
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-functionalized polydopamine-coated polyurethane foam: a readily prepared and highly reusable structured catalyst for selective alkyne semi-hydrogenation and Suzuki coupling under air†
Han
Peng
a,
Xiong
Zhang
b,
Vasiliki
Papaefthimiou
 b,
Cuong
Pham-Huu
*b and
Vincent
Ritleng
*a
b,
Cuong
Pham-Huu
*b and
Vincent
Ritleng
*a
aUniversité de Strasbourg, Ecole européenne de Chimie, Polymères et Matériaux, CNRS, LIMA, UMR 7042, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: vritleng@unistra.fr
bUniversité de Strasbourg, CNRS, Institute of Chemistry and Processes for Energy, Environment and Health (ICPEES), UMR 7515, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: cuong.pham-huu@unistra.fr
First published on 29th November 2022
Abstract
A polyurethane foam (PUF) was coated with a thin layer of polydopamine (PDA), and Pd strongly anchored onto PDA via simple immersion of PDA@PUF in a hydro-alcoholic solution of Pd(NH3)4Cl2·H2O at room temperature under air to afford a Pd@PDA@PUF foam. XPS showed the absence of initial reduction of palladium despite the redox properties of PDA, and HR-SEM and SEM-EDX suggested a coordination of evenly dispersed Pd(II) ions by the catechol moieties of the PDA layer, thus resulting in a single atom-type dispersion of the active phase. The “dip-and-play” catalytic pattern of Pd@PDA@PUF and high dispersion of Pd on the surface guaranteed high activity, selectivity and reusability in alkyne semi-hydrogenation and in Suzuki coupling under air without prior reduction procedures. Self-enhancing properties, observed in recycling and control experiments, were explained by XPS as the chelated Pd(II) ions were gradually reduced to isolated coordinated Pd(0) atoms under hydrogenation or Suzuki coupling reaction conditions.
Introduction
Continuous processes based on structured catalytic supports (SCSs) have gained widespread application in industry. Structured supports possess a large surface-to-volume ratio, and enable efficient mass transfers along with a small pressure drop. The high voidage and open structure along with the macroscopic shape allow the intimate mixing of the reagents, and the high accessibility of the reactants to the active sites, while avoiding tedious separation of the products from the catalyst.1 Open cell foams account for a large proportion among the variety of SCSs. Of ceramic or metallic constitution, these host architectures are ideal supports for catalytically active metallic nanoparticles (NPs).2–4 However, the preparation of these foams from polymeric foams serving as templates requires multiple energy- and material-intensive steps, including physisorption processes for the deposition of the catalytic phase on the support surface.3 Furthermore, these expensive structures have several drawbacks: (i) their high rigidity brings out micro-cracks and makes them more brittle, (ii) the presence of many randomly distributed closed cells significantly reduces the reproducibility of the reactions, and (iii) numerous chemical treatments in highly corrosive media are often necessary for recovering the catalyst adsorbed on the foam before landfill disposal.5Open cell polyurethane foam (PUF), as a kind of commercially available, inexpensive, mechanically flexible, and easily engineered material,6 presenting the same structural properties as ceramic and metallic foams, represents an interesting alternative to the latter. The absence of microporosity to deposit an active phase is however a challenge to overcome. Inspired by the mussel-like adhesive principle,7 some of us recently reported the successful coating of PUF with an adhesive layer of polydopamine (PDA) by simple immersion in a buffered aqueous solution of dopamine, its further functionalization with both inorganic and molecular catalysts, and its application in catalysis or dye degradation.8–14 PDA's strongly adhesive properties ensured the robust anchoring of the catalysts, leading to high activity and stability over a large number of cycling tests and to the absence of catalyst leaching in the liquid medium. The high versatility of PDA, which can interact with virtually any kind of surface and can be easily functionalized, is the critical point. The catechol and amine functions of PDA indeed confer both strong adhesion properties and the possibility of surface modification by means of different chemical interactions, ranging from weak dispersion forces to covalent bonds, such as hydrogen bonding, π–π stacking, coordination bonds, and Michael-type addition (via internal imine group formation).15
The semi-hydrogenation of alkynes to alkenes is of paramount importance both in industrial and in organic synthetic domains,16,17 but the selective production of olefins from alkynes requires catalysts that are able to both hydrogenate the triple bond and hamper overhydrogenation toward alkanes. In addition, these catalysts should limit the undesired double bond isomerization.18 In this respect, supported Pd nanoparticle-based catalysts19–23 are among the best candidates in terms of activity, but they often suffer from moderate selectivity.24 Typical approaches to improve it25 rely on the modification of the Pd nanoparticles (NPs) by the addition of N-, P-, or S-containing ligands,26–28 their use in combination with nanomicelles,29 or ‘doping’ with a less active metal.26,30,31 A classic example in this domain is the famous Lindlar catalyst, in which Pd/CaCO3 is partially poisoned by the addition of Pb and quinoline. However, the use of Pb requires a precise control of its location with respect to the Pd active phase and represents a serious limitation for the eco-sustainable use of the Lindlar catalyst. A more recent approach relies on the strategy of active site isolation, which includes the development of single atom-based catalysts.25,32 These attractive strategies with, in some cases, selectivity up to 99% and reuse up to 10 times may however be not so valuable if one cannot scale up the process for industrial implementation in a practical and highly operable manner with cost competitiveness and in an eco-friendly mode. For instance, Lu et al.33 reported an unsupported nanoporous palladium (PdNPore) while Kuwahara et al.34 introduced a yolk–shell nanostructured composite composed of Pd nanoparticles (NPs), aminopolymers, and poly(ethylenimine) (PEI) confined in hollow silica spheres, and Shen et al.35 recently described a two-dimensionality (2D) enhanced interface-confined effect that led to a strong interaction between Pb species and ultrathin 2D palladium nanosheets (Pd NSs). These various catalysts exhibited high conversion and chemoselectivity in semi-hydrogenation of phenylacetylene and other alkynes as well as superior recyclability ensuring catalyst reuse for several times. However, the adopted catalyst designs are tedious, time-consuming and energy-intensive. A simpler and easier to scale-up catalyst production strategy is thus still desirable but remains challenging.
This challenge prompted us to consider palladium-driven heterogeneous catalysis with PDA-coated open cell polyurethane foam (PDA@PUF) as a non-innocent support for the semi-hydrogenation of alkynes and possibly other organic transformations. Indeed, polydopamine is known to act as an efficient chelating agent towards a variety of metals, such as Ag+, Au+, Pd2+, Pt2+ salts, etc.,36,37 forming stable complexes, and moreover to be able to reduce these metal ions thanks to the reductive properties of the catechol moieties. Accordingly, the generation of such PDA-supported palladium complexes can be regarded as an “open gate” for the convenient and rapid preparation of heterogeneous Pd(0) catalysts. Inspired by this concept, catalytic composites based on polydopamine-supported palladium have been successfully prepared and applied in ammonia borane hydrolysis, various cross-couplings and nitroarene transfer hydrogenation,38–40 providing this topic a promising future.
In this context, we put forward an effective and straightforward approach for the preparation of Pd-functionalized polydopamine-coated polyurethane foam starting from a commercial PUF, dopamine, and [Pd(NH3)4]Cl2·H2O. The whole process leaves no doubt that the preparation of the macroscopic catalytic object is unprecedently facile and efficient with only two successive operations of dipping, stirring and rinsing at room temperature. This simple route lowers the threshold for catalyst preparation by several orders of magnitude compared to current site isolation strategies. Furthermore, the catalytic performance of the resulting Pd@PDA@PUF catalytic material in a “dip-and-play” mode reaches a high standard in efficiency, versatility and stability. Thus, the as-prepared catalyst displays high catalytic performance in the selective semi-hydrogenation of various alkynes as well as in Suzuki coupling reactions, and moreover shows remarkable stability for 15 cycling tests. Noteworthily, the XPS, HR-SEM and SEM-EDX data suggest an initial chelation of the Pd(II) salt by the catechol/o-quinone moieties of the PDA layer, which would result in a “single atom” nature of the active phase that would be at the origin of Pd@PDA@PUF's high alkene selectivity in alkyne hydrogenation catalysis. Furthermore, in the absence of the necessity of a prior reduction procedure to generate an active phase, self-enhancing catalytic properties have been observed along the recycling experiments, both in the semi-hydrogenation reactions and in the Suzuki couplings under air. These were explained by XPS that showed a progressive reduction of the immobilized Pd(II) salt to an active Pd(0) phase along the catalytic tests.
Experimental section
Materials and methods
Unless otherwise stated, all reagents and solvents were used as provided by commercial suppliers without any further purification or treatment. Polyurethane open cell foam (20 pores per inch, TCL 40100 soft white reticulated) was given by FoamPartner. Dopamine hydrochloride (008896) and 1,2-diphenylethyne (95% – 464554) were purchased from Fluorochem. Tris base (99.9+% – T1503), styrene (99% – 24086-9), 4-bromotoluene (98% – B82200), trans-stilbene (96% – 13993-9), heptane (99% – 650536) and [PdCl2(NH3)4]·H2O (≥99.9% – 323438) were purchased from Sigma-Aldrich. 1-Heptyne (99% – A11130), ethyl phenylpropiolate (98% – L00603), 2-methyl-3-butyn-2-ol (98% – L07593), phenylacetylene (98% – A12139), benzeneboronic acid (98% – A14257), 4-bromobenzonitrile (98% – A10914), 1-bromo-4-tert-butylbenzene (97% – A15924), 4-bromobenzotrifluoride (99% – A15942), methyl 2-bromobenzoate (99% – A12435) and 2-bromobiphenyl (98% – B21840) were purchased from Alfa Aesar. Bromobenzene (99% – 106682500), 4-bromoanisole (98% – 106630050) and 2-bromonaphthalene (99% – 179710250) were purchased from Acros Organics. Chlorobenzene (>98% – C1948), 4-phenyl-1-butene (>98% – P0161), 4-phenyl-1-butyne (P0358), 4-octyne (>99% – O0127), cis-4-octene (>95% – O0135), 3-hexyne (>97% – H0430), 4-octane (>97% – O0022), 2-methyl-3-buten-2-ol (>97% – M0178), trans-3-hexene (>99% – H0399), trans-4-octene (>99% – O0136), cis-3-hexene (>97% – H0398), ethyl (E)-cinnamate (>99% – C0359) and 3-bromo-4-methoxytoluene (>97% – B3032) were provided by TCI. Ethyl 3-phenylpropanoate (98% – 56675) was provided by Fluka. Ethyl 3-phenylpropionate (98% – 11885.51) and cis-stilbene (97% – 13277.85) were obtained from Janssen. 18.2 MΩ deionized water (TOC < 1 ppb), supplied by a Q20 Millipore system, was used for the preparation of aqueous solutions and washing procedures with water. Purifications by column chromatography were carried out with Macherey silica gel (Kielselgel 60). TraceMetal grade HCl 37% (Fisher Chemical) and Rotipuran Supra HNO3 69% (Roth) were used for the ICP-AES analyses. Element standards were purchased from CPI international.High-resolution scanning electron microscopy (HR-SEM) analyses were carried out on a high-resolution JEOL JSM-7900F SEM-FEG working at 2–7 kV accelerated voltage and at a distance of 6 to 10 mm. Images were obtained with a secondary electron detector. In some cases, samples were coated with a fine layer of carbon (ca. 10 nm) using a Balzer SCF004 coater to ensure good electric conduction for EDX analyses. EDX analyses were carried out at a 7 kV accelerated voltage using a QUANTAX energy dispersive spectrometer equipped with two Bruker XFlash6-30 detectors.
X-ray photoelectron spectroscopy (XPS) was carried out in an ultrahigh vacuum (UHV) spectrometer equipped with a RESOLVE 120 MCD5 hemispherical electron analyzer and a dual anode Al Kα X-ray source (1486.6 eV). Survey and high-resolution spectra were recorded in constant pass energy mode (100 and 20 eV, respectively). Binding energies were calibrated by referring to the C 1s peak at 284.4 eV. Shirley-type background subtraction and fitting of the spectra were done with the software package Casa XPS vs. 2.3. High resolution XPS peaks deconvolution was accomplished with mixed Gaussian–Lorentzian curves minimizing the mean squared error. The Pd 3d peak was fitted using two doublets (i.e. Pd0 and PdII) representing metallic and oxidized Pd species. Asymmetric and symmetric peak shapes were used to fit the metallic and oxidized Pd components, respectively. For the fitting of the C 1s peaks, five components were used corresponding to the various chemical states of the C atoms in the PDA support layer.
Inductively coupled plasma-atomic emission spectrometry (ICP-AES) measurements were performed by the Plateforme Analytique des Inorganiques of the Institut Pluridisciplinaire Hubert Curien (UMR CNRS 7178), Strasbourg, France. For Pd loading determination, Pd@PDA@PUF samples were mineralized with aqua regia (2 mL HCl, 1 mL HNO3) at 185 °C for 50 minutes under pressure (Multiwave ECO, Anton Paar). A blank test was carried out in parallel under the same conditions. Pd quantification in the clear obtained solutions was carried out by ICP-AES (Varian 720ES) at two wavelengths: 340.458 nm and 360.955 nm.
Powder X-ray diffraction (PXRD) measurements were carried out on a Bruker D-8 Advance diffractometer equipped with a Vantec detector (Cu Kα radiation) working at 45 kV and 40 mA. X-ray diffractograms were recorded in the 10–80° 2θ region at room temperature in air, using a step size and step time of 0.05° and 80 s, respectively.
Gas chromatographic (GC) analyses were performed on a GC Agilent with FID detectors using an Agilent HP1 column (30 m, 0.35 mm, 0.25 μm), with hydrogen as the gas carrier and with tetradecane as the internal standard. The retention times and GC responses in terms of area versus concentration of the reagents and products were calibrated using pure components diluted in an acetone solution at various concentrations. The conversion and product distribution were calculated from the GC results.
NMR spectra were recorded at 298 K on a Bruker Avance III HD 400 MHz spectrometer operating at 400.13 MHz for 1H and at 100.61 MHz for 13C. The chemical shifts are referenced to the residual deuterated or 13C solvent peaks. Chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz, respectively (see the ESI†).
Open cell polyurethane foam (PUF) coating with polydopamine (PDA)
The process was adapted from our published procedure.8 Dopamine hydrochloride (2 mg mL−1) was dissolved in an aqueous solution (500 mL) of Tris base (10 mM) buffered to pH 8.5 with aqueous HCl (1 M). Four cubic samples of PUF (ca. 2 cm × 2 cm × 2 cm) were immersed in the stirred solution for 17 h at room temperature. The solution turned quickly orange, then slowly black. The resulting PDA-coated foam (PDA@PUF) was then taken out of the solution, dried in an oven at 80 °C for 1.5 h, washed in vigorously stirred water (3 × 10 min in 500 mL) and dried again in an oven at 80 °C.Functionalization of PDA@PUF with [PdCl2(NH3)4]·H2O
A cubic sample of PDA@PUF (ca. 2 cm × 2 cm × 2 cm) was washed in vigorously stirred (725 rpm) H2O/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) (120 mL) at room temperature for 1 h, and air-dried prior to functionalization.
:5) (120 mL) at room temperature for 1 h, and air-dried prior to functionalization.
The cubic sample of PDA@PUF was then immersed in a solution of [Pd(NH3)4Cl2]·H2O (20 mg, 0.076 mmol) in H2O/EtOH (1:5) (120 mL) at room temperature. After 19 h of stirring at 725 rpm, the resulting Pd-functionalized foam, Pd@PDA@PUF, was removed from the reaction medium, washed in vigorously stirred water (3 × 120 mL) and ethanol (2 × 100 mL), and then dried in an oven at 80 °C for 1 h.
Pd@PDA@PUF was characterized by HR-SEM (Fig. 2), SEM-EDX (Fig. 3 and S1†), XPS (Fig. 4 and S2†), XRD (Fig. S3†), TGA (Fig. S4†) and ICP-AES revealing a mean Pd content of 2.437 ± 0.323 g kg−1 (0.244 ± 0.032 wt%) assessed from the analysis of six samples.
General procedure for the catalytic alkyne-to-alkene hydrogenation with Pd@PDA@PUF
A stirring bar and a cubic sample of Pd@PDA@PUF of ca. 8 cm3, whose mass was adjusted in relation to its Pd content established by ICP-AES to reach a Pd loading of 7 μmol, were introduced in a Schlenk tube. Ethanol (20 mL) was then added to immerse the foam totally, and H2 was bubbled through the solution at room temperature for 30 min under a constant flow rate (13 mL min−1). Afterwards, the selected alkyne (1.52, 3 or 6 mmol) (Pd content: 0.46, 0.23 or 0.12 mol% vs. substrate) was added in one portion by syringe to the stirred solution (625 rpm), and T0 reaction time was set. During the reaction course, H2 was fed under ambient conditions (room temperature and atmospheric pressure) at a constant flow rate (2 or 13 mL min−1). The reaction was kinetically monitored by regularly sampling the reaction medium, and analysing the removed aliquots (50 μL) by GC or 1H NMR. The aliquots were diluted with acetone or ethanol (3 mL) for GC analysis or dried under high vacuum and re-dissolved in a deuterated solvent (CDCl3 or (CD3)2CO) for 1H NMR spectroscopy.General procedure for the Suzuki cross-coupling of aryl bromides and phenylboronic acid with Pd@PDA@PUF under aerobic conditions
A stirring bar and a cubic sample of Pd@PDA@PUF of ca. 8 cm3, whose mass was adjusted in relation to its Pd content established by ICP-AES to reach a Pd loading of 7 μmol, were introduced in a tube in a similar fashion to the hydrogenation experiments. Ethanol/water (1:1) (8 mL) was then added to immerse the foam. This was followed by the addition of phenylboronic acid (0.6 or 3.6 mmol), K2CO3 (0.75 or 4.5 mmol), and aryl bromide (0.5 or 3 mmol) (Pd content: 1.4 or 0.23 mol% vs. aryl bromide). The reaction mixture was then heated under stirring (625 rpm) by putting the tube in an oil bath at 65 °C. After 4 h, the reaction medium was poured into water (2 mL) and the product was extracted with ethyl acetate (2 × 10 mL). The combined extracts were then washed with brine (10 mL) and dried over anhydrous MgSO4. At this point, an aliquot was removed by syringe to determine the conversion of aryl bromide by GC analysis, and the solvent was removed under reduced pressure. The crude material was then purified by column chromatography over silica with petroleum ether/ethyl acetate (20:1) as eluent to give the desired product, whose identity was confirmed by 1H and 13C NMR in CDCl3.
Pd@PDA@PUF recovery and reuse in successive semi-hydrogenation and Suzuki reaction
The above-described general procedures for the catalytic alkyne-to-alkene hydrogenation and Suzuki coupling were performed using phenylacetylene (3 mmol) and a H2 flow rate of 13 mL min−1 for the semi-hydrogenation, and bromobenzene (3 mmol) and phenylboronic acid (3.6 mmol) for the Suzuki coupling, and a single cubic sample of Pd@PDA@PUF of ca. 8 cm3 (270 mg, 0.276 wt% Pd, i.e.: 7.00 × 10−3 mmol) as the catalyst for 15 successive runs. Five initial runs of semi-hydrogenation were conducted, then five runs of Suzuki coupling, then again 5 final runs of semi-hydrogenation. After each semi-hydrogenation run, the Pd@PDA@PUF foam was washed by immersing it in EtOH (20 mL × 3) in an ultrasonic bath (80 W, 50/60 Hz) at 25 °C for 10 min, and dried under vacuum. After each Suzuki run, the Pd@PDA@PUF foam was first washed with H2O (10 mL × 1) and ethyl acetate (10 mL × 2) (collecting all washing liquors and adding them to the reaction mixture for analysis), then washed with EtOH (10 mL × 3) in an ultrasonic bath (80 W, 50/60 Hz) at 25 °C for 10 min, and dried under vacuum.Control experiments
:40 mixture of 3-hexyne (1.8 mmol) and (Z)-2-hexene (1.2 mmol), as reagents at T0. The course of the reaction was monitored by regularly sampling the reaction medium and analysing the removed aliquots (50 μL) by GC after dilution in acetone (3 mL).
Results and discussion
Preparation and characterization of Pd@PDA@PUF
The Pd@PDA@PUF material was readily prepared by following a two-step dipping procedure involving: (i) the coating of a thin layer of PDA on a cubic sample of PUF by simple immersion in an aqueous solution of dopamine at room temperature buffered to pH of 8.5, typical of a marine environment, followed by washing with water,9,12 (ii) the subsequent immersion of the resulting PDA@PUF foam into a well stirred hydro-alcoholic solution of [Pd(NH3)4Cl2]·H2O (0.63 mM) at room temperature for 19 h (Fig. 1), followed by thorough washing with water and ethanol. The successful anchoring of Pd on the foam surface was confirmed by ICP-AES, XPS and SEM-EDX (vide infra).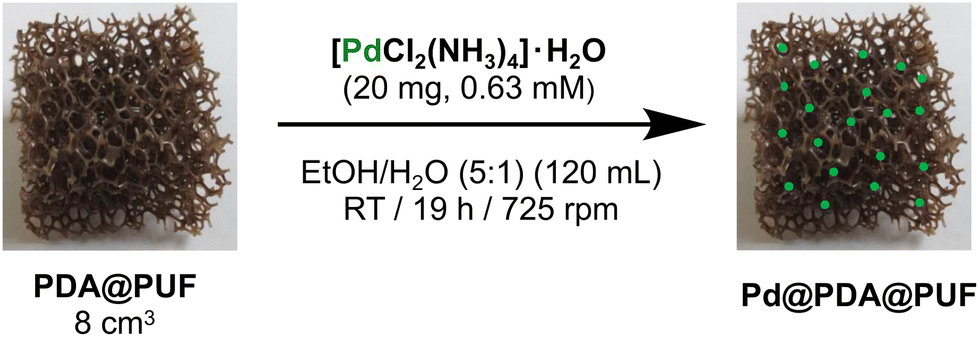 | ||
| Fig. 1 Functionalization of PDA@PUF by a simple one-step dipping procedure in a hydro-alcoholic solution containing a Pd salt. | ||
It is noteworthy that due to the easily engineered nature of PUF, the size and shape of the Pd@PDA@PUF can be easily tuned by simple cutting to produce catalysts in the form of cubes, cylinders or thin threads, that can fit any reactor's dimension and shape. This property should greatly facilitate the scale-up of the process for an industrial implementation. In this study, we used cubic samples of 2 cm × 2 cm × 2 cm for the semi-hydrogenation reactions in order to insert the foam into our 100 mL Schlenk tube tightly and ensure that the needle conveying H2 could reach the bottom of the flask without compressing the foam and causing structural deformation (Fig. S5†).
ICP-AES measurements on several samples of Pd@PDA@PUF revealed a mean Pd loading of 2.437 ± 0.323 g kg−1 (0.244 ± 0.032 wt%). High-resolution scanning electron microscopy (HR-SEM) images of the as-synthesized Pd@PDA@PUF are displayed in Fig. 2. Low magnification images (top) show that the PDA was coated on the whole surface of PUF as a rough film at the macroscopic level, while higher magnification images (bottom) reveal that the rough film structure is constituted by numerous scattered polymer aggregates typical of PDA coating on a PUF.8–10 No Pd particles could be identified, suggesting a relatively small size for the latter or, more likely, a molecular nature of the immobilized palladium species as the catechol moieties of the PDA layer may simply act as a chelating ligand substituting two chloride or NH3 ligands of [Pd(NH3)4Cl2]·H2O,15,41 which could result in a single atom-type distribution of Pd over the surface. No TEM characterization could however be carried out to confirm or reject this assumption. The elastic character of the PUF host substrate indeed prevented it to be cut into thin slices for observation, even after a freezing step in liquid N2 had been attempted to enhance its rigidity.
 | ||
| Fig. 2 HR-SEM images with different magnifications of the as-synthesized Pd@PDA@PUF. | ||
SEM micrographs combined with energy dispersive X-ray spectroscopy (EDX) revealed a quasi-uniform distribution of Pd over the foam surface (Fig. 3), without apparent aggregates or segregation areas, ensuring a relatively good structural homogeneity of the material. Without surprise, a similarly homogeneous distribution of C, O and N was also observed, highlighting the homogeneous PDA coating on PUF (Fig. S1†). It is noteworthy that no Cl was detected and that the mapping of the O atoms coincides almost perfectly with that of Pd (Fig. 3), further highlighting a possible simple coordination of the Pd complex by the catechol moieties.
 | ||
| Fig. 3 Elemental mapping of Pd and O obtained by SEM-EDX spectroscopy on the as-synthesized Pd@PDA@PUF and corresponding EDX spectrum. | ||
X-ray photoelectron spectroscopy (XPS) was performed to obtain information on the oxidation state of Pd as well as on the nature of its interaction with the PDA layer. In agreement with the results of EDX spectroscopy, the XPS survey scan spectrum of the as-synthesized Pd@PDA@PUF indicated the presence of elements O, N, Pd and C (Fig. S2A†). The high-resolution Pd 3d XPS core level region (Fig. 4) could be clearly attributed to Pd(II) species with a binding energy of the 3d5/2 peak at 337.2 eV (Fig. 4A). In contrast to the standard redox potential of PDA that lies at about +0.3 V/NHE while that of the Pd2+/Pd0 redox couple lies at +0.99 V/NHE42 and with some literature precedents, such as the immobilization of K2PdCl4 on PDA-coated cotton microfiber which resulted in the partial reduction of Pd(II) salt to Pd(0) NPs,43 but in agreement with other reports where no reduction of Pd(II) to Pd(0) upon immobilization of a Pd salt on PDA was observed,44 the PDA layer coated on PUF seems here to initially serve as a pure adhesive layer without reduction of the Pd(II) precursor. Again, this highlights a possible simple coordination of the Pd(II) species by the catechol moieties of the PDA layer.
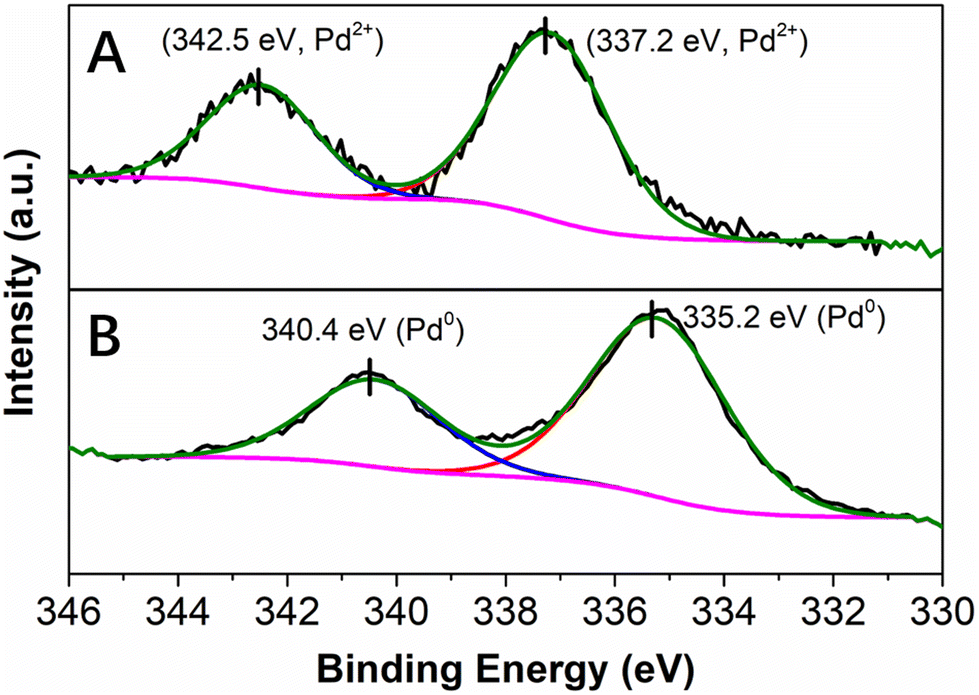 | ||
| Fig. 4 High resolution XPS spectra of the Pd 3d core level region of Pd@PDA@PUF: (A) as-synthesized, (B) spent (after 15 cycling tests). | ||
XRD failed to show evident diffraction peaks corresponding to palladium phases for the as-synthesized Pd@PDA@PUF (Fig. S3†), most likely because of the low palladium loading on the support (0.244 ± 0.032 wt%), but probably also because of the small size of the palladium particles or molecular species. For the sake of completeness, thermogravimetric analysis (TGA) of PUF, PDA@PUF and the as-synthesized Pd@PDA@PUF materials was also recorded. The polymeric materials all presented similar results (Fig. S4†), thereby showing the absence of obvious variations of the material composition which was in agreement with the low loadings of PDA and Pd onto pristine PUF.
Semi-hydrogenation of alkynes
Pd@PDA@PUF (0.244 ± 0.032 wt%), in the form of cubic samples of ca. 8 cm3 whose masses were adjusted in relation to their Pd content to have a Pd loading of 7 μmol, was investigated as the catalyst for the selective semi-hydrogenation of aliphatic, aromatic, terminal and internal alkynes (0.076 to 0.3 M, giving Pd loadings of 0.46 to 0.12 mol%) in ethanol at room temperature under a constant flow rate of H2 (2 to 13 mL min−1) into the solution to maintain its concentration constant during the whole duration of the catalytic processes.Optimization studies (see the ESI†) conducted with this substrate at various concentrations (0.076 to 0.3 M) and under various H2 flow rates (2 to 13 mL min−1) allowed us to establish an optimal H2 flow rate of 13 mL min−1 and alkyne concentration of 0.3 M (Pd loading of 0.12 mol%). Under these optimized conditions, the selectivity for ST as high as 95% at 98% conversion could be observed after 4.25 h of reaction, giving a TON of 776 (expressed as the number of (mol of PA converted to ST)/(molPd)) and a TOF of 183 h−1 (Fig. 5). Using an initial PA concentration of 0.15 M (Pd loading of 0.23 mol%) under otherwise similar conditions, led to a comparable TOF (188 h−1) and ST selectivity (95% at 91% conversion) but of course to a lower TON (376) (Table S1 and Fig. S6†). Satisfyingly, in both cases, ethylbenzene (EB) started to be produced appreciably only after the full conversion of PA into ST. Such results may be explained by the fact that PA displays high adsorption properties on Pd@PDA@PUF, which could prevent ST to be adsorbed before all PA was consumed. The formation rate of EB is indeed almost similar to that of ST, which tends to show that the two successive hydrogenation reactions are initiated on similar active sites.
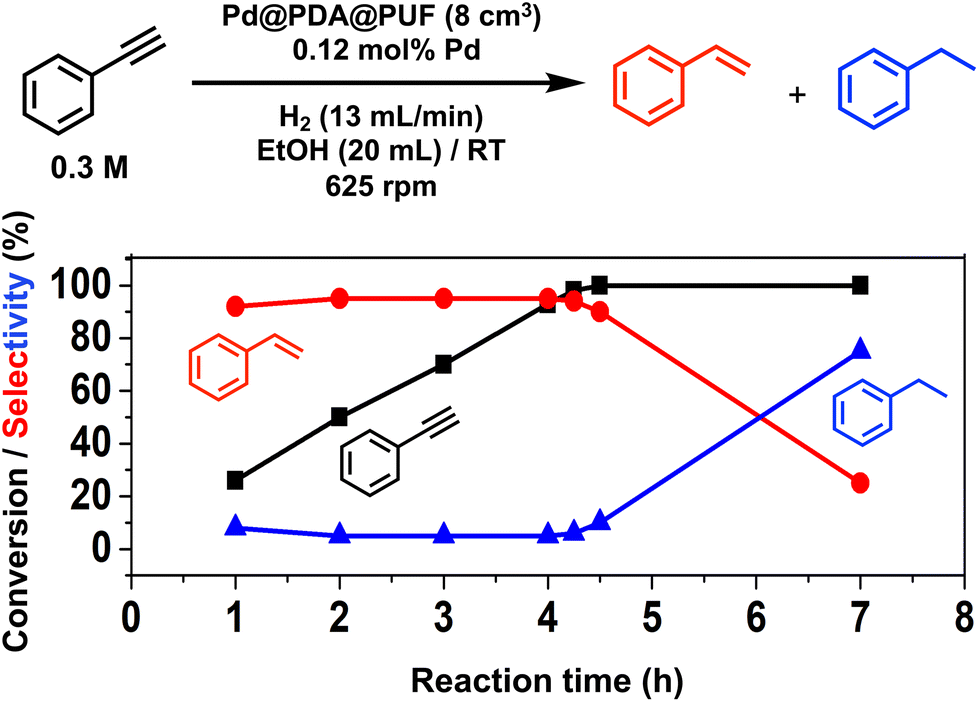 | ||
| Fig. 5 Hydrogenation of phenylacetylene with Pd@PDA@PUF (8 cm3); black squares: PA conversion; red circles, ST selectivity; blue triangles: EB selectivity. Reaction conditions: PA (6 mmol), Pd (7 × 10−3 mmol, 0.12 mol%), EtOH (20 mL), H2 flow rate (13 mL min−1), room temperature, 625 rpm. | ||
For comparison, the catalysis based on the unsupported precursor, [Pd(NH3)4Cl2]·H2O (0.23 mol% Pd), was investigated under similar conditions: PA (3 mmol), EtOH (20 mL), RT, H2 (13 mL min−1). 91% PA conversion with 95% ST selectivity was observed after 1 h of reaction (TON = 376, TOF = 376 h−1), which is just twice as fast as what was observed with Pd@PDA@PUF under the same conditions (vide supra and Table S1†). This shows that PDA@PUF, as a support, retained the selectivity of the homogeneous Pd catalyst, which is very rare,45 while rendering it easily recyclable (vide infra) in contrast to the latter, which yielded a suspension of a black solid at the end of the hydrogenation reaction (Fig. S7†), most likely Pd(0) particles.
These interesting results prompted us to compare the catalytic performance of our easily prepared Pd@PDA@PUF catalyst with state-of-the-art Pd-systems. As can be seen in Table 1, Pd@PDA@PUF compares well with or even outperforms, in terms of reaction conditions, selectivity and/or TON, many catalytic systems, whose preparation typically implies much more complex synthetic paths aimed at the fine tailoring of the active phases (entries 2–10) or at the production of highly dispersed, size- and shape-controlled nanocomposites (entries 9–12) up to single atom catalysts (entries 13–15). The only notable exception is the graphdiyne-supported Pd single-atom catalyst PdS–GDY, which can achieve TON as high as 12586 with an exceptional selectivity of 99.3 (entry 14). However, the latter necessitates a tedious synthetic process compared to Pd@PDA@PUF, which precludes its use on an industrial scale.
| Entry | Catalyst | Solvent | T (K) | P (bar) | t (min) | Conv.a (%) | Select.a (%) | TONb |
|---|---|---|---|---|---|---|---|---|
| a PA conversion and ST selectivity determined by GC analysis. b Turnover number expressed as (mol of PA converted to ST)/(molPd). c Pd loading in the catalytic material (and in the reaction medium): 0.244 wt% (0.12 mol%). d Pd loading in the catalytic material (and in the reaction medium): 0.924 wt% (0.57 mol%). e Pd loading in the catalytic material (and in the reaction medium): 4.15 wt% (0.43 mol%). f Pd loading in the catalytic material (and in the reaction medium): 2.6 wt% (0.175 mol%). g Pd loading in the catalytic material (and in the reaction medium): 8 wt% (10 mol%). h Pd loading in the catalytic material (and in the reaction medium): 5 wt% (0.43 mol%). i Pd loading in the catalytic material (and in the reaction medium): 1 wt% (0.016 mol%). j Pd loading in the catalytic material (and in the reaction medium): 5 wt% (0.26 mol%). k Pd loading in the catalytic material (and in the reaction medium): 5.64 wt% (0.091 mol%). l Pd loading in the catalytic material (and in the reaction medium): 1.67 wt% (0.26 mol%). m Pd loading in the catalytic material (and in the reaction medium): 1 wt% (0.094 mol%). n Pd loading (in the reaction medium): (0.5 mol%). o Pd loading in the catalytic material (and in the reaction medium): 0.42 wt% (0.0079 mol%). p Pd loading in the catalytic material: 0.18 at%. q Pd loading in the catalytic material (and in the reaction medium): 0.12 wt% (0.1 mol%). | ||||||||
| 1 | Pd@PDA@PUFc | EtOH | 298 | 1 | 255 | 98 | 95 | 776 |
| 246 | 0.9Pd/Cd | EtOH | 298 | 1 | 55 | 98 | 96 | 165 |
| 347 | SiO2@CuFe2O4–Pde | n-Hexane | — | 1 | 150 | 98 | 98 | 224 |
| 435 | Pd–Pb NSsf | EtOH | 313 | 4 | 30 | 100 | 95.8 | 549 |
| 548 | Pd/Ag@CeO2-1.5g | EtOH | 313 | 15 | 720 | 97 | 99 | 9.6 |
| 647 | Lindlar cat. (Aldrich)h | n-Hexane | — | 1 | 150 | 82 | 92 | 176 |
| 749 | Lindlar cat. (NHU Co.)i | EtOH | 303 | 1 | 270 | 5 | >99 | 309 |
| 850 | Lindlar cat. (TCI)j | CH3CN | 303 | 1 | 60 | 86 | 99 | 327 |
| 949 | Pd/mpg-C3N4k | EtOH | 303 | 1 | 85 | 100 | 94 | 1037 |
| 1050 | MWCNTs–Fe3O4–Cu2O–Pdl | CH3CN | 303 | 1 | 90 | 100 | 98 | 377 |
| 1151 | PyC12S–Pd/VCm | CH2Cl2 | 303 | 1 | 300 | 98 | 94 | 980 |
| 1234 | Pd + PEI(L)@HSSn | MeOH/1,4-dioxane | 303 | 1 | 240 | 97 | 87 | 169 |
| 1352 | PdS–GDYo | EtOH | 298 | 2 | 120 | 100 | 99.3 | 12586 |
| 1453 | Pd0.18Cu15/Al2O3p | n-Hexane | 298 | 6.9 | 480 | 90 | 94 | — |
| 1554 | Pd1/SBA-15@N–Cq | EtOH | 323 | 1 | 10 | 96 | 93 | 893 |
| Entry | Substrate | Time (h) | Conv.b (%) | Alkene sel.b (%) | Z/E ratiob |
|---|---|---|---|---|---|
| a Reaction conditions: alkyne (3 mmol), Pd@PUF@PDA (8 cm3, Pd: 0.23 mol%), EtOH (20 mL), room temperature, H2 flow (13 mL min−1), 625 rpm. b Determined by GC or 1H NMR. c Reaction conducted at 10 °C. d No (E)-3-hexene detected by GC. | |||||
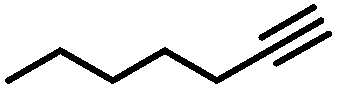
| 1 |
|
1.8 | 97 | 98 | — |
| 2c |

|
3.3 | >99 | 99 | — |
| 3 |
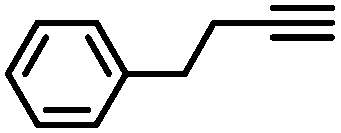
|
1.8 | 97 | 96 | — |
| 4 |
|
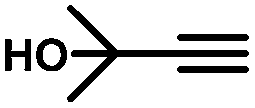
1.3 | 99 | 94 | — |
| 5 |

|
1.5 | >99 | 99 | 1/0d |
| 6 |
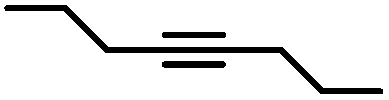
|
1.5 | >99 | 99 | 37/1 |
| 7 |

|
3 | 98 | 85 | 16/1 |
| 8 |
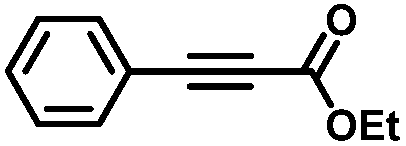
|
4 | 99 | 88 | 7/1 |
More specifically, excellent results were obtained for all investigated terminal alkynes (1-heptyne, 4-phenyl-1-butyne, 2-methylbut-3-yn-2-ol) with alkene selectivity ranging from 94 to 98% at conversions of 97 to 99% (Table 2, entries 1, 3 and 4), and over-hydrogenation to the corresponding alkane or double-bond isomerization being only appreciable after full alkyne consumption (Fig. 6A–D). In the case of 1-heptyne, the alkene selectivity could even be increased to 99% at full alkyne conversion by lowering the reaction temperature to 10 °C (entry 2 and Fig. 6B).
 | ||
| Fig. 6 Hydrogenation with Pd@PDA@PUF of 1-heptyne at 25 °C (A), 1-heptyne at 10 °C (B), 4-phenyl-1-butyne at 25 °C (C), 2-methylbut-3-yn-2-ol at 25 °C (D); black squares: alkyne conversion; red circles, 1-alkene selectivity; blue triangles: alkane selectivity; pink triangle: 1-alkene isomerization products. Reaction conditions: alkyne (3 mmol), Pd (7 × 10−3 mmol, 0.23 mol%), EtOH (20 mL), H2 flow rate (13 mL min−1), 25 or 10 °C, 625 rpm. | ||
The semi-hydrogenation of aliphatic internal alkynes such as 3-hexyne and 4-octyne proved even more selective with selectivity as high as 99% to the (Z)-alkene at full alkyne conversion (Table 2, entries 5 and 6). Most impressively, this selectivity was maintained at very high levels even several hours after all the alkyne had been consumed as isomerization to the (E)-alkene or over-hydrogenation to the fully saturated hydrocarbon proved to be very slow (Fig. 7A and B). To further highlight the aptitude of Pd@PDA@PUF to selectively hydrogenate 3-hexyne to (Z)-3-hexene, we performed a competitive experiment starting from a mixture of 60% 3-hexyne and 40% (Z)-3-hexene. During the first 50 min of the catalytic test, we observed a totally selective conversion of the alkyne to (Z)-3-hexene without any appreciable competition of the hydrogenation of the latter to hexane. Remarkably, Pd@PDA@PUF maintained complete chemoselectivity even after complete consumption of 3-hexyne (after ca. 50 min) (Fig. 7C). Such a gain of selectivity compared to the semi-hydrogenation of terminal alkynes (vide supra) might be attributed, on one side, to the less favourable double-bond isomerization of the resulting alkene, and on the other side, to the increased steric hindrance around the double bond, which renders its further hydrogenation to the alkane harder.
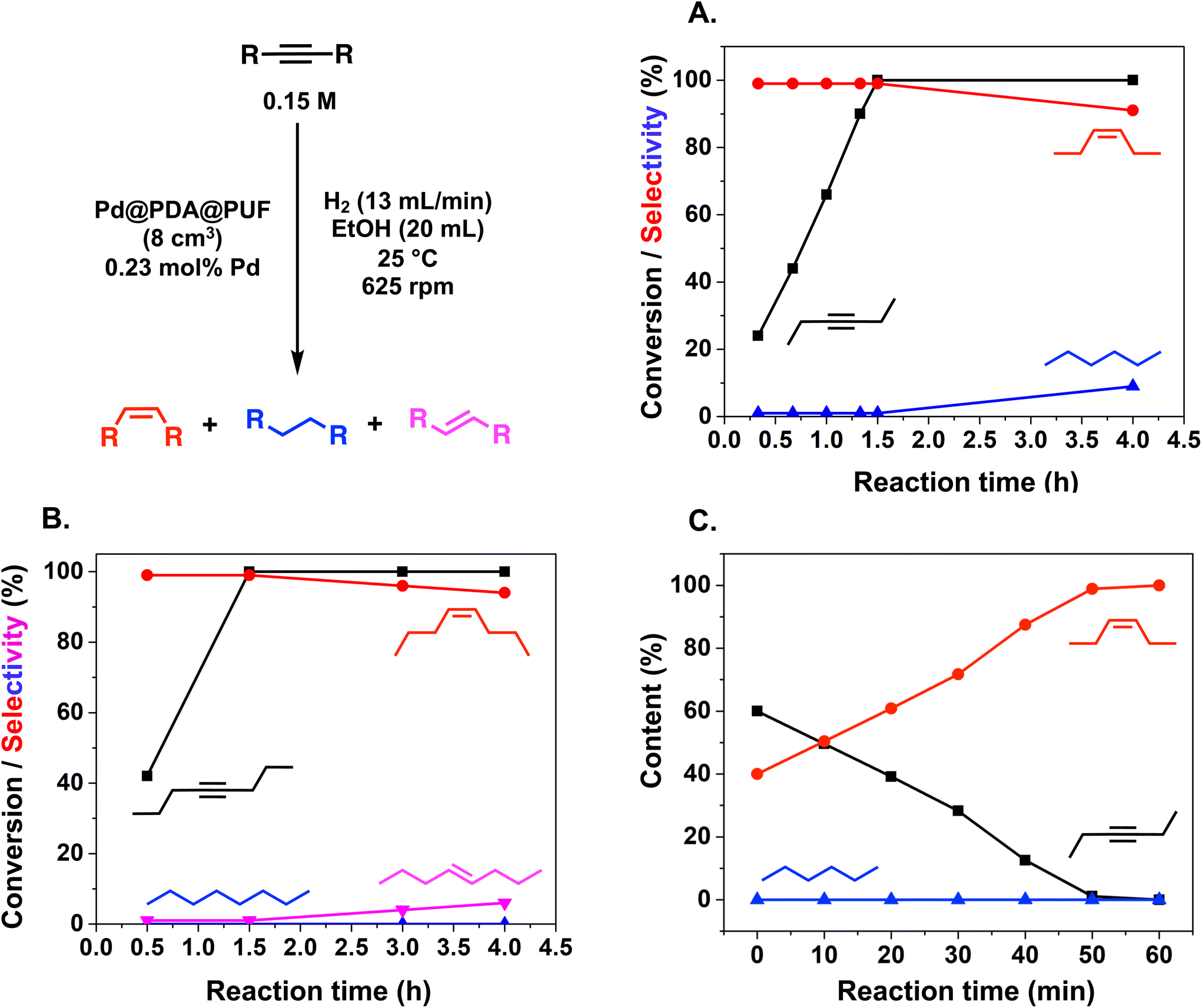 | ||
| Fig. 7 Hydrogenation with Pd@PDA@PUF (8 cm3) of 3-hexyne (A), 4-octyne (B), a 60:40 mixture of 3-hexyne and (Z)-3-hexene (C); black squares: alkyne conversion (A and B) or content (C); red circles, (Z)-alkene selectivity (A and B) or content (C); blue triangles: alkane selectivity; pink triangle: (E)-alkene selectivity. Reaction conditions: alkyne (3 mmol) (A and B) or alkyne (1.8 mmol) and alkene (1.2 mmol) (C), Pd (7 × 10−3 mmol, 0.23 mol%), EtOH (20 mL), H2 flow rate (13 mL min−1), 25 °C, 625 rpm. | ||
In the case of the bulky aromatic alkyne 1,2-diphenylacetylene, the alkene selectivity (Z/E ratio of 16:1) was only 85% at full alkyne conversion (Table 2, entry 7), as 1,2-diphenylethane started to form from the beginning of the reaction. Furthermore, (E)-1,2-diphenylethylene was detected in amounts varying between 10 and 5% throughout the whole process, while the amount of (Z)-1,2-diphenylethylene varied between 85 and 80% during the first 3 h before diminishing gradually when all the starting alkyne had been consumed, i.e., after 3 h reaction (Fig. 8). Such a result might be attributed to the aromatic rings, which could provide easier access to the double-bond after the initial alkyne-to-alkene hydrogenation through π–π-interaction with the PDA layer,15 compared to saturated alkenes which readily decoordinate after the initial alkyne-to-alkene hydrogenation.
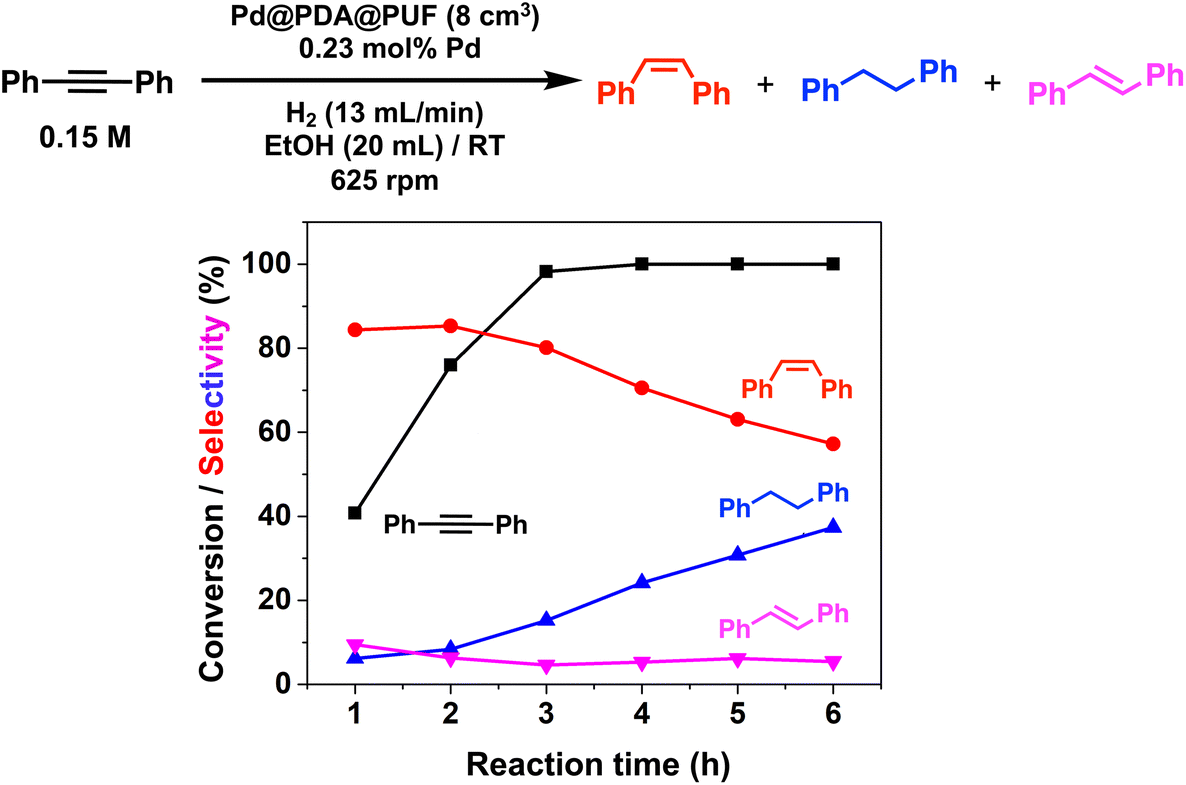 | ||
| Fig. 8 Hydrogenation with Pd@PDA@PUF (8 cm3) of 1,2-diphenylacetylene; black squares: alkyne conversion; red circles, (Z)-alkene selectivity; blue triangles: alkane selectivity; pink triangle: (E)-alkene selectivity. Reaction conditions: 1,2-diphenylacetylene (3 mmol), Pd (7 × 10−3 mmol, 0.23 mol%), EtOH (20 mL), H2 flow rate (13 mL min−1), 25 °C, 625 rpm. | ||
Finally, in the case of ethyl-3-phenylpropynoate (Table 2, entry 8), it is worth noting that the ester functionality was not affected by the hydrogenation, but similar to what was observed with 1,2-diphenylacetylene, full consumption of the alkyne took a longer time (4 h) than with aliphatic alkynes or PA, and non-negligible amounts of the alkane (12%) and (E)-alkene (in a 1:7 ratio with the Z-isomer) were observed at this point.55
Among all the studied substrates, aliphatic alkynes clearly give higher alkene selectivity than aromatic alkynes, particularly in the case of symmetric internal alkynes with which over-hydrogenation to alkanes is hardly observed, even after prolonged reaction times. In agreement with the HR-SEM and SEM-EDX data, which suggest a very small size of the immobilized Pd species, a possible explanation of Pd@PDA@PUF high alkene selectivity may be related to the possible chelation of Pd(II) ions by the catechol moieties of the PDA layer, which would result in a “single atom”-type dispersion of the active phase.
Suzuki coupling of aryl bromides and phenylboronic acid under mild aerobic conditions
The Pd-catalyzed Suzuki cross-coupling, as one of the most powerful method for the construction of biaryl compounds, is extensively utilized in the synthesis of natural products or pharmaceuticals, as well as in the preparation of organic electronics.56–58 It thus aroused our curiosity to check whether our catalyst system was efficient in catalyzing this type of reactions.Initial studies focused on the coupling under air of phenylboronic acid with chloro- or bromobenzene in EtOH/H2O (1:1) (8 mL) at 65 °C in the presence of K2CO3 as the base and cubic samples of Pd@PDA@PUF (0.244 ± 0.032 wt%) of ca. 8 cm3 as the catalytic material, whose masses were adjusted to have a Pd loading of 7 μmol, i.e.: of 1.4 or 0.23 mol% depending on the amount of aryl bromide (0.5 or 3.0 mmol).
With a catalytic loading of 1.4 mol% Pd, only 20% conversion of chlorobenzene to biphenyl was observed after 23 h of reaction (Table 3, entry 1). However, when bromobenzene was used instead of chlorobenzene, 88% conversion to biphenyl was observed after 3 h and 96% after 4 h (entries 2 and 3). Satisfyingly, a similar conversion was observed after the same reaction time when increasing the amount of bromobenzene 6-fold (conversely diminishing the Pd loading to 0.23 mol%), thus making it possible to reach a TON of 426 and a TOF of 107 h−1 (entry 4). In this case also, Pd@PDA@PUF displayed a high catalytic efficiency without any pre-activation procedures, and moreover, under aerobic conditions in aqueous medium in the absence of a phosphine ligand.59–62
|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Pd loading (mol%) | Time (h) | Conv.b (%) | TOF (h−1) |
|
a Reaction conditions: halobenzene (0.5–3 mmol), phenylboronic acid (0.6–3.6 mmol), K2CO3 (0.75–4.5 mmol), Pd@PUF@PDA (8 cm3, Pd: 1.4–0.23 mol%), EtOH/H2O (1:1) (8 mL), 65 °C, 625 rpm.
b Determined by GC or NMR.
|
|||||
| 1 | Cl | 1.4 | 23 | 20 | 0.62 |
| 2 | Br | 1.4 | 3 | 88 | 21 |
| 3 | Br | 1.4 | 4 | 96 | 17 |
| 4 | Br | 0.23 | 4 | 98 | 107 |
With these optimized conditions in hand, we then briefly examined the scope of the coupling reaction by varying the nature of the aryl bromide (Fig. 9). The reaction proceeded with high efficiency for all five para-substituted bromobenzene substrates examined, whether they bore electron-donating or electron-withdrawing groups, and isolated yields ranging from 90 to 97% were obtained after 4 h reaction. The reaction of 2-bromonaphtalene with phenylboronic acid, in contrast, resulted in a slightly lower activity with 83% isolated yield after 4 h of reaction. The sterically crowded 2-bromobiphenyl and 3-bromo-4-methoxytoluene were coupled efficiently, giving isolated yields of 86 and 94%, respectively. Finally, the ortho-ester-substituted aryl bromide, methyl 2-bromobenzoate, gave the expected biaryl compound in 64% isolated yield after 4 h of reaction. These results show that both sensitive functionalities and sterically crowded substrates are well tolerated by Pd@PDA@PUF.
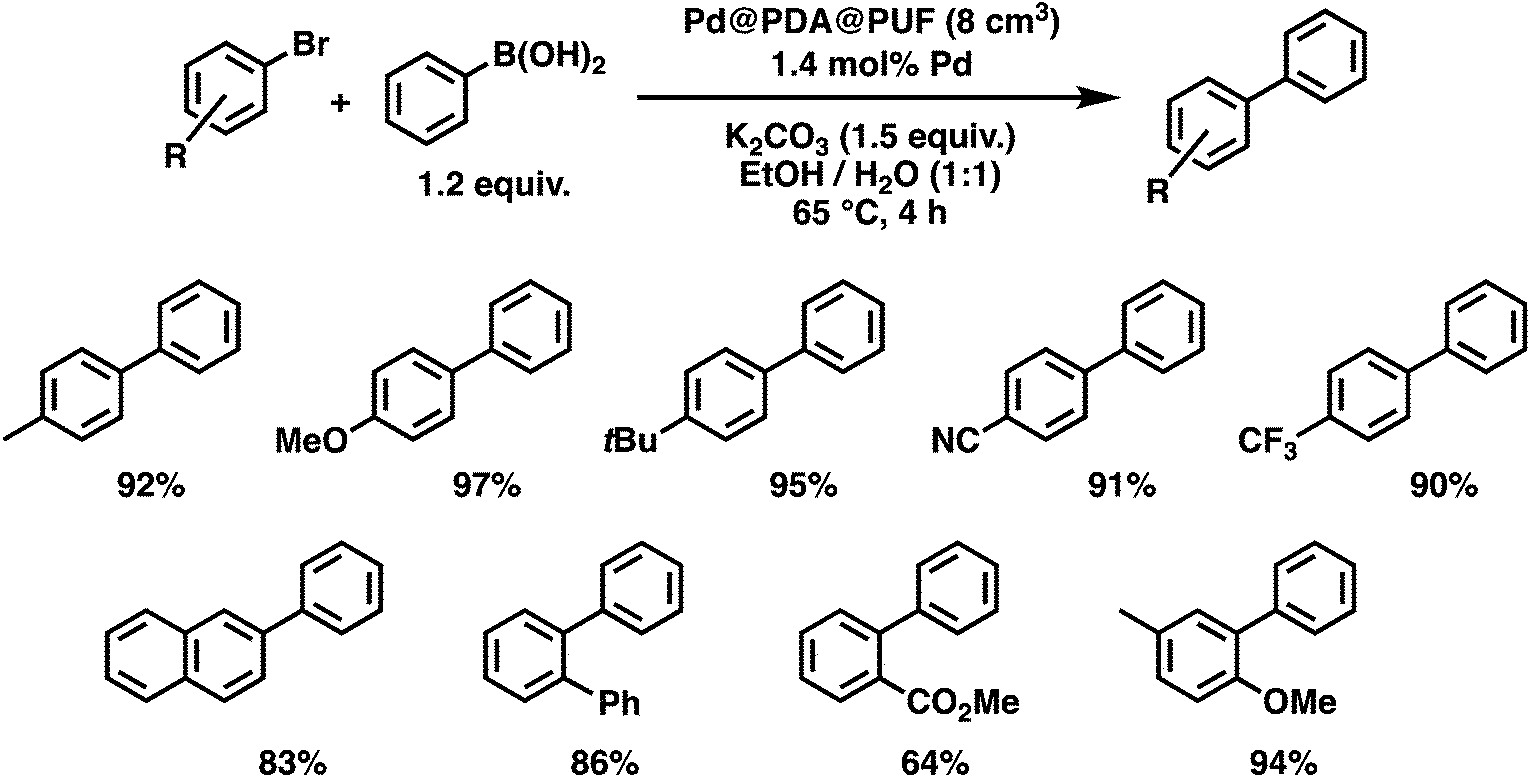 | ||
| Fig. 9 Scope of the Pd@PDA@PUF-catalyzed Suzuki cross-coupling. | ||
Catalyst recycling, leaching test and evidence of progressive Pd(II) reduction
Reusability, a criterion of great significance in evaluating heterogeneous catalytic systems operated in the liquid-phase medium, is normally measured as the relative activity of the catalyst maintained in successive runs of a given catalytic reaction. In the case of powdered catalyst, the product–catalyst separation is generally carried out via physical methods, such as filtration or centrifugation. Such methods are tedious in practical use and time-consuming, along with problems linked to catalyst loss. The macroscopic catalyst used in the present work allows one to operate the product–catalyst separation in a much easier way as the catalyst can be just taken out from the reaction medium followed by a short washing step to remove the adsorbed liquid on its surface before reuse.To shed more light on the recycling properties of Pd@PDA@PUF, 15 consecutive catalytic runs were conducted with a single cubic sample of Pd@PDA@PUF (8 cm3, Pd: 7 μmol, i.e.: 0.23 mol% for all reactions as 3 mmol of substrate were used in each case), consisting of 5 runs of semi-hydrogenation of PA, followed by 5 runs of Suzuki coupling of bromobenzene with phenylboronic acid, and then again 5 runs of PA semi-hydrogenation (Fig. 10).
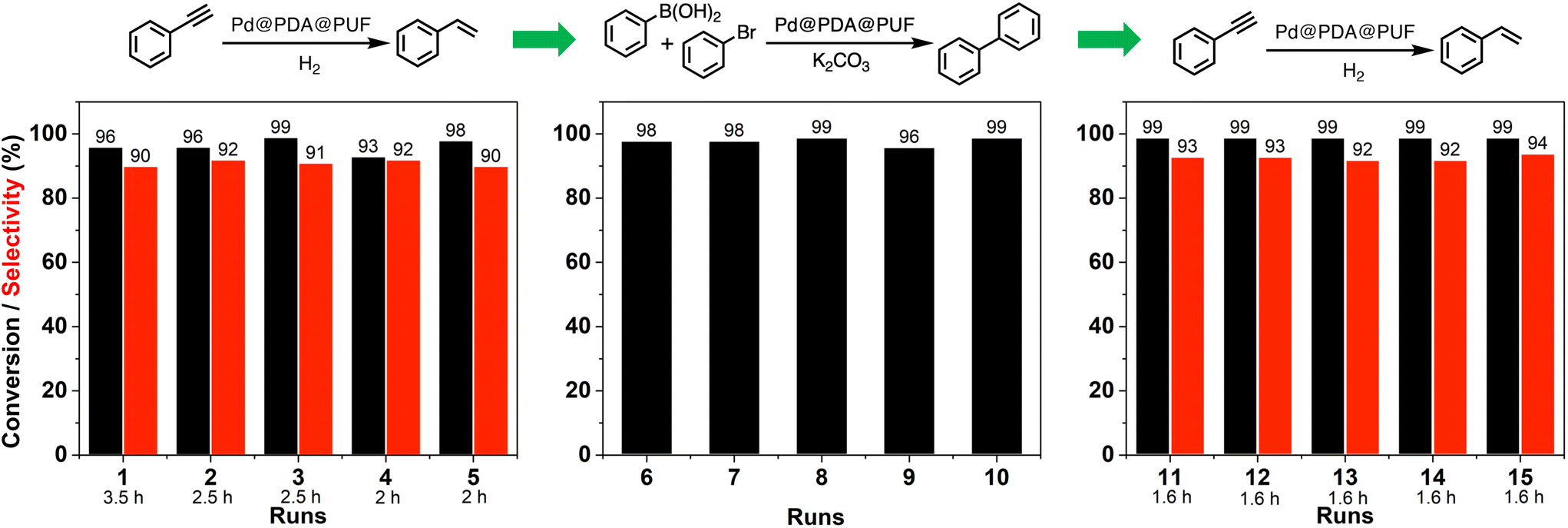 | ||
| Fig. 10 Conversion and selectivity measured over the 15 successive runs of semi-hydrogenation of phenylacetylene (runs 1 to 5 and 11 to 15) and Suzuki coupling of bromobenzene with phenylboronic acid (runs 6 to 10) catalysed by a single cubic sample of Pd@PDA@PUF (8 cm3, Pd: 7 μmol). Hydrogenation conditions: phenylacetylene (3 mmol), EtOH (20 mL), H2 flow rate (13 mL min−1), 25 °C, 625 rpm. Suzuki coupling's conditions: bromobenzene (3 mmol), phenylboronic acid (3.6 mmol), K2CO3 (4.5 mmol), EtOH/H2O (1:1) (8 mL), 65 °C. | ||
During the first 5 runs of semi-hydrogenation, the time necessary to consume nearly all PA decreased from 3.5 h in run 1 to 2 h in run 5, while the ST selectivity was maintained at 91 ± 1%, thereby suggesting a gradual improvement of the catalytic activity of Pd@PDA@PUF over these first 5 runs. The next 5 runs of Suzuki coupling each resulted in 97.5 ± 1.5% conversion in 4 h of reaction, showing no apparent variation of catalytic activity. Finally, the 5 last runs of semi-hydrogenation demonstrated the extremely high stability of the foam-based catalyst, as both the reaction rate and the selectivity remained unchanged for the whole set of tests with 99% conversion and 93% ± 1% of ST selectivity after 1.6 h of reaction for run 11 to 15. Noteworthily, 45 mmol of substrate (PA and bromobenzene) was efficiently converted over these 15 runs with just 7 μmol of Pd, which corresponds to a Pd loading 0.0156 mol% if one takes into consideration the total amount of converted substrates.
It must be highlighted here that despite the fact that Pd@PDA@PUF experienced 65 °C for 4 h in a basic environment during the coupling reactions and that it was washed with water and organic solvents (ethanol and ethyl acetate) in an ultrasonic bath after each coupling reaction, the activity in the subsequent semi-hydrogenation tests (i.e.: runs 11 to 15) did not drop, but even slightly increased. A determining requirement to establish whether Pd@PDA@PUF was competent in accomplishing such a recycling experiment was that the catalyst could endure prolonged rinsing steps like ultrasonic bath washings. To our delight, Pd@PDA@PUF achieved this goal, maintaining both high conversion and selectivity from the beginning to the end. PUF (structured catalytic support), PDA (adhesive layer), and Pd precursor (active species), after a facile combination, thus constitute a very stable heterogeneous structured catalyst for both semi-hydrogenation and Suzuki coupling reactions. Even more satisfyingly, the performance of semi-hydrogenation catalysis showed an interesting development during the first runs of catalysis due, most probably, to the progressive reduction of the immobilized Pd, as shown by XPS analyses (vide infra).
ICP-AES analyses of the Pd content of both foams after these 15 cycling tests and the reaction medium confirmed the robust anchoring of Pd. No Pd was indeed detected in the reaction solution after the first run of semi-hydrogenation reaction, although the most important catalyst leaching is typically expected during the first run. Moreover, the Pd content of the Pd@PDA@PUF foam after the cycling tests showed only negligible deviation (2.205 mg g−1) compared to the value measured for the as-synthesized foam (2.758 mg g−1). This absence or quasi-absence of leaching was further demonstrated by a stop-and-go experiment that was carried out for the hydrogenation of 4-octyne with a fresh Pd@PDA@PUF foam. The latter indeed showed the quenching of the reaction as soon as Pd@PDA@PUF was removed from the reaction medium, and the restart of the reaction when the foam was re-immersed (Fig. S8†).
Furthermore, the surface of the spent catalyst after these 15 cycling tests remained remarkably similar compared to that of the as-synthesized Pd@PDA@PUF foam, as revealed by HR-SEM (Fig. S9†). In particular, high magnification images still show numerous scattered polymer aggregates typical of PDA coating, and again, no Pd particles could be identified. This further suggests an initial simple coordination of the Pd(II) ions, and moreover shows the absence of mobility of the active phase even after reduction to Pd(0). Accordingly, SEM micrographs combined with EDX spectroscopy again showed a quasi-uniform distribution of C, O, N and Pd without notable aggregates or segregations area (Fig. S10†).
To gain more information about the absence of need for pre-catalysis time-and-energy consuming reduction procedures and the possible reduction of the immobilized Pd(II) species under the catalytic conditions, XPS measurements were performed: (i) on the Pd@PDA@PUF foam that had been used for the cycling tests (Fig. 4B), and (ii) on foams that had been used for a single run of semi-hydrogenation of PA or of Suzuki coupling of bromobenzene and phenylboronic acid (Fig. 11). In contrast to what was observed for the as-synthesized foam (Fig. 4A), only Pd(0) was observed on the spent Pd@PDA@PUF foam after 15 catalytic runs of alkyne hydrogenation and Suzuki coupling (Fig. 4B). Remarkably, this zero-oxidation state could be observed even after letting the foam standing in air for weeks, revealing a stabilizing effect of the PDA layer through surface interaction with the anchored metal (vide infra). It is noteworthy, however, that this full reduction of the immobilized Pd(II) species to Pd(0) is not the result of a ‘one-shot-process’. Thus, as can be seen in Fig. 11, Pd(II) was only partially reduced to Pd(0) after a single catalytic run, either in semi-hydrogenation or Suzuki coupling. This result explains the observed activity of the as-synthesized Pd@PDA@PUF without prior reduction, the reaction mixtures being sufficiently reductive to activate the Pd@PDA@PUF catalytic material, as well as the gradual improvement of the catalytic activity of Pd@PDA@PUF observed over the first 5 runs of the cycling tests and in the optimization study (Fig. S6†). Furthermore, it is worth mentioning that the Pd(II) immobilized species was reduced at varying degrees depending on the nature of the reaction. Thus, approximately 25% Pd(II) was reduced to Pd(0) after one run of the semi-hydrogenation reaction63 while ca. 57% Pd(0) was formed after one Suzuki coupling, suggesting (i) that the Suzuki coupling reaction medium was more reductive than the alkyne hydrogenation one, and (ii) that a classical Pd(0)/Pd(II) mechanism64 was occurring, even though the cross-coupling was carried out in an open vessel under air. Furthermore, the easy reduction of Pd(II) to Pd(0) observed in our experiments may be attributed, at least in part, to the small dimension, or even the coordinated ion state, of the active phase. Indeed, for larger Pd nanoparticles, the reduction could be more tedious at low temperature due to the slow diffusion of oxygen out and of dihydrogen in, throughout the oxide particle.
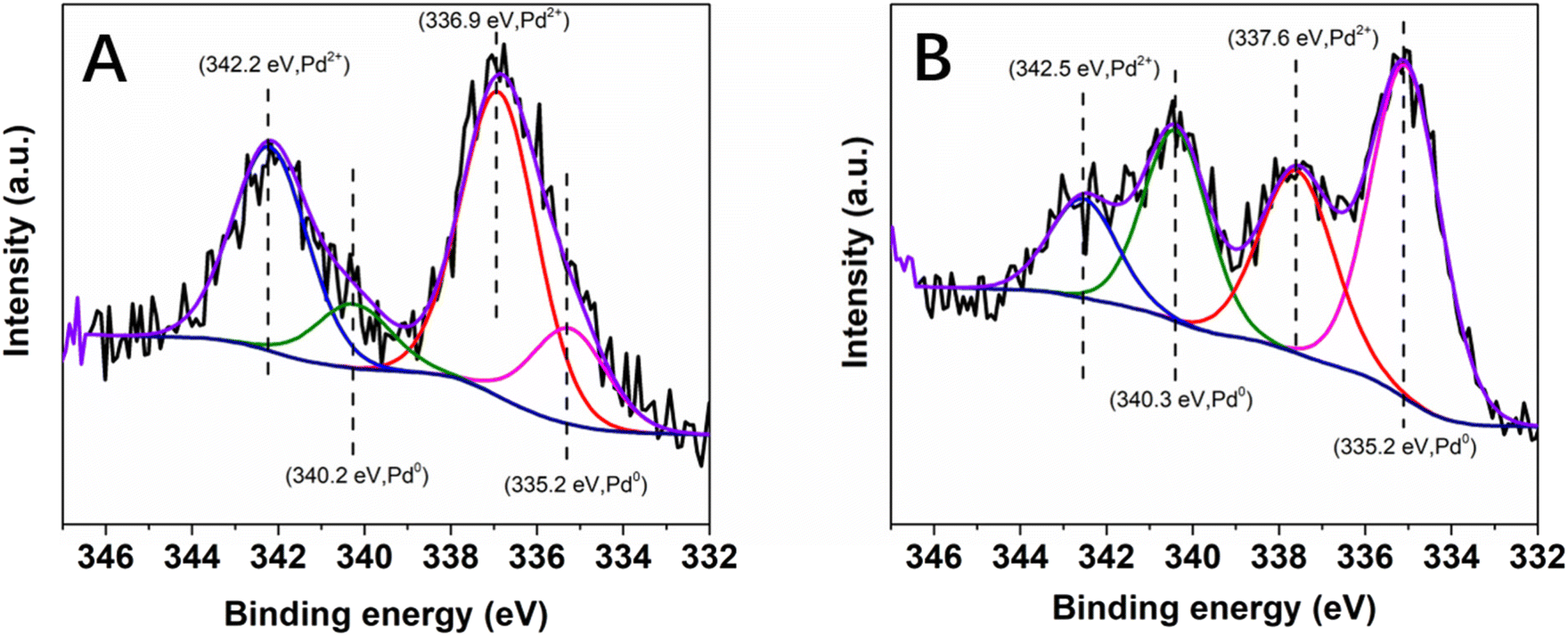 | ||
| Fig. 11 High-resolution XPS spectra of the Pd 3d core level region of Pd@PDA@PUF: (A) after one run of semi-hydrogenation of phenylacetylene, (B) after one run of Suzuki coupling of bromobenzene and phenylboronic acid. | ||
The high-resolution XPS spectra of the C 1s core level region (Fig. 12) of the as-synthesized and spent Pd@PDA@PUF catalyst after 15 cycling tests suggest a non-innocent behaviour of the PDA layer. In the C 1s spectrum of the pristine Pd@PUF@PDA foam, four peaks at 284.4, 285.8 eV, 287.2 eV and 289.2 eV can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C/C–N/C–O, CO, and O–CO groups of polydopamine, respectively (Fig. 12A).63 The C 1s spectrum of the spent foam shows the same four peaks, but their intensity varied significantly compared to that of the as-synthesized catalyst, due to a probable reductive alteration of the catechol/o-quinone ratio of the PDA layer. Specifically, the area of the CC peak increased by 15% while that of the CO peak decreased by 8%, which suggests that a non-negligible proportion of o-quinone-like groups has been reduced to catechols after the 15 cycling tests. We suspect that this partial reduction of the PDA layer helps to prevent the re-oxidation of Pd(0) to Pd(II) when the foam is exposed to air (vide supra), and explain the formidable stability of the Pd@PDA@PUF catalyst over time.
C, C–C/C–N/C–O, CO, and O–CO groups of polydopamine, respectively (Fig. 12A).63 The C 1s spectrum of the spent foam shows the same four peaks, but their intensity varied significantly compared to that of the as-synthesized catalyst, due to a probable reductive alteration of the catechol/o-quinone ratio of the PDA layer. Specifically, the area of the CC peak increased by 15% while that of the CO peak decreased by 8%, which suggests that a non-negligible proportion of o-quinone-like groups has been reduced to catechols after the 15 cycling tests. We suspect that this partial reduction of the PDA layer helps to prevent the re-oxidation of Pd(0) to Pd(II) when the foam is exposed to air (vide supra), and explain the formidable stability of the Pd@PDA@PUF catalyst over time.
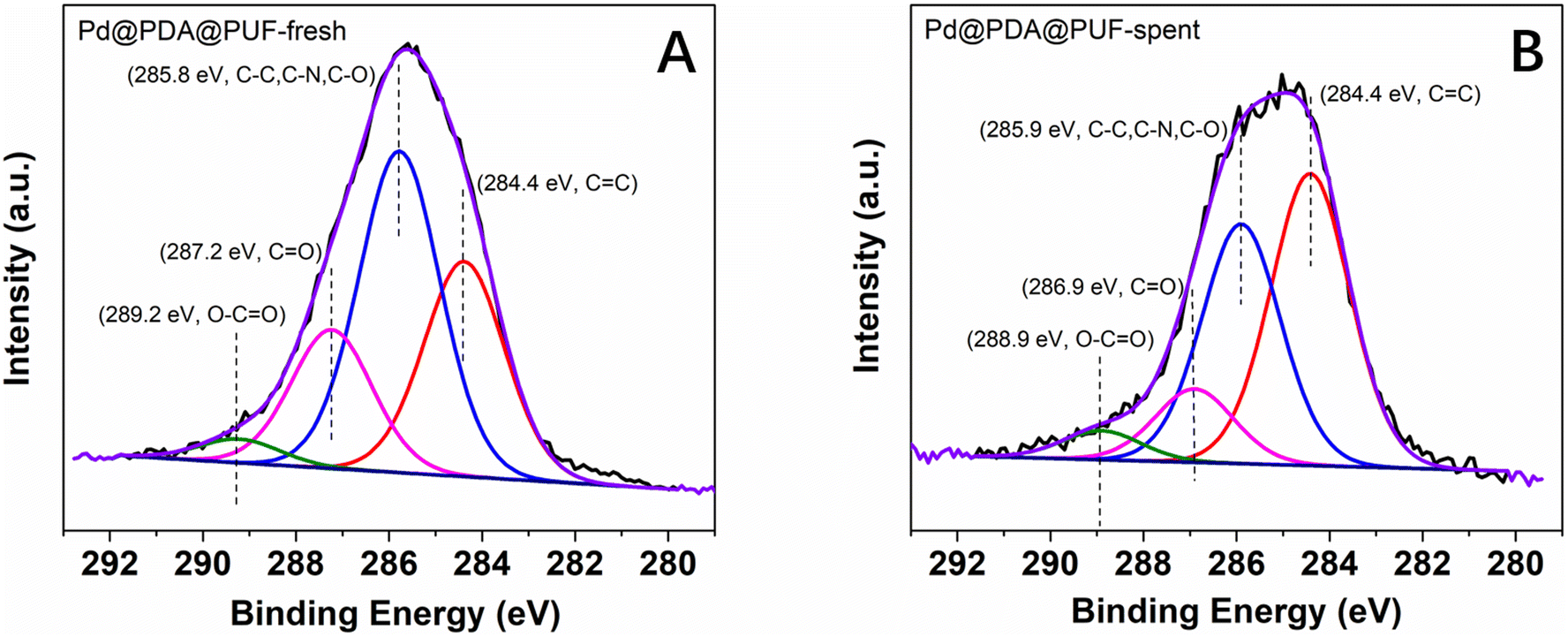 | ||
| Fig. 12 High-resolution XPS spectra of the C 1s core level region of Pd@PDA@PUF: (A) as-synthesized, (B) spent (after 15 cycling tests). | ||
Conclusion
In summary, we have developed an effective and general strategy to readily prepare Pd@PDA@PUF foam that can operate as a versatile and highly reliable heterogeneous catalyst without a prior-reduction procedure. The Pd@PDA@PUF foam was prepared by a simple dip-coating protocol of PDA-coated PUF in a hydro-alcoholic solution of Pd(NH3)4Cl2·H2O at room temperature, and this easy-to-make composite acts as an efficient and reusable heterogeneous catalyst for the semi-hydrogenation of alkynes under 1 atm H2 at ambient temperature, giving remarkably high alkene selectivity (up to 99%) at full alkyne conversion, as well as for the Suzuki coupling of aryl bromides under aerobic conditions.Moreover, the Pd@PDA@PUF catalyst is extremely stable and showed decay neither in activity nor in selectivity when running the hydrogenation and coupling reactions for 15 cycles altogether with thorough washing procedures in an ultrasonic bath between each catalytic run. The as-recovered catalyst even displayed improved catalytic performance due to both the progressive in situ reduction of the Pd(II) immobilized species into Pd(0), as demonstrated by XPS studies, and the strong interaction between Pd and the non-innocent adhesive PDA layer, which prevents re-oxidation of the Pd active phase during air exposure between two cycling tests, as well as Pd leaching into the medium.
Furthermore, the very small size of the Pd species, as shown by HR-SEM and SEM-EDX studies, suggests an initial simple coordination of the Pd(II) ions by the catechol moieties of the PDA layer. The resulting “single atom”-type dispersion of the active phase and the absence of aggregation of the reduced Pd over time may be the reason for the very high selectivity observed in alkyne semi-hydrogenation.
The easy-to-engineer character of the size and shape of the Pd@PDA@PUF catalyst and its efficient mass transfer and transport properties, are expected to enable relatively easy scale-up procedures and adaptation to continuous flow processes, and studies in this direction are currently underway in our laboratories.
All in all, we believe this “dip-and-play” catalytic mode and easy adaptation to any kind of reactor will stimulate the design of new generations of heterogeneous catalysts and processes, and open the gate for a wider range of applications by virtue of its unprecedently facile preparation and excellent catalytic and reusable performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Strasbourg (IdEx Unistra 2019). H. P. thanks the international doctoral program of the Université de Strasbourg for his PhD fellowship. M. Thierry Romero is gratefully acknowledged for acquiring SEM images. The authors thank the NMR Platform and Analytical Platform managers of LIMA, Dr Emeric Wasielewski and Mr. Matthieu Chesse, who contributed, by their valuable technical support, to the completion of this research project.Notes and references
- J. J. Bakker, W. J. Groendijk, K. M. de Lathouder, F. Kapteijn, J. A. Moulijn, M. T. Kreutzer and S. A. Wallin, Ind. Eng. Chem. Res., 2007, 46, 8574–8583 Search PubMed.
- L. Giani, G. Groppi and E. Tronconi, Ind. Eng. Chem. Res., 2005, 44, 4993–5002 CrossRef CAS.
- J. Richardson, Y. Peng and D. Remue, Appl. Catal., A, 2000, 204, 19–32 CrossRef CAS.
- M. Lacroix, P. Nguyen, D. Schweich, C. P. Huu, S. Savin-Poncet and D. Edouard, Chem. Eng. Sci., 2007, 62, 3259–3267 CrossRef CAS.
- M. Lacroix, L. Dreibine, B. de Tymowski, F. Vigneron, D. Edouard, D. Bégin, P. Nguyen, C. Pham, S. Savin-Poncet and F. Luck, Appl. Catal., A, 2011, 397, 62–72 CrossRef CAS.
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 Search PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 Search PubMed.
- D. Edouard, V. Ritleng, L. Jierry and N. T. T. Chau Dalencon, WO2016012689A2, 2016.
- E. Pardieu, N. T. T. Chau, T. Dintzer, T. Romero, D. Favier, T. Roland, D. Edouard, L. Jierry and V. Ritleng, Chem. Commun., 2016, 52, 4691–4693 RSC.
- L. Lefebvre, J. Kelber, L. Jierry, V. Ritleng and D. Edouard, J. Environ. Chem. Eng., 2017, 5, 79–85 CrossRef CAS.
- L. Lefebvre, J. Kelber, X. Mao, F. Ponzio, G. Agusti, C. Vigier-Carrière, V. Ball, L. Jierry, V. Ritleng and D. Edouard, Environ. Prog. Sustainable Energy, 2018, 38, 329–335 Search PubMed.
- A. Ait Khouya, M. L. Mendez Martinez, P. Bertani, T. Romero, D. Favier, T. Roland, V. Guidal, V. Bellière-Baca, D. Edouard, L. Jierry and V. Ritleng, Chem. Commun., 2019, 55, 11960–11963 Search PubMed.
- L. Birba, V. Ritleng, L. Jierry, G. Agusti, P. Fongarland and D. Edouard, Int. J. Energy Res., 2020, 44, 10612–10627 CrossRef CAS.
- F. Ponzio, J. Kelber, L. Birba, K. Rekab, V. Ritleng, L. Jierry and D. Edouard, Environ. Technol. Innovation, 2021, 23, 101618 CrossRef CAS.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- H. U. Blaser, A. Schnyder, H. Steiner, F. Rössler and P. Baumeister, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3284–3308 Search PubMed.
- A. M. Kluver and C. J. Elsevier, in Handbook of homogeneous hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, vol. 3, pp. 375–394 Search PubMed.
- C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 Search PubMed.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam and A. Wootsch, J. Catal., 2006, 242, 26–37 CrossRef CAS.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, B. K. Pradhan, Á. Linares-Solano and D. Cazorla-Amorós, J. Phys. Chem. C, 2008, 112, 3827–3834 Search PubMed.
- R. Venkatesan, M. H. Prechtl, J. D. Scholten, R. P. Pezzi, G. Machado and J. Dupont, J. Mater. Chem., 2011, 21, 3030–3036 RSC.
- P. M. Uberman, N. J. Costa, K. Philippot, R. C. Carmona, A. A. Dos Santos and L. M. Rossi, Green Chem., 2014, 16, 4566–4574 Search PubMed.
- J. A. Delgado, O. Benkirane, C. Claver, D. Curulla-Ferré and C. Godard, Dalton Trans., 2017, 46, 12381–12403 RSC.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Havecker, A. Knop-Gericke, S. D. Jackson and R. Schlogl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2019, 120, 683–733 Search PubMed.
- F. P. da Silva, J. L. Fiorio and L. M. Rossi, ACS Omega, 2017, 2, 6014–6022 CrossRef CAS PubMed.
- Y. Zhao, G. Fu and N. Zheng, Catal. Today, 2017, 279, 36–44 CrossRef CAS.
- M. Zhao, Y. Ji, M. Wang, N. Zhong, Z. Kang, N. Asao, W.-J. Jiang and Q. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 34804–34811 CrossRef CAS PubMed.
- E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051–14054 CrossRef CAS PubMed.
- P. V. Markov, G. O. Bragina, A. V. Rassolov, G. N. Baeva, I. S. Mashkovsky, V. Y. Murzin, Y. V. Zubavichus and A. Y. Stakheev, Mendeleev Commun., 2016, 26, 502–504 CrossRef CAS.
- C. M. Kruppe, J. D. Krooswyk and M. Trenary, ACS Catal., 2017, 7, 8042–8049 Search PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- Y. Lu, X. Feng, B. S. Takale, Y. Yamamoto, W. Zhang and M. Bao, ACS Catal., 2017, 7, 8296–8303 CrossRef CAS.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Catal., 2019, 9, 1993–2006 Search PubMed.
- C. Shen, Y. Ji, P. Wang, S. Bai, M. Wang, Y. Li, X. Huang and Q. Shao, ACS Catal., 2021, 11, 5231–5239 CrossRef CAS.
- Z. Wang, Y. Zou, Y. Li and Y. Cheng, Small, 2020, 16, 1907042 CrossRef CAS PubMed.
- A. Kunfi and G. London, Synthesis, 2019, 2829–2838 Search PubMed.
- J. Manna, S. Akbayrak and S. Özkar, Appl. Catal., B, 2017, 208, 104–115 Search PubMed.
- Y. Li, L. Xu, B. Xu, Z. Mao, H. Xu, Y. Zhong, L. Zhang, B. Wang and X. Sui, ACS Appl. Mater. Interfaces, 2017, 9, 17155–17162 Search PubMed.
- A. Kunfi, Z. May, P. Németh and G. London, J. Catal., 2018, 361, 84–93 Search PubMed.
- B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS PubMed.
- V. Ball, Catal. Today, 2018, 301, 196–203 Search PubMed.
- J. Xi, J. Xiao, F. Xiao, Y. Jin, Y. Dong, F. Jing and S. Wang, Sci. Rep., 2016, 6, 21904 CrossRef CAS PubMed.
- A. Kunfi, V. Szabó, Á. Mastalir, I. Bucsi, M. Mohai, P. Németh, I. Bertóti and G. London, ChemCatChem, 2017, 9, 3236–3244 CrossRef CAS.
- S. Huebner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef CAS.
- A. Ait Khouya, H. Ba, W. Baaziz, J.-M. Nhut, A. Rossin, S. Zafeiratos, O. Ersen, G. Giambastiani, V. Ritleng and C. Pham-Huu, ACS Appl. Nano Mater., 2021, 4, 2265–2277 Search PubMed.
- K. H. Lee, B. Lee, K. R. Lee, M. H. Yi and N. H. Hur, Chem. Commun., 2012, 48, 4414–4416 RSC.
- S. Song, K. Li, J. Pan, F. Wang, J. Li, J. Feng, S. Yao, X. Ge, X. Wang and H. Zhang, Adv. Mater., 2017, 29, 1605332 CrossRef PubMed.
- D. Deng, Y. Yang, Y. Gong, Y. Li, X. Xu and Y. Wang, Green Chem., 2013, 15, 2525–2531 RSC.
- S. Yang, C. Cao, L. Peng, J. Zhang, B. Han and W. Song, Chem. Commun., 2016, 52, 3627–3630 RSC.
- T. Yoshii, D. Umemoto, Y. Kuwahara, K. Mori and H. Yamashita, ACS Appl. Mater. Interfaces, 2019, 11, 37708–37719 Search PubMed.
- X.-P. Yin, S.-F. Tang, C. Zhang, H.-J. Wang, R. Si, X.-L. Lu and T.-B. Lu, J. Mater. Chem. A, 2020, 8, 20925–20930 RSC.
- M. B. Boucher, B. Zugic, G. Cladaras, J. Kammert, M. D. Marcinkowski, T. J. Lawton, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2013, 15, 12187–12196 Search PubMed.
- Z. Li, Q. Ren, X. Wang, W. Chen, L. Leng, M. Zhang, J. H. Horton, B. Liu, Q. Xu and W. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 2530–2537 CrossRef CAS PubMed.
- Overlaps of the 1H NMR peaks of the reaction products with those of the starting material prevented to carry out precise kinetic monitoring of the reaction.
- F. Diederich and P. J. Stang, Metal-catalyzed cross-coupling reactions, John Wiley & Sons, 2008 Search PubMed.
- T. J. Colacot, New trends in cross-coupling: theory and applications, Royal Society of Chemistry, 2015 Search PubMed.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- B. Tao and D. W. Boykin, J. Org. Chem., 2004, 69, 4330–4335 Search PubMed.
- A. N. Marziale, S. H. Faul, T. Reiner, S. Schneider and J. Eppinger, Green Chem., 2010, 12, 35–38 RSC.
- C. Liu, X. Rao, Y. Zhang, X. Li, J. Qiu and Z. Jin, Eur. J. Org. Chem., 2013, 4345–4350 CrossRef.
- G. Ding, L. Hao, H. Xu, L. Wang, J. Chen, T. Li, X. Tu and Q. Zhang, Commun. Chem., 2020, 3, 1–8 CrossRef.
- It can however not be excluded that the low BE tale in spectrum A may also be due to differential charging.
- M. C. D'Alterio, È. Casals-Cruañas, N. V. Tzouras, G. Talarico, S. P. Nolan and A. Poater, Chem. – Eur. J., 2021, 27, 13481–13493 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03283j |
| This journal is © The Royal Society of Chemistry 2023 |