
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-based catalysts for CO2 hydrogenation: a perspective on active sites
Yun-Fei
Shi
a,
Sicong
Ma
 *b and
Zhi-Pan
Liu
*ab
*b and
Zhi-Pan
Liu
*ab
aCollaborative Innovation Center of Chemistry for Energy Material, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Key Laboratory of Computational Physical Science, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: zpliu@fudan.edu.cn
bKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: scma@mail.sioc.ac.cn
First published on 14th August 2023
Abstract
CO2 hydrogenation is regarded as a revolutionized field in heterogeneous catalysis, not only mitigating environmental problems caused by greenhouse gases but also producing valuable chemicals. This Perspective, going over both theoretical and experimental advances, aims to bridge Cu-based catalyst structures, the most important type of CO2 hydrogenation catalyst, and their catalysis applications with varied activity and selectivity. We provide a systematic overview of the catalytic active sites, the reaction mechanism, and their impact on the reaction selectivity, stability, and activity for CO2 hydrogenation. There is a particular focus on the nature of the industrial Cu/ZnO/Al2O3 catalyst, where a large volume of literature is available exploring the reaction energetics on the possible reaction sites, including Cu metal, CuZn alloy, and ZnOxHy overlayers. The recent advances in designing better catalytic active sites, such as the Cu single-atom catalyst, supported Cu cluster catalyst, and bimetallic Cu–M, are then followed to illustrate how the activity and selectivity vary upon changing the active sites. Our perspectives on the future research directions are finally provided, which should benefit the understanding of complex catalytic active sites and the design of better CO2 hydrogenation catalysts.
 Yun-Fei Shi | Yun-Fei Shi graduated from Fudan University in 2019. He is currently doing a PhD degree at Fudan University under the supervision of Prof. Zhi-Pan Liu. His research interests include reaction mechanisms of heterogeneous catalysis and machine learning accelerated automated reaction network exploration. |
 Sicong Ma | Sicong Ma graduated from the China University of Petroleum (Beijing, China). He received his PhD degree in 2019 at Fudan University under the supervision of Prof. Zhi-Pan Liu. He then continued to stay in Fudan University to carry out post-doctoral research. In 2021 he joined the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, where he currently holds a position as an assistant research fellow. His current research interests are mainly focused on the machine learning applications in constructing the homogeneous/heterogeneous catalyst database (e.g. zeolite-related database and phosphine ligand database). |
 Zhi-Pan Liu | Zhi-Pan Liu graduated in Chemistry at Shanghai Jiao Tong University (Shanghai, China). He received his PhD degree in 2003 at the Queens University of Belfast under the supervision of Prof. Peijun Hu. He then moved to the University of Cambridge, where he joined the surface science group supervised by Prof. Sir David King. In 2005 he joined Fudan University, where he has since stayed, with a position of a Changjiang professor in Physical Chemistry. His current research interests include methodology development for potential energy surface exploration, theory on heterogeneous catalysis, and machine-learning-based atomic simulation. |
Broader contextCO2 hydrogenation is regarded as a revolutionized field in heterogeneous catalysis, not only mitigating environmental problems caused by greenhouse gas but also producing valuable chemicals. This Perspective, going over both theoretical and experimental advances, aims to bridge Cu-based catalyst structures, the most important type of CO2 hydrogenation catalyst, and their catalysis applications with varied activity and selectivity. We believe that this Perspective can arouse the interest of researchers in related fields and provide some directions for the future research in energy and environmental catalysis. |
1. Introduction
The conversion of carbon dioxide (CO2) into value-added products via heterogeneous catalysis is a promising approach to reducing greenhouse gas emissions with great economic benefits. To date, the hydrogenation of CO2 and CO mixed gas has already been realized in industry through the low-pressure methanol synthesis process with a Cu-based catalyst, i.e. a Cu/ZnO/Al2O3 (CZA) catalyst.1,2 The global CH3OH production via this technology is massive, reaching 106.89 million metric tons in 2021.3 This industrial achievement demonstrates the great value of CO2 conversion in making renewable fuels and chemical compounds. In recent years, significant efforts have been devoted to developing new types of catalysts for CO2 hydrogenation (without the presence of CO), where the Cu element continues to play a pivotal role in these endeavors. This Perspective focuses on the recent theoretical and experimental advances in the structural characteristics of Cu-based catalysts and their linkages to the product selectivity in CO2 hydrogenation from a fundamental point of view.The industrial CZA catalyst serves as a textbook example for understanding how CO2 is converted on Cu-based catalysts.4 Despite tremendous efforts, there is a huge debate regarding the active site of the CZA catalyst, not least because of the disparity between experimental characterization setups and industrial conditions. It was once accepted that metallic Cu serves as the active site while ZnO acts as a support,5 although, apparently, the synergy between Cu and Zn components is critical to this technology. Several hypotheses have been further proposed to rationalize the synergy effects, including Cu+ in the ZnO lattice,6 lattice strain,7 Schottky junctions,8etc. However, most of these hypotheses lack clear support from the latest experimental and theoretical results. In 2012, Behrens et al.9 reported the experimental evidence for the enrichment of Zn on the Cu surface using X-ray photoelectron spectroscopy (XPS) and high-resolution transmission electron microscopy (HRTEM) methods. Since then, researchers focused on two models of active sites: the CuZn surface alloy model,10 in which Zn is reduced to form a CuZn alloy phase, and the ZnOx/Cu interface model,11 which grows in situ due to the strong metal–support interaction (SMSI) effect.
In addition to the fundamental efforts to clarify the reaction mechanism on the CZA catalyst, the exploration of the possibility to apply Cu single-atom catalysts (SACs) as low-temperature CO2 hydrogenation catalysts is mainstream in heterogeneous catalysis.12,13 The Cu SACs refer to catalysts with Cu cations stabilized by the support material, where the valence state of Cu can be tuned by the coordination environment and thus depends on the synthetic routes. Recent literature shows that these Cu SACs can exhibit distinctive product selectivity due to the delicately designed Cu coordination environment.
Another active research direction relates to the alloying between Cu and the second metal element, for example, the bimetallic Cu–M system (M = Pd,14 Ni,15 Fe,16–18etc.). Introducing a second metal into the Cu-catalyst can result in the formation of non-methanol products, such as methane,15 long-chain hydrocarbons,18 or ethanol.16 For example, the Cu–Ni catalyst can lead to high selectivity on CH4,15 while the Cu–Fe catalyst tends to produce long-chain hydrocarbons.17 However, the active site and reaction mechanism of these new catalytic systems are much less studied—whether the alloy metal is indeed the active site remains skeptical. As pointed out by Hwang et al.16 in the Cu–Fe catalyst, although the CuFe alloy phase is regarded as the active site for the Cu–Fe bimetallic catalyst, FeOx and FeCx also exist in the catalyst and thus may still be the active centers.
With encouraging progress in Cu-based catalysts for CO2 hydrogenation achieved in recent years, we here aim to overview the current understanding of the active sites of different types of Cu-based catalysts, with a particular emphasis on the atomic structure as revealed by joint theoretical calculations and experimental characterization. This Perspective is organized as follows. The thermodynamics and reaction networks for CO2 hydrogenation are first summarized. This follows sequentially the recent advancements in probing the catalytic active sites using advanced theoretical and experimental techniques, including CZA catalysts, Cu SAC catalysts, and their closely related supported Cu cluster catalysts, and bimetallic Cu–M catalysts. Finally, an outlook for future research directions is provided.
2. Thermodynamics and the kinetics of CO2 hydrogenation on Cu surfaces
CO2 hydrogenation can in principle yield a diverse range of products, including primary reduction products (such as CO, HCOOH, and HCHO), alcohols (CH3OH and C2H5OH), and hydrocarbons (CH4, C2H4, and even long-chain alkanes). It is noteworthy that while methanol is a commonly targeted product in industrial CO2 hydrogenation, it is not the most thermodynamically favorable product at a typical operating temperature of 500 K. Fig. 1 illustrates the Gibbs free energy changes (ΔG) for these gas-phase reactions.19 It shows that CO and CH3OH, the most frequently observed products, have similar ΔG at 500 K (0.21 and 0.39 eV, respectively), both slightly above zero. When the feed gas is compressed to increase the input pressure the reaction can be driven forward, resulting in a more favorable equilibrium conversion of CH3OH compared to the economically undesirable CO. However, the maximum one-pass conversion of CH3OH is still limited to 15–35%,20 which can be increased by decreasing the temperature and increasing the pressure. Consequently, the methanol synthesis industry tends to favor lower reaction temperatures as long as the reaction rate remains acceptable.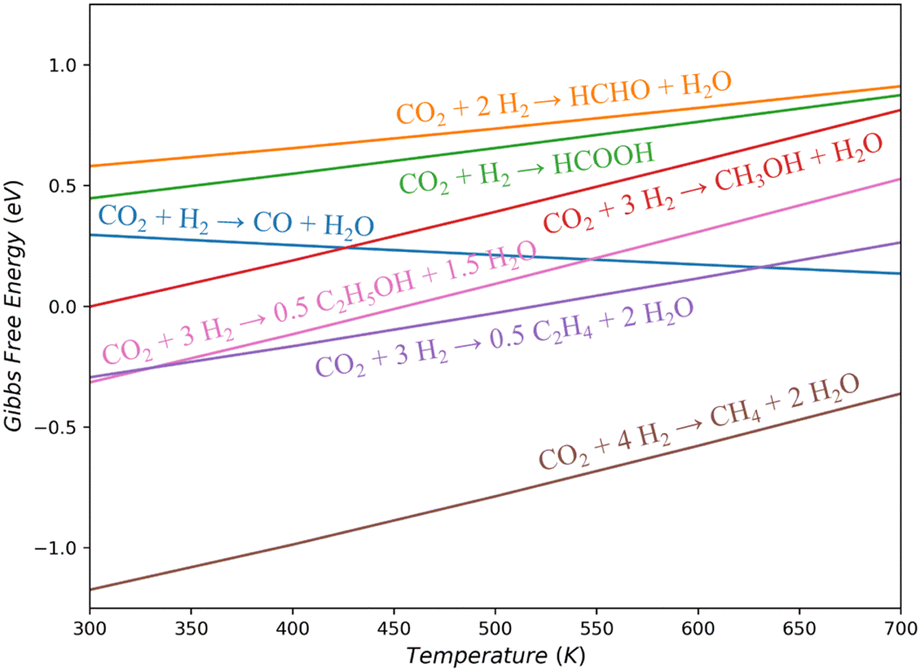 | ||
| Fig. 1 Gas-phase Gibbs free energy variation against temperature for CO2 hydrogenation to low-carbon products. All gas pressures set to 1 bar. | ||
Products other than CH3OH and CO are less frequently reported on Cu-based catalysts. HCOOH and HCHO are thermodynamically unfavorable than CH3OH across the entire temperature range (ΔG is 0.26/0.34 eV higher than that of CH3OH at 500 K, respectively). They exhibit low selectivity, likely due to their instability and susceptibility to further hydrogenation into CH3OH. Products like CH4, C2H4, and C2H5OH are consistently thermodynamically more favorable than CH3OH (ΔG is 1.18/0.42/0.30 eV lower at 500 K), but the hydrocarbon and multi-carbon products are rarely reported in the products using Cu-based catalysts. On Cu metal, a model system, CO2 hydrogenation only produces CO and CH3OH.21
The answer to the conflict of product selectivity with thermodynamics preference lies in the reaction kinetics in catalysis. Fig. 2 summarizes the generally-regarded CO2 hydrogenation mechanism on Cu surfaces obtained from density functional theory (DFT) studies.22–27 Three general findings are outlined in the following.
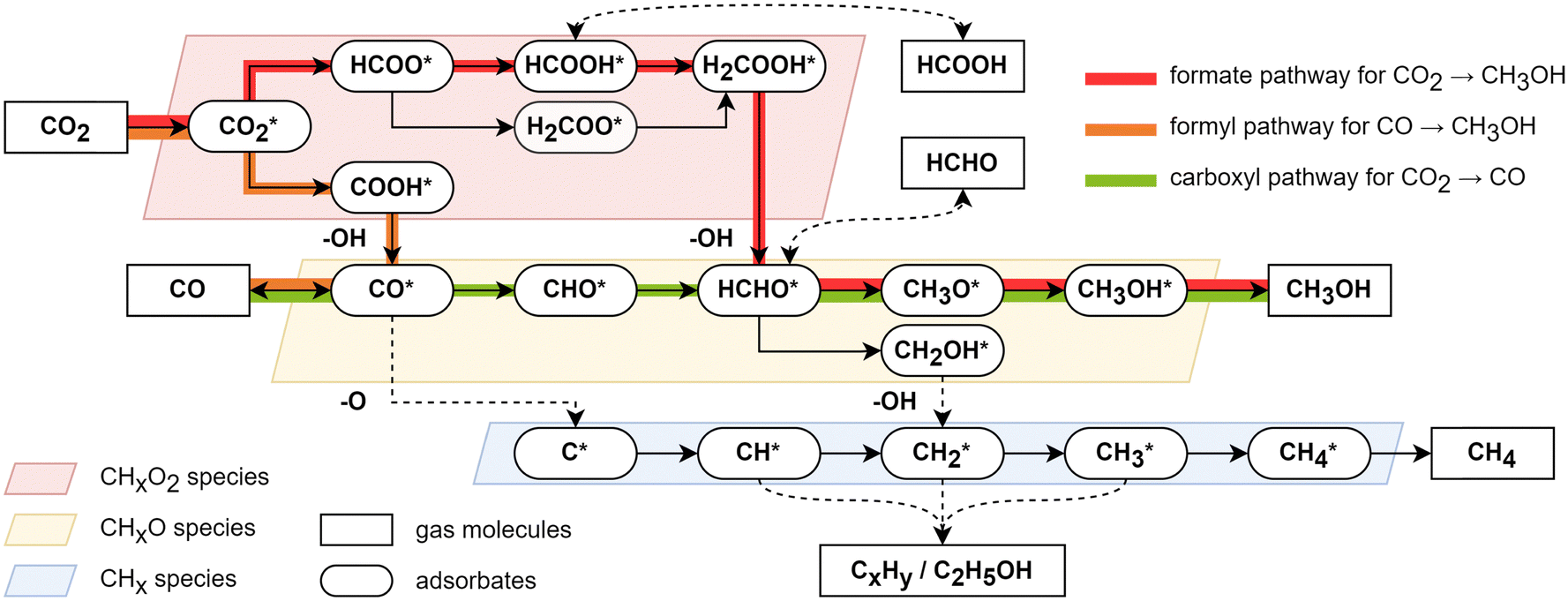 | ||
| Fig. 2 The possible reaction network and reaction pathway on the Cu surface for CO2 hydrogenations. | ||
(i) A most-mentioned CO2 hydrogenation pathway to CH3OH follows the sequence: CO2* (superscript * indicates the adsorbed state) → HCOO* → HCOOH* → H2COOH* → HCHO* → CH3O* → CH3OH*, termed the formate pathway.22 The rate-determining step occurs in the first half of the reaction before CH2O formation, involving the hydrogenation of CHxO2 (x = 0–3) species, such as CO2 to HCOO*, HCOOH* to H2COOH*, or H2COOH to HCHO*. If the overall barrier for the further hydrogenation of HCOOH* and HCHO* is not higher than that for the formation of HCOO*, the reaction will show low product selectivity to HCOOH or HCHO, which is the case in most Cu surfaces. For example, the Gibbs free energy barriers on Cu(211) surfaces is 1.4 eV with the rate-determining steps (RDS) being CO2 and HCOO* hydrogenation.25
(ii) Another mechanism for CO2 to CH3OH is the reverse water–gas shift (r-WGS) coupling with CO hydrogenation.28 In the pathway, CO2 is first hydrogenated to CO through CO2* → COOH* → CO*, termed the carboxyl pathway, and then CO* is further hydrogenated to CH3OH* through CO* → CHO* → HCHO* → CH3O* → CH3OH*, termed the formyl pathway. This pathway on metal Cu surfaces appears to be kinetically unfavorable compared to the formate pathway due to the higher Gibbs energy barrier of CO2* → COOH*, e.g. >1.8 eV for Cu(111),22 Cu(100)23 and Cu(211).27 Additionally, COOH* is less stable than HCOO* on Cu surfaces, leading to a lower rate of subsequent reactions.
(iii) Hydrogenation toward hydrocarbons or multi-carbon products is challenging on pure Cu catalysts, but appears to be likely on Cu-based multi-metallic catalysts such as CuFe and CuNi catalysts.29 This is likely due to the high barrier associated with the C–O bond breaking to produce CHx (x = 0–4) species, which is essential for subsequent C–C coupling.25 The direct dissociation of CO* to form C* and O*, known to be feasible on Fe catalysts, encounters a high energy barrier (>2 eV) on Cu surfaces, making it unlikely to occur.22 Another possible pathway of CHx generation is via the HCHO* → CH2OH* → CH2* route, but its overall barrier is higher than that of HCHO* → CH3O* → CH3OH, leading to a low selectivity.25
Quantitatively, the low coordination number (CN) of Cu (the number of Cu atoms neighboring the centering Cu) is generally more active in CO2 hydrogenation. Table 1 summarizes the overall energy barriers on different Cu surfaces. For the same reaction step, as listed in entries 1–3, and entries 4–8 in Table 1,22,23,30 generally the barrier is lower as long as the CN is reduced. When converting to the Gibbs free energy, the close-packed Cu(111) is found to be inert, as evidenced by the very high Gibbs free energy barrier of 1.98 eV for CO2 hydrogenation, where the rate-determining step is the HCHO* + H* step.22,25 To allow CO2 hydrogenation, the introduction of step and point defects is thus a must.25,30–32 The Cu(211) surface exhibits a significantly lower Gibbs free barrier of 1.40 eV for methanol production. These findings indicate the importance of manipulating the surface structure for enhancing the catalytic activity of Cu-based catalysts in CO2 hydrogenation.
| Cu surface | RDS | CN | E a (eV) | Ref. |
|---|---|---|---|---|
| Cu(111) | HCOO* → H2COOH* | 9 | 1.27 | 22 |
| Cu(100) | HCOO* → H2COOH* | 8 | 1.25 | 23 |
| Cu(110) | HCOO* → H2COOH* | 7 | 1.19 | 23 |
| Cu(111) | CO2 → HCOO* | 9 | 1.15 | 30 |
| Cu(100) | CO2 → HCOO* | 8 | 1.03 | 30 |
| Cu(111) point defect | CO2 → HCOO* | 8 | 1.02 | 30 |
| Cu(100) point defect | CO2 → HCOO* | 7 | 0.92 | 30 |
| Cu(211) | CO2 → HCOO* | 7 | 0.92 | 30 |
3. CZA industrial catalyst
As the active site of the CZA catalyst remains highly controversial, we first overview the basic facts of the CZA catalyst. The industrial CZA catalyst is prepared by the traditional co-precipitation method. The precipitate with a molar ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:Al = 7:3:1 turns to composites containing at least nanoparticles (5–10 nm) of metallic Cu, wurtzite ZnO, and amorphous Al2O3 after air calcination and H2 activation. When operated under conditions such as 500–550 K and 50–100 bar, the CZA catalyst exhibits high selectivity for methanol production (e.g., 99.8%) and exceptional long-term stability (design life >3 years).33 It is worth noting that a mixed CO, CO2, and H2 feed gas, instead of pure CO2 or pure CO with H2, is utilized in the fixed-bed catalysis, where the CO2:CO ratio range is 0.5–4.1,34,35 Interestingly, the isotopic-labeling experiments revealed that CO2 is the dominant carbon source for methanol production, accounting for more than 90% of the carbon in methanol.24 On the other hand, the CO-free CO2/H2 feed gas leads to a more oxidizing atmosphere.
:Zn:Al = 7:3:1 turns to composites containing at least nanoparticles (5–10 nm) of metallic Cu, wurtzite ZnO, and amorphous Al2O3 after air calcination and H2 activation. When operated under conditions such as 500–550 K and 50–100 bar, the CZA catalyst exhibits high selectivity for methanol production (e.g., 99.8%) and exceptional long-term stability (design life >3 years).33 It is worth noting that a mixed CO, CO2, and H2 feed gas, instead of pure CO2 or pure CO with H2, is utilized in the fixed-bed catalysis, where the CO2:CO ratio range is 0.5–4.1,34,35 Interestingly, the isotopic-labeling experiments revealed that CO2 is the dominant carbon source for methanol production, accounting for more than 90% of the carbon in methanol.24 On the other hand, the CO-free CO2/H2 feed gas leads to a more oxidizing atmosphere.
There are two main vying views regarding the active sites of CZA catalysts, both of which have garnered experimental and theoretical evidence, fueling an ongoing debate. The first view posits that ZnO oxide is partially reduced and the Zn migrates to the Cu surface, leading to intermixing with the Cu lattice and the formation of a surface alloy. According to DFT calculations, the reducing potential of the feed gas is sufficient to reduce ZnO to form a CuZn surface alloy with moderate Zn coverage (0.35 ML36 or 0.22 ML25), but not enough to form a CuZn bulk alloy or metallic Zn. In experiments also high content of the CuZn surface alloy is observed under strong reduction conditions, e.g., CO2-free CO and H2 atmospheres.37–39 Kuld et al.40 found an increasing Zn coverage with an increasing CO/CO2 ratio in the feed gas, which led to a higher methanol reaction rate, indicating that stronger reducing potential in the feed gas resulted in higher Zn coverage. Nakamura et al.41 also found that the maximum turnover frequency (TOF) on a Zn deposited on a Cu(111) model catalyst with 0.19 ML Zn coverage was 10 times faster compared to that on a clean Cu(111) surface. While earlier DFT studies reported that the Zn-doped Cu(211) surface shows a lower energy barrier of CO2 hydrogenation to methanol than that without Zn doping, following the same formate pathway,9,24 the later ab initio microkinetics study reveals a negative effect of Zn alloying on CO2 hydrogenation activity.42,43
Recently, we developed a microkinetic-guided machine learning pathway search (MMLPS) method to explore the reaction network for CO2 and CO hydrogenations on thermodynamically favorable Cu–Zn alloy surface structures.25 Taking Cu(211) as an example, Fig. 3a plots 14958 reaction pairs (IS, TS, FS) on the Cu(211) surface through automated reaction sampling based on global neural network potential (G-NN) calculations, from which the CO2 and CO hydrogenation reaction pathways are resolved and the reaction mechanism is identified to be the formate pathway rather than the r-WGS pathway for CO2 hydrogenation on the Cu(111) and Zn-alloyed Cu(211) surface (Fig. 3b). The key intermediates along the pathways on 0.11 ML and 0.22 ML Zn–Cu(211) surfaces are shown in Fig. 3c and d. Interestingly, we found that the Zn decorates at the step-edge of the Cu(211) surface with a coverage of up to 0.22 ML under reaction conditions, and the Zn–Zn dimeric site is thermodynamically unfavorable. CO2 and CO hydrogenations occur exclusively at the step-edge of the (211) surface with a Zn coverage of up to 0.11 ML, where the low coverage of Zn (0.11 ML) does not much affect the reaction kinetics (Fig. 3b), demonstrating no activity improvement on the CuZn surface alloy. The microkinetics simulations based on DFT calculation reproduced nicely the experimental finding that CO2 hydrogenation dominates methanol synthesis, instead of CO hydrogenation.
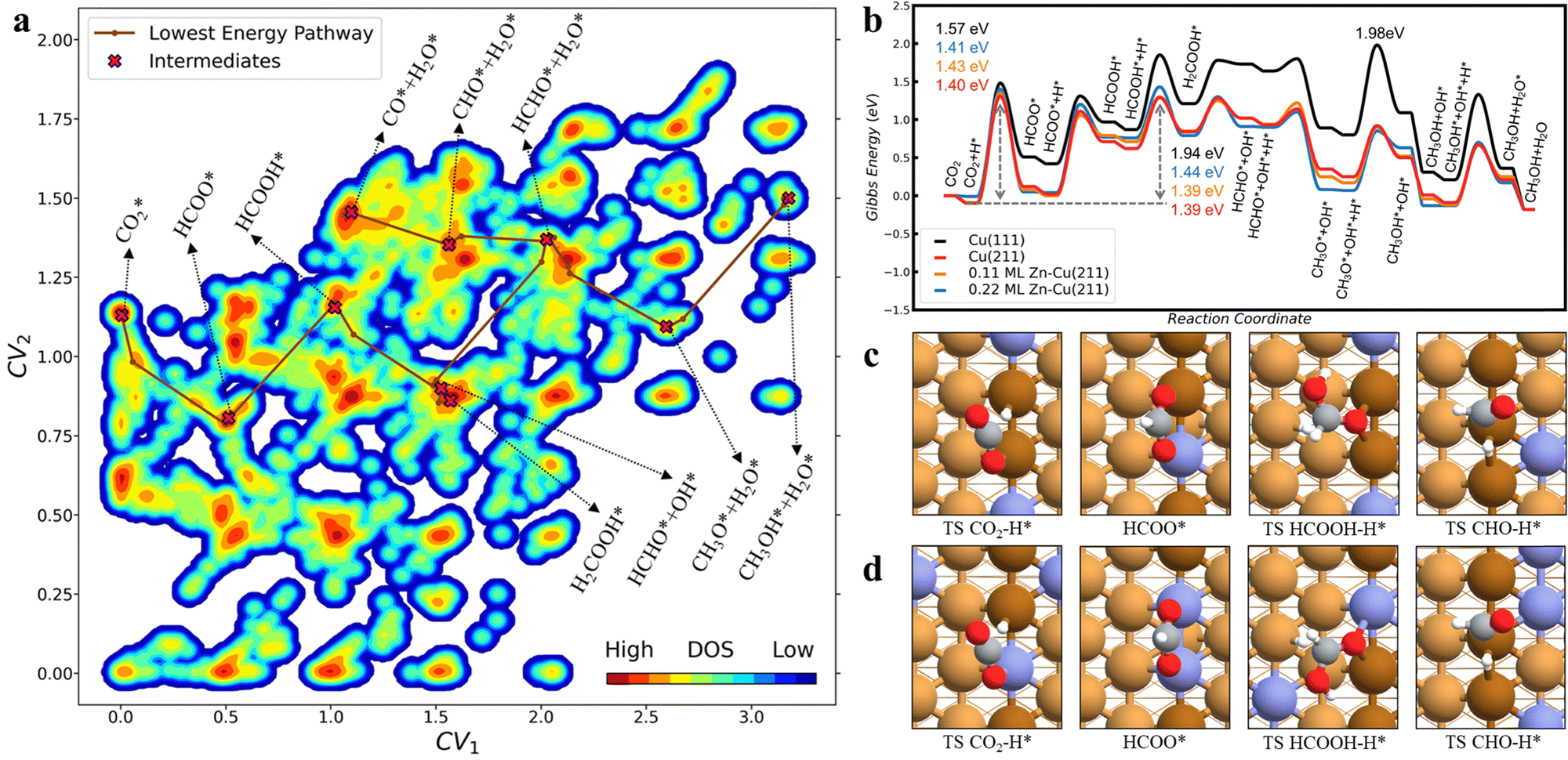 | ||
| Fig. 3 (a) Contour plot for 14958 reaction pairs (IS, TS, FS) obtained by MMLPS on the Cu(211) surface. The color indicates the occurrence frequency of the state in reaction pair collection (density of state, DOS). All structures are projected to the plot with two collective variables (CV). Key intermediates along the lowest-energy pathway are highlighted by brown lines. (b) CO2 hydrogenation Gibbs energy profile on Cu(211) and Zn alloyed Cu(211) surfaces. (c) and (d) The structure of the key reaction step for CO2 hydrogenation on (c) 0.11 ML Zn–Cu(211) and (d) 0.22 ML Zn–Cu(211). The color scheme for atoms is as follows: H: white; O: red; Cu: yellow; and Zn: blue. Reprinted with permission from ref. 25. Copyright 2022 American Chemical Society. | ||
Another popular view proposes that Zn species form a ZnOx(Hy)–Cu interface, where Zn atoms are primarily present as ZnOx(Hy) clusters of layers on the Cu surface. It was once suggested that the reduced Zn ions migrate to the Cu surface and undergo re-oxidation, forming a few layers of ZnOx on the Cu surface and leading to an SMSI effect.44 This phenomenon is indeed observed in HRTEM photos under ex situ conditions, revealing the formation of a graphite-like ZnO overlayer on the surface of Cu nanoparticles after H2 activation (Fig. 4).11 This phenomenon was also observed by other research groups.9,45,46 The presence of cationic Zn as the active site was also suggested by Laudenschleger et al.,47 who conducted a high-pressure pulse experiment in which NH3 is injected into a high-pressure methanol synthesis. They observed a temporary decrease in methanol yield due to NH3 poisoning, but the co-fed C2H4 hydrogenates to C2H6 on the Cu surface at a normal rate. Based on these observations, they proposed that a positively charged Zn on Cu is responsible for the active sites in CO2 hydrogenation, which is poisoned by NH3. They also reported that the higher CO2 content, a more oxidizing condition, may lead to more positively charged Zn on the Cu surface, resulting in more obvious NH3 poisoning.
 | ||
| Fig. 4 (A)–(C) HRTEM images of Cu/ZnO/Al2O3 after different times of electron beam exposure. Red-colored sites correspond to Cu particles. Yellow indicates graphitic-like ZnOx. Green highlights rock salt ZnO, and blue regions correspond to the wurtzite ZnO structure. Reprinted with permission from ref. 11. Copyright 2015 John Wiley & Sons, Inc. | ||
As for the phase of Zn cations in catalysts, growing experimental and theoretical results support that the presence of H species may be critical in the SMSI process and also in the active site, suggesting the relevance of the ZnOxHy phase rather than the ZnOx phase. Beck et al.36 first showed that the formation of active sites is influenced by the H2 pressure. They observed significant changes in the Zn K-edge X-ray absorption near edge structure (XANES) signals during temperature-programmed reduction around operating temperature when the H2 pressure was kept beyond 1 bar. However, no significant changes were observed in the zinc (+2) oxidation state until 500 °C under 1 mbar H2. In addition, theoretical studies also found the strong adsorption of H on the ZnOx cluster supported on Cu(111), stabilizing the ZnOx cluster on Cu under the H2-rich reaction conditions.46,48–50 The presence of H may further alter the reaction kinetics. Reichenbach et al.51 showed that the energy barrier of CO2 hydrogenation on Zn7O3/Cu(111) is 1.33 eV, which decreases to 1.30 eV when H adsorbs on the ZnO cluster. Kattel et al. proposed another Zn6O7H7/Cu(111) model whose CO2 hydrogenation overall energy barrier is only 1.05 eV (HCOO*→ H2COOH*), much lower than that of ZnCu(211) (1.49 eV).52 It should be noted that due to the complexity of the potential energy surface (PES), the structures of ZnOxHy clusters on Cu surfaces are not explored systematically yet and thus the reported energetics on the manually constructed ZnOxHy clusters may not be representative.
To date, it is apparent that the catalytic active site of the CZA catalyst, although still in debate, should involve low-coordinated Cu sites. The presence of Zn, as both the CuZn alloy and ZnOH are likely forms, at least helps to stabilize the active site. The advent of advanced characterization and theoretical methods has brought the understanding of active sites much closer to the truth.
4. Cu SACs
Cu SACs are promising low-temperature catalysts for CO2 hydrogenation with high selectivity towards desirable products. Table 2 summarizes the recent advances in Cu SACs, highlighting the Cu coordination environment, valence state, reaction intermediates, and the computed reaction barriers, if available. As shown, three types of elements, namely O, N, and C, were explored as the anionic coordination, which can alter the valence state of Cu and also the product selectivity.| Cu SAC | Temp. (K) | Product (selectivity) | CNCu | Cuδ+ | Reaction intermediates | E a (eV) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| 1 | Cu1@FAU zeolite | 513 | MeOH (89.5%) | [CuO3.8] | +2 | HCOO, HCOOH, CH3O | ∼2.30 | 13 |
| 2 | Cu1/ZnO | 443 | MeOH (99.1%) | [CuO3.5] | 1 < δ < 2 | COOH, CH2O | 1.94 | 53 |
| 3 | Cu1/amorphous-ZrO2 | 453 | MeOH (100%) | [CuO3] | +1.4 | HCOO, CH3O | 1.46 | 12 |
| 4 | Cu1/Zr12-MOF | 358 | EtOH (>99%) | [CuO3] | +1 | CHO, CH2O, CH3O | 54 | |
| 5 | Cu1/C2N | HCOOH | [CuN2] | HCOO | 0.57 | 55 | ||
| 6 | Cu1/C3N4 | HCOOH | [CuN4] | HCOO | 0.86 | 56 | ||
| 7 | Cu1/C3N4 | 423 | CO (94.3%) | [CuN3] | +1.64 | COOH | 57 | |
| 8 | Cu1/C3N4 | 423 | MeOH (95.5%) | [CuN4] | +1.05 | HCOO, CH3O | 57 | |
| 9 | Cu1/graphine | HCOOH | [CuC3] | HCOO | 1.47, 1.18 | 58 and 59 | ||
Entries 1–4 in Table 3 exhibit an interesting trend in Cu SACs, that is, methanol selectivity decreases with the increase of the Cu–O coordination number. For example, Chai et al.13 reported 89.5% selectivity towards methanol on a faujasite-encaged mononuclear Cu center catalyst that has a higher [CuO3.8] coordination environment with a Cu valence state of +2 (Table 2, entry 1). Wu et al.53 conducted a study on Cu1/ZnO catalysts and found that CO2 hydrogenation to methanol had a selectivity of 99.1% with Cu coordination of [CuO3.5] and the valence state of Cuδ+ (1 < δ < 2) (Table 2, entry 2). Interestingly, the study revealed that water present at the optimal levels acts as an active chemical reagent, opening the reaction pathways of CO2 → COOH → HCOOH.
| Alloy | Active site | Alloy composition | Temp (K) | Pressure (Mpa) | Conversion (%) | Product (selectivity) | Ref. |
|---|---|---|---|---|---|---|---|
| CuPd | Pd–Cu/SiO2 | Cu:Pd = 0.34:0.66 |
523 | 4.1 | 6.60 | CO (66%) | 14 |
| CH3OH (34%) | |||||||
| Pd–Cu/Ti0.1Zr0.9O2 | Cu:Pd =3:1 |
523 | 4.1 | 10.10 | CO (55.4%) | 78 | |
| CH3OH (44.6%) | |||||||
| 1Pd–10Cu/CeO2 | Cu:Pd = 1:0.06 |
543 | 3 | 17.8 | CH3OH (23.7%) | 75 | |
| CuNi | Ni-in-Cu | Cu:Ni =4.14:1 |
673 | 0.1 | 26 | CO (>99.9%) | 81 |
| CuNi-rGO | Cu:Ni = 2:1 |
498 | 4.0 | 7.87 | CH3OH (98.7%) | 82 | |
| Cu–Ni/CeO2-nanotubes | Cu:Ni = 2:1 |
533 | 4 | 17.8 | CH3OH (76%) | 84 | |
| CO (∼25%) | |||||||
| Cu–Ni/MgAl2O4 | Cu:Ni = 15.5:1 |
623 | — | 86 | CH4 (>98%) | 15 | |
| CuFe | sp-CuFeZn | Cu:Fe = 15.9:5.7 |
613 | 3.0 | 33.40 | CO (18%) | 18 |
| CH4 (∼14%) | |||||||
| C2+ parafin (∼49.5%) | |||||||
| C2+ olefin (15.2%) | |||||||
| im-CuFeZn | Cu:Fe = 15.7:5.7 |
613 | 3.0 | 28.60 | CO (26.5%) | 18 | |
| CH4 (∼10%) | |||||||
| C2+ paraffin (∼10%) | |||||||
| C2+ olefin (46.1%) | |||||||
| Cs-Cu0.8Fe1.0Zn1.0 | Cu:Fe = 28.9:35.7 |
603 | 5.0 | 36.60 | C2+OH (19.8%) | 88 | |
| CO (∼20%) | |||||||
| C2+ alkane (∼43%) | |||||||
| 4.6K–Cu–Mg–Zn–Fe | Cu:Fe = 1:0.98 |
593 | 5 | 30.40 | C2+OH (15.7%) | 89 | |
| CO (30.6%) | |||||||
| C2+ alkane (∼30%) | |||||||
| Fe–Cu–K | Cu:Fe = 8.6:69.8 |
573 | 2.5 | 35 | C2–C4 HC (∼26%) | 16 | |
| C5+ HC (50.7%) | |||||||
| C2+ olefin (72.7%) | |||||||
| CuFeO2-24 | Cu:Fe = 1:1 |
573 | 1 | 16.70 | CO (31.4%) | 17 | |
| C5+ HC (44.5%) | |||||||
| HEA | CoNiCuRuPd/TiO2 | Co:Ni:Cu:Ru:Pd = 1:1:1:1:1 |
673 | 0.1 | ∼46 | CH4 (68.3%) | 92 |
| CO (31.7%) | |||||||
| Zr0.5(NiFeCuMnCo)0.5Ox | Zr:Ni:Fe:Cu:Mn:Co = 5:1:1:1:1:1 |
673 | 0.1 | ∼29 | CO (90%) | 93 | |
| Co3MnNiCuZnOx | Cu:Co:Ni = 1:3:1 |
773 | 0.1 | 48 | CO (94%) | 94 | |
Recently, CO2 hydrogenation to methanol with 100% selectivity was achieved on an amorphous ZrO2 surface (Cu1/ZrO2) Cu SAC, as shown by Zhao and colleagues12 (Table 2, entry 3). Not only a high selectivity, the catalyst also exhibits exceptional stability over 100 hours under 453 K reaction conditions (Fig. 5a). The authors employed in situ XANES and SSW-NN global PES exploration to identify the Cu coordination environment and found that the Cu single atom in the catalyst adopts a quasi-planar three-oxygen coordination and a valence state of +1.4, as depicted in Fig. 5b–d. Furthermore, DFT calculations prove that the amorphous ZrO2 plays a key role in stabilizing the Cu single atom. The formation of Cu SACs is exothermic on amorphous ZrO2 but endothermic on ZrO2 flat (111) and terrace (112) surfaces, indicating that the amorphous ZrO2 support is instrumental in maintaining the high stability of the Cu atom. The CO2 hydrogenation on the isolated Cuδ+ cation site follows the CO2 → HCOO → H2COO → H2COOH → H2CO → H3CO → H3COH pathway with the rate-determining step being the HCOO* species hydrogenation (Fig. 5e). The calculated TOF for CO2 hydrogenation to CH3OH on the isolated Cuδ+ cation is approximately 2.89 h−1, consistent with the experimental TOF of 1.37 h−1 and being about two orders of magnitude larger than the CO2 hydrogenation to the CO product (0.03 h−1).
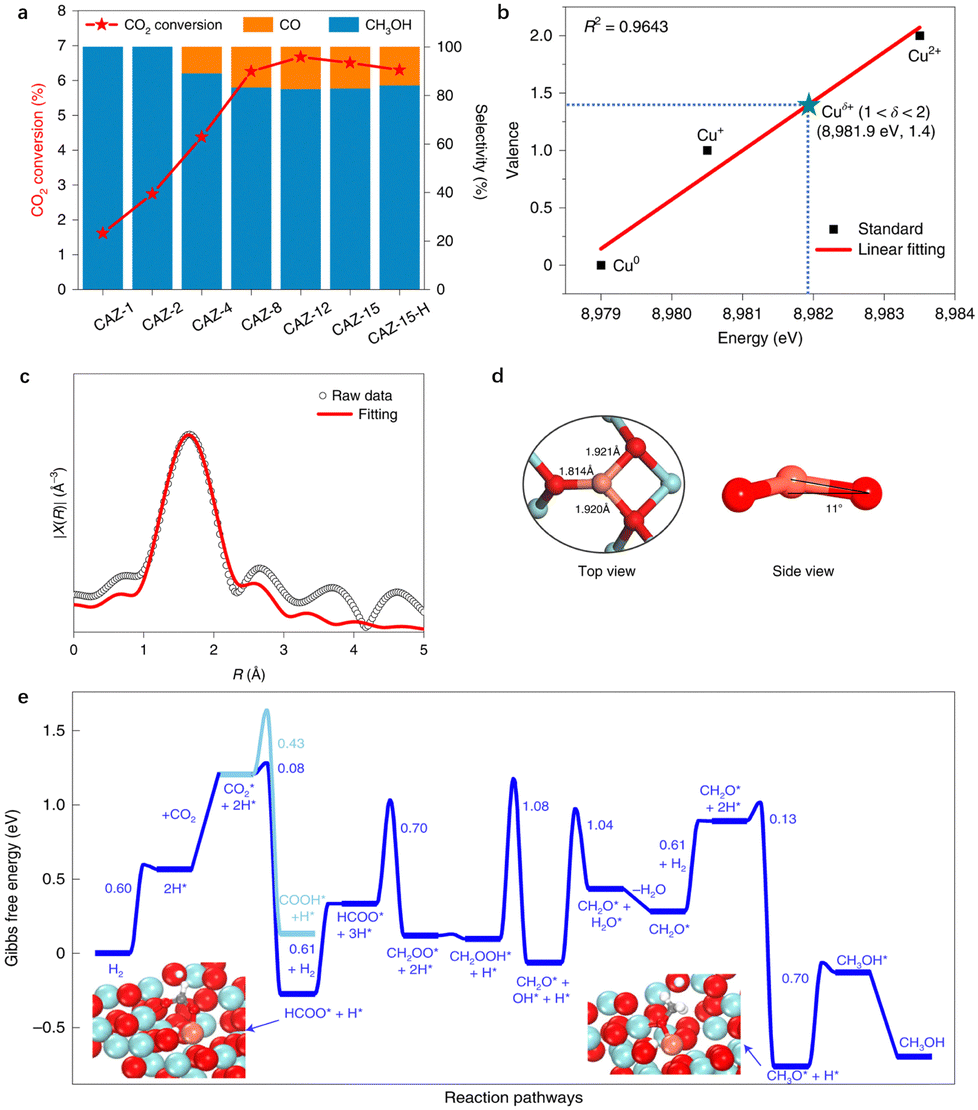 | ||
| Fig. 5 The structure and catalytic performance of CO2 hydrogenation to methanol on Cu SAC supported on the amorphous ZrO2 surface (CAZ-1). (a) The catalytic performance of CO2 conversion and selectivity. (b) The mean chemical valence of Cuδ+ species under in situ test conditions detected using XANES spectra. (c) Fitting of k2-weighted in situ extended X-ray absorption fine structure (EXAFS) data. (d) The identified [CuO3] configuration from SSW-NN simulation. (e) Gibbs free energy profile of CO2 hydrogenation to CH3OH/CO. Reprinted with permission from ref. 12. Copyright 2022 Springer Nature. | ||
By coupling neighboring [CuO3], CO2 hydrogenation can also produce ethanol with high selectivity, as shown by An et al.54 (Table 2, entry 4). They synthesized Cu single atoms at a [CuO3] coordination supported on a Zr12 cluster in a metal–organic framework (MOF). With the assistance of a Cs+ alkali cation, the catalyst exhibited >99% selectivity towards EtOH in tetrahydrofuran solution at 358 K. The proposed mechanism involved the coupling of CHO/CH3O on two cooperative Cu1+ sites for ethanol synthesis.
Compared to the common [CuOx], the [CuNx] and the [CuCx] patterns in Cu SACs are much less reported in the literature. The DFT calculations first reveal the low reaction barriers (<0.9 eV) for CO2 hydrogenation to HCOOH on the [CuNx] pattern (Table 2, entries 5–8).55–59 The energy barrier on the [CuCx] pattern is higher than that on the [CuNx] pattern, but still lower than that on the [CuOx] pattern (Table 2, entry 9).58,59 This suggests that the [CuNx] and [CuCx] sites may exhibit even higher CO2 hydrogenation activity than the [CuOx] sites. Indeed, Yang et al.57 demonstrated the high selectivity and activity of a C3N4-supported Cu SAC with [CuN4] and [CuN3] coordinations for CO2 hydrogenation at low temperatures (<150 °C). The [CuN3] site with the valence state of Cu+1.64 tends to catalyze CO2 hydrogenation to CO, while the [CuN4] site with the valence state of Cu+1.05 favors producing methanol via the formate pathway. This catalyst achieves a methanol selectivity of 95.5% at a CH3OH productivity of 4.2 mmol g−1 h−1 (Table 2, entries 7 and 8), which surpasses that of the state-of-the-art CZA catalyst by 3.2 times (1 mmol g−1 h−1). Overall, the Cu SAC exhibits excellent methanol selectivity in CO2 hydrogenation, but the conversion rate is still too low due to the low concentration of the active site. New synthetic approaches are pursued to precisely disperse Cu single sites and inhibit the subsequent catalyst sintering.
5. Supported/confined Cu nanoclusters
Utilization of metal clusters is a common strategy to enhance the catalytic performance, not only because of the large surface area of metal particles but also due to the availability of low-coordinating surface atoms. For CO2 hydrogenation, theoretical calculations indeed show that Cu4 clusters possess a lower activation barrier of 1.18 eV for formate formation.60 In practice, it is a major concern on how to stabilize these Cu nanoclusters to achieve long-term catalyst stability.Various supports, such as SiO2, Al2O3, ZrO2, and CeO2, have been tested and demonstrated to have good catalytic performances. Newly-formed Cu–O bonds were detected, which leads to the electron transfer between Cu and supports, giving rise to various ionic Cu species, including Cu2+, Cu+, and Cuδ+ that coexist with Cu0 of nanoparticles. It was believed that Cu0 is responsible for dissociating H2, while Cu+ polarizes the C–O bond to promote intermediate conversion. The product selectivity can thus be tuned by adjusting the Cu0/Cu+ ratio.61–63 For example, Yu et al.64 demonstrated that a Cu cluster catalyst supported on inert SiO2 prepared using flame spray pyrolysis, results in five times more Cu+ species than the traditional catalyst made using ammonia evaporation, and increases the methanol selectivity by inhibiting CO desorption and promoting CO* hydrogenation to CH3OH. DFT calculations by Sun et al.65 also showed that CO adsorption on Cu+ sites is stronger than that on Cu0, and CO can be further hydrogenated to CHO* species.
The support may also participate in the reaction cycle by acting as the reaction site for key elementary steps. For the Cu/ZnO catalyst, Valant et al.66 investigated the effect of different Zn contents on CO2 hydrogenation and found that the catalyst with Zn:(Zn + Cu) = 0.62 shows the highest methanol activity of 4.6 mol h−1 kg−1 at 523 K. The adsorbed H amount on ZnO shows a volcano-like profile against the Zn:(Zn + Cu) content, consistent with the activity variation, pointing to the synergetic effect between Cu and ZnO. Song et al. unveiled the remarkable activity (11.3%) and methanol STY (242 gCH3OH kgcat−1 h−1) of 8 wt% Cu/ZnAl2O4 at 220 °C and 3 MPa, far surpassing that of conventional 8 wt% Cu/ZnO/Al2O3 composite oxides (6.8% for CO2 conversion and 144 gCH3OH kgcat−1 h−1 for methanol yield). They believed that the ZnAl2O4 dispersed the Cu cluster with a small particle size of ∼3.2 nm, quite smaller than that on ZnO/Al2O3 (∼6.4 nm). And the higher H2 dissociation ability on ZnAl2O4 strongly enhanced the CO2 conversion and methanol yield.67 The Cu/ZrO2 catalyst has been widely reported with high methanol yield, where ZrO2 can facilitate the formation and transformation of formate, thereby enhancing methanol synthesis.68–71 Especially, the crystalline phase of ZrO2 can significantly affect the catalytic performance. As reported by Tada et al., the active sites on Cu/a-ZrO2 (a: amorphous) with a TOF of 29–39 h−1 were more suitable for CO2-to-methanol than those on Cu/t-ZrO2 (t: tetragonal) with a TOF of 16–23 h−1 and Cu/m-ZrO2 (m: monoclinic) with a TOF of 6–8 h−1.70
Confining Cu nanoclusters in small cages is another approach to stabilizing nanoparticles and achieving high catalytic performance. Zhu et al. have demonstrated that the Cu nanoclusters encapsulated into MOF materials with Zr6 oxide nodes show a high methanol yield. By subtly tuning the Zr–O–Cu interface, it is found that the Zr–O–Cu interface is at least part of the active site that strongly adsorbs CO2. As illustrated in Fig. 6, the binding of CO2 is not energetically favored on the Cu metal particle with slightly positive adsorption energy. The adsorption of CO2 to the O2− or Zr4+–O2− sites of the Zr6O8 node to form a bidentate or tridentate carbonate is considerably stronger, resulting in adsorption energies of −42.7 and −51.0 kJ mol−1, respectively. The adsorption of CO2 at the Cu–Zr4+ interfacial sites is significantly stronger (−80.8 kJ mol−1) than the adsorption on the Cu nanoparticles and ZrO2 nodes.72 Cui et al. also reported a series of zeolite-fixed Cu/ZnOx@Na-ZSM-5 catalysts. The ultrasmall Cu/ZnOx nanoparticles (∼2 nm) in the Na-ZSM-5 zeolite exhibit the space-time yield of methanol of 44.88 gMeOH gCu−1 h−1, much more higher than those of the supported Cu/ZnOx/Na-ZSM-5 catalyst (13.32 gMeOH gCu−1 h) and industrial Cu/ZnO/Al2O3 catalyst (8.46 gMeOH gCu−1 h) under identical conditions. The zeolite was suggested to prevent the separation of Cu–ZnOx interfaces.73 Ding et al. prepared a high-performance Cu@Na-Beta catalyst which shows a high ethanol yield of ∼14% at 300 °C ∼12000 mL gcat−1 h−1, and 2.1 MPa, corresponding to a space-time yield of ∼398 mg gcat−1 h−1. They proposed the reaction key step as the reaction of CO2* with the surface methyl species at step sites of Cu nanoparticles to form CH3COO*, which finally leads to ethanol after the hydrogenation steps.74
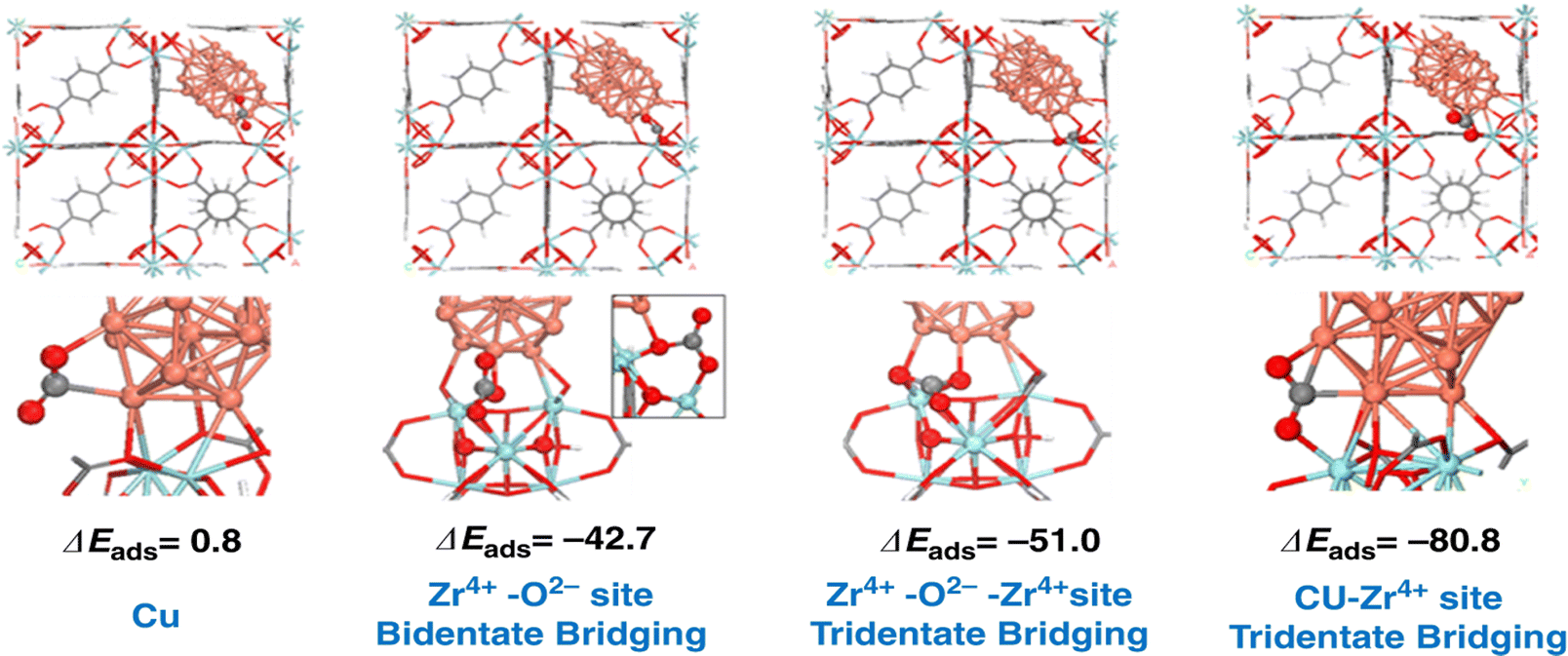 | ||
| Fig. 6 Adsorption energies (ΔEads in kJ mol−1) of CO2 at different Cu/UiO-66-a interface sites. Reprinted with permission from ref. 72. Copyright 2020 Springer Nature. | ||
The supported/confined Cu nanocluster offers convenient ways to access rich chemical environments of Cu, leading to low-coordination Cu, positively charged Cuδ+ and different interface-contacted Cu. While more adjustable variables in synthesis enable the fine tune of the catalytic performance for catalytic reactions such as CO2 hydrogenation, it becomes increasingly difficult to resolve the catalytic active site and understand the catalytic mechanism. The higher yield and the desirable selectivity continue to be the main goals in this area of catalysis research.
6. Bimetallic Cu-based and high entropy alloy catalysts
Introducing other transition metal components into Cu can significantly modify the electronic and geometric structures of the catalyst and thus alter the catalytic performance. Table 3 summarizes the catalytic performance of representative Cu-based bimetallic catalysts and high entropy alloy (HEA) catalysts. At elevated temperatures (mostly 500–700 K) and pressures (mostly 1–5 MPa), the main products of these catalysts span a large range, including not only common CO and CH3OH but also CH4, long-chain hydrocarbon (HC) and even alcohols.CO2 hydrogenation on the Pd–Cu system produces methanol with the CO byproduct.75 Song et al.14,76 investigated an amorphous silica-supported Pd–Cu catalyst with a Pd/(Pd + Cu) ratio of 0.25–0.34 and found that it exhibits twice the methanol selectivity compared to the sum of the selectivities of the two corresponding monometallic catalysts. Further screening of catalyst supports led to an improved methanol selectivity of 44.6% on the Pd–Cu/Ti0.1Zr0.9O2 catalyst.77,78 Catalyst characterization studies by using X-ray diffraction (XRD) and TEM confirm the formation of a Pd–Cu alloy nanoparticle comprising bcc PdCu and fcc PdCu3 phases.14 Nie et al.79 suggested that the catalytically active site is the bcc PdCu(111) surface with unsaturated Pd, as shown in Fig. 7a. The reaction proceeds through a water-assisted formate pathway with an overall Gibbs energy barrier of 1.23 eV, which is more active than the fcc PdCu3(111) surface. Microkinetics simulations show that CO formation from CO2 through the r-WGS pathway is faster than CH3OH synthesis, consistent with the experimental results of a high CO selectivity (>70%).
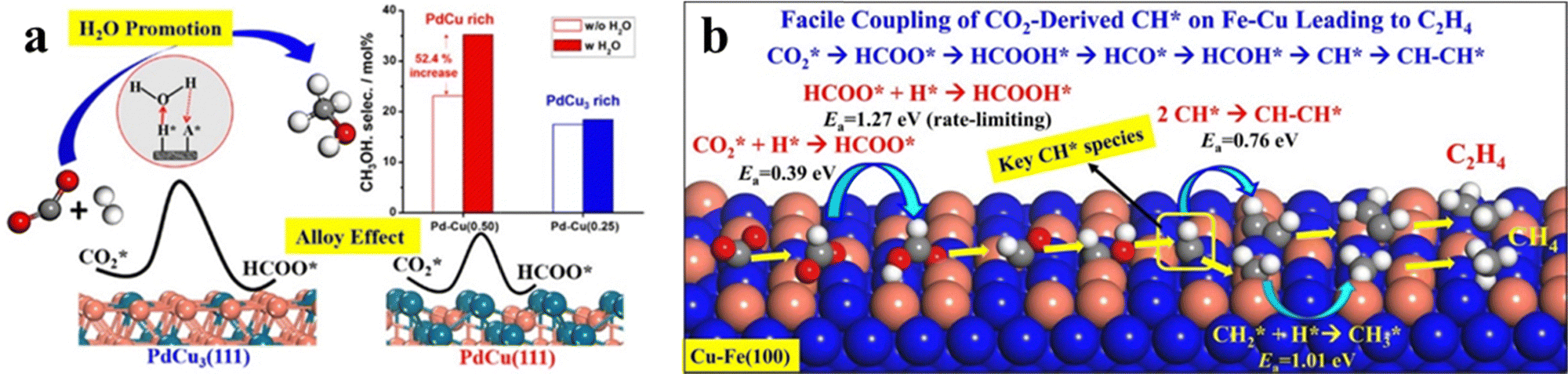 | ||
| Fig. 7 (a) Methanol selectivity of different Pd–Cu bimetallic catalysts with and without H2O. The in situ produced water can accelerate CO2 conversion to methanol. The stepped bcc PdCu(111) active site shows a lower barrier than fcc PdCu3(111) and shows higher methanol selectivity. Reprinted with permission from ref. 79. Copyright 2018 American Chemical Society. (b) Cu-alloyed Fe(100) active sites for the Cu–Fe bimetallic catalyst and its reaction mechanism of CO2 hydrogenation toward C2H4. Reprinted with permission from ref. 90. Copyright 2017 American Chemical Society. | ||
Cu–Ni alloys can catalyze CO2 hydrogenation to CO,80,81 CH4,15 or CH3OH.82–86 Originally, monometallic Ni is known as an active CO2 methanation catalyst. Wang et al.81 demonstrated that a Cu–Ni catalyst synthesized through a hydrothermal method (with a Cu/Ni ratio of 4.14) exhibits a high selectivity (>99.9%) for CO production during the r-WGS reaction at 1 bar pressure, due to the high dispersion of Ni on the Cu–Ni alloy as detected by high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Indeed, DFT calculations confirmed that the H-assisted CO2 dissociation has a lower energy barrier of 1.30 eV on Ni-doped Cu(111) compared with plain Cu(111). Tan et al.84,86 synthesized a Cu–Ni alloy on CeO2-nanotubes and achieved CO2 hydrogenation to CH3OH with a maximum CH3OH space-time yield at a Cu:Ni ratio of 2 and a CH3OH selectivity of 76%. Summa et al.15 used a Cu promoted Ni–Mg–Al hydrotalcite-derived catalyst with a low loading of 1.4 wt% for CO2 hydrogenation to methane, achieving selectivity above 98% and CO2 conversion up to 86%. XRD and XANES suggest the formation of the Cu–Ni alloy during the reduction step. Furthermore, compared with the Cu-free catalyst, promotion with Cu strongly increased the number of medium-strength basic sites revealed by CO2 temperature programmed desorption, which is crucial for methanation.
Catalysts with Cu and Fe can lead to a distribution of multi-carbon products in CO2 hydrogenation. Fe alone serves as a famous Fischer–Tropsch catalyst that converts CO to hydrocarbon, and its combination with Cu allows direct conversion of CO2 to a multi-carbon product.17,87–89 Their product and selectivity are listed in Table 3. For example, Si et al.18 developed an FeCu nano-alloy catalyst supported on ZnO by a sputtering method (sp-CuFeZn), which demonstrated a high paraffin selectivity of 63.5% (containing ∼14% of CH4). In situ XRD reveals that in sp-CuFeZn the metal mainly exists as an FeCu4 nanoalloy under the reaction conditions. Hwang et al.16 synthesized an Fe–Cu–K catalyst that exhibited superior selectivity towards liquid hydrocarbons (50.7% toward C5+ hydrocarbons and 72.7% toward olefin), with a 1.7-fold increase in CO2 conversion and higher chain growth probability compared to the Fe–K catalyst. The observed phases include the Cu–Fe alloy, Fe3O4, and a small amount of iron carbide (Fe5C2) after the reaction.
Theoretical calculations have been utilized to provide insights into the activity of the Cu–Fe alloy. Nie et al.90,91 utilized Fe(100) and Cu-alloyed Fe(100) to investigate the reaction mechanism of CO2 hydrogenation towards C1 and C2 hydrocarbons. On the Cu-alloyed Fe(100) surface, the adsorption strength of CO2 decreases as the surface Cu coverage increases, increasing the CO2 dissociation barrier.91 At medium Cu coverage (θCu = 0.4 ML), the CO2 hydrogenation to HCOO* occurs with a low hydrogenation barrier of 0.39 eV. The HCOO* species then undergoes a series of steps, including HCOO* → HCOOH* → HCO* → HCOH* → CH* → CHCH* → CH2CH* → CH2CH2*, to produce ethylene with a low energy barrier of 1.27 eV (Fig. 7b).90 Since these theoretical studies generally utilize single-crystal surfaces, the active site of Cu–Fe catalysts requires further detailed characterization.
While HEA emerges as a new direction in catalysis, several Cu-based HEA catalysts have also been developed for CO2 hydrogenation. Mori et al.92 synthesised a TiO2 supported CoNiCuRuPd nanoparticle catalyst, which is found to be both active and extremely durable during CO2 hydrogenation. The main product leads to CH4 (selectivity 68.3%) and CO (31.7%) at 673K, 1 bar with a conversion of ∼46%, which retained 96% of its original activity after 72 h. Hou et al.93 reported a high entropy cubic Zr0.5(NiFeCuMnCo)0.5Ox, where the configuration entropy leads to observation of in situ reversible exsolution–dissolution of supported metallic species in multi redox cycles. In CO2 hydrogenation, the CO2 conversion is 29% and CO selectivity is over 90%, with no obvious activity loss during the 500 h reaction, affording ultrahigh thermal stability. It is worth noting that although the HEA catalysts do not show the desirable selectivity to high-value products (other than CO and CH4), their ultrahigh thermal stability in long-term CO2 hydrogenation may provide useful hints to improve catalyst durability in general. As experimental reports of HEA's applications in CO2 hydrogenation are limited, more research work is needed to identify how Cu cooperates with other metals during the HEA catalysis.92,94
In general, Cu-based multi-metallic alloys feature the ability of making diverse products, but the activity and selectivity are still not satisfactory to meet the requirement of the industrial application. To optimize the catalyst performance, more fundamental research studies are required for probing the active site structure and resolving the reaction intermediates during CO2 hydrogenation.
7. Perspectives
This perspective provides an overview of the current understanding of active sites in copper-based catalysts for CO2 hydrogenation, including commercial CZA, Cu SACs, supported/confined Cu clusters, and Cu-based alloy catalysts. Generally speaking, despite the variation of the valence state and the coordination environment of Cu, the CO2 hydrogenation main product of a mono-metallic Cu catalyst, e.g. CZA, Cu SACs and supported/confined Cu clusters, is either CO or methanol. Once the second metal is introduced to form a Cu–X alloy catalyst, the CO2 hydrogenation product can switch to long-chain hydrocarbons such as alkanes and olefins. Therefore, how to improve the catalysts’ selectivity and long-term stability is a general concern in research, which requires a deep understanding of the active sites and reaction mechanisms. There are still many challenges in the field and are highlighted as follows.The SMSI effect observed during CZA catalyst activation is long believed to be the key reason for creating the active site and resulting in a high activity of the CZA catalyst. There is growing interest in the in situ atomic structure of ZnOxHy on Cu surfaces. As mentioned in Section 3, the traditional techniques of structure characterization face great difficulties due to the essentiality of reaction conditions (H2/CO pressure) and the great complexity of the potential energy surface.
The state-of-the-art machine-learning atomic simulations emerge as a promising approach to determining the overlayer structure. Recently, we utilized the G-NN potential based grand canonical global search method, namely automatic search of optimal phase95 (ASOP) to explore the stable ZnxOyHz/Cu(111) structure.25Fig. 8a and b plot the PES of ZnxOyHz/Cu(111) under typical reaction conditions (500 K, pH2 = 40 bar, pCO = pCO2 = 10 bar), where the x- and y-axes are the coverage of O and Zn/H and the contour is the free energy of the minimum from G-NN. The figure contains 1751 minimum structures at different periodic supercells of Cu(111) (ncell = 4–12) with varied ZnxOyHz composition (θZn = x/ncell = 0–1, the same for θO and θH). This shows that besides clean Cu and CuZn surface alloys (where θO ≈ 0), a stable region is located where the coverage is θZn:θO:θH ≈ 1:1:1. After further validation by DFT for low-lying minima, a number of energetically nearly degenerate most stable structures are identified. The most stable one (GM) is (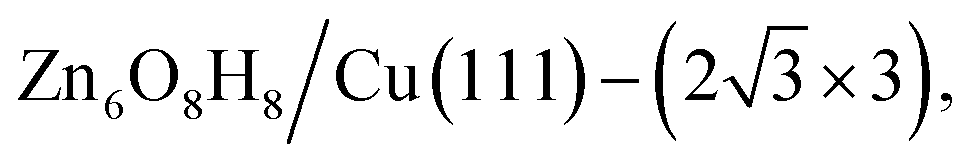 ) whose free energy is −0.043 eV per Cu(111) unit cell, being more stable than clean Cu(111) and bulk ZnO. Many other structures with a similar energy are available, including
) whose free energy is −0.043 eV per Cu(111) unit cell, being more stable than clean Cu(111) and bulk ZnO. Many other structures with a similar energy are available, including 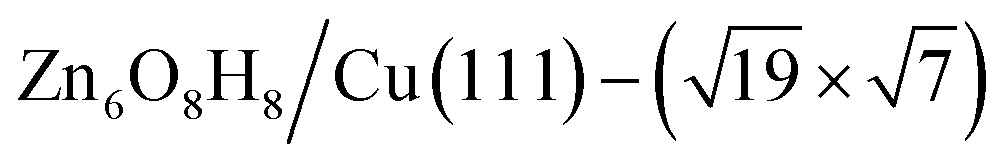 (denoted as I, free energy −0.038 eV per unit cell),
(denoted as I, free energy −0.038 eV per unit cell), 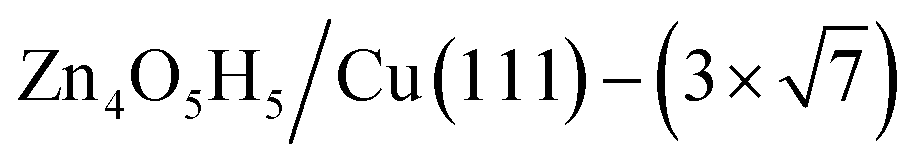 (denoted as II, free energy −0.029 eV per unit cell), as well as
(denoted as II, free energy −0.029 eV per unit cell), as well as 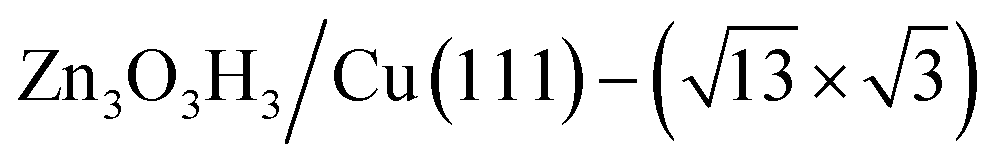 (−0.042 eV per unit cell). Apparently, these structures share a similar [–Zn–OH–Zn–] repeating unit and the multiple degenerate structures suggest the amorphous nature of the ZnOxHy overlayer on the Cu(111) surface (Fig. 8c). Further work is required to clarify the ZnOxHy structures on the stepped Cu surfaces and determine the CO2 hydrogenation mechanism over these overlayer structures.
(−0.042 eV per unit cell). Apparently, these structures share a similar [–Zn–OH–Zn–] repeating unit and the multiple degenerate structures suggest the amorphous nature of the ZnOxHy overlayer on the Cu(111) surface (Fig. 8c). Further work is required to clarify the ZnOxHy structures on the stepped Cu surfaces and determine the CO2 hydrogenation mechanism over these overlayer structures.
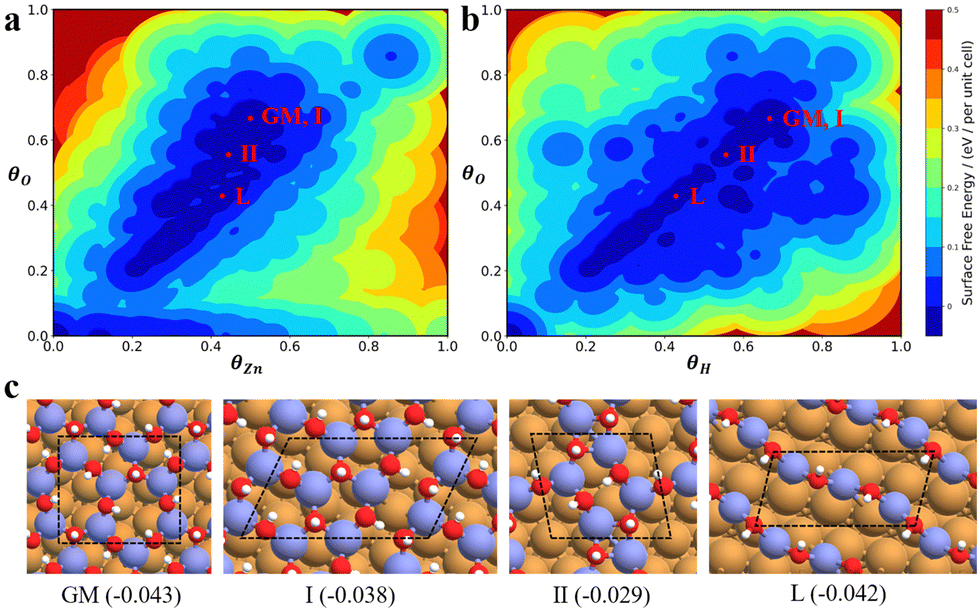 | ||
Fig. 8 The PES contour map for ZnxOyHz/Cu(111) from ASOP simulation, using (a) coverage of O and Zn as axes, or (b) coverage of O and H as the axes. (c) The identified stable ZnxOyHz/Cu(111) structures. From left to right, GM: 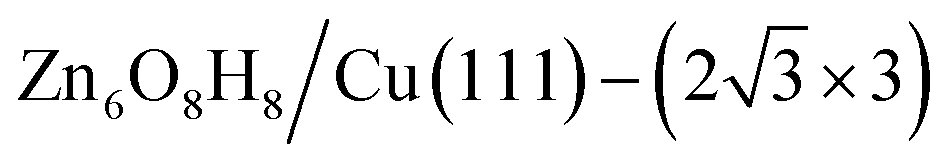 , I: , I: 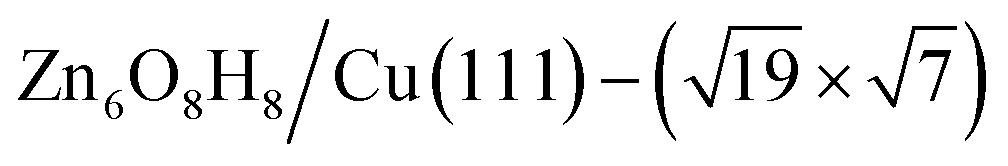 , II: , II: 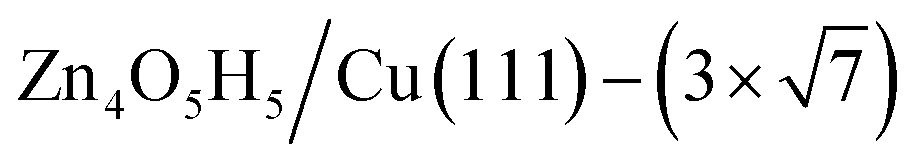 , L: , L: 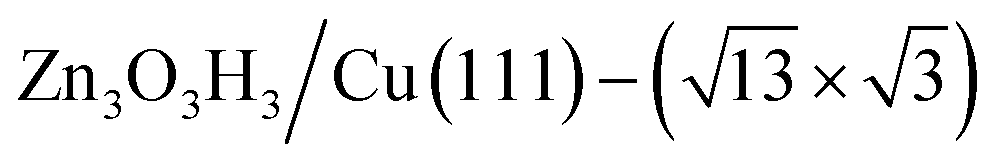 . The color scheme for atoms is as follows: H: white; O: red; Cu: yellow; and Zn: blue. . The color scheme for atoms is as follows: H: white; O: red; Cu: yellow; and Zn: blue. | ||
In addition, the role of the Al2O3 component in the CZA catalyst needs to be revisited. While Al2O3 was traditionally viewed as a support material, it may still act as an active component in methanol synthesis. In particular, the presence of the ZnAl2O4 spinel has been mentioned in recent literature as a factor for promoting CO hydrogenation.96,97
For the Cu SAC catalyst, the low conversion rate (for example, <2%12,53) limits its practical applications, which calls for increasing the metal loading and the concentration of active Cu SAC sites. Moreover, as the [CuNx] active sites have shown lower reaction barriers, it is fascinating to increase the concentration of [CuNx] active sites by immobilizing Cu SACs on nitrides such as GaN or Ta2N3, which may achieve better catalysis performance for CO2 hydrogenation than [CuOx] and [CuCx] active sites.
For the supported/confined Cu cluster catalyst, optimizing the Cu-oxide interface is an effective approach to improving the activity of CO2 hydrogenation. Recent studies have shown that the inverse oxide/metal configuration can enhance interfacial reactivity in well-defined CeOx/Cu(111), ZnO/Cu(111), and ZrOx/Cu models used as catalysts for the water–gas shift and methanol synthesis reactions.98–100 In these models, the oxide species are grown as a thin layer on top of the copper surface, creating an interface that promotes catalytic activity by improving the electron transfer and adsorption of CO2 and H2 molecules. The active site structures for these inverse catalysts are however unclear.
Overcoming the above challenges in the future should benefit greatly from the rapid progress of machine learning methodology and artificial intelligence (AI) applications to catalysis. First, machine learning atomic simulations that can speed up significantly the complex PES exploration are powerful new tools to identify unknown structures,72 and to search for unknown reactions,25,101 as practiced by our group using SSW-NN simulations in recent years. By combining these two features of SSW-NN, it is possible to resolve the active site centers using first principles. Second, machine learning techniques hold great promise to guide the design of new materials/catalysts. A high-throughput screening scheme can be built via a machine learning database of materials with essential catalytic properties and then utilized to predict the activity. Recent years have seen efforts to establish a database for key catalytic properties,102 such as the adsorption energy of key intermediates, the morphology and acidity of zeolites.103 These approaches can be applied in the design of HEA (Cu, Co, Ni, Zn, and Sn) catalysts for CO2 hydrogenation.104,105
We note that electrochemical CO2 hydrogenation has attracted more and more attention in recent years, which is not the focus of the current perspective, and there has been significant and interesting progress. For example, the electrochemical C–N coupling of CO2 and nitrogenous small molecules (e.g. NO and NO2) can simultaneously eliminate greenhouse gas emissions and environmental pollutants;106 the CO2 electrochemical reduction on Cu-based catalysts can produce other high-value products (e.g. ethylene or ethanol) under ambient conditions.107,108 These new reaction routes together with the thermal approach of CO2 hydrogenation are going to be the key catalysis hotspots towards greener and more sustainable future.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work received financial support from the National Natural Science Foundation of China (12188101, 22033003, and 22203101), the Tencent Foundation for XPLORER PRIZE, Youth Innovation Promotion Association CAS (No. 2023265), Science & Technology Commission of Shanghai Municipality (23ZR1476100) and the Fundamental Research Funds for the Central Universities (20720220011).References
- ICI LTD, UK Pat., GB1159035A, 1969.
- ICI LTD, US Pat., US3326956A, 1967.
- Global methanol production 2022, https://www.statista.com/statistics/1323406/methanol-production-worldwide/, (accessed 25 June 2023).
- G. Ertl and D. R. Burgess, Handbook of heterogeneous catalysis, VCH, Weinheim, 1997, p. 1856 Search PubMed.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- K. Klier, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1982, vol. 31, pp. 243–313 Search PubMed.
- M. M. Günter, T. Ressler, B. Bems, C. Büscher, T. Genger, O. Hinrichsen, M. Muhler and R. Schlögl, Catal. Lett., 2001, 71, 37–44 CrossRef.
- J. C. Frost, Nature, 1988, 334, 577–580 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- T. Fujitani, I. Nakamura, T. Uchijima and J. Nakamura, Surf. Sci., 1997, 383, 285–298 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- Y. Chai, B. Qin, B. Li, W. Dai, G. Wu, N. Guan and L. Li, Natl. Sci. Rev., 2023, 10, nwad043 CrossRef PubMed.
- X. Jiang, N. Koizumi, X. Guo and C. Song, Appl. Catal., B, 2015, 170–171, 173–185 CrossRef CAS.
- P. Summa, B. Samojeden, M. Motak, D. Wierzbicki, I. Alxneit, K. Świerczek and P. Da Costa, Catal. Today, 2022, 384–386, 133–145 CrossRef CAS.
- S.-M. Hwang, S. J. Han, J. E. Min, H.-G. Park, K.-W. Jun and S. K. Kim, J. CO2 Util., 2019, 34, 522–532 CrossRef CAS.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- Z. Si, C. C. Amoo, Y. Han, J. Wei, J. Yu, Q. Ge and J. Sun, J. Energy Chem., 2022, 70, 162–173 CrossRef CAS.
- D. R. Burgess Jr., in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 2021 Search PubMed.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- M. D. Higham, M. G. Quesne and C. R. A. Catlow, Dalton Trans., 2020, 49, 8478–8497 RSC.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- Y.-F. Shi, P.-L. Kang, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2022, 144, 13401–13414 CrossRef CAS PubMed.
- P. Wu and B. Yang, ACS Catal., 2017, 7, 7187–7195 CrossRef CAS.
- X. Sun, P. Wang, Z. Shao, X. Cao and P. Hu, Sci. China: Chem., 2019, 62, 1686–1697 CrossRef CAS.
- Y. Yang, D. Mei, C. H. F. Peden, C. T. Campbell and C. A. Mims, ACS Catal., 2015, 5, 7328–7337 CrossRef CAS.
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Appl. Catal., B, 2021, 291, 120073 CrossRef CAS.
- L. Ma, W. Zhao, B. Wang, L. Ling and R. Zhang, Fuel, 2022, 313, 122686 CrossRef CAS.
- W. Liao and P. Liu, ACS Catal., 2020, 10, 5723–5733 CrossRef CAS.
- X. Zhang, J. X. Liu, B. Zijlstra, I. A. W. Filot, Z. Y. Zhou, S. G. Sun and E. J. M. Hensen, Nano Energy, 2018, 43, 200–209 CrossRef CAS.
- D. Sheldon, Johns. Matthey Technol. Rev., 2017, 61, 172–182 CrossRef CAS.
- S. S. Iyer, T. Renganathan, S. Pushpavanam, M. Vasudeva Kumar and N. Kaisare, J. CO2 Util., 2015, 10, 95–104 CrossRef CAS.
- K. Girod, H. Lohmann, S. Schlüter and S. Kaluza, Processes, 2020, 8, 1673 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjaer, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- P. Amann, B. Klötzer, D. Degerman, N. Köpfle, T. Götsch, P. Lömker, C. Rameshan, K. Ploner, D. Bikaljevic, H.-Y. Wang, M. Soldemo, M. Shipilin, C. M. Goodwin, J. Gladh, J. Halldin Stenlid, M. Börner, C. Schlueter and A. Nilsson, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- A. Beck, M. Zabilskiy, M. A. Newton, O. Safonova, M. G. Willinger and J. A. van Bokhoven, Nat. Catal., 2021, 4, 488–497 CrossRef CAS.
- R. Dalebout, L. Barberis, G. Totarella, S. J. Turner, C. La Fontaine, F. M. F. de Groot, X. Carrier, A. M. J. van der Eerden, F. Meirer and P. E. de Jongh, ACS Catal., 2022, 12, 6628–6639 CrossRef CAS PubMed.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., 2014, 126, 6051–6055 CrossRef.
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- S. Wang, M. Jian, H. Su and W. Li, Chin. J. Chem. Phys., 2018, 31, 284–290 CAS.
- W. Janse van Rensburg, M. A. Petersen, M. S. Datt, J.-A. van den Berg and P. van Helden, Catal. Lett., 2015, 145, 559–568 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CAS.
- T. Lunkenbein, F. Girgsdies, T. Kandemir, N. Thomas, M. Behrens, R. Schlögl and E. Frei, Angew. Chem., 2016, 128, 12900–12904 CrossRef.
- D. Li, F. Xu, X. Tang, S. Dai, T. Pu, X. Liu, P. Tian, F. Xuan, Z. Xu, I. E. Wachs and M. Zhu, Nat. Catal., 2022, 5, 99–108 CrossRef CAS.
- D. Laudenschleger, H. Ruland and M. Muhler, Nat. Commun., 2020, 11, 3898 CrossRef PubMed.
- Y. Xu, Z. Dai, Y. Ding and L. Zhang, J. Chem. Phys., 2022, 157, 221101 CrossRef CAS PubMed.
- K. Mondal, Megha, A. Banerjee, A. Fortunelli, M. Walter and M. Moseler, J. Phys. Chem. C, 2022, 126, 764–771 CrossRef CAS.
- X.-K. Wu, G.-J. Xia, Z. Huang, D. K. Rai, H. Zhao, J. Zhang, J. Yun and Y.-G. Wang, Appl. Surf. Sci., 2020, 525, 146481 CrossRef CAS.
- T. Reichenbach, K. Mondal, M. Jäger, T. Vent-Schmidt, D. Himmel, V. Dybbert, A. Bruix, I. Krossing, M. Walter and M. Moseler, J. Catal., 2018, 360, 168–174 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- W. Wu, Y. Wang, L. Luo, M. Wang, Z. Li, Y. Chen, Z. Wang, J. Chai, Z. Cen, Y. Shi, J. Zhao, J. Zeng and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202213024 CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- J. Ma, H. Gong, T. Zhang, H. Yu, R. Zhang, Z. Liu, G. Yang, H. Sun, S. Tang and Y. Qiu, Appl. Surf. Sci., 2019, 488, 1–9 CrossRef CAS.
- K. Homlamai, T. Maihom, S. Choomwattana, M. Sawangphruk and J. Limtrakul, Appl. Surf. Sci., 2020, 499, 143928 CrossRef CAS.
- T. Yang, X. Mao, Y. Zhang, X. Wu, L. Wang, M. Chu, C.-W. Pao, S. Yang, Y. Xu and X. Huang, Nat. Commun., 2021, 12, 6022 CrossRef CAS PubMed.
- D. N. Sredojević, Ž. Šljivančanin, E. N. Brothers and M. R. Belić, ChemistrySelect, 2018, 3, 2631–2637 CrossRef.
- S. Ali, R. Iqbal, A. Khan, S. U. Rehman, M. Haneef and L. C. Yin, ACS Appl. Nano Mater., 2021, 4, 6893–6902 CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CAS.
- J. Zhang, X. Sun, C. Wu, W. Hang, X. Hu, D. Qiao and B. Yan, J. Energy Chem., 2023, 77, 45–53 CrossRef CAS.
- Y. Gu, C. Han, J. Huang, V. A. Vinokurov and W. Huang, Fuel, 2022, 322, 124111 CrossRef CAS.
- J. Yang, N. Gong, L. Wang, Y. Wu, T. Zhang, H. Xie, G. Yang and Y. Tan, New J. Chem., 2021, 45, 20832–20839 RSC.
- J. F. Yu, M. Yang, J. X. Zhang, Q. J. Ge, A. Zimina, T. Pruessmann, L. Zheng, J. D. Grunwaldt and J. Sun, ACS Catal., 2020, 10, 14694–14706 CrossRef CAS.
- L. Sun, J. Han, Q. Ge, X. Zhu and H. Wang, RSC Adv., 2022, 12, 19394–19401 RSC.
- A. Le Valant, C. Comminges, C. Tisseraud, C. Canaff, L. Pinard and Y. Pouilloux, J. Catal., 2015, 324, 41–49 CrossRef CAS.
- L. Song, H. Wang, S. Wang and Z. Qu, Appl. Catal., B, 2023, 322, 122137 CrossRef CAS.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Mol. Catal. A: Chem., 2011, 345, 60–68 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- S. Tada, S. Kayamori, T. Honma, H. Kamei, A. Nariyuki, K. Kon, T. Toyao, K. Shimizu and S. Satokawa, ACS Catal., 2018, 8, 7809–7819 CrossRef CAS.
- K. Samson, M. Śliwa, R. P. Socha, K. Góra-Marek, D. Mucha, D. Rutkowska-Zbik, J.-F. Paul, M. Ruggiero-Mikołajczyk, R. Grabowski and J. Słoczyński, ACS Catal., 2014, 4, 3730–3741 CrossRef CAS.
- Y. Zhu, J. Zheng, J. Ye, Y. Cui, K. Koh, L. Kovarik, D. M. Camaioni, J. L. Fulton, D. G. Truhlar, M. Neurock, C. J. Cramer, O. Y. Gutiérrez and J. A. Lercher, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- W.-G. Cui, Y.-T. Li, L. Yu, H. Zhang and T.-L. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 18693–18703 CrossRef CAS PubMed.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
- E. J. Choi, Y. H. Lee, D.-W. Lee, D.-J. Moon and K.-Y. Lee, Mol. Catal., 2017, 434, 146–153 CrossRef CAS.
- X. Jiang, X. Wang, X. Nie, N. Koizumi, X. Guo and C. Song, Catal. Today, 2018, 316, 62–70 CrossRef CAS.
- F. Lin, X. Jiang, N. Boreriboon, Z. Wang, C. Song and K. Cen, Appl. Catal., A, 2019, 585, 117210 CrossRef.
- F. Lin, X. Jiang, N. Boreriboon, C. Song, Z. Wang and K. Cen, Catal. Today, 2021, 371, 150–161 CrossRef CAS.
- X. Nie, X. Jiang, H. Wang, W. Luo, M. J. Janik, Y. Chen, X. Guo and C. Song, ACS Catal., 2018, 8, 4873–4892 CrossRef CAS.
- K. P. Reddy, D. Kim, S. Hong, K.-J. Kim, R. Ryoo and J. Y. Park, ACS Appl. Mater. Interfaces, 2023, 15, 9373–9381 CrossRef CAS PubMed.
- L.-X. Wang, E. Guan, Z. Wang, L. Wang, Z. Gong, Y. Cui, Z. Yang, C. Wang, J. Zhang, X. Meng, P. Hu, X.-Q. Gong, B. C. Gates and F.-S. Xiao, ACS Catal., 2020, 10, 9261–9270 CrossRef CAS.
- C. Wang, Y. Fang, G. Liang, X. Lv, H. Duan, Y. Li, D. Chen and M. Long, J. CO2 Util., 2021, 49, 101542 CrossRef CAS.
- F. Zhao, M. Gong, Y. Zhang and J. Li, J. Porous Mater., 2016, 23, 733–740 CAS.
- Q. Tan, Z. Shi and D. Wu, Ind. Eng. Chem. Res., 2018, 57, 10148–10158 CrossRef CAS.
- F. Zhao, M. Gong, K. Cao, Y. Zhang, J. Li and R. Chen, ChemCatChem, 2017, 9, 3772–3778 CAS.
- Q. Tan, Z. Shi and D. Wu, Int. J. Energy Res., 2019, 43, 5392–5404 CAS.
- J. Liu, A. Zhang, X. Jiang, M. Liu, Y. Sun, C. Song and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 10182–10190 CrossRef CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CrossRef CAS.
- D. Xu, M. Ding, X. Hong and G. Liu, ACS Catal., 2020, 10, 14516–14526 CrossRef CAS.
- X. Nie, H. Wang, M. J. Janik, Y. Chen, X. Guo and C. Song, J. Phys. Chem. C, 2017, 121, 13164–13174 CAS.
- X. Nie, H. Wang, M. J. Janik, X. Guo and C. Song, J. Phys. Chem. C, 2016, 120, 9364–9373 CAS.
- K. Mori, N. Hashimoto, N. Kamiuchi, H. Yoshida, H. Kobayashi and H. Yamashita, Nat. Commun., 2021, 12, 3884 CrossRef CAS PubMed.
- S. Hou, X. Ma, Y. Shu, J. Bao, Q. Zhang, M. Chen, P. Zhang and S. Dai, Nat. Commun., 2021, 12, 5917 CrossRef CAS PubMed.
- J. Zhao, J. Bao, S. Yang, Q. Niu, R. Xie, Q. Zhang, M. Chen, P. Zhang and S. Dai, ACS Catal., 2021, 11, 12247–12257 CrossRef CAS.
- D. Chen, C. Shang and Z.-P. Liu, J. Chem. Phys., 2022, 156, 094104 CrossRef CAS PubMed.
- J. Su, L. Zhang, H. Zhou, Y. Ye, X. Zheng, C. Liu, S. Liu, W. Jiao, X. Liu, C. Wang, Y. Wang and Z. Xie, ACS Catal., 2023, 13, 2472–2481 CrossRef CAS.
- W.-D. Hu, C.-M. Wang, Y.-D. Wang, J. Ke, G. Yang, Y.-J. Du and W.-M. Yang, Appl. Surf. Sci., 2021, 569, 151064 CrossRef CAS.
- J. A. Rodriguez, J. Graciani, J. Evans, J. B. Park, F. Yang, D. Stacchiola, S. D. Senanayake, S. Ma, M. Pérez, P. Liu, J. F. Sanz and J. Hrbek, Angew. Chem., Int. Ed., 2009, 48, 8047–8050 CrossRef CAS PubMed.
- S. D. Senanayake, D. Stacchiola and J. A. Rodriguez, Acc. Chem. Res., 2013, 46, 1702–1711 CrossRef CAS PubMed.
- C. Wu, L. Lin, J. Liu, J. Zhang, F. Zhang, T. Zhou, N. Rui, S. Yao, Y. Deng, F. Yang, W. Xu, J. Luo, Y. Zhao, B. Yan, X.-D. Wen, J. A. Rodriguez and D. Ma, Nat. Commun., 2020, 11, 5767 CrossRef CAS PubMed.
- P.-L. Kang, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2019, 141, 20525–20536 CrossRef CAS PubMed.
- A. Kolluru, M. Shuaibi, A. Palizhati, N. Shoghi, A. Das, B. Wood, C. L. Zitnick, J. R. Kitchin and Z. W. Ulissi, ACS Catal., 2022, 12, 8572–8581 CrossRef CAS.
- S. Ma and Z.-P. Liu, Chem. Sci., 2022, 13, 5055–5068 RSC.
- D. Roy, S. C. Mandal and B. Pathak, J. Phys. Chem. Lett., 2022, 13, 5991–6002 CrossRef CAS PubMed.
- D. Roy, S. C. Mandal and B. Pathak, ACS Appl. Mater. Interfaces, 2021, 13, 56151–56163 CrossRef CAS PubMed.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
| This journal is © The Royal Society of Chemistry 2023 |