
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Under pressure: offering fundamental insight into structural changes on ball milling battery materials†
Laura L.
Driscoll
 ac,
Elizabeth H.
Driscoll
*ac,
Bo
Dong
ac,
Farheen N.
Sayed
bc,
Jacob N.
Wilson
ad,
Christopher A.
O’Keefe
b,
Dominic J.
Gardner
abc,
Clare P.
Grey
bc,
Phoebe K.
Allan
ac,
Adam A. L.
Michalchuk
ad and
Peter R.
Slater
*ac
ac,
Elizabeth H.
Driscoll
*ac,
Bo
Dong
ac,
Farheen N.
Sayed
bc,
Jacob N.
Wilson
ad,
Christopher A.
O’Keefe
b,
Dominic J.
Gardner
abc,
Clare P.
Grey
bc,
Phoebe K.
Allan
ac,
Adam A. L.
Michalchuk
ad and
Peter R.
Slater
*ac
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: e.h.driscoll@bham.ac.uk; p.r.slater@bham.ac.uk
bYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
cThe Faraday Institution, Harwell Science and Innovation Campus, Didcot OX11 0RA, UK
dFederal Institute for Materials Research and Testing (BAM), Richard-Willsaetter-Strasse, 12489 Berlin, Germany
First published on 29th August 2023
Abstract
Synthesis of Li ion battery materials via ball milling has been a huge area of growth, leading to new high-capacity electrode materials, such as a number of promising disordered rocksalt (DRS) phases. In prior work, it was generally assumed that the synthesis was facilitated simply by local heating effects during the milling process. In this work, we show that ball milling Li2MoO4 leads to a phase transformation to the high pressure spinel polymorph and we report electrochemical data for this phase. This observation of the formation of a high pressure polymorph shows that local heating effects alone cannot explain the phase transformation observed (phenakite to spinel) and so indicates the importance of other effects. In particular, we propose that when the milling balls collide with the material, the resulting shockwaves exert a localised pressure effect, in addition to local heating. To provide further support for this, we additionally report ball milling results for a number of case studies (Li2MnO3, Li2SnO3, Nb2O5) which reinforces the conclusion that local heating alone cannot explain the phase transformations observed. The work presented thus provides greater fundamental understanding of milling as a synthetic pathway and suggests potential strategies to prepare such samples without milling (e.g., doping to create internal chemical pressure). In addition, we suggest that further research is needed into the effect of the use of milling as a route to smaller particles, since we believe that such milling may also be affecting the surface structure of the particles through the influence of the shockwaves generated.
Broader contextResearch into lithium ion battery materials has grown substantially over the last decade following their introduction into hybrid and all electric vehicles (EV). This growth is forecast to increase as governments worldwide look to transition towards zero emission transport, as well to introduce large scale stationary energy storage to complement renewable energy expansion. The EV market has particularly stimulated research into new electrode materials, with a strong growth in research into new high capacity cathode materials containing earth abundant elements, such as Mn. A number of promising materials have been identified, yet many rely on synthesis via high energy ball milling. In this prior research it has been largely assumed that the synthesis is simply achieved through local heating during the milling process. In this paper, we use a number of case studies to illustrate that this is not correct and we provide for the first time evidence to support the importance of local pressure effects in the synthesis of such battery materials, which will aid researchers in future studies to synthesise such phases both through milling and other routes. In addition, the work has significance for researchers using milling as a route to produce smaller particle sizes, including the possibility such milling may also be affecting the surface structure of the particles. Given the growing interest of mechanochemical synthesis as an alternative synthesis route in a wide range of other areas, from inorganic materials to organic compounds, the work also has widespread significance to the scientific community. |
Introduction
Li-ion batteries will play a key role in decarbonising the global economy infrastructure, both in terms of transport as well as for grid storage of energy from renewable sources. Current Li-ion technology relies predominantly on cationic redox, in which a transition metal will vary its oxidation state with the (de-)intercalation of lithium into the host structure in order to maintain charge neutrality. Common metals include Fe, Mn, Ni and Co, although there is a move to eliminate the latter, due to cost and ethical reasons. However, although reliable, cationic redox is limited to the accessible oxidation states of the transition metal cations selected to form the electrode material, e.g., Mn cannot be oxidised beyond Mn4+ in traditional cathodes used in Li-ion batteries. Therefore, in order to increase the potential capacity of cathodes, complementary anionic redox is perceived to be one of the next or “an important” developmental step for increased electrode capacity. In this respect, disordered rocksalt (DRS) cathodes have been identified as a valuable family of materials due to the ability to display both anionic and cationic redox with relatively small volume expansion on (de-)intercalation of Li.1 Notable early examples reported in the literature are summarised in Table 1.2–5 The synthesis of these particular phases has relied extensively on high impact ball milling, particularly for the F containing systems, although there has been growing research showing that some lower F content (usually less than 10%) DRS systems can be prepared by standard high temperature synthesis.6–10| Material | Synthesis route | Precursors | Cell parameters (DRS) (Å) | Testing conditions | Capacity (mA h g−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| 1st charge | 1st discharge | ||||||
| Li4Mn2O5 | High impact milling. 700 rpm, 20 h | LiMnO2, Li2O | 4.1732(9) | 1.2–4.8 V rate: C/20 | — | 355 | 2 |
| LiMnO2 | High impact milling. 600 rpm, 36 h | LiMnO2 | 4.153(5) | 1.5–4.8 V rate: 10 mA g−1 | — | 250 | 3 |
| Li2MnO2F | High impact ball milling. 750 rpm, 18 h | LiF, Li2O and Mn2O3 | 4.1176(5) | 2.0–4.8 V rate: C/10 (22.4 mA g−1) | 291 | 283 | 4 |
| Li2NiO2F | High impact milling. 750 rpm, 12 h | LiNiO2, LiF | 4.0748(2) | 2.0–4.8 V | 300 | 200 | 5 |
In this respect, past work has shown that Li4Mn2O5 can be synthesised via extensive milling of LiMnO2, Li2O and carbon black for 20 h. The system delivers a large capacity (∼350 mA h g−1) due to the utilisation of both cationic and anionic redox.2 Literature has also shown that by just milling LiMnO2 to synthesise the disordered rocksalt equivalent phase at 600 rpm for 36 h can also deliver high capacities of 200–250 mA h g−1.3 Indeed, to further boost the performance of such systems, a common modification is to incorporate fluorine in an attempt to increase the operating voltage and to reduce the valence state of the transition metal, therefore increasing the materials capacity by granting greater access to potential transition metal redox. While a few reports exist where fluorinated DRS have been synthesised using high temperatures, those reported to be successful possess F contents significantly lower than materials achievable through mechanochemical routes (e.g. Li1.3Ti0.3Mn0.4O1.7F0.3 (high temperature synthesis) versus Li2MnO2F (mechanochemical synthesis)).11 Notable examples of materials made through mechanochemical routes include Li2MnO2F, Li2NiO2F and LixMn2−xO2−yFy, where x = 1.167–1.33 and y = 0–0.667, can all be synthesised using high milling speeds and long milling times (12–55 h at 500–750 rpm).4,5,12 While the synthesis of such battery materials through ball milling has been a growing research area, the mechanism behind the synthesis is still rather poorly understood. Traditionally, this synthesis has been perceived to proceed through the introduction of localised heat (from the balls striking the powder); however, there has been acknowledgement in the research community that the products of such milling reactions are difficult to predict based on known thermodynamic variables.13 This is emphasised by the fact that many of these samples cannot be prepared by standard solid state synthesis at high temperatures, suggesting that there must be another variable involved.
Our work has been focused on trying to develop new Li-ion battery materials by ball milling, as well as aiming to understand the mechanistic aspects of this process, so that we can improve our targeted synthesis of such new materials. The work presented here was focused on an initial study into the effect of ball milling on Li2MoO4. This system was selected in an attempt to produce a cation deficient DRS phase containing Mo6+ (d0), such that any electrochemical activity observed during delithiation must be due to anionic rather than cationic redox, therefore providing a model system for further characterisation to understand the mechanism of anionic redox. However, as we will show, instead of a DRS phase, a new spinel phase was identified, which previous reports suggest could only be achieved when using high pressures.14,15 This observation of forming a high pressure phase raised important questions regarding the features of the ball milling process, and so led us to select additional systems to examine. To this effect, DRS systems synthesised from milling Li-rich layered phases (such as Li2MnO3 and Li2SnO3) and Nb2O5 were selected for further study. In each of these cases, we show that the observed changes on milling cannot be simply attributed to local heating effects and present supporting arguments that local pressure effects are also important, such that we propose that the impact of the balls creates shockwaves that not only deliver local heat, but crucially generate local pressure, and it is this latter contribution that explains why many of the reported phases cannot be prepared by a standard solid state high temperature synthesis route. Thus, the work provides crucial information that helps to rationalise the factors dictating the success of the mechanochemical process in the synthesis of a number of new high capacity cathode materials (in particular DRS systems) reported in the literature, and why these materials cannot be prepared by standard high temperature synthesis.
Experimental
The initial starting material, Li2MoO4, was synthesised through traditional solid-state methods. Stoichiometric amounts of Li2CO3 and MoO3 were ground together in a pestle and mortar and heated in air at 600 °C for 12 h at a heating rate of 100 °C h−1. In the follow-on additional case study work on Li2MO3 (M = Mn, Sn), samples were similarly prepared by solid state synthesis. Li2MnO3 was prepared using stoichiometric amounts of Li2CO3 and Mn2O3 before heating the sample in air at 700 °C for 12 h. Li2SnO3 was prepared by mixing stoichiometric amounts of Li2CO3 and SnO2 and heated at 950 °C in air for 24 h (with an intermediate regrind). The samples (packed inside an Ar filled glovebox) were then milled under an inert atmosphere, using a Fritsch premium line 7 planetary ball mill, for 2–16 h at 450 rpm (the pots were loaded with approximately 2 g of sample into 20 mL silicon nitride grinding bowls containing 10 silicon nitride grinding balls each of a 10 mm diameter). After milling, samples were transferred and stored in the glovebox for further characterisation. A second set of ball milling experiments were performed using a Fritsch Pulverisette P23 vibratory ball mill to understand variables of frequency/ball size/time. For these measurements, a sample of Li2MoO4 (50 mg) was loaded into custom-built milling jars (ca. 4.5 mL volume), which comprise hemispherical stainless steel ends and a polymethylmethacrylate (PMMA) shaft.16 For each milling reaction, a single stainless steel milling ball of either 7 mm diameter (1.41 g) or 10 mm diameter (3.86 g) was added, and the sample was milled at a frequency of either 40 or 50 Hz. For all mechanochemical reactions, data analysis was performed ex situ, with a fresh sample used for each time point.For the study of the effect of ball milling on Nb2O5, T-Nb2O5 (orthorhombic phase; Alfa Aesar) was used as-purchased, while H-Nb2O5 (monoclinic; high temperature phase) was prepared by heating the T-phase up to 1100 °C for 12 h. The Nb2O5 phases were then ball milled for 2 h at 700 rpm in air (sample mass of 2 g in 20 mL silicon nitride grinding bowls containing 10 silicon nitride grinding balls in each bowl).
Sample purity and unit cell parameters were determined from powder X-ray diffraction using a Bruker D8/PANalytical Emperean (Cu Kα radiation) diffractometer operating in reflection mode. Air sensitive samples (ball milled Li2MoO4 on Fritsch premium line 7 planetary ball mill) were sealed in Kapton tape within the glovebox prior to measurement. The frequency-time study conducted for Li2MoO4 on the P23 mill made use of a D2 Bruker (Co Kα radiation) diffractometer. Variable temperature PXRD was conducted using a Bruker D8 (Cu Kα radiation) in reflection mode fitted with an Anton Parr heating stage in order to determine whether there were any phase changes on subsequently heating the milled samples. Samples were heated in air in the region of 100 to 800 °C with PXRD data collected every 25 °C.
For imaging the particles, a Zeiss EVO15 SEM in secondary electron mode was used to understand the morphological changes in: Li2MoO4, Li2MnO3 and Nb2O5, while a ThermoFisher Scientific Apreo 2 SEM was used for the Li2SnO3 (due to instrument availability). The respective voltages and current used in the imaging are included on the associated figures. For TEM imaging of particles, a ThermoFisher Scientific (FEI) Talos F200X G2 operated in the scanning mode at 200 kV was used.
Thermogravimetric analysis studies were conducted using a Netzsch STA 449 F1 Jupiter thermogravimetric analyser coupled with a Netzsch 403 C mass spectrometer (heating rate of 0.5 °C min−1 under an oxygen atmosphere).
To investigate the difference in local environments of Mo in as-prepared (phenakite structure) Li2MoO4 and the spinel-like Li2MoO4 phase prepared by milling, 95Mo (nuclear spin I = 5/2) solid state nuclear magnetic resonance (ssNMR) spectra were recorded. The 95Mo (N.A. = 15.87%) nuclide is preferred over 97Mo (N.A. = 9.58%) due to its higher natural abundance and its lower quadrupole moment (Q = −0.022 × 10−28 m2), which results in narrower lineshapes. Both samples were packed into 4 mm zirconia rotors (with Kel-F cap) inside Ar filled glovebox and spectra were recorded on Bruker Avance III HD spectrometer (B0 = 16.4 Tesla, ν0(1H) = 700 MHz) with ν0(95Mo) = 45.627 MHz. A Bruker 4 mm triple channel MAS probe was used with a spinning speed of 10 kHz. A single pulse (d1-π/2-de-ACQ) experiment was used with an optimised 3 μs π/2 pulse, 10 s recycle delay (d1), ACQ time of 0.655 s and 640 co-added transients. 95Mo chemical shifts were referenced to a secondary reference of 2 M solution of Na2MoO4 in D2O. Line broadening of 20 Hz and 100 Hz were applied to the obtained spectra of parent Li2MoO4 and BM-Li2MoO4, respectively. For 7Li (nuclear spine I = 3/2, N.A. = 92.41%, Q = −0.041 × 10−28 m2) MAS ssNMR spectra were recorded on a Bruker Avance III spectrometer (B0 = 4.7 Tesla, ν0(1H) = 200 MHz) with ν0(7Li) = 77.778 MHz. A Bruker 1.3 mm double channel probe at 50 kHz MAS speed was used with single pulse (d1-π/2-de-ACQ) experiment, a 0.5 s recycle delay (d1), 5 μs pre-scan delay, ACQ time of 0.02666 s and 16 co-added transients. 7Li chemical shifts were calibrated using a secondary reference of solid 7Li2CO3 (δiso = 1.1 ppm, relative to a 1 M aqueous solution of LiCl at δiso = 0 ppm). Line broadening of 500 Hz was added to the spectra of both the samples. For 7Li, spin–lattice relaxation time constant (T1) measurements were done using a saturation recovery experiment. Fitting of the experimental spectra was performed using the SOlid Lineshape Analysis (SOLA) tool in TopSpin. The crystal structures were visualised using the CrystalMaker software package.
Given that ball milling Li2MoO4 led to a structural transformation to spinel, and spinel-type Li2MoO4 has not been investigated before as a Li-ion battery electrode, we decided to electrochemically characterise this spinel phase. Samples were milled with carbon black (ratio of 75% AM and 25% CB) for 2 h at 300 rpm under an inert atmosphere (20 mL silicon nitride grinding bowl, ca. 2 mL of sample, 10 silicon nitride grinding balls). Lithium half-cells (Swagelok configuration) were assembled using pre-cut lithium disks (11 mm) and 85 μL of 1.0 M LiPF6 in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) ethylene carbonate and dimethyl carbonate (Sigma) as the electrolyte, with a glass fibre separator (grade GF/D; 12.7 mm). 3–10 mg of the ball milled material were loaded into the cell. The electrochemical performance of these cells containing BM-Li2MoO4 (spinel) was characterised over different voltage ranges between 0.5–4.8 V vs. Li/Li+ at a current density of 10 mA g−1.
:50 (v/v) ethylene carbonate and dimethyl carbonate (Sigma) as the electrolyte, with a glass fibre separator (grade GF/D; 12.7 mm). 3–10 mg of the ball milled material were loaded into the cell. The electrochemical performance of these cells containing BM-Li2MoO4 (spinel) was characterised over different voltage ranges between 0.5–4.8 V vs. Li/Li+ at a current density of 10 mA g−1.
Results and discussion
Effect of ball milling of Li2MoO4
Due to the position of the Mo6+/5+ and Mo5+/4+ redox couples, Li2MoO4 has mostly been considered as an anode for Li-ion batteries, demonstrating extremely high capacities when charged to lower voltages.17–19 This material adopts the phenakite structure and the original aim of the milling study was an attempt to generate a cation deficient disordered rocksalt containing a d0 cation that could show solely oxygen redox on delithiation, in order to provide a potential system to develop a deeper understanding of anionic redox processes. Although Li2MoO4 does not have a layered structure, there is evidence in the literature that suggests a disordered rocksalt phase may be feasible in the Li–Mo–O system. In particular, Ehrenberg et al. demonstrated the formation of (Li3Mo)O4 from Li2MoO4via chemical lithiation using n-butyllithium in hexane in a glovebox, albeit taking 4 weeks to achieve a single phase.20The milling approach was taken, as prior work has shown that high-impact milling of layered Li-rich phases, such as Li2MnO3, produces DRS phases.3,21 The results of our milling study showed, however, that a DRS phase was not produced. Rather, after a couple of hours of milling, the X-ray diffraction data showed that the peaks from the phenakite polymorph were greatly reduced and, instead, broad peaks corresponding to a spinel phase appeared. This spinel phase then became the predominant phase after 8 h of milling, reaching full conversion after 16 h (Fig. 1). Using an initial starting structural model for a related spinel Ag2MoO4, Rietveld refinement produced a good fit to the X-ray diffraction data, confirming this phase transformation to spinel after milling (Fig. 2). Cell parameters and structural data for the spinel phase are shown in Table 2. The milled material adopts the traditional spinel structure where the Li occupies the octahedral site (16d) and the Mo is found on the tetrahedral site (8a). This differs from the original phenakite host in which all cations are tetrahedrally coordinated.
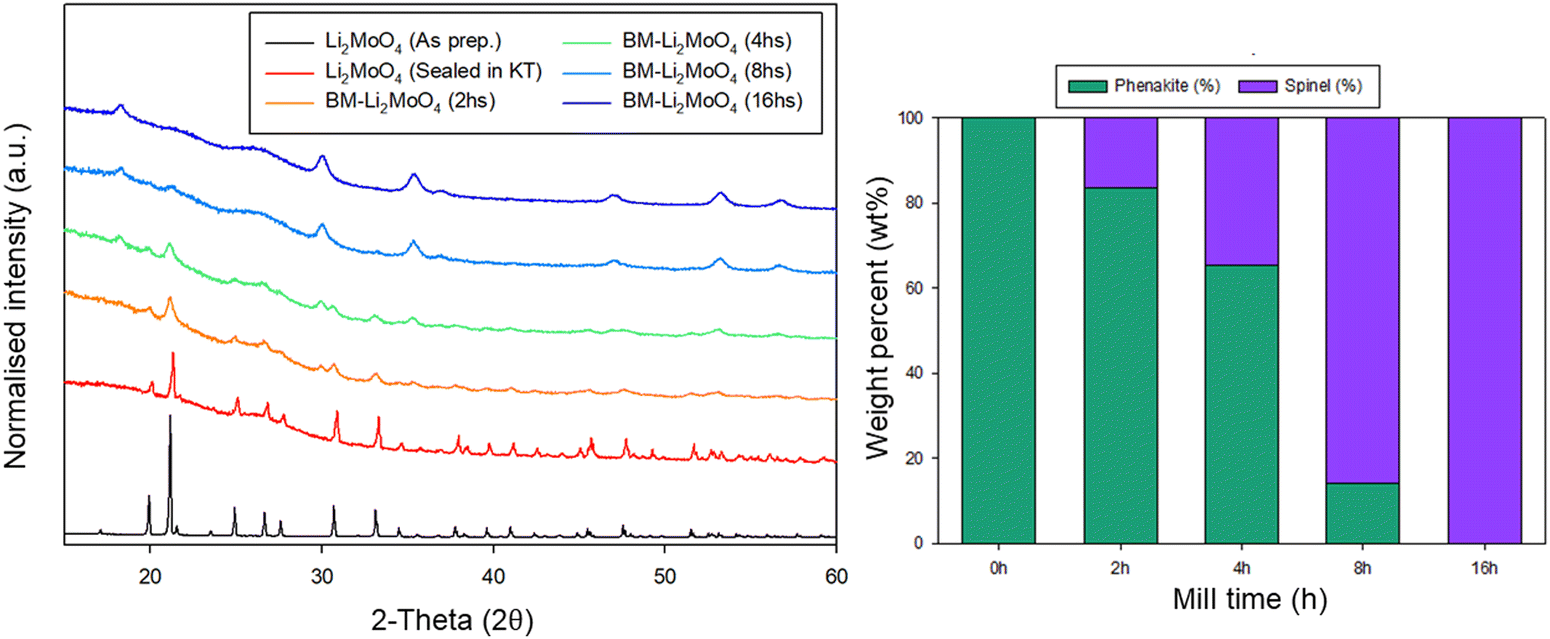 | ||
| Fig. 1 PXRD of samples prior to and after milling illustrating a transformation from phenakite to spinel structure (left), with the calculated phase fractions from Rietveld refinement (right). (Cu Kα). Note: the high background in samples after milling can be attributed to the Kapton tape (KT) used to measure the material to prevent air exposure. | ||
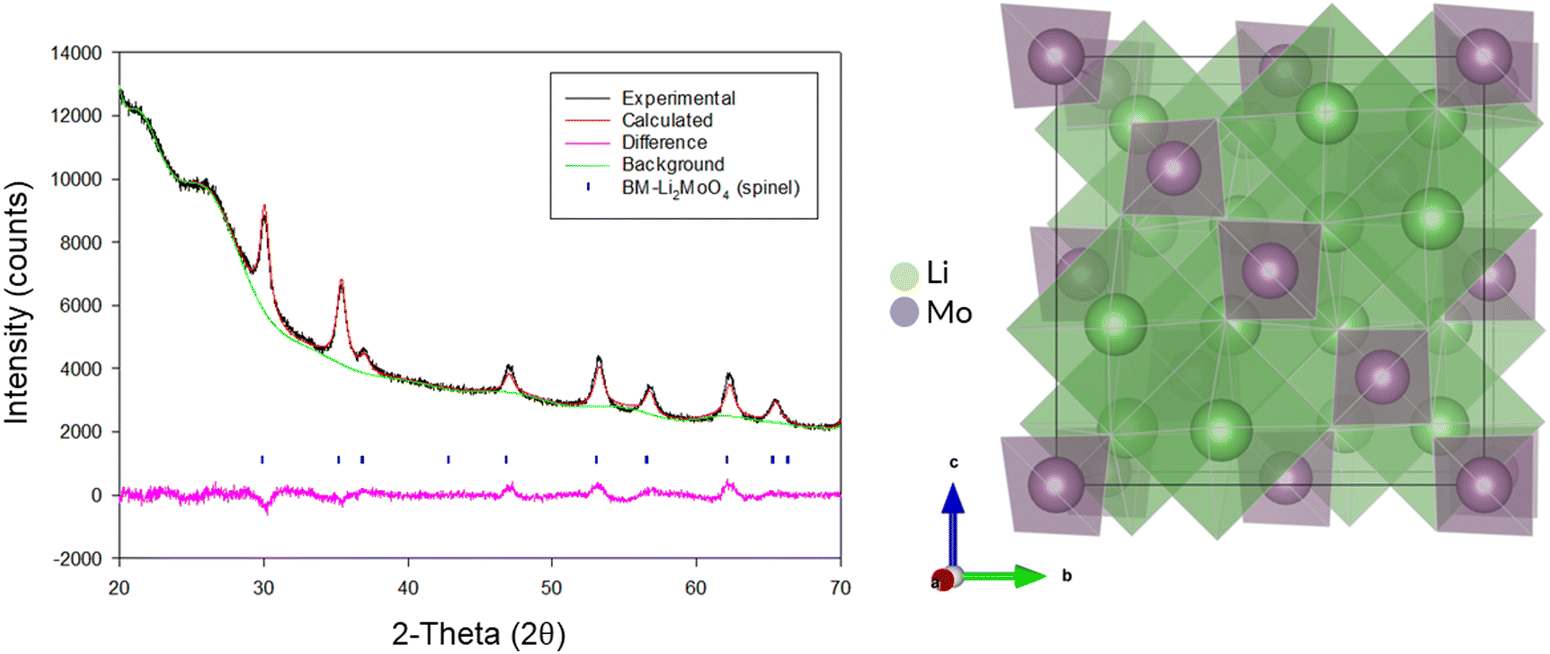 | ||
| Fig. 2 Observed, calculated and difference profiles for Rietveld refinement of BM-Li2MoO4 (16 h) (left) and an image of the spinel structure adopted by this phase (right). (Cu Kα). | ||
Spinel-Li2MoO4, cell parameters: 8.45(16) Å, space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Z = 8 m, Z = 8 |
||||||
|---|---|---|---|---|---|---|
| Site | Multiplicity | x | y | z | Occupancy | Parameter of fit |
| Li | 16 | 0.625 | 0.625 | 0.625 | 1.0 | R wp – 2.52% |
| Mo | 8 | 0 | 0 | 0 | 1.0 | R exp – 1.44% |
| O | 32 | 0.364 | 0.364 | 0.364 | 1.0 | GOF – 1.74 |
When the site occupancies are refined, the occupancies do not deviate significantly from 1.0, which suggests a highly cation-ordered phase.
In order to gather structural information, specifically relating to the local structural environment, 95Mo and 7Li NMR spectra were collected. In Fig. 3 the experimental 95Mo NMR spectra are shown, where a significant difference between as prepared (phenakite-type) and ball milled (spinel-type) Li2MoO4 is observed. The obtained NMR parameters from fitting the data are summarised in the ESI† (Fig S1).
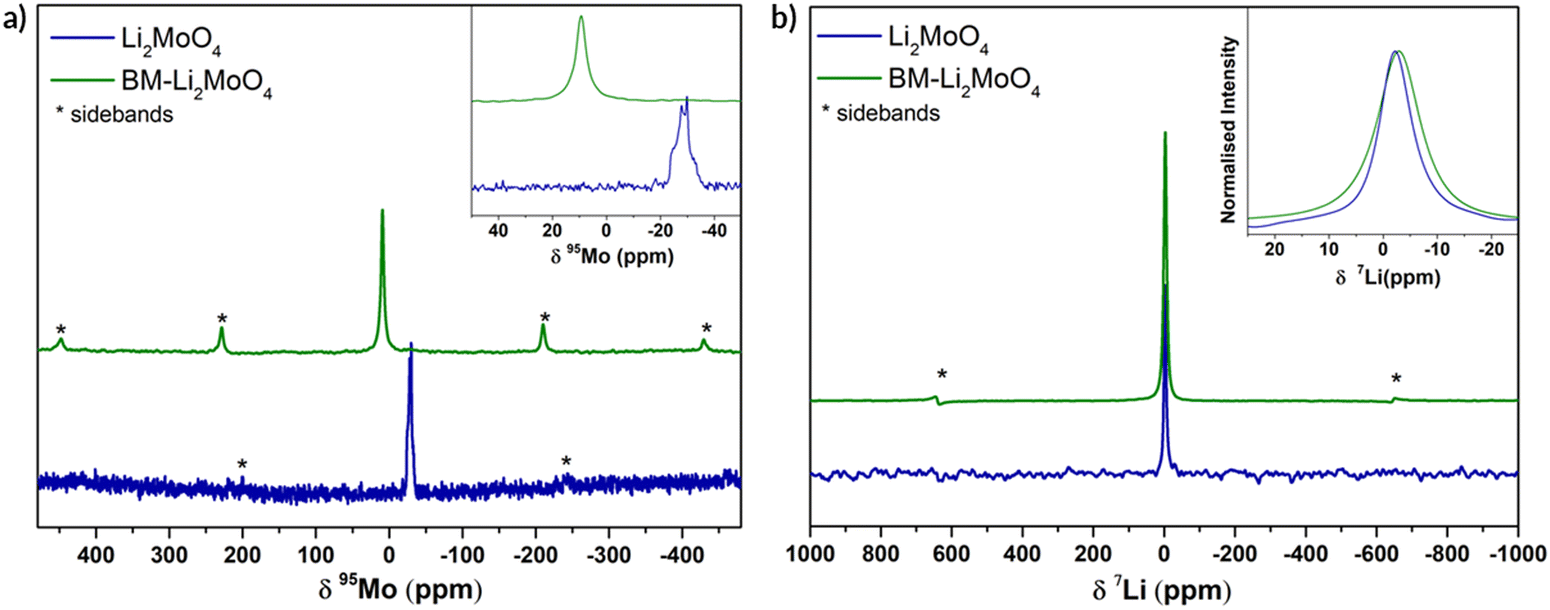 | ||
| Fig. 3 (a) 95Mo NMR for Li2MoO4 and BM-Li2MoO4 at 700 MHz (16.4 Tesla), 10 kHz MAS, single pulse spectra. (b) 7Li NMR at 200 MHz (4.7 Tesla), 50 kHz MAS, with 1 μs pulse for Li2MoO4 and BM-Li2MoO4. In both figures, inset focuses on the central transition peak. | ||
For the parent Li2MoO4 (space group R) sample, a typical second-order central transition quadrupolar (nuclear spin I = 5/2) line shape is clearly seen at −29.9 ppm, whereas for the BM-Li2MoO4 sample a symmetric line shape centred at 9.7 ppm is observed. In the samples under investigation, a change in the oxidation state of Mo upon ball milling is not expected (and subsequent TGA studies confirmed that there was no oxygen loss and hence no change in Mo oxidation state) and is unlikely to account for the differences in isotropic chemical shifts (δiso) observed in the 95Mo spectra (Fig. 3a). However, any deviation in the MoO4 tetrahedra from the ideal tetrahedral symmetry can influence the chemical shift and the quadrupolar coupling constants (CQ). The 95Mo chemical shifts observed for both the samples in our study lies within the reported chemical shifts range of AxMoO4 compositions (108 to −150 ppm).22
The extracted CQ value (ca. 1270 (± 10) kHz) and asymmetry parameter, η of 0.66, for the Mo environment (18f, site symmetry = 1) in the parent trigonal structure is consistent with the distorted tetrahedral environment with three short and one long Mo–O bonds. By contrast, the Mo (8a, site symmetry = −43 m) environment in the BM-Li2MoO4 sample is highly symmetrical (CQ is <125 (± 70) kHz) in agreement with the perfect tetrahedral environment, with four identical Mo–O bond lengths expected for a spinel; the small residual non-zero CQ is ascribed to disorder and defects in the BM sample. The lowest previously reported 95Mo CQ (340 kHz) value is for Cs2MoO4 (Mo on 4c site, symmetry =.m. in space group pmcn) where a sharp peak with no discernible second-order line shape was seen. The small CQ value was ascribed to a symmetric MoO4 tetrahedron environment. Hence, the value of CQ for BM-Li2MoO4 is lower than that observed for Cs2MoO4 and the lowest of any of Mo containing samples reported so far.23
In the 7Li NMR spectra, central transition peak positions are −2.38 and −2.97 ppm for Li2MoO4 and BM-Li2MoO4 phases. The inset in Fig. 3b shows a broadening in peak width for the ball milled sample, implying that the Li environment is more disordered. Since peak broadening can also be influenced by both Li mobility, spin–lattice relaxation time constant (T1) measurements were performed. From fitting of the data, T1s of 2500 s and 690 ms were obtained for the parent and BM-Li2MoO4 samples, respectively, suggesting that motion may contribute to the linewidth of the Li site in BM-Li2MoO4 sample.
To confirm the crystalline nature of the samples before and after ball milling, TEM measurements were conducted (Fig. 4). The observed HR-TEM fringes, not only showed both samples to be crystalline, but also allowed the interplanar distances to be indexed. These distances were found to be 7.34 Å (110) and 5.20 Å (111) for the pristine and ball-mill sample, respectively.
 | ||
| Fig. 4 TEM images of pristine (a) and ball milled (c) Li2MoO4 particles, with inset images showing the SAD pattern recorded on the particles, and the respective HR-TEM images (b and d) clearly showing the lattice fringes. The observed interplanar distance from the fringes have been indexed. | ||
SEM images were also conducted on both samples (Fig. 5). The pristine Li2MoO4 shows generally a uniform shape with an average particle size of 11 (3) μm, while the ball-milled version loses this uniformity and presents a mixture of sizes – likely due to fragmentation of particles to produce finer particles which agglomerate into larger entities.
 | ||
| Fig. 5 SEM images of Li2MoO4 – pristine (a) and ball milled (b). | ||
This phase conversion was also reproduced by processing in an alternative mill: P23 vibratory ball mill; this mill operates at an oscillating amplitude of 0.9 mm with a maximum velocity of 50 Hz (<1 ms−1). Remarkably, despite the significantly reduced input energy per impact offered by the P23, we found that the phase transformation of Li2MoO4 from its phenakite to spinel form occurs much more quickly as compared with high energy planetary ball milling (Fig. 6). When milling at 50 Hz with a 10 mm ball, first signs of the phase transformation appear in the PXRD profiles within 5 min of ball milling, with no signs of the phenakite phase being present after only 10 min milling. The conversion rate was reduced slightly by reducing either the frequency of the mill (to 40 Hz) or the size of the milling ball (to 7 mm). However, even at the lowest energy (40 Hz with 7 mm ball), complete conversion to the spinel phase was observed with 120 min of milling (see ESI,† Table S1).
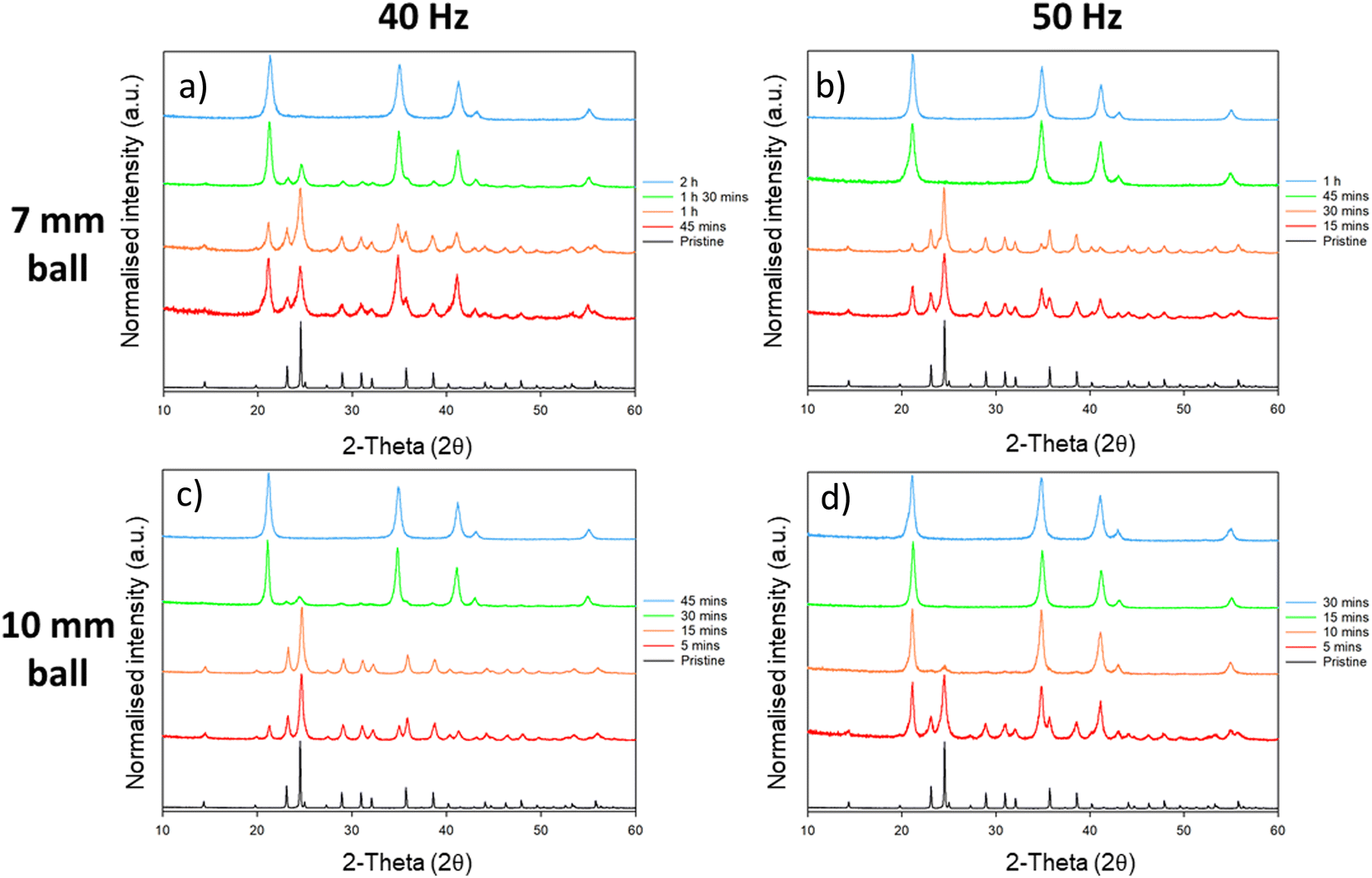 | ||
| Fig. 6 Resulting PXRD patterns from the study of ball mill frequency, time and ball size on the milling of Li2MoO4. Patterns (a) and (b) made use of a 7 mm ball at 40 Hz and 50 Hz, respectively. Patterns (c) and (d) made use of a 10 mm ball at 40 Hz and 50 Hz respectively. The conversion rate was shown to significantly reduce with lower frequency or ball size. (Co Kα). 50 mg of material was loaded into the pots in each case. | ||
We suspect that the unexpected rate enhancement offered by the P23 results from the low level of bulk heating that accompanies this low energy milling process. Our previous studies on P23 show that even for higher energy experiments (i.e. using a 15 mm ball at 50 Hz), milling jar temperatures do not exceed 320 K, even when milling is sustained for hours.24 We therefore propose that the slow transformation observed by planetary ball milling results from the competition between the mechanically-drive forward transformation and the thermally driven reverse transformation. In contrast, the latter process does not occur in the low energy P23.
Given the successful structural transformation by ball milling, as illustrated by the XRD and NMR data, electrochemical characterisation of this new spinel-type Li2MoO4 was then performed using Swagelok powder cells to evaluate its performance. A cell was initially cycled in the region of 4.8–1.0 V to assess where, if any, redox processes were likely to occur. The cell shows a relatively small charge capacity (ca. 70 mA h g−1) when charged to 4.8 V (Fig. 7a). On discharging to the lower voltages (1 V), additional capacity (up to an additional ca. 160 mA h g−1 (Fig. 7a) giving a total discharge capacity of ca. 230 mA h g−1) is obtained with two plateau regions in the voltage profile, which correspond to two distinct peaks in the dQ/dV data (occurring at 1.55 and 1.26 V respectively; see Fig. 7b). These data suggest additional lithium incorporation leading to Mo reduction (from Mo6+ → Mo4+) with decreasing voltage. Dismantling the cells discharged to these low voltages showed additional peaks attributed to a rocksalt phase, consistent with the work of Ehrenberg et al., which demonstrated the formation of a DRS-type (Li3Mo)O4 from Li2MoO4via chemical lithiation using n-butyllithium in hexane in a glovebox.20
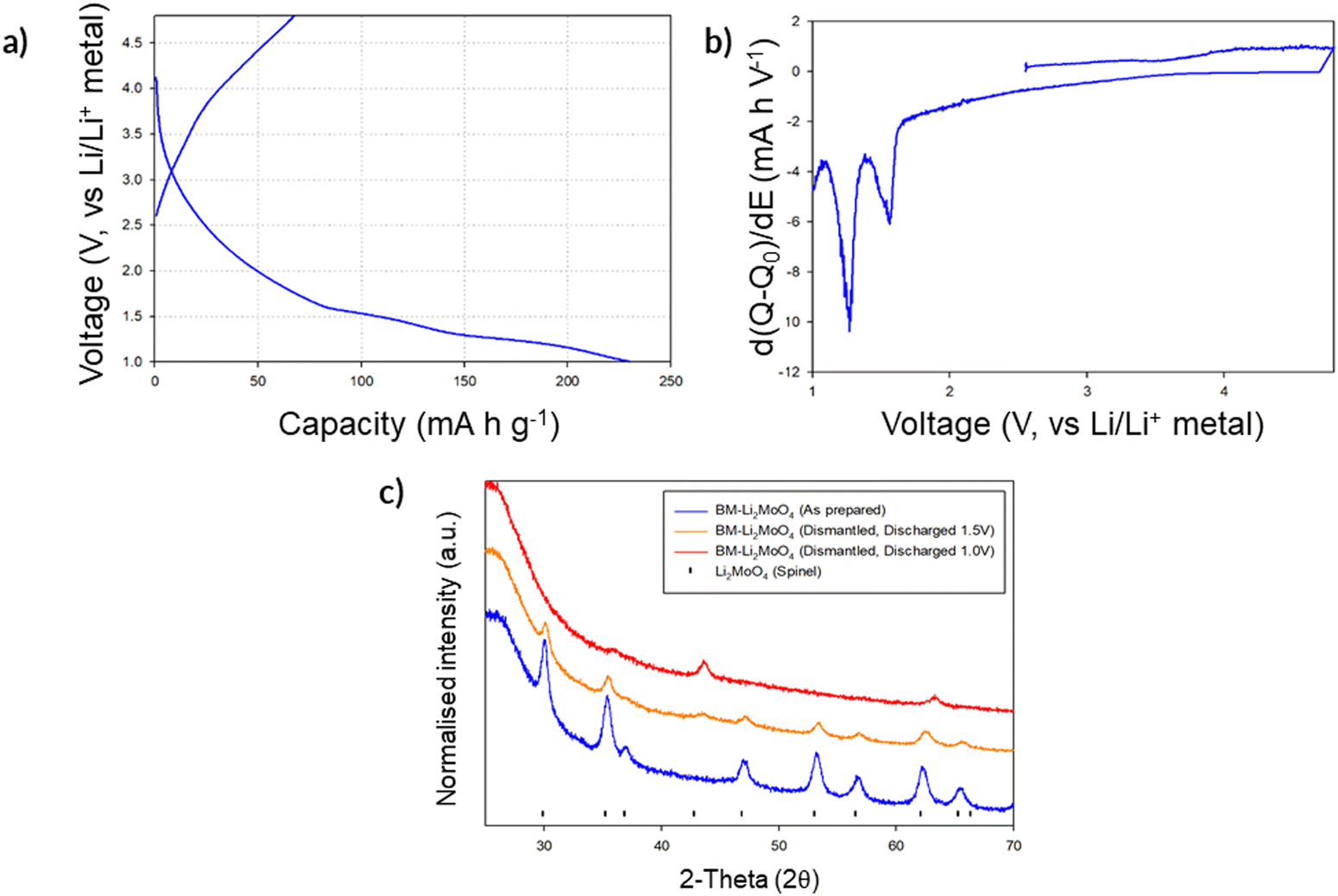 | ||
| Fig. 7 (a) First cycle voltage profile for BM-Li2MoO4 cycled in the region of 4.8–1.0 V at 10 mA g−1; (b) associated dQ/dV plot of the first charge/discharge cycle; (c) PXRD of dismantled swagelok cells showing phases formed once discharged down to 1.5 and 1.0 V (Cu Kα). | ||
Due to the appearance of this secondary DRS phase at low voltages (Fig. 7c), additional cell tests were performed, both examining reducing the voltage range to eliminate its occurrence and expanding the voltage range to examine the maximum extra Li that could be inserted: thus, cells were also tested in the ranges of 4.8–2.0 V, 4.8–1.5 V, and 3.0–1.0 V to evaluate the performance of the spinel vs. DRS phases. The corresponding cell data are shown in Fig. 8. Cells cycled with the upper cut off voltage maintained the spinel structure and achieved a small reversible charge–discharge capacity of ca. 60–70 mA h g−1. This equates to a reversible extraction of ∼0.5 mole of Li from BM-Li2MoO4, which can be attributed to anion redox (as Mo will not oxidise beyond Mo6+). Further lowering of the discharge voltage increases the discharge capacity of the material (due to conversion reactions); however, the cells performance degrades substantially as this cut off voltage was lowered, e.g. cells discharged to 0.5 V deliver an initial high capacity ca. 500 mA h g−1; however, only 180 mA h g−1 can be achieved after 5 cycles (see ESI,† Fig. S2). Regardless of the selected voltage range, all cells show a significant decrease in performance with repeated cycling. In some cases when the cells were dismantled, the separator appeared discoloured (pale blue), which may suggest that dissolution of the cathode material may be the origin of some of this capacity fade.
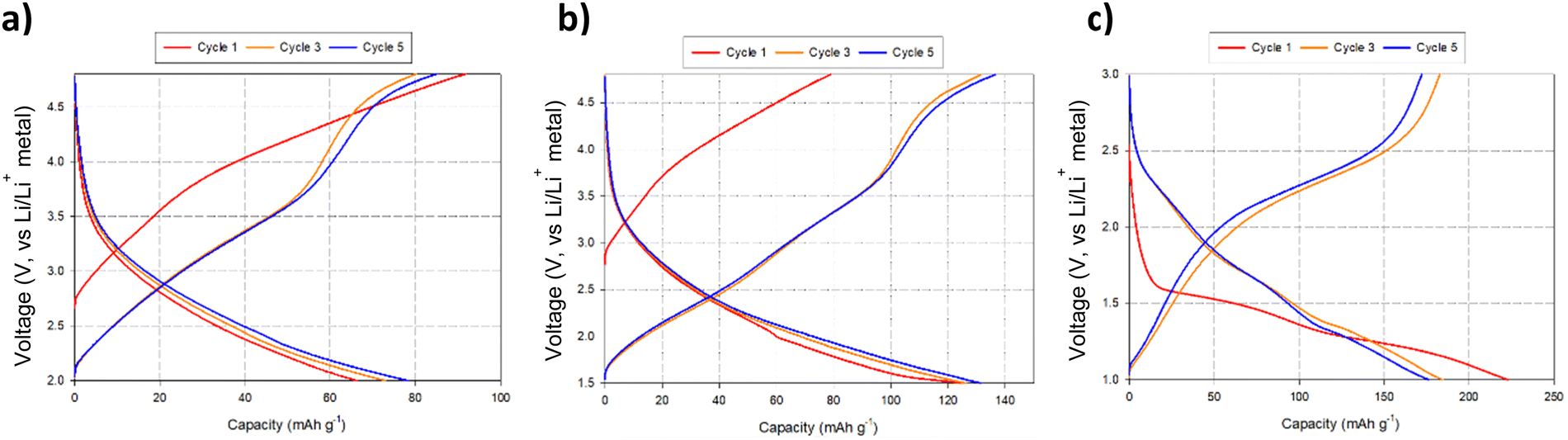 | ||
| Fig. 8 Voltage profiles for BM-Li2MoO4 using varying voltage ranges: (a) 4.8–2.0 V, (b) 4.8–1.5 and (c) 3.0–1.0 V. Note: cell c was discharged first before charging to higher voltages. | ||
A further interesting observation regarding this newly observed spinel phase was that for the planetary ball milled sample it was shown to have limited stability once removed from the glovebox. ESI,† Fig. S3 illustrates the spinel phase gradually reverting back to the original phenakite host phase on exposure to an ambient air atmosphere, the start of the conversion back to phenakite happens with minutes, although the rate of degradation then slows. Somewhat surprisingly the P23 ball milled sample appeared to be stable towards air. This fact, and the fact that the rate of degradation of the planetary ball milled sample slows with time would suggest that the transformation is not a bulk process, but perhaps a moisture-surface catalysed process which is more rapid for the planetary ball milled sample.
Overall, the work therefore shows that milling Li2MoO4 leads to a structural transformation from phenakite to a spinel phase; the electrochemistry of the spinel is rather complex, with a small capacity (ca. 60–70 mA h g−1) on delithiation attributed to O redox in the higher voltage ranges (above 2 V) and additional capacity at lower voltages attributed to Mo redox, which also drives a transformation towards a DRS structure. Due to the instability and the significant capacity fade observed on cycling, it is unlikely that the spinel Li2MoO4 will have a practical use within electrochemical storage. However, given the interesting fact that the milling process was able to form this new metastable spinel variant of Li2MoO4, we decided to investigate further the possible reasons for the phase transformation on milling. In this respect, we compared the cell volume per formula unit for as prepared phenakite Li2MoO4 to the spinel phase to see whether this might provide some information as to why the transformation occurred on milling.
This comparison shows a significant change in the volume per formula unit between the two structures. The original phenakite phase possesses a volume per formula unit of 94.7 Å3, whereas the new spinel phase equates to a value of 74.4 Å3, indicating a volume reduction of 21%, thus illustrating that the milling process leads to the formation of a denser phase. In order to try to explain this observation, the literature was explored to find any references to a spinel polymorph of Li2MoO4. This search indicated that the spinel phase was a high-pressure polymorph. In this respect, Liebertz et al. theorised in 1967 that spinel-type Li2MoO4 cannot be obtained at normal pressures up to the melting point of phenakite (∼700 °C).25 The authors suggested that a pressure of 10 kbar and a temperature of 400 °C were required to induce the transition from phenakite to spinel. The cell parameter and the subsequent calculated volume reduction reported in this work (a = 8.434 Å, 21% volume reduction) are in good agreement with the results from our milling study.16 A later study from the 80's, conducted by Yamaoka et al., successfully synthesised the spinel Li2MoO4 phase using pressures of 4 GPa at temperatures of 500 °C.15 Here it was shown that the ΔS for the transition from phenakite to spinel is negative, which indicates that the spinel cannot be a stable high temperature polymorph of Li2MoO4. Thus, both these prior studies support the postulate that the transition induced by ball milling is not simply caused by local heating as it has been suggested in the literature on ball milling of Li-ion battery materials. Rather this work suggests that local pressure effects are vital. These local pressure effects can effectively be produced from the impacts generated as the balls strike the powder.26 Moreover, the resultant “high pressure” phase then can readily transform back to the parent phase in ambient air, presumably facilitated by the presence of moisture (hence the greater stability within a glove box environment). While it is unlikely that local pressures reach ca. 4 GPa under ball milling conditions – especially within the low energy Pulverisette mill – it is known that phase transitions can occur at significantly lower pressures under uniaxial compression.27 This owes to the additional shear component associated with anisotropic compression, and is consistent with the force loading expected under ball milling conditions. Moreover, the presence of structural defects (including surface energy associated with particle comminution) accumulated during ball milling is also known to depress thermodynamic transition conditions.26,28,29 We therefore expect that dedicated studies on uniaxial compression will offer exceptional new insight into ball milling transformations and allow us to better understand how to design mechanochemical methods to enhance battery material behaviour.
While the electrochemical performance of spinel-type Li2MoO4 was insufficient for Li-ion battery cathode applications, the fact that ball milling led to the formation of this high-pressure polymorph is a fascinating result.28,30 In particular, it raises important information on the milling process. Specifically, it suggests that the milling process is giving rise to both local heating and pressure effects, and that the latter may be the missing variable that accounts for why some systems (e.g. many DRS Li-ion battery materials) can only be prepared by this route. In order to attempt to provide further evidence for this postulate, we decided to study other phase transformations by ball milling. Two additional systems were studied: (1). The layered to DRS phase transition in Li2MO3 (M = Mn, Sn); (2). Possible phase transformation from milling Nb2O5.
Milling of Li2MnO3 and Li2SnO3
Given the large interest in DRS electrode materials formed by ball milling, we first investigated the phase transformation of Li2MnO3 from layered structure into DRS. Freire et al. have previously demonstrated that milled Li2MnO3 exhibits improved electrochemical performance (delivering an initial discharge capacity of 290 mA h g−1), which is credited to nanosizing of the material and the conversion to the DRS structure.31,32 Therefore, in order to investigate whether similar local pressure effects may apply to the formation of this DRS phase on milling, we examined the effect of ball milling Li2MnO3 (layered structure: C2/m – space group 12). This phase was therefore ball milled at 450 rpm for a total of 16 h. After each step, a small amount of powder was removed in order to characterise the phases formed (Fig. 9). Although there is extensive overlap between the two phases, the loss of the intense (001) peak at ∼18 (2θ) as well as the loss of other peaks pertaining to the layered phase, in conjunction with the relative increase in the intensity of the peak at ∼45 (2θ), suggests the successful formation of a DRS phase between 8–16 h of milling.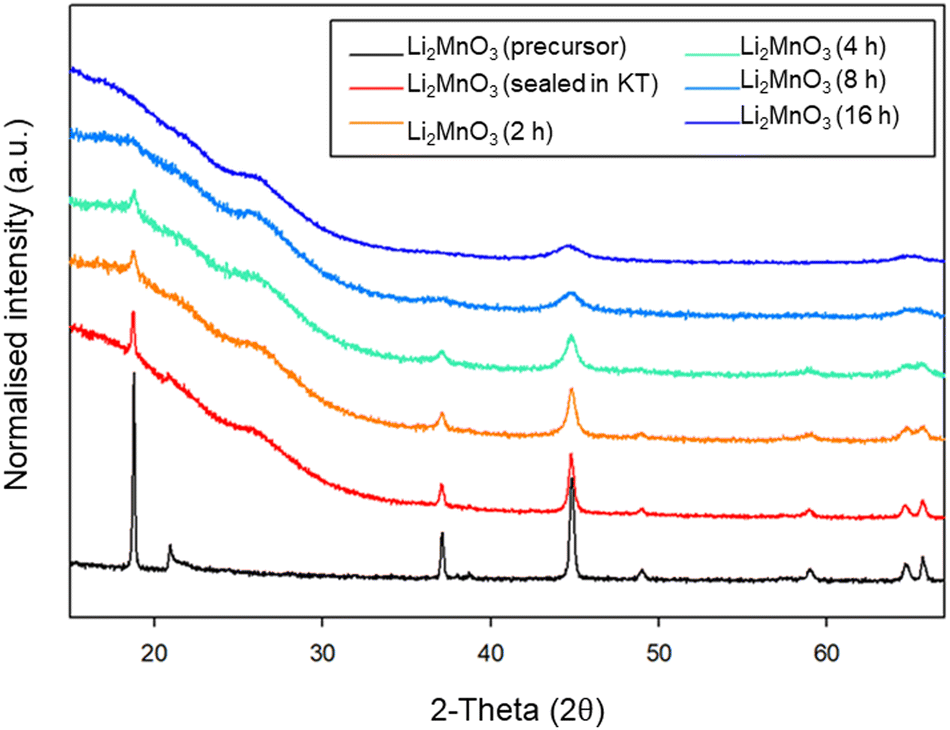 | ||
| Fig. 9 PXRD data for the timed milling study of Li2MnO3 (2–16 h at 450 rpm) showing a transition from a layered to DRS structure (Cu Kα). | ||
Due to the extremely weak and broad diffraction data for the produced DRS, Rietveld refinement was only performed to confirm the overall structure and determine cell parameters (Fig. 10). The occupancies for each of the sites was fixed based on the initial stoichiometry of the parent Li2MnO3 phase. Structural refinement resulted in a cell parameter of 4.06(4) Å (Table 3), which is consistent with other reports of DRS phases synthesised through this route in the literature.3,5,21
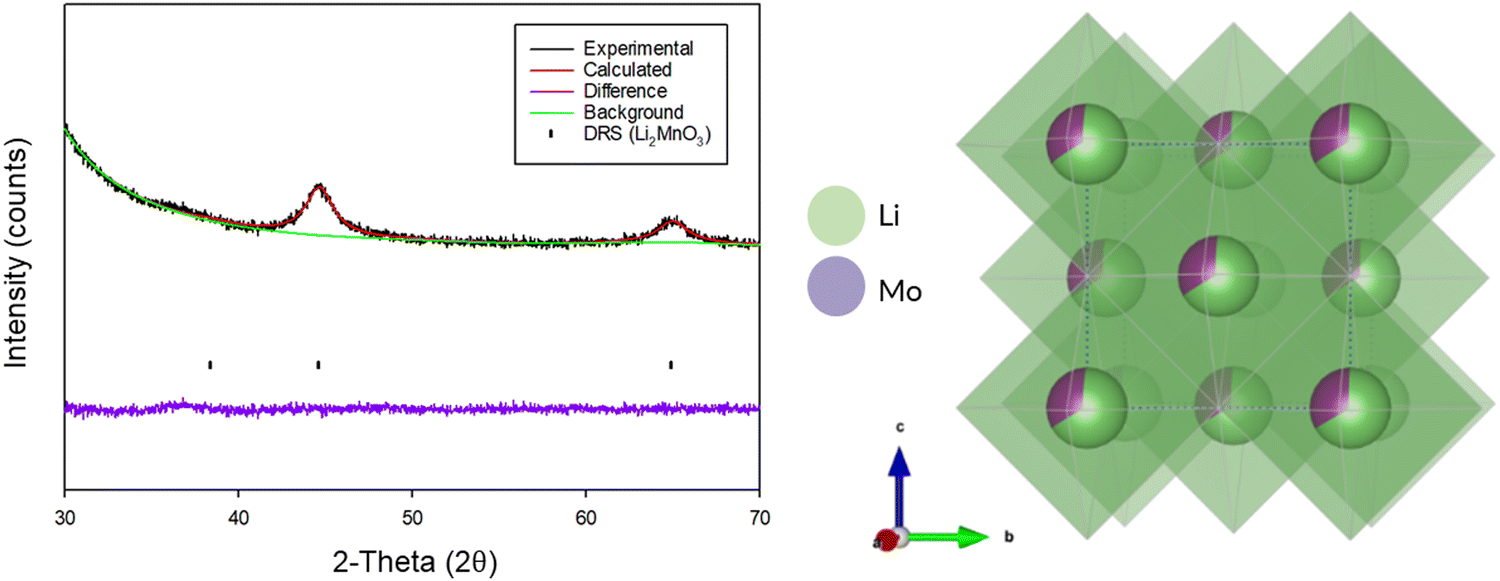 | ||
| Fig. 10 Observed, calculated and difference plot from the Rietveld refinement of BM-Li2MnO3 after 16 h (Cu Kα) and an image of the crystal structure. | ||
| DRS-Li2MnO3, cell parameters: 4.06(4) Å, space group: Fmm, Z = 2 |
|||||
|---|---|---|---|---|---|
| Site | x | y | z | Occupancy | Parameter of fit |
| Li | 0 | 0 | 0 | 0.67 | R exp – 1.179% |
| Mn | 0 | 0 | 0 | 0.33 | R wp – 3.897% |
| O | 0.5 | 0.5 | 0.5 | 1.0 | GOF – 3.303 |
In order to gain further evidence regarding the stability of the observed structural change to DRS, this DRS-Li2MnO3 was studied by variable temperature (VT) XRD. Should only local heating effects be occurring during the milling process, it should be predicted that no change in structure is observed on heating. The VT diffraction data from this study are shown in Fig. 11. When heated, the DRS structure is mostly maintained until ca. 500 °C. However, above this temperature, the intensity attributed to the (001) reflection for layered Li2MnO3 starts to grow with increasing temperature, along with the appearance of additional peaks pertaining to the original layered phase, albeit with some intensity differences compared to the original sample prior to milling (most likely related to different levels of Li/Mn order). This suggests that, although the long range layered structure has been restored, some disorder remains. This conversion of DRS-Li2MnO3 back to layered-Li2MnO3 on heating further reinforces that additional effects must be occurring in the milling process alongside local heating.
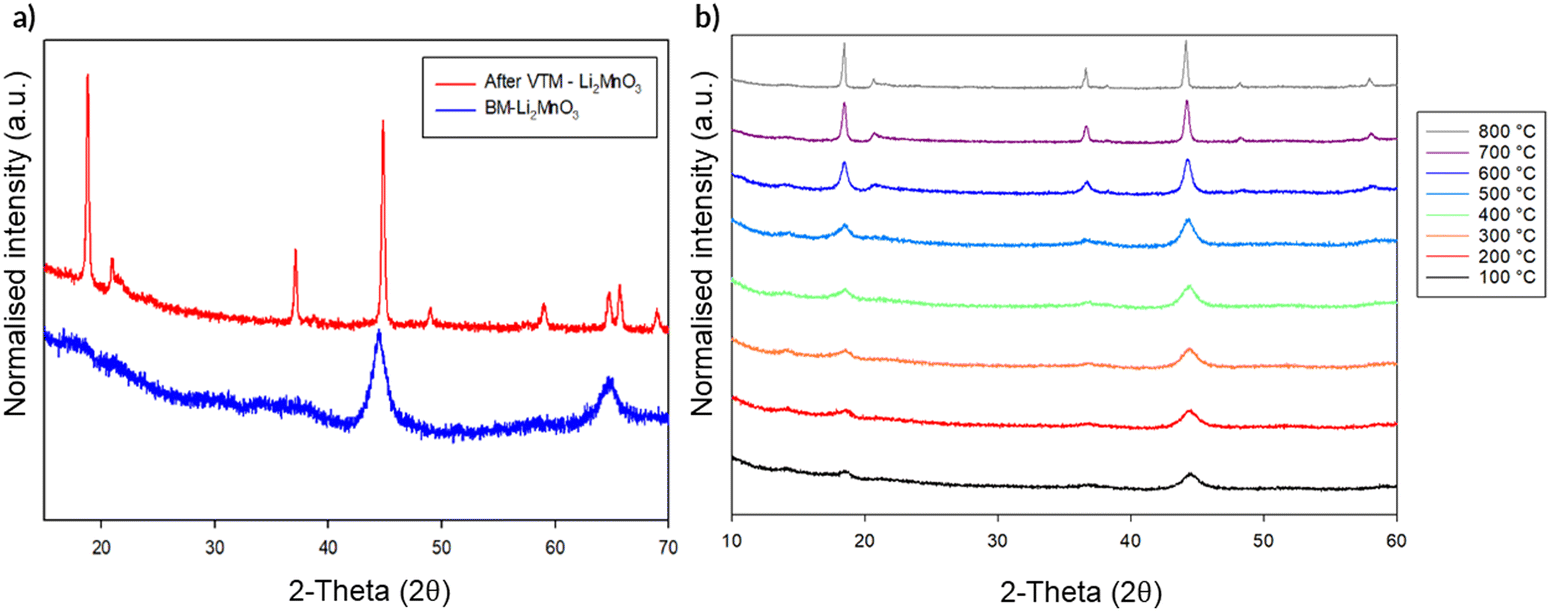 | ||
| Fig. 11 (a) PXRD of BM-Li2MnO3 sample before (DRS phase) and after VT-PXRD (layered phase) (b) VT-PXRD experiment in the region of 100–800 °C showing the change in the relative intensity of peaks with increasing temperature indicating a transformation from DRS to layered on heating. (Cu Kα). | ||
In order to assess whether densification of the material, similar to that observed in Li2MoO4, has occurred, the volume per formula unit for Li2MnO3 was calculated for the phase prior to and after heating. The DRS (25 °C) phase gave a value of 50.24(15) Å3 per formula unit, whereas the resultant Li2MnO3 after the VT experiment (25 °C) was 49.81 (1)Å3 per formula unit, which was somewhat surprisingly lower than the milled phase, thus opposite to what was expected. However, given that Mn could be reduced on milling, a TGA study was conducted on the sample milled for 16 h (see ESI,† Fig. S4). When this sample was heated in oxygen (700 °C), the sample shows a large mass gain of ca. 8%, suggesting that the original sample was oxygen deficient: Li2MnO3−x. Thus, the results suggest that in this case milling leads to oxygen loss (and hence the reduction in oxidation state of the Mn). Given the different oxygen contents between as-synthesised Li2MnO3 and the ball milled sample, and the fact that the latter therefore contains some Mn3+, which is larger than Mn4+, it is therefore not possible to confirm the postulated local pressure effects on milling as for Li2MoO4. Therefore, a further system, Li2SnO3, where no change in oxidation state was expected, was studied. The phases formed during milling and the resultant VT-PXRD data are shown in Fig. 12. As for Li2MnO3, Li2SnO3 shows a structural transformation from layered to DRS. For this Sn system, the volume per formula unit of the layered phase equates to 60.11(4) Å3, whereas the newly formed DRS yields a value of 59.54(8) Å3, indicating that a slight densification has occurred. As for the Mn system, the thermal stability of the DRS Li2SnO3 phase was investigated by VT-XRD. The results showed that the DRS is maintained for a wide temperature range until ∼650 °C, when peaks attributed to the precursor layered phase start to reappear. This observation, therefore, confirms that the DRS phase is not stable on heating to elevated temperatures. Thus, the observation of the reappearance of the layered phase in ball milled Li2MnO3 and Li2SnO3 after heating adds further evidence that the ball milling process is not simply inducing local heating of the material. In this case, the exact mechanism for phase transformation may be more complicated, owing to potential oxidation state variation and oxygen loss, particularly for the Mn system. However, the preliminary results suggest that thermal effects alone cannot be responsible for the production of these particular DRS phases through milling and the data for the Li2SnO3 system are consistent with the transformation to a denser structure.
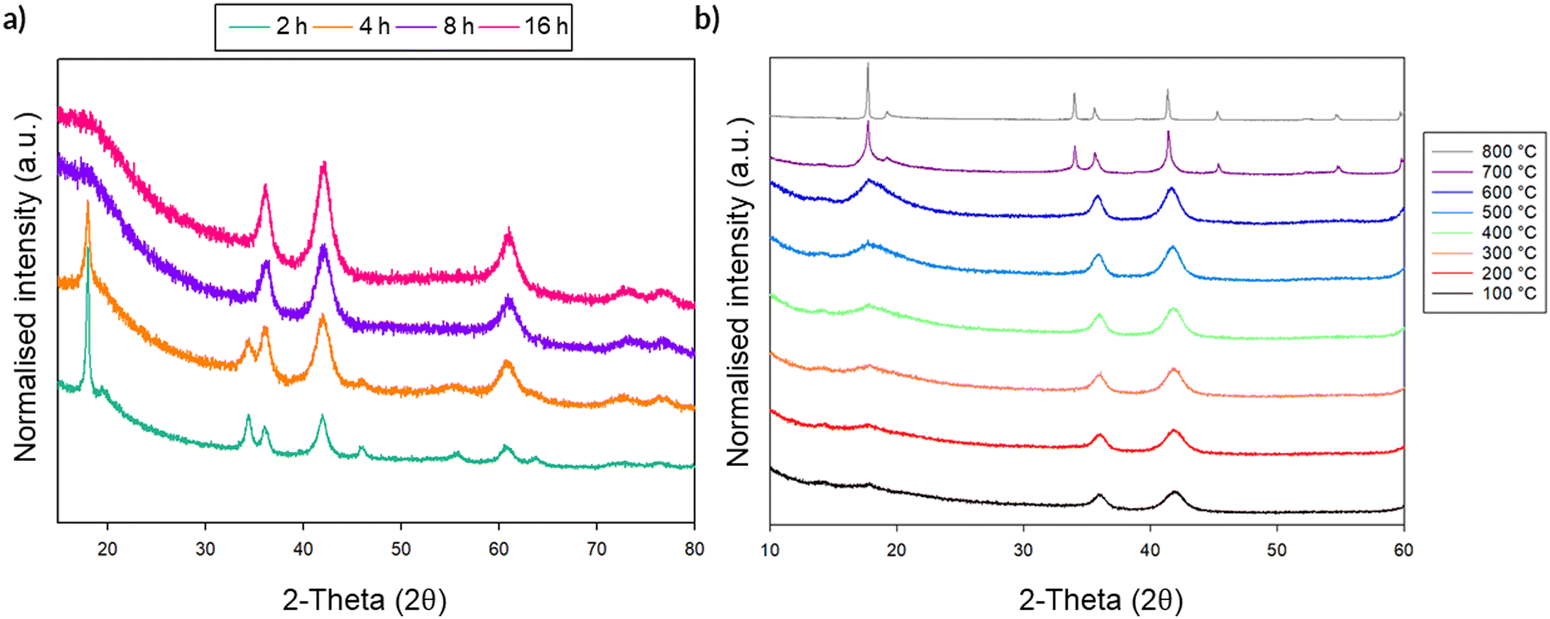 | ||
| Fig. 12 (a) PXRD of Li2SnO3 with increasing mill time. Conversion to DRS occurs at ∼8 h (b) Plot showing the changes in PXRD pattern with increasing temperature, showing the transformation from DRS to layered on heating. (Cu Kα). | ||
As with the Li2MoO4 system to confirm the crystallinity and evaluate the morphology during the ball-mill process, imaging techniques were conducted for Li2MnO3 and Li2SnO3, which are presented ESI,† Fig. S5–S7. SEM images were taken for the pristine and ball-milled samples, and again the uniform particles are shown to be lost to produce fine particles and agglomerates on milling. TEM imaging was carried out on the ball-milled samples only; a mixture of amorphous and crystalline regions are noted – thus indexing the diffraction fringes was found to be quite challenging. The electrochemical data for the Mn system, and comments on the Sn, can be found in (ESI,† Fig. S8).
Milling of Nb2O5
Given the previous results on the DRS Li2MnO3−x systems, where oxygen loss features complicated the interpretation of the mechanistic features of the milling process, another system, Nb2O5, which has an open structure like phenakite-type Li2MoO4, was investigated. Nb-based oxides have come under the spotlight recently for application as high-powered anode materials, with significant capacity retention at high charge rates, along with observed capacities being higher than other high-power anodes, such as lithium titanate. Undoped Nb2O5 was chosen as it has numerous polymorphs,20–23 and so was expected to yield a similar phase transformation on milling.The focus for this case study was T-Nb2O5 (with orthorhombic symmetry: the low temperature polymorph) and H-Nb2O5 (with monoclinic symmetry; the high temperature polymorph).
The T-Nb2O5 structure is composed of highly distorted octahedral and pentagonal bipyramidal Nb–O sites, while the H-Nb2O5 phase consists of ReO3-like blocks of octahedra and adopts one of the Wadsley–Roth family of crystallographic shear structures (3 × 4 block) (Fig. 13).33
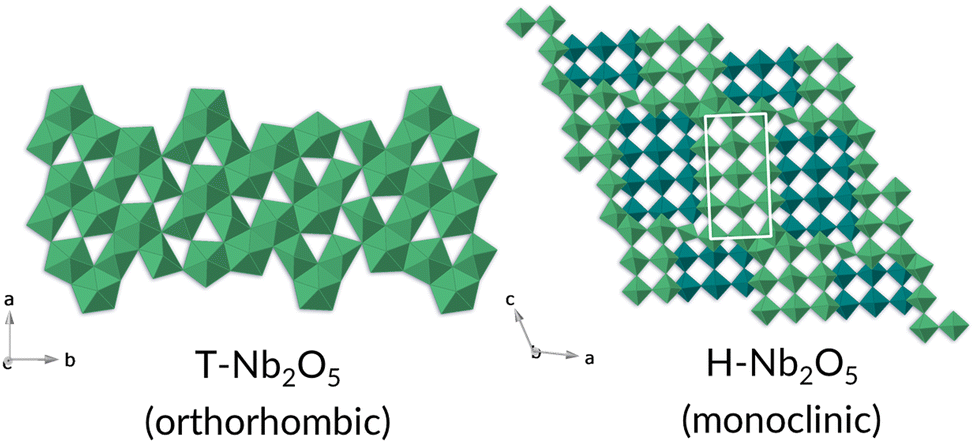 | ||
| Fig. 13 Crystal structures of T- and H-Nb2O5 phases, left and right respectively. The Nb is represented by the polyhedra; for simplicity the oxygens have been omitted. | ||
The H-Nb2O5 phase can be synthesised through heating of the T-Nb2O5 to high temperature and is very stable once formed; it does not transform back to T-Nb2O5 on cooling or by annealing at low temperatures.24
The initial milling studies using the Fritsch premium line 7 planetary ball mill showed that, while there was no change in structure on milling T-Nb2O5, a noticeable phase change was observed on milling H-Nb2O5 (Fig. 14a). After ball milling for a relatively brief period (700 rpm, 2 h), the high temperature polymorph of H-Nb2O5 reverts back to the low temperature T-Nb2O5 polymorph. The unit cell parameters are shown in Table 4 for these phases and for the ball-milled phase.
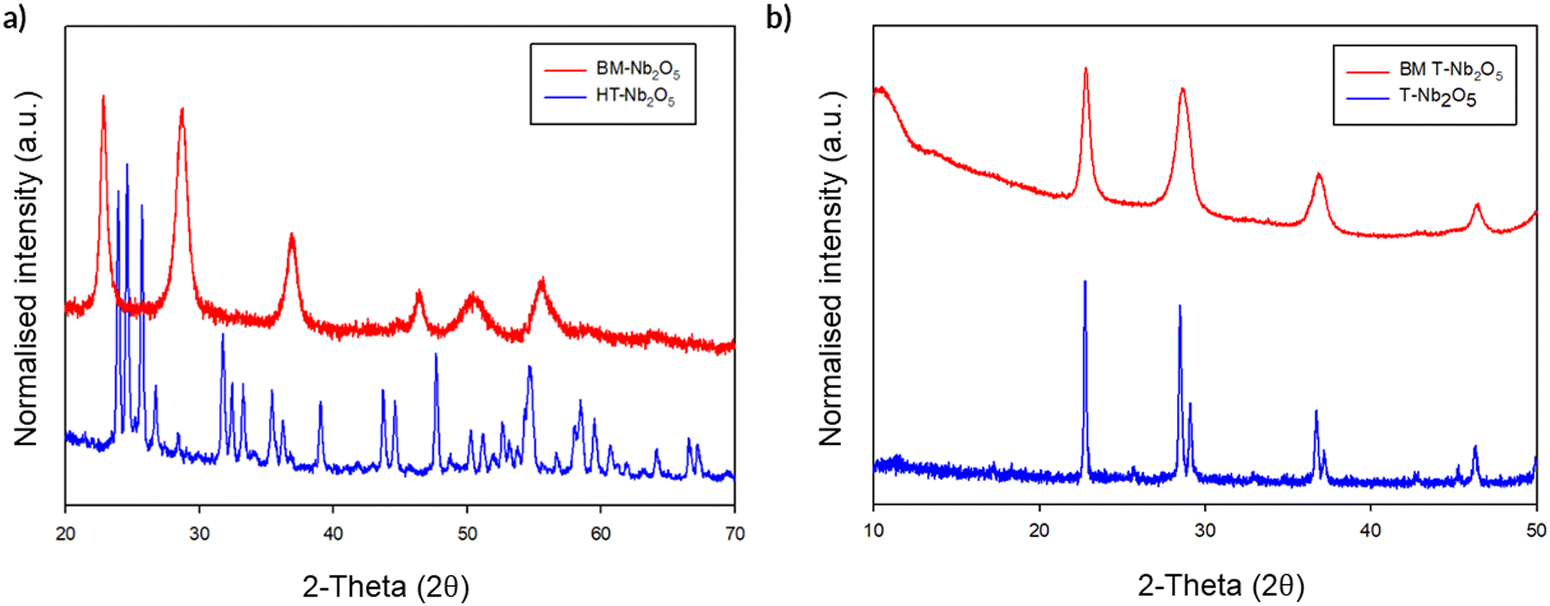 | ||
| Fig. 14 (a) High temperature (HT) phase – H-Nb2O5 before and after milling (BM) showing a phase conversion to T-Nb2O5. (b) Low temperature phase (T-Nb2O5) before and after milling showing no change in structure, just broadening of the peaks due to the smaller particle size after milling. (Cu Kα). | ||
| Composition | Crystal symmetry | a (Å) | b (Å) | c (Å) | β (°) | Cell volume (Å3) |
|---|---|---|---|---|---|---|
| T-Nb2O5 | Orthorhombic (Pbma) | 6.220(2) | 29.254(7) | 3.948(2) | 90.0 | 718.3(5) |
| H-Nb2O5 | Monoclinic (P 1 2 1) | 21.166(7) | 3.8229(9) | 19.148(5) | 118.03(1) | 1367.7(7) |
| 700 rpm | Orthorhombic (Pbma) | 6.24(1) | 29.19(8) | 3.93(1) | 90.0 | 716(3) |
This transition from H-Nb2O5 to T-Nb2O5 on milling is surprising, since previous detailed studies on the transitions between different polymorphs of Nb2O5 suggest that once the H-Nb2O5 structure has formed by heating to high temperatures, the change should be irreversible.34 Furthermore, the fact that T-Nb2O5 is not converted to H-Nb2O5 on milling reinforces that the milling process is not simply causing local heating, as otherwise such a phase transformation might be expected. Given these observations, we then considered our local pressure postulate and what might be the effect of such local pressure effects during the milling process. In this respect, if one investigates the effect of pressure on these phase transitions, there are reports that H-Nb2O5 can transform to either B-Nb2O5 or T-Nb2O5 with increasing temperature and pressure, with the T-phase being the more stable phase under high pressure/temperature conditions.35 With regards to this, Zibrov et al. conducted a temperature/pressure study which suggests that H-Nb2O5 converts T-Nb2O5 at either 1250 °C/7.5 GPa or 1300 °C/8.0 GPa.36 The driving force for this process is the fact that the T-Nb2O5 phase is denser than H-Nb2O5 (the volume per formula unit for the as-prepared H-Nb2O5 and the resultant T-Nb2O5 phase after milling are 97.7 Å3 and 85.5 Å3, respectively, corresponding to a 14% reduction on milling). Thus, for these examples, both the fact that H-Nb2O5 (high temperature polymorph) converts to T-Nb2O5 (low temperature polymorph) on milling, and under the same conditions the reverse (T to H phase) does not happen, provides strong evidence for local pressure effects during the milling process.
This phase transition phenomenon was also reproduced on P23 mill, with a time-frequency study presented in the (ESI,† Fig. S9 and S10). The full conversion to the T-phase was observed to occur within 30 mins and 1 h for frequencies 50 Hz and 40 Hz, respectively. The morphology change of the rod-like character of H-Nb2O5 to agglomerates for the 50 Hz study are also presented in ESI,† Fig. S11.
Discussion
Prior work on ball milling of Li-ion battery materials, e.g. the formation of DRS systems, have generally assumed that the process is simply facilitated by local heating effects when the milling balls strike the powders. Following our observation of the transformation of Li2MoO4 to the high-pressure spinel polymorph, and subsequent studies on Li2MO3 (Mn, Sn) and Nb2O5 systems, we have provided clear evidence that local heating alone cannot explain these structural changes, but rather other effects are important. In particular, we provide evidence in support of the importance of local pressure effects, which is clearly supported by the observed changes for materials with open structures (as in the Li2MoO4 and Nb2O5 examples), where a transformation to a much denser structure is observed. Our theory regarding this is that when the milling balls collide with the material, shockwaves are generated, exerting a localised pressure, in addition to local heating. For the layered material to DRS examples (Li2MnO3 and Li2SnO3), the results for Li2MnO3 are complicated by the milling process leading to oxygen loss, while for Li2SnO3 the results are consistent with the above conclusions, with the DRS phase having a slightly smaller volume per formula unit than the layered phase. In both cases, there is confirmation that local heating effects alone cannot explain the observed phase transformation, since we have shown that heating these samples to higher temperatures leads to a transformation from DRS back to the layered structure. For these layered to DRS changes, we believe that the denser structure for the starting materials means that the effect of local pressure is more subtle, but nevertheless present.Given the growing interest in this route as a way of synthesising new high-capacity Li-ion battery electrode materials, these observations are significant for helping to clarify why these phase transitions occur. In particular, there is a need to design routes to make such high-capacity materials via more scalable routes (the ball milling process is typically limited to small sample size, and long milling times), so that they could potentially be commercialised. The new knowledge on the importance of local pressure effects leads to the potential to design strategies to make such materials via more scalable solid state reaction routes: for example, we can induce such transformations by introducing dopants that create internal strain (chemical pressure), and indeed this could be a factor in some of the DRS materials that can be prepared at elevated temperature by standard solid state synthesis. With respect to this, a classic example can be found in the superconductivity field for Nd2CuO4 which can be transformed from the T′ structure to the denser T/O structure on application of pressure (21.5 GPa), while this phase transition can be also achieved through replacing Nd with La to create internal chemical pressure: here the introduction of the larger La in place of Nd increases the internal strain within the fluorite layers (T′ structure), such that at a critical value, the structure transforms to the T/O phase to relieve this strain.37,38 As an example of the potential in the battery field, we have recently reported the synthesis, structure and electrochemical performance of the phase Li1.2Ni0.4Mg0.2Mo0.2O2, which adopts a cation ordered layered structure.39 Preliminary studies have since shown that partial doping with F to partially replace Ni3+ with the larger Ni2+ leads to a transformation from layered to DRS structure (as shown in ESI,† Fig. S12).
Another significant consequence of this work is the potential effect of shorter time milling on electrode performance. Commonly milling is used to reduce electrode particle size, so as to improve performance by reducing Li-ion diffusion lengths. While such shorter milling times may not lead to complete transformation of the material, it is possible that denser surface layers/partial transformation may result, and so some of the performance changes observed may be indeed due to similar local pressure effects on the particle surfaces. This is an area that warrants further study.
Conclusions
In this study, using examples from the battery literature, it has been shown that high impact ball milling can lead to the appearance of either the equivalent high-pressure polymorphs of the milled material or to the production of metastable phases that can only be synthesised through this route, which is attributed to both local heating and pressure effects from the shockwaves generated when the milling balls strike the powder. The results have two important consequences; the first is that it provides greater fundamental understanding of milling as a synthetic pathway, leading to potential routes to preparing such samples without milling (e.g. doping to create internal chemical pressure). The second relates to the use of milling as a route to smaller particle and the consequent possibility that such milling may also be affecting the surface structure of the particles. Nevertheless, the new knowledge about the importance of local pressure effects represents an exciting opportunity to exploit this route further both within the battery sector and beyond. Further characterisation and study are required to fully understand all the variables involved; however, the results clearly show that local pressure effects must be considered as a variable going forward.Author contributions
Laura L. Driscoll: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. Elizabeth H. Driscoll: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. Bo Dong: data curation, formal analysis, writing – original draft. Farheen N. Sayed: data curation, formal analysis, writing – original draft, writing – review & editing. Jacob N. Wilson: data curation. Christopher A. O’Keefe: data curation, formal analysis. Dominic J. Gardner: data curation. Clare P. Grey: supervision, resources, writing – original draft. Phoebe K. Allan: funding acquisition, supervision. Adam M. L. Michalchuk: methodology, formal analysis, supervision, writing – review & editing. Peter R. Slater: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Faraday Institution CATMAT (FIRG016) project for funding. We would also like to thank EPSRC Underpinning Multi-User Equipment Call (EP/P030467/1) for the TEM facility and Dr Heather Greer for assistance with data collection.References
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Unlocking the Potential of Cation-Disordered Oxides for Rechargeable Lithium Batteries, Science (80-.), 2014, 343, 519–522, DOI:10.1126/science.1246432.
- M. Freire, N. V. Kosova, C. Jordy, D. Chateigner, O. I. Lebedev, A. Maignan and V. Pralong, A new active Li-Mn-O compound for high energy density Li-ion batteries, Nat. Mater., 2016, 15, 173–177, DOI:10.1038/nmat4479.
- T. Sato, K. Sato, W. Zhao, Y. Kajiya and N. Yabuuchi, Metastable and nanosize cation-disordered rocksalt-type oxides: revisit of stoichiometric LiMnO2 and NaMnO2, J. Mater. Chem. A, 2018, 6, 13943–13951, 10.1039/c8ta03667e.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Fo, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Lithium manganese oxyfluoride as a new cathode material exhibiting oxygen redox, Energy Environ. Sci., 2018, 926–932, 10.1039/c7ee03195e.
- X. Xu, J.-J. Marie, G. J. Rees, L. Pi, C. Gong, S. Pu, R. A. House, A. W. Robertson and P. G. Bruce, Li2NiO2F a New Oxyfluoride Disordered Rocksalt Cathode Material, J. Electrochem. Soc., 2021, 2–8, DOI:10.1149/1945-7111/ac1be1.
- J. Lee, J. K. Papp, R. J. Clément, S. Sallis, D. H. Kwon, T. Shi, W. Yang, B. D. McCloskey and G. Ceder, Mitigating oxygen loss to improve the cycling performance of high capacity cation-disordered cathode materials, Nat. Commun., 2017, 8, 981, DOI:10.1038/s41467-017-01115-0.
- Z. Lun, B. Ouyang, D. A. Kitchaev, R. J. Clément, J. K. Papp, M. Balasubramanian, Y. Tian, T. Lei, T. Shi, B. D. McCloskey, J. Lee and G. Ceder, Improved Cycling Performance of Li-Excess Cation-Disordered Cathode Materials upon Fluorine Substitution, Adv. Mater., 2019, 9, 21802959, DOI:10.1002/aenm.201802959.
- M. J. Crafton, Y. Yue, T. Y. Huang, W. Tong and B. D. McCloskey, Anion Reactivity in Cation-Disordered Rocksalt Cathode Materials: The Influence of Fluorine Substitution, Adv. Energy Mater., 2020, 10, 2001500, DOI:10.1002/aenm.202001500.
- J. Ahn, D. Chen and G. Chen, A Fluorination Method for Improving Cation-Disordered Rocksalt Cathode Performance, Adv. Energy Mater., 2020, 10, 2001671, DOI:10.1002/aenm.202001671.
- J. Ahn, Y. Ha, R. Satish, R. Giovine, L. Li, J. Liu, C. Wang, R. J. Clement, R. Kostecki, W. Yang and G. Chen, Exceptional Cycling Performance Enabled by Local Structural Rearrangements in Disordered Rocksalt Cathodes, Adv. Energy Mater., 2022, 12, 2200426, DOI:10.1002/aenm.202200426.
- D. Chen, J. Ahn, E. Self, J. Nanda and G. Chen, Understanding cation-disordered rocksalt oxyfluoride cathodes, J. Mater. Chem. A, 2021, 9, 7826–7837, 10.1039/d0ta12179g.
- Z. Lun, B. Ouyang, Z. Cai, R. J. Clément, D.-H. Kwon, J. Huang, J. K. Papp, M. Balasubramanian, Y. Tian, B. D. McCloskey, H. Ji, H. Kim, D. A. Kitchaev and G. Ceder, Design Principles for High-Capacity Mn-Based Cation-Disordered Rocksalt Cathodes, Chem, 2020, 6, 153–168, DOI:10.1016/j.chempr.2019.10.001.
- D. A. Kitchaev, Z. Lun, W. D. Richards, H. Ji, R. J. Clément, M. Balasubramanian, D. H. Kwon, K. Dai, J. K. Papp, T. Lei, B. D. McCloskey, W. Yang, J. Lee and G. Ceder, Design principles for high transition metal capacity in disordered rocksalt Li-ion cathodes, Energy Environ. Sci., 2018, 11, 2159–2171, 10.1039/c8ee00816g.
- J. Liebertz and C. J. M. Rooymans, Phase behaviour of Li2MoO4 at high pressures and temperatures, Solid State Commun., 1967, 5, 405–409, DOI:10.1016/0038-1098(67)90785-5.
- E. Takayama-Muromachi, A. Navrotsky and S. Yamaoka, Calorimetric study of high-pressure polymorphs of Li2WO4 and Li2MoO4, J. Solid State Chem., 1986, 65, 241–250, DOI:10.1016/0022-4596(86)90059-9.
- G. I. Lampronti, A. A. L. Michalchuk, P. P. Mazzeo, A. M. Belenguer, J. K. M. Sanders, A. Bacchi and F. Emmerling, Changing the game of time resolved X-ray diffraction on the mechanochemistry playground by downsizing, Nat. Commun., 2021, 12, 1–9, DOI:10.1038/s41467-021-26264-1.
- X. Liu, Y. Lyu, Z. Zhang, H. Li, Y. S. Hu, Z. Wang, Y. Zhao, Q. Kuang, Y. Dong, Z. Liang, Q. Fan and L. Chen, Nanotube Li2MoO4: A novel and high-capacity material as a lithium-ion battery anode, Nanoscale, 2014, 6, 13660–13667, 10.1039/c4nr04226c.
- H. Y. Wang, B. K. Zou, Z. F. Tang, Z. Y. Wen and C. H. Chen, Enhancing cyclability and rate performance of Li2MoO4 by carbon coating, Mater. Lett., 2016, 177, 54–57, DOI:10.1016/j.matlet.2016.04.162.
- J. Zhang, R. Li, Q. Chen, G. Zhao and J. Jia, Porous carbon-coated Li2MoO4 as high-performance anode materials for lithium-ion batteries, Mater. Lett., 2018, 233, 302–305, DOI:10.1016/j.matlet.2018.09.032.
- D. Mikhailova, A. Voss, S. Oswald, A. A. Tsirlin, M. Schmidt, A. Senyshyn, J. Eckert and H. Ehrenberg, Lithium Insertion into Li2MoO4: Reversible Formation of (Li3Mo)O4 with a Disordered Rock-Salt Structure, Chem. Mater., 2015, 27, 4485–4492, DOI:10.1021/acs.chemmater.5b01633.
- M. N. Obrovac, O. Mao and J. R. Dahn, Structure and electrochemistry of LiMO2 (M = Ti, Mn, Fe, Co, Ni) prepared by mechanochemical synthesis, Solid State Ionics, 1998, 112, 9–19 CrossRef CAS.
- V. M. Mastikhin, O. B. Lapina and R. I. Maximovskaya, 95Mo Solid-State NMR Spectra of Molybdates, Chem. Physic Lett., 1988, 148, 413–416 CrossRef CAS.
- M. A. M. Forgeron and R. E. Wasylishen, Molybdenum magnetic shielding and quadrupolar tensors for a series of molybdate salts: A solid-state 95Mo NMR study, Phys. Chem. Chem. Phys., 2008, 10, 574–581, 10.1039/b713276j.
- K. Linberg, B. Röder, D. Al-Sabbagh, F. Emmerling and A. A. L. Michalchuk, Controlling polymorphism in molecular cocrystals by variable temperature ball milling, Faraday Discuss., 2022, 241, 178–193, 10.1039/d2fd00115b.
- J. Liebertz and C. J. M. Rooymans, Phase behaviour of li2moo4 at high pressures and temperatures, Solid State Communications, 1967, 5, 405–409 CrossRef CAS.
- A. P. Amrute, Z. Lodziana, H. Schreyer, C. Weidenthaler and F. Schüth, High-surface-area corundum by mechanochemically induced phase transformation of boehmite, Science, 2020, 366, 485–489, DOI:10.1126/science.aaw9377.
- E. E. McBride, A. Krygier, A. Ehnes, E. Galtier, M. Harmand, Z. Konôpková, H. J. Lee, H. P. Liermann, B. Nagler, A. Pelka, M. Rödel, A. Schropp, R. F. Smith, C. Spindloe, D. Swift, F. Tavella, S. Toleikis, T. Tschentscher, J. S. Wark and A. Higginbotham, Phase transition lowering in dynamically compressed silicon, Nat. Phys., 2019, 15, 89–94, DOI:10.1038/s41567-018-0290-x.
- P. N. Kuznetsov, L. I. Kuznetsova, A. M. Zhyzhaev, V. I. Kovalchuk, A. L. Sannikov and V. V. Boldyrev, Investigation of mechanically stimulated solid phase polymorphic transition of zirconia, Appl. Catal. A Gen., 2006, 298, 254–260, DOI:10.1016/j.apcata.2005.10.002.
- I. J. Lin and S. Nadiv, Review of the phase transformation and synthesis of inorganic solids obtained by mechanical treatment (mechanochemical reactions, Mater. Sci. Eng., 1979, 39, 193–209, DOI:10.1016/0025-5416(79)90059-4.
- Y. S. Kwon, P. P. Choi, J. S. Kim, D. H. Kwon and K. B. Gerasimov, Decomposition of intermetallics during high-energy ball-milling, Mater. Sci. Eng. A, 2007, 449–451, 1083–1086, DOI:10.1016/j.msea.2006.02.267.
- M. Freire, O. I. Lebedev, A. Maignan, C. Jordy and V. Pralong, Nanostructured Li2MnO3: A disordered rock salt type structure for high energy density Li ion batteries, J. Mater. Chem. A, 2017, 5, 21898–21902, 10.1039/c7ta07476j.
- M. Diaz-Lopez, P. A. Chater, Y. Joly, O. Proux, J. L. Hazemann, P. Bordet and V. Pralong, Reversible densification in nano-Li2MnO3cation disordered rock-salt Li-ion battery cathodes, J. Mater. Chem. A, 2020, 8, 10998–11010, 10.1039/d0ta03372c.
- K. J. Griffith, A. C. Forse, J. M. Griffin and C. P. Grey, High-Rate Intercalation without Nanostructuring in Metastable Nb2O5 Bronze Phases, J. Am. Chem. Soc., 2016, 138, 8888–8899, DOI:10.1021/jacs.6b04345.
- K. J. Griffith, A. C. Forse, J. M. Griffin and C. P. Grey, High-Rate Intercalation without Nanostructuring in Metastable Nb2O5 Bronze Phases, J. Am. Chem. Soc., 2016, 138, 8888–8899, DOI:10.1021/jacs.6b04345.
- J. L. Waring, R. S. Roth and H. S. Parker, Temperature-pressure phase relationships in niobium pentoxide, J. Res. Natl. Bur. Stand. Sect. A Phys. Chem., 1973, 77A, 705, DOI:10.6028/jres.077a.042.
- I. P. Zibrov, V. P. Filonenko, P. E. Werner, B. O. Marinder and M. Sundberg, A New High-Pressure Modification of Nb2O5, J. Solid State Chem., 1998, 141, 205–211, DOI:10.1006/jssc.1998.7954.
- H. Wilhelm, C. Cros, F. Arrouy and G. Demazeau, Pressure induced structural transition in the solid-solution La2-xNdxCuO4 for x = 0.6, 0.7, 1.2, and 1.5, J. Solid State Chem., 1996, 126, 88–94, DOI:10.1006/jssc.1996.0314.
- H. Wilhelm, C. Cros, E. Reny, G. Demazeau and M. Hanfland, Influence of pressure on the crystal structure of Nd2CuO4, J. Mater. Chem., 1998, 8, 2729–2732, 10.1039/a805886e.
- B. Dong, J. Castells-Gil, P. Zhu, L. L. Driscoll, E. Kendrick, P. K. Allan and P. R. Slater, Synthesis, structure and electrochemical properties of a new cation ordered layered Li-Ni-Mg-Mo oxide, Mater. Adv., 2023, 4, 1021–1029, 10.1039/d2ma00981a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee00249g. The raw data can be accessed here: https://doi.org/10.25500/edata.bham.00000998. |
| This journal is © The Royal Society of Chemistry 2023 |