
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of the recent progress on direct heterogeneous catalytic CO2 hydrogenation to gasoline-range hydrocarbons
Xin
Shang
 ab,
Guodong
Liu
a,
Xiong
Su
*a,
Yanqiang
Huang
*a and
Tao
Zhang
ab
ab,
Guodong
Liu
a,
Xiong
Su
*a,
Yanqiang
Huang
*a and
Tao
Zhang
ab
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, No. 457 Zhongshan Road, Dalian 116023, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China. E-mail: suxiong@dicp.ac.cn; yqhuang@dicp.ac.cn
First published on 26th April 2023
Abstract
Since climate change and the energy crisis have imposed a growing threat to the living environment, the chemical conversion of carbon dioxide (CO2) with green hydrogen into gasoline-range hydrocarbons is promising for the mitigation of CO2 emissions and the storage of renewable energy. This review provides a comprehensive view of recent advances in the production of gasoline-range hydrocarbons from the direct hydrogenation of CO2. The typical catalytic systems are addressed first with the corresponding thermodynamic analysis. Then, the design and optimization of active components in different catalytic systems are discussed in detail, with perspectives on tandem catalysis and reaction coupling involving the Fischer–Tropsch (FT) process, methanol synthesis and acid catalysis. We also provide an overview of the recent progress in industrial process design and highlight the great achievements in practical industry applications. A summary and prospects will also be provided based on the discussion eventually.
 Xin Shang | Xin Shang received his BS degree from Tianjin University in 2016. He is currently pursuing his PhD degree in University of Chinese Academy of Sciences (UCAS), Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS), under the supervision of Prof. Yanqiang Huang. His research interests include C1 chemistry, synthesis and application of molecular sieves and nanostructured materials. |
 Guodong Liu | Guodong Liu received his BS degree in 1998 and PhD degree in 2019 from Dalian University of Technology. He has been working at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS) since 2019 in charge of developing heterogeneous catalysts of industrial scale. His research interests include the synthesis of zeolite materials and their applications in aromatization of alkanes, alkylation of arenes, and epoxidation of alkenes. |
 Xiong Su | Xiong Su received his BS degree from Dalian University of Technology and PhD degree in industrial catalysts from Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences (CAS). He continued his academic career in DICP as an assistant professor and was promoted to associate professor in 2017. His research interests include C1 chemistry, synthesis and application of molecular sieves and nanostructured materials. |
 Yanqiang Huang | Yanqiang Huang received his BSc (2002) from Dalian University of Technology, and PhD (2008) from Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS). After that he joined DICP as a staff scientist and was promoted to full professor in 2016. His active research interests include propellant catalytic decomposition, CO2 capture and utilization and C1 chemistry. |
 Tao Zhang | Tao Zhang received his PhD in physical chemistry in 1989 from Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS), under the supervision of Prof. Liwu Lin. He was the director of DICP from 2007 to 2017, selected as an Academician by the CAS in 2013, and promoted as a vice president of CAS in 2017. His main research interests include single atom catalysis and biomass conversion. |
Broader contextTo meet the challenges of climate and the environment and achieve the goal of “carbon neutrality”, the production of value-added chemicals via alternative non-oil routes, especially with carbon dioxide (CO2) and green hydrogen as raw materials, has become an appealing subject for the alleviation of CO2 emission and the storage of renewable energy. In recent years, many efforts have been devoted to realize the direct conversion of CO2 into gasoline-range hydrocarbons, including paraffin, olefins, naphthenes and aromatics in a range of C5–11, owing to their versatility in industry. There have also been achievements with the assistance of coupling reactions over catalytic systems involving multiple different active components. In this review, we present a comprehensive discussion on recent advances in direct heterogeneous catalytic CO2 hydrogenation to the gasoline-range hydrocarbons. We also expound on the design and optimization of active components in different catalytic systems, highlight the critical scientific problems, and introduce the progress in terms of industrial application, which can provide great inspiration for future research on high value-added CO2 utilization. |
1. Introduction
The use of fossil fuels, a non-renewable resource, has been instrumental in driving human productivity and civilization. However, the growing need for human development has led to a sharp increase in CO2 emissions, causing severe climate and environmental problems.1,2 Although the application of renewable energy, such as wind, solar, tidal, and biomass energy, might alleviate these problems, its massive utilization is still restricted by intermittent fluctuation and difficulty in storage. Therefore, the chemical conversion of CO2 with such ‘green’ hydrogen into value-added products would blaze a promising trail in mitigating CO2 emissions and also provide an efficient way for energy storage.3–8Gasoline-range hydrocarbons, mainly including paraffins, olefins, naphthene and aromatics in a range of C5–11, provide important fuels in daily life and versatile platform chemicals in industry. The majority of gasoline-range hydrocarbons are produced through the extraction of oil resources, while plenty of alternative routes have been proposed to synthesize gasoline-range hydrocarbons from raw materials other than oil. Several studies have been reported on the successful conversion of biomass feed into gasoline via the pyrolysis of waste resources, such as plant straw and grain husk, followed by the hydrogenation and separation process.9,10 Researchers are also working on producing aromatics-dominated hydrocarbons from methane and other light alkanes by dehydrogenation and aromatization reactions,11,12 while the high energy consumption and insufficient catalyst stability still constrain their massive application. Excellent results have been achieved by transforming methanol into aromatics and other gasoline-range hydrocarbons, and researchers are still making efforts to gain insights into the discrimination and evolution of intermediates, which are believed to be the key to the optimization of catalysts13,14 and further industrial production. Fischer–Tropsch (FT) synthesis is a typical process for syngas conversion to long-chain hydrocarbons for decades, and studies are still devoted to the regulation of product distribution for more value-added hydrocarbons, in which the introduction of acid catalysts could tailor the carbon chains, thus increasing the proportion of gasoline-range hydrocarbons.15–19 Recently, composite catalysts containing oxide and zeolite components (Ox-Zeo) have been applied to syngas conversion to olefins, aromatics and fuel.20–29 In comparison with the modified FT process, the Ox-Zeo systems show advantages in target product selectivity contributed by the suppression of methanation and facile control of chain growth.
Owing to the great significance of CO2 utilization and the versatility of hydrocarbons in industry, many efforts have been made on CO2 conversion to hydrocarbons. Alternative approaches featuring various energy inputs have been proposed, including photochemical,30–33 electrochemical34–37 and thermochemical3,5,38 routes. However, obtaining long-chain products like gasoline-range hydrocarbons from CO2 with acceptable yield efficiency is still in the infant stage for most photochemical and electrochemical processes. In contrast, the thermochemical route to long-chain hydrocarbons above methane has been studied for decades39–42 and has achieved remarkable progress. Therefore, the hydrogenation of CO2via the thermochemical route might have potential for practical massive production in industry at the current stage and will be discussed in the following.
There have been recent reports on efficient CO2 hydrogenation to C2–4 hydrocarbons, showing appreciable yields,43–46 but the reaction conditions and performance can be quite different as with CO2 hydrogenation into gasoline-range hydrocarbons. A vital challenge is simultaneously promoting the activation of relatively inert CO2 molecules and accurately regulating the complex process of carbon chain growth. Hence, the option of a catalyst component for CO2 hydrogenation depends on the inherent activity and also the coupling effect with additional components for controlling chain growth. Zeolites, featuring abundant acid sites and ordered channels, are commonly incorporated into such catalytic systems to regulate the selectivity for specific hydrocarbons.47–50 Besides imposing a crucial effect on product distribution, the chain growth also affords the consumption of certain intermediates, which might contribute to enhanced kinetics, thereby propelling the whole conversion reaction.47
Aside from extensive studies on improving the yield of total hydrocarbon products, considerable efforts have also been made to enhance the selectivity for more valuable chemicals such as benzene, toluene, and xylene (BTX) from CO2 hydrogenation due to their wide application in industry as compared to fuel.48,51,52 The high cost of the separation process makes the direct selective synthesis of these aromatic products a more promising prospect. Nevertheless, the desired targets are not easy to achieve because of the complex reaction mechanism and subtle catalyst design. The steric effect induced by different topologies or morphologies as well as the optimization of acid strength and density could also contribute to improved selectivity for specific hydrocarbons. Recently, CO2 hydrogenation with the introduction of benzene or toluene for the selective synthesis of xylene or other valuable products has been reported.53–56 Different reaction routes and dynamics may take effect since the CO2 hydrogenation process may provide reaction intermediates in situ as the alkylation reagents.
To date, there is still a lack of specific reviews on the highly selective hydrogenation of CO2 to value-added gasoline-range hydrocarbons involving the complicated control of carbon chain growth. This article reviews recent advances regarding direct catalytic CO2 hydrogenation to gasoline-range hydrocarbons and focuses on the subtle catalyst design and enhanced reaction coupling with the cooperation of different active components. Prospects and challenges are also discussed with expectations to bring about new insights and strategies for CO2 conversion.
2. Catalytic systems and thermodynamic analysis
In general, the tandem conversion of CO2 into gasoline-range hydrocarbons involves two fundamental steps based on the reaction coupling strategy: the hydrogenation of CO2 to active intermediates for chain growth and the controlled C–C coupling reactions. Therefore, at least two types of active sites should be contained in the catalytic systems for the simultaneous realization of these two functions, and the integration of various active components might play a vital role in the whole reaction. In the past decade, many advances have been made in developing efficient catalytic systems for the direct conversion of CO2 into gasoline-range hydrocarbons, and the typical systems are summarized in Fig. 1. The strategies based on iron-based FT catalysts have been proposed to realize primary chain propagation, while the active intermediate is believed to be preliminary CO via the reverse water gas shift (RWGS) and secondary olefins through the traditional FT synthesis process.50 To subtly control product selectivity beyond the limit of the Anderson–Schulz–Flory (ASF) distribution57,58 (Fig. 2), zeolites have been introduced into the systems for subsequent reactions, such as further chain growth, (de)alkylation, (de)hydrogenation and cyclization, leading to an increasing proportion of gasoline-range hydrocarbons.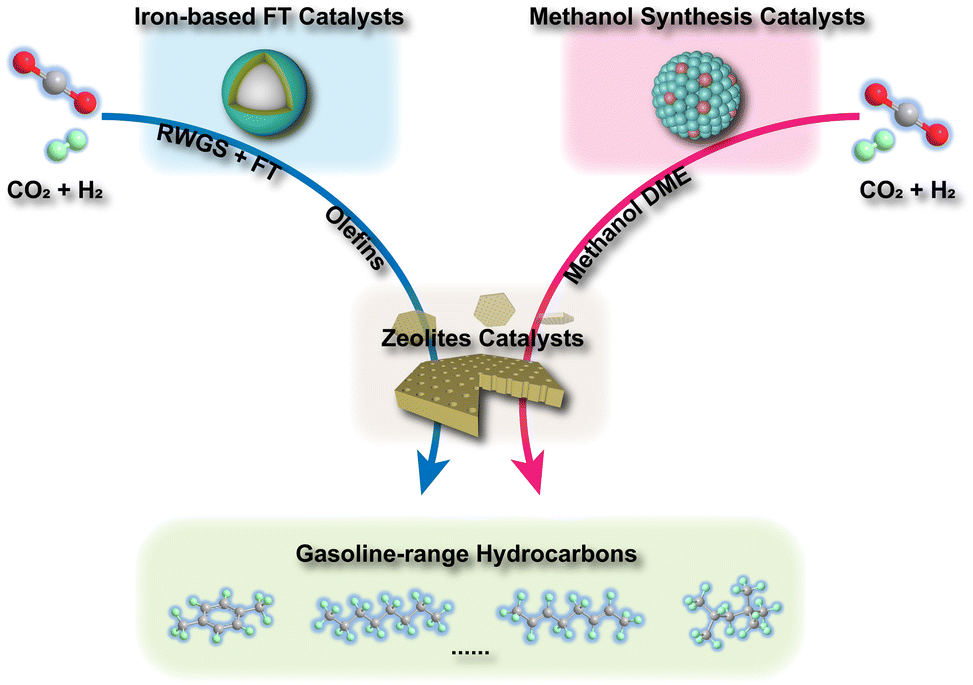 | ||
| Fig. 1 Two typical catalytic systems for direct CO2 hydrogenation into gasoline-range hydrocarbons. | ||
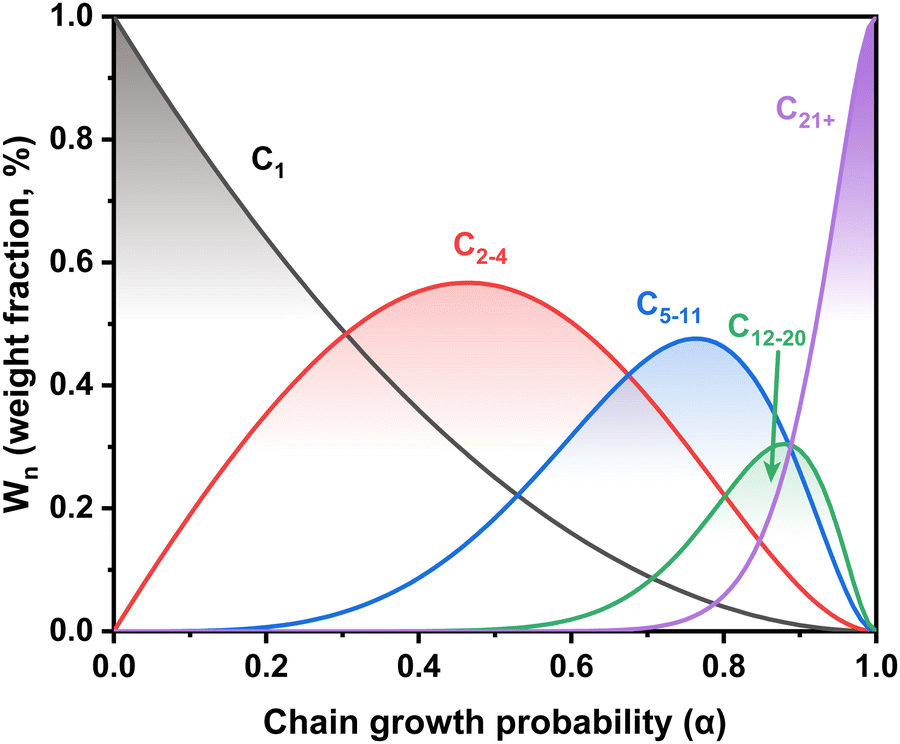 | ||
| Fig. 2 The dependence of the weight fraction of hydrocarbon products in a typical Fischer–Tropsch (FT) process on the chain growth probability according to the Anderson–Schulz–Flory (ASF) distribution. | ||
Another typical system is based on methanol synthesis catalysts. This has been proposed to suppress the significant side methanation reaction and inherent unmanageable chain growth reaction over iron-based catalysts since the C–O bond activation and C–C bond formation could take place over separate sites in the methanol synthesis catalysts and zeolites, respectively. Therefore, the superior selectivity for gasoline-range hydrocarbons, particularly aromatics, could be obtained over the catalytic systems based on the methanol synthesis route, with the formation of less methane and other alkanes. However, the conventional Cu-based catalysts for methanol synthesis are always operated at relatively low temperatures (<573 K),59–62 much lower than the reaction temperature (>673 K) for C–C coupling in zeolites.63–67 Higher temperatures would lead to a significantly higher fraction of CO due to the intrinsic thermodynamic equilibrium (Fig. 3(a)). Some metal oxide catalysts could be feasibly operated at higher temperatures, showing desirable methanol selectivity.63,68–70 Thus, they have been widely applied in the gasoline-range hydrocarbon production from CO2 hydrogenation via Ox-Zeo composite catalysts, whereas the poor hydrogen activation ability still limits the yield of hydrocarbons. The methanol synthesis reaction is nearly blocked thermodynamically at high temperatures, while the formation of representative hydrocarbons is still possible according to the Gibbs free energy change as shown in Fig. 3(b). The transformation of methanol into hydrocarbons might even shift the reaction equilibrium to propel the generation of the methanol intermediate, making Ox-Zeo a feasible system for the conversion of CO2 to hydrocarbons.47
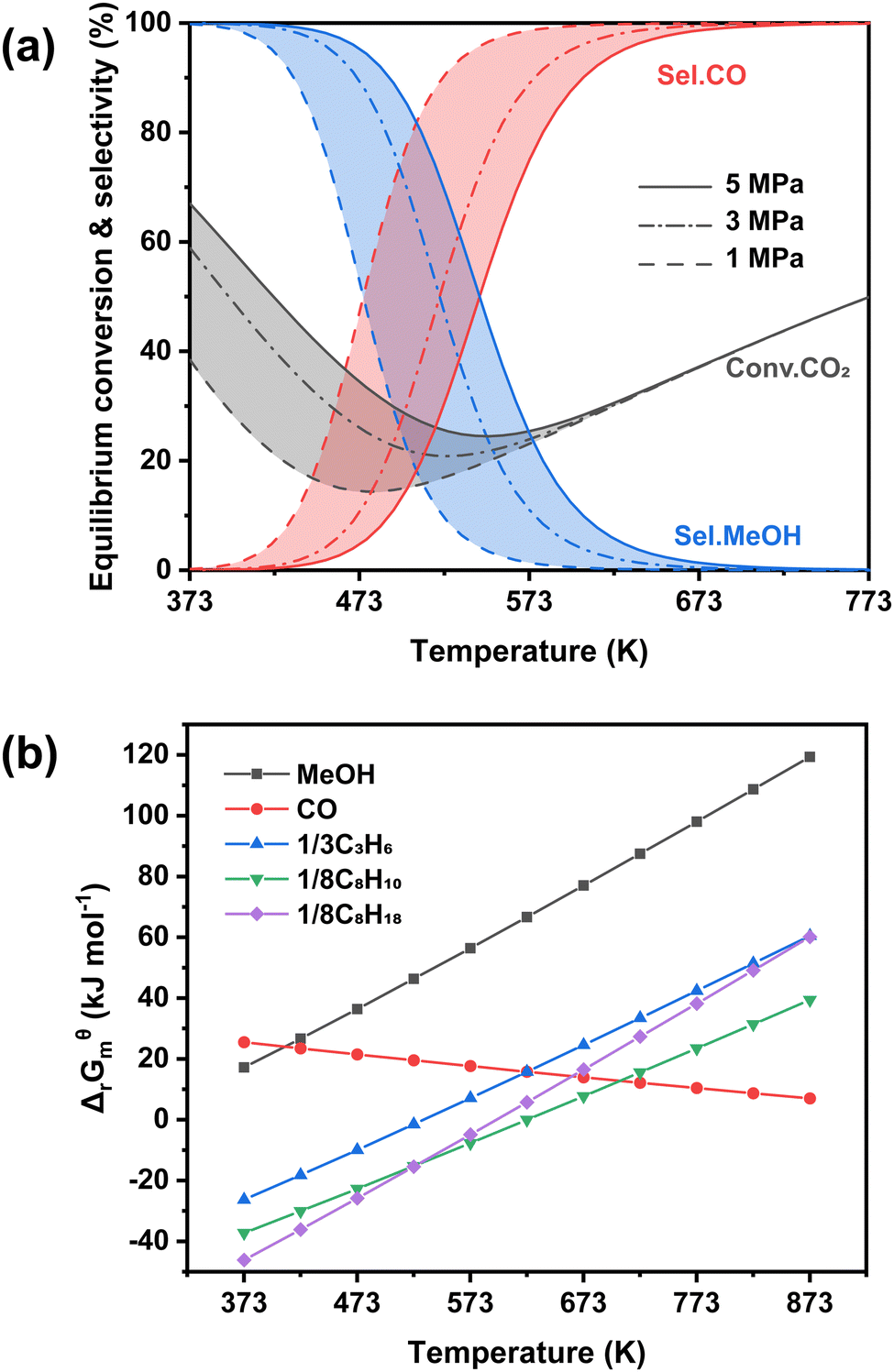 | ||
| Fig. 3 Thermodynamics analysis. (a) Equilibrium conversion and selectivity in CO2 hydrogenation to methanol and CO, (b) Gibbs free energy of CO2 hydrogenation to different products. | ||
To date, for the realization of direct CO2 hydrogenation into gasoline-range hydrocarbons, the majority of studies have been focused on the aforementioned two types of catalytic systems. Therefore, we will provide a comprehensive summary of recent cases of these two systems in the following, especially concentrating on the catalyst design and reaction mechanism. Representative catalysts and their performances are summarized in Table 1.
| Pathway | Composition | Organization mannera | H2/CO2 | T (K) | P (MPa) | Space velocity (mL g−1 h−1) | Conv. (%) | Primary productsb | Sel.c (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Organization manner mainly includes the sole-component mode, dual-bed mode, granule-mixing mode and powder-mixing mode. Layer-by-layer arrangement of different components is denoted as dual-bed mode (zeolite components are at the bottom). Granule-mixing mode is constructed by mixing and stacking the particles of different components in certain meshes (200–400 μm). Powder-mixing mode is fabricated by mixing components with an agate mortar (particle size is 0.5–1 μm), followed by transformation into larger granules for stacking. b Primary products are referred to as the major organic products among the effluent hydrocarbons. c The calculation of selectivity excludes CO. d Benzene is introduced into the system for toluene and xylene synthesis. e Toluene is introduced into the systems for xylene synthesis. f Benzene is introduced into the system, P modification shifts the product from ethylbenzene to propylbenzene. | ||||||||||
| RWGS + FT | Fe–Co EDTA–2Na | Sole | 1 | 623 | 3.0 | 6000 | 32.8 | Gasoline | 46.3 | 73 |
| FeAlOx | Sole | 1 | 603 | 3.5 | 2000 | 20.2 | C5+ | 77.0 | 75 | |
| K0.016Pd0.12Fe | Sole | 3 | 613 | 3.0 | 6000 | 41.8 | C5+ | 70.6 | 76 | |
| K–Zn–Fe | Sole | 3 | 593 | 3.0 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
57.4 | C5+ | 77.2 | 74 | |
| RWGS + FT + zeolite | Na–Fe3O4/HZSM-5 | Granule-mixing | 3 | 593 | 3.0 | 4000 | 34.0 | Gasoline | 73.0 | 50 |
| Na–Fe3O4/HZSM-5 | Dual-bed | 3 | 593 | 3.0 | 4000 | ∼34 | Gasoline | 67 | 50 | |
| Na–Fe3O4/HZSM-5 | Powder-mixing | 3 | 593 | 3.0 | 4000 | 13 | Methane | 60 | 50 | |
| NaFe/ZSM-5 | Granule-mixing | 3 | 593 | 3.0 | 4000 | ∼27 | Aromatics | ∼40 | 51 | |
| ZnFeOx–nNa/HZSM-5 | Granule-mixing | 3 | 593 | 3.0 | 1000 | 41.2 | Aromatics | 75.6 | 83 | |
| Na–Fe@C/H-ZSM-5-0.2 M | Granule-mixing | 3 | 593 | 3.0 | 9000 | ∼30 | Aromatics | 50.2 | 91 | |
| Na–Fe3O4/HMCM-22 | Granule-mixing | 3 | 593 | 3.0 | 4000 | 26 | C4+ isoparafin | ∼60 | 92 | |
| Fe–Zn–Zr@HZSM-5–Hbeta(4:1) |
Core–shell | 3 | 613 | 5.0 | 3000 | 14.9 | C4+ isoparafin | 81.3 | 95 | |
| K–CoFeOx(1:5)/HZSM-5-Si |
Granule-mixing | 3 | 593 | 3.0 | 4000 | 51.2 | C5+ | 58.8 | 88 | |
| Na–FeMnOx + HZSM-5 | Granule-mixing | 3 | 593 | 3.0 | 1000 | 44.5 | Aromatics | 64.2 | 89 | |
| MeOH + zeolite | In2O3/HZSM-5 | Granule-mixing | 3 | 613 | 3.0 | 9000 | 13.1 | Gasoline | 78.6 | 49 |
| In2O3/HZSM-5 | Dual-bed | 3 | 613 | 3.0 | 9000 | ∼10 | Gasoline | ∼70 | 49 | |
| In2O3/HZSM-5 | Powder-mixing | 3 | 613 | 3.0 | 9000 | 8 | Methane | 94.3 | 49 | |
| ae-ZnO–ZrO2/H-ZSM-5 | Powder-mixing | 3 | 613 | 4.0 | 7200 | 16 | Aromatics | 76 | 98 | |
| ZnZrO/ZSM-5 | Powder-mixing | 3 | 593 | 4.0 | 1200 | ∼15 | Aromatics | 78 | 47 | |
| ZnZrO/ZSM-5 | Dual-bed | 3 | 593 | 4.0 | 1200 | ∼12 | Non-aromatic C5+ | ∼50 | 47 | |
| ZnZrO/ZSM-5 | Granule-mixing | 3 | 593 | 4.0 | 1200 | ∼9 | Non-aromatic C5+ | ∼60 | 47 | |
| ZnCr2O4/Sbx-HZSM-5 | Powder-mixing | 1 | 548 | 2.0 | 300 | ∼13 | Aromatics | ∼80 | 97 | |
| ZnAlOx&H-ZSM-5 | Powder-mixing | 3 | 593 | 3.0 | 6000 | ∼6 | Aromatics | 73.9 | 48 | |
| Co–In2O3/ZrO2 + HZSM-5 | Powder-mixing | 3 | 573 | 3.0 | 3600 | 5.5 | C5+ | 64.5 | 104 | |
| ZnZrOx&Zn/ZSM-5 | Powder-mixing | 3 | 623 | 3.0 | 12000 |
∼21 | Toluene & xylened | 93 | 53 | |
| ZnZrO-Z5 | Powder-mixing | 3 | 633 | 3.0 | 12000 |
∼7 | Xylenee | 92.4 | 56 | |
| ZnCrOx-ZSM5 | Powder-mixing | 3 | 663 | 4.0 | 15000 |
20.9 | Xylenee | ∼90 | 54 | |
| ZnZrOx, SAPO-34, P-ZSM-5 | Dual-bed | 673 | 3.0 | 6000 | ∼7 | Ethylbenzene or propylbenzenef | 83.5 or 64.7 | 55 | ||
3. Iron-based FT catalysts
Iron-based catalysts, well known as FT catalysts for syngas conversion to hydrocarbons, always suffer from the water gas shift (WGS) reaction with the significant production of CO2. However, this feature is in favor of CO2 conversion to long-chain hydrocarbons since CO could be formed via the RWGS process over iron-based catalysts followed by a typical FT process for C–C coupling. The lack of RWGS activity over other known FT catalysts such as cobalt71 and ruthenium72 makes them unsuitable for CO2 hydrogenation to long-chain hydrocarbons due to their higher methanation activity than carbon chain growth. The two-step mechanism via the CO intermediate for CO2 hydrogenation over iron catalysts has been widely accepted, while the true active components are still the subject of debate in terms of the RWGS and chain propagation. There is a prevailing view that iron oxides play a vital role in the initial hydrogenation of CO2 to CO, and iron carbide species are responsible for the subsequent carbon chain propagation.3.1 Sole iron-based catalysts
Considerable efforts have been made toward the modification of iron-based catalysts to adjust the product distribution toward more gasoline-range hydrocarbons or other value-added chemicals for promising applications in industry.73–76 Promotors, such as alkali, alkaline earth, transition metals, or certain non-metallic elements, and supports with tuned structure and chemical nature, are often introduced to facilitate the adsorptive activation of CO2, the formation of iron carbide active sites for C–C coupling and carbon chain growth, realizing the selective synthesis of specific hydrocarbons.77–82Dai et al. prepared a series of iron–cobalt (Fe–Co)-supported catalysts modified by ethylenediaminetetraacetic acid disodium salt (EDTA–2Na).73 The addition of a small amount of Co and EDTA–2Na suppresses the sintering of Fe, and the reduction of Fe species is promoted by the decomposed EDTA moiety. The synergistic effect of Fe–Co bimetals enhances CO2 adsorption, while Na+ improves the product distribution of long-chain hydrocarbons by modifying the basicity of the surface. Khan et al. reported a bifunctional iron aluminium oxide (FeAlOx) catalyst that directly converted CO2 into C5+ hydrocarbons with a selectivity of 77.0% and a CO2 conversion of 20.2%.75 The amorphous AlOx phase plays a vital role in both the adsorption of reactants and the formation of active intermediates as well as subsequent C–C coupling reactions, leading to the generation of long-chain linear olefins. Xiong et al. reported that Fe catalysts promoted by a combination of K and Pd showed a three-synergistic catalytic effect to achieve the high selectivity for C5+ hydrocarbons in the CO2 hydrogenation reaction.76 K addition to the optimized catalyst promoted the formation of active iron carbide species for the carbon chain growth of olefins. Meanwhile, both CO2 conversion and C5+ hydrocarbon selectivity could be boosted by the generation of active H from the PdFe alloy. The maximum yield of C5+ hydrocarbons reached 20.0% under a high space velocity of 6000 mL gcat−1 h−1.
3.2 Composite iron-based catalysts with zeolites
Although some approaches have been used to adjust the product distribution, the highly selective synthesis of hydrocarbons in a certain range remains challenging over sole iron-based catalysts due to ASF distribution (Fig. 2), based on which the proportion of C5–11 hydrocarbons cannot go beyond 50%. The other component of zeolites with abundant acid sites and ordered channels, well known as a group of materials for petroleum cracking and reforming and separation engineering, have been proposed to be incorporated into iron-based catalyst systems. As such, the carbon chain growth might be under accurate control, thereby providing enhanced C5–11 hydrocarbon selectivity.Sun and Ge's group reported a composite catalyst containing Na–Fe3O4 and HZSM-5 for the direct conversion of CO2 to gasoline-range hydrocarbons with a selectivity of up to 78%.50 The multifunctional catalyst provides three types of active sites, i.e., Fe3O4 for CO2 to CO via RWGS, Fe5C2 for CO to the α-olefin intermediate through the FT process, and acid sites for the formation of gasoline-range hydrocarbons via the acid-catalyzed oligomerization, isomerization and aromatization reactions (Fig. 4(a)). The appropriate proximity of different active sites plays a crucial role in the tandem reaction. The granule-mixing of iron components and zeolite components at the micrometer scale gave the best performance, while the powder-mixing style with closer contact showed a much higher methane selectivity due to Na+ migration to acid sites. The hydrocarbon distribution in gasoline-range products is also highly influenced by proximity, although further research disclosed a significant relationship between the product distribution and Brønsted acid sites (BAS) in HZSM-551 (Fig. 4(b)). The formation of aromatics is promoted by the high Brønsted acidity, while the fraction of light aromatics could be further increased by the passivation of external BAS over HZSM-5.
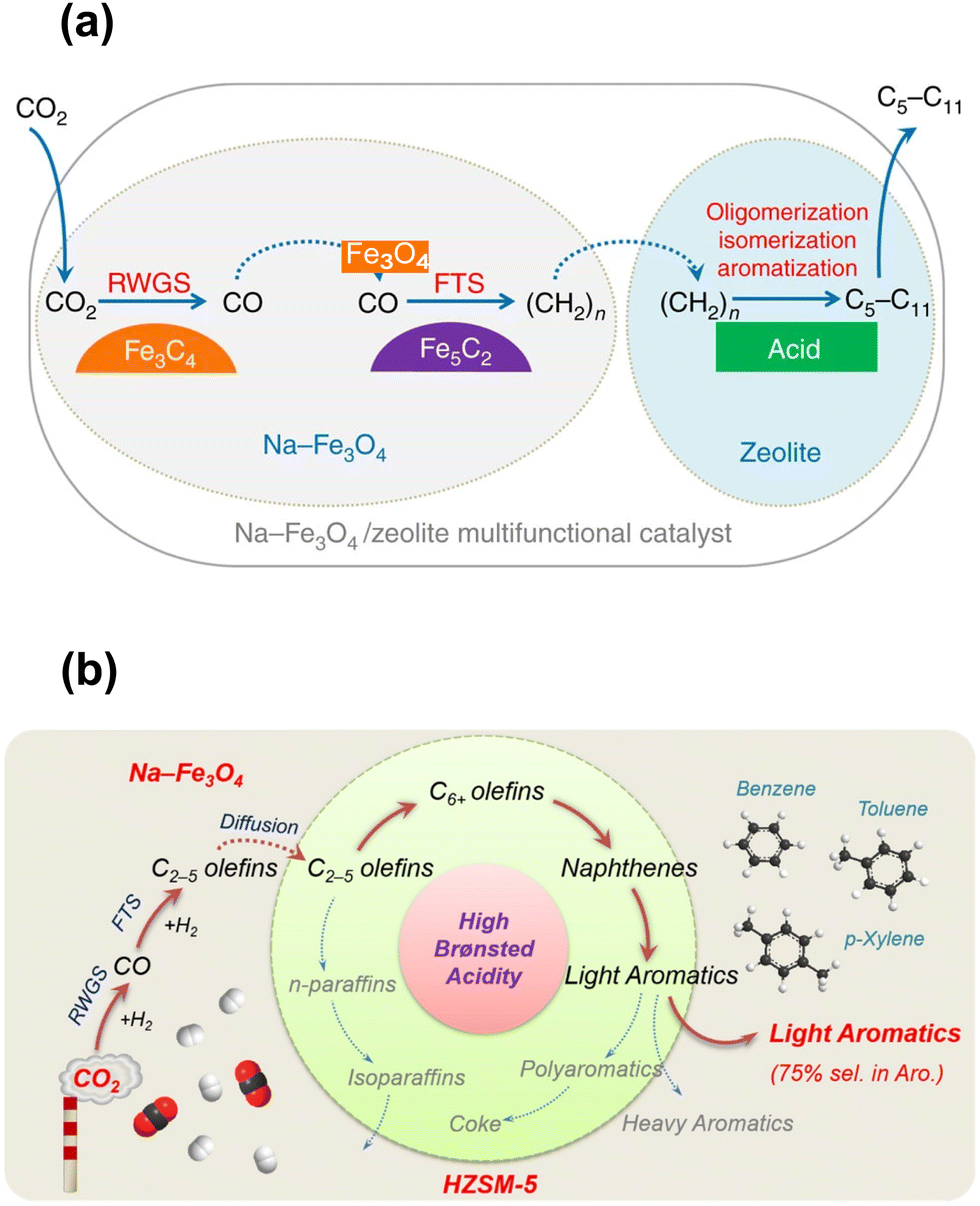 | ||
| Fig. 4 (a) Active sites and reaction routes over the Na–Fe3O4/zeolite composite catalyst.50 (b) Reaction scheme of light aromatic synthesis via CO2 hydrogenation over the NaFe/HZSM-5 composite catalyst.51 Reproduced from ref. 50 and 51 with permission from Springer Nature and Elsevier, copyright 2017 and 2021. | ||
Current studies have demonstrated the equal importance of regulating the iron-based components and zeolite components for the optimization of catalytic performance in CO2 hydrogenation to gasoline-range hydrocarbons. Therefore, we have summarized the recent achievements regarding the design of these two components as well as the proximity effect.
Gao and Sun et al. reported that the combination of ZnFeOx–nNa spinel oxide and hierarchical nanocrystalline HZSM-5 aggregates could realize aromatics selectivity of up to 75.6% at 41.2% CO2 conversion.83 The addition of zinc significantly promotes the reduction of iron oxide, which is in favor of the formation of Fe3O4 and metallic Fe, resulting in more exposed active sites. A suitable amount of residual sodium is conducive to the formation of light olefin intermediates and further aromatization reactions, while excessive sodium would inhibit the reduction of iron oxide (Fig. 5(a) and (b)).
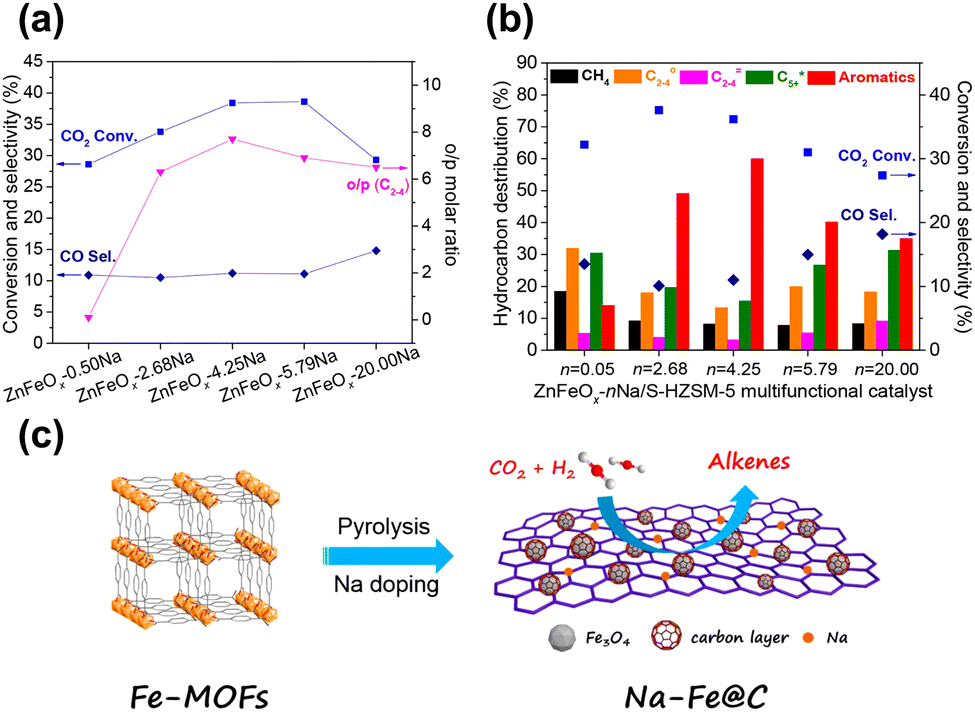 | ||
| Fig. 5 Catalytic performance of (a) ZnFeOx–nNa and (b) ZnFeOx–nNa/S-HZSM-5 in CO2 hydrogenation.83 (c) Illustration of the fabrication of Na–Fe@C by pyrolysis of Fe-based MOFs.91 Reproduced from ref. 83 and 91 with permission from the American Chemical Society and Elsevier, copyright 2019 and 2020. | ||
Potassium-doped iron catalysts in combination with ZSM-5 were also reported to be effective for CO2 hydrogenation into isoparaffin-dominated gasoline-range hydrocarbons84 and aromatics.85–87 Well-dispersed K species inhibit the adsorption of active H species, leading to a higher yield of olefins formed over iron-based components. Consequently, these intermediate products could be readily transformed into long-chain hydrocarbons or aromatics in the zeolite components.
There are also reports on transition metal-doped iron-based components,73,74,76,88,89 featuring a synergistic effect on the promoted CO2 activation. Song et al. achieved an excellent CO2 conversion of 57.3% contributed by a CO-reduced Cu–Fe2O3 with the beneficial synergism between Cu and Fe and the distinctive CO reduction prior to reaction. This contribution is favorable to the formation of oxygen vacancies for CO2 adsorption and iron carbide for FT synthesis.90
To increase the proportion of exposed active sites and their accessibility, researchers have exploited some novel preparation methods and designed distinct precursors for the synthesis of iron-based components.74,91 Tsubaki and Yang et al. developed a multifunctional catalyst composed of Na–Fe@C, which was derived from metal–organic frameworks (MOF), and hollow HZSM-5 with short diffusional channels for the conversion of CO2 into aromatic hydrocarbons91 (Fig. 5(c)). The active sites over iron-based components are highly accessible and appropriate for intermediate adsorption, owing to the abundant porous structures and high specific area obtained by the pyrolysis of MOFs. As a result, superior CO2 conversion and alkene selectivity were achieved, facilitating the subsequent aromatization reaction. This result further suggests the significance of the subtle design and preparation of iron-based components.
Ramirez et al. reported a zeolite topology effect on the product distribution in the direct conversion of CO2 with composite iron-based catalysts. The zeolite component with MOR topology exhibited a high selectivity for olefins, while that with MFI topology demonstrated aromatic-dominated products. The remarkably discriminative selectivity was further rationalized by first-principles simulation, which showed different reactivities for carbonium ion intermediates in MOR and ZSM-5. The proportion of isoparaffin, playing a role in determining the octane value of gasoline, could be increased to 70% in C4+ hydrocarbons by using HMCM-22 and H-Beta instead of HZSM-592 (Fig. 6). The higher fraction of isoparaffins might be attributed to the differences in both topological and acidic properties, since the stronger Brønsted acidity with suited channels in HZSM-5 favors the aromatization reaction, while the isomerization reaction would be dominant over HMCM-22 or H-Beta.
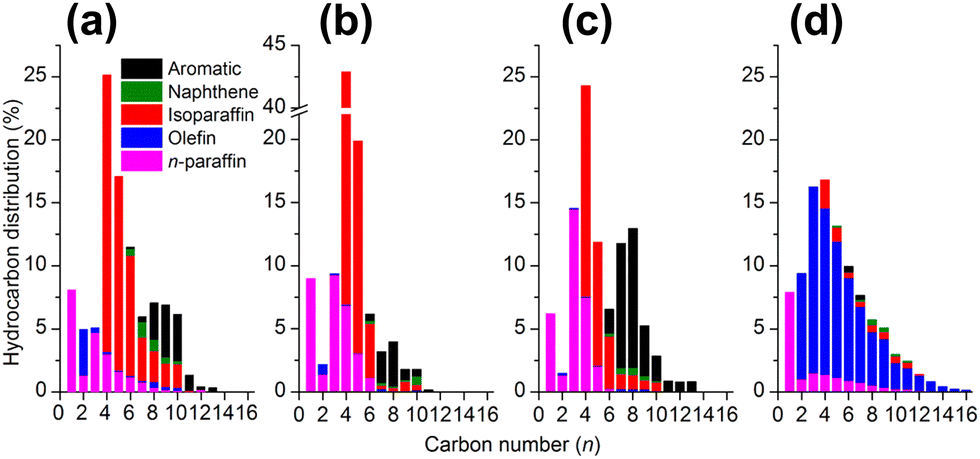 | ||
| Fig. 6 Detailed hydrocarbon product distribution from CO2 hydrogenation over Na–Fe3O4 combined with (a) HMCM-22, (b) Hbeta, (c) HZSM-5, and (d) sole Na–Fe3O4.92 Reproduced from ref. 92 with permission from the American Chemical Society, copyright 2018. | ||
The diffusion behavior of hydrocarbons in zeolites is also crucial for both stability and selectivity, which is influenced by the channels and structures. HZSM-5 with a hierarchical pore structure derived from nanocrystalline aggregates is considered to be favorable for the highly efficient aromatization reaction.83 The alkali-treated H-ZSM-5, with suitable acid properties, is also beneficial for aromatic production through the aromatization of alkenes.91 Garcia-Hurtado et al. investigated the influence of the zeolite structure and crystal size on the composite iron-based catalyst system for CO2 hydrogenation.87 Shorter intracrystalline diffusion paths facilitate the escape of light olefins in nanosized MFI zeolites, while prolonged paths in larger crystals favor the formation of more aromatics.
Tuning the acid properties of zeolites is another efficient way to readily adjust the product distribution, which could be achieved by the chemical deposition of silica or heteroatomic doping. Selectively shielding the surface acid sites by silica could suppress the consecutive alkylation reactions resulting in a higher proportion of BTX,51,83,89,91,93 while the introduction of elements such as zinc93 and phosphorous89,94 is also conducive to the formation of light aromatics by neutralizing the strong acid sites.
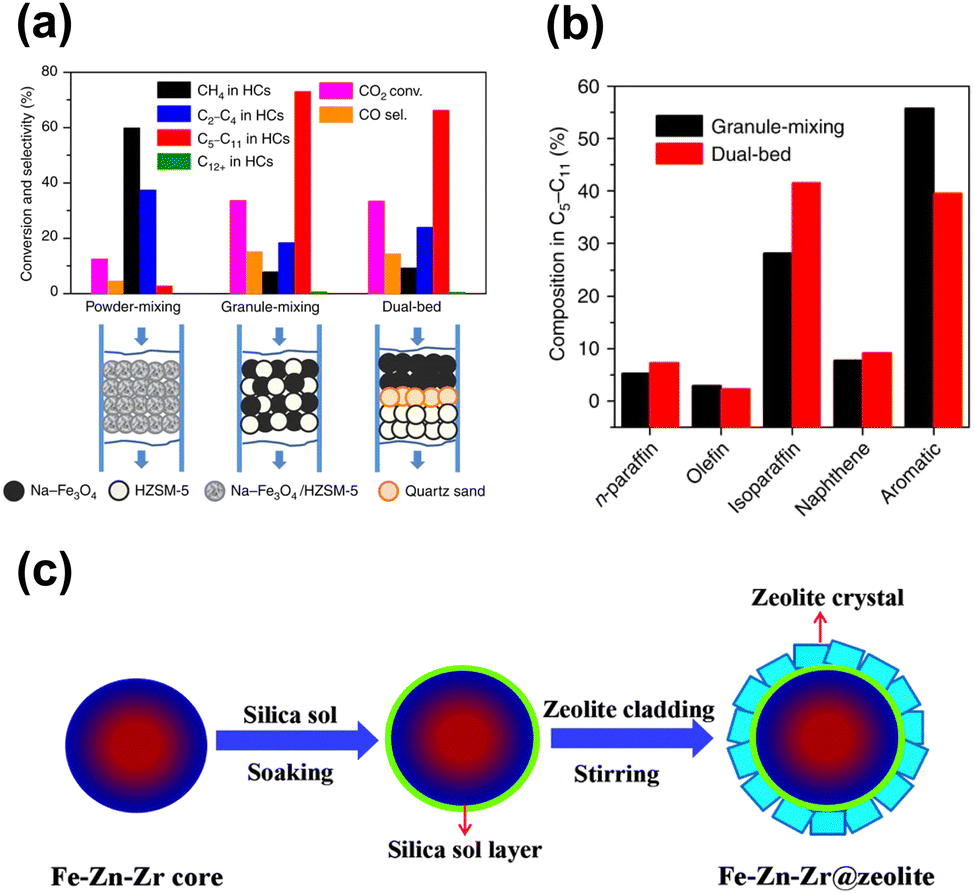 | ||
| Fig. 7 (a) and (b) Proximity effect on the catalytic performance of the Na–Fe3O4/zeolite composite catalyst.50 (c) Preparation of the Fe–Zn–Zr@zeolite catalyst.95 Reproduced from ref. 50 and 95 with permission from the Springer Nature and Royal Society of Chemistry, copyright 2017 and 2016. | ||
In contrast, Tan et al. synthesized an Fe–Zn–Zr@zeolite core–shell catalyst with a single-zeolite or a double-zeolite shell by a simple cladding method for CO2 to isoalkane-dominated C5+ hydrocarbons95,96 (Fig. 7(c)), as the core–shell structure is widely applied for the tandem reaction due to its distinct spatial features. The hydrocarbon distribution is related to the various types of zeolite shells and the ratios of different zeolites, while high isoalkane selectivity is contributed by an improved confinement effect from the core–shell structure.
4. Methanol synthesis catalysts
Although an excellent gasoline-range hydrocarbon yield could be obtained through an FT route, the proportion of aromatics, a group of value-added chemicals in the hydrocarbon products, is always inferior to the results over oxide-plus-zeolite (Ox-Zeo) catalytic systems, which is believed to occur through a methanol intermediate route. The inherent chain growth mechanism over FT catalysts ruled by ASF distribution still limits the aromatic yield. The efficiency of the gasoline-range hydrocarbons also suffers from remarkable methanation reactions. In contrast, the Ox-Zeo system could achieve superior aromatic selectivity for C5–11 hydrocarbons contributed by the facile control of chain growth in confined zeolites framework through a methanol-to-gasoline (MTG) or aromatic (MTA) route.47–49,52,97,98 Furthermore, there have been some reports on the CO2 conversion to aromatics via a tandem reaction involving methanol synthesis and the subsequent alkylation with benzene or toluene to xylene, ethylbenzene and other specific aromatic hydrocarbons.53–56,99,1004.1 Tandem reactions of methanol synthesis and MTG or MTA
Given that substantial achievements have been made in both the fields of methanol synthesis and methanol transformation, an intuitive idea has emerged, that is, the merging of these two processes into an integrated system for direct CO2 transformation into gasoline-range hydrocarbons. To realize such goals, the multifunctional active components for highly efficient methanol synthesis and the subsequent conversion into target products, along with the coupling effect and interactions between the different components should be of critical concern.In2O3-based components have been applied in CO2 hydrogenation recently with excellent methanol selectivity due to their abundant oxygen vacancies.49,68,101 Sun and Zhong et al. reported an In2O3/HZSM-5 system for the direct conversion of CO2 into liquid fuels, in which the formation of the methanol intermediate occurred through the activation of CO2 and H2 over the oxygen vacancies on the In2O3 surface.49 The undesired RWGS reaction was further inhibited by regulating the proximity of the two components, leading to a high-efficiency CO2 transformation to hydrocarbons (Fig. 8). With the combination of HZSM-5, a high selectivity of 78.6% for gasoline-range hydrocarbons with a low methane selectivity of 1% was achieved by this composite catalyst. The possible mechanism involving the conversion of CO2 over the In2O3 catalyst surface and the formation of hydrocarbons inside HZSM-5 was further investigated by DFT calculation. The oxygen vacancy sites on In2O3 are considered the key active sites for the transformation of CO2 into methanol, which enters the hydrocarbon pool and subsequently leads to the formation of aromatics and isoparaffins through a series of complex consecutive reactions.
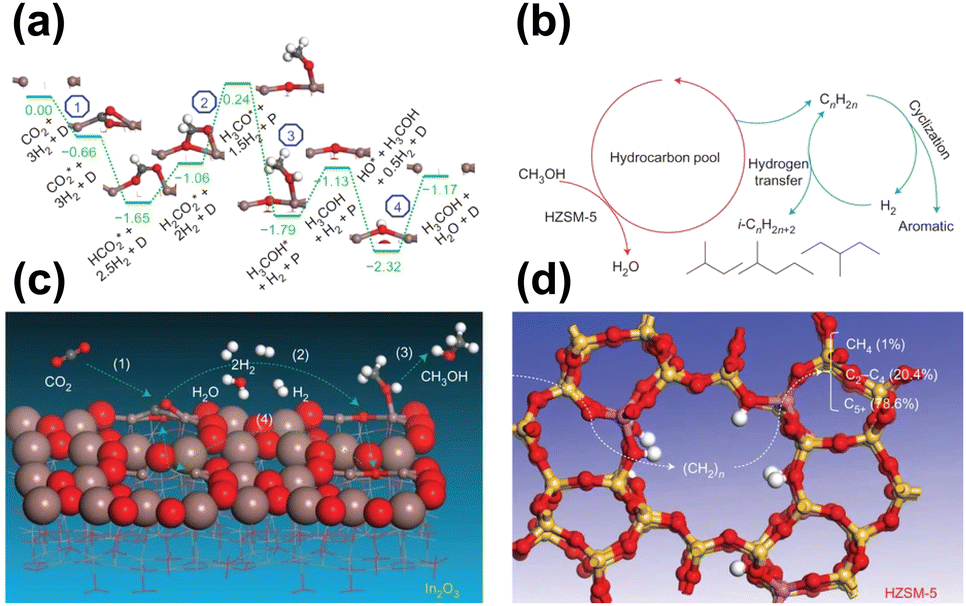 | ||
| Fig. 8 (a) Energy profile from DFT calculations for CO2 hydrogenation to CH3OH on the In2O3 (110) surface. (b) Schematic of the hydrocarbon-pool mechanism for CH3OH conversion into hydrocarbons inside HZSM-5. (c) Schematic of the formation of CH3OH from CO2 at the oxygen-vacancy site on the In2O3 catalyst surface. (d) Schematic of hydrocarbon formation from CH3OH at the acidic site inside the pores of the HZSM-5 catalyst.49 Reproduced from ref. 49 with the permission from Springer Nature, copyright 2017. | ||
ZnZrOx solution components exhibit remarkable performances for CO2 hydrogenation to methanol with a selectivity of up to 90% and high stability even in the sulfur-containing stream.63 The synergistic effect between Zn and Zr contributes to the excellent performance, while the enrichment of Zn clusters on the surface plays a vital role.63,102 Li et al. achieved a high selectivity for aromatics up to 73% at CO2 conversion of 14% with the combination of ZnZrO with HZSM-5 through a CHxO intermediate.47 The introduction of zeolite propels the tandem reactions, rendering a more favorable transformation of CO2 toward active intermediate species in terms of thermodynamics, due to the coupling of CO2 hydrogenation and aromatics formation (Fig. 9(a)). The H2O formed from CO2 hydrogenation over ZnZrO at a moderate content can effectively regulate the adsorption and conversion of ethene, thus obviously increasing the selectivity for aromatics (Fig. 9(b)). The presence of H2O and CO2 also suppresses the generation of polycyclic aromatics, resulting in enhanced stability of the composite catalyst. It is speculated that the ethene and H2O tend to adsorb on the weak acid sites, while the strong acid sites are more efficient for aromatization reactions of ethene. Once the H2O is adsorbed on the weak acid sites, the adsorption of ethene could be significantly inhibited, which would facilitate the aromatization of ethene over strong acid sites.
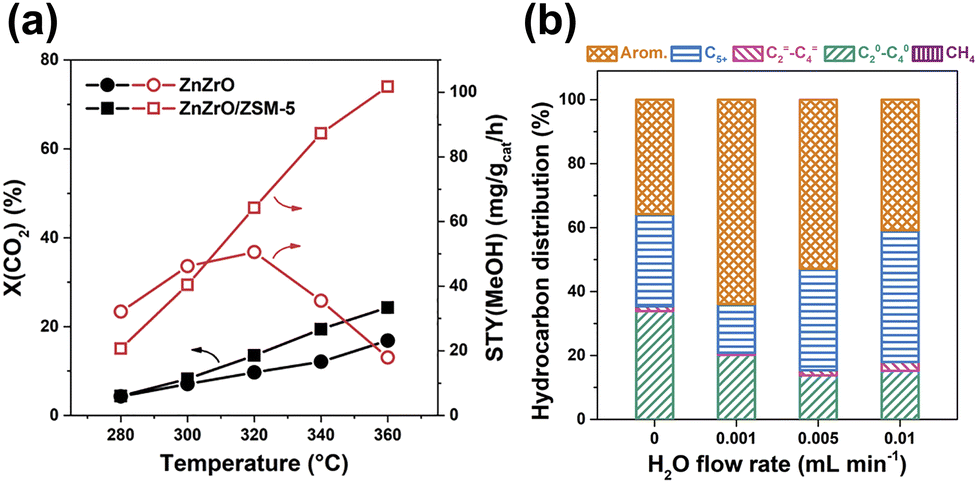 | ||
| Fig. 9 (a) The hydrogenation of CO2 over ZnZrO and ZnZrO/HZSM-5. (b) Ethene conversion over HZSM-5 with co-feeding different contents of H2O.47 Reproduced from ref. 47 with permission from Elsevier, copyright 2019. | ||
Zhou et al. fabricated a highly active ZnO–ZrO2 aerogel by a combined sol–gel and subsequent supercritical drying method, then it was integrated with HZSM-5 for aromatics production from CO2. This catalyst system obtained a much higher space–time yield (0.24 g goxide−1 h−1) of aromatic hydrocarbons than previous reports.98 The combination of electron paramagnetic resonance (EPR) and CO2 temperature-programmed desorption (CO2-TPD) technology confirmed that the ZnO–ZrO2 aerogel component with high surface area provides abundant oxygen vacancies in favor of CO2 adsorption, activation and subsequent conversion (Fig. 10). The molar ratio of Zn and Zr also has a great influence on the concentration of oxygen vacancies and capability of H2 activation. The well-matched CO2 and H2 activation derived from the synergy between ZrO2 and ZnO plays a crucial role in the excellent conversion of CO2 into aromatics. Tian et al. applied a ZnZrO component prepared with ZIF-8 as the precursor via a simple calcination process for CO2 conversion.103 The positive correlation between the concentration of oxygen vacancies and the adsorption and activation capacity of CO2 was disclosed by the generalized gradient approximation (GGA) of density functional theory (DFT) simulation results. A CO2 conversion of 15.2% and aromatic selectivity of 63.9% can be achieved under optimal reaction conditions.
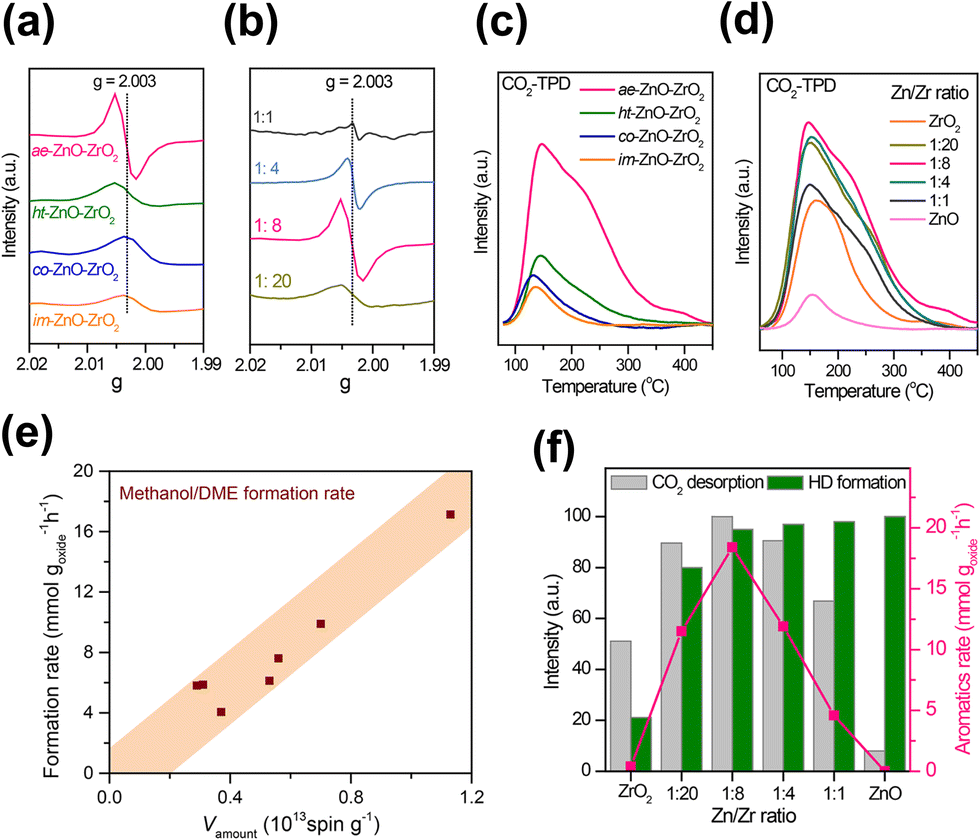 | ||
| Fig. 10 EPR spectra of ZnO–ZrO2 components with (a) different preparation methods and (b) Zn/Zr molar ratios, CO2-TPD curves of ZnO–ZrO2 components with (c) different preparation methods, and (d) Zn/Zr molar ratios. (e) The dependence of the CH3OH/DME formation rate on the total number of oxygen vacancies. (f) Normalized CO2 desorption amounts and HD formation rates against the Zn/Zr molar ratio and the effect of the Zn/Zr molar ratio on the formation rate of aromatics.98 Reproduced from ref. 98 with the permission from American Chemical Society, copyright 2019. | ||
Ni et al. reported a composite catalyst consisting of a nano-scaled spinel structural ZnAlOx component and HZSM-5 zeolite, exhibiting a 73.9% selectivity for aromatics and only 0.4% selectivity for methane.48 Methanol and dimethyl ether, as intermediates transmitted to H-ZSM-5 and subsequently converted into olefins and finally aromatics, are synthesized by the hydrogenation of formate species formed on the ZnAlOx surface. The ZnAlOx component is not only responsible for the activation of CO2 hydrogenation by Zn2+, but also plays a vital role in promoting aromatization by the shield effect on the external Brønsted acid.
Although the dependence of the CO2 conversion performance on the oxygen vacancies and unsaturated coordination sites has been acknowledged by researchers, the CO2 hydrogenation dynamics is still limited by the insufficient activation of hydrogen. In a recent study, Li et al. added an appropriate amount of cobalt to the In2O3–ZrO2/HZSM-5 tandem catalyst, by which a significant increase in CO2 conversion was obtained.104 This result could be due to the hydrogen spillover from metallic Co to the surface of ZrO2, which could significantly accelerate the formation of intermediates and subsequent hydrogenation steps.
The diffusion properties could be regulated by adjusting the morphology of zeolites, leading to distinct product distribution in some cases. By employing the sheet-like HZSM-5 with short channels along the b-axis and nano-sized ZnCr2O4, Arslan et al. obtained tetramethylbenzene-dominated aromatic products97 (Fig. 11(a)). The specific morphology of HZSM-5 allows aromatics such as benzene, toluene, ethylbenzene, and xylenes to quickly diffuse through the short straight channels along the b-axis. These aromatics are transformed into multi-methylbenzene via surface methylation over the external acidic sites of the catalyst. The excellent activity and remarkable selectivity could also be attributed to the asymmetric desorption of oxygenate species and aromatic hydrocarbons induced by the interactions of oxides and zeolites. Coincidentally, the ultrathin nanosheet HZSM-5 has also been applied in CO2 hydrogenation by Li et al. to produce gasoline-range hydrocarbons.104 With a suitable thickness of nanosheets along the b-axis, improved C5+ selectivity and slower carbon deposition could be achieved. In contrast, the extened b-axis of the chain-like nanocrystal HZSM-5 reported by Wang et al. was conducive to improve the selectivity for light aromatics such as para-xylene (PX), while a hollow structure with low diffusion resistance facilitates the formation of tetramethylbenzene.52 These results are closely related to the diverse diffusion properties of molecules with different sizes in specific channels.
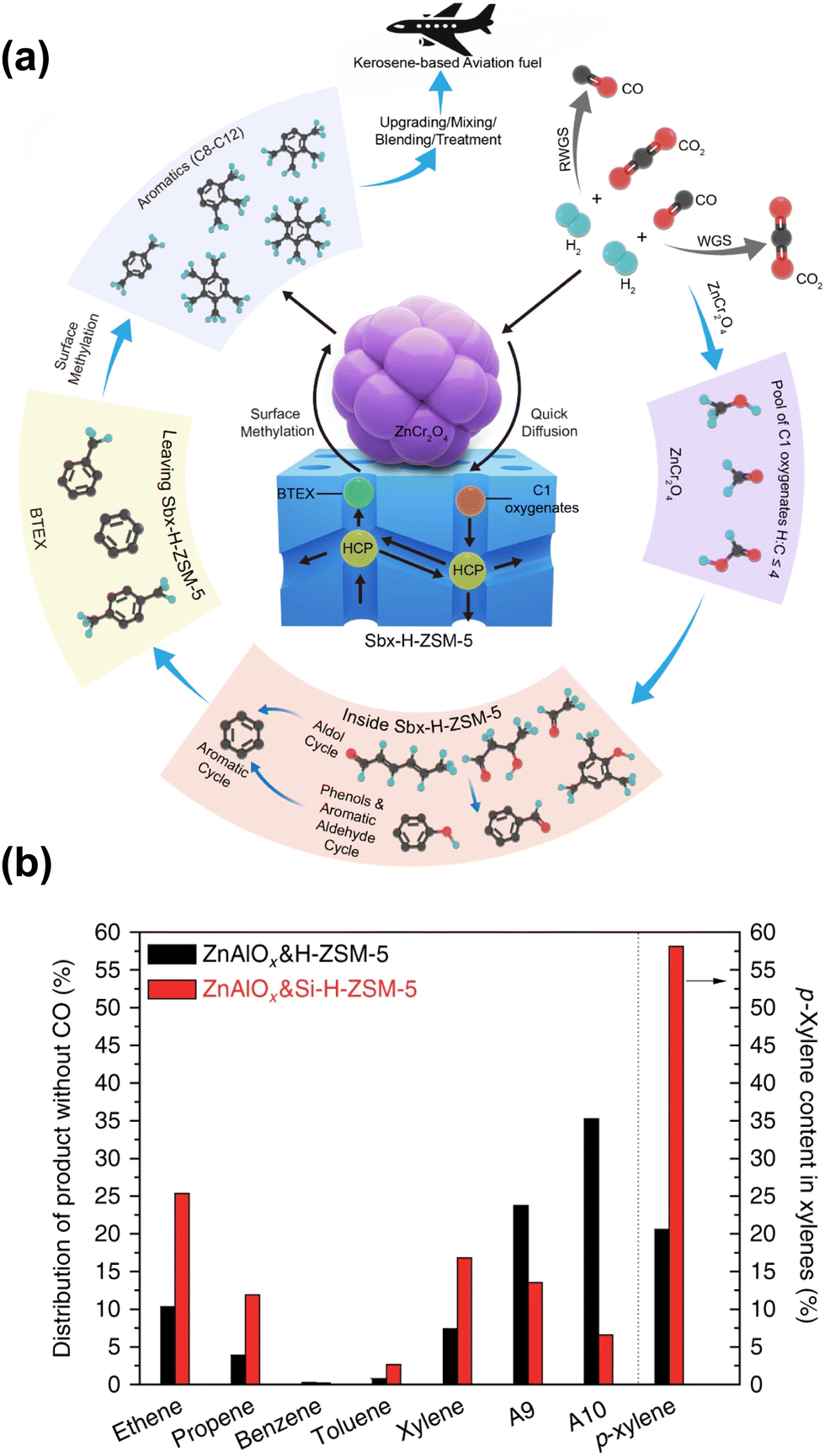 | ||
| Fig. 11 (a) The proposed reaction mechanism for the highly selective conversion of CO2 or syngas into aviation fuel precursors over ZnCr2O4 combined with short b-axis HZSM-5.97 (b) A comparison of the catalytic behaviors of ZnAlOx&H-ZSM-5 and ZnAlOx&Si-H-ZSM-5.48 Reproduced from ref. 97 and 48 with permission from the American Chemical Society and Springer Nature, copyright 2022 and 2017. | ||
Similar to the cases of composite iron-based catalysts, the SiO2 deposition and the heteroatomic doping could help shift the product distribution to a higher fraction of BTX. The SiO2 deposition on HZSM-5 by Ni et al. significantly improved the PX proportion in xylene from a thermodynamic equilibrium value of around 20% to above 58%, while the ethene and propene selectivity also increased48 (Fig. 11(b)). The catalytic behavior of ZnAlOx&Si-H-ZSM-5 would be interesting to industry because ethene, propene and PX are the most important commodity hydrocarbon chemicals. Modified nano-hierarchical-pore HZSM-5 zeolite (Zn/Z5) prepared via steam-assisted crystallization using ZIF-8 as a sacrificial template and Zn source was incorporated with ZnZrO to realize the CO2 conversion to aromatics.103 The Zn/Z5 zeolite with a smaller particle size, larger specific surface area, mesopore volume and suitable acidic distribution, was in favor of the improved physical transport of intermediates and further aromatization.
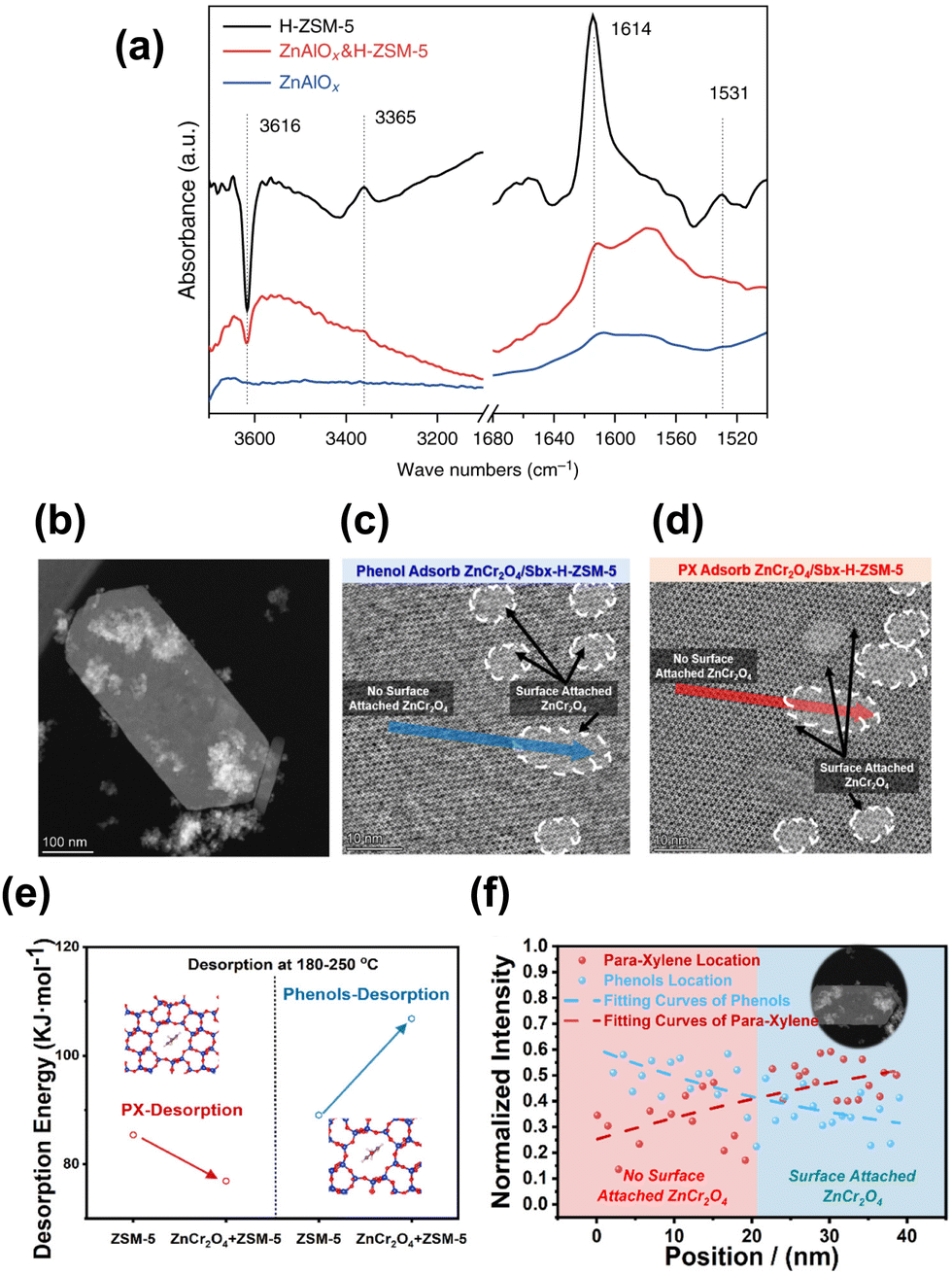 | ||
| Fig. 12 (a) FTIR subtraction spectra relative to the adsorption of DTBPy.48 (b) STEM images of the physically mixed composite catalyst of ZnCr2O4 and HZSM-5. (c) iDPC-STEM images of phenol adsorption on ZnCr2O4/HZSM-5. (d) iDPC-STEM images of PX adsorption on ZnCr2O4/HZSM-5. (e) Calculated activation energies of the phenol and PX desorption from HZSM-5 with and without ZnCr2O4. (f) Intensity profile of phenols and PX in HZSM-5 with and without ZnCr2O4.97 Reproduced from ref. 48 and 97 with the permission from American Chemical Society and Springer Nature, copyright 2022 and 2017. | ||
4.2 Tandem reactions of methanol synthesis and alkylation
Although 70–80% selectivity for aromatic hydrocarbons could be obtained in some cases through the tandem reactions of methanol synthesis and MTA, it is still a challenge to acquire some specific aromatics with high selectivity and facility investments for the subsequent separation process. Therefore, tandem conversion reactions of CO2 to methanol and alkylation with oversupplied benzene and toluene for the production of value-added aromatics such as PX and ethylbenzene have been proposed to break the limitations of the current aromatization processes. Although CO2 provides only a part of the carbon source for the final products, these strategies still make sense in the present stage based on their possible high efficiency for industrial production. Yuan's group applied ZnZrO and SiO2-modified HZSM-5 in the methylation of toluene with CO2 and H2, reaching a high xylene selectivity of up to 90% and a PX proportion in xylene of up to 70%.56 In comparison with the process involving methanol, which exhibits inferior PX selectivity (∼30%) and carbon usage, in situ CO2 hydrogenation provides more active intermediates as methylation species and more appropriate reaction dynamics under similar operation conditions (Fig. 13(a)). Miao et al. also achieved a high PX selectivity in xylene of up to 82.8% through CO2 hydrogenation in the presence of toluene by using a ZnCrOx-P/ZSM-5 catalyst.54 Light olefins in gas products could also be significantly increased with phosphorous modification.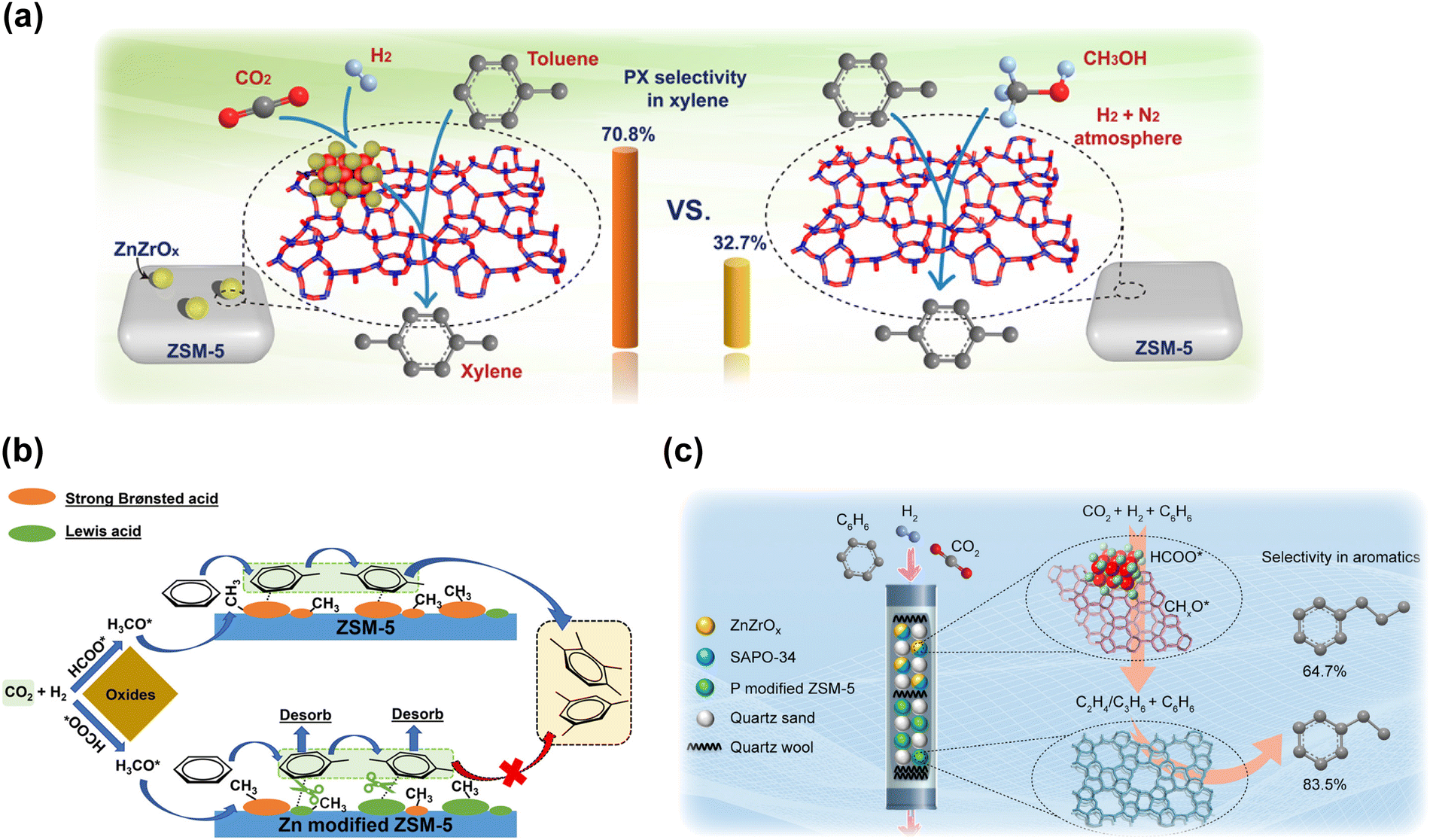 | ||
| Fig. 13 (a) A comparison of toluene methylation with CO2 and H2 to xylene and that with methanol.56 (b) A schematic of benzene alkylation with CO2 and H2 to toluene and xylene over Zn-modified ZSM-5 compared with normal ZSM-5.53 (c) Schematic of benzene alkylation with CO2 and H2 to produce ethylbenzene and propylbenzene through a dual-bed system.55 Reproduced from ref. 56, 53 and 55 with permission from AAAS, American Chemical Society and Elsevier, copyright 2020, 2022 and 2022. | ||
Shang et al. reported a ZnZrOx&Zn/ZSM-5 catalyst for CO2 hydrogenation to toluene and xylene with the introduction of benzene.53 A total selectivity for toluene and xylene of up to 93% was achieved at the CO2 conversion of 21%. Abundant Zn-induced moderate acid sites play a vital role in the enhanced coupling effect, which is considered to be beneficial for the product desorption and catalytic cycle as well as the suppression of consecutive alkylation reactions, resulting in an elevation in both reactant conversion and specific aromatic selectivity (Fig. 13(b)). Yuan et al. also developed a dual-bed system for direct benzene alkylation with CO2 and H2 toward ethylbenzene and propylbenzene.55 ZnZrO combined with the SAPO-34 zeolite was arranged as the top layer for the production of ethene and propene, followed by alkylation with benzene over phosphorous-modified ZSM-5 zeolite at the bottom layer. With the rational spatial distribution of these different components, 83.5% selectivity for ethylbenzene or 64.7% selectivity for propylbenzene in total aromatics could be obtained (Fig. 13(c)).
5. Process design and industrial application
To blaze a trail for the technology of direct CO2 hydrogenation to gasoline-range hydrocarbons from laboratory to industrial production, we still need to gain insights into the design and optimization of catalysts and also the control of the reaction process as well as the reactor design, since the CO2 hydrogenation involves collaboration among various components due to the coexistence of multiple phases. The arrangement of the process must take into account the advantageous operation zone of each component, the enhanced coupling effect, the evolution of catalysts with time on stream, and so on.18,108,109Early in the research of Gao et al. in 2017, a pilot-scale fixed-bed reactor was equipped for operation under industrial working conditions with recycled tail gas (Fig. 14(a)). This setup includes a recycling compressor with an inlet port connected to the cold trap outlet. The tail gas was first cooled down and the liquid-phase products were collected in a cold trap (273 K). The remaining gas was recycled to mix with the fresh feed gas, and then the total mixture entered the fixed bed reactor.
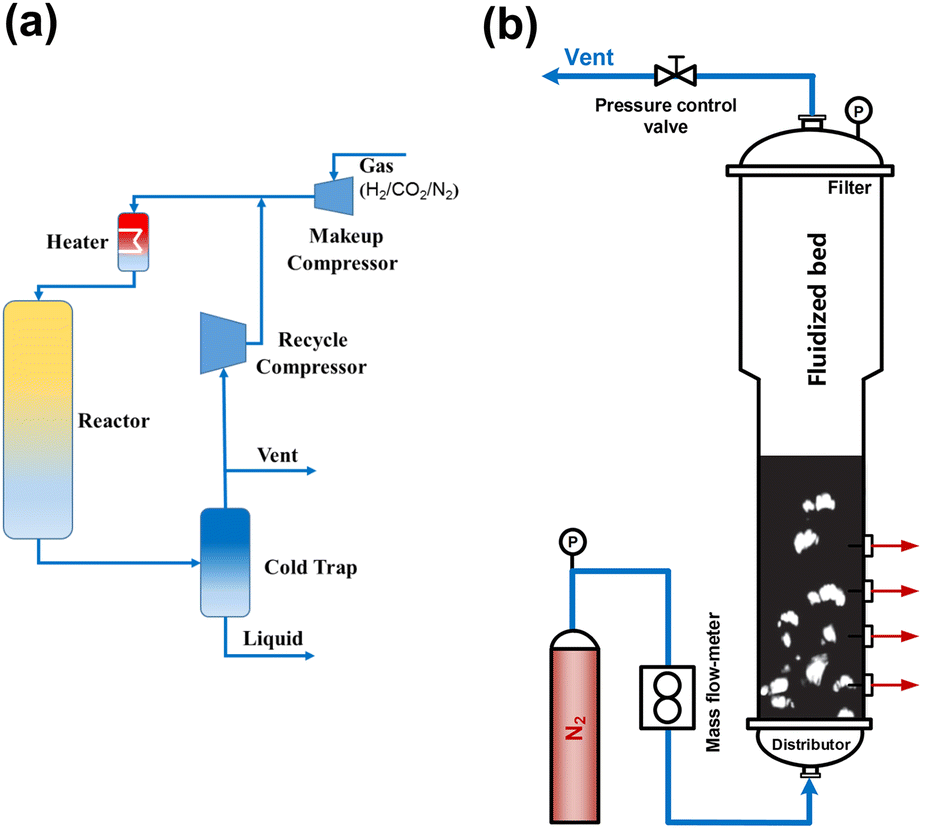 | ||
| Fig. 14 (a) Schematic flow diagram of the pilot-scale fixed-bed reactor,49 (b) schematic diagram of the cold-flow apparatus.110 Reproduced from ref. 49 and 110 with the permission from Springer Nature and Wiley, copyright 2017 and 2022. | ||
Wei and Lu et al. conducted both theoretical analysis and experimental study to obtain a phase diagram to guide the design and operation of an efficient fluidized-bed reactor (Fig. 14(b)), which might be applied in CO2 conversion to aviation fuel.97,110 They carried out a pilot scale test with an annual CO2 consumption of 105 m3 based on their previous studies, in which a blueprint of CO2 (from waste gas) hydrogenated with green hydrogen into jet fuel was drawn. Furthermore, in 2021, a 10000 ton scale industrial project of COx to aromatics was constructed in Inner Mongolia, China, with the cooperation of Wei's group and some energy companies. The technology developed by Wei et al. for syngas conversion to aromatics realized a selectivity of tetramethylbenzene-dominated aromatic products of up to 80% according to their previous studies.107,111 With a suitable adaptation in some procedures, the system for CO is expected to also be used in CO2 hydrogenation.
On the other hand, in March 2022, the world's first pilot unit of CO2 hydrogenation to gasoline with a scale of 1000 tons per year, developed by the Dalian Institute of Chemical Physics (DICP), was successfully launched in Shandong Province, China. The quality and components of gasoline products conformed to the Chinese VI standard as expected, with the total conversion of CO2 and H2 up to 95% and the selectivity of gasoline up to 85% in all carbon-containing products. Although several problems regarding the stability and economy need to be further solved, all these exciting results would inspire more efforts and achievements in the direct CO2 conversion to gasoline-range hydrocarbons in industry.
6. Conclusions
6.1 Summary
As a promising strategy for both the suppression of CO2 emission and the utilization of green hydrogen energy, the direct conversion of CO2 into gasoline-range hydrocarbons has attracted significant attention worldwide. Iron-based catalysts, well known as FT catalysts for syngas conversion to hydrocarbons, could realize the CO2 to long-chain hydrocarbons via the CO intermediate through a RWGS reaction. By further combining with zeolites, the limit of ASF distribution could be broken to obtain a high selectivity for gasoline-range hydrocarbons. The modification of iron-based components is crucial for the improvement of CO2 activation, while the topology and acid properties of zeolite components play a vital role in product distribution by regulating the diffusion or reaction dynamics. On the other hand, tandem reactions involving methanol synthesis could exhibit superior selectivity in comparison to iron-based catalysts due to the separate C–O activation and C–C coupling reactions. The transition metal oxides are applied in most cases instead of Cu-based catalysts because of the higher selectivity for methanol at high temperatures. The oxygen vacancy sites are believed to be the key active sites for CO2 activation, while hydrogen activation is equally important for the formation of intermediates. By combining the zeolite components with the tuned channel structure and acid sites, the product distribution could be shifted to more BTX or other value-added hydrocarbons. Moreover, the tandem reactions of methanol synthesis and alkylation provide an efficient strategy for the synthesis of high-value aromatics through coupling reactions from CO2 hydrogenation.Both the modified FT route and methanol-mediated route afford appreciable efficiency for direct CO2 hydrogenation to gasoline-range hydrocarbons, despite their significant differences. Generally, the FT-based process always contributes to higher CO2 conversion and the corresponding higher hydrocarbon yield due to the superior efficiency of C–C coupling over modified Fe-based catalysts. In contrast, methanol synthesis tends to be limited thermodynamically, but favors the production of specific hydrocarbons, e.g., aromatics, owing to the controllable C–C coupling in zeolites with distinct topology and acidic properties. Fortunately, such a gap between FT-based and methanol-based systems is supposed to be narrowed by optimizing the active components and operating conditions.
Notably, the interactions between different components make a great difference in both FT-based and methanol-based systems. Such interactions are always inevitably present under harsh redox conditions and can be regulated to some extent by controlling the proximity of two components. Strong interactions can even lead to substantial element migration, exhibiting various effects via the modification of the acidic properties and channel structure.
Besides laboratory research, the practical industrial application of CO2 hydrogenation to gasoline-range hydrocarbons has been exemplified by a batch of pilot test projects with considerable achievements, suggesting the great significance of intensive research on these catalytic systems in the future.
6.2 Outlook
As summarized above, the whole process of gasoline-range hydrocarbon production from CO2 hydrogenation involves complicated coupling reactions including the efficient activation of C–O and the controllable formation of C–C bonds, as well as the fine-tuned generation and transfer of various intermediates. Therefore, the common adjustments of the catalyst structure and active sites make a big difference in the ultimate efficiency, and the integrated manner and matched operating conditions of different components also play a critical role in practical production. However, deep insights into fundamental mechanisms involving the interactions between different active components are still lacking, especially under realistic reaction conditions, bringing challenges to the further optimization of catalysts. Hence, it is important to gain a comprehensive understanding of the relationship between the active parts for the optimized design of catalysts and processes with high reliability and long-term economy. Another strategy for improving economic efficiency is to selectively synthesize specific target products of high commercial value since the extensive cost of the necessary separation process can significantly limit the realization of industrial production. By subtly regulating the topology of zeolites or steering varied reaction routes, it could be possible to preferentially synthesize particular products in direct CO2 hydrogenation, which is of great significance to future practical application. Consequently, the optimization of catalysts and processes to achieve outstanding efficiency and excellent product selectivity is the most important direction for the development of further research, inspiring promising prospects in the scaling up of industrial production in the future.Abbreviation
| FT | Fischer–Tropsch |
| Ox-Zeo | Oxide and zeolite components |
| BTX | Benzene, toluene, and xylene |
| WGS | Water-gas shift |
| RWGS | Reverse water-gas shift |
| ASF | Anderson–Schulz–Flory |
| MOF | Metal–organic frameworks |
| PX | para-Xylene |
| MTG | Methanol to gasoline |
| MTA | Methanol to aromatic |
| XPS | X-ray photoelectron spectroscopy |
| TEM | Transmission electron microscopy |
| iDPC | Integrated differential phase contrast |
| TGA | Thermo-gravimetric analysis |
| DTBPy-FTIR | 2,6-Di-tert-butyl-pyridine absorption |
| EPR | Electron paramagnetic resonance |
| CO2-TPD | CO2 temperature-programmed desorption |
| GGA | Generalized gradient approximation |
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors gratefully acknowledge the Strategic Priority Research Program of the Chinese Academy of Sciences (no. XDA29040600 and no. XDB36030200), the National Natural Science Foundation of China (21978286, 21925803, U19A2015), the Youth Innovation Promotion Association CAS, and the Young Top-notch Talents of Liaoning Province (XLYC2007082).References
- S. I. Seneviratne, M. G. Donat, A. J. Pitman, R. Knutti and R. L. Wilby, Nature, 2016, 529, 477–483 CrossRef CAS
.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC
- Z. Zhou and P. Gao, Chin. J. Catal., 2022, 43, 2045–2056 CrossRef CAS
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC
- E. M. Rubin, Nature, 2008, 454, 841–845 CrossRef CAS
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC
- P. Tan, J. Catal., 2016, 338, 21–29 CrossRef CAS
- M. V. Luzgin, V. A. Rogov, S. S. Arzumanov, A. V. Toktarev, A. G. Stepanov and V. N. Parmon, Angew. Chem., Int. Ed., 2008, 47, 4559–4562 CrossRef CAS PubMed
- S. Ilias and A. Bhan, ACS Catal., 2012, 3, 18–31 CrossRef
- W. Zhang, J. Chen, S. Xu, Y. Chu, Y. Wei, Y. Zhi, J. Huang, A. Zheng, X. Wu, X. Meng, F. Xiao, F. Deng and Z. Liu, ACS Catal., 2018, 8, 10950–10963 CrossRef CAS
- J. Kang, K. Cheng, L. Zhang, Q. Zhang, J. Ding, W. Hua, Y. Lou, Q. Zhai and Y. Wang, Angew. Chem., Int. Ed., 2011, 50, 5200–5203 CrossRef CAS
- Y. Xu, G. Ma, J. Bai, Y. Du, C. Qin and M. Ding, ACS Catal., 2021, 11, 4476–4485 CrossRef CAS
- J. Sun, X. G. Li, A. Taguchi, T. Abe, W. Q. Niu, P. Lu, Y. Yoneyarna and N. Tsubaki, ACS Catal., 2014, 4, 1–8 CrossRef CAS
- X. Yang, R. Wang, J. Yang, W. Qian, Y. Zhang, X. Li, Y. Huang, T. Zhang and D. Chen, ACS Catal., 2020, 10, 3797–3806 CrossRef CAS
- K. Cheng, J. Kang, S. Huang, Z. You, Q. Zhang, J. Ding, W. Hua, Y. Lou, W. Deng and Y. Wang, ACS Catal., 2012, 2, 441–449 CrossRef CAS
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS
- N. Li, F. Jiao, X. L. Pan, Y. X. Chen, J. Y. Feng, G. Li and X. H. Bao, Angew. Chem., Int. Ed., 2019, 58, 7400–7404 CrossRef CAS
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS
- C. Liu, J. J. Su, S. Liu, H. B. Zhou, X. H. Yuan, Y. C. Ye, Y. Wang, W. Q. Jiao, L. Zhang, Y. Q. Lu, Y. D. Wang, H. Y. He and Z. K. Xie, ACS Catal., 2020, 10, 15227–15237 CrossRef CAS
- Y. Wang, W. Zhan, Z. Chen, J. Chen, X. Li and Y. Li, ACS Catal., 2020, 10, 7177–7187 CrossRef CAS
- P. Zhang, L. Tan, G. Yang and N. Tsubaki, Chem. Sci., 2017, 8, 7941–7946 RSC
- Z. Huang, S. Wang, F. Qin, L. Huang, Y. Yue, W. Hua, M. Qiao, H. He, W. Shen and H. Xu, ChemCatChem, 2018, 10, 4519–4524 CrossRef CAS
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Chem. Sci., 2018, 9, 4708–4718 RSC
- F. Jiao, X. Pan, K. Gong, Y. Chen, G. Li and X. Bao, Angew. Chem., Int. Ed., 2018, 57, 4692–4696 CrossRef CAS PubMed
- W. Zhou, J. Kang, K. Cheng, S. He, J. Shi, C. Zhou, Q. Zhang, J. Chen, L. Peng, M. Chen and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12012–12016 CrossRef CAS PubMed
- M. Tahir and N. S. Amin, Renewable Sustainable Energy
Rev., 2013, 25, 560–579 CrossRef CAS
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef
- C. B. Hiragond, N. S. Powar, J. Lee and S.-I. In, Small, 2022, 18, 2201428 CrossRef CAS
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS
- S. Liu, H. Yang, X. Su, J. Ding, Q. Mao, Y. Huang, T. Zhang and B. Liu, J. Energy Chem., 2019, 36, 95–105 CrossRef
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC
- W. W. Russell and G. H. Miller, J. Am. Chem. Soc., 1950, 72, 2446–2454 CrossRef CAS
- J. W. Niemantsverdriet and A. M. van der Kraan, J. Catal., 1981, 72, 385–388 CrossRef CAS
- D. B. Bukur, K. Okabe, M. P. Rosynek, C. P. Li, D. J. Wang, K. R. P. M. Rao and G. P. Huffman, J. Catal., 1995, 155, 353–365 CrossRef CAS
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS
- X. L. Liu, M. H. Wang, H. R. Yin, J. T. Hu, K. Cheng, J. C. Kang, Q. H. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS
- S. Wang, L. Zhang, P. Wang, X. Liu, Y. Chen, Z. Qin, M. Dong, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2022, 8, 1376–1394 CAS
- S. Wang, L. Zhang, P. Wang, W. Jiao, Z. Qin, M. Dong, J. Wang, U. Olsbye and W. Fan, Nat. Catal., 2022, 5, 1038–1050 CrossRef CAS
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019 CrossRef CAS PubMed
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed
- J. Wei, R. Yao, Q. Ge, D. Xu, C. Fang, J. Zhang, H. Xu and J. Sun, Appl. Catal., B, 2021, 283, 119648 CrossRef CAS
- T. Wang, C. Yang, P. Gao, S. Zhou, S. Li, H. Wang and Y. Sun, Appl. Catal., B, 2021, 286, 119929 CrossRef CAS
- X. Shang, G. Liu, X. Su, Y. Huang and T. Zhang, ACS Catal., 2022, 12, 13741–13754 CrossRef CAS
- D. Miao, X. Pan, F. Jiao, Y. Ji, G. Hou, L. Xu and X. Bao, Catal. Sci. Technol., 2021, 11, 4521–4528 RSC
- J. Zuo, C. Liu, X. Han, D. Wen, X. Liu, L. Ye, W. Zhuang and Y. Yuan, Chem. Catal., 2022, 2, 1223–1240 CrossRef CAS
- J. Zuo, W. Chen, J. Liu, X. Duan, L. Ye and Y. Yuan, Sci. Adv., 2020, 6, eaba5433 CrossRef CAS PubMed
- G. Henrici-Olivé and S. Olivé, Angew. Chem., Int. Ed. Engl., 1976, 15, 136–141 CrossRef
- R. A. Friedel and R. B. Anderson, J. Am. Chem. Soc., 1950, 72, 1212–1215 CrossRef CAS
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 CrossRef CAS
- M. Yang, J. Yu, A. Zimina, B. B. Sarma, L. Pandit, J.-D. Grunwaldt, L. Zhang, H. Xu and J. Sun, Angew. Chem., Int. Ed., 2022, e202216803 Search PubMed
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed
- K. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed
- N. Y. Chen, W. W. Kaeding and F. G. Dwyer, J. Am. Chem. Soc., 1979, 101, 6783–6784 CrossRef CAS
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed
- X. Zhang, G. Zhang, W. Liu, F. Yuan, J. Wang, J. Zhu, X. Jiang, A. Zhang, F. Ding, C. Song and X. Guo, Appl. Catal., B, 2021, 284, 119700 CrossRef CAS
- W. H. Feng, M. M. Yu, L. J. Wang, Y. T. Miao, M. Shakouri, J. Q. Ran, Y. F. Hu, Z. Y. Li, R. Huang, Y. L. Lu, D. Q. Gao and J. F. Wu, ACS Catal., 2021, 11, 4704–4711 CrossRef CAS
- R. W. Dorner, D. R. Hardy, F. W. Williams, B. H. Davis and H. D. Willauer, Energy Fuels, 2009, 23, 4190–4195 CrossRef CAS
- E. Zaǧli and J. L. Falconer, J. Catal., 1981, 69, 1–8 CrossRef
- C. Y. Dai, X. Zhao, B. R. Hu, X. B. Zhang, Q. X. Luo, X. W. Guo and X. X. Ma, J. CO2 Util., 2021, 43, 101369 CrossRef CAS
- Z. Y. Cai, F. L. Zhang, S. B. Yu, Z. P. He, X. J. Cao, L. Zhang and K. Huang, Catal. Sci. Technol., 2022, 12, 3826–3835 RSC
- M. K. Khan, P. Butolia, H. Jo, M. Irshad, D. Han, K. W. Nam and J. Kim, ACS Catal., 2020, 10, 10325–10338 CrossRef CAS
- L. M. Xiong, S. Liu, Y. Men, L. Li, X. D. Niu, K. L. Guo, J. Z. Xu, W. An, J. G. Wang and Y. Cong, J. Environ. Chem. Eng., 2022, 10, 108407 CrossRef CAS
- X. Zhou, J. Ji, D. Wang, X. Duan, G. Qian, D. Chen and X. Zhou, Chem. Commun., 2015, 51, 8853–8856 RSC
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed
- G. Yu, B. Sun, Y. Pei, S. Xie, S. Yan, M. Qiao, K. Fan, X. Zhang and B. Zong, J. Am. Chem. Soc., 2010, 132, 935–937 CrossRef CAS PubMed
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed
- S. Y. Hong, D. H. Chun, J. I. Yang, H. Jung, H. T. Lee, S. Hong, S. Jang, J. T. Lim, C. S. Kim and J. C. Park, Nanoscale, 2015, 7, 16616–16620 RSC
- P. Wang, W. Chen, F. K. Chiang, A. I. Dugulan, Y. J. Song, R. Pestman, K. Zhang, J. S. Yao, B. Feng, P. Miao, W. N. Xu and E. J. M. Hensen, Sci. Adv., 2018, 4, eaau2947 CrossRef CAS PubMed
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS
- S. S. Geng, F. Jiang, Y. B. Xu and X. H. Liu, ChemCatChem, 2016, 8, 1303–1307 CrossRef CAS
- C. Y. Dai, X. Zhao, B. R. Hu, J. X. Zhang, Q. Q. Hao, H. Y. Chen, X. W. Guo and X. X. Ma, Ind. Eng. Chem. Res., 2020, 59, 19194–19202 CrossRef CAS
- A. Ramirez, A. D. Chowdhury, A. Dokania, P. Cnudde, M. Caglapn, I. Yarulina, E. Abou-Hamad, L. Gevers, S. Ould-Chikh, K. De Wispelaere, V. van Speybroeck and J. Gascon, ACS Catal., 2019, 9, 6320–6334 CrossRef CAS
- E. García-Hurtado, A. Rodríguez-Fernández, M. Moliner and C. Martínez, Catal. Sci. Technol., 2020, 10, 5648–5658 RSC
- K. Y. Qin, Y. Men, S. Liu, J. G. Wang, Z. P. Li, D. D. Tian, T. L. Shi, W. An, X. L. Pan and L. Li, J. CO2 Util., 2022, 65, 102208 CrossRef CAS
- G. Y. Song, M. Z. Li, L. Xu, X. P. Yang, M. A. Nawaz, H. M. Yuan, Z. X. Zhang, X. M. Xu and D. H. Liu, Ind. Eng. Chem. Res., 2022, 61, 6820–6830 CrossRef CAS
- G. Y. Song, M. Z. Li, P. K. Yan, M. A. Nawaz and D. H. Liu, ACS Catal., 2020, 10, 11268–11279 CrossRef CAS
- Y. Wang, S. Kazumi, W. Gao, X. Gao, H. Li, X. Guo, Y. Yoneyama, G. Yang and N. Tsubaki, Appl. Catal., B, 2020, 269, 118792 CrossRef CAS
- J. Wei, R. Yao, Q. Ge, Z. Wen, X. Ji, C. Fang, J. Zhang, H. Xu and J. Sun, ACS Catal., 2018, 8, 9958–9967 CrossRef CAS
- M. G. Sibi, M. K. Khan, D. Verma, W. Yoon and J. Kim, Appl. Catal., B, 2022, 301, 120813 CrossRef CAS
- C. Y. Dai, X. Zhao, B. R. Hu, J. X. Zhang, Q. Q. Hao, H. Y. Chen, X. W. Guo and X. X. Ma, Ind. Eng. Chem. Res., 2020, 59, 19194–19202 CrossRef CAS
- X. Wang, G. Yang, J. Zhang, S. Chen, Y. Wu, Q. Zhang, J. Wang, Y. Han and Y. Tan, Chem. Commun., 2016, 52, 7352–7355 RSC
- X. Wang, C. Zeng, N. Gong, T. Zhang, Y. Wu, J. Zhang, F. Song, G. Yang and Y. Tan, ACS Catal., 2021, 11, 1528–1547 CrossRef CAS
- M. T. Arslan, G. Tian, B. Ali, C. Zhang, H. Xiong, Z. Li, L. Luo, X. Chen and F. Wei, ACS Catal., 2022, 12, 2023–2033 CrossRef CAS
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2019, 10, 302–310 CrossRef
- D. Wen, J. Zuo, X. Han, J. Liu, L. Ye and Y. Yuan, Catal. Sci. Technol., 2022, 12, 2555–2565 RSC
- B. Yu, C. Ding, J. Wang, Y. Zhang, Y. Meng, J. Dong, H. Ge and X. Li, J. Phys. Chem. C, 2019, 123, 18993–19004 CrossRef CAS
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS
- S. Tada, N. Ochiai, H. Kinoshita, M. Yoshida, N. Shimada, T. Joutsuka, M. Nishijima, T. Honma, N. Yamauchi, Y. Kobayashi and K. Iyoki, ACS Catal., 2022, 12, 7748–7759 CrossRef CAS
- H. F. Tian, J. P. Jiao, F. Zha, X. J. Guo, X. H. Tang, Y. Chang and H. S. Chen, Catal. Sci. Technol., 2022, 12, 799–811 RSC
- W. H. Li, J. X. Zhang, X. Jiang, M. C. Mu, A. F. Zhang, C. S. Song and X. W. Guo, Ind. Eng. Chem. Res., 2022, 61, 6322–6332 CrossRef CAS
- Y. Li, M. Wang, S. Liu, F. Wu, Q. Zhang, S. Zhang, K. Cheng and Y. Wang, ACS Catal., 2022, 12, 8793–8801 CrossRef CAS
- Y. Wang, G. Wang, L. I. van der Waals, K. Cheng, Q. Zhang, K. P. de Jong and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 17735–17743 CrossRef CAS PubMed
- M. T. Arslan, B. A. Qureshi, S. Z. A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9, 2203–2212 CrossRef CAS
- X. Yang, X. Su, D. Chen, T. Zhang and Y. Huang, Chin. J. Catal., 2020, 41, 561–573 CrossRef CAS
- X. Yang, T. Sun, J. Ma, X. Su, R. Wang, Y. Zhang, H. Duan, Y. Huang and T. Zhang, J. Energy Chem., 2019, 35, 60–65 CrossRef
- F. Lu, C. Zhang, Y. Wang, W. Qian and F. Wei, AlChE J., 2022, 68, e17530 CAS
- M. T. Arslan, B. Ali, S. Z. A. Gilani, Y. Hou, Q. Wang, D. Cai, Y. Wang and F. Wei, ACS Catal., 2020, 10, 2477–2488 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |