
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroreforming injects a new life into solid waste
Yingxin
Ma
a,
Yu
Zhang
 *b,
Wenfang
Yuan
a,
Mengmeng
Du
a,
Sailei
Kang
a and
Bocheng
Qiu
*a
*b,
Wenfang
Yuan
a,
Mengmeng
Du
a,
Sailei
Kang
a and
Bocheng
Qiu
*a
aDepartment of Chemistry, College of Sciences, Nanjing Agricultural University, Nanjing 210095, China. E-mail: bochengqiu@njau.edu.cn
bDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China. E-mail: yu_zhang1225@163.com
First published on 29th August 2023
Abstract
The drive to upgrade the system capacity for renewable electricity, coupled with relieving our reliance on the finite fossil resources, promotes the exploration for economically competitive and environmentally friendly technologies that can steer the conversion of the renewable feedstocks into fuels, chemicals, and materials. An appealing remedy is to utilize ubiquitous solid waste (e.g., biomass and plastics) as platform precursors to synthesize valuable chemicals used globally on a daily basis. Although the defined functionality of biomass differs from that of plastics, they share considerable structural similarities in terms of the polymeric nature and the type of bonds connecting the constituent monomers, thereby establishing an intimate correlation between their valorization routes. Electroreforming methodology towards upgrading of biomass and plastic wastes into commodity chemicals coupled with hydrogen evolution is thus viable and meanwhile remains intriguing. In this review, we draw parallels between electrochemical valorization of biomass and plastics, with a focus on elucidating the state-of-the-art catalysts for each documented reaction and evaluating their corresponding techno-economy. In parallel, the pretreatment methodologies for raw solid waste and the progress in computational simulations and operando spectroscopies are reviewed in detail. We conclude with a comprehensive discussion of the emerging challenges for catalyst and reactor optimization, large-scale operation, and technology flexibility and compatibility.
Broader contextThe relentless growth of biomass and plastic solid waste in the environment is causing an ecological crisis, which promotes the exploration of new approaches to reclaim the value of biomass and plastics. Electroreforming driven by renewable electricity has inherent abilities to initiate redox via a small electric potential difference between electrodes and to reduce negative impacts of solid waste on the environment, thereby circumventing the harsh conditions required to overcome the energy barrier using thermal inputs. This review draws similarities between electrocatalysis on biomass and plastics as both substrates are of low-cost and recalcitrant, and meanwhile highlights the innovation in catalyst design and mechanism investigation of solid waste deconstruction and upcycling. By side-by-side comparisons, we reveal the strengths and limitations of electroreforming of biomass and plastic waste streams and how lessons drawn from previous literature can be applied to improve biomass and plastic reclamation. Taken together, we aim to reduce the severe global burden of biomass and plastics of the ecosystem and further investigate the impact of electroreforming on a carbon-neutral future. |
1. Introduction
Rapid population explosion and global economic growth over the last few decades have elicited a conceivable discrepancy between energy supply and demand.1–4 Fossil fuel resources, including coal, crude oil, and natural gas, have accounted for a large fraction of global energy demand and their excessive depletion contributes to the increasing levels of carbon footprint, as witnessed by the improved frequency and intensity of climate catastrophes over the past decade.5–8 In this context, how to develop a global-scale sustainable energy supply system, while imposing no net CO2 emission remains a huge challenge faced by humanity today. Currently, new insights are gained to diversify our energy sources and reduce our dependence on fossil fuels by turning to renewable electricity resources (e.g., solar, tidal, wind, and hydroelectric energy), which play an increasingly pivotal role in decarbonization of the industrial and agricultural sectors.9–13 Furthermore, their production methods are environmentally friendly and economically competitive with conventional power evolution.14,15 However, their temporal fluctuations and spatial separation of source and sink enable a contribution to global energy supply of less than 10%, thereby inviting the exploration of feasible storage and conversion technologies.16 Hydrogen, a fuel extensively utilized in the petroleum, chemical, and fertilizer industries, has been frequently considered as a promising candidate to bridge the gap between petrochemical resources and intermittent renewable energies.17–20 At present, hydrogen as an energy carrier is mainly produced using carbon-intensive energy technologies (i.e., natural gas steam reforming and coal gasification are also known methods for producing gray and black hydrogen, respectively). In spite of their low-cost and well-developed features, they are CO2 emitting processes (830 Mt CO2 produced annually) and suffer from high temperature requirements and a large energy input.21–23 In other words, hydrogen fuel is environmentally friendly only when utilizing low-carbon power technologies for its generation. To this end, benign-by-design techniques with an emphasis on instinctively attaining environmental sustainability have been developed to realize H2 generation in a cost-effective manner.24Electrochemical water splitting with net-zero greenhouse gas emission provides a way to harness intermittent renewable energies to produce green hydrogen, which thus offers a prospect for the widespread deployment of H2 during the global energy revolution.25–27 Moreover, one of the benefits of industrial-scale water electrolysis is that hydrogen obtained from water electrolysis has a high purity (>99.9%), thereby avoiding further purification treatment.28,29 Thermodynamically, water electrolysis requires a voltage of 1.23 V to drive the anodic oxygen evolution reaction (OER) and cathodic hydrogen evolution reaction (HER),30,31 whereas, as a matter of fact, the input voltage for water electrolysis is substantially higher than 1.23 V (even beyond 1.8 V) owing to the sluggish kinetics of the OER.32–34 Moreover, the gaseous oxygen produced at the expense of high electric energy consumption only offers a lower market value relative to the hydrogen fuel, which is deemed as a major hindrance to economic viability of water electrolysis.35–37 Therefore, a question has been raised as to whether there exists an alternative to the OER step.
Nearly half of greenhouse gas emissions are relevant to the production of industrial and agricultural goods, among which solid waste (e.g., plastic and biomass) is one contributor to CO2 emission.38–40 Taking post-consumer plastics as an example, 260 megatons (Mts) of plastic waste produced annually brings about significant damage to the natural ecosystem, agricultural production and human health once discarded into the environment,41–43 and given the fact that the mass of CO2 footprint from plastic incineration is three times higher than that of plastics, the potential CO2 emission embodied in plastic waste accounts for 2% of global CO2 emission.44,45 Moreover, if the current trend continues, greenhouse gas emissions from irresponsible consumption of plastics are projected to account for approximately 15% of the global carbon budget by 2050.46 Beyond plastics, biomass wastes, including animal feed, crop straws, and wood, are also recognized as renewable and earth-abundant resources and their annual global production is as high as 100–140 gigatons (Gts).47 Nevertheless, most of the biomass resources always ends up with incineration or decomposition by microorganisms, which is typically a CO2-emitting process and also there is a waste of carbon resources. As a matter of fact, both plastics and biomass are utilized as feedstocks for production of valuable chemicals via chemocatalytic processes,48 thereby reclaiming the carbon resource present in plastics and biomass. Note that raw plastics and biomass need be converted into their derivatives with water-solubility via pretreatment prior to electrochemical upgrading. In particular, assuming that the inherent carbon content in solid waste is nearly 50% (on a dry basis, 47–51 wt% carbon within biomass and 60–90 wt% carbon within plastics), 140 Gts of solid waste produced annually (i.e., at least 70 Gts of the carbon content) are sufficient to meet the global demand of gasoline, diesel, and jet fuel by factors of 45, 63, and 185 times, respectively.49 It is therefore advisable, if not imperative, to exploit and devise reliable economically viable strategies for upcycling of solid waste, which contributes to reducing carbon footprint while creating economic values. A less-ambitious alternative, but deemed more feasible, is electroreforming of solid waste derivatives, namely cathodic HER from water splitting paired with anodic oxidation of organics over electrocatalysts. Thus, electroreforming of solid waste derivatives produces valuable commodity chemicals and H2 fuel, while offering a favorable thermodynamics of oxidation relative to water electrolysis.50–52 Moreover, the high anodic potential requirements of the OER result in a wide potential window to implement organic oxidation prior to water oxidation. For example, the electrooxidation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) and ethylene glycol (polyethylene terephthalate derivative) to formic acid has been incorporated as the anode reaction with lower potentials of 0.118 and 0.128 V versus the reversible hydrogen electrode (vs. RHE) under standard conditions, respectively, compared to that of the OER (1.23 V).53,54 On the other hand, electrochemical valorization of waste streams can complement the widely adopted mechanical recycling, avoiding the requirement of post-consuming polymers that can be only converted to lower quality products. In light of this, solid waste electroreforming with the objective of reducing energy consumption and adding product value can be a viable alternative to substitute the energy-intensive water electrolysis or industrially implemented technologies required to produce commodity chemicals.
Reforming of solid waste derivatives has been promoted electrochemically with the advantage of the potential use of renewable electricity as the energy input compared to the thermal route, thus receiving remarkable attention of the scientific community followed by witnessing the rapidly increased number of studies in this domain. Accordingly, a series of reviews focusing on electroreforming of biomass waste have been published recently, covering different aspects of biomass electrochemical valorization: emerging electrocatalysts, strategies to improve the productivity and selectivity, and summing up the impressive achievements made thus far.55 In parallel to this, a number of reviews concerning upcycling of plastic waste using photocatalysis, electrocatalysis, and mechanochemistry have been published, with a focus on catalyst design and structure–activity interaction analysis.56–60 Regrettably, despite a great deal of structural similarities between biomass and plastics, there are no traceable published reviews concerning electrochemical upcycling of both biomass and plastic waste. Most notable is the missing link between biomass and plastic upgrading processes, especially pretreatment methodology that is a prerequisite for the electrochemical conversion of raw biomass and plastics, which remains less-mentioned in previous reviews.61–63 In this regard, a comprehensive and critical review concerning electroreforming of biomass and plastics is extremely needed, with a focus on the shared design of the state-of-the-art electrocatalysts for each documented reaction (Fig. 1). In parallel, composition deconstruction of solid waste, the state-of-the-art pretreatment methodologies, technical superiority and economic evaluation of electroreforming technology are also discussed in this review. Moreover, we discuss how to investigate the reaction mechanism using operando spectroscopy combined with computational simulations and how some lessons learnt from previous literature can be applied to further study. We conclude with a thoughtful discussion of technoeconomic challenges for industrial implementation of this still-nascent technology and further directions of fundamental research that remain crucial to boost the productivity and selectivity via an economically competitive way.
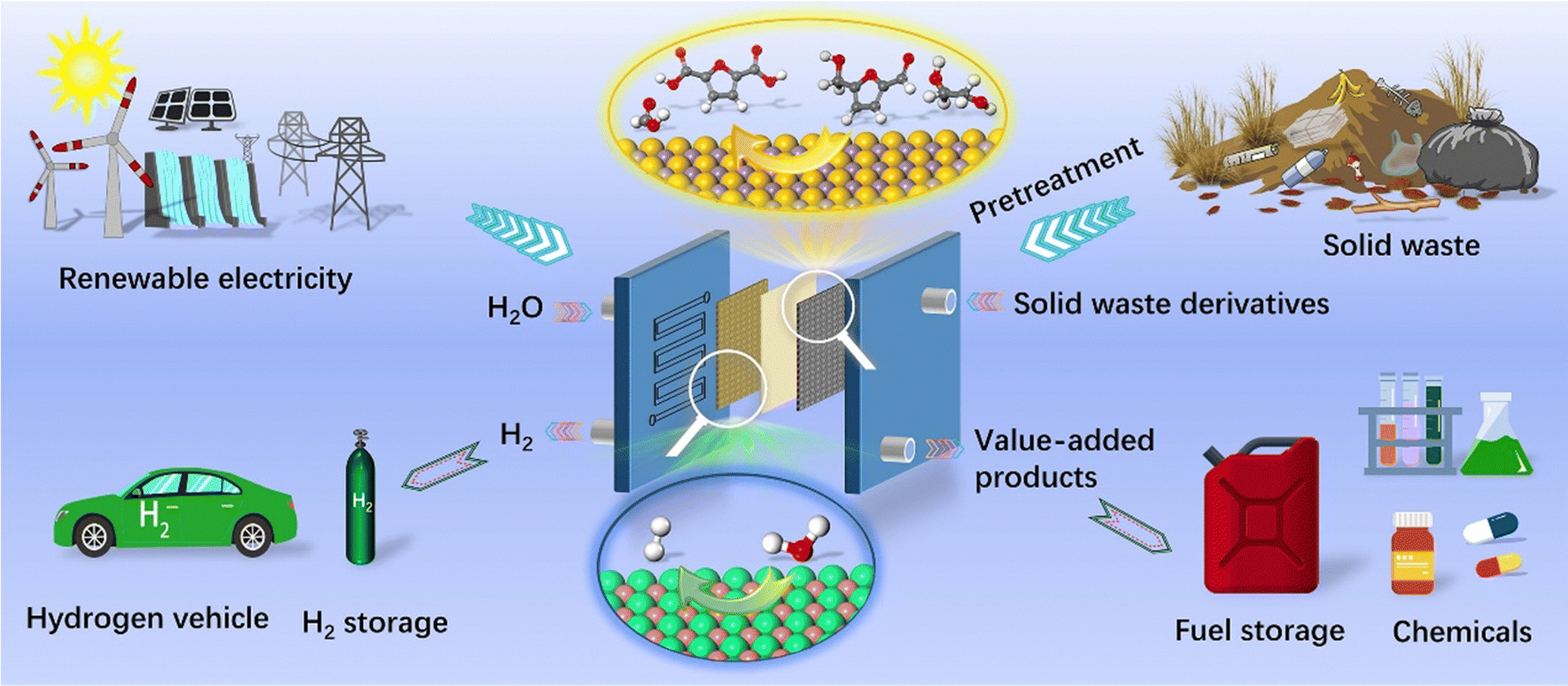 | ||
| Fig. 1 Illustration of electroreforming of solid waste (e.g. biomass and plastic) using renewable electricity. | ||
2. Solid waste deconstruction and pretreatment
2.1 Composition and availability of solid waste
The major compositions of solid waste involve biomass, plastic, and other wastes (e.g., paper, cardboard, glass, metal, and rubber), of which the remaining residues except for biomass and plastic are out of the scope of this review to be elucidated in detail and the readers may refer to review articles specializing on these wastes.64–67 Here, we aim to provide a description of the structure and sources of biomass and plastic (Fig. 2). Although biomass and plastic present substantial structural similarities in terms of the polymeric nature and the type of bonds connecting the constituent units, for clarity, their chemical compositions and the availability of each documented ingredient for electroreforming are separately presented in this section.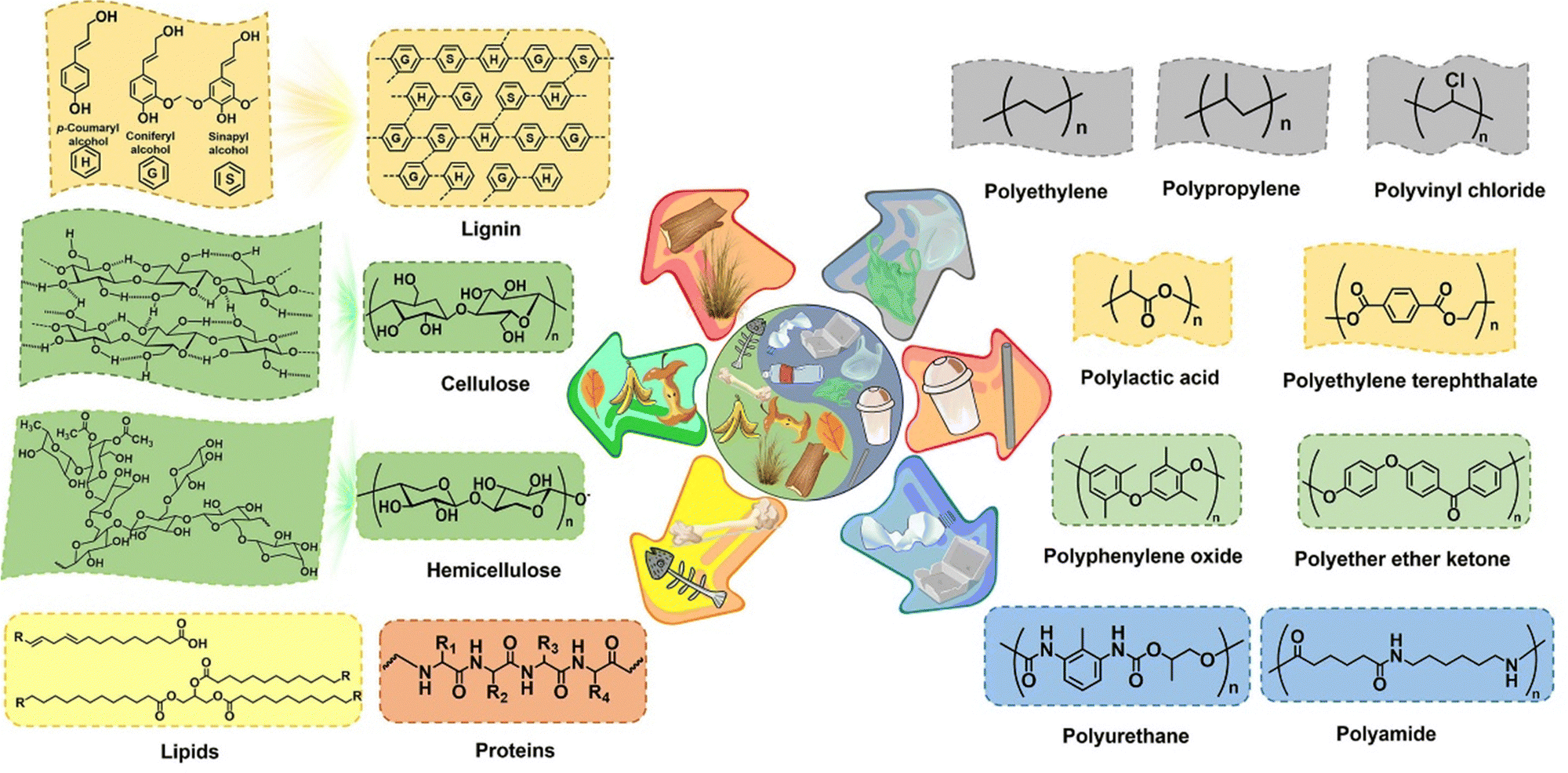 | ||
| Fig. 2 Chemical composition of solid waste that can be potentially valorized via electroreforming, mainly including plastics and biomass. Plastics involve polyolefin (e.g., PE and PP), polyester (e.g., PLA and PET), polyether, and polyamide, and biomass is a source of protein, lipid, and lignocellulose (i.e., cellulose, hemicellulose, and lignin). | ||
Biomass evolving from biological photosynthesis using available atmospheric CO2 and water as feedstocks and solar light as primary energy is acknowledged as the only sustainable source of organic carbon in Earth.68 As for the chemical constituent of biomass, their elemental distribution varies significantly even among the vegetal biomass (e.g., food wastes and agriculture residues). It is estimated that vegetal source on a dry basis comprises of 47–51 wt% carbon, 41–43 wt% oxygen, 6 wt% hydrogen, and the remaining sulfur and ash accounting for less than 10 wt%.69 In general, lignocellulosic biomass, including hardwood, softwood, grass, and agriculture waste, represents the most abundant form of terrestrial biomass.70,71 The lignocellulosic biomass serves as a feedstock consisting of three major biopolymers: cellulose (35–50%), hemicellulose (20–35%), and lignin (10–25%), as well as a small fraction of proteins, lipids, and ash.72
Cellulose, a major ingredient of lignocellulosic biomass, is constructed by the extensively repeated unit of disaccharide cellobiose (namely the dimer of β-1,4-linked glucose) cross-linked via the intramolecular and intermolecular hydrogen bond interactions that tightly bind each constituent unit, which renders cellulose to be rigid, semicrystalline, and even insoluble in most common solvents.73 The degree of polymerization for cellulose, in the case of woody biomass, is up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, which is deemed as a huge challenge for its depolymerization.74 Complimentarily, cellulosic biomass features many polar hydroxyl groups and constitutes the largest source of the organic carbon (>50%) in the biosphere, which offers opportunity to obtain a large amount of intermediate resources once it is depolymerized into water-soluble oligomers or constituent monomers.75 The second most abundant ingredient, hemicellulose, appears to be a random and amorphous heteropolymer, which comprises six sugar monomers, including galactomannan, xylan, glucuronoxylan, arabinoxylan, xyloglucan and glucomannan, with xylan occupying the largest proportion.76 Accordingly, hemicellulose contains many polar groups (e.g., hydroxyl and aldehyde groups) and its degree of polymerization ranges from 50 to 300, which is much lower than that of cellulose, indicating a reduced difficulty in depolymerization.77 Lignin, a major non-carbohydrate component, is recognized as a hydrophobic polyether with a complex and stable three-dimensional configuration that renders it recalcitrant.78 A notable advantage of lignin is its higher heating value, which is attributed to the lower O content of 30 wt% than that of cellulose (49 wt%).79 Finally, we extend further into the remaining small fraction: lipids and proteins. Lipids extracted from biomass are mainly made of saturated and unsaturated long-chain fatty acids (
000, which is deemed as a huge challenge for its depolymerization.74 Complimentarily, cellulosic biomass features many polar hydroxyl groups and constitutes the largest source of the organic carbon (>50%) in the biosphere, which offers opportunity to obtain a large amount of intermediate resources once it is depolymerized into water-soluble oligomers or constituent monomers.75 The second most abundant ingredient, hemicellulose, appears to be a random and amorphous heteropolymer, which comprises six sugar monomers, including galactomannan, xylan, glucuronoxylan, arabinoxylan, xyloglucan and glucomannan, with xylan occupying the largest proportion.76 Accordingly, hemicellulose contains many polar groups (e.g., hydroxyl and aldehyde groups) and its degree of polymerization ranges from 50 to 300, which is much lower than that of cellulose, indicating a reduced difficulty in depolymerization.77 Lignin, a major non-carbohydrate component, is recognized as a hydrophobic polyether with a complex and stable three-dimensional configuration that renders it recalcitrant.78 A notable advantage of lignin is its higher heating value, which is attributed to the lower O content of 30 wt% than that of cellulose (49 wt%).79 Finally, we extend further into the remaining small fraction: lipids and proteins. Lipids extracted from biomass are mainly made of saturated and unsaturated long-chain fatty acids (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C12) and glycerides, both of which are hydrophobic and chemically inert, thus being prone to form an oily phase that makes the separation easier but the electroreforming more challenging.80 Proteins are composed of long chain amino acid residues cross-linked in the form of a three-dimensional network.81 Compared with lipids, the protein components in biomass have not attracted wide attention thus far as there is only a little traceable literature concerning protein upgrading,82 whereas the amino acid constituent in protein often functions as a feedstock for production of higher-value commodity chemicals.
C12) and glycerides, both of which are hydrophobic and chemically inert, thus being prone to form an oily phase that makes the separation easier but the electroreforming more challenging.80 Proteins are composed of long chain amino acid residues cross-linked in the form of a three-dimensional network.81 Compared with lipids, the protein components in biomass have not attracted wide attention thus far as there is only a little traceable literature concerning protein upgrading,82 whereas the amino acid constituent in protein often functions as a feedstock for production of higher-value commodity chemicals.
Switching focus to the synthetic polymers, plastic packaging features convenience and practicality as well as major environmental concerns. It is estimated that post-consumer plastic packaging accounts for a small portion of global municipal solid waste (12%), while the durability makes plastic waste an increasing concern owing to its negative impact on the environment.83,84 In contrast to the lignocellulosic biomass, most of the post-consumer plastic fails to be decomposed by microorganisms and thus leads to their accumulation in the environment. Plastic waste can be divided into four major streams in terms of the type of bonds linking the monomeric units: polyolefins with C–C linkages, polyesters and polyethers with C–O linkages, polyurethanes with N–C–O linkages, and polyamides with C–N linkages. Polyolefin plastic with long hydrocarbon chains, including polyethylene (PE), polypropylene (PP), polystyrene (PS), and polyvinyl chloride (PVC), occupies a major portion of overall plastic production (>70%).85 However, these plastics show unprecedented and severe challenges to upgrade them economically owing to their robust C–C linkages that have extreme resistance to chemical/enzymatic decomposition under mild conditions. Note that the C–C bond is also the dominant linkage in phenol formaldehyde resin (PF) that does not fall into the polyolefin category. Polyester, typified by polyethylene terephthalate (PET) and polylactic acid (PLA), is relatively easy to fully depolymerize into versatile platform chemicals or their monomeric units that can be further chemically valorized, thereby indicating the potential possibility for electroreforming.86,87 For example, PET is susceptible to hydrolysis into ethylene glycol and terephthalate in a basic medium, of which ethylene glycol as a platform molecule well favors the chemical valorization and terephthalate can be recycled back to PET for reuse via acidification and purification processes.56 Of special note is PLA, which is well accepted as a biodegradable polyester, but its natural degradation process requires a long period of time in seawater and agricultural soil.88–90 Polyether plastic is constructed using phenolic monomers via ether linkages, including epoxy resins (ERs), polyoxymethylene (POM) and poly(phenylene oxide) (PPO). Taking ERs as an example,91 benefiting from their macromolecular nature, they function as anticorrosive coating materials and engineering adhesives, with an annual global production of up to 3.5 Mt. Polyurethane, a widely used polymeric plastic linked by the carbamate ester groups, is synthesized from polyols and isocyanates. The global polyurethane market size was estimated at US$ 70.67 billion in 2020, with a projected increase to US$ 95.24 billion in 2028.92 The remaining polyamides in plastic streams are characterized by C–N linkages and produced via the condensation reaction between amines and carboxylic acids. The representative one in polyamide streams may be Nylon-66 that is a highly crystalline polymer widely used in the automobile industry, instrument manufacturing, and electronic engineering.93,94 Such a high degree of crystallinity for Nylon-66 endows it with high heat resistance, high mechanical strength, and good ductility, but gives rise to its insolubility in most solvents, which is a huge challenge for electroreforming.
2.2 Pretreatment
The first challenge faced in upcycling of waste streams stems from their chemical diversity and complexity (e.g., plastics, biomass, and food residues), which impedes in many cases the valorization process and meanwhile indicates the necessity of pretreatment prior to electroreforming. Generally, developing ideal pretreatment strategies requires knowledge on the composition of the mixed solid waste streams, and pretreatment strategies should isolate the biomass and plastics from the mixed waste and should be capable of depolymerizing the sorted waste into its corresponding constituent units. On this basis, some physicochemical pretreatment processes are first carried out to separate plastics, cellulose, hemicellulose, and lignin. For example, sink-float density separation that features robust apparatus of low complexity and high capacity is capable of selective isolation of biomass and plastics from mixed waste.109 The industrially adopted technologies, including sorting, washing, and grinding steps, in combination with ultrasonic and supercritical fluid extraction used to remove certain impurities, are then employed to recycle and further purify biomass and plastics collected from density separation.110The next obstacle for raw biomass and plastic electroreforming in an aqueous medium arises from their ultralow solubility induced by the rigid polymeric nature. Taking lignocellulosic biomass as an example, a prerequisite for lignocellulose valorization is the depolymerization of its three major constituents: cellulose and hemicellulose to water-soluble oligomers and monosaccharides and lignin to water-soluble oligomers and phenolic units. In light of this, following the mechanical separation, the sorted biomass and plastics need to be further depolymerized and solubilized via chemical or biological routes, with the goal to obtain oligomers with improved solubility or carbon-neutral monomers that can be electrochemically upcycled.95,111,112 By having a close-up view of biomass and plastics, their substantial compositional and structural similarities can be identified (Fig. 3). Specifically, the overall structural frameworks of both biomass and plastics are mainly based on the C–C, C–O, and C–N linkages, which means that their cleavages are a prerequisite for obtaining the soluble monomeric units. More to the point, biomass and plastics are potentially capable of sharing the depolymerization methodology (Table 1). For example, C–C linkages appear widely in several common plastics, including PE, PP, PS, PVC, and PF, while they play a crucial role in the construction of lignin and fatty acid chains.113 At present, the main depolymerization methods for these waste streams are based on chemical cleavage of C–C bonds using elevated levels of acids and bases, costly solvents (e.g., ionic liquids), organocatalysts, high temperature, and high pressure, which inevitably increase the product cost.114–116 An economically viable alternative, while deemed more effective, has been reported by Lin et al.117 who claimed that PS could be rapidly degraded in a frozen aqueous solution. More specifically, PS particles were converted into the excited state at the liquid interface between ice crystals, which were further combined with dioxygen to form singlet oxygen (1O2) that contributed to accelerating the degradation of PS.
 | ||
| Fig. 3 Illustration of the structural similarities between plastic and biomass in terms of the type of bonds connecting the constituent monomers. | ||
| Polymer | Linkage | Pretreatment methodology | Ref. |
|---|---|---|---|
| Polyethylene | C–C | Hydrothermal treatment using 6% HNO3 at 180 °C | 95 |
| Polyethylene | C–C | Microwave (1200 W and 180 °C) heating in diluted H2SO4 | 96 |
| Lignocellulosic biomass | C–C | Ionic-liquid-assisted deconstruction process | 97 |
| Polystyrene | C–C | Freeze-accelerated depolymerization reaction | 98 |
| Polypropylene | C–C | Ru/C-catalyzed hydrogenolysis at 200–250 °C under 20–50 bar H2 | 99 |
| Polyethylene terephthalate | C–O | Alkali-induced hydrolysis (concentrated KOH/NaOH ranging from 1 to 10 M) | 100 |
| Polyethylene terephthalate | C–O | Microwave-induced alcoholysis using a ZnO catalyst | 101 |
| Polylactic acid | C–O | Organocatalyzed reductive depolymerization | 102 |
| Cellulose | C–O | Ball-milling processing in the presence of kaolinite catalysts | 103 |
| Lignin | C–O | Using cellulose-derived solvent (cyrene) at 120 °C | 104 |
| Polyurethane | N–C–O | Hydrogenation using Ru catalysts in tetrahydrofuran solvent | 105 |
| Polyurethane | N–C–O | Marine-microorganisms-assisted depolymerization | 106 |
| Chitin | C–N | Using acidified molten salt hydrates (40 mM HCl and 60 wt% LiBr) at 120 °C | 107 |
| Polyamide | C–N | Hydrogenation using a Ru pincer catalyst at 150 °C under 70 bar H2 | 108 |
Analogous to C–C linkages, C–O bonds are also prevalent in solid waste and serve as the major linkages not only in polyester and polyether but also in cellulose, hemicellulose, and triglycerides. Among these, polyesters and triglycerides dominated by ester bonds easily implement hydrolysis into their corresponding constituent monomers with an aid of dilute alkali or acid treatment.118–120 Additionally, thermochemical approaches are widely employed to depolymerize polyester into constituent monomers. Very recently, Hu et al.121 developed a catalyst-free thermochemical depolymerization route that can selectively steer the transformation from post-consumer PET plastics to monomers by means of pyrolysis. The remaining polyether, cellulose, and hemicellulose feature ether bonds. Note that although glycosidic bonds in cellulose belong to a special acetal-type ether bond, the nature of both the C–O bond in lignocellulose and the ether bond in polyether is similar.122 The dissociation energy of the ether bond is generally higher than that of the ester bond, which is one reason why polyether and cellulose undergo depolymerization in the presence of a mass of concentrated and corrosive acidic/basic solutions.123 Such a depolymerization process normally involves hydrolytic, oxidative, and reductive steps, resulting in a widely distributed feedstocks that make the subsequent separation operation difficult. Alternatively, ball-milling processes in the presence of mineral acids and solid acids have been proposed to convert cellulose to water-soluble oligomers.103,124
Shifting the focus to C–N bonds, in general, C–N linkages are mainly distributed in protein and chitin, as well as polyurethane and polyamide. The methods developed currently towards pretreatment of polyurethane and polyamide are based on pyrolysis and glycolysis.125–127 As for chitin, the pretreatment method typically includes two separate steps: deacetylation and depolymerization, of which deacetylation frequently involves the use of a large quantity of concentrated and corrosive alkali, thus resulting in environmental concerns.107 Alternative routes, such as microwave-assisted treatment and freeze–pump–thaw cycles, have been explored to promote the deacetylation process.128 Following the deacetylation process, depolymerization is carried out with the assistance of acid, oxidizer, ultrasound, or plasma treatment.129 Compared with the separate operations for deacetylation and depolymerization, mechanochemistry is a better option to pretreat the recalcitrant raw waste, with the ability to integrate deacetylation and depolymerization process into one step. Moreover, another notable advantage of mechanochemical methodology is its ability to proceed in a solvent-free way, which avoids the subsequent handling of wastewater. For instance, Yan et al.130 developed a solid-state mechanochemical strategy to convert chitin to low molecular weight chitosan with enhanced solubility, in which chitin underwent concurrent base-catalysed deacetylation and depolymerization processes with the aid of ball-milling.
Basically, pretreatment is an essential first step towards solid waste valorization, which remains as important as the subsequent electroreforming, whereas pretreatment and electroreforming in previous literature concerning waste upgrading are separately investigated, a fact which is relevant to the direct use of simple water-soluble molecules (e.g., alcohols and aldehydes) rather than raw waste in a rich body of literature. Future studies should focus on the development of energy-saving and cost-effective chemical pretreatment strategies that are compatible with electroreforming processes, which is of paramount importance in moving forward to a circular consumption of solid waste and generation of renewable carbon-neutral platform chemicals. Indeed, enzyme engineering has offered a blueprint for depolymerization of the sorted polymers, especially polyester plastic, which provides solutions where mechanical pretreatment is not possible and meanwhile contributes to the production of water-soluble platform molecules that are chemically valorized via electroreforming.143–145 Last, but not least, benefitting from a number of structural similarities between biomass and plastics, the lessons learnt from developing pretreatment technologies for biomass depolymerization can be applied to process plastic waste and vice versa.
3. Comprehensive evaluation of electroreforming technology
Hydrogen fuel, and, more broadly, commodity chemicals produced via electroreforming of solid waste using renewable electricity can serve as a uniquely dense and portable form of decarbonized transportation fuel and an energy storage medium to overcome the bottleneck of intermittent renewable energy. However, for commercialization, electroreforming technology using solid waste as the feedstock and renewable electricity as the energy input has to be competitive with conventional energy evolution processes. Consequently, the technical superiority, energetic analysis, and economic assessment of solid waste electroreforming are proposed in detail in this section, which is envisioned to provide critical guidance for the future development of this still-nascent technology.3.1 Technical superiority
Before assessing the technical superiority of electroreforming for waste valorization, the strengths and limitations of several conventional approaches (e.g. pyrolysis and fermentation) and emerging technologies (e.g. photoreforming) need to be emphasized. The industrially implemented pyrolysis technology is capable of converting mixed waste streams into crude oil that can be further valorized into monomers for resynthesis of virgin-grade resins in cooperation with polymer manufacturing, whereas the pyrolysis process relies heavily on harsh conditions (e.g., elevated temperature, high pressure, and use of toxic/costly catalysts) required to deliver activation energy, thereby rendering the whole concept energy intensive.138 Another technology implemented in industry is microbial fermentation that utilizes microorganisms to metabolize lignocellulosic biomass for concurrent production of bioethanol and hydrogen in a low temperature range (25–65 °C),140 but such a bio-based technology is currently incapable of processing the mixed waste or plastics.146 Analogous to fermentation, photoreforming is implemented by harnessing photonic energy under mild conditions, such as ambient temperature and in an aqueous reaction environment.147 Although photoreforming allows complete mineralization of waste to CO2 and concurrent production of hydrogen, it suffers from low solar-to-chemical conversion efficiency, which is considered a large obstacle for large-scale application.148In comparison to the aforementioned three conventional methodologies towards waste-to-chemical conversion, the electrochemical process presents unique strengths for solid waste valorization (Table 2). To begin with, electrochemical synthesis proceeds in an aqueous feedstock under ambient conditions (i.e., low energy input, room temperature, and atmospheric pressure), while enabling oxidative or reducing equivalent evolution without external H2 and chemical oxidants (water can be utilized as a hydrogen and oxygen source in electroreforming of solid waste). Secondly, electrochemistry can drive direct synthesis of target products via a lower number of reaction steps by using inexpensive starting materials and electrocatalysts and meanwhile high activity and selectivity can be realized via tailoring the electronic structure of catalytic sites. Thirdly, through tuning the electric potential differences between electrodes, the driving force for solid waste oxidation half-reaction is maneuvered accordingly, thereby contributing to boosting the productivity and optimizing the product distributions. Finally, in the particular case of integrating electricity from renewable energy, one notable advantage of electroreforming is its huge economic feasibility and environmental push for decarbonization of the global energy economy.
| Electroreforming | Photoreforming | Pyrolysis | Fermentation | |
|---|---|---|---|---|
| Feedstocks | Biomass, plastic | Biomass, plastic | Biomass, plastic | Biomass |
| Temperature | ∼25 °C | ∼25 °C | 300–900 °C | 25–65 °C |
| Products | H2, organics | H2, organics, and CO2 | Crude oil | H2, CH4, NH3, CO2, and organics |
| Market prices (products) | 1.0–580 $ per kg | 0.6–100 $ per kg | 0.67 $ per kg | 0.5–6.8 $ per kg |
| CO2 emission | 0.97–4 kg CO2eq kg−1 H2 | 0.8–9.0 kg CO2eq kg−1 H2 | 10–85 g CO2eq MJ−1 crude oil | 3.4–5.6 kg CO2eq kg−1 H2 |
| Strengths | Utilization of renewable-electricity; coproduction of high-purity H2 and high-value chemicals; and high yield and selectivity | Utilization of solar-energy; high-purity H2 evolution; simple setup; and mild conditions | Direct reforming of raw waste; production of high-energy-density bio-oil | Environment-friendly; cost-effective; substrate specificity; and low energy consumption |
| Limitations | Pretreatment required for raw wastes; lab-scale operation | Low solar-to-chemical conversion efficiency; lab-scale implementation | Harsh conditions; high energy input; purification of crude oil; and CO2 emission | Not compatible with plastic or complex waste; CO2 emission |
| Ref. | 63 and 131–133 | 16, 134 and 135 | 136–138 | 139–142 |
Thanks to the aforementioned advantages over pyrolysis, fermentation, and photoreforming, electrochemical manufacturing has a well-established track record for upgrading biomass-/plastic-derived platform chemicals on an industrial scale. Electrosynthesis has made a breakthrough in aluminium production via the Hall–Héroult process as early as the 1880s and the chlor-alkali process has been industrialized since the 1890s.149,150 The most representative one that has been commercialized is the up-scaling electrosynthesis of adiponitrile using acrylonitrile as a feedstock via electro-oxidative decarboxylation and Kolbe coupling reactions, which has been realized by Asahi Chemical, BASF, and Monsanto.151 Note that the adiponitrile mentioned here is a key intermediate used to synthesize polyadiohexylenediamine (Nylon-66).152 Other impressive cases of industrial electrosynthesis processes include the production of acetoin from butanone, 1,4-dihydronaphthalene from furan, succinic acid from maleic acid, and arabinose from gluconate.153 Of note, in spite of their clear-cut competitive advantages over conventional routes that have gained more research focus, most of the electrosynthesis processes are still in the lab-scale stage (maybe proof-of-concept) and there is a long way to go for electrochemical synthesis for its widespread use.
3.2 Energetic analysis
Shifting focus to the energetic analysis of electroreforming, the thermodynamics plays a pivotal role in the industrial-scale deployment of the solid waste electroreforming reaction. Taking the OER as an example, the energetically demanding and kinetically sluggish OER only contributes to O2 with little value (Fig. 4a).166 By contrast, thermodynamically more favorable solid waste oxidation at the anode aims to substantially improve the energy efficiency of an electrolyzer and meanwhile offers high-value products.167 In this regard, providing fresh insights from energetic perspectives is of vital importance for the future advances of solid waste electroforming.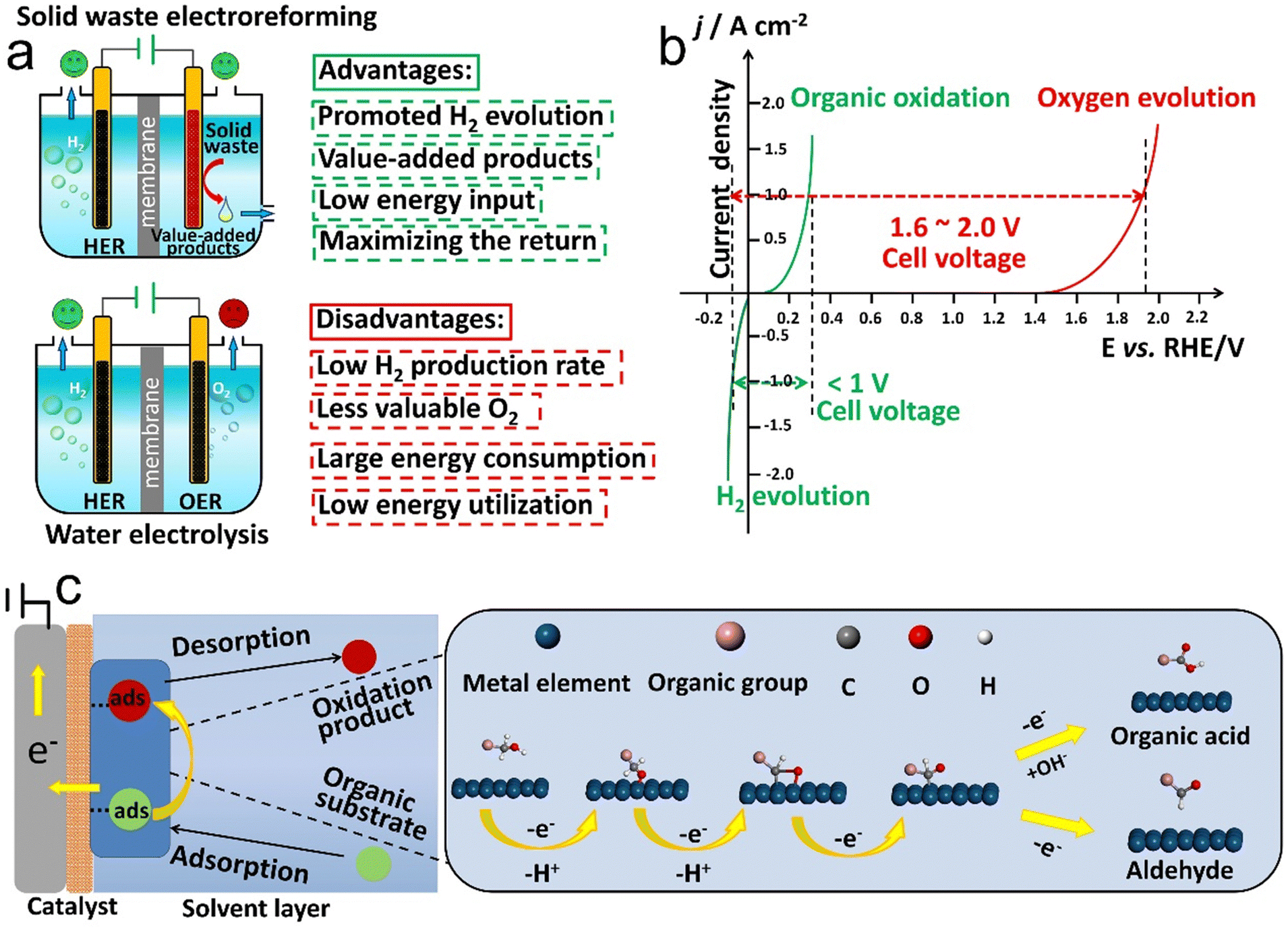 | ||
| Fig. 4 (a) Comparison of water electrolysis and solid waste electroreforming. (b) Theoretical i–v plot for water electrolysis and organic electroreforming according to the Butler–Volmer kinetics law. (c) Schematic illustration of a typical electrooxidation reaction. | ||
Regarding water electrolysis and solid waste electroreforming, the chemical evolution induced by the synchronous transfer of equivalent protons and electrons requires a rapid charge transfer to realize a high-efficiency performance during water and solid waste electrolysis. Consequently, solid electrocatalysts are always regarded as a driving force towards high-activity and high-selectivity electrochemical processes. Besides, it should be noted that the practical operational voltage for electrocatalysis is higher than the thermodynamic potential (i.e., theoretical cell voltage, Ucell) owing to the resistance loss in the cell. The surplus of potential is termed as overpotential, which manifests the voltage required to surmount the activation barrier from the anode (ηa) and cathode (ηc), as well as the electrolyte's inherent resistance (ηIR).168 Accordingly, the applied voltage can be described as:
| U = Ucell + ηa + ηc + ηIR. |
For electroreforming of solid waste, the operation principle involves the HER and organic oxidation at the cathode and anode, respectively, which indicates that the Ucell value varies along with the thermodynamics of platform molecule oxidation. Of note, owing to the composition complexities and a high degree of polymerization of solid waste, pretreatment is required to generate water-soluble oligomer or monomeric units that are more malleable for catalytic conversion with regard to raw solid waste. Here, we list 5 common platform chemicals (i.e. methanol, ethanol, glycerol, ethylene glycol, and 5-hydroxymethylfurfural) as building blocks that are typical biomass and plastic derivatives and provide their oxidation thermodynamics parameters (Table 3). Regardless of water electrolysis or organic electroreforming, an external energy input is required, as evidenced by their corresponding Gibbs free energy change greater than zero (ΔG > 0). However, the energy required to generate one mole of hydrogen from electroreforming of organic molecules is far lower than that from water electrolysis, which is a sign of the favorable thermodynamics of solid waste electroreforming. For the actual operation with the multielectron transfer and the solution ohmic losses, to reach a current density of 1 A cm−2, water electrolysis requires a thermodynamic potential ranging from 1.6 to 2.0 V according to the Butler–Volmer kinetics law.191 For comparison, electroreforming of organics derived from solid waste needs a cell potential of less than 1.0 V (Fig. 4b).165 Consequently, electroreforming of solid waste derivatives offers the additional benefit of valuable chemicals at the anode paired with cathodic hydrogen generation, which endows it with energy-saving, value-addition, and cost-effectiveness advantages compared to water electrolysis for high-purity hydrogen production.
| Feedstock | Products | Equation | ΔrH⊖m and ΔrG⊖m (kJ mol−1) | Cell voltage | Electron transfer | Power (kW h kgH2−1) | Ref. |
|---|---|---|---|---|---|---|---|
| Notes: for the overall process under standard conditions, the reaction enthalpy (ΔrH⊖m) and Gibbs free energy (ΔrG⊖m) are calculated using the following equations: ΔrH⊖m = νBΔrH⊖f(products) – νBΔrH⊖f(reactants) and ΔrGmθ = νBΔrG⊖f(products) – νBΔrG⊖f(reactants), respectively, where νB, ΔrH⊖f, and ΔrG⊖f indicate the stoichiometric number, standard enthalpy of formation, and standard Gibbs free energy of formation, respectively.154,165 | |||||||
| Water | H2/O2 | H2O → H2 + 1/2O2 | 285.8 and 237.2 | 1.229 | 2 | 48.2–64.3 | 154 and 155 |
| Methanol | CO2 | CH3OH + H2O → 3H2 + CO2 | 131.5 and 9.3 | 0.016 | 6 | 13.4 | 156 and 157 |
| Formic acid | CH3OH + H2O → 2H2 + HCOOH | 100 and 41.7 | 0.11 | 4 | |||
| Ethanol | CO2 | C2H5OH + 3H2O → 6H2 + 2CO2 | 348 and 97.3 | 0.84 | 12 | 19.6–28.1 | 157–159 |
| Acetic acid | C2H5OH + 3H2O → C2H4O2 + 2H2 | 79.1 and 22 | 0.057 | 4 | |||
| Glycerol | CO2 | C3H8O3 + 3H2O → 3CO2 + 7H2 | 342.8 and 3.9 | 0.003 | 14 | 20.4–33 | 157, 159 and 160 |
| Formic acid | C3H8O3 + 3H2O → 3CH2O2 + 4H2 | 248.3 and 102.9 | 0.13 | 8 | |||
| Glyceral-dehyde | C3H8O3 → C3H6O3 + H2 | 61.1 and 9.7 | 0.206 | 2 | |||
| Glyceric acid | C3H8O3 + H2O → C3H6O4 + 2H2 | 88.7 and 35.1 | 0.091 | 4 | |||
| HMF | FDCA | C6H6O3 + 2H2O → C6H4O5 + 3H2 | 468.4 and 65.6 | 0.118 | 6 | 7.2–40.1 | 161–163 |
| Ethylene glycol | CO2 | C2H6O2 + 2H2O → 2CO2 + 5H2 | 238.9 and 8.1 | 0.008 | 10 | 21.4 | 164 |
| Formic acid | C2H6O2 + 2H2O → 2CH2O2 + 3H2 | 175.9 and 74.1 | 0.128 | 6 | |||
| Glycolic acid | C2H6O2 + H2O → C2H4O3 + 2H2 | 157.1 and 54.9 | 0.142 | 4 | |||
3.3 Economic assessment
Following the discussion on the energetic analysis, technoeconomic evaluation for hydrogen evolution is carried out to provide a basis for comparison between solid waste electroreforming and water electrolysis. To begin with, electroreforming technology requires a lower electricity compared to water electrolysis (Table 3) to produce 1 kg H2. Taking glycerol electroreforming as an example, it has been proved that electrooxidation of glycerol as an alternative to the OER contributes to reducing the electricity input by up to 53%.192 Another notable case from Deng's group reported that the electricity consumption to obtain 1 m3 H2via electroreforming of native biomass at 0.2 A cm−2 was as low as 0.69 kW, which was 16.7% of the energy consumed for water electrolysis under the same conditions.193 However, despite the advantages of electroreforming over water electrolysis in terms of energy consumption, it is estimated that the average cost of green hydrogen ($4.5 kgH2−1) generated from electroreforming of solid waste is currently far higher compared to the steam reforming technology ($1.4 kgH2−1), a fact that is linked to the high investment required for grid electricity ($0.07 kW h−1) and an electrolyzer. Moreover, even with using photovoltaic electricity instead of alternative fossil-fuel-derived grid electricity, the cost of hydrogen ($2.2 kgH2−1) still goes beyond that of fossil-derived hydrogen.194 Of note, it is well acknowledged that the hydrogen generation pathway should be comprehensively evaluated based on overall cost as well as the environmental impact, that is, if the environmental burdens from steam reforming are taken into account, the price gap of hydrogen between electroreforming and steam reforming will be further narrowed.195 Apart from the reduction of operating costs and carbon footprint, electroreforming of solid waste also facilitates economical production of valuable chemicals at anodes. Under this scenario, the economic feasibility of electroreforming technology should be assessed more profitably and reasonably if both the cathodic and anodic reactions are taken into consideration.Here, the market prices of some typical raw materials derived from biomass and plastics and their corresponding products obtained by the partial electrooxidation reaction are displayed in Table 4. Apparently, the difference in the market values between raw materials and their corresponding products leads to economic feasibility of electroreforming technology. Particularly, the most representative example is the conversion from inexpensive HMF ($10 kg−1) to FDCA with a high market price ($32–580 kg−1). In addition, solid waste valorization via anodic oxidation involves the adsorption of reactant molecules, the breakage and formation of chemical bonds, and the final desorption of products, of which chemical-bond-formation/-cleavage are associated with multi-electron transfer as well as production of various chemicals (Fig. 4c). Moreover, product selectivity over electrocatalysts plays a vital role in the economic feasibility of electroreforming, as the product overoxidation results in not only a lower economic value but also a negative environmental impact (overoxidation of organics to evolve CO2). Using two benzyl alcohol oxidation processes as an example, with two-electron transfer, benzyl alcohol is electro-oxidized into a value-added benzaldehyde product, while further oxidation contributes to the formation of benzoic acid with even lower value than raw benzyl alcohol (Table 4). Overall, the aforementioned analysis highlights that the HER in combination with solid waste upgrading is capable of holding significant economic feasibility.
| Raw materials (market price, $ kg−1) | Oxidation half-reaction | Products (market price, $ kg−1) | Ref. |
|---|---|---|---|
| H2O | 2H2O → O2 + 4H+ + 4e− | O2 (0.02–0.04) | 169 |
| HMF (10.0) | HMF + H2O → FDCA + 6H+ + 6e− | FDCA (32–580) | 170 and 171 |
| Ethylene glycol (0.8–1.0) | Ethylene glycol + H2O → glycolic acid + 4H+ + 4e− | Glycolic acid (1.9) | 172 |
| Ethylene glycol + H2O → oxalic acid + 8 H+ + 8 e− | Oxalic acid (1.4) | 173 | |
| Furfuryl alcohol (1.5) | Furfuryl alcohol + H2O → 2-furoic acid + 4H+ + 4e− | 2-Furoic acid (4.0) | 174 and 175 |
| Furfural (1.0–2.7) | Furfuryl + H2O → 2-furoic acid + 2H+ + 2e− | 2-Furoic acid (4.0) | 176 and 177 |
| Ethanol (0.1) | Ethanol → acetaldehyde + 2H+ + 2e− | Acetaldehyde (1.0) | 178 and 179 |
| 2ethanol → ethyl acetate + 4H+ + 4e− | Ethyl acetate (1.1) | ||
| Benzyl alcohol (2.0–2.5) | Benzyl alcohol → benzaldehyde + 2H+ + 2e− | Benzaldehyde (2.9) | 169, 180 and 181 |
| Benzyl alcohol → benzoic acid + 4H+ + 4e− | Benzoic acid (1.8) | ||
| Glycerol (0.04–0.6) | Glycerol → lactic acid + 2 H+ + 2 e− | Lactic acid (1.5) | 182–184 |
| 1,3-Propanediol (2.2) | 1,3-Propanediol → acrylic acid + 2H+ + 2e− | Acrylic acid (1.6–2.9) | 185–187 |
4. Electroreforming of solid-waste-derived platform molecules
As mentioned in Section 2.2, direct upcycling of solid waste in an aqueous electrolyte via electroreforming still remains a huge challenge as a consequence of the incredible constituent complexity and a high polymerization degree of solid waste. Accordingly, it necessitates separation and depolymerization of biomass or plastic streams prior to electroreforming, especially polyolefin and lignin with C–C linkages, to obtain monomeric units that are regarded as platform chemicals towards the electroreforming reaction. Note that direct electroreforming of raw waste in a non-aqueous electrolyte has been implemented on a lab-scale,196–198 which is however out of the scope of this review. In view of this, a variety of platform molecules, including glycerol, ethylene glycol, HMF, furfural, and glucose, have been used as feedstocks in electrocatalysis for conversion into a wide range of commodity chemicals with potential applications as pharmaceutical precursors, polymers and cosmetic ingredients.199–201 In this section, several common platform chemicals utilized as building blocks in electroreforming and their corresponding oxidation mechanisms towards formation of high-value products are elucidated in detail.4.1 Ethylene glycol
Ethylene glycol, a versatile chemical intermediate required in the production of perfumes, cosmetics, and resins, is currently obtained from petroleum-derived olefin via three successive steps (i.e., cracking, epoxidation, and hydration).100,202,203 Alternatively, ethylene glycol monomers can be produced from solid waste via following two routes with economically competitive and environmentally friendly advantages: (1) hydrolysis of PET waste into ethylene glycol and terephthalic acid (the terephthalic acid recycled to synthesize PET);144 (2) direct conversion from renewable biomass or its derivatives (e.g., sorbitol).207 Note that PET is an abundant polyester plastic with nearly 70 million tons produced annually for utilization in textiles and packaging, which is indicative of a massive untapped resource of ethylene glycol in PET waste.208Electroreforming of ethylene glycol allows generation of both valued chemicals (e.g., glycolaldehyde, glycolic acid, oxalate, and formic acid) (Scheme 1) and hydrogen, thereby attracting considerable attention recently.54 Following this research line, coproduction of formate and H2via electroreforming of ethylene glycol derived from hydrolysis of PET in an alkaline electrolyte was realized by Zhao et al. who utilized CuO nanowires as electrocatalysts (Fig. 5a and b).204 Moreover, 86.5% selectivity and 88% faradaic efficiency to formate could be obtained over CuO (Fig. 5c) and the theoretical simulations revealed that the favorable pathway of formate generation on CuO was to generate a glyoxal intermediate and ultimately formate via C–C bond scission (Fig. 5d). Despite the production of valuable formate from PET via electroreforming, an issue concerning the design of an electrolyzer was overlooked, that is, ohmic resistance of the electrolyzer based on a single cell would elicit an energy loss, especially at high current density, thereby lowering down the faradaic efficiency and productivity.209 To this end, a zero-gap reactor equipped with anion exchange membranes was employed to convert ethylene glycol to formate in parallel with H2 evolution over CoNi0.25P/Ni foam (CoNi0.25P/NF) catalysts (Fig. 5e), as reported by Duan et al.205 Furthermore, the formate productivity over CoNi0.25P/NF was determined to be 3.6 and 5.1 mmol cm−2 h−1 at the high current densities of 300 and 500 mA cm−2, respectively, with high faradaic efficiencies (Fig. 5f). Besides, a proof-of-concept production of potassium diformate (KDF) and H2via electroreforming of real-world PET plastics was demonstrated (Fig. 5g). Note that KDF, a safe growth promoter for animals, was prepared using the formate product as a precursor via acidification, condensation, and crystallization processes, which further improved the economic feasibility of ethylene electroreforming.
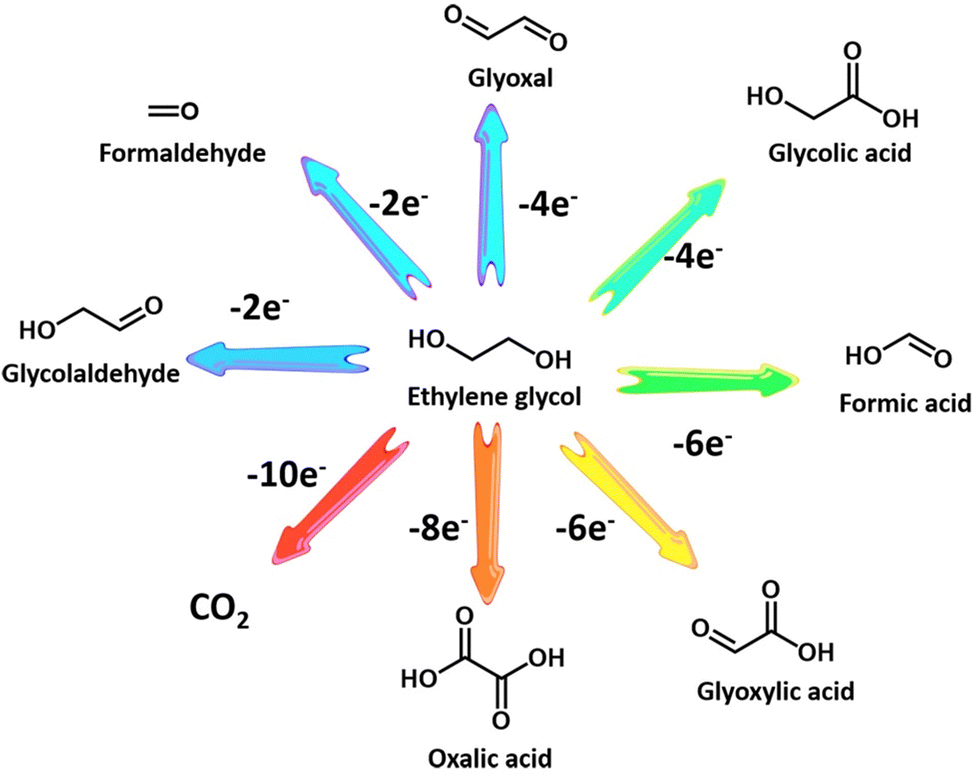 | ||
| Scheme 1 Catalytic conversion pathway for ethylene glycol electrooxidation. | ||
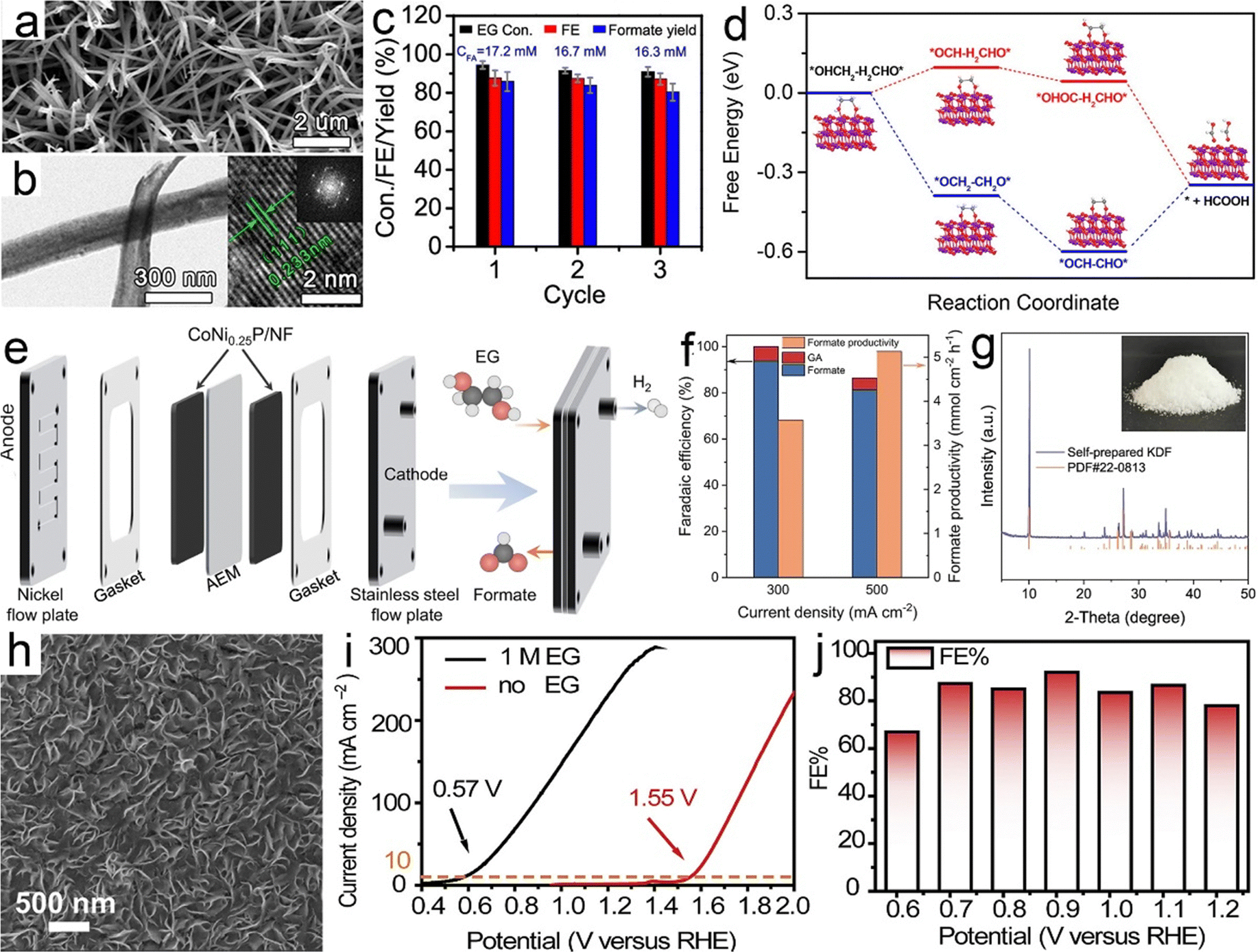 | ||
| Fig. 5 (a) SEM and (b) TEM images of CuO nanowires (the inset is its corresponding fast Fourier transform (FFT) image). (c) Ethylene glycol conversion, faradaic efficiency and formate yield over CuO nanowires and (d) the corresponding possible reaction pathways on CuO. Reprinted with permission.204 Copyright 2022, American Chemical Society. (e) Membrane-electrode assembly setup for electroreforming of ethylene glycol, (f) faradaic efficiency and productivity at the constant current densities of 300 and 500 mA cm−2 for ethylene glycol oxidation, and (g) XRD pattern and photograph (inset) of self-prepared potassium diformate (KDF). Reprinted with permission.205 Copyright 2021 Nature Publishing Group. (h) SEM image of PdAg/NF, (i) the comparison of LSV curves with the presence/absence of ethylene glycol, and (j) faradaic efficiencies (FEs) determined at different potentials. Reprinted with permission.206 Copyright 2021 Elsevier Inc. | ||
The interest in glycolic acid product, compared with formic acid, arises from its wider market size in metal cleaning, leather processing, and food packaging, thus enabling high value addition.210 However, glycolic acid generation with high selectivity remains challenging due to the uncontrollable and severe C–C bond cleavage followed by the formation of formic acid during ethylene glycol oxidation. Currently, the selective conversion of ethylene glycol to glycolic acid is only enabled by noble metals, such as Pt,211 Pd,212 and Au.213 A recent work from Shi et al.206 reported PdAg/NF electrocatalysts to evoke the transformation from ethylene glycol to glycolic acid with high selectivity and faradaic efficiency (Fig. 5h). As demonstrated in Fig. 5i, glycolic acid could be produced massively even at a high current density of 300 mA cm−2 without triggering the OER and the highest faradaic efficiency was obtained when the operational potential was controlled at 0.91 V vs. RHE (Fig. 5j).
Overall, electroreforming of ethylene glycol is one topic in the forefront of biomass and plastic upcycling, with the pursuit of concomitantly producing valuable chemicals and H2. Additionally, anodic oxidation of ethylene glycol has also been paired with CO2 reduction that evolves formic acid at the cathode,54 which however is beyond the scope of our review. The fly in the ointment is that most of the currently reported electrocatalysts only favor the production of formic acid with less value relative to glycolic acid, which indicates that the biggest challenge for reforming of ethylene glycol is the selectivity for the target product with high value (e.g., glycolic acid). Given the fact that precious metal-based catalysts at an anode have been stated to be capable of incomplete oxidation of ethylene glycol to glycolic acid via a four-electron pathway at a low overpotential in an alkaline medium,214,215 noble metals or their alloy electrocatalysts may be therefore a good choice to obtain glycolic acid. More efforts should be made to explore advanced catalysts with a low noble metal loading density, with the aim of obtaining a high selectivity to glycolic acid via impeding the undesired reaction pathways (i.e., avoiding the cleavage of the C–C bond).
4.2 Glycerol
Glycerol is a major by-product (i.e., biomass waste) in biodiesel preparation, where production of 1 ton biodiesel yields approximately 110 kg raw crude glycerol, thereby producing in high amounts.216 The recent report from Organization for Economic Cooperation and Development (OECD) pointed out that the global glycerol produced annually was estimated at 3.6 billion litres in 2016 and projected to increase to 4.0 billion litres in 2026, which brought about a huge surplus across the market over the last few decades.217 Hence, glycerol serves as a highly versatile platform molecule that can be further electrochemically valorized, and considerable attention has therefore been paid to exploring electrocatalysts to convert glycerol into a plethora of fine chemicals and materials. The electrochemical oxidation of glycerol involves multi-electron transfer via a broad pathway (Scheme 2), thereby resulting in a battery of downstream products, such as glyceraldehyde, glyceric acid, dihydroxyacetone, glycolic acid, and formic acid.218 Of note, the product selectivity can be tuned through the choice of electrocatalyst, potential, reaction medium, and electrolyte pH.219,220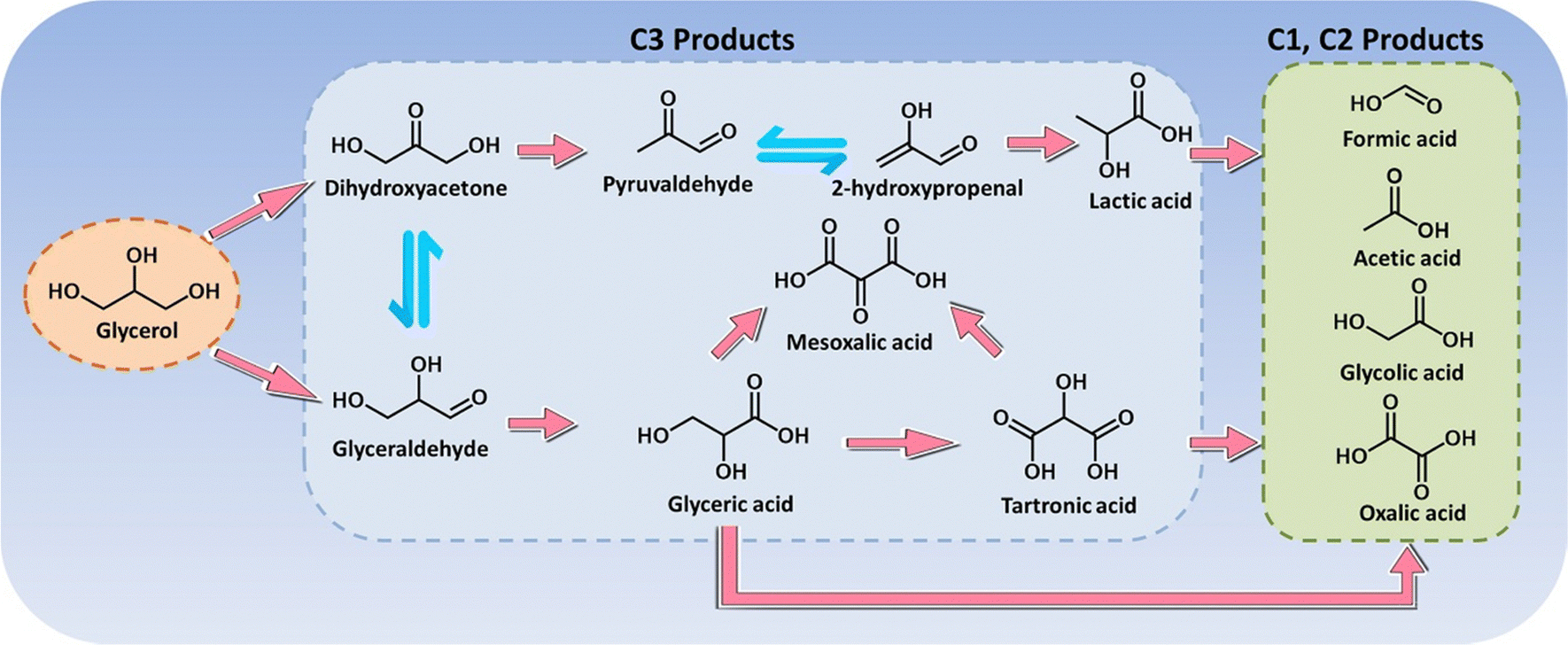 | ||
| Scheme 2 Reaction scheme illustration of the general pathway for glycerol electrooxidation. | ||
In electrooxidation of glycerol, formic acid (formate in alkaline medium) is the most commonly observed prevalent C1 product that is effective as a preservative, a liquid hydrogen carrier, and a fuel for direct/indirect fuel cells.221,222 Typically, for oxidation of one mole of glycerol without any carbon dissipation, an ideal scenario includes the selective C–C cleavage and deep oxidation into three moles of formic acid, which is accompanied by the production of four moles of hydrogen. In this regard, exploring electrocatalysts with tunable oxidative ability and a precisely steered pathway is extremely encouraged. Recently, a comparative study of cobalt-based spinel oxides as electrocatalysts towards reforming of glycerol was carried out by Jin et al.223 and CuCo2O4 was found to be the best-performing candidate with a high selectivity to formic acid (80.6%) and an overall faradaic efficiency of 89.1%. In another case, Johnsson et al.226 reported CoMoO4 nanorods as low-cost electrocatalysts to convert glycerol into formate in an alkaline electrolyte (Fig. 6a) and they systemically studied the effects of reaction parameters (i.e., potentials, reaction times, and temperatures) on the product selectivity. As revealed in Fig. 6b–d, formate was the dominant product with a high selectivity of roughly 90% within the potential ranging from 1.08 to 1.28 V, the reaction time ranging from 30 to 120 min, and the temperature ranging from 25 to 60 ×C. Electrooxidation of glycerol in the aforementioned cases was typically carried out using a conventional alkaline electrolyzer that is comprised of an anode with an alkaline electrolyte containing 0.1–1.0 M glycerol in parallel with a cathode in an alkali catholyte. Distinct from such an alkaline electrolyzer, a hybrid alkali/acid electrolytic cell, namely an alkaline anode paired with an acidic cathode, has a remarkably large voltage (0.059 × pH) owing to the pH gradient between the anode and cathode chamber. Inspired by this, Wen et al.225 used a high entropy alloy as an anode in a 1.0 M KOH electrolyte containing 0.1 M glycerol and the commercial RuIr/Ti as a cathode in 0.5 M H2SO4 catholyte to convert glycerol to formate (Fig. 6e and f). Notably, to reach equivalent current density, the operational voltage required in a hybrid electrolyzer is far lower than that in a conventional cell (Fig. 6g), thus indicating an energy-saving process in the asymmetric-electrolyte. Shifting the focus to the higher-value C3 products from glycerol oxidation, glycerol with three hydroxyl groups facilitates the formation of various C3 acids. Well-accepted concepts of a consecutive reaction mechanism for the formation, for instance, of glyceric acid have been theoretically investigated in previous reports.227 However, from experimental perspectives, engineering selectivity to glyceric acid over electrocatalysts still remains a huge challenge. Pt is a well-acknowledged effective catalyst towards both alcohol partial oxidation and hydrogen evolution, thus serving as a dominant catalyst in glycerol electroreforming.228 For example, Johnsson et al.224 designed an advanced catalyst based on Pt nanoparticles (NPs) confined into oxygen vacancies of molybdenum oxide (MoOx/Pt) nanosheets (Fig. 6h) to initiate selective electrolysis of glycerol into glycerate. As displayed in Fig. 6i, the peak current of MoOx/Pt at 0.78 V enabled a maximum specific mass activity for glycerol oxidation, much higher than that for bare Pt NPs and Pt/C electrodes. Furthermore, theoretical simulations were further carried out to understand the intrinsic mechanism (Fig. 6j). Apparently, the first two deprotonation steps from glycerol to glycerate were more energetically favorable on MoOx/Pt. Likewise, regarding the potential-determining step (i.e., the fourth deprotonation step), a smaller free energy change on MoOx/Pt could be observed, agreeing with its high specific mass activity. Apart from glyceric acid, tartronic acid is also one of the dominant products in glycerol oxidation, and it always serves as a pharmaceutical and anti-corrosive protective agent.229 The conversion from glycerol to tartronic acid with high selectivity has been realized over Au-based catalysts.230
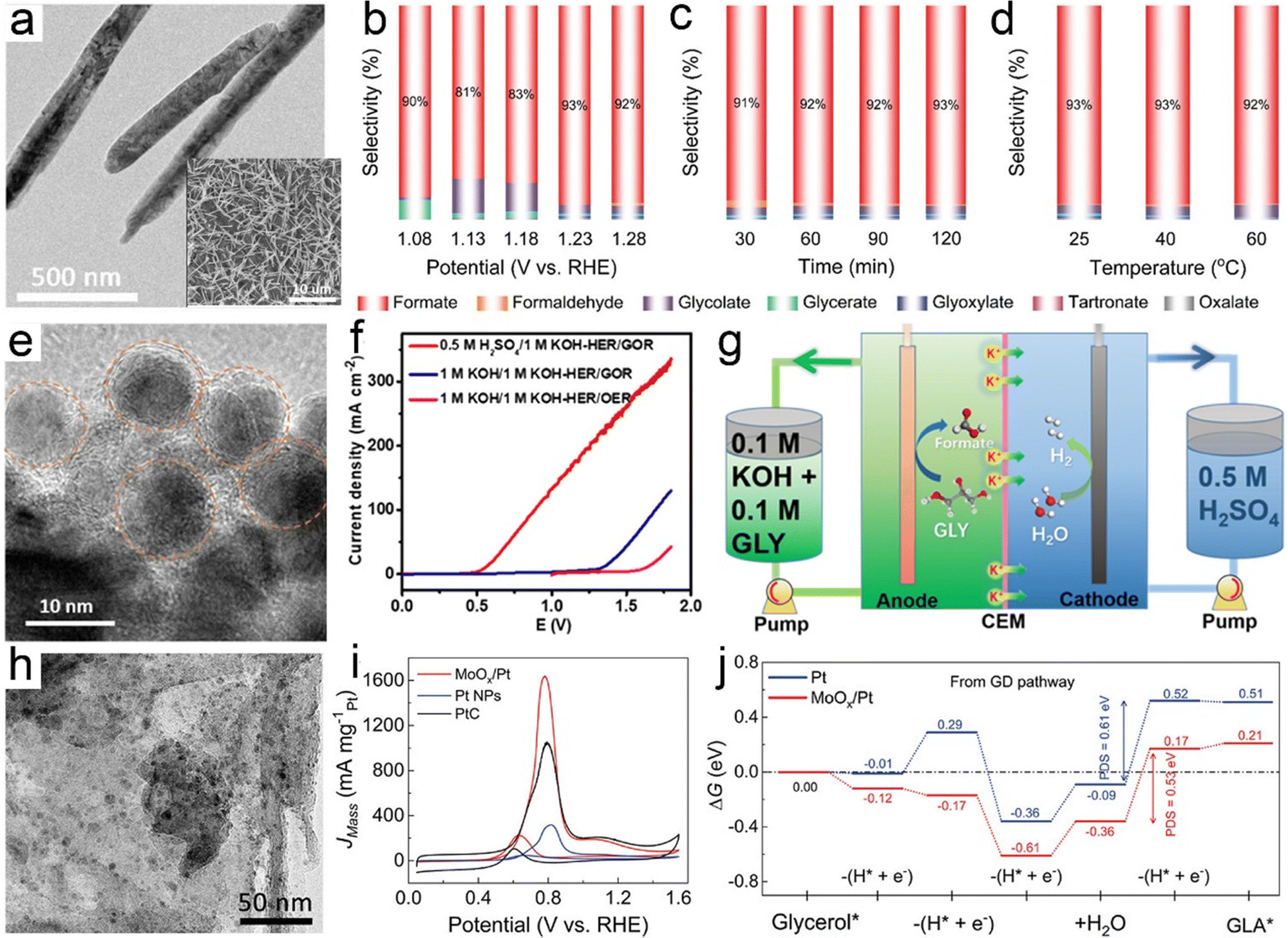 | ||
| Fig. 6 (a) TEM image of a CoMoO4/NF electrode and the inset is its SEM image. (b)–(d) Product selectivity on the CoMoO4/NF electrodes based on different reaction parameters. Reprinted with permission.224 Copyright 2022 John Wiley and Sons. (e) TEM image of a high entropy alloy, (f) the activity comparison of a hybrid electrolyzer and a traditional electrolyzer towards electroreforming of glycerol, and (g) illustration of a hybrid electrolytic cell. Reprinted with permission.225 Copyright 2022, American Chemical Society. (h) TEM image of MoOx/Pt catalysts, (i) LSV curves normalized by mass loading of Pt, and (j) calculated free energy diagrams through the glycerol oxidation pathway over Pt and MoOx/Pt. Reprinted with permission.224 Copyright 2021 John Wiley and Sons. | ||
Broadly, currently for electroreforming of glycerol with the primary goal to evolve C3 products with a large market size, especially glyceric acid and tartronic acid, rely heavily on the use of catalysts based on noble metals and their alloys (e.g., Pt, Pd, Au, PtBi, and PdRu), which successfully achieve the glycerol valorization at a relatively low potential, while suffering from high cost that render them less compatible with the cost-effective hydrogen evolution process.231–233 Moreover, another challenge for glycerol electrooxidation is the poor durability of Pt-based electrocatalysts, which is primarily attributed to their poisoned surface with adsorbed acyl and CO species.234 Such bottlenecks may be overcome by some well-established strategies, one of which is composition engineering, such as construction of electrocatalysts that comprise active catalytic-sites hybridized with poisoning-resistant materials.235
4.3 HMF
HMF derived from lignocellulosic biomass via dehydration of edible C6 carbohydrates has drawn much attention owing to its potential transformation into various furan derivatives, including 2,5-diformylfuran (DFF), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furancarboxylic acid (FFCA), and FDCA (Scheme 3).236 Among them, most notably FDCA, one of the “top 10 + 4” building block chemicals listed by the US Department of Energy, has been advocated as a viable substitute for terephthalic acid for sustainable production of polyesters, polyurethanes, and polyamides.237 Bio-plastic polyethylene furandicarboxylate (PEF) has been found to possess comparable mechanical properties to petroleum-derived PET, and has therefore been regarded as a promising alternative to PET.238 The general oxidation pathway from HMF to FDCA is illustrated in Scheme 3 and FDCA can be synthesized via the oxidation of an alcohol group to DFF or the oxidation of aldehyde to a HMFCA intermediate and their subsequent conversion to FFCA followed by the further oxidation to FDCA. Although there are well-defined lab-scale mehtods to selectively oxidize HMF to FDCA via non-electrochemical routes, they are carried out under harsh reaction conditions, such as elevated temperatures (>100 °C) and high-pressures O2 (0.3–2.4 MPa), which are potentially unfavorable for industrial scale-up processes and further lowering down the atom economy of the process.239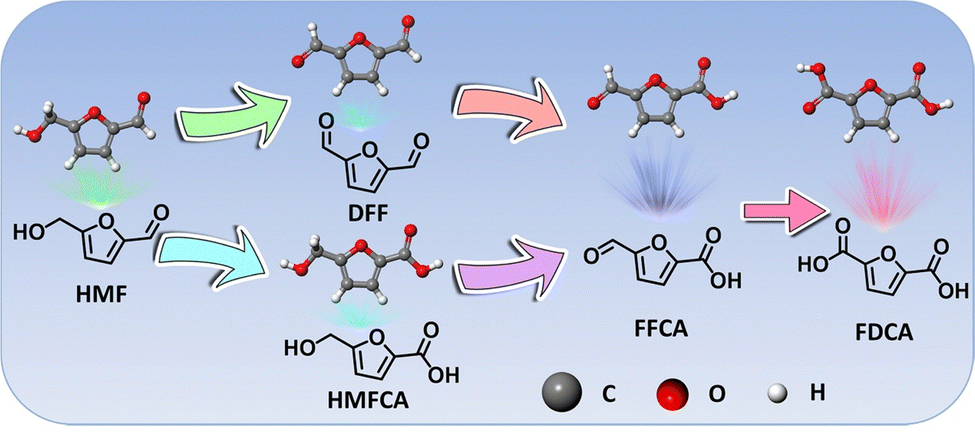 | ||
| Scheme 3 Schematic illustration of the general pathway for HMF electrooxidation. | ||
As early as 1991, Grabowski et al.240 reported electrooxidation of HMF using NiOOH and obtained a 71% product yield of FDCA after 4 h operation at 0.6 VSHE. Regrettably, continuous attention has not been paid to this topic. Until 2016, Sun et al.241 fabricated a three-dimensional (3D) Ni2P array on Ni foam (Ni2P/NF) to catalyze the oxidation of HMF to FDCA combined with hydrogen evolution from water splitting (Fig. 7a). In this case, the voltage required to reach the same current density is at least 200 mV smaller than that required in pure water electrolysis in the presence of HMF (10 mM) (Fig. 7b), and HMF electrolysis enables a high yield and selectivity to FDCA, and a H2 production rate of 120 μmol h−1 (Fig. 7c and d). In addition to Ni2P, other Ni-based materials, including Ni/NiO,242 Ni(OH)2/Cu(OH)2,243 and Au@Ni,244 have been shown to effectively transform HMF into FDCA paired with hydrogen generation. Despite the high faradaic efficiencies obtained for FDCA catalyzed by these Ni-based catalysts, the H2 production over Ni sites at the cathode remains below 150 μmol h−1. Compared with Ni, Co-based materials show a higher H2 production activity (>250 μmol h−1), as validated by Zhou et al. who utilized Co3O4 nanowires to co-produce FDCA and H2 from HMF electroreforming with a high H2 production of 270 μmol h−1.245 In another case, a significantly higher H2 evolution activity of 500 μmol h−1 from electroreforming of HMF was realized over the CoP electrocatalyst, while the selectivity to FDCA at the anode fell to 80%.246 In view of this, it is of vital importance to achieve a balance between achieving high current efficiency of the FDCA product and seeking to increase the H2 production rate.
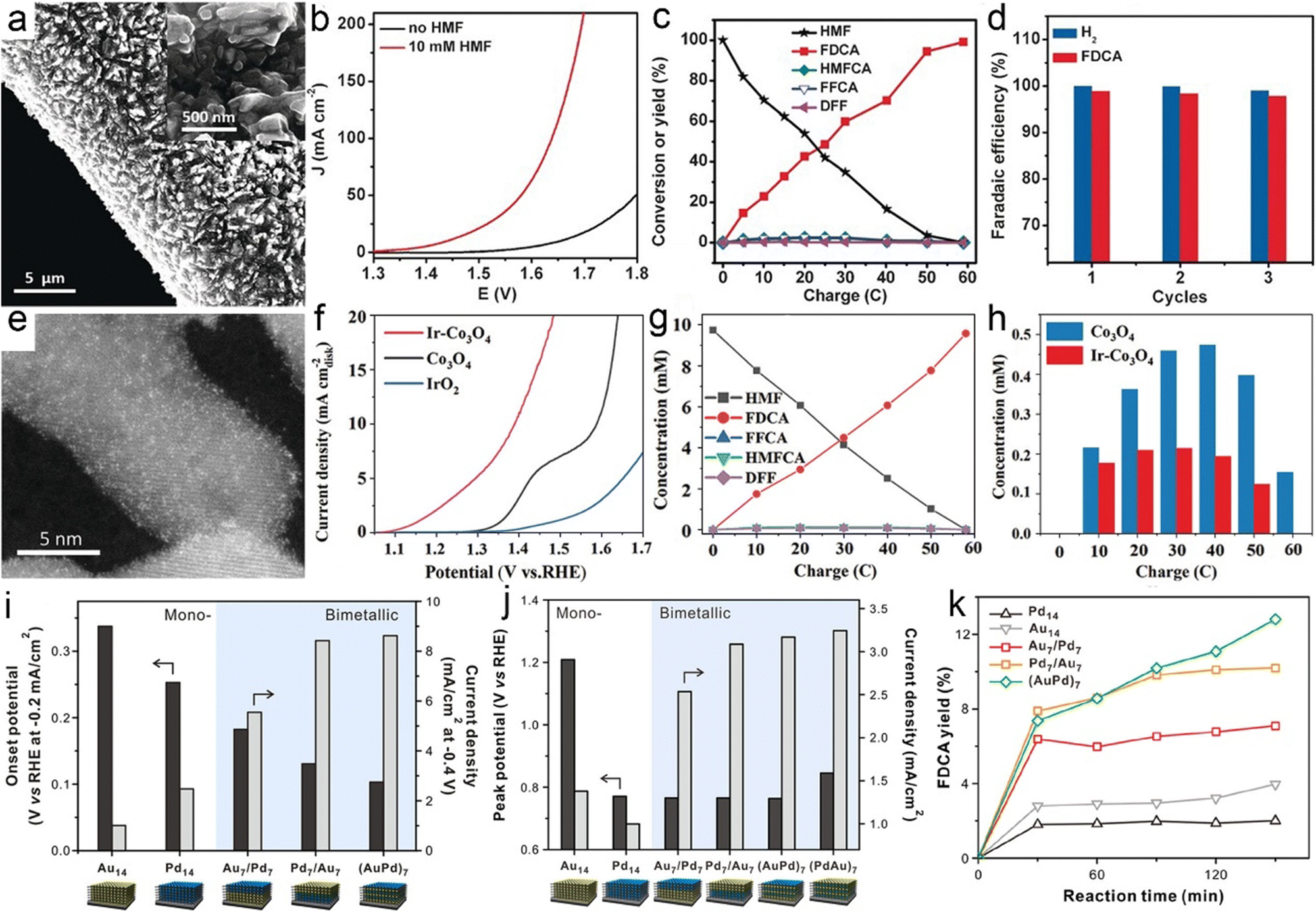 | ||
| Fig. 7 (a) SEM images of Ni2P/NF with different magnifications. (b) LSV curve comparison. (c) Conversion of HMF and the yields of the corresponding oxidation products during the electrochemical oxidation of HMF. (d) Faradaic efficiencies of the HER and FDCA evolution. Reprinted with permission.241 Copyright 2016 John Wiley and Sons. (e) STEM image of Ir-Co3O4. (f) LSV curve comparison. (g) The concentrations of substrates, intermediates, and products during electroreforming of HMF over Ir-Co3O4. (h) The intermediate concentrations over Ir-Co3O4 and Co3O4. Reprinted with permission.247 Copyright 2021 John Wiley and Sons. (i and j) Comparison of HER (i) and HMF oxidation activities (j) over different 3D hybrid electrodes. (k) Yields of FDCA products. Reprinted with permission.248 Copyright 2020, American Chemical Society. | ||
The next covered cases mainly concentrate on the oxidation half-reaction from HMF to FDCA rather than the HER half-reaction. To deeply understand the intrinsic mechanism of FDCA generation, the adsorption behavior of HMF and the identification of the main intermediate species need to be investigated. Zou et al.247 incorporated Ir single atoms on Co3O4 (Ir-Co3O4) to uncover the adsorption mechanism of HMF (Fig. 7e). As shown in Fig. 7f, the cooperative adsorption of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups of HMF on both Ir and Co catalytic sites of Ir-Co3O4 brought about a significantly lower onset potential with respect to pristine Co3O4, which was beneficial to the following FDCA formation. As expected, Ir-Co3O4 exhibited a high yield (98%) and faradaic efficiency (98%) of FDCA via the HMFCA intermediate pathway (the detected concentration of HMFCA is dramatically higher than that of DFF) (Fig. 7g and h). In parallel with the adsorption behavior of HMF molecules, the adsorbed OH− (OHad−) plays a pivotal role in HMF oxidation due to the fact that the H* derived from the dehydrogenation of hydroxyl groups needs to be combined with OHad− in an alkaline medium, that is, the competitive adsorption between OH− and HMF contributes to poor yield and selectivity to FDCA.249 To this end, tunable competitive adsorption sites based on Co3O4 with oxygen vacancies (OV-Co3O4) were constructed by Wang et al.250 From experimental studies and theoretical simulations, it was observed that the nucleophilic OH− ions were inclined to fill into OV sites prior to in combination with HMF molecules via a lattice oxygen oxidation mechanism, which was conducive to the dehydrogenation behavior of the HMFCA intermediate. As a consequence, OV-Co3O4 exhibited a high activity towards HMF electrooxidation with a low overpotential of 90 mV a current density of 10 mA cm−2 in a basic medium.
C groups of HMF on both Ir and Co catalytic sites of Ir-Co3O4 brought about a significantly lower onset potential with respect to pristine Co3O4, which was beneficial to the following FDCA formation. As expected, Ir-Co3O4 exhibited a high yield (98%) and faradaic efficiency (98%) of FDCA via the HMFCA intermediate pathway (the detected concentration of HMFCA is dramatically higher than that of DFF) (Fig. 7g and h). In parallel with the adsorption behavior of HMF molecules, the adsorbed OH− (OHad−) plays a pivotal role in HMF oxidation due to the fact that the H* derived from the dehydrogenation of hydroxyl groups needs to be combined with OHad− in an alkaline medium, that is, the competitive adsorption between OH− and HMF contributes to poor yield and selectivity to FDCA.249 To this end, tunable competitive adsorption sites based on Co3O4 with oxygen vacancies (OV-Co3O4) were constructed by Wang et al.250 From experimental studies and theoretical simulations, it was observed that the nucleophilic OH− ions were inclined to fill into OV sites prior to in combination with HMF molecules via a lattice oxygen oxidation mechanism, which was conducive to the dehydrogenation behavior of the HMFCA intermediate. As a consequence, OV-Co3O4 exhibited a high activity towards HMF electrooxidation with a low overpotential of 90 mV a current density of 10 mA cm−2 in a basic medium.
The aforementioned cases with a focus on tuning the electronic structure of electrocatalysts to boost the activity and selectivity suggest that exploring new materials for HMF oxidation has become a subject of intensive investigations. In stark contrast to this, the nanoarchitecture of the electrode (e.g., electrode thickness and location of catalytic sites) has been rarely studied thus far. A traceable work from Kim et al.248 reported a 3D hybrid electrode that consisted of negatively charged graphene oxide and two electroactive components of Au and Pd NPs via layer-by-layer assembly for coproduction of FDCA and H2 from electroreforming of HMF. As indicated in Fig. 7i–k, the 3D electrodes showed a tailorable electrocatalytic performance towards HMF electroreforming relying on composition and position of catalytic sites. Even with an identical composition of electrocatalysts (i.e., Au7/Pd7 and Pd7/Au7), the yield of FDCA over Pd7/Au7 (Au NPs at the outer layer) was higher with respect to that for Au7/Pd7.
The aforementioned cases have demonstrated the successful transformation of HMF into FDCA, with high selectivity and productivity. Nevertheless, selective conversion of HMF into DFF is barely investigated, which may be attributed to the fact that the use of alkaline electrolyte inhibits the DFF production.251 In this context, a neutral medium seems to be a better choice for the realization of HMF-to-DFF conversion, as evidenced by Wang's report,250 where the oxidation of the aldehyde group of HMF was significantly suppressed in a neutral electrolyte. Encouraged by this finding, Duan et al.252 constructed single-atom Ru supported on NiO (Ru/NiO) to selectively steer the conversion of HMF into DFF under neutral conditions, with a selectivity of up to 90%. The experimental observation combined with theoretical calculations collectively revealed that the neutral conditions played a vital role in DFF production and single-atom Ru promoted the HMF oxidation via accelerating water dissociation for OH* evolution. In spite of the successful implementation of HMF-to-DFF conversion under neutral conditions, the activity towards HMF oxidation in a neutral medium is much lower than that in an alkaline medium,250 which is deemed as a large hindrance for future industrial applications. In light of this, it is extremely desirable to explore new strategies that enable selective production of DFF under alkaline conditions. Currently, some promising approaches, including the construction of a liquid–liquid–solid system and the introduction of the salting-out effect, have shown the selective transformation of benzyl alcohol into benzaldehyde in alkali,253,254 which could be expected to extend to the DFF production. Except for DFF, there is no available literature study concerning electrochemical access to HMFCA, and HMFCA typically exists as an intermediate in FDCA synthesis.255 Current traceable work concerning HMFCA synthesis is mainly based on photocatalytic or thermocatalytic oxidation of HMF.256,257
Overall, electroreforming of HMF with the main aim of obtaining the most sought oxidation product FDCA has been successfully implemented over noble-metal and base-metal compounds. Of note, most reported HMF electrooxidation reactions over well-established electrocatalysts are carried out in an alkaline electrolyte, which is attributed to the fact that high alkalinity of the electrolyte favors the dehydrogenation of hydroxyl groups, which thus benefits the faradaic efficiency and the yield of FDCA. However, it has been reported that HMF converts into a dark brown-colored insoluble solid caused by strong alkalinity of the electrolyte (pH ≥ 13).258 Moreover, Choi et al.259 pointed out that a strongly basic solution (pH = 14) reduced the concentration of HMF (0.5 M) by more than half after 8 h, while the degradation of HMF at pH 13 remained at a low level at only 15% under the same conditions. Given the fact that the FDCA product may be partly insoluble in an electrolyte with the pH lower than 10, pH 13 seems to be a viable choice for both lab-scale electrolysis and industrial implementation.
4.4 Furfural
Similar to HMF, furfural is also one of the furanic derivatives and presents a synthetic platform for a variety of value-added chemicals (Scheme 4).260 On the other hand, furfural with a global production of up to 300 KT year−1 is mainly derived from xylose, one product of the low-cost hemicellulose, whereas HMF production relies almost entirely on the conversion of edible C6 sugars with added market value and is still on the pilot plant scale.261 In view of this, furfural seems to be a substrate of heightened interest with respect to HMF. One of the most sought products from conversion of furfural is furoic acid with broad applications in the agrochemical, cosmetic, and pharmaceutical industries. Furthermore, furoic acid has been well-acknowledged as a key intermediate to produce FDCA via carboxylation, which has been proved to be a scalable route by the preliminary technoeconomic analysis.262,263 Currently, the industrial access to furoic acid from furfural is implemented in concentrated basic solution via a Cannizzaro disproportionation mechanism, which frequently suffers from a low yield (<50%) and results in large amounts of by-products (e.g., furfural alcohol). Generally, from the viewpoint of economic viability, electroreforming of furfural into furoic acid may be the preferred option due to its substantial advantages over the conventional methods, most critically its amenability to implement under benign conditions and to pair with hydrogen evolution. For instance, Wang et al.264 implemented the electrooxidation of furfural into furoic acid under basic conditions over metallic Cu nanowires at a low potential (Fig. 8a and b). Combining with the theoretical simulations, a viable reaction pathway towards electrooxidation of furfural with a low potential was proposed based on the intermediate gem-diolate anions (GDA) that were derived from the reversible hydration of furfural (Fig. 8c). Of note, owing to being in an alkaline electrolyte, in which the Cannizzaro disproportionation reaction is known to occur, a small amount of furfuryl alcohol was accordingly generated (Fig. 8b). In another case, a furoic acid yield of up to 80% was obtained over a Ni electrode under a basic electrolyte265 and what was worth mentioning in this case was that no furfural alcohol was generated, which was ascribed to the substantially higher rate for the oxidation of furfuryl alcohol relative to that of the Cannizzaro reaction. | ||
| Scheme 4 Illustration of the reaction pathway for electrooxidation of furfural. | ||
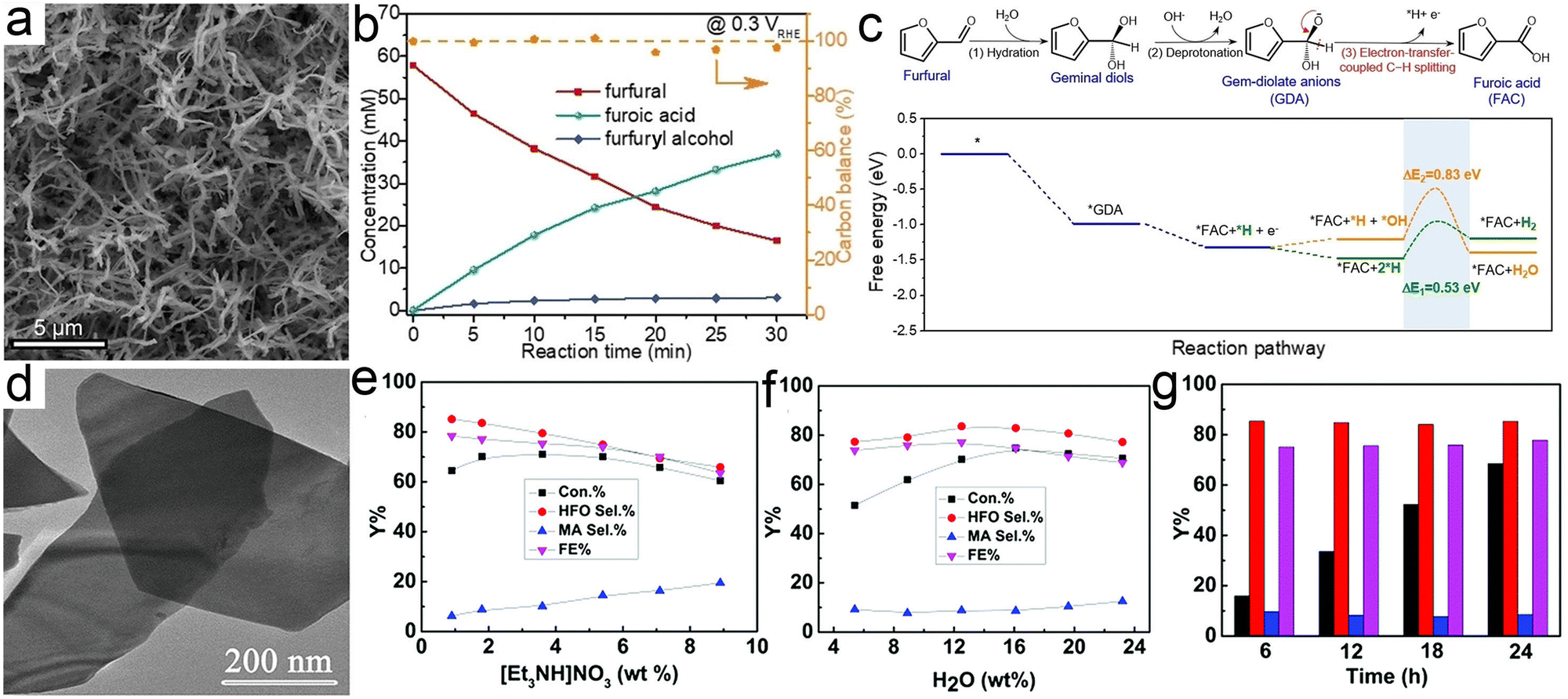 | ||
| Fig. 8 (a) TEM image of Cu nanowires. (b) Concentration variation of feedstocks and products as a function of time for chronoamperometric tests at 0.3 V vs. RHE over Cu nanowires. (c) Illustration of the reaction pathway from furfural to furoic acid and the corresponding free-energy diagram. Reprinted with permission.264 Copyright 2022 John Wiley and Sons. (d) TEM image of CuS nanosheets. (e) and (f) The effects of the concentrations of [Et3NH]NO3 (e) and water (f) on furfural conversion. (g) The long-term operation for furfural electrooxidation over CuS nanosheets. Reprinted with permission.266 Copyright 2019 Royal Society of Chemistry. | ||
Electrooxidation of furfural into furoic acid has been widely investigated and with the exception of one recent report,261 each case has involved the use of either a nonaqueous or an alkaline electrolyte that is less representative of biomass hydrolysis feedstocks. As a matter of fact, compared with basic conditions, one of the major advantages for acidic environments is their ability to inhibit the disproportionation product from the Cannizzaro reaction. Among the various metallic electrocatalysts, Pt stands out owing to its corrosion-resistant nature in acid as well as the high activity and stability within ethanol or dimethyl ether oxidation reaction, especially in the case of the electrooxidation process on the (111) plane of Pt.267 One systematic study from Holewinski et al. reported that the high selectivity to furoic acid from furfural electrooxidation was achieved on the metallic Pt sites below 1.2 V vs. RHE,261 while selectivity shifts towards 5-hydroxy-2(5H)-furanone (HFO) above 1.2 V vs. RHE, with a parallel pathway for maleic acid production as well. The low selectivity to CO2 over HFO and maleic acid at a high potential (>1.2 V) in this case was attributed primarily to the high surface coverage of organic intermediates that restricted the adsorption of oxygen from dissociation of water molecules.
The next most desired product from electrooxidation of furfural is 5-hydroxy-2(5H)-furanone (HFO), which is an essential component of many bioactive compounds.260 The general route to HFO is implemented via direct thermal-oxidation of furfural, which however suffers from a poor selectivity to HFO owing to its amenability to further oxidation into maleic acid under thermocatalytic conditions.268 Benefiting from the easy access to the active oxygen species from water dissociation, electrocatalysis is thus recognized as a promising approach to generate HFO from electrooxidation of furfural. Unfortunately, scanty attention has been paid to investigate the electrochemical pathway from furfural to HFO and only one traceable study reported by Han et al.266 demonstrated the transformation of furfural into HFO over CuS nanosheets using H2O as the oxygen source (Fig. 8d). In this case, the composition of the electrolyte had a significant impact on the selectivity to the HFO product. As shown in Fig. 8e and f, the high concentrations of [Et3NH]NO3 and H2O well favored the generation of hydroxyl radicals, which nevertheless contributed to the further oxidation of HFO to maleic acid. On the other hand, the low concentration of [Et3NH]NO3 failed to provide a good electric conductivity, while the dissociation of ion pairs in [Et3NH]NO3 mediated by water remained elusive when the concentration of water was significantly reduced. With optimal concentrations of [Et3NH]NO3 and water, high selectivity (83.6%) and conversion (70.2%) to HFO were realized via multistep oxidation of furfural, including C–C cleavage, ring opening and oxidation, and intramolecular isomerization (Fig. 8g).
The interest in maleic acid, with respect to HFO, arises from its greater market demand, with a global production of up to 25 KT year−1 that are mainly used to fabricate polyester resins and flavorants.269 Ring-opening of furan is the key step across the reaction pathway from furfural to maleic acid, which is accelerated by the use of a basic medium. For example, Choi et al.270 chose three typical catalysts: Pt, MnO2, and PbO2, all of which maintained durability in acidic electrolytes. Nonetheless, through the experimental investigations, PbO2 was found to be the only catalyst that was capable of converting furfural to maleic acid with a yield of 65.1%. The reaction pathway over PbO2 proceeded via 2-furanol as an intermediate. Maleic acid can also be electro-synthesized via a tandem reaction (i.e., production of HFO followed by its further conversion to maleic acid).271 In another case, Han et al.272 used metal-free Se-doped C3N4 to convert furfural to maleic acid in 0.5 M KHCO3 solution, with a yield of up to 84.1%. Of particular note in this case is the effect of the substituents on the furan ring on the selectivity to maleic acid, following the order: carboxyl group > aldehyde group > hydroxyl group (the furan ring with a –COOH group enabled the highest selectivity to maleic acid). In spite of several advances in electrosynthesis of maleic acid achieved recently, there is not yet consensus on the exact mechanism of maleic acid production from electroreforming of furfural, indicating more efforts (e.g., computer simulations and in situ spectroscopy) should be made to reveal the mechanism of maleic acid formation.
One outstanding advantage of electroreforming of furfural into commodity chemicals (e.g., furoic acid, HFO, and maleic acid) is the use of lower-cost furfural feedstock, compared with electroreforming of the other waste substrates, which can be directly produced from corncobs. Regrettably, the impurities involved in raw furfural may result in the deactivation of electrocatalysts, and the extra distillation of low-grade furfural to pure furfural is detrimental to the economic viability of furfural electroreforming. Thus, more attention should be paid to focus on the development of impurity-resistant electrocatalysts, especially metal-free materials, with robust performance in concentrated furfural.
4.5 Glucose
Glucose, a product of photosynthesis in plants, is known as a main energy source in living organisms. At present, glucose derived from chemical sectors, especially pulp and paper processing industries, has served as a major biomass waste.273 Furthermore, it has been shown that nearly all glucose molecules in aqueous solution exist in the form of pyranose rings that feature chemically active hemiacetal groups. In light of this, glucose offers convenient access to various commodity chemicals via electrolysis, including gluconic acid (GNA), glucaric acid, and levulinic acid (Scheme 5). Among these compounds, GNA needs to be highlighted in particular as a vital intermediate with wide uses in production of food, beverages, and pharmaceuticals. Wen et al.156 used iron-doped cobalt diselenide (Fe-CoSe2) in an alkaline–acidic asymmetric electrolytic cell to initiate glucose oxidation at the anode paired with cathodic HER, with a high electrocatalytic performance in alkali for yielding GNA and hydrogen evolution in acid (∼90 μmol h−1). In another study, Breugelmans et al.274 evaluated the electrooxidation of glucose to GNA on a wide range of metallic electrodes (Cu, Pt, and Au) and observed the highest selectivity up to 86.6% for the Au electrode at 0.55 VRHE, while Pt as a benchmark electrocatalyst for alcohol oxidation only enabled 78.4% selectivity. The factors governing the high selectivity to GNA over Au have been deeply understood and summarized by the following four points: (1) more negative potential of Au for glucose oxidation as compared to other metals; (2) poor activity of Au in C–C bond breakage or CC bond isomerization to avoid undesired reaction pathways; (3) low competitive adsorption of the ionic species (e.g., OH−) and the glucose substrate over Au sites; and (4) extraordinary chemoselective inertness of Au for the oxidation of secondary alcohols. Of particular note in this case was the role of Cu in glucose electrooxidation, and Cu played a crucial role in selective oxidation of the alcohol group via avoiding competition of the OER owing to its poor catalytic performance for the OER as reported previously,259 whereas the Cu electrode in this case well favored the C–C bond scission, thus resulting in a mixture of gluconic, glucaric, and formic acid, and a poor selectivity to GNA.
 | ||
| Scheme 5 Schematic illustration for glucose electrooxidation pathways. | ||
What is more valuable than the GNA product is the access to glucaric acid (GRA) with an immense market value of USD 550.4 million in 2016 and estimated to reach USD 1.3 billion by 2050, which was identified as a top value-added chemical derived from biomass by US Department of Energy as early as 2004.275 GRA and its derivatives (e.g., GRA salts and GRA-1,4-lactone) are not only viewed as the building blocks for the production of detergents, polymers, and anticorrosion reagents, but also serve as drugs for cancer prevention.276 The industrial production of GRA is currently carried out by thermal oxidation of glucose using HNO3 with a low yield of nearly 40% or selective oxidation of cyclic nitroxides in the presence of ClO−/Br− with a high yield of approximately 90%.277 Notably, these two methodologies involve the use of corrosive and toxic reagents, thereby giving rise to a mass of hazardous side-products. In this context, realization of the conversion from glucose to GRA via electroreforming seems to be economically competitive and environmentally friendly relative to the conventional catalytic oxidation. In general, GRA is electrochemically converted from glucose via a two-step process: oxidation of glucose to GNA and oxidation of GNA to GRA (Scheme 5). Of the two steps, the latter is more difficult because the desorption capability of GRA on metallic sites remains poor, thus blocking catalytic sites to adsorb new generated GNA required to be further oxidized, which is actually a significant problem that is nonetheless seldom mentioned in previous reports.278 Regarding the electrooxidation of glucose to GRA, a notable case recently reported by Yu and Huber has to be emphasized.279 They fabricated Ni foam supported NiFe oxide (NiFeOx-NF) and nitrides (NiFeNx-NF) as an anode and a cathode, respectively, to motivate the GRA production from glucose oxidation combined with the HER in a basic medium (Fig. 9a). The constructed electrolyzer delivered a small overpotential of 160 mV to reach 100 mA cm−2 for glucose oxidation, outstanding long-term stability, and a faradaic efficiency and yield of 87% and 83% for GRA, respectively (Fig. 9b and c). To be more rigorous, they presented a techno-economic analysis to evaluate the economic feasibility of glucose electroreforming via currently used chemical routes. As displayed in Fig. 9d, compared to non-electrocatalytic routes, the glucose electroreforming strategy enabled a higher GRA yield, lower downstream separation cost, shorter reaction time, smaller E-factor (i.e., the mass ratio of the generated waste to target products), and less negative environmental impact.
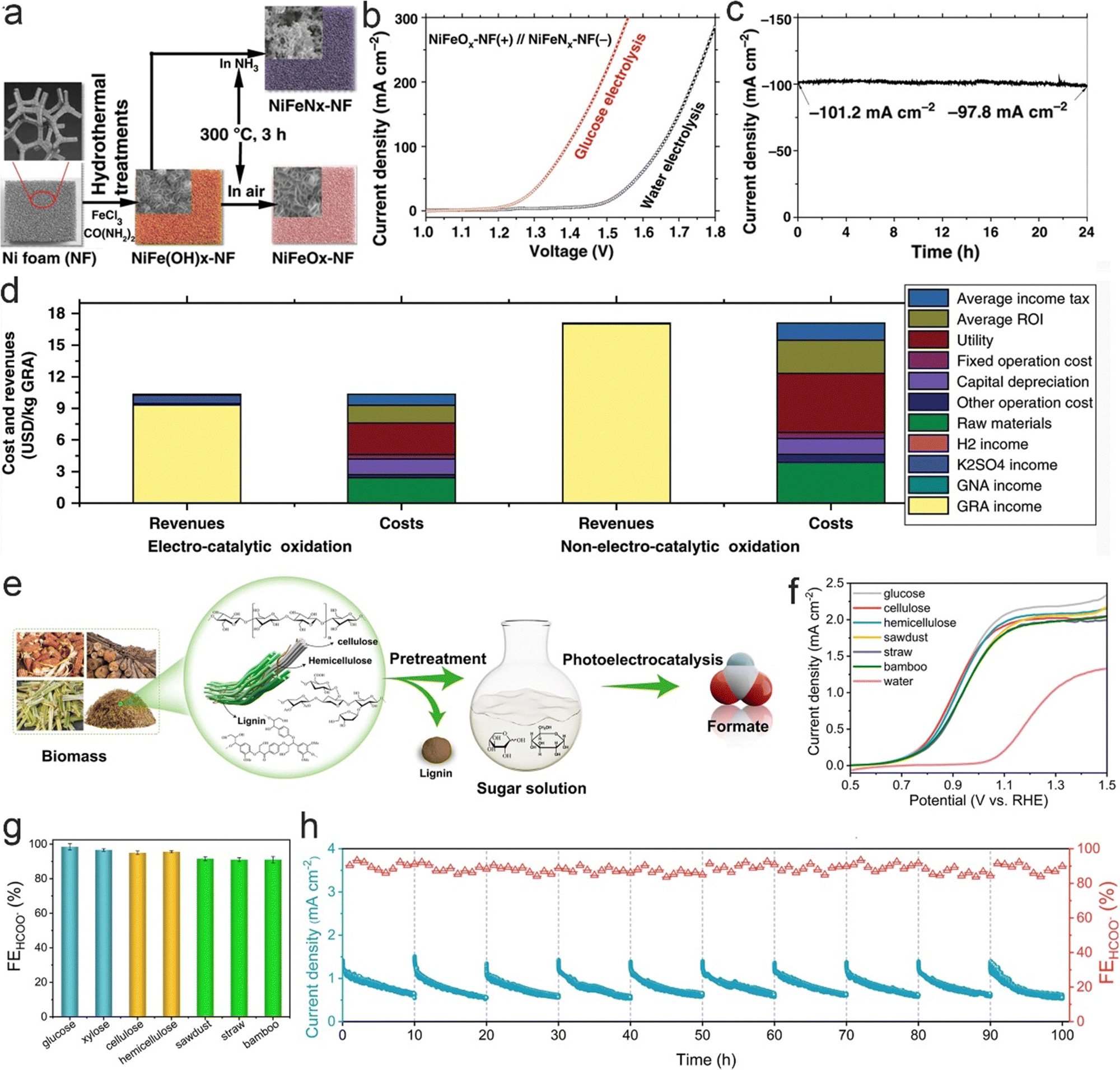 | ||
| Fig. 9 (a) Schematic illustration for the preparation of NiFeNx-NF and NiFeOx-NF catalysts. (b) Comparison of glucose electroreforming and water electrolysis over cathodic NiFeNx-NF and anodic NiFeOx-NF catalysts. (c) Long-term durability tests of glucose electrolysis. (d) Evaluation of revenues and costs between electrocatalysis and non-electrolysis routes towards the conversion from glucose to GRA. Reprinted with permission.279 Copyright 2020 Nature Publishing Group. (e) Schematic illustration for the formate production from lignocellulosic glucose. (f) LSV curves of different substrates over NiOOH/α-Fe2O3. (g) Faradaic efficiencies to formate at 1V for 2h over NiOOH/α-Fe2O3 using different feedstocks. (h) Chronoamperometry data and faradaic efficiencies for formate evolution at 1V in sawdust-derived sugar solution over NiOOH/α-Fe2O3 for 100h. Reprinted with permission.280 Copyright 2023 Nature Publishing Group. | ||
Instead of pure glucose, the use of a low-cost source of lignocellulosic glucose as a feedstock seems to be more compatible, especially in the case of more inexpensive sawdust, straw, and bamboo, which further reduce the production cost of GRA and GNA, however remains challenging. Currently, the electrochemical valorization of raw lignocellulosic glucose into formic acid has been realized by Chu et al. who utilized a facile acidic pretreatment strategy to depolymerize the real-word cellulosic glucose into water-soluble sugar fragments and then carried out the electrochemical methods for formic acid evolution (Fig. 9e).280 A series of raw-biomass-derived glucose solutions could be selectively converted into formate over NiOOH/α-Fe2O3, with high productivity and faradaic efficiency (Fig. 9f–h). The depolymerization step in this case was carried out using dilute sulfuric acid via a thermal treatment, which increased the energy input and the environmental risk. In this regard, it necessitates exploration of bifunctional electrocatalysts with acidic and metallic sites capable of processing raw lignocellulosic biomass directly.
As a whole, electroreforming of glucose into hydrogen and commodity chemicals (e.g., GNA, GRA, and formate) has been already implemented on the lab-scale, showing its potential to replace the conventional thermal routes currently industrialized. However, much effort is still needed to tackle some drawbacks identified in aforementioned investigations, such as the leaching of active components in electrocatalysts. Taking example of Au-based electrocatalysts,284 although Au-based electrodes are less prone to deactivation by product adsorption, the leaching of Au is substantial (up to 70 wt% with 6 cycles), which is responsible for low glucose conversion and high selectivity to desirable products, thus necessitating the catalyst reutilization via minimizing the leaching of active components.
5. Mechanism investigation
In view of the above-mentioned cases, transition-metal-based materials have been deemed as catalysts for electrooxidation of organics. The surface or even bulk reconstruction in catalysts during electroreforming has been widely accepted. For instance, the in situ generated CoO2 motifs over CoOOH is regarded as active species for biomass oxidation via “electrochemical–chemical”285 or “hydride transfer”286 mechanisms, whereas the direct evidence for Co4+ ions remains lacking. Therefore, deciphering the real catalytic sites is vitally important to establish a clear structure–activity relationship within electroreforming of solid waste.5.1 Operando spectroscopy
In situ/operando spectroscopy is recognized as a powerful tool to investigate catalyst structural adaption during electrocatalysis, with well-documented studies reported concerning potential-dependent atomic configuration of materials for electrooxidation of organics.287–289 Duan et al.281 employed in situ Raman tests and corroborated with operando X-ray absorption spectroscopy, to reveal the real active sites for glucose oxidation. As revealed in Fig. 10a, the Co–O mode in CoOOH occurred when the potential increased from 1.1 to 1.3 V, which then disappeared once the potential exceeded 1.3 V. Meanwhile, the high-valency Co4+–O species (i.e., CoO2 motifs) were formed when the potential ranged from 1.4 to 1.6 V. Interestingly, once glucose was introduced, Co4+–O species disappeared and the COOH mode re-appeared, showing that the CoO2 motifs were the real catalytic sites for glucose oxidation. Operando XAS spectroscopy, as a complement to the Raman results, was further carried out, with the aim of investigating the oxidation state of Co in CoOOH under catalytic conditions. By comparing the potentiodynamic Co oxidation state in the electrolyte with/without glucose (Fig. 10b and c), it was deduced that the Co4+–O species and partially reduced Co3+–O sites were available for glucose oxidation. In another case, Chen et al.282 studied systematically that how the proton and oxygen anion (de)intercalation processes in hydroxide catalysts govern the elementary reaction steps in glycerol electrooxidation. As demonstrated in Fig. 10d–f, the Raman band for NiOOH appeared on the surface of Ni hydroxide throughout the potential windows in KOH electrolyte but could be not observed when glycerol was injected. By contrast, the Raman modes, corresponding to NiOOH or CoOOH, were observed when the electrochemical measurements were carried out over both NiCo and Co hydroxide, regardless of being in KOH electrolyte with/without glycerol. These results showed that the proton deintercalation from the lattices of the NiCo and Co hydroxide was preferred relative to the glycerol dehydrogenation process.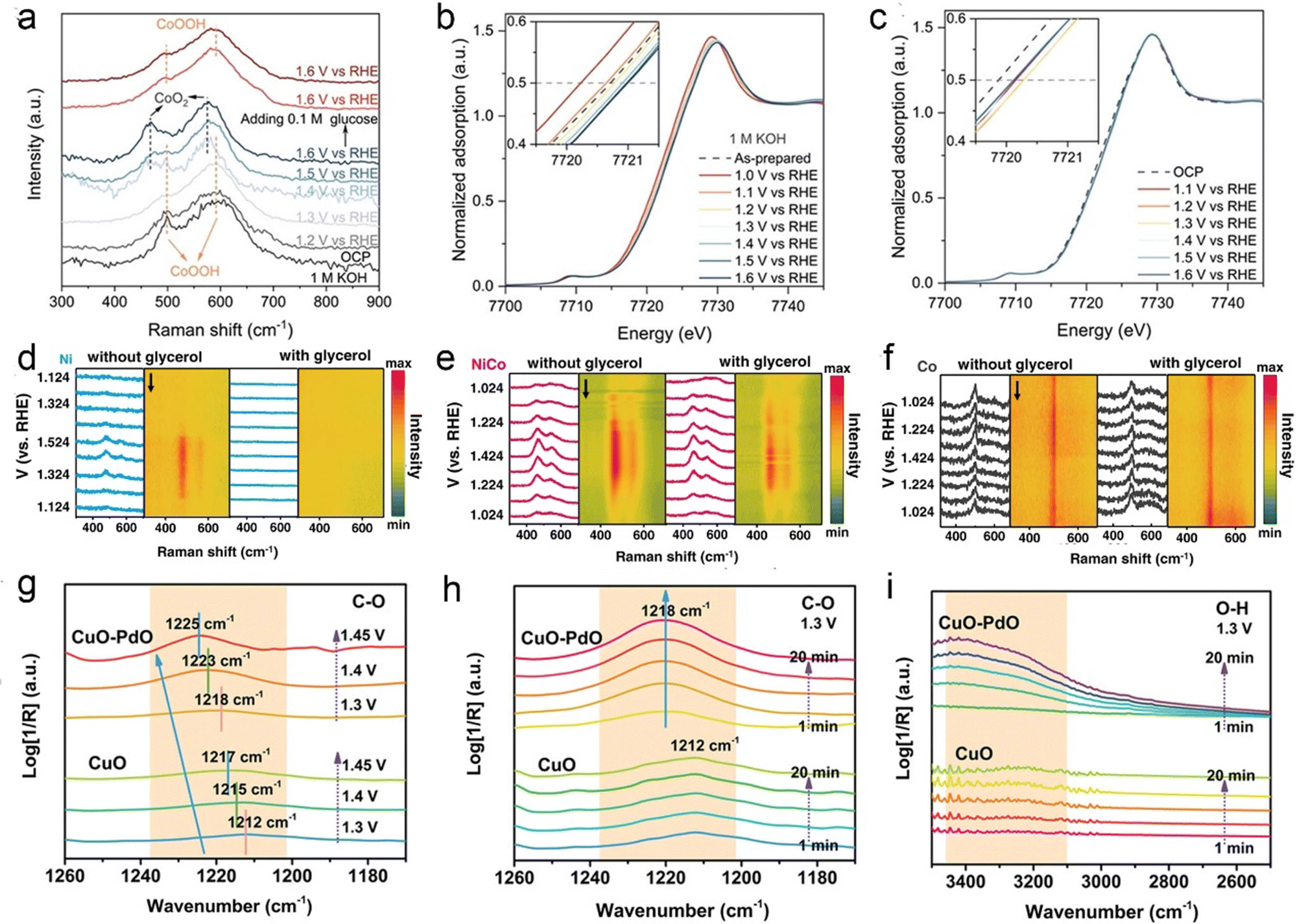 | ||
| Fig. 10 (a) In situ Raman spectra of CoOOH during the OER and glycerol oxidation. (b,c) XANES profiles of CoOOH in 1 M KOH without (b)/with (c) 0.1 M glycerol. Reprinted with permission.281 Copyright 2023 John Wiley and Sons. (d)–(f) In situ Raman spectra for (d) Ni hydroxide, (e) NiCo hydroxide, and (f) Co hydroxide in 1 M KOH without (left) and with (right) 0.1 M glycerol. Reprinted with permission.282 Copyright 2022 Nature Publishing Group. (g) In situ ATR-FTIR spectra of the CuO and CuO–PdO catalysts at different potentials during HMF electrooxidation. (h) In situ ATR-FTIR spectra of the CuO and CuO–PdO catalysts at 1.3 VRHE. (i) In situ ATR-FTIR spectra of the CuO and CuO–PdO catalysts. Reprinted with permission.283 Copyright 2022 John Wiley and Sons. | ||
Apart from elucidating the catalyst structural evolution under reaction conditions, understanding in depth the reaction mechanism is essential to design a high-performance catalyst, but challenging owing to a broad spectrum of reaction intermediates, products, and by-products involved in plastic-/biomass-derived feedstock electrooxidation. In situ Fourier transform infrared (FTIR) spectroscopy is a widely used means to shed light on the adsorption ability of the intermediates and the reaction pathway towards oxidation of organics. Zou et al.283 utilized in situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy to investigate the adsorption properties of the reaction intermediates over CuO before and after introduction of PdO (Fig. 10g–i). With the increase of the potential, the intensity of the peak at 1212 cm−1, corresponding to the C–O stretching vibration peak of the HMF substrate and intermediates, increased more obviously over CuO–PdO relative to bare CuO (Fig. 10g and h), implying that more HMF molecules were enriched on CuO–PdO. Meanwhile, the CuO–PdO interface allowed a stronger adsorption capability of OH species than CuO, as evidenced by its higher O–H vibration peak intensity (Fig. 10i). It should be noted that if the reaction intermediates share a lot of structural similarities, they are hard to distinguish using in situ FTIR spectroscopy. Taking the glycerol oxidation as an example,290 aldehydes and carbonyl acids as intermediate products contain carbonyl groups, which makes it difficult to identify them via only observing the typical peak at around 1740 cm−1 (carbonyl group).
Broadly, operando or in situ spectroscopic techniques have definitely provided useful information on the catalyst structure dynamics and reaction mechanisms. However, there are some limitations present in the in situ spectroscopy study. A major issue is that the spectra are recorded at an ensemble level, that is, an average signal of all responsive reaction species is collected, which may hide the key information on the real catalytic species and thus be misleading.291 Only when each species on the catalyst is identical and clearly defined will the obtained spectra reflect the real information on each individual site. Given all this, the rational design of electrocatalysts with a well-defined structure is extremely crucial for deciphering the real active species and reaction mechanism using in situ spectroscopy.
5.2 Theoretical simulations
In parallel to significant efforts made for reaction mechanism investigation by using advanced in situ spectroscopy, computational simulations as a supplementary means are carried out in an orderly manner. In our recent work, to highlight the advantages of a Pt/NiOOH heterostructure for the selective transformation of ethylene glycol to glycolic acid with respect to bare Pt and NiOOH, the adsorption ability and the reaction pathway are calculated.211 As shown in Fig. 11a, the Pt/NiOOH interface enabled the lowest adsorption energy for ethylene glycol among all the models, indicating the favorable reactant adsorption process over Pt/NiOOH. Meanwhile, the reaction pathway on Pt/NiOOH, with bare Pt as a reference model, was studied (Fig. 11b). The results showed that the free energy change of the potential-determining step (PDS) was thermodynamically uphill over bare Pt, but was downhill over Pt/NiOOH, revealing that the process for glycolic acid evolution was promoted by the design of the Pt/NiOOH interface. In a similar vein, Hu et al.292 utilized density functional theory (DFT) calculations to confirm the reaction pathway from HMF to FDCA over InOOH and oxygen-vacancy-rich InOOH (InOOH-OV). As displayed in Fig. 11c, both the InOOH and InOOH-OV models preferred the path that yields the HMFCA intermediate rather than that forms DFF. In the favorable pathway, the free energy change of the PDS, namely the step from HMFCA to FFCA, was determined to be 0.54 and –0.41 eV for InOOH and InOOH-OV, respectively, which imply the promoted oxidation process from the hydroxyl into aldehyde group over InOOH-OV. In addition to the reaction pathway study, the d-band center of the catalyst has always been regarded as a descriptor to explain the organic oxidation performance, especially the product selectivity.293 For instance, the d-band center of Pd for Pd/Ni(OH)2 allowed a downshift as compared to that for bare Pd, suggesting a weaker binding strength of carbonyl intermediates over Pd/Ni(OH)2 during ethylene glycol oxidation.212 Such a desorption ability prevented the over-oxidation of the glycolic acid product, thus gaining better selectivity to glycolic acid over Pd/Ni(OH)2.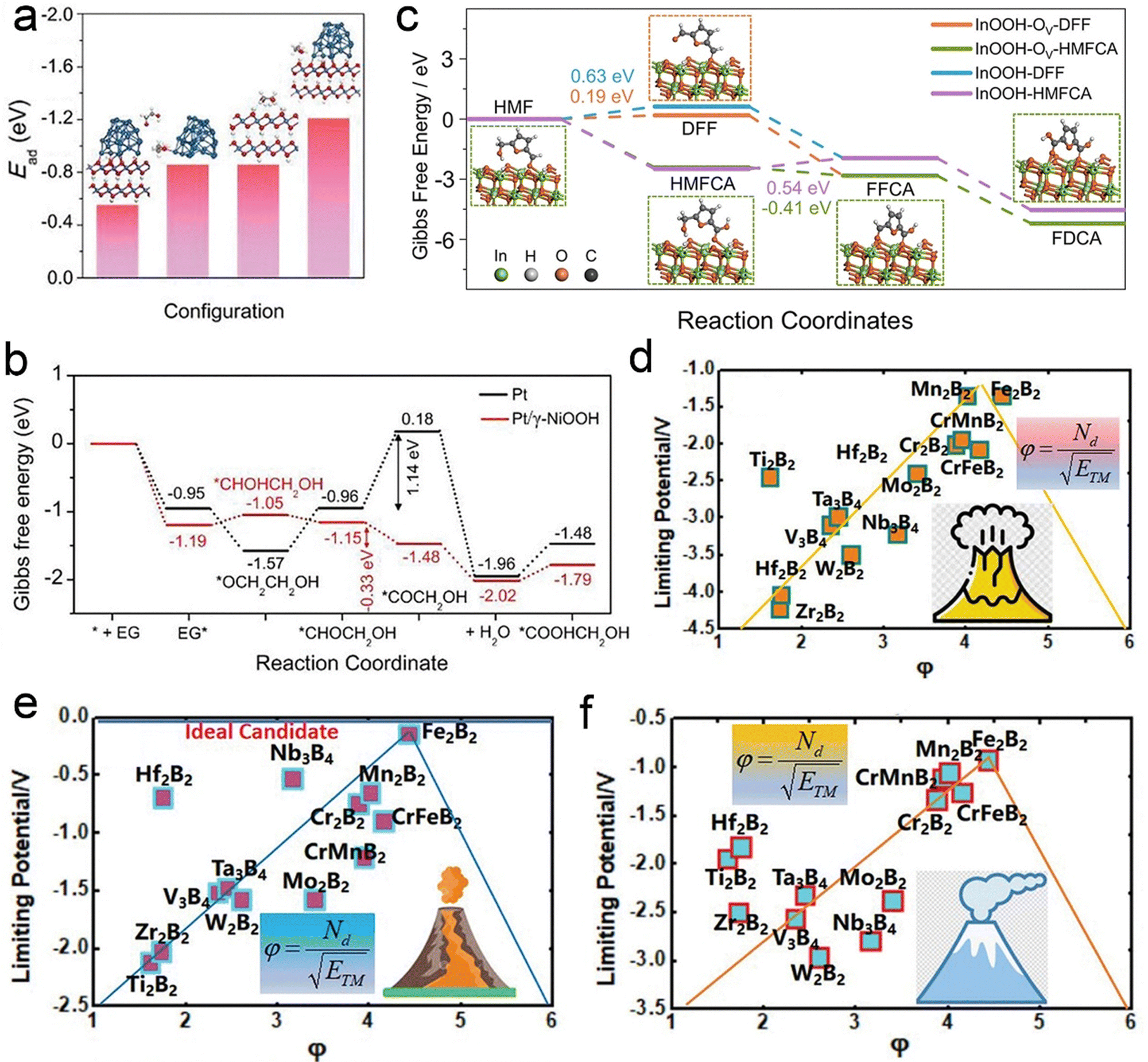 | ||
| Fig. 11 (a) The calculated adsorption energy for ethylene glycol over various catalysts. (b) The reaction pathways from ethylene glycol to glycolic acid over Pt/NiOOH and bare Pt. Reprinted with permission.211 Copyright 2023 John Wiley and Sons. (c) Free energy diagrams for HMF oxidation over InOOH and InOOH-OV. Reprinted with permission.292 Copyright 2023 Nature Publishing Group. (d–f) Scaling relation plot for furfural-upgrading limiting potentials of MBenes as a function of the descriptor ϕ along the FAC (d), HDPO (e), and HFO formation (f), and the insets are the formulas for ϕ calculation. Reprinted with permission.294 Copyright 2023 John Wiley and Sons. | ||
Theoretical simulations are also used to realize a high-throughput screening of catalysts towards electrooxidation of solid-waste-derived feedstocks. Li et al.294 calculated the limiting potentials for furoic acid (FAC), 6-hydroxy-2,3-dihydro-6H-pyrano-3-one (HDPO), and 5-hydroxy-2(5H)-furanone (HFO) over all the transition-metal boride (MBene) catalysts and established three scaling relations (the limiting potential as a function of ϕ) based on Sabatier's principle (Fig. 11d–f). The calculation results demonstrated that Mn2B2, Fe2B2, and Nb3B4 were well-suited for FAC generation, while Mn2B2, Fe2B2, and CrMnB2 enabled high activity for the oxidation of furfural to HDPO with low limiting potentials. It should be noted that the ϕ value in this case is considered as a descriptor of material properties developed for screening catalysts. Currently, the emergence of artificial intelligence (AI) may accelerate the screening process, significantly improving algorithms and developments in data science. Actually, machine learning (ML), a simple and practical AI framework based on computer and statistical science, has been used to develop electrocatalysts for CO2 reduction.295 The application of ML in solid waste upgrading will be expected to present a paradigm shift, in which the next-generation electrocatalysts will be designed and synthesized.
Conclusions and prospects
Throughout this review, we have summarized recent advances in applying electroreforming strategies to transform solid waste derivatives into value-added commodities. Meanwhile, pretreatment methods for raw plastics and biomass are discussed in detail, and the techno-economic evaluation of electroreforming, compared with conventional overall water electrolysis, is carried out. In spite of the continuous progress and the vast potential opportunities, a wide array of challenges need to be tackled to make electrochemical upcycling of solid waste into a reality (Fig. 12), ranging from the technology flexibility for converting complex feedstocks to the design of catalysts with stability and impurity tolerance. | ||
| Fig. 12 Research perspective for valorization of solid waste. | ||
To begin with, the chemical diversity and complexity of raw solid waste present a large barrier to realize electroreforming, that is, pretreatment is of great necessity. Taking plastics as an example, post-consumer plastics are physically mixed and include complex materials (e.g., multilayer packages), and most of the pretreatment methods, including chemical and biological processes, only favor the selective depolymerization of single plastic stream. Therefore, developing a pretreatment strategy that allows the deconstruction of mixed plastics or biomass into water-soluble platform molecules without sorting seems to be desirable. Currently, depolymerization methods for biomass are widely studied, but remain rare for mixed plastics. One traceable study by Beckham et al.296 reported that metal-catalyzed oxidation depolymerized comingled plastics into oxygenated small molecules that could aid catalytic conversion. Essentially, such a strategy for processing the mixed solid waste can be paired with subsequent electrochemical valorization, which may be revolutionary in the domain of chemical electrosynthesis.
Second, the pretreated solid waste contains not only organic feedstocks but also inorganic ion impurities inherent in the solid waste, such as K, Ca, Zn, Fe, Al, and S,69 and these alkali ions may be inert as they are components of commonly used electrolytes. Other metallic ions, namely Zn, Fe, and Al, are detrimental to the electrochemical reactions, in the particular case of their deposition on the electrocatalyst surface. The remaining sulfur compounds are usually present at below the 200 ppm level, but maybe lead to catalyst poisoning when they exist as a form of thiols or sulfide ions. Thus, exploring catalysts with long-term durability and impurity tolerance and high-performance membrane is needed, with the aim of realizing a transition from single-component system to a more realistic system.
Third, the electrocatalysts currently developed are in general not feedstock-flexible, but are operable over a specific feedstock (e.g., alcohol, aldehyde, and carboxylic acid). By contrast, the feedstocks derived from solid waste, including raw biomass and plastic streams, are inherently multi-component and heterogeneous. On the other hand, reactors favor the operation over a range of potentials, whereas the networks of the renewable energy derived from intermittent power are not well developed and conventional electrochemical systems seem to perform poorly. In light of this it necessitates the exploration of next-generation catalysts and reactors that are tailored for complex feedstocks and intermittent power.
Last, but not least, although electroreforming strategies achieve the upcycling of solid waste into commodity chemicals and concurrent hydrogen evolution via cathode water reduction, there is immense research space for further broadening the utilization prospect. For instance, anodic organics oxidation can be paired with CO2 or CO reduction reaction,297,298 which aims to generate higher-value C1 or C2+ chemicals at the cathode relative to hydrogen from the HER. Moreover, co-production of the same valued chemicals from the two half-cells using different cathode and anode feeds or even the sole feed seems to be economically competitive and appealing as it avoids the extra system complexity and separation cost.299–301 Overall, it is conceivable that the integration of the abundant available solid waste, a broad range of catalysts and reactors, and renewable electricity will undoubtedly make a breakthrough in the chemical supply chain.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFC2103600), the Fundamental Research Funds for the Central Universities, and the Natural Science Foundation of Jiangsu Province (BK20210382).Notes and references
- D. M. Kammen and D. A. Sunter, Science, 2016, 352, 922–928 CrossRef CAS PubMed.
- N. S. Diffenbaugh and M. Burke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9808–9813 CrossRef CAS PubMed.
- J. A. Dowling, K. Z. Rinaldi, T. H. Ruggles, S. J. Davis, M. Yuan, F. Tong, N. S. Lewis and K. Caldeira, Joule, 2020, 4, 1907–1928 CrossRef.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- M. Grätzel, Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 26–32 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- P. Erickson, M. Lazarus and G. Piggot, Nat. Clim. Change, 2018, 8, 1037–1043 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- P. Denholm, D. J. Arent, S. F. Baldwin, D. E. Bilello, G. L. Brinkman, J. M. Cochran, W. J. Cole, B. Frew, V. Gevorgian and J. Heeter, Joule, 2021, 5, 1331–1352 CrossRef.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- S. Kong, X. Lv, X. Wang, Z. Liu, Z. Li, B. Jia, D. Sun, C. Yang, L. Liu and A. Guan, Nat. Catal., 2023, 6, 6–15 CrossRef CAS.
- W. Liu, P. Fu, Y. Zhang, H. Xu, H. Wang and M. Xing, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218813120 CrossRef CAS PubMed.
- B. Qiu, C. Zhong, M. Xing and J. Zhang, RSC Adv., 2015, 5, 17802–17808 RSC.
- J. P. Lemmon, Nature, 2015, 525, 447–449 CrossRef CAS PubMed.
- K. M. Jablonka, C. Charalambous, E. Sanchez Fernandez, G. Wiechers, J. Monteiro, P. Moser, B. Smit and S. Garcia, Sci. Adv., 2023, 9, eadc9576 CrossRef PubMed.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustainability, 2021, 4, 383–391 CrossRef.
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2011, 100, 410–426 Search PubMed.
- B. Qiu, Y. Zhang, X. Guo, Y. Ma, M. Du, J. Fan, Y. Zhu, Z. Zeng and Y. Chai, J. Mater. Chem. A, 2022, 10, 719–725 RSC.
- B. Qiu, M. Du, Y. Ma, Q. Zhu, M. Xing and J. Zhang, Energy Environ. Sci., 2021, 14, 5260–5288 RSC.
- G. Chen, X. Tu, G. Homm and A. Weidenkaff, Nat. Rev. Mater., 2022, 7, 333–334 CrossRef.
- L. Santamaria, G. Lopez, E. Fernandez, M. Cortazar, A. Arregi, M. Olazar and J. Bilbao, Energy Fuels, 2021, 35, 17051–17084 CrossRef CAS PubMed.
- A. M. Ranjekar and G. D. Yadav, Ind. Eng. Chem. Res., 2021, 60, 89–113 CrossRef CAS.
- S. Chen, C. Pei and J. Gong, Energy Environ. Sci., 2019, 12, 3473–3495 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu and L. Zheng, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- Y. Zhang, Y. Ma, W. Yuan, L. Cai, Y. Chai and B. Qiu, Mater. Horiz., 2023 10.1039/D3MH00814B.
- C. Zhang, Z. Guo, Y. Tian, C. Yu, K. Liu and L. Jiang, Nano Res. Energy, 2023, 2, e9120063 CrossRef.
- X. Zhou, T. Yang, T. Li, Y. Zi, S. Zhang, L. Yang, Y. Liu, J. Yang and J. Tang, Nano Res. Energy, 2023, 2, e9120086 CrossRef.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- Z. Su, Q. Huang, Q. Guo, S. J. Hoseini, F. Zheng and W. Chen, Nano Res. Energy, 2023, 2, e9120078 CrossRef.
- Q. Yun, Q. Lu, X. Zhang, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 626–646 CrossRef CAS.
- Y. Ma, Y. Zhang, M. Xing, S. Kang, M. Du, B. Qiu and Y. Chai, Chem. Commun., 2022, 58, 6642–6645 RSC.
- B. Qiu, L. Cai, Y. Wang, S. Ma, Y. H. Tsang and Y. Chai, Mater. Today Energy, 2019, 11, 89–96 CrossRef CAS.
- C. Deng, C. Y. Toe, X. Li, J. Tan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2201047 CrossRef CAS.
- K. Zhang, J. Liu, L. Wang, B. Jin, X. Yang, S. Zhang and J. H. Park, J. Am. Chem. Soc., 2020, 142, 8641–8648 CrossRef CAS PubMed.
- B. Qiu, L. Cai, N. Zhang, X. Tao and Y. Chai, Adv. Sci., 2020, 7, 1903568 CrossRef CAS PubMed.
- I. Vollmer, M. J. Jenks, M. C. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 132, 15524–15548 CrossRef.
- G. Ruoppolo, P. Ammendola, R. Chirone and F. Miccio, Waste Manage., 2012, 32, 724–732 CrossRef CAS PubMed.
- S. O. Adegoke, A. A. Adeleke, P. P. Ikubanni, C. T. Nnodim, A. O. Balogun, O. A. Falode and S. O. Adetona, Cogent Eng., 2021, 8, 1994106 CrossRef.
- I. Agenda, in The World Economic Forum: Geneva, Switzerland, 2016, p. 36 Search PubMed.
- T. Hundertmark, M. Mayer, C. McNally, T. J. Simons and C. Witte, How plastics waste recycling could transform the chemical industry, McKinsey & Company, 2018 Search PubMed.
- M. Du, M. Xing, S. Kang, Y. Ma, B. Qiu and Y. Chai, EcoMat, 2022, 4, e12259 CrossRef CAS.
- J. G. Olivier, K. Schure and J. Peters, Trends in Global CO2 and Total Greenhouse Gas Emissions, 2020 Report, PBL Netherlands Environmental Assessment Agency, 2020 Search PubMed.
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 19857–19861 CrossRef CAS PubMed.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- E. J. Cho, L. T. P. Trinh, Y. Song, Y. G. Lee and H.-J. Bae, Bioresour. Technol., 2020, 298, 122386 CrossRef CAS.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef.
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51–55 CrossRef CAS PubMed.
- G.-F. Chen, Y. Luo, L.-X. Ding and H. Wang, ACS Catal., 2018, 8, 526–530 CrossRef CAS.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J. G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- P. Prabhu, Y. Wan and J.-M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- X. Jiao, K. Zheng, Z. Hu, S. Zhu, Y. Sun and Y. Xie, Adv. Mater., 2021, 33, 2005192 CrossRef CAS.
- R. Shi, K.-S. Liu, F. Liu, X. Yang, C.-C. Hou and Y. Chen, Chem. Commun., 2021, 57, 12595–12598 RSC.
- X. Guo, D. Xiang, G. Duan and P. Mou, Waste Manage., 2010, 30, 4–10 CrossRef PubMed.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- F. W. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- D. J. Kang, Y. Jüttke, L. González-García, A. Escudero, M. Haft and T. Kraus, Small, 2020, 16, 2000928 CrossRef CAS PubMed.
- L. Lyu, J. Pei, D. Hu, G. Sun and E. H. Fini, J. Cleaner Prod., 2022, 345, 131151 CrossRef.
- Y. Chen, H. Shang, Y. Ren, Y. Yue, H. Li and Z. Bian, ACS ES&T Engg., 2022, 2, 1039–1052 Search PubMed.
- Y. Chen, M. Xu, J. Wen, Y. Wan, Q. Zhao, X. Cao, Y. Ding, Z. L. Wang, H. Li and Z. Bian, Nat. Sustainability, 2021, 4, 618–626 CrossRef.
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- E. Peduzzi, G. Boissonnet and F. Maréchal, Fuel, 2016, 181, 207–217 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 RSC.
- S. Cheng, I. D’cruz, M. Wang, M. Leitch and C. Xu, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- X. Zhou, W. Li, R. Mabon and L. J. Broadbelt, Energy Environ. Sci., 2018, 11, 1240–1260 RSC.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 CrossRef CAS.
- S.-H. Li, S. Liu, J. C. Colmenares and Y.-J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- J. Luterbacher, D. M. Alonso and J. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- Y. S. Lai, F. De Francesco, A. Aguinaga, P. Parameswaran and B. E. Rittmann, Green Chem., 2016, 18, 1319–1326 RSC.
- F. De Schouwer, L. Claes, A. Vandekerkhove, J. Verduyckt and D. E. De Vos, ChemSusChem, 2019, 12, 1272–1303 CrossRef CAS PubMed.
- P. O'donoghue, J. Ling, Y.-S. Wang and D. Söll, Nat. Chem. Biol., 2013, 9, 594–598 CrossRef PubMed.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de Los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- J. Zheng, M. Arifuzzaman, X. Tang, X. C. Chen and T. Saito, Mater. Horiz., 2023, 10, 1608–1624 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- R. Wei, D. Breite, C. Song, D. Gräsing, T. Ploss, P. Hille, R. Schwerdtfeger, J. Matysik, A. Schulze and W. Zimmermann, Adv. Sci., 2019, 6, 1900491 CrossRef PubMed.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustainability, 2023, 6, 965–973 CrossRef.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- B. Sun, J. Zhang, M. Wang, S. Yu, Y. Xu, S. Tian, Z. Gao, D. Xiao, G. Liu, W. Zhou, M. Wang and D. Ma, Nat. Sustainability, 2023, 6, 712–719 CrossRef.
- R. Mi, L. Zeng, M. Wang, S. Tian, J. Yan, S. Yu, M. Wang and D. Ma, Angew. Chem., Int. Ed., 2023, 62, e202304219 CrossRef CAS PubMed.
- C. Verma, L. O. Olasunkanmi, E. D. Akpan, M. Quraishi, O. Dagdag, M. El Gouri, E.-S. M. Sherif and E. E. Ebenso, React. Funct. Polym., 2020, 156, 104741 CrossRef CAS.
- X. Jin, J. Dong, X. Guo, M. Ding, R. Bao and Y. Luo, Polym. Int., 2022, 71, 1384–1392 CrossRef CAS.
- D. L. Francisco, L. B. Paiva and W. Aldeia, Polym. Compos., 2019, 40, 851–870 CrossRef CAS.
- Y. Wu, J. Zhao, C. Wang, T. Li, B.-H. Zhao, Z. Song, C. Liu and B. Zhang, Nat. Commun., 2023, 14, 3057 CrossRef CAS PubMed.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef.
- D. C. Dibble, C. Li, L. Sun, A. George, A. Cheng, Ö. P. Çetinkol, P. Benke, B. M. Holmes, S. Singh and B. A. Simmons, Green Chem., 2011, 13, 3255–3264 RSC.
- J. Lv, W. Zuo, C. Tian, M. Wang, Q. Liao and Z. Lin, Cell Rep. Phys. Sci., 2023, 4, 101456 CrossRef CAS.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- Y. Luo, E. Selvam, D. G. Vlachos and M. Ierapetritou, ACS Sustainable Chem. Eng., 2023, 11, 4209–4218 CrossRef CAS.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS PubMed.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- X. Meng, Y. Pu, M. Li and A. J. Ragauskas, Green Chem., 2020, 22, 2862–2872 RSC.
- W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2021, 14, 4176–4180 CrossRef CAS PubMed.
- N. R. Gunawan, M. Tessman, D. Zhen, L. Johnson, P. Evans, S. M. Clements, R. S. Pomeroy, M. D. Burkart, R. Simkovsky and S. P. Mayfield, Sci. Total Environ., 2022, 850, 158761 CrossRef CAS PubMed.
- G. Gözaydın, S. Song and N. Yan, Green Chem., 2020, 22, 5096–5104 RSC.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- G. D. Mumbach, R. de Sousa Cunha, R. A. F. Machado and A. Bolzan, J. Environ. Manage., 2019, 243, 453–462 CrossRef CAS PubMed.
- S. Ügdüler, K. M. Van Geem, M. Roosen, E. I. Delbeke and S. De Meester, Waste Manage., 2020, 104, 148–182 CrossRef PubMed.
- S. Behera, R. Arora, N. Nandhagopal and S. Kumar, Renewable Sustainable Energy Rev., 2014, 36, 91–106 CrossRef CAS.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai and Z. Zheng, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- C. Marquez, C. Martin, N. Linares and D. De Vos, Mater. Horiz., 2023, 10, 1625–1640 RSC.
- M. Davaritouchaee and S. Chen, Biomass Bioenergy, 2018, 116, 249–258 CrossRef CAS.
- A. Zheng, Z. Zhao, Z. Huang, K. Zhao, G. Wei, X. Wang, F. He and H. Li, Energy Fuels, 2014, 28, 5804–5811 CrossRef CAS.
- M. Arifuzzaman, B. G. Sumpter, Z. Demchuk, C. Do, M. A. Arnould, M. A. Rahman, P.-F. Cao, I. Popovs, R. J. Davis, S. Dai and T. Saito, Mater. Horiz., 2023, 10, 3360–3368 RSC.
- C. Tian, J. Lv, W. Zhang, H. Wang, J. Chao, L. Chai and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202206947 CrossRef CAS PubMed.
- J. Zou, G. Zhang and X. Xu, Appl. Catal., A, 2018, 563, 73–79 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- Q. Dong, A. D. Lele, X. Zhao, S. Li, S. Cheng, Y. Wang, M. Cui, M. Guo, A. H. Brozena, Y. Lin, T. Li, L. Xu, A. Qi, I. G. Kevrekidis, J. Mei, X. Pan, D. Liu, Y. Ju and L. Hu, Nature, 2023, 616, 488–494 CrossRef CAS PubMed.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- V. Zubar, A. T. Haedler, M. Schütte, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2022, 15, e202101606 CrossRef CAS PubMed.
- R. Donadini, C. Boaretti, A. Lorenzetti, M. Roso, D. Penzo, E. Dal Lago and M. Modesti, ACS Omega, 2023, 8, 4655–4666 CrossRef CAS PubMed.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2021, 122, 385–441 CrossRef PubMed.
- N. Pacheco, M. Garnica-Gonzalez, M. Gimeno, E. Bárzana, S. Trombotto, L. David and K. Shirai, Biomacromolecules, 2011, 12, 3285–3290 CrossRef CAS PubMed.
- X. Ma, G. Gözaydın, H. Yang, W. Ning, X. Han, N. Y. Poon, H. Liang, N. Yan and K. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7719–7728 CrossRef CAS PubMed.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- N. Govindarajan, G. Kastlunger, H. H. Heenen and K. Chan, Chem. Sci., 2022, 13, 14–26 RSC.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628 CrossRef CAS PubMed.
- E. Cetinkaya, I. Dincer and G. F. Naterer, Int. J. Hydrogen Energy, 2012, 37, 2071–2080 CrossRef CAS.
- F. Wang and H. Duan, Chem. Catal., 2022, 2, 644–646 CrossRef CAS.
- M. Rumayor, J. Corredor, M. J. Rivero and I. Ortiz, J. Cleaner Prod., 2022, 336, 130430 CrossRef CAS.
- Z. Chen, M. Wang, E. Jiang, D. Wang, K. Zhang, Y. Ren and Y. Jiang, Trends Biotechnol., 2018, 36, 1287–1298 CrossRef CAS PubMed.
- X. Hu and M. Gholizadeh, J. Energy Chem., 2019, 39, 109–143 CrossRef.
- Z. Tian, X. Guo, D. Wang, D. Sun, S. Zhang, K. Bu, W. Zhao and F. Huang, Adv. Funct. Mater., 2020, 30, 2001286 CrossRef CAS.
- E. Kwietniewska and J. Tys, Renewable Sustainable Energy Rev., 2014, 34, 491–500 CrossRef CAS.
- R. Łukajtis, I. Hołowacz, K. Kucharska, M. Glinka, P. Rybarczyk, A. Przyjazny and M. Kamiński, Renewable Sustainable Energy Rev., 2018, 91, 665–694 CrossRef.
- S. K. Bhatia, S. S. Jagtap, A. A. Bedekar, R. K. Bhatia, K. Rajendran, A. Pugazhendhi, C. V. Rao, A. Atabani, G. Kumar and Y.-H. Yang, Sci. Total Environ, 2021, 765, 144429 CrossRef CAS PubMed.
- S. N. Djomo and D. Blumberga, Bioresour. Technol., 2011, 102, 2684–2694 CrossRef CAS PubMed.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole and Y. Zhang, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- V. Tournier, C. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier and S. Gavalda, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- C.-C. Chen, L. Dai, L. Ma and R.-T. Guo, Nat. Rev. Chem., 2020, 4, 114–126 CrossRef PubMed.
- T. Uekert, F. Dorchies, C. M. Pichler and E. Reisner, Green Chem., 2020, 22, 3262–3271 RSC.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- M. Kan, M. Tao, W. Zhuang, S. Wu, Z. Ye and J. Zhang, Sol. RRL, 2023, 7, 2300411 CrossRef CAS.
- W. Xiao and D. Wang, Chem. Soc. Rev., 2014, 43, 3215–3228 RSC.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- D. E. Blanco and M. A. Modestino, Trends Chem., 2019, 1, 8–10 CrossRef CAS.
- J. J. Dai, Y. B. Huang, C. Fang, Q. X. Guo and Y. Fu, ChemSusChem, 2012, 5, 617–620 CrossRef CAS PubMed.
- G. G. Botte, Electrochem. Soc. Interface, 2014, 23, 49 CrossRef.
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef PubMed.
- H. J. Kim, J. Lee, S. K. Green, G. W. Huber and W. B. Kim, ChemSusChem, 2014, 7, 1051–1056 CrossRef CAS PubMed.
- D. Zheng, J. Li, S. Ci, P. Cai, Y. Ding, M. Zhang and Z. Wen, Appl. Catal., B, 2020, 277, 119178 CrossRef CAS.
- C. Lamy, T. Jaubert, S. Baranton and C. Coutanceau, J. Power Sources, 2014, 245, 927–936 CrossRef CAS.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Lavacchi, A. Marchionni, F. Vizza and P. K. Shen, ChemSusChem, 2010, 3, 851–855 CrossRef CAS PubMed.
- M. Bellini, M. Bevilacqua, M. Innocenti, A. Lavacchi, H. Miller, J. Filippi, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, J. Power Sources, 2014, 161, D3032 CAS.
- H. J. Kim, Y. Kim, D. Lee, J.-R. Kim, H.-J. Chae, S.-Y. Jeong, B.-S. Kim, J. Lee, G. W. Huber and J. Byun, ACS Sustainable Chem. Eng., 2017, 5, 6626–6634 CrossRef CAS.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X.-Z. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2022, 5, 66–73 CrossRef CAS.
- R. S. Assary, P. C. Redfern, J. R. Hammond, J. Greeley and L. A. Curtiss, Chem. Phys. Lett., 2010, 497, 123–128 CrossRef CAS.
- Y. Xie, Z. Zhou, N. Yang and G. Zhao, Adv. Funct. Mater., 2021, 31, 2102886 CrossRef CAS.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036 CrossRef CAS PubMed.
- N. Tuleushova, Y. Holade, D. Cornu and S. Tingry, Electrochem. Sci. Adv., 2023, 3, e2100174 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842–846 CrossRef CAS PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- M. Götz, A. Rudi, R. Heck, F. Schultmann and A. Kruse, GCB Bioenergy, 2022, 14, 447–462 CrossRef.
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Green Chem., 2021, 23, 3154–3171 RSC.
- Q. Yang, G. Chu, L. Zhang, D. Zhang and J. Yu, Energy Convers. Manage., 2022, 258, 115529 CrossRef CAS.
- X. Zhou, M. Zha, J. Cao, H. Yan, X. Feng, D. Chen and C. Yang, ACS Sustainable Chem. Eng., 2021, 9, 10948–10962 CrossRef CAS.
- J. Byun, D. Kim, D. Lee, H. J. Kim and J. Han, J. Ind. Eng. Chem., 2019, 73, 268–275 CrossRef CAS.
- A. Al Ghatta, R. C. Aravenas, Y. Wu, J. M. Perry, J. Lemus and J. P. Hallett, ACS Sustainable Chem. Eng., 2022, 10, 8846–8855 CrossRef CAS PubMed.
- D. T. Win, Au J. Technol., 2005, 8, 185–190 Search PubMed.
- S. H. Krishna, K. Huang, K. J. Barnett, J. He, C. T. Maravelias, J. A. Dumesic, G. W. Huber, M. De Bruyn and B. M. Weckhuysen, AIChE J., 2018, 64, 1910–1922 CrossRef CAS.
- S. D. Phillips, S. B. Jones, P. A. Meyer and L. J. Snowden-Swan, Biofuels, Bioprod. Biorefin., 2022, 16, 640–652 CrossRef CAS.
- P. Khamhaeng, N. Laosiripojana, S. Assabumrungrat and P. Kim-Lohsoontorn, Int. J. Hydrogen Energy, 2021, 46, 30891–30902 CrossRef CAS.
- S. Pugh, R. McKenna, I. Halloum and D. R. Nielsen, Metab. Eng. Commun., 2015, 2, 39–45 CrossRef PubMed.
- J. A. Satrio and L. Doraiswamy, Chem. Eng. J., 2001, 82, 43–56 CrossRef CAS.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- R. Ciriminna, C. D. Pina, M. Rossi and M. Pagliaro, Eur. J. Lipid Sci. Technol., 2014, 116, 1432–1439 CrossRef CAS.
- C. Lee, M. K. Aroua, W. M. A. W. Daud, P. Cognet, Y. Pérès-Lucchese, P.-L. Fabre, O. Reynes and L. Latapie, Renewable Sustainable Energy Rev., 2015, 42, 963–972 CrossRef CAS.
- B. Tabah, A. Varvak, I. N. Pulidindi, E. Foran, E. Banin and A. Gedanken, Green Chem., 2016, 18, 4657–4666 RSC.
- S. S. Bhagwat, Y. Li, Y. R. Cortés-Peña, E. C. Brace, T. A. Martin, H. Zhao and J. S. Guest, ACS Sustainable Chem. Eng., 2021, 9, 16659–16669 CrossRef CAS.
- A. J. Straathof, S. Sie, T. T. Franco and L. A. Van der Wielen, Appl. Microbiol. Biotechnol., 2005, 67, 727–734 CrossRef CAS PubMed.
- J. Joo, T. Kim, J. Lee, S. I. Choi and K. Lee, Adv. Mater., 2019, 31, 1806682 CrossRef PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- Y. Chen, A. Lavacchi, H. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036 CrossRef CAS PubMed.
- S. Verma, S. Lu and P. J. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- W. Liu, Y. Cui, X. Du, Z. Zhang, Z. Chao and Y. Deng, Energy Environ. Sci., 2016, 9, 467–472 RSC.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- T. Terlouw, C. Bauer, L. Rosa and M. Mazzotti, Energy Environ. Sci., 2021, 14, 1701–1721 RSC.
- T. Cui, L. Ma, S. Wang, C. Ye, X. Liang, Z. Zhang, G. Meng, L. Zheng, H.-S. Hu and J. Zhang, J. Am. Chem. Soc., 2021, 143, 9429–9439 CrossRef CAS PubMed.
- C. Yang, S. Maldonado and C. R. Stephenson, ACS Catal., 2021, 11, 10104–10114 CrossRef CAS.
- K. Yan, Y. Zhang, M. Tu and Y. Sun, Energy Fuels, 2020, 34, 12703–12709 CrossRef CAS.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- S. Kandasamy, S. P. Samudrala and S. Bhattacharya, Catal. Sci. Technol., 2019, 9, 567–577 RSC.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622–627 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem. Catal., 2021, 1, 941–955 CrossRef CAS.
- X. Guo, J. Guan, B. Li, X. Wang, X. Mu and H. Liu, Sci. Rep., 2015, 5, 16451 CrossRef PubMed.
- I. Taniguchi, S. Yoshida, K. Hiraga, K. Miyamoto, Y. Kimura and K. Oda, ACS Catal., 2019, 9, 4089–4105 CrossRef CAS.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae and D. Sinton, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- P. K. Samantaray, A. Little, D. M. Haddleton, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055–4081 RSC.
- M. Du, Y. Zhang, S. Kang, C. Xu, Y. Ma, L. Cai, Y. Zhu, Y. Chai and B. Qiu, Small, 2023 DOI:10.1002/smll.202303693.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 135, e202300094 CrossRef.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong and M. Shao, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- X. Yang, K. X. Yao, J. Y. Ye, Q. Yuan, F. Zhao, Y. Li and Z. Zhou, Adv. Funct. Mater., 2021, 31, 2103671 CrossRef CAS.
- S. Bai, Y. Xu, K. Cao and X. Huang, Adv. Mater., 2021, 33, 2005767 CrossRef CAS PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- C. C. Chong, A. Aqsha, M. Ayoub, M. Sajid, A. Z. Abdullah, S. Yusup and B. Abdullah, Environ. Technol. Innovation, 2020, 19, 100859 CrossRef.
- I. Velázquez-Hernández, M. T. Oropeza-Guzmán, M. Guerra-Balcázar, L. Álvarez-Contreras and N. Arjona, ChemNanoMat, 2019, 5, 68–78 CrossRef.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- N. Logeshwaran, I. R. Panneerselvam, S. Ramakrishnan, R. S. Kumar, A. R. Kim, Y. Wang and D. J. Yoo, Adv. Sci., 2022, 9, 2105344 CrossRef CAS PubMed.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- J. r Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- X. Yu, E. C. Dos Santos, J. White, G. Salazar-Alvarez, L. G. Pettersson, A. Cornell and M. Johnsson, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos dos Santos, A. Anil, A. Cornell, L. G. Pettersson and M. Johnsson, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS.
- X. Zhou, H. Yan, X. Feng, H. Zhao, Y. Liu, X. Chen, D. Chen and C. Yang, Green Chem., 2021, 23, 3664–3676 RSC.
- H. Xu, H. Shang, C. Wang and Y. Du, Adv. Funct. Mater., 2020, 30, 2000793 CrossRef CAS.
- T. Zhan, W. Liu, J. Teng, C. Yue, D. Li, S. Wang and H. Tan, Chem. Commun., 2019, 55, 2620–2623 RSC.
- J. Cai, H. Ma, J. Zhang, Z. Du, Y. Huang, J. Gao and J. Xu, Chin. J. Catal., 2014, 35, 1653–1660 CrossRef CAS.
- S. Hirasawa, H. Watanabe, T. Kizuka, Y. Nakagawa and K. Tomishige, J. Catal., 2013, 300, 205–216 CrossRef CAS.
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed.
- M. Zhao, Z. Lyu, M. Xie, Z. D. Hood, Z. Cao, M. Chi and Y. Xia, Small Methods, 2020, 4, 1900843 CrossRef CAS.
- C. Y. Wang, Z. Y. Yu, G. Li, Q. T. Song, G. Li, C. X. Luo, S. H. Yin, B. A. Lu, C. Xiao and B. B. Xu, ChemElectroChem, 2020, 7, 239–245 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem. A, 2021, 9, 20164–20183 RSC.
- R.-J. van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- F. A. Kucherov, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Angew. Chem., Int. Ed., 2017, 129, 16147–16151 CrossRef.
- D. K. Mishra, H. J. Lee, J. Kim, H.-S. Lee, J. K. Cho, Y.-W. Suh, Y. Yi and Y. J. Kim, Green Chem., 2017, 19, 1619–1623 RSC.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- J. Wang, Z. Zhao, C. Shen, H. Liu, X. Pang, M. Gao, J. Mu, F. Cao and G. Li, Catal. Sci. Technol., 2021, 11, 2480–2490 RSC.
- J. Zhang, P. Yu, G. Zeng, F. Bao, Y. Yuan and H. Huang, J. Mater. Chem. A, 2021, 9, 9685–9691 RSC.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cychy, S. Cheong, P. V. Kumar, T. M. Benedetti, C. Deng, K. H. Wu and C. E. Marjo, Angew. Chem., Int. Ed., 2020, 59, 15487–15491 CrossRef CAS PubMed.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- M. Park, M. Gu and B.-S. Kim, ACS Nano, 2020, 14, 6812–6822 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, 2107185 CrossRef CAS PubMed.
- Y. Kwon, S. C. Lai, P. Rodriguez and M. T. Koper, J. Am. Chem. Soc., 2011, 133, 6914–6917 CrossRef CAS PubMed.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 134, e202200211 CrossRef.
- H. Huang, X. Song, C. Yu, Q. Wei, L. Ni, X. Han, H. Huang, Y. Han and J. Qiu, Angew. Chem., Int. Ed., 2023, 135, e202216321 CrossRef.
- L. Xu, Z. Huang, M. Yang, J. Wu, W. Chen, Y. Wu, Y. Pan, Y. Lu, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210123 CrossRef CAS PubMed.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- D. Gao, F. Han, G. I. N. Waterhouse, Y. Li and L. Zhang, Biomass Convers. Biorefin., 2022, 1–13, DOI:10.1007/s13399-022-03512-y.
- T. Xia, W. Gong, Y. Chen, M. Duan, J. Ma, X. Cui, Y. Dai, C. Gao and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204225 CrossRef CAS PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- D.-H. Nam, B. J. Taitt and K.-S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- K. A. Payne, S. A. Marshall, K. Fisher, M. J. Cliff, D. M. Cannas, C. Yan, D. J. Heyes, D. A. Parker, I. Larrosa and D. Leys, ACS Catal., 2019, 9, 2854–2865 CrossRef CAS PubMed.
- G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966–2972 RSC.
- T. Wang, Z. Huang, T. Liu, L. Tao, J. Tian, K. Gu, X. Wei, P. Zhou, L. Gan and S. Du, Angew. Chem., Int. Ed., 2022, 134, e202115636 CrossRef.
- P. Parpot, A. Bettencourt, G. Chamoulaud, K. Kokoh and E. Belgsir, Electrochim. Acta, 2004, 49, 397–403 CrossRef CAS.
- H. Wu, J. Song, H. Liu, Z. Xie, C. Xie, Y. Hu, X. Huang, M. Hua and B. Han, Chem. Sci., 2019, 10, 4692–4698 RSC.
- L. Gong, N. Agrawal, A. Roman, A. Holewinski and M. J. Janik, J. Catal., 2019, 373, 322–335 CrossRef CAS.
- X. Li, B. Ho, D. S. Lim and Y. Zhang, Green Chem., 2017, 19, 914–918 RSC.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustainable Chem. Processes, 2015, 3, 9 CrossRef.
- S. R. Kubota and K.-S. Choi, ACS Sustainable Chem. Eng., 2018, 6, 9596–9600 CrossRef CAS.
- S. Thiyagarajan, D. Franciolus, R. J. Bisselink, T. A. Ewing, C. G. Boeriu and J. Van Haveren, ACS Sustainable Chem. Eng., 2020, 8, 10626–10632 CAS.
- X. Huang, J. Song, M. Hua, B. Chen, Z. Xie, H. Liu, Z. Zhang, Q. Meng and B. Han, Chem. Sci., 2021, 12, 6342–6349 RSC.
- A. Haile, G. G. Gelebo, T. Tesfaye, W. Mengie, M. A. Mebrate, A. Abuhay and D. Y. Limeneh, Bioresour. Bioprocess., 2021, 8, 1–22 CrossRef.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef PubMed.
- X. Bai, Q. Hou, H. Qian, Y. Nie, T. Xia, R. Lai, G. Yu, M. L. U. Rehman, H. Xie and M. Ju, Appl. Catal., B, 2022, 303, 120895 CrossRef CAS.
- Y. Wan and J.-M. Lee, ACS Catal., 2021, 11, 2524–2560 CrossRef CAS.
- J. Lee, B. Saha and D. G. Vlachos, Green Chem., 2016, 18, 3815–3822 RSC.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao, M. Shen, Z. Li, Z. Zou, R. Xiao and S. Chu, Nat. Commun., 2023, 14, 1013 CrossRef CAS PubMed.
- Y. Q. Zhu, H. Zhou, J. Dong, S. M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li and M. Shao, Angew. Chem., Int. Ed., 2023, 135, e202219048 CrossRef.
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed.
- P. Zhou, X. Lv, S. Tao, J. Wu, H. Wang, X. Wei, T. Wang, B. Zhou, Y. Lu and T. Frauenheim, Adv. Mater., 2022, 34, 2204089 CrossRef CAS PubMed.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 133, 20698–20705 CrossRef.
- M. K. Goetz, M. T. Bender and K.-S. Choi, Nat. Commun., 2022, 13, 5848 CrossRef CAS PubMed.
- X. Zhang, J. Zhan, Z. Yu, J. Deng, M. Li and Y. Shao, Chin. J. Chem., 2023, 41, 214–224 CrossRef CAS.
- Q. F. He, Y. J. Zhang, Z. L. Yang, J. C. Dong, X. M. Lin and J. F. Li, Chin. J. Chem., 2023, 41, 355–369 CrossRef CAS.
- J. Xu, Q. He, Z. Xiong, Y. Yu, S. Zhang, X. Hu, L. Jiang, S. Su, S. Hu and Y. Wang, Energy Fuels, 2020, 35, 2870–2913 CrossRef.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 133, 21634–21642 CrossRef.
- M. J. Hülsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- F. Ye, S. Zhang, Q. Cheng, Y. Long, D. Liu, R. Paul, Y. Fang, Y. Su, L. Qu, L. Dai and C. Hu, Nat. Commun., 2023, 14, 2040 CrossRef CAS PubMed.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang and X. Li, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- Y. Xiao, C. Shen, W. Zhang, M. Zhang, H. Zhang, T. Shao, Z. Xiong, Y. Ding, S. Hao and L. Liu, Small, 2023, 19, 2205876 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss and X. Dong, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- H. Yadegari, A. Ozden, T. Alkayyali, V. Soni, A. Thevenon, A. Rosas-Hernández, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 3538–3544 CrossRef CAS.
- H. Sheng, A. N. Janes, R. D. Ross, H. Hofstetter, K. Lee, J. Schmidt and S. Jin, Nat. Catal., 2022, 5, 716–725 CrossRef CAS.
- G. Han, G. Li and Y. Sun, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang and C. Xie, Nat. Catal., 2022, 5, 66–73 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |