., 2020, 24, 274–278 CrossRef CAS.
- G. Martelli, P. Cantelmi, C. Palladino, A. Mattellone, C. Corbisiero, T. Fantoni, A. Tolomelli, M. Macis, A. Ricci, W. Cabri and L. Ferrazzano, Green Chem., 2021, 23, 8096–8107 RSC.
- P. H. G. Egelund, S. Jadhav, V. Martin, H. Johansson Castro, F. Richner, S. Thordal Le Quement, F. Dettner, C. Lechner, R. Schoenleber and D. S. Pedersen, ACS Sustainable Chem. Eng., 2021, 9, 14202–14215 CrossRef CAS.
- M. Alhassan, O. Al Musaimi, J. M. Collins, F. Albericio and B. G. de la Torre, Green Chem., 2020, 22, 2840–2845 RSC.
- B. G. de la Torre, S. Ramkisson, F. Albericio and J. Lopez, Org. Process Res. Dev., 2021, 25, 1047–1053 CrossRef.
- J. Pawlas and J. H. Rasmussen, ChemSusChem, 2021, 14, 3231–3236 CrossRef CAS PubMed.
- C. L. Scremin, L. Zhou, K. Srinivasachar and S. L. Beaucage, J. Org. Chem., 1994, 59, 1963–1966 CrossRef CAS.
- Z. Guo, H. M. Pfundheller and Y. S. Sanghvi, Org. Process Res. Dev., 1998, 2, 415–417 CrossRef CAS.
- A. H. Krotz, R. L. Carty, M. N. Moore, A. N. Scozzari, D. L. Cole and V. T. Ravikumar, Green Chem., 1999, 1, 277–282 RSC.
- A. H. Krotz, R. L. Carty, A. N. Scozzari, D. L. Cole and V. T. Ravikumar, Org. Process Res. Dev., 2000, 4, 190–193 CrossRef CAS.
- Z. S. Cheruvallath, R. L. Carty, M. Andrade, M. N. Moore, Q. Song, C. Rentel, D. L. Cole and V. T. Ravikuma, Org. Process Res. Dev., 2003, 7, 917–920 CrossRef CAS.
- Y. S. Sanghvi, V. T. Ravikumar, A. N. Scozzari and D. L. Cole, Pure Appl. Chem., 2001, 73, 175–180 CrossRef CAS.
- C. Vargeese, J. Carter, J. Yegge, S. Krivjansky, A. Settle, E. Kropp, K. Peterson and W. Pieken, Nucleic Acid Res., 1998, 26, 1046–1050 CrossRef CAS PubMed.
- J. Yang, J. A. Stolee, H. Jiang, L. Xiao, W. F. Kiesman, F. D. Antia, Y. A. Fillon, A. Ng and X. Shi, J. Org. Chem., 2018, 83, 11577–11585 CrossRef CAS PubMed.
- A. F. Sandahl, T. J. D. Nguyen, R. A. Hansen, M. B. Johansen, T. Skydstrup and K. V. Gothelf, Nat. Commun., 2021, 12, 2760–2766 CrossRef CAS PubMed.
- J. Kundu, A. Ghosh, U. Ghosh, A. Das, D. Nagar, S. Pattanayak, A. Ghose and S. Sinha, J. Org. Chem., 2022, 87, 9466–9478 CrossRef CAS PubMed.
- H. Krishna, K. Jastrzebska and M. Caruthers, FEBS Lett., 2019, 593, 1459–1467 CrossRef CAS PubMed.
- C. Li, A. J. Callahan, M. D. Simon, K. A. Totaro, A. J. Mijalis, K.-S. Phadke, G. Zhang, N. Hartrampf, C. K. Schissel, M. Zhou, H. Zong, G. J. Hanson, A. Loas, N. L. B. Pohl, D. E. Verhoeven and B. L. Pentelute, Nat. Commun., 2021, 12, 4396 CrossRef CAS PubMed.
- N. Oka, T. Wada and K. Saigo, J. Am. Chem. Soc., 2002, 124, 4962–4963 CrossRef CAS PubMed.
- N. Oka, T. Wada and K. Saigo, J. Am. Chem. Soc., 2003, 125, 8307–8317 CrossRef CAS PubMed.
- D. A. DiRocco, Y. Ji, E. C. Sherer, A. Klapars, M. Reibarkh, J. Dropinski, R. Mathew, P. Maligres, A. M. Hyde, J. Limanto, A. Brunskill, R. T. Ruck, L.-C. Campeau and I. W. Davies, Science, 2017, 356, 426–430 CrossRef CAS PubMed.
- M. Wang, L. Zhang, X. Huo, Z. Zhang, Q. Yuan, P. Li, J. Chen, Y. Zou, Z. Wu and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20814–20819 CrossRef CAS PubMed.
- A. L. Featherston, Y. Kwon, M. M. Pompeo, O. D. Engl, D. K. Leahy and S. J. Miller, Science, 2021, 371, 702–707 CrossRef CAS PubMed.
- K. W. Knouse, J. N. deGruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. Mcdonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Science, 2018, 361, 1234–1238 CrossRef CAS PubMed.
- D. Butler, N. Iwamoto, M. Meena, N. Svrzikapa, G. L. Verdine and I. Zlatev, US Patent10590413, 2017 (Priority n.61671,656, 2012) Search PubMed.
- (a) P. Kandasamy, G. McClorey, M. Shimizu, N. Kothari, R. Alam, N. Iwamoto, J. Kumarasamy, G. R. Bommineni, A. Bezigian, O. Chivatakarn, D. C. D. Butler, M. Byrne, K. Chwalenia, K. E. Davies, J. Desai, J. D. Shelke, A. F. Durbin, R. Ellerington, B. Edwards, J. Godfrey, A. Hoss, F. Liu, K. Longo, G. Lu, S. Marappan, J. Oieni, I.-H. Paik, E. Purcell-Estabrook, C. Shivalila, M. Tischbein, T. Kawamoto, C. Rinaldi, J. Rajão-Saraiva, S. Tripathi, H. Yang, Y. Yin, X. Zhao, C. Zhou, J. Zhang, L. Apponi, M. J. A. Wood and C. Vargeese, Nucleic Acids Res., 2022, 50, 5443–5466 CrossRef CAS PubMed; (b) P. Kandasamy, Y. Liu, V. Aduda, S. Akare, R. Alam, A. Andreucci, D. Boulay, K. Bowman, M. Byrne, M. Cannon, O. Chivatakarn, J. D. Shelke, N. Iwamoto, T. Kawamoto, J. Kumarasamy, S. Lamore, M. Lemaitre, X. Lin, K. Longo, R. Looby, S. Marappan, J. Metterville, S. Mohapatra, B. Newman, I.-H. Paik, S. Patil, E. Purcell-Estabrook, M. Shimizu, P. Shum, S. Standley, K. Taborn, S. Tripathi, H. Yang, Y. Yin, X. Zhao, E. Dale and C. Vargeese, Nucleic Acids Res., 2022, 50, 5401–5423 CrossRef CAS PubMed.
- N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kemenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotec., 2017, 35, 845–851 CrossRef CAS PubMed.
- M. A. Schmidt, B. Zheng, K. Knouse, J. deGruyter, M. D. Eastgate, P. Baran, W. R. Ewing, R. E. Olson and I. M. Mcdonald, US Patent201862657551P, 2018 Search PubMed.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
- Y. Huang, K. W. Knouse, S. Qiu, W. Hao, N. M. Padial, J. C. Vantourout, B. Zheng, S. E. Mercer, J. Lopez-Ogalla, R. Narayan, R. E. Olson, D. G. Blackmond, M. D. Eastgate, M. A. Schmidt, I. M. McDonald and P. S. Baran, Science, 2021, 373, 1265–1270 CrossRef CAS PubMed.
- M. Ociepa, K. W. Knouse, D. He, J. C. Vantourout, D. T. Flood, N. M. Padial, J. S. Chen, B. B. Sanchez, E. J. Sturgell, B. Zheng, S. Qiu, M. A. Schmidt, M. D. Eastgate and P. S. Baran, Org. Lett., 2021, 23, 9337–9342 CrossRef CAS PubMed.
- M. Catani, C. De Luca, J. Medeiros Garcia Alcântara, N. Manfredini, D. Perrone, E. Marchesi, R. Weldon, T. Müller-Späth, A. Cavazzini, M. Morbidelli and M. Sponchioni, Biotechnol. J., 2020, 1900226 CrossRef CAS PubMed.
- (a) K. Chiba, S. Kim and Y. Kono, US Patent8633298B2, 2014 (Priority n. JP2005-272905, 2005) Search PubMed; (b) K. Chiba, S. Kim and Y. Kono, US Patent8293948B2, 2012 (Priority n. JP2006084019, 2006) Search PubMed.
- (a) D. Takahashi, US Patent9353148B2, 2016 (Priority n. JP2011-122943, 2011) Search PubMed; (b) d. Takahashi, US Patent9334302B2, 2016 (Priority n. JP2011/274901, 2011) Search PubMed; (c) D. Takahashi, US Patent2016060198A1, 2016 (Priority n. JP2009-083579, 2009) Search PubMed; (d) D. Takahashi, US Patent9206230B2, 2015 (Priority n. JP2009-296366, 2009) Search PubMed; (e) D. Takahashi, US Patent9029504B2, 2015 (Priority n. JP2009060291, 2009) Search PubMed.
- (a) Technology – Exactmer. https://exactmer.com/technology/ (accessed on 22nd November 2022); (b) C. Noti, M. Cristau, J. Riegler, V. d. l. P. d. M. Castro Pinzon, H. M. Rodriguez Cabrera, F. Albericio Palomera, W. Chen and A. G. Livingston, WO Patent2016188835A1, 2016 (Priority n. US201562165324P, 2015) Search PubMed; (c) A. G. Livingston, J. Kim, I. B. Valtcheva, P. R. J. Gaffney and M. Schaepertoens, WO Patent2016020708A1, 2016 (Priority n. GBGB1414120.4A, 2014) Search PubMed; (d) A. G. Livingston, J. Kim, I. B. Valtcheva and P. R. J. Gaffney, WO Patent2016024105A1, 2016 (Priority n. GBGB1414213.7A, 2014) Search PubMed; (e) A. G. Livingston, S. Kumbharkar, L. Peeva and J. Da Silva Burgal, US Patent20170007963A1, 2017 (Priority n. GB1401392.4, 2014) Search PubMed; (f) M. Mechelhoff, P. Marchetti, A. G. Livingston and Z. Karina, US Patent10239024B2, 2019 (Priority n. EP14169648, 2014) Search PubMed; (g) M. Schaepertoens, P. R. J. Gaffney, G. Szekely and A. G. Livingston, US Patent10239996B2, 2019 (Priority n. GBGB1413954.7A, 2014) Search PubMed; (h) M. Cook and A. G. Livingston, US Patent, 20200122094A1, 2020 (Priority n. GB1710912.5, 2017) Search PubMed; (i) A. G. Livingston, P. Marchetti, P. Gaffney and R. Liu, US Patent10913033B2, 2021 (Priority n. GBGB1609873.3A, 2016) Search PubMed; (j) A. G. Livingston and M. F. Jimenez Solomon, US Patent11117104B2, 2021 (Priority n. GB1117950, 2011) Search PubMed; (k) A. G. Livingston, Z. Jiang, R. Dong, J. Xu and S. Li, WO Patent2021205141A1, 2021 (Priority n. GB2005106.6, 2020) Search PubMed.
- (a) C. Seifert, US Patent20210079036A1, 2021 (Priority n. US201862678564P, 2018) Search PubMed; (b) C. Seifert, WO Patent2020159837A1, 2020 (Priority n. Priority to US201962800142P, 2019) Search PubMed; (c) C. Seifert, WO Patent2019217116A1, 2019 (Priority n. Priority to US201862667591P, 2018) Search PubMed; (d) C. Seifert, WO Patent2019231760A1, 2019 (Priority n. Priority to US201862678564P, 2018) Search PubMed; (e) C. Seifert and G. Li, US Patent10947267B2, 2021 (Priority n. US201562270432P, 2015) Search PubMed.
- K. Murray and A. Livingston, Chem. Today, 2022, 39, 32–34 Search PubMed.
- X. Zhou, W. F. Kiesman, W. Yan, H. Jiang, F. D. Antia, J. Yang, Y. A. Fillon, L. Xiao and X. Shi, J. Org. Chem., 2022, 87, 2087–2110 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |