
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in perovskite oxide electrocatalysts for Li–O2 batteries
Lulu
Lyu†
a,
Seonyong
Cho†
a and
Yong-Mook
Kang
 *abc
*abc
aDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: dake1234@korea.ac.kr
bKU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul, 02841, Republic of Korea
cEnergy Storage Research Center, Clean Energy Research Division, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
First published on 18th March 2023
Abstract
Lithium–oxygen batteries (LOBs) have been anticipated as promising energy-storage devices; however, their practical application is plagued by low energy efficiency, small capacity, and the short cycle life. When applied as air cathodes for LOBs, perovskite oxides exhibit an enormous potential for favorable battery performance due to their catalytic activity for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). Their tunable compositions, diverse structures and unique electronic properties allow flexible manipulation of their catalytic activity. This mini-review comprehensively describes recent advances in perovskite oxide electrocatalysts for LOBs. First, the energy-storage mechanism of LOBs and crucial catalytic descriptors of perovskites for the ORR/OER are analyzed. Then, several ingenious tactics to promote the catalytic activity of perovskite oxides are highlighted. A discussion of synthetic protocols for the preparation of perovskite oxides follows, including how the nanostructure, morphology and composition of perovskite oxides can be controlled by different methods and the effect of these on catalytic performance. Additionally, A-/B-/O-site regulation, oxygen vacancies, and the composites of perovskites and functional materials are thoroughly investigated. Finally, we summarize the challenges and outlook of perovskite oxide electrocatalysts for LOBs.
 Lulu Lyu | Lulu Lyu received her PhD (2021) from the Department of Convergence Science and Technology at Seoul National University. She is currently a research professor in Department of Materials Science and Engineering at Korea University. Her research focuses on nanomaterials and their applications for electrocatalysts and energy-storage materials. |
 Seonyong Cho | Seonyong Cho is a PhD student in the department of materials science and engineering at Korea University under the supervision of Prof. Yong-Mook Kang. His research area is catalysts and system design for lithium–air batteries. |
 Yong-Mook Kang | Yong-Mook Kang completed his BS (1999), MS (2001), and PhD (2004) at Korea Advanced Institute of Science and Technology. He has been a senior researcher at Samsung SDI Co., Ltd. He is currently a professor at Department of Materials Science and Engineering in Korea University, Republic of Korea. His research area covers electrode or catalyst materials for rechargeable batteries. He has been appointed as RSC (Royal Society of Chemistry) fellow and representative of Korea from 2015 and a member of Y-KAST (Young Korean Academy of Science and Technology) from 2020. |
Broader contextThe transition from fossil fuels to renewable energy is a pivotal approach to realizing a low-carbon economy, which stimulates the exploration of electrochemical energy storage devices. Lithium–oxygen batteries (LOBs) have been regarded as promising energy-storage systems, while their inferior energy efficiency, low capacity, and poor cycle life greatly hinder their practical applications. Owing to their catalytic activity for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), perovskite oxides with a formula of ABO3 have been investigated as air cathodes for LOBs. Their catalytic activity can be flexibly adjusted by different cation/anion species, distinct nanostructures and unique electronic properties. This mini-review aims to comprehensively provide recent advances in perovskite oxide electrocatalysts for LOBs. It begins with introducing the fundamental energy-storage mechanism of LOBs, and catalytic descriptors of perovskites for the ORR/OER. Next, strategies to improve the catalytic activity of perovskite oxides are highlighted, including the size effect, nanostructure engineering, A-/B-/O-site regulation, oxygen vacancies, and composites of perovskites with other functional materials. Finally, the challenges and outlook of perovskite oxide electrocatalysts for LOBs are given. |
1. Introduction
World energy consumption is expected to be 50% higher than 2018 levels (576 EJ) by 2050 (864 EJ). To achieve net zero fossil fuel use by 2050, the annual growth rate of renewable energy production must expand by a factor of 6–8.1 The soaring demand for electrical energy storage and conversion systems has driven exploration into metal–ion batteries, metal–air batteries, and redox–flow batteries.2–6 Lithium–air batteries have a superior theoretical energy density of above 5000 W h kg−1 (Fig. 1), and a large theoretical open circuit voltage of 2.96 V (eqn (1)).6–8 Electric vehicles using these batteries may have a driving range of more than 550 kilometers.9 As shown in Fig. 2(a), lithium–oxygen batteries (LOBs) are composed of a metallic lithium anode, an air cathode and a Li+-containing aprotic electrolyte.10–14 The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) occurring at the cathode dominate the discharging and charging processes of LOBs, respectively.15–19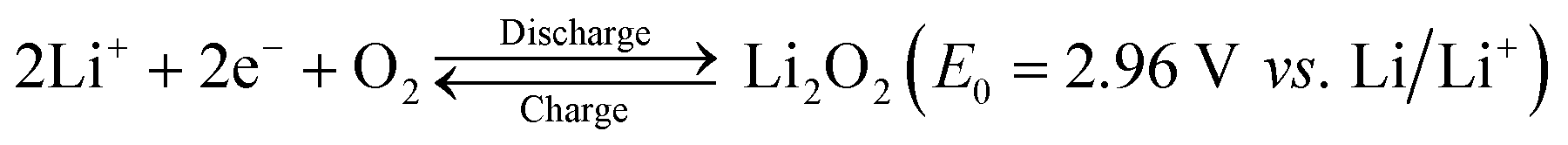 | (1) |
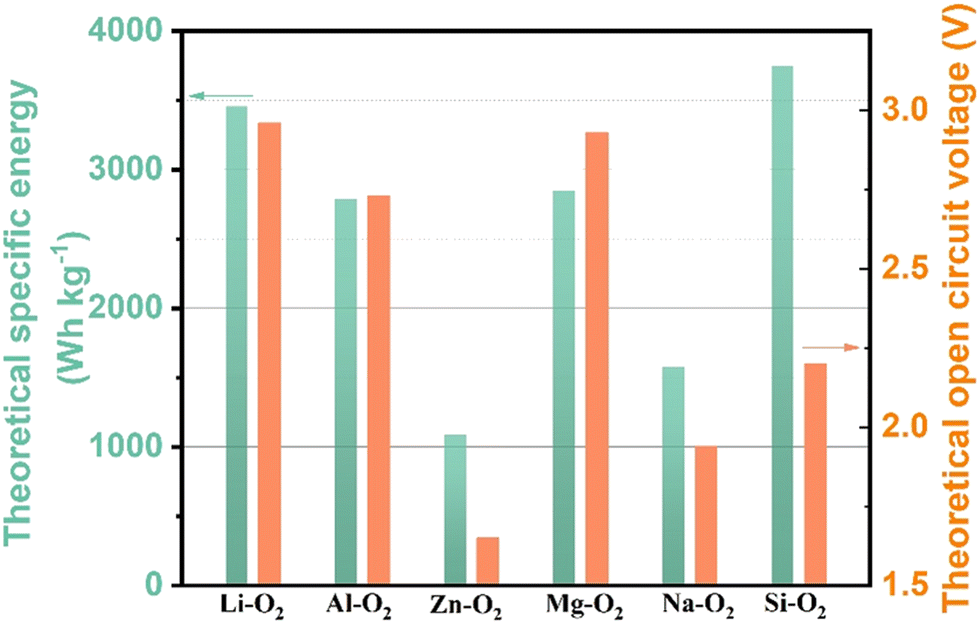 | ||
| Fig. 1 The theoretical specific energy and open circuit voltage of various metal–air batteries. The specific energy is calculated by multiplying the theoretical specific capacity by the voltage. | ||
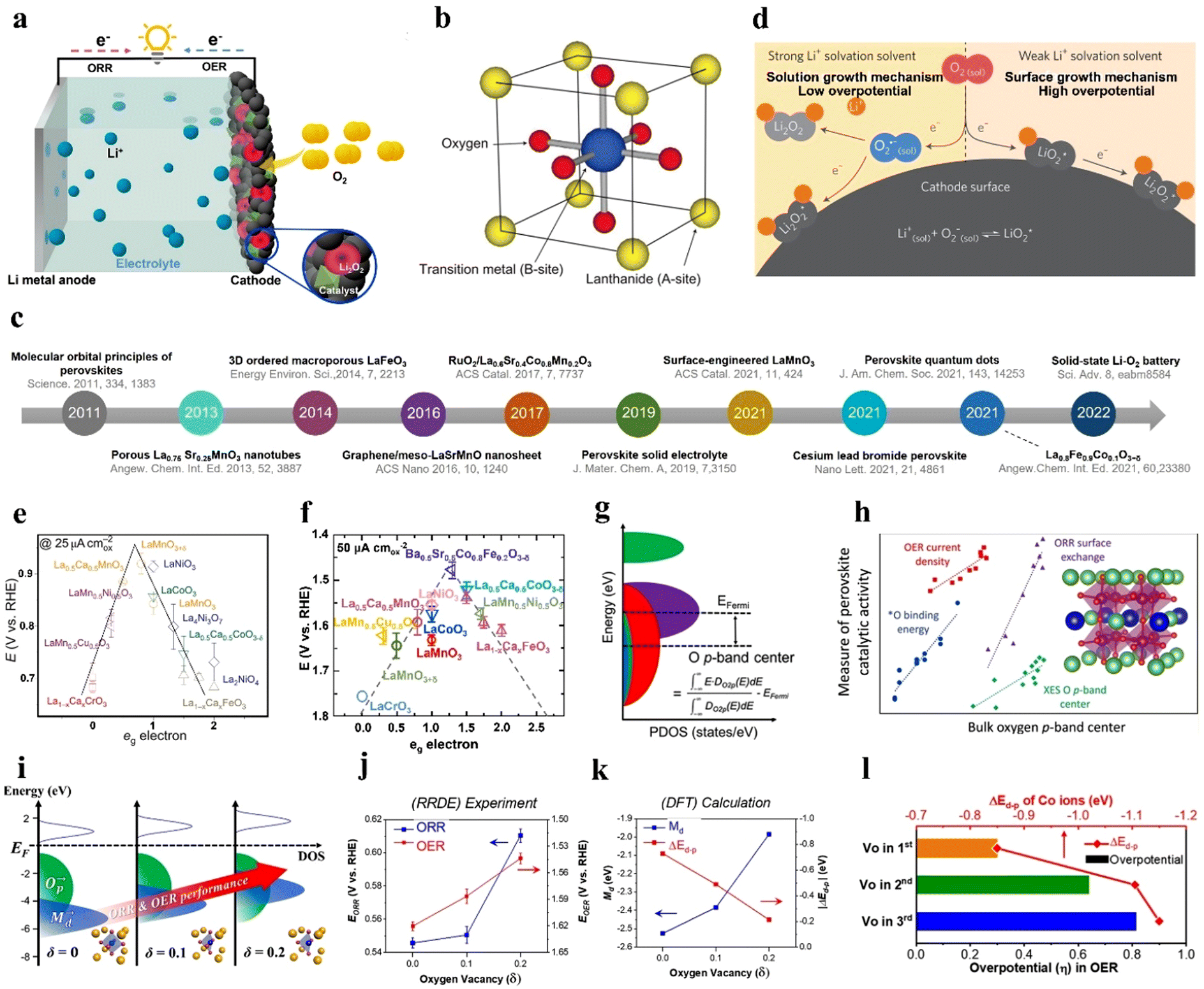 | ||
| Fig. 2 Overview and mechanisms of LOBs, and descriptors for the catalytic activity of perovskite oxides. (a) Illustration of LOBs. (b) ABO3 perovskite structure. (c) Timeline of research findings of perovskite oxides for LOBs. (d) Solution growth and surface-growth mechanisms in the LOB. Reproduced with permission.49 Copyright 2016, Springer Nature. (e) Potentials as a function of eg orbitals in perovskites. (b) and (e) Reproduced with permission.33 Copyright 2011, Springer Nature. (f) OER performance as a function of the eg orbital in perovskites. (g) Projected density of states (PDOS) of the O 2p orbit. (h) Catalytic activity as a function of the oxygen p-band center. (g) and (h) Reproduced with permission.50 Copyright 2019, American Chemical Society. (i) The relationship between the electronic structure of Sm0.5Sr0.5CoO3−δ and ORR/OER performance. (j) The relationship between ORR/OER activity and Vo based on experimental results. (k) The relationship between ΔEd–p, Md, and Vo based on DFT results. (l) Overpotential and ΔEd–p values of the OER according to the Vo position in the 1st, 2nd, and 3rd layer of perovskites. (i)–(l) Reproduced with permission.51 Copyright 2020, American Chemical Society. | ||
Though LOBs hold great promise due to their high theoretical energy density, several crucial issues should be overcome to realize their real-world application. Specifically, the insulating discharged product of Li2O2 covered on the cathode surface blocks the diffusion pathway for oxygen and electrolyte ions, and leads to electrical passivation on the cathode, resulting in a large voltage hysteresis and limiting the capacity of LOBs, especially at high rates.16 Reactive species generated during cycling give rise to electrolyte decomposition and electrode corrosion, and consequently terminate the cell life.5
The electrochemical performance and reaction pathways of LOBs are critically linked to the cathode design. Oxygen cathode materials should have a porous framework for O2 diffusion and Li2O2 storage, superior catalytic activity, favorable ionic/electronic conductivity, electrochemical stability and nontoxicity.20 Currently, noble metal materials such as Pt, Pd, IrO2, and RuO2 stand out as active electrocatalysts to address the sluggish kinetics of either the ORR or OER;5,21 however, their high cost, scarcity, and inferior cycling stability hamper practical applications. Various nonprecious electrocatalysts like transition metal oxides,11,13,16,17 metal carbides,22–24 and perovskite oxides25–27 have been investigated as cathode catalysts for LOBs. It should be noted that transition metal oxides possess relatively low electrical conductivity and poor stability,28 and metal carbides have limited active sites because of the aggregation of metal particles. By contrast, perovskite oxides, a class of mixed-metal oxides, have a structural formula of ABO3, where A is a lanthanide, alkaline or rare-earth cation, and B is a transition metallic element from the 3d, 4d or 5d configurations.6,29–32 An ideal perovskite structure is a cubic structure, where the A cation has 12-fold coordination with O, while the B site resides in a corner-sharing octahedron of O anions (Fig. 2(b)).33 More than 90% of metal cations in the periodic table can be accommodated in the framework of perovskites.34 The crystal structure of perovskites is slightly different from that of spinel oxides (AB2O4), where A and B cations occupy tetrahedral and octahedral sites, respectively.35 Owing to their distinct electronic, catalytic, and magnetic properties, perovskites have been applied in batteries and solar cells.36–39 Their tunable compositions, crystalline structures, low cost and facile synthesis make them promising electrocatalysts for the ORR/OER. However, perovskite electrocatalysts, when applied in non-aqueous LOBs, generally exhibit low Brunauer–Emmett–Teller (BET) surface areas, few morphologies and large particle sizes, which compromises their catalytic activity and the performance of LOBs.
The catalytic activity of perovskites can be flexibly tuned by several strategies. First, nanosizing bulk perovskites can expose enriched active sites and facilitate the diffusion of reactive species.40,41 Second, substituting A-, B- and O-sites with foreign elements can change the electronic and crystal structures of the perovskites, thereby tuning the binding strength with oxygen intermediates.42–44 Third, oxygen vacancies (Vo) can change the valence state of B-site cations and thus modulate the interaction between the metal ion and oxygen molecules.45 Lastly, heterostructures combining perovskites with carbon materials, metal–organic frameworks (MOFs), and MXenes possess large surface areas, porous structures, and enhanced charge transfer efficiencies, which accommodate more LOB discharge products and increase the utilization of active sites.
The anticipated high energy efficiency, superior capacity, and long cycle life of LOBs are highly governed by the physicochemical properties of perovskite-based air cathodes. Even though review papers related to perovskite-based catalysts in aqueous solutions have been reported,46–48 reviews that thoroughly outline recent advances in aprotic solutions for LOBs are rare. Hence, this review provides a timely and comprehensive understanding of recent advances towards perovskite catalysts for LOBs. Scheme 1 shows an overview of the perovskite-based electrocatalysts for LOBs in this review. Fig. 2(c) illustrates the timeline of the most impactful research findings related to perovskite oxides used for LOBs. Initially in this mini-review, the energy-storage mechanisms of LOBs and the activity descriptors of perovskite oxides for the ORR/OER are demonstrated. Next, key strategies to boost the catalytic activity of perovskite oxides are described, including morphology and nanostructure control, A-/B-/O-site regulation, oxygen vacancies, and composites with other functional materials. Finally, a summary of the challenges and outlook of perovskite oxide electrocatalysts for LOBs is provided.
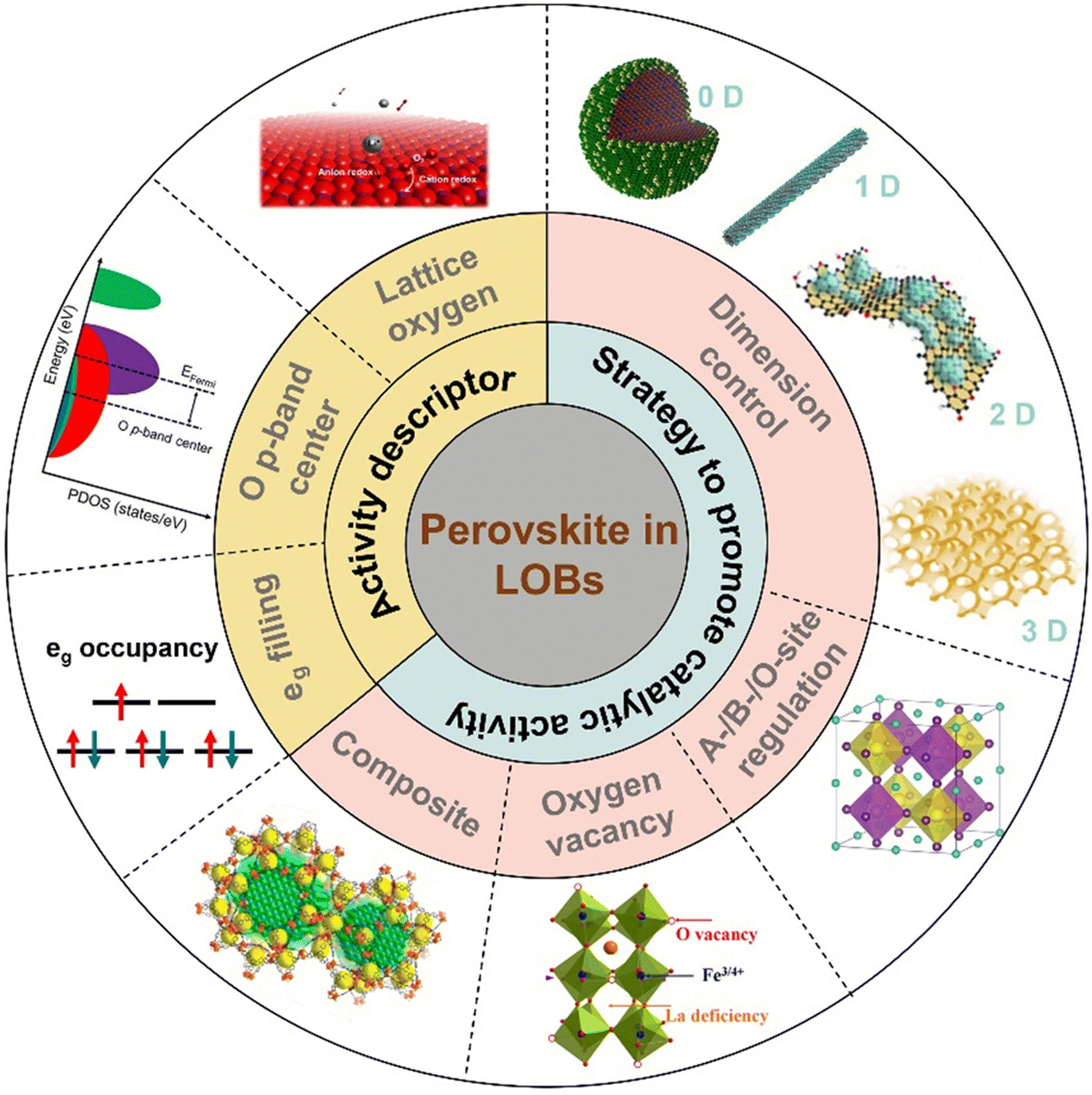 | ||
| Scheme 1 Schematic illustration of perovskite oxides as cathodes for LOBs. | ||
2. Mechanisms of LOBs
The growth and distribution of Li2O2 influences the reaction overpotential of an LOB. The electronic structures and morphologies of Li2O2 depend on solution growth (eqn (2)–(4)) or surface-growth mechanisms (eqn (5)–(7)), as shown in Fig. 2(d).52 The two growth mechanisms are associated with the solubility of the LiO2 intermediate. It has been reported that the donor number of the solvent can influence the solubility of LiO2 and thus manipulates O2 reduction pathways.53Solution growth mechanism: O2 molecules are reduced to superoxide (O2−), which dissolves into the electrolyte (expressed as O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sol−). Li+ ions react with O2sol− to generate soluble LiO2sol. The LiO2sol intermediate in the electrolyte becomes Li2O2 after a disproportionation reaction.
sol−). Li+ ions react with O2sol− to generate soluble LiO2sol. The LiO2sol intermediate in the electrolyte becomes Li2O2 after a disproportionation reaction.
| O2 + e− → O2sol− | (2) |
| Li+ + O2sol− → LiO2sol | (3) |
| 2LiO2sol → Li2O2 + O2 | (4) |
The solution-growth mechanism can be promoted by enhancing LiO2 solubility in a high-donor-number-based electrolyte, generating toroidal Li2O2 made of lamellae and leading to a high discharge capacity. Due to the loose contact between Li2O2 and the catalyst and the low conductivity of the Li2O2 toroid, an LOB based on the solution-growth model exhibits a low round-trip efficiency and high charge overpotential despite its high specific capacity.52,54 Moreover, a solvent with high polarity is easily affected by nucleophilic attack by O2−.55 Efforts have been made to realize surface growth mechanisms by adding 2,5-di-tert-butyl-1,4-benzoquinone (DBBQ) in a low-donor-number solvent (LiTFSI in ether), where DBBQ avoided the direct formation of Li2O2 on the cathode surface.55
Surface growth mechanism: superoxide (O2−) adsorbs on the cathode surface to produce O2sur− and then reacts with Li+ ions from the electrolyte to generate LiO2sur. LiO2sur is further reduced to generate Li2O2 by a 1e− reduction step. The lower LiO2 solubility in the surface growth pathway results in the generation of thin-film Li2O2 coating the surface, corresponding to a low capacity.
| O2 + e− → O2sur− | (5) |
| Li+ + O2sur− → LiO2sur | (6) |
| LiO2sur + Li+ + e− → Li2O2 | (7) |
3. Descriptors for the catalytic activity of perovskite oxides
3.1 eg filling
Shao-Horn et al.33 established an eg-filling theory to disclose the fundamental mechanism of the ORR in ABO3-type perovskites in alkaline solution. The d orbital of the central metal possesses two energy levels, namely, eg and t2g orbitals. Assuming that the surface transition metal center (B site) directly adsorbs O2 molecules, the eg orbital is predicted to overlap the O–2pσ orbital, and this overlap is stronger than that between the t2g and O–2pπ orbitals. Hence, compared to t2g, eg-filling of the B site is considered a more suitable indicator for the bonding strength between the B site and oxygen. Fig. 2(e) shows a volcano plot demonstrating the ORR activity of perovskites as a function of eg-filling. A moderate amount of eg-filling close to 1 promises the highest ORR performance. Low eg-filling is predicted to result in strong B–O2 bonding, and high eg-filling in weak O2 interaction. Likewise, the same group proposed that an eg occupation close to 1.2 and a high metal–oxygen covalency in perovskites contribute to optimized OER activity.32Fig. 2(f) depicts OER activity as a function of the occupancy of the eg-symmetry electron site of the transition metal cation. Shao–Horn's principle highlights that optimizing eg filling is a feasible approach for the design of highly active perovskite catalysts, such as LaCoO3,40,56 SrNb0.1Co0.7Fe0.2O3−δ,57 PrBa0.5Sr0.5Co1.5Fe0.5O5−δ,41 and CaCu3Fe4O12.583.2 Oxygen p-band center
The oxygen p-band center (Op) refers to the centroid of the PDOS of the O 2p orbit relative to the Fermi level (Fig. 2(g)), and is considered to be a bulk electronic structure descriptor for the evaluation of the catalytic activity of perovskites, based on density functional theory (DFT) calculations (Fig. 2(h)).50 The gap value between the metal d-band center (Md) and Op, ΔEd–p, is qualitatively associated with the bond hybridization of oxygen and the transition metal (TM). The strong hybridization between the metal 3d and O 2p orbitals positively affects the ORR/OER activity.50 One study suggests that the concentration of Vo can influence both Md and ΔEd–p in Sm0.5Sr0.5CoO3−δ.51 The increased Vo in Sm0.5Sr0.5CoO3−δ decreases the coordination number and valence state of Co (<4+), further affecting the magnetic moment of the perovskite. The relationship between δ in Sm0.5Sr0.5CoO3−δ and its electronic structure was revealed as an association between the Vo concentration and ORR/OER performance (Fig. 2(i)). Both experimental and DFT results suggest that the ORR can be enhanced by lifting the Md close to the Fermi level, and OER can be boosted by decreasing ΔEd–p, which can be tuned by the generation of Vo in the perovskite (Fig. 2(j) and (k)). Altogether, ORR and OER activity is strengthened by reducing ΔEd–p, which can be realized by increasing Md. More interestingly, the closer Vo is to the perovskite surface, the greater the promotion effect on catalytic activity (Fig. 2(l)). However, the fundamental mechanism of the ORR/OER of perovskites in a nonaqueous solution has not yet been fully understood.3.3 Lattice oxygen
In addition to the B-site metal cationic redox contribution to the catalytic activity of perovskites, lattice oxygen on or near the perovskite surface is regarded as another redox pair that participates in the OER.59–62 Grimaud et al.60 predicted that when the O 2p orbit center moves up towards the Fermi level, the oxidation of lattice oxygen in perovskite oxides is thermodynamically favorable. Perovskite oxides with lattice-oxygen participation also demonstrate pH-dependent OER activity, implying a non-cooperative proton–electron transfer process.60 Pan et al.63 prepared Si-doped SrCoO3−δ (Si-SCO), which showed better OER performance at different pH conditions than SCO (Fig. 3(a) and (b)). The lattice-oxygen oxidation mechanism (LOM) together with the adsorbate evolution mechanism (AEM) reaction pathways on Si-SCO are illustrated in Fig. 3(c), in which the LOM indicates that oxygen atoms on perovskites directly serve as active sites in the OER, and that AEM occurs through a concerted proton-coupled electron-transfer process on transition metal moieties. The contribution from the LOM to the OER activity of bulk perovskites was superior to that from the AEM. Notably, the influence of lattice oxygen from the perovskite surface resulted in a surface vacancy, and the generated vacant site was refilled by oxygen anions moving from the bulk material. The intrinsic OER activity was highly related to the diffusion rate of oxygen ions. A higher replenishment speed of surface lattice oxygen was responsible for a fast ion-diffusion rate, which benefited high OER activity (Fig. 3(d)). Si-SCO based on the LOM mechanism underwent surface reconstruction after cycling, as revealed by high-resolution transmission electron microscopy (HR-TEM) images (Fig. 3(e)), verifying the possible participation of lattice-oxygen redox.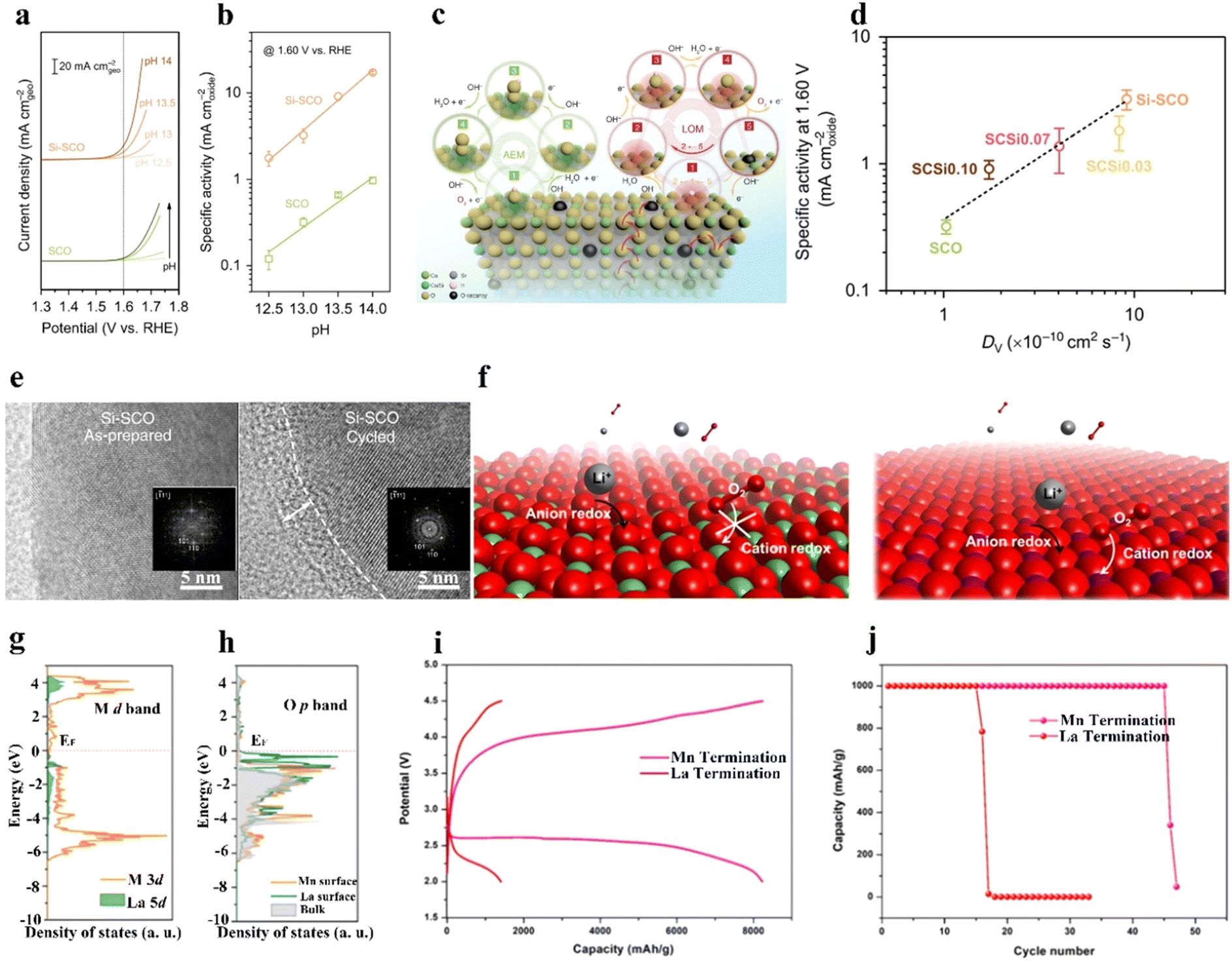 | ||
| Fig. 3 Lattice oxygen on or near the perovskite surface to participate in the OER. (a) OER current density, and (b) specific activity of Si-doped SCO and SCO under different pH conditions. (c) Schematic illustration of the AEM and LOM mechanisms on Si-doped SCO. (d) The relationship between OER and the diffusion rate of Vo. (e) HR-TEM images of Si-SCO before and after cycling. Insets: Corresponding fast Fourier transformed patterns. (a)–(e) Reproduced with permission.63 Copyright 2020, Springer Nature. (f) Illustration of reactions on La- and Mn-terminated LaMnO3 surfaces. (g) The density of states of the Mn 3d and La 5d bands. (h) Oxygen 2p bands of Mn- and La-terminated LaMnO3. (i) Discharge/charge curves at 200 mA g−1. (j) Cycling stability of Mn- and La-terminated LaMnO3 catalysts in a nonaqueous solution under air. (f)–(j) Reproduced with permission.61 Copyright 2021, American Chemical Society. | ||
In nonaqueous solutions, oxygen sites on the catalyst surface can directly interact with Li+ ions from the electrolyte to generate surface Li sites, promoting the growth of Li2O2.17 To illustrate the influence of both cation and anion redox on ORR/OER activity, our group prepared LaMnO3 (LMO) perovskites with different surface atomic arrangements, namely, La-terminated or Mn-terminated surfaces (Fig. 3(f)).61 The d band of the TM approached the Fermi-level in Mn-terminated LMO, while the O p band was near the Fermi-level in La-terminated LMO (Fig. 3(g) and (h)). Mn-terminated LMO involvement in both anion redox (surface lattice-O redox) and cation redox (TM redox) reactions resulted in the generation of a film-like discharge product. La-terminated LMO was inactive toward O2 adsorption because of the nearly void d bands of the La sites, while the Mn-terminated surface actively adsorbed both O2 and Li ions. Owing to the exposed lattice O from the stretched metal–oxygen bond and the presence of Mn3+ with a half-filled d band, the Mn-terminated LMO showed higher activity toward adsorbed O2 than La-terminated LMO. Under air, the assembled LOB using an Mn-terminated LMO cathode exhibited superior performance than that using a La-terminated LMO (Fig. 3(i) and (j)).
Dynamic surface oxygen-involved reaction leads to an unstable surface, especially when the surface oxygen cannot be refilled by the bulk oxygen.63–65 Hence, it is important to stabilize the perovskite surface to achieve high OER/ORR activity. Constant dissolution and deposition of catalysts during operando conditions may be a possible way to stabilize the catalyst surface, referred to as a self-healing process.63
4. Strategies for enhancing the catalytic activity of perovskites
Several tactics to promote the catalytic activity of perovskites are summarized in this mini-review, including dimensional control by synthetic methods, site substitution, oxygen vacancies, and perovskite-based composite catalysts. Table 1 lists the electrochemical performance and highlights of LOBs with perovskite cathodes.| Perovskite | Synthetic method | Structure | Specific capacity (mA h g−1 @mA g−1) | Overpotential (V@mA h g−1@mA g−1) | Cycle life (cycles@mA h g−1@mA g−1) | Highlights | Ref. |
|---|---|---|---|---|---|---|---|
| Note: rGO: reduced graphene oxide. | |||||||
| CePbBr3 | Hot-injection method | Cubic nanocrystal | 7648@100 | 400@1000@500 | 66 | ||
| LaMnO3 | Electrospinning | Nanofiber | 18000@200 |
100@1000@200 | Collective anion and TM redox | 61 | |
| La0.7MnO3−δ | Sol–gel | Nanoparticle | 29286@50 |
0.38@1000@200 | 375@1000@300 | A-site cationic defects | 67 |
| S-doped LiNaO3 | Sol–gel | Nanoparticle | 24067@100 |
0.37@1000@200 | 347@1000@100 | Oxygen vacancy | 68 |
| LaF3/La0.8Fe0.9Co0.1O3−δ | Sol–gel | Nanoparticle | 7373.5@100 | 1.29@500@200 | 157@500@200 | F dopant | 69 |
| Ni-doped LaNiO3 | Sol–gel | Nanoparticle | 5910@50 | 0.9@500@250 | 50@500@250 | Oxygen vacancy | 70 |
| Oxygen-defective CaMnO3 | Sol–gel | Interconnected nanoparticle | 3600@250 | 1.0@500@500 | 114@500@500 | Redox mediator LiI; defects | 45 |
| LaMn0.7Co0.3O3−δ | Electrospinning | Nanofiber | 0.81@1000@200 | 161@1000@200 | 1D nanotube structure | 71 | |
| La0.6Ca0.4Fe0.8Ni0.2O3 | Electrospinning | Nanotube | 13019@400 |
137@500@400 | 1D porous double-doped perovskite | 72 | |
| RuO2@La0.6Sr0.4Co0.8Mn0.2O3 | Electrospinning | RuO2 nanosheets on nanofiber | 12741.7@50 | 1.32@500@50 | 100@500@50 | RuO2/perovskite composite | 73 |
| CoO/Sr0.9Y0.1CoO3−δ | Electrospinning | Nanorod | 3100@50 | 130@500@50 | CoO/perovskite composite | 25 | |
| La0.8Sr0.2VO3 | Electrospinning | Porous nanotube | 5800@2000 | 1.16@1000@100 | 253@1000@2000 | High reversibility under high current densities | 42 |
| LaSrCoO/Ti3C2Tx | Hydrothermal | Perovskite nanoparticle on 2D nanosheet | 11340@500 |
1.02@1000@500 | 80@1000@500 | MXene/perovskite composite | 27 |
| LaNi0.9Co0.1O3 | Hydrothermal | Nanosheet | 1.5@1000@0.1 mA cm−2 | 100@1000@0.1 mA cm−2 | A binder-free electrode | 74 | |
| La0.8Sr0.2Co0.8Fe0.2O3−δ | Sol–gel | Nanoparticle | 26833@50 |
0.55@1000@100 | 200@1000@300 | Adjust metal–O covalency | 43 |
| LaCo0.75Mn0.25O3−δ | Hydrothermal | Hollow spherical structure | 10301@200 |
1.15@500@200 | 60@500@200 | Tunable defect and surface structure | 75 |
| LaFeO3−δ | Sol–gel | 2D nanosheet | 14983@100 |
1.2@1000@100 | 60@1000@100 | Oxygen defects | 76 |
| LaNi0.25Co0.75O3−δ | Sol–gel | Interconnected nanoparticle | 7720@0.1 mA cm−2 | 49@1000 | Ni dopant | 77 | |
| Ni3S2/PrBa0.5Sr0.5Co2O5+δ | Electrospinning & ALD | Ni3S2 nanoparticle on hollow perovskite nanofiber | 12874@100 |
0.68@1000@100 | 120@1000@100 | Metal sulfide/perovskite composite | 78 |
| Fe2O3/LaNiO3 | Microwave & ALD | 2D nanosheet | 10419@100 |
0.77@1000@100 | 90@500@100 | Iron oxide/perovskite composite | 79 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ@Pd | Wet chemistry | Nanoparticle | 16912@200 |
38@500@200 | Pd/perovskite composite | 80 | |
| La0.8Fe0.9Co0.1O3−δ | Sol–gel | Nanoparticle | 7270.1@100 | 0.7 V@500@200 | 215@500@500 | Metal and alloy exsolution | 81 |
| La0.75Sr0.25MnO3 | Electrospinning | Porous nanotube | 11000@0.025 mA cm−2 |
124@1000@0.15 mA cm−2 | Hollow 1D channels beneficial for electron transport and O2 diffusion | 82 | |
| Sr-doped-La2NiO4/NiO | Spray pyrolysis | Spherical particle | 131380@200 |
0.66@500@500 | 188@500@200 | Sr dopant | 83 |
| LaNi0.5Co0.5O3 | Sol–gel | Spherical nanoparticle | 7.96 mA h cm−2@0.1 mA cm−2 | 0.05@0.5 mA cm−2@0.1 mA cm−2 | 100@0.5 mA cm−2@0.1 mA cm−2 | 98.2% energy efficiency LOB | 84 |
| LaFeO3−δ | Emulsion polymerization & Annealing | 3D ordered macroporous honeycomb | 15000@0.025 |
124@1000@0.15 mA cm−2 | 3D ordered macroporous structure | 85 | |
| La0.6Sr0.4CoO3−δ | Hydrothermal & template method | Macrosphere | 4895@100 | 1.06@100(non-capacity limit) | 30@500@0.1 mA cm−2 | Catalytic activity in both aqueous and non-aqueous solution | 86 |
| RuO2/La2LiRuO6−δ | Ball milling | Nanoparticle | 0.9@100@10 mA gcarbon−2 | 665@200@100 | Solid-state LOB | 38 | |
| Ag@La0.6Sr0.4Fe0.9Mn0.1O3 | Sol–gel & UV irradiation | Nanoparticle | 12477 |
1.3@500@400 | 147@500@400 | Ag/perovskite composite | 87 |
| Ni-La0.9Mn0.6Ni0.4O3−δ | Electrospinning | Nanoparticles on nanofiber | 16656@400 |
95@500@400 | Exsolution of Ni nanoparticles | 88 | |
| LaCo0.8Fe0.2O3@rGO | Hydrothermal | Nanowire on rGO sheet | 7088.2@200 | 0.98@500@200 | 56@500@200 | rGO/perovskite composite | 89 |
| α-Fe2O3/La0.8FeO3−δ | Sol–gel | Nanoparticle | 7183@100 | 1.0@500@100 | 108@500@100 | Fe2O3/perovskite composite | 90 |
4.1 Dimensional control by synthetic methods
Bulk perovskites show inferior stability and insufficient bifunctionality for the ORR/OER, giving rise to low specific capacities, poor rate capability, inferior round-trip efficiencies, and cycling instability. Nanosizing the bulk perovskites effectively enlarges the interfacial contact between the electrolyte and catalyst and exposes more active sites for the ORR/OER.11,14,15,91 Various nanostructures and morphologies of perovskites have been fabricated, including nanocrystals,66,92 0D nanospheres,75,83,86,93 1D nanofibers/nanotubes,25,71–73,78,82,94 2D nanosheets,76 3D ordered structures,85,95 and nanocubes.96,97 This mini-review summarizes the research on small- and large-sized perovskite catalysts for LOBs in recent years.CsPbBr3 perovskite cubic nanocrystals (∼8.5 nm) were prepared as catalysts for LOBs by a typical hot-injection strategy (Fig. 4(a) and (b)).66 During charging, the decomposition of Li2O2 in the CsPbBr3 nanocrystals followed two steps: (1) Li2O2 = LiO2 + Li+ + e− and (2) LiO2 = O2 + Li+ + e− (rate-determining step), as shown in Fig. 4(c). The (100) facet displayed the smallest adsorption energy toward LiO2 compared to the (110), (210), (211), and (321) facets, resulting in a small overpotential for the OER. For the ORR process, LiO2 preferentially dissolved in the electrolyte and produced the toroid-type Li2O2 based on the solution growth mechanism (Fig. 4(d)). Additionally, CsPbBr3 perovskite quantum dots (4–5 nm) were deposited into an iron-based metal–organic framework in organic solvent via a sequential deposition method, named CsPbBr3@PCN-333(Fe) (Fig. 4(e) and (f)).92 Oleic acid and oleylamine were added to the mixture as organic stabilizers. The composite was applied as a photoelectric cathode for a light-assisted LOB. The LOB using the CsPbBr3@PCN-333(Fe) cathode retained a higher discharge voltage (2.5 V at 0.5 mA cm−2) than CsPbBr3 (<2 V) and PCN-333(Fe) (2.4 V) (Fig. 4(g)).
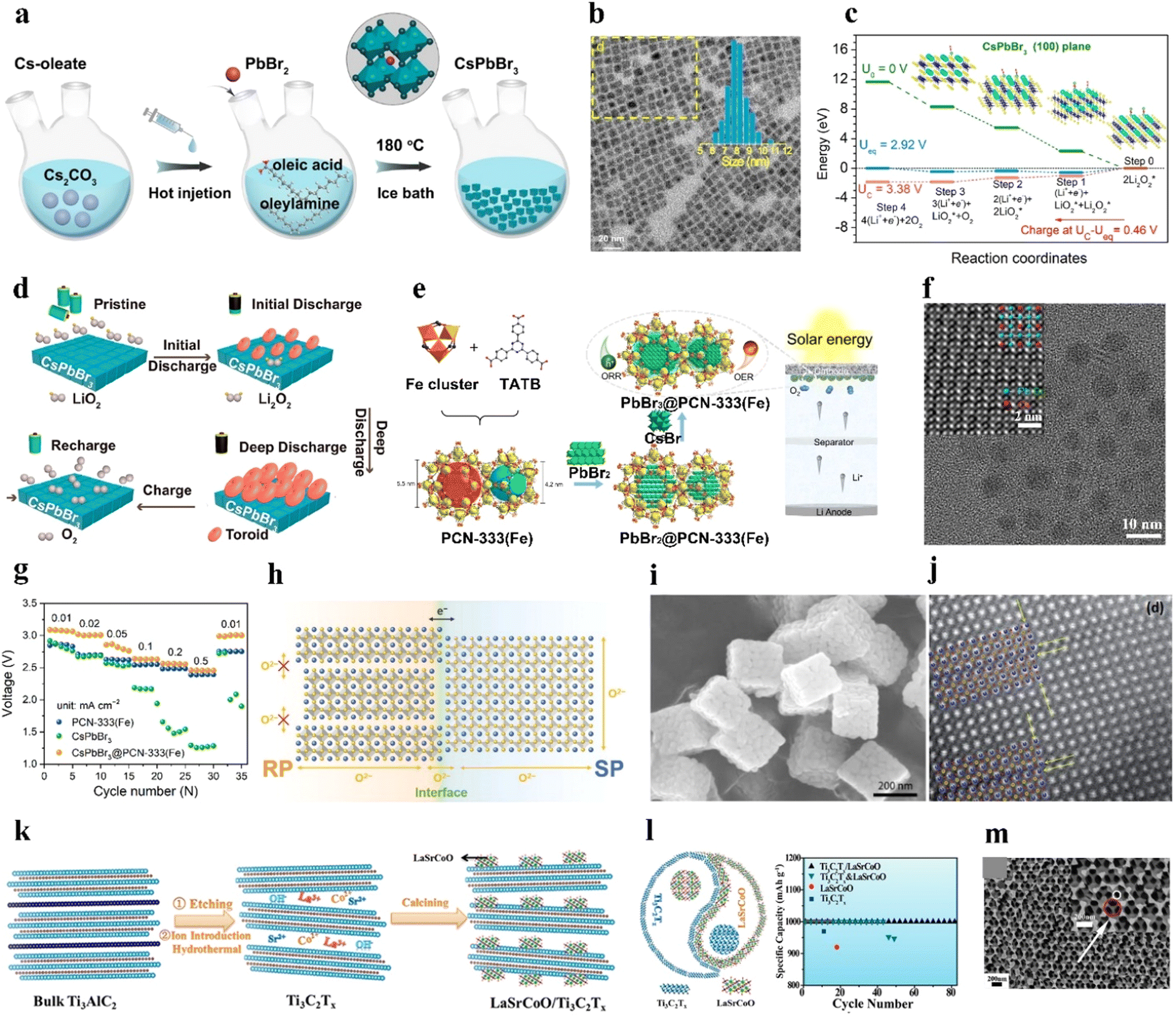 | ||
| Fig. 4 Synthesis of large perovskites. (a) Illustration of the synthesis process of CsPbBr3 nanocrystals. (b) Transmission electron microscopy (TEM) image of CsPbBr3 nanocrystals. (c) Gibbs free energy at different potentials of CsPbBr3. (d) Demonstration of discharging/charging processes for LOBs with CsPbBr3. (a)–(d) Reproduced with permission.66 Copyright 2021, American Chemical Society. (e) Preparation process of CsPbBr3@PCN-333(Fe). (f) TEM image of CsPbBr3@PCN-333(Fe). (g) Variation of discharge voltage at distinct current densities with CsPbBr3@PCN-333(Fe) and control samples under illumination. (e)–(g) Reproduced with permission.92 Copyright 2021, American Chemical Society. (h) Demonstration of the possible electron and oxygen–ion transfer at the interface of dual phases in LaSr3−y. Reproduced with permission.101 Copyright 2021, Wiley-VCH. (i) Scanning electron microscopy (SEM) image of La0.9Co0.8Ni0.2O3−x nanocubes. Reproduced with permission.97 Copyright 2019, Elsevier. (j) Scanning TEM image of LaNi0.9Cu0.1O3 with crystal strains. Reproduced with permission.74 Copyright 2017, Royal Society of Chemistry. (k) Illustration of the synthesis process of the LaSrCoO/Ti3C2Tx composite. (l) Cycling stability of LaSrCoO/Ti3C2Tx. Reproduced with permission.27 Copyright 2019, American Chemical Society. (m) SEM image of macroporous LaMnO3. Reproduced with permission.95 Copyright 2019, Elsevier. | ||
Sol–gel. Sol–gel is a facile synthetic method used to prepare perovskite nanomaterials based on the inorganic polymerization reaction of molecular precursors.26,43,45,67–70,84,98–100 First, different metal nitrates or acetates with specific stoichiometric ratios are dissolved in deionized water, followed by adding chelating agents (e.g., ethylenediamine tetraacetic acid and citrate acid) and binding agents (like ethylene glycol). An NH4OH solution is added to the above mixture to adjust the pH of the mixture to 6–8, allowing complete complexation. Then, the mixture is aged at 80–120 °C to form a gel. The gel is further dried below 300 °C to generate a fluffy resultant, followed by high-temperature annealing (above 600 °C) under air to remove organic moieties. The sol–gel approach combined with ultraviolet light was used to place Ag nanoparticles on La0.6Sr0.4Fe0.9Mn0.1O3 with a BET specific surface area (SSA) of 20.83 m2 g−1. The derived LOB exhibited a superior capacity (12
477 mA h g−1 at 400 mA g−1) and a long cycle life (147 cycles under a limited capacity of 500 mA h g−1 at 400 mA g−1).87
Vo can be created by annealing the perovskite product derived from the sol–gel under a reducing atmosphere such as an H2/Ar mixed atmosphere.45 Zhu et al.100 prepared La1−xFeO3−δ perovskites with surface Vo and a small portion of Fe4+ species, as bifunctional ORR/OER catalysts by an EDTA–citrate complexing sol–gel process. In addition, Xu et al.101 used a sol–gel method to prepare cation-deficient LaSr3−y with dual phases of Ruddlesden–Popper perovskite (RP) and single perovskite (SP) with controllable structures and compositions. The interfacial interaction of the perovskite composite elevated the oxygen and ionic transport, which enhanced the lattice-O participation in the OER process in an alkaline electrolyte (Fig. 4(h)).
Oxygen in sol–gel derived perovskite oxides can be replaced by N, S, and F, which is realized by placing anion precursors and the perovskite oxide in two separated porcelain boats in a tube furnace, followed by heating at around 300–400 °C under an Ar atmosphere.68,69 In short, the sol–gel method allows flexibility in the construction of various perovskites. However, perovskite oxides prepared by this method generally possess a low BET surface area (<50 m2 g−1), unfavorable for electrolyte diffusion and limiting the number of exposed active sites.
Electrospinning. 1D perovskite nanofibers with high surface-to-volume ratios show a relatively large surface area and well-distributed porosity, which maximizes the electrolyte-accessible active sites, facilitates mass transport, accommodates reaction products, and mitigates the clogging issue on the catalyst surface. An electrospinning method has been widely adopted to prepare 1D porous perovskite nanomaterials, such as La0.6Ca0.4Fe0.8Ni0.2O3, LaMn0.7Co0.3O3–x nanotubes, PrBa0.5Sr0.5Co2−xFexO5+δ nanofibers, La0.6Sr0.4Co0.8Mn0.2O3 nanofibers, Sr0.9Y0.1CoO3−δ nanorods, and La0.8Sr0.2VO3 nanofibers.25,42,71–73,102 First, one or more metal precursors are dissolved in organic solvent and mixed with polymer solutions like polyacrylonitrile and polyvinylpyrrolidone (PVP) to generate the mother solution. Then, the mixture is filled into a plastic syringe tube equipped with a stainless-steel nozzle. The solution is electrospun onto a rotating drum, as a collector, under a high voltage. Thereafter, the collected nanofibers are stabilized and pyrolyzed in a muffle oven or tube furnace.
The morphology and porous structure of the electrospinning products can be tailored using different amounts of polymer precursors during electrospinning. As such, Wang et al.72 demonstrated that adjusting the concentration of PVP changed the morphology of La0.6Ca0.4Fe0.8Ni0.2O3 from nanofiber to nanotube. A low concentration of PVP, around 0.13 g mL−1, resulted in the formation of hollow nanotubes with a diameter of 100–200 nm, while a higher-concentration of PVP of around 0.18 g mL−1 generated solid nanofibers after calcination. When PVP was present in a low amount, it preferentially lied at the perovskite core to form a hollow structure. Although, the metal composition, morphology and porous structure of the resultant can be tuned by adopting various precursor components and annealing temperatures, the distribution of multi-scale pores cannot be realized by the electrospinning technique.
Hydrothermal. A hydrothermal method has been used to prepare perovskites or their composites as effective catalysts for LOBs.27,74,97 Sun et al.97 prepared porous La0.9Co0.8Ni0.2O3−x nanocubes by a hydrothermal method in the presence of PVP and glycine. The nanocubes had a larger BET surface area (23.45 m2 g−1) than that of the sample prepared by the sol–gel process (4.82 m2 g−1) (Fig. 4(i)). PVP and glycine, as a potential crystal face inhibitor and shape-control agent, respectively, were applied to produce the cube-like framework.
A binder-free electrode is conducive for effective electron transfer. As such, LaNi0.9M0.1O3 (M: Cu or Co) perovskite nanosheets were deposited on 3D Ni foam by a hydrothermal method as self-standing air electrodes for LOBs, and the derived sample exhibited an SSA of 240 m2 g−1.74 Substituting Ni3+ with Cu2+ increased the crystalline plate distance and generated Vo. Crystal strain and dislocation defects can be observed in LaNi0.9Cu0.1O3 (Fig. 4(j)), attributed to the substitution with a TM ion of a different size, as well as the formation of Vo induced by the different chemical valence of the metal ions. Due to the microporous structure of the LaNi0.9Cu0.1O3 catalyst and the use of a redox mediator (tetrathiafulvalene), the derived LOB exhibited a small overpotential of 0.72 V and high round-trip efficiency. Li2O2 was uniformly deposited on the catalyst nanosheet during the discharge process and then decomposed after the charging process. Moreover, La0.5Sr0.5CoO3−δ (LaSrCoO) nanoparticles were deposited in Ti3C2Tx nanosheets by hydrothermal and post-thermal treatment (Fig. 4(k)).27 The LaSrCoO/Ti3C2Tx composite showed longer cycle life compared to LaSrCoO and Ti3C2Tx due to the synergistic effect of the dual components (Fig. 4(l)).
Since solid discharge products deposit on the surface of the catalysts, a porous structure of catalyst is essential for improving the electrochemical performance of LOBs. Macropores provide sufficient space for efficient mass diffusion (Li+ ions and O2), allowing fast ORR/OER kinetics. Mesoporous channels can trap the Li2O2 discharge product and prevent it from diffusing into the electrolyte, ensuring favorable interfacial contact between Li2O2 and the catalyst and thus lowering the reaction overpotentials.54 Hard template synthesis,83,95 a soft-template route,76 and wet-chemistry80 have been used to prepare porous perovskites. Ordered macroporous LaMnO3 with an SSA of 20.3 m2 g−1 was prepared using PMMA colloidal crystals as a hard template, facilitating both oxygen and electrolyte diffusion (Fig. 4(m)).95 However, only a few reports investigate how the porous structure and porosity influence the catalytic activity of perovskites for LOBs. Hence, more efforts are needed to investigate the property–activity relationship of perovskite catalysts.
4.2 A-/B-/O-site regulation in ABO3
Perovskites with A-site vacancies can be prepared by a nonstoichiometric method. Du et al.67 produced La defects in La0.7MnO3−δ (L0.7MO) as a bifunctional catalyst for the OER/ORR by decreasing the stoichiometric ratio of La(NO3)3·6H2O in the sol–gel synthesis. The defective sample possessed a larger BET surface area (34.7 m2 g−1) and pore size (20.6 nm) than that of the defective-free sample (11.9 m2 g−1, 8.9 nm), beneficial for oxygen and Li+ transport and the storage of Li2O2. The generated La defects effectively elevated the Mn–O bond covalency, which optimized the eg electron filling state of the Mn ions and improved the overlapping status of Mn 3d and O 2p orbitals. Furthermore, La defects on the L0.7MO surface served as multiple unsaturated active spots to adsorb oxygen intermediates, leading to strong interaction between the catalyst and LiO2. Thereafter, the adsorbed LiO2 underwent a one-electron transfer reaction to generate a film-like Li2O2 uniform covering on the catalyst surface, which corresponded to a surface growth mechanism. As a result, lattice-oxygen redox reactions and electron transfer in La0.7MnO3−δ were accelerated between Mn and oxygen adsorbates. The LOB using L0.7MO delivered a superior capacity of 29286 mA h g−1 at a current density of 50 mA g−1 (compared with 13709 mA h g−1 from the cell using a defective-free sample), a small voltage gap (0.38 V) and stable cycling life (375 cycles under a confined capacity of 1000 mA h g−1 at 300 mA g−1).
Owing to the overlap between B-site ions and the O2− orbital, the electronic conductivity is governed by B–O–B bonds and the valence transition of the B cations.47 A shorter B–B distance is conducive to the formation of O–O bonds between the neighboring adsorbates, beneficial for OER kinetics.104 A shorter B–B bond in ABO3 and a smaller B–O–B angle can be realized by foreign-cation doping.105 As such, doping a Co element into LaMn0.7Co0.3O3−x nanotubes shortened the Mn–Mn distance and promoted the formation of O–O bonds, favorable for promoting the OER performance.71 The assembled LOB could be operated for 161 cycles with a confined capacity of 1000 mA h g−1 at 200 mA g−1.
Furthermore, the oxidation state of the B-site TM and the amount of Vo can be regulated by foreign-cation doping, providing a versatile way to correlate physicochemical features with catalytic activity. The presence of aliovalent dopants results in the generation of a positively charged Vo to maintain charge neutrality based on Coulomb's law. Defective perovskite structures assist in the migration, emission, and intercalation of lattice oxygen, thereby enhancing the redox capability of B-site cations. For example, the introduction of Sr decreases the valence state of the B-site metal and the crystallinity structure.42 Sung et al.42 induced a phase transition from La0.8Sr0.2VO4 monazite (LSV4) to La0.8Sr0.2VO3 perovskite (LSV3) under a reducing atmosphere (Fig. 5(a)). LSV3 showed a defective and porous structure, strain and lattice shrinkage induced by mismatching of VO6 octahedra (Fig. 5(b)). The reduced oxidation state from V5+ to V3+ induced by the Sr dopant allowed the generation of enriched Vo active sites. LSV3 exhibited better ORR and OER performance (Fig. 5(c) and (d)), conducive to the formation and decomposition of Li2O2. Accordingly, the assembled LOB demonstrated high reversibility over 253 cycles with a limited capacity of 1000 mA h g−1 at 2000 mA g−1 and stable rate capabilities at different current densities from 100 to 2000 mA g−1 (Fig. 5(e)).
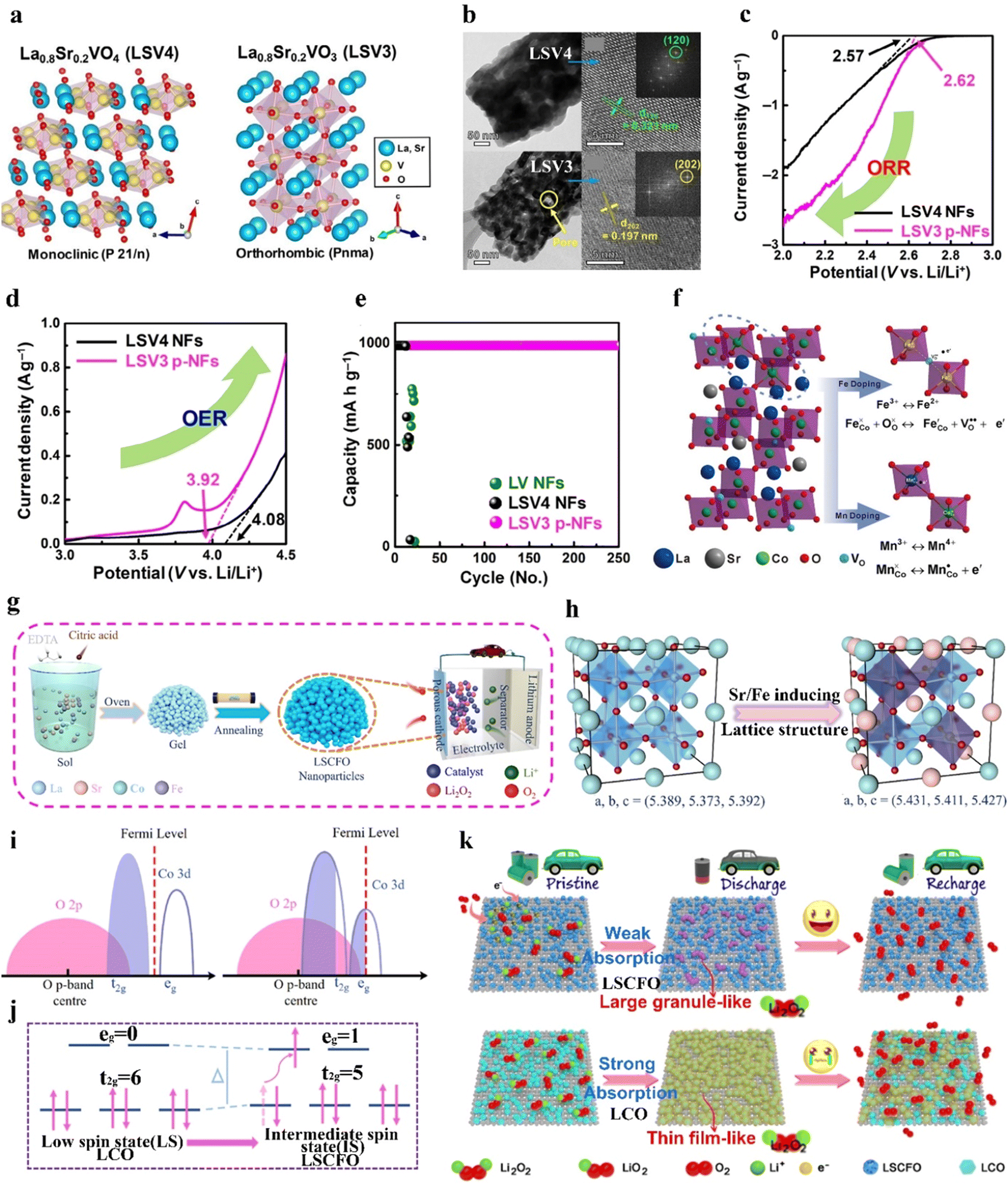 | ||
| Fig. 5 The impact of B-site substitution on perovskites. (a) Crystal structures of LSV4 and LSV3. (b) Low- and high-resolution TEM images of LSV4 and LSV3. (c) ORR and (d) OER linear sweep voltammetry of LSV4 and LSV3 at 5 mV s−1. (e) Cycling stability of LOBs with LSV4 and LSV3 catalysts. (a)–(e) Reproduced with permission.42 Copyright 2021, Wiley-VCH. (f) Demonstration of the Fe and Mn dopants in La0.6Sr0.4CoO3. Reproduced with permission.106 Copyright 2018, Wiley-VCH. (g) Schematic illustration of the preparation of LSCFO. (h) Crystal structures, and (i) the overlapping state of the Co 3d–O 2p bonds of LCO and LSCFO. (j) Schematic illustration of the spin state transformation from LCO to LSCFO. (k) Illustration of the oxygen electrode reaction pathways of LCO and LSCFO. (g)–(k) Reproduced with permission.43 Copyright 2021, American Chemical Society. | ||
In LaCoO3, partial replacement of the Co site in the B-site with Mn or Fe can boost the rate kinetics and coulombic efficiency and elongate the cycle life of the resulting LOB. Abundant Vo in mesoporous LaCo0.75Mn0.25O3−σ (LCMO) effectively bound the intermediate product of Li2−xO2, accelerated electron transfer, and lowered the reaction energy barrier.75 Accordingly, the LOB using an LCMO cathode exhibited a low potential of 1.12 V and a high specific capacity of 10301 mA h g−1 at 200 mA g−1. In another example, the doping effect of different cations like Mn and Fe in the catalytic activity of La0.6Sr0.4CoO3 was compared (Fig. 5(f)).106 The Fe dopant was effective in reducing the grain size of the perovskite nanoparticles, leading to a higher SSA (10.69 m2 g−1) than that of the pristine sample (6.68 m2 g−1) and thus exposing more active sites. Moreover, the introduced Fe2+ contributed to generating more Vo, according to 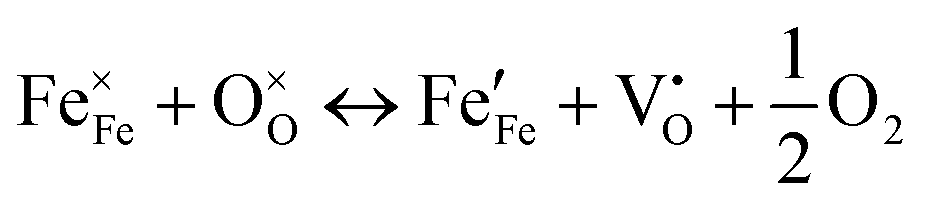 . Mn possessed a mixed valence between 2+/3+ and 3+/4+, and exhibited an electron-donating feature. Thus, the electron-rich Mn-doped sample increased the electrical conductivity, which was beneficial for electron transfer during the ORR/OER. Mn and Fe dopants with larger radii than Co can expand the crystalline structure, and thus create more pathways for Li+ transport. In addition to a TM dopant, a magnesium dopant in LaNi1−xMgxO3 has been introduced to replace Ni, and this suppressed the formation of low-valence Ni2+ ions (eg > 1). The presence of Ni3+ (eg = 1) in the Mg-doped sample ensured high ORR/OER activity.107 The derived LOB with the Mg-doped cathode demonstrated a higher discharge capacity than that using the pristine sample.
. Mn possessed a mixed valence between 2+/3+ and 3+/4+, and exhibited an electron-donating feature. Thus, the electron-rich Mn-doped sample increased the electrical conductivity, which was beneficial for electron transfer during the ORR/OER. Mn and Fe dopants with larger radii than Co can expand the crystalline structure, and thus create more pathways for Li+ transport. In addition to a TM dopant, a magnesium dopant in LaNi1−xMgxO3 has been introduced to replace Ni, and this suppressed the formation of low-valence Ni2+ ions (eg > 1). The presence of Ni3+ (eg = 1) in the Mg-doped sample ensured high ORR/OER activity.107 The derived LOB with the Mg-doped cathode demonstrated a higher discharge capacity than that using the pristine sample.
Dual cation dopants in ABO3 perovskites can synergistically regulate the adsorption capability of catalysts toward the LiO2 intermediate and the morphology of the discharge product. LaCoO3 (LCO) is a nonmagnetic insulator due to the low-spin state of Co3+ with an atomic configuration of t62ge0g.43 Sr and Fe cations doped in porous LaCoO3 perovskite nanoparticles (LSCFO) by a sol–gel method resulted in the formation of abundant Vo and enhanced Co 3d–O 2p covalency bonds, and induced the transformation of Co3+ from a low-spin state to an intermediate-spin state (Fig. 5(g)–(j)).43 The enhanced Co 3d–O 2p covalency facilitated electron transfer between the surface TM and adsorbed intermediates, and the optimized spin state transformed the perovskite from a nonconductor state to a metallic state. Hence, the produced granule-like Li2O2 in LSCFO can be more effectively decomposed during the charging process, compared to LCO (Fig. 5(k)). This LOB device demonstrated long-term cycling stability over 200 cycles at 300 mA g−1, and an ultrahigh specific capacity of 26833 mA h g−1 at 50 mA g−1. Moreover, Cu with a low valence state and Co with a favorable 4e− process were also co-doped to LaMnO3 by gel combustion to promote ORR/OER activity.108
067 mA h g−1 at 100 mA g−1, low overpotential (0.37 V), favorable rate capability, and long cycle life (347 cycles with a limited capacity of 1000 mA h g−1 at 100 mA g−1) (Fig. 6(d) and (f)).
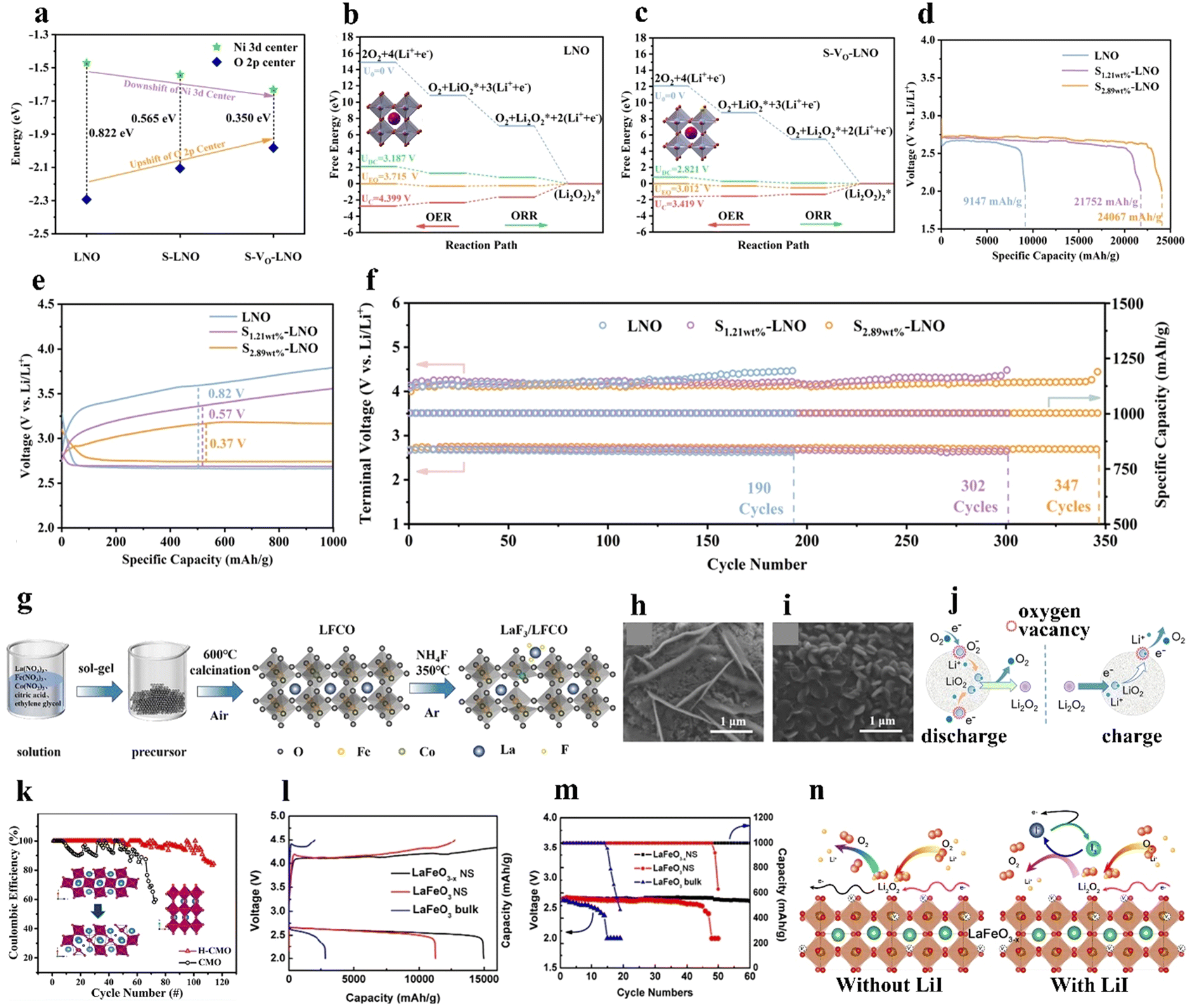 | ||
| Fig. 6 The impact of B-site substitution and oxygen vacancies on perovskites. (a) Illustration of Ni 3d and O 2p band centers S-Vo-LNO and control samples. Free energy diagram of the ORR on (b) LNO and (c) S-Vo-LNO. (d) Discharge curves at 100 mA g−1, (e) discharge/charge curves with a confined capacity of 1000 mA h g−1 at 100 mA g−1. (f) Cycling stability of the LOBs with LNO and S-Vo-LNO cathodes. (a)–(f) Reproduced with permission.68 Copyright 2022, Elsevier. (g) Illustration of the synthesis process of LaF3/LFCO. SEM images of (h) LFCO and (i) LaF3/LFCO cathodes after the first discharge. (g)–(i) Reproduced with permission.69 Copyright 2022, Elsevier. (j) Formation and decomposition processes of Li2O2 on N-doped LNO during the discharge/charge process. Reproduced with permission.70 Copyright 2018, American Chemical Society. (k) Coulombic efficiency of H-CMO and CMO. Insets: Their crystal structures. Reproduced with permission.45 Copyright 2020, American Chemical Society. (l) The initial capacity at 100 mA g−1 and (m) cycling stability of LaFeO3 and LaFeO3−x. (n) ORR and OER mechanisms on the LaFeO3−x surface with/without LiI. (k)–(n) Reproduced with permission.76 Copyright 2020, Elsevier. | ||
Regulating the morphology and composition of the discharge product Li2O2 is a feasible way to improve the performance of LOBs, and can be realized by anion doping in perovskites. Using fluorine, the dopant with the highest electronegativity (4.0), can improve the electronic conductivity of electrocatalysts and generate Vo by forming metal–F bonds. As shown in Fig. 6(g), Hou et al.69 introduced an F dopant into La0.8Fe0.9Co0.1O3−δ (LFCO), reconstructing the surface property, manipulating the electronic structure, and tailoring the discharge reaction pathway of the LOB. The F dopant distorted the crystalline structure, increased the disorder degree in the adjacent environment of the Fe–O bond, and tailored the Fe/Co–O orbital hybridization. Based on theoretical calculations, due to the strong LiO2 adsorption energy in the derived LaF3/LFCO composite, the discharge reaction process on the cathode followed the surface growth mechanism. In this context, LiO2 may not transport to the electrolyte and may instead favor the formation of small-sized Li2O2 (0.2 μm) in LaF3/LFCO, while the sample without F demonstrates larger-sized Li2O2 (2 μm) that leads to a larger overpotential (Fig. 6(h) and (i)). The generated petal-like F-doped Li2O2 shows better electrical conductivity and smaller charge transfer resistance, beneficial for lowering the overpotential (1.29 V) and enhancing the cycling stability of LOB (157 cycles with a limited capacity of 500 mA h g−1 at 200 mA g−1).
A nitrogen dopant with an analogous atomic size to O and lower ionization energy could induce the formation of Vo because of charge balancing. Zhang et al.70 synthesized N-doped LaNiO3 (LNO) with an increased amount of Vo and Ni3+. Ni3+ with its optimized eg-filling orbital (t62ge1g) is more favorable toward ORR/OER catalytic activity. Furthermore, the generated Vo enhanced the formation and further decomposition of Li2O2 and hampered side reactions between the cathode and electrolyte (Fig. 6(j)). As a result, the LOB using the LNO cathode reached a high discharge capacity of up to 5910 mA h gcathode−1 at 50 mA gcathode−1. Though anion dopants in the O-site have been investigated in semiconductors to adjust the bandgap energy, few studies of anion-doped perovskites as catalysts for LOBs have been reported.
4.3 Oxygen vacancy
The role of oxygen defects in perovskites cannot be neglected because they can tune the electronic structure, surface chemistry and crystalline nanostructures, thereby modifying the physicochemical properties of catalysts.111The creation of Vo modulates the interaction between the B-site TM and oxygen molecules. The eg-filling of the B-site cation is a decisive factor in determining oxygen adsorption/desorption energy. A lower eg-filling results in strong B–O2 bonds, and a higher eg-filling contributes to weak bonding.45 As well as the cation doping discussed above, annealing materials under a reducing atmosphere is another viable approach to create Vo in perovskites. For example, Vo was introduced to CaMnO3 through thermal treatment under mixed H2/Ar gas at 300 °C. The presence of Vo in CaMnO3 created mixed-valence Mn3+/Mn4+ states, modifying the electronic structure of CaMnO3.45 The mixed Mn4+ (t2g3e0g) and Mn3+ (t32ge1g) exhibited an intermediate eg occupation, facilitating the formation of superoxide and Li2O2 and resulting in better ORR activity (a smaller Tafel slope of −0.756 V dec−1). Multiple valences of the B-site cation and unique structural arrangements, along with the good ionic conductivity of perovskite oxides, collectively promote the B(n+1)+/Bn+ redox reaction. Accordingly, the LOB using defective CaMnO3 (H-CMO) provided an improved cycling capability of 114 cycles with a limited capacity of 500 mA h g−1 at 500 mA g−1 (Fig. 6(k)). The LOB using H-CMO achieved a higher capacity of around 3600 mA h g−1 at 250 mA g−1, outperforming the cell using CMO (2750 mA h g−1).
The presence of Vo in perovskites optimizes the surface adsorption energy, provides a large body of active sites and promotes Li+ or e− conductivity. Gao et al.76 produced Vo in LaFeO3−x by calcination of the pristine LaFeO3 under Ar gas, which distorted the FeO6 octahedra and changed the valence state of Fe. Theoretical calculation results suggested that O2 molecules tended to adsorb on the surface-defective sample more than on LaFeO3. The existence of Fe with different valence states can increase ionic and electronic conductivity. Accordingly, the LOB with oxygen-defective LaFeO3−σ showed a higher initial capacity, better rate capacity, and longer cycle life than that with LaFeO3 (Fig. 6(l) and (m)). Moreover, an LiI redox mediator was used to further reduce the overpotential due to the I3− to I− redox reaction (Fig. 6(n)). However, the lack of accurate control of the Vo content continues to hamper efforts to quantitatively investigate the structure–activity relationship between Vo and catalytic activity.
4.4 Perovskite-based composite catalysts
Combining perovskite oxides with conductive substances such as carbon, MOFs, MXene, and metal compounds is a feasible way to enhance their electrochemical activity and overcome their intrinsic low conductivity. The intimate interfacial interplay between dual materials gives rise to unique electronic structures and improved structural stability.In addition, carbon nanofibers or nanosheets have been adopted as ideal frameworks to load with perovskites as conductive and highly porous free-standing cathodes.74,113,114 Yang et al.113 prepared sandwich-like graphene/mesoporous LaSrMnO nanosheets as free-standing cathodes for LOBs. Mesopores offered a high surface area for loading discharged products and easy access channels for the electrolyte, and served as reservoirs of O2 to feed the ORR/OER reactions. Macropores between nanosheets expedited Li+ diffusion and O2 transport into the inner space of the catalyst. In the composite, the perovskite phase was responsible for decreasing the reaction overpotential, and the graphene foam acted as a conductive medium to promote electron transport, provided sufficient room for the deposition/decomposition of Li2O2 and prevented the clogging of the discharged resultants. Accordingly, the assembled LOB demonstrated a high specific capacity, rate capability, and long cycling life.
Unfortunately, the electrochemical oxidation of carbon materials under high potentials during the OER process deactivates active sites. Highly active oxygen radicals like O2−, LiO2 and Li2O2 corrode carbon materials, and decompose carbons and organic electrolytes, shortening the cycle life of LOBs.84 Highly durable frameworks for loading perovskite oxides are thus required.
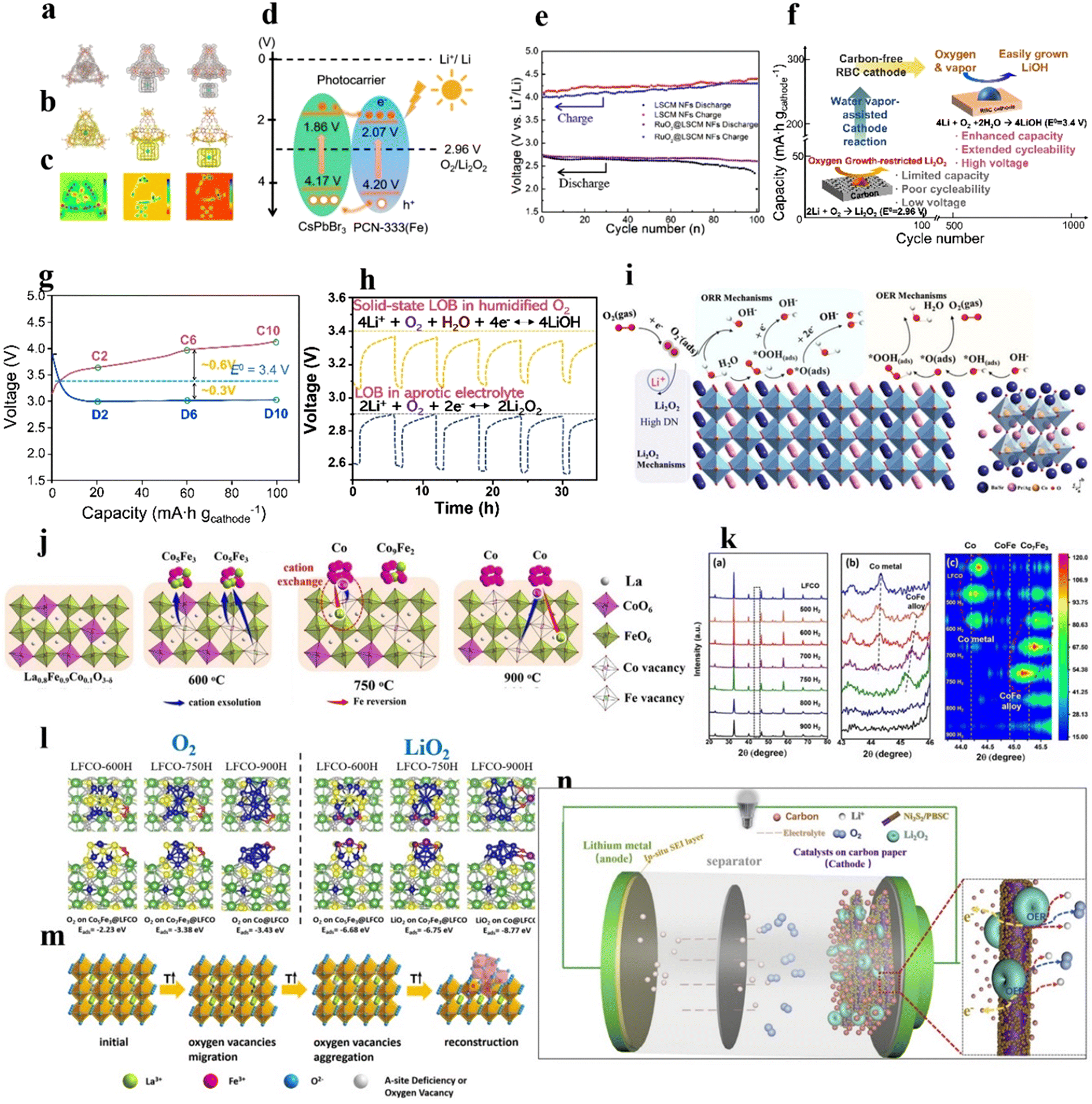 | ||
| Fig. 7 Perovskite-based composite catalysts. (a) Charge density of CsPbBr3 inside the cage, outside the cage, and isolated. (b) Corresponding charge density difference. (c) Corresponding 2D deformation charge density. (d) Demonstration of the VB and CB energy levels and the redistribution of photocarriers between PCN-333(Fe) and CsPbBr3 under light. (a)–(d) Reproduced with permission.92 Copyright 2021, American Chemical Society. (e) Discharge/charge terminal voltages of LOBs with RuO2@LSCM NF and LSCM NF cathodes. Reproduced with permission.73 Copyright 2021, American Chemical Society. (f) Schematic illustration of a solid-state LOB. (g) Discharge–charge curves of LOBs in humidified O2. (h) Galvanostatic intermittent titration technique (GITT) results of the LOB tested in liquid electrolyte without water vapor. (i) Schematic illustration of the ORR/OER at the Ag/PBSC heterostructure surface. Reproduced with permission.118 Copyright 2022, Wiley-VCH. (j) Dual-exsolution of LFCO controlled by temperature. (k) XRD patterns, enlarged XRD pattern at 43–46°, and depth view of the XRD pattern at 43.4–45.8° of LFCO. (l) Binding energy of O2 and LiO2 on LFCO obtained at different temperatures. (j)–(l) Reproduced with permission.81 Copyright 2022, Wiley-VCH. (m) Schematic illustration of the preparation of the α-Fe2O3/LaFeO3−x composite. Reproduced with permission.90 Copyright 2018, American Chemical Society. (n) Schematic illustration of the LOB employing Ni3S2/PBSC cathodes, and schematic of the charging process. Reproduced with permission.78 Copyright 2019, Elsevier. | ||
742 mA h g−1 at 50 mA g−1, a low voltage range of 2.6–4.3 V, and a stable cycle life (≥100 cycles with a limited capacity of 500 mA h g−1 at 50 mA g−1), as shown in Fig. 7(e).
Interestingly, water vapor as an additive was introduced to oxygen gas in the solid-state LOB, which transformed the discharge product from growth-limited Li2O2 to easily formed LiOH, and improved the specific capacity of the LOB.38,117 Kim et al.38 hybridized electron conducting RuO2 and La2LiRuO6−δ as a highly active electrocatalyst for a LiOH-related reaction in a solid-state LOB to increase capacity and cycle life (665 cycles with a limited capacity of 200 mA h gcathode−1 at 100 mA gcathode−1) (Fig. 7(f)). Humidity increased the discharge voltage of the LOB to 3.4 V, surpassing the cell operated under dry oxygen (2.96 V) (Fig. 7(g) and (h)). LiOH was capable of absorbing water vapor and became hydrated LiOH as a Li+-ion conductor, boosting the reaction kinetics.
Furthermore, due to its cheap price, good conductivity, and catalytic activity, metallic Ag has also been combined with perovskites as a cathode for LOBs.87,118 Zou et al.118 took advantage of the heterostructural interface between the conductive Ag nanoparticles and a PrBa0.5Sr0.5Co2O5+δ (PBSC) perovskite, and demonstrated that Ag could nucleate Li2O2 with a low overpotential (Fig. 7(i)).
The segregation of TM oxides in A-site defective perovskites can promote LOB performance. For example, the formation of Vo during the thermal treatment of La0.85FeO3−δ at 900 °C resulted in the segregation of α-Fe2O3 (Fig. 7(m)).90 In the composite, the interaction between LaFeO3−x and α-Fe2O3 led to structural distortion, exposing ample active sites for the OER/ORR. The LOB with the composite catalyst exhibited a longer cycle life (108 cycles) than that with α-Fe2O3 (21 cycles) and La0.85FeO3−δ (51 cycles) catalysts. In another example, Ni nanoparticles were exsolved on the La0.9Mn0.6Ni0.4O3−δ nanofiber surface during thermal treatment under a H2/Ar atmosphere.88 The interaction between metal nanoparticles and perovskite and the formation of Vo synergistically increased the number of active sites, and improved electrical conductivity and O2 adsorption.
Ni, Co, and Fe-based compounds with favorable catalytic activity have been combined with perovskites to prepare hybrid catalysts with the collective superiorities of dual-functional materials. Zhang et al.78 used an atomic-layer-deposition approach to deposit a layer of Ni3S2 (10 nm) on 1D PrBa0.5Sr0.5Co2O5+δ (PBSC) nanofibers as an integrated catalyst. The Li2O2 discharge product could be effectively decomposed to Li+ and O2 by virtue of the high OER activity of the PBSC and Ni3S2. The peroxide combined with Vo in the PBSC and then oxidized to oxygen, and the nanosized Ni3S2 and unique 1D porous PBSC could rapidly transfer electrons from active sites to the current collector. The assembled LOB (Fig. 7(n)) showed a low overpotential of 0.68 V at 1000 mA h g−1), large capacity (12874 mA h g−1 at 100 mA g−1), and long cycle life (>120 cycles with a limited capacity of 1000 mA h g−1 at 100 mA g−1). Another example shows that FeOOH clusters on the LaNiO3 surface enhanced OH− adsorption and weakened the Ni–O bond in an alkaline electrolyte, which optimized the OH− filling barrier to boost the lattice O-involved OER and decreased the reaction barrier of the LOM.59 Co3O4 nanoparticles on LaCo0.6Ni0.4O3 facilitated O2 adsorption on the catalyst surface and O2/Li2O2 conversion.119 In addition to the above-mentioned materials, Ti3C2Tx nanosheets with low Li ionic diffusion barriers (<0.07 eV) have been used as substrates to load La0.5Sr0.5CoO3−δ perovskite as a catalyst for LOBs.27 The derived LOB could be operated over 80 cycles at 500 mA g−1 with a restriction of 1000 mA h g−1, superior performance to that of La0.5Sr0.5CoO3−δ (15 cycles) and Ti3C2Tx (9 cycles).
5. Conclusion
Perovskite oxides have been studied as promising ORR/OER electrocatalysts due to their favorable catalytic activity, tunable compositions and nanostructures. This mini-review analyzes the recent advances in perovskite oxide electrocatalysts for LOBs, and highlights the synthetic techniques, fundamental catalytic mechanisms, and various strategies for the enhancement of catalytic performance. There is ample space to advance this technology by exploring the ORR/OER mechanisms, porous structure development, composition regulation, defect engineering, and composite synthesis for perovskite oxides in nonaqueous LOBs. Below is a summary of the existing challenges in the use of perovskite oxides as catalysts.(i) Notwithstanding massive endeavors to study the catalytic mechanisms of perovskite oxides for the ORR/OER in aqueous electrolytes, the investigation of fundamental catalytic mechanisms in the nonaqueous electrolyte scenario is still in its infancy. The relationship between the composition, crystal structure, and catalytic performance needs to be clarified and deeply investigated, as this knowledge is critical for guiding the design of highly active catalysts for LOBs.
It should be noted that in aqueous alkaline electrolytes, in situ surface reconstruction or amorphization of perovskites could occur by anodic polarization during the OER, which has been proved to be beneficial for enhancing the OER performance.120–122 A-site cations have high solubility in alkaline electrolytes, and BO2 metal oxides also suffer from chemical dissolution during the lattice-involved OER process. The dissolved B-site cations either release to the bulk electrolyte or react with OH− ions and then deposit on the material surface to form an oxyhydroxide phase.123 The processes can be described in eqn (8)–(11):
| 2ABO3 ↔ 2BO2 + 2A2+ + O2 + 4e− | (8) |
| 2ABO3 + 2H2O ↔ 2BO2 + 2A2+ + 4KOH | (9) |
| BO2 ↔ O2 + B4+ + 4e− | (10) |
| BO2 + 2H2O ↔ B4+ + 4OH− | (11) |
Of particular note, the surface reconstruction of perovskites in nonaqueous electrolytes has rarely been studied. It is speculated that it could occur when the surface lattice oxygen participates in the catalytic process. More explicitly, the active lattice oxygen of perovskites can assist in adsorbing Li+ ions from the aprotic electrolyte due to its shallower O 2p band adjacent to the Fermi level to donate electrons.61 After that, the generated electrophilic oxyl groups on the surface serve as initial growth species to react with nucleophilic moieties like LiO2, triggering the formation of the discharge product.17,61 Even though the surface lattice oxygen as catalytic sites has been proved to improve the ORR/OER kinetics, it may give rise to the generation of surface oxygen defects. Oxygen defects will be compensated by generating cationic vacancies, leading to the dissolution of surface cations and eventually resulting in the instability of the catalyst surface.61,124 As a result, the surface composition and nanostructure of perovskites are altered due to the formation of oxygen defects and the dissolution of surface cations. In this regard, the change in the surface electronic structure, crystallinity, valence state, and coordination environment of cations, and morphology of perovskite oxides upon cycling can be investigated by in situ or ex situ characterizations such as in situ TEM, Raman, X-ray absorption spectroscopy, etc. These techniques help to understand how these changes influence the catalytic activity and durability of cathodes for LOB. The combination of results from advanced characterization, experiments, and theoretical calculations is required for a comprehensive understanding of the underlying mechanisms of the reaction occurring at specific active sites under operando status.
(ii) Myriad cations or anions can be accommodated at the A-, B-, and O-sites of perovskites, ensuring the versatile adjustment of their properties for optimized catalytic performance. Screening and synthesis of ideal perovskite oxide catalysts are challenging and tedious processes due to the multitude of choices of elements. Therefore, artificial intelligence such as machine learning and high-throughput calculations can be used to gain unique insights and to screen highly active catalysts based on data sets. This will enable researchers to design materials with optimized compositions and ideal experimental parameters.29,125,126
(iii) High specific surface areas and controllable porosities are required to provide ample catalytically active spots and allow easy transport of reactants. It is known that the large-area triple boundaries, where oxygen, electrolyte, and oxygen cathodes exist, play crucial roles in effectively transporting mass and accommodating the discharged products. However, perovskite oxide catalysts prepared by the current synthetic methods (e.g., sol–gel and solid-state reaction) show a small SSA (<300 m2 g−1), uncontrollable pore structures, and irregular morphology in some cases, impeding mass transport. Novel 3D-printed ordered perovskite,127 self-assembled anti-perovskites,128 and 3D structured perovskitoids39 with large SSAs, controllable morphologies and tailorable porous architectures may overcome these challenges, and are promising cathode candidates for LOBs.
(iv) It is imperative to pay attention to the activity-stability tradeoff of perovskites. Long cycling stability of energy-storage devices is a prerequisite for real-world applications, while LOBs with perovskite cathodes can be operated for only several hundred cycles (<200 cycles in many cases), which is unfavorable for practical applications. The poor cycling stability of LOB batteries with perovskite cathodes can be attributed to the following reasons. First, the insoluble Li2O2 product covered on the perovskite surface blocks oxygen diffusion pathways, leading to severe volume expansion of the air cathode, and inducing side reactions at the cathode.20 Due to limited active sites at the Li2O2/catalyst interface, voltage polarization could occur and the reaction kinetics for the ORR/OER become sluggish.20 Second, metal elements in perovskites facilitate the generation of metal–O bonds in an O2-rich environment, which may gradually degrade the material structure. Third, some perovskites can be partially decomposed in aprotic solvents. For example, due to the reaction between positive Pb2+ ions and polar aprotic solvents, the PbI6 octahedral framework collapsed in a trihalide perovskite.129 Moreover, the improved catalytic activity of surface lattice oxygen of perovskites is at the expense of surface stability, where surface cations will be dissolved in the electrolyte, and bulk cation ions migrate to the surface area, resulting in instability of perovskite structures. To circumvent these obstacles, a porous structure of the cathode is highly required to increase oxygen diffusion paths and the number of nucleation sites for the discharged product, effectively decreasing the overpotential during charging/discharging processes of LOBs. Furthermore, soluble redox mediators could be introduced to the electrolyte to tune the formation and decomposition pathways of Li2O2 for further decreasing the overpotential of the ORR and OER. On top of that, nanostructured perovskite oxides (nanocrystals or quantum dots) can be combined with large-surface-area materials, such as graphene, porous carbon, MOFs, and covalent organic frameworks to harness the collaborative benefits of multiple active components. Meanwhile, a high loading of perovskite oxide on the substrates is required to achieve high catalytic performance.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (NRF-2020M3D1A1110522 and NRF-2022R1A2B5B03001781).References
- J. L. Holechek, H. M. E. Geli, M. N. Sawalhah and R. Valdez, Sustainability, 2022, 14, 4792 CrossRef.
- R. Zhang, H. Zhou, P. Sun, Q. Ma, M. Lu, H. Su, W. Yang and Q. Xu, Battery Energy, 2022, 1, 20220023 CrossRef CAS.
- C. Y. J. Lim and Z. W. Seh, Battery Energy, 2022, 1, 20220008 CrossRef CAS.
- Y. Guo, Y. Zhang and H. Lu, Battery Energy, 2022, 1, 20210014 CrossRef CAS.
- C. Shu, J. Wang, J. Long, H. K. Liu and S. X. Dou, Adv. Mater., 2019, 31, 1804587 CrossRef PubMed.
- M. D. Radin and D. J. Siegel, Energy Environ. Sci., 2013, 6, 2370–2379 RSC.
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS PubMed.
- W. J. Kwak, R. Sharma, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y. K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- K. Yoo, S. Banerjee, J. Kim and P. Dutta, Energies, 2017, 10, 1748 CrossRef.
- M. Park, C. Liang, T. H. Lee, D. A. Agyeman, J. Yang, V. W. Hei Lau, S. Il Choi, H. W. Jang, K. Cho and Y. M. Kang, Adv. Energy Mater., 2020, 10, 1903225 CrossRef CAS.
- Y. K. Jo, W. Tamakloe, X. Jin, J. Lim, S. B. Patil, Y. M. Kang and S. J. Hwang, Appl. Catal., B, 2019, 254, 523–530 CrossRef CAS.
- Y. Bae, H. Park, Y. Ko, H. Kim, S. K. Park and K. Kang, Batteries Supercaps, 2019, 2, 311–325 CrossRef CAS.
- T. H. Gu, D. A. Agyeman, S. J. Shin, X. Jin, J. M. Lee, H. Kim, Y. M. Kang and S. J. Hwang, Angew. Chem., Int. Ed., 2018, 130, 16216–16221 CrossRef.
- K. Song, J. Jung, M. Park, H. Park, H. J. Kim, S. Il Choi, J. Yang, K. Kang, Y. K. Han and Y. M. Kang, ACS Catal., 2018, 8, 9006–9015 CrossRef CAS.
- D. A. Agyeman, M. Park and Y. M. Kang, J. Mater. Chem. A, 2017, 5, 22234–22241 RSC.
- K. Song, E. Cho and Y. M. Kang, ACS Catal., 2015, 5, 5116–5122 CrossRef CAS.
- Y. Zheng, K. Song, J. Jung, C. Li, Y. U. Heo, M. S. Park, M. Cho, Y. M. Kang and K. Cho, Chem. Mater., 2015, 27, 3243–3249 CrossRef CAS.
- D. A. Agyeman, K. Song, S. H. Kang, M. R. Jo, E. Cho and Y. M. Kang, J. Mater. Chem. A, 2015, 3, 22557–22563 RSC.
- H. Song, R. Choi, J. Jung, G. Kim, K. Song, Y. Il Kim, S. C. Jung, Y. K. Han and Y. M. Kang, Energy Environ. Sci., 2014, 7, 1362–1368 RSC.
- Z. Chang, J. Xu and X. Zhang, Adv. Energy Mater., 2017, 7, 1700875 CrossRef.
- W. Bai, Z. Zhang, K. Wang and J. Chen, Battery Energy, 2022, 1, 20220019 CrossRef CAS.
- Y. Lai, Y. Jiao, J. Song, K. Zhang, J. Li and Z. Zhang, Mater. Chem. Front., 2018, 2, 376–384 RSC.
- Y. Yang, X. Xue, Y. Qin, X. Wang, M. Yao, Z. Qin and H. Huang, J. Phys. Chem. C, 2018, 122, 12665–12672 CrossRef CAS.
- Y. Liu, J. Cai, J. Zhou, Y. Zang, X. Zheng, Z. Zhu, B. Liu, G. Wang and Y. Qian, eScience, 2022, 2, 389–398 CrossRef.
- J. Wang, X. Cheng, Z. Li, M. Xu, Y. Lu, S. Liu, Y. Zhang and C. Sun, ACS Appl. Energy Mater., 2018, 1, 5557–5566 CAS.
- J. Cheng, Y. Jiang, M. Zhang, L. Zou, Y. Huang, Z. Wang, B. Chi, J. Pu and J. Li, Phys. Chem. Chem. Phys., 2017, 19, 10227–10230 RSC.
- Z. Sun, M. Yuan, L. Lin, H. Yang, C. Nan, G. Sun, H. Li and X. Yang, ACS Appl. Energy Mater., 2019, 2, 4144–4150 CrossRef CAS.
- W. Xiong, H. Yin, T. Wu and H. Li, Eur. J. Chem., 2022, 29, e202202872 Search PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- N. I. Kim, Y. J. Sa, T. S. Yoo, S. R. Choi, R. A. Afzal, T. Choi, Y. S. Seo, K. S. Lee, J. Y. Hwang, W. S. Choi, S. H. Joo and J. Y. Park, Sci. Adv., 2018, 4, eaap9360 CrossRef PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough, Y. Shao-horn, F. Calle-vallejo, A. D. Oscar, M. J. Kolb, M. T. M. Koper, J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef PubMed.
- J. X. Flores-Lasluisa, F. Huerta, D. Cazorla-Amorós and E. Morallón, Environ. Res., 2022, 214, 113731 CrossRef CAS PubMed.
- K.-L. Wang, Y.-G. Yang, Y.-H. Lou, M. Li, F. Igbari, J.-J. Cao, J. Chen, W.-F. Yang, C. Dong, L. Li, R.-Z. Tai and Z.-K. Wang, eScience, 2021, 1, 53–59 CrossRef.
- X. Li, W. Ma, D. Liang, W. Cai, S. Zhao and Z. Zang, eScience, 2022, 2, 646–654 CrossRef.
- M. Kim, H. Lee, H. J. Kwon, S. Bak, C. Jaye, D. A. Fischer, G. Yoon, J. O. Park, D. Seo, S. B. Ma and D. Im, Sci. Adv., 2022, 2, eabm8584 CrossRef PubMed.
- Y. Miao, X. Wang, H. Zhang, T. Zhang, N. Wei, X. Liu, Y. Chen, J. Chen and Y. Zhao, eScience, 2021, 1, 91–97 CrossRef.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- M. Sung, G. Lee and D. Kim, InfoMat, 2021, 3, 1295–1310 CrossRef CAS.
- D. Du, R. Zheng, X. Chen, W. Xiang, C. Zhao, B. Zhou, R. Li, H. Xu and C. Shu, ACS Appl. Mater. Interfaces, 2021, 13, 33133–33146 CrossRef CAS PubMed.
- X. Liu, H. Gong, T. Wang, H. Guo, L. Song, W. Xia, B. Gao, Z. Jiang, L. Feng and J. He, Chem. – Asian J., 2018, 13, 528–535 CrossRef CAS PubMed.
- G. S. Hegde, A. Ghosh, R. Badam, N. Matsumi and R. Sundara, ACS Appl. Energy Mater., 2020, 3, 1338–1348 CrossRef CAS.
- H. Wang, M. Zhou, P. Choudhury and H. Luo, Appl. Mater. Today, 2019, 16, 56–71 CrossRef CAS.
- K. Wang, C. Han, Z. Shao, J. Qiu, S. Wang and S. Liu, Adv. Funct. Mater., 2021, 31, 2102089 CrossRef CAS.
- M. Zhang, G. Jeerh, P. Zou, R. Lan, M. Wang, H. Wang and S. Tao, Mater. Today, 2021, 49, 351–377 CrossRef CAS.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- R. Jacobs, J. Hwang, Y. Shao-Horn and D. Morgan, Chem. Mater., 2019, 31, 785–797 CrossRef CAS.
- H. Lee, O. Gwon, K. Choi, L. Zhang, J. Zhou, J. Park, J. W. Yoo, J. Q. Wang, J. H. Lee and G. Kim, ACS Catal., 2020, 10, 4664–4670 CrossRef CAS.
- X. Wu, W. Yu, K. Wen, H. Wang, X. Wang, C. W. Nan and L. Li, J. Energy Chem., 2021, 60, 135–149 CrossRef CAS.
- A. Khetan, A. Luntz and V. Viswanathan, J. Phys. Chem. Lett., 2015, 6, 1254–1259 CrossRef CAS PubMed.
- Y. Zhou, Y. Zhao, Z. Liu, Z. Peng, L. Wang and W. Chen, J. Energy Chem., 2021, 55, 55–61 CrossRef CAS.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- Q. Li, J. Wu, T. Wu, H. Jin, N. Zhang, J. Li, W. Liang, M. Liu, L. Huang and J. Zhou, Adv. Funct. Mater., 2021, 31, 2102002 CrossRef CAS.
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897–3901 CrossRef CAS PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- J.-W. Zhao, C.-F. Li, Z.-X. Shi, J.-L. Guan and G.-R. Li, Research, 2020, 2020, 1–15 Search PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- D. A. Agyeman, Y. Zheng, T. H. Lee, M. Park, W. Tamakloe, G. H. Lee, H. W. Jang, K. Cho and Y. M. Kang, ACS Catal., 2021, 11, 424–434 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. P. M. Veder, D. Guan, R. O’Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- C. Yang and A. Grimaud, Catalysts, 2017, 7, 149 CrossRef.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- Y. Zhou, Q. Gu, Y. Li, L. Tao, H. Tan, K. Yin, J. Zhou and S. Guo, Nano Lett., 2021, 21, 4861–4867 CrossRef CAS PubMed.
- D. Du, R. Zheng, M. He, C. Zhao, B. Zhou, R. Li, H. Xu, X. Wen, T. Zeng and C. Shu, Energy Storage Mater., 2021, 43, 293–304 CrossRef.
- R. Li, J. Long, M. Li, D. Du, L. Ren, B. Zhou, C. Zhao, H. Xu, X. Wen, T. Zeng and C. Shu, Mater. Today Chem., 2022, 24, 100889 CrossRef CAS.
- H. Hou, Y. Cong, Q. Zhu, Z. Geng, X. Wang, Z. Shao, X. Wu, K. Huang and S. Feng, Chem. Eng. J., 2022, 448, 137684 CrossRef CAS.
- J. Zhang, C. Zhang, W. Li, Q. Guo, H. Gao, Y. You, Y. Li, Z. Cui, K. C. Jiang, H. Long, D. Zhang and S. Xin, ACS Appl. Mater. Interfaces, 2018, 10, 5543–5550 CrossRef CAS PubMed.
- H. Gong, T. Wang, H. Guo, X. Fan, X. Liu, L. Song, W. Xia, B. Gao, X. Huang and J. He, J. Mater. Chem. A, 2018, 6, 16943–16949 RSC.
- Z. Wang, L. Zou, S. Guo, M. Sun, Y. Chen, B. Chi, J. Pu and J. Li, J. Power Sources, 2020, 468, 228362 CrossRef CAS.
- X. Zhang, Y. Gong, S. Li and C. Sun, ACS Catal., 2017, 7, 7737–7747 CrossRef CAS.
- T. V. Pham, H. P. Guo, W. Bin Luo, S. L. Chou, J. Z. Wang and H. K. Liu, J. Mater. Chem. A, 2017, 5, 5283–5289 RSC.
- X. Li, Z. Qian, G. Han, B. Sun, P. Zuo, C. Du, Y. Ma, H. Huo, S. Lou and G. Yin, ACS Appl. Mater. Interfaces, 2020, 12, 10452–10460 CrossRef CAS PubMed.
- R. Gao, Q. Chen, W. Zhang, D. Zhou, D. Ning, G. Schumacher, D. Smirnov, L. Sun and X. Liu, J. Catal., 2020, 384, 199–207 CrossRef CAS.
- R. S. Kalubarme, G.-E. Park, K.-N. Jung, K.-H. Shin, W.-H. Ryu and C.-J. Park, J. Electrochem. Soc., 2014, 161, A880–A889 CrossRef CAS.
- Z. Zhang, K. Tan, Y. Gong, H. Wang, R. Wang, L. Zhao and B. He, J. Power Sources, 2019, 437, 226908 CrossRef CAS.
- C. Gong, L. Zhao, S. Li, H. Wang, Y. Gong, R. Wang and B. He, Electrochim. Acta, 2018, 281, 338–347 CrossRef CAS.
- M. Y. Oh, J. H. Kim, Y. W. Lee, K. J. Kim, H. R. Shin, H. Park, K. T. Lee, K. Kang and T. H. Shin, ACS Appl. Energy Mater., 2019, 2, 8633–8640 CrossRef CAS.
- Y. Cong, Z. Geng, Q. Zhu, H. Hou, X. Wu, X. Wang, K. Huang and S. Feng, Angew. Chem., Int. Ed., 2021, 60, 23380–23387 CrossRef CAS PubMed.
- J. J. Xu, D. Xu, Z. L. Wang, H. G. Wang, L. L. Zhang and X. B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3887–3890 CrossRef CAS PubMed.
- H. Kim, Y. S. Lim and J. H. Kim, Chem. Eng. J., 2022, 431, 134278 CrossRef CAS.
- Q. Qiu, Z. Pan, P. Yao, J. Yuan, C. Xia and Y. Zhao, Chem. Eng. J., 2023, 452, 139608 CrossRef CAS.
- J. J. Xu, Z. L. Wang, D. Xu, F. Z. Meng and X. B. Zhang, Energy Environ. Sci., 2014, 7, 2213–2219 RSC.
- P. Sennu, V. Aravindan, K. S. Nahm and Y. S. Lee, J. Mater. Chem. A, 2017, 5, 18029–18037 RSC.
- Z. Wang, X. Peng, S. Guo, M. Sun, J. Cheng, L. Zou, B. Chi and J. Pu, ACS Appl. Energy Mater., 2021, 4, 9376–9383 CrossRef CAS.
- S. Guo, L. Zou, M. Sun, Z. Wang, S. Han, B. Chi, J. Pu and J. Li, ACS Appl. Energy Mater., 2020, 3, 10015–10022 CrossRef CAS.
- J. G. Kim, Y. Kim, Y. Noh, S. Lee, Y. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 5429–5439 CrossRef CAS PubMed.
- Y. Cong, Z. Geng, Y. Sun, L. Yuan, X. Wang, X. Zhang, L. Wang, W. Zhang, K. Huang and S. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 25465–25472 CrossRef CAS PubMed.
- J. Jung, K. Song, Y. Bae, S. Il Choi, M. Park, E. Cho, K. Kang and Y. M. Kang, Nano Energy, 2015, 18, 71–80 CrossRef CAS.
- G. Y. Qiao, D. Guan, S. Yuan, H. Rao, X. Chen, J. A. Wang, J. S. Qin, J. J. Xu and J. Yu, J. Am. Chem. Soc., 2021, 143, 14253–14260 CrossRef CAS PubMed.
- J. Kim, H. Kim, S. Shin, H. W. Lee and J. H. Kim, Electrochim. Acta, 2022, 412, 140097 CrossRef CAS.
- Y. Bu, G. Nam, S. Kim, K. Choi, Q. Zhong, J. H. Lee, Y. Qin, J. Cho and G. Kim, Small, 2018, 14, 1802767 CrossRef PubMed.
- H. Lin, P. liu, S. Wang, Z. Zhang, Z. Dai, S. Tan and D. Chen, J. Power Sources, 2019, 412, 701–709 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Zhao, Z. Liu and W. Chen, Sci. Rep., 2014, 4, 2–7 Search PubMed.
- M. Sun, L. Zou, Z. Wang, S. Guo, Y. Chen, B. Chi, J. Pu and J. Li, Electrochim. Acta, 2019, 327, 135017 CrossRef CAS.
- X. Li, T. Zhu, C. Wen, Y. Yang, S. Ma, X. Huang, H. Li and G. Sun, Electrochim. Acta, 2019, 317, 367–374 CrossRef CAS.
- X. Han, Y. Hu, J. Yang, F. Cheng and J. Chen, Chem. Commun., 2014, 50, 1497–1499 RSC.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- X. Xu, Y. Pan, L. Ge, Y. Chen, X. Mao, D. Guan, M. Li, Y. Zhong, Z. Hu, V. K. Peterson, M. Saunders, C. Te Chen, H. Zhang, R. Ran, A. Du, H. Wang, S. P. Jiang, W. Zhou and Z. Shao, Small, 2021, 17, 2101573 CrossRef CAS PubMed.
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594–11601 CrossRef CAS PubMed.
- V. Celorrio, L. Calvillo, G. Granozzi, A. E. Russell and D. J. Fermin, Top. Catal., 2018, 61, 154–161 CrossRef CAS PubMed.
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Adv. Mater., 2017, 29, 1603004 CrossRef PubMed.
- U. A. Palikundwar, V. B. Sapre, S. V. Moharil and K. R. Priolkar, J. Phys.: Condens. Matter, 2009, 21, 235405 CrossRef PubMed.
- J. Cheng, Y. Jiang, M. Zhang, Y. Sun, L. Zou, B. Chi, J. Pu and L. Jian, ChemCatChem, 2018, 10, 1635–1642 CrossRef CAS.
- Z. Du, P. Yang, L. Wang, Y. Lu, J. B. Goodenough, J. Zhang and D. Zhang, J. Power Sources, 2014, 265, 91–96 CrossRef CAS.
- Y. Lv, Z. Li, Y. Yu, J. Yin, K. Song, B. Yang, L. Yuan and X. Hu, J. Alloys Compd., 2019, 801, 19–26 CrossRef CAS.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- R. B. Wexler, G. S. Gautam, E. B. Stechel and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 13212–13227 CrossRef CAS PubMed.
- S. Biniazi, H. Asgharzadeh, I. Ahadzadeh, O. Aydin and M. Farsak, Dalton Trans., 2022, 51, 18284–18295 RSC.
- Y. Yang, W. Yin, S. Wu, X. Yang, W. Xia, Y. Shen, Y. Huang, A. Cao and Q. Yuan, ACS Nano, 2016, 10, 1240–1248 CrossRef CAS PubMed.
- L. Zou, S. Guo, Z. Wang, M. Sun, F. Yu, B. Chi and J. Pu, Electrochim. Acta, 2021, 381, 138219 CrossRef CAS.
- Y. Gong, X. Zhang, Z. Li, Z. Wang, C. Sun and L. Chen, ChemNanoMat, 2017, 3, 485–490 CrossRef CAS.
- K. R. Yoon, D. S. Kim, W. H. Ryu, S. H. Song, D. Y. Youn, J. W. Jung, S. Jeon, Y. J. Park and I. D. Kim, ChemSusChem, 2016, 9, 2080–2088 CrossRef CAS PubMed.
- Q. Cui, L. Ma, P. Zhang, Y. Cao and J. Wang, J. Energy Chem., 2021, 57, 401–405 CrossRef CAS.
- L. Zou, Z. Lu, Z. Wang, B. Chi and J. Pu, J. Am. Ceram. Soc., 2022, 105, 2690–2701 CrossRef CAS.
- M. Sun, S. Guo, Z. Wang, L. Zou, B. Chi, J. Pu and J. Li, Electrochim. Acta, 2020, 363, 137235 CrossRef CAS.
- Y. Fang, Y. Fang, R. Zong, Z. Yu, Y. Tao and J. Shao, J. Mater. Chem. A, 2022, 10, 1369–1379 RSC.
- H. Li, Y. Chen, J. Z. Y. Seow, C. Liu, A. C. Fisher, J. W. Ager and Z. J. Xu, Small Sci., 2022, 2, 2100048 CrossRef CAS.
- B. Bao, Y. Liu, M. Sun, B. Huang, Y. Hu, P. Da, D. Ji, P. Xi and C. H. Yan, Small, 2022, 18, 2201131 CrossRef CAS PubMed.
- C. E. Beall, E. Fabbri and T. J. Schmidt, ACS Catal., 2021, 11, 3094–3114 CrossRef CAS.
- A. Grimaud, A. Demortiere, M. Saubanere, W. Dachraoui, M. Duchamp, M. L. Doublet and J. M. Tarascon, Nat. Energy, 2017, 2, 16189 CrossRef CAS.
- J. A. Esterhuizen, B. R. Goldsmith and S. Linic, Nat. Catal., 2022, 5, 175–184 CrossRef.
- A. Chen, X. Zhang and Z. Zhou, InfoMat, 2020, 2, 553–576 CrossRef CAS.
- X. Huang, Q. Guo, D. Yang, X. Xiao, X. Liu, Z. Xia, F. Fan, J. Qiu and G. Dong, Nat. Photonics, 2020, 14, 82–88 CrossRef CAS.
- Q. Liang, Y. Zhao, J. De Chen, J. J. Dai, X. Ding, Z. Tong, S. J. Xie, J. Zhang, Z. H. Zhou, J. T. Li, J. F. Li and Y. Zhou, Chem. Mater., 2022, 34, 5607–5620 CrossRef CAS.
- B. J. Kim, D. H. Kim, S. L. Kwon, S. Y. Park, Z. Li, K. Zhu and H. S. Jung, Nat. Commun., 2016, 7, 11735 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |