
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable synthesis of azobenzenes, quinolines and quinoxalines via oxidative dehydrogenative couplings catalysed by reusable transition metal oxide–Bi(III) cooperative catalysts†
Marianna
Kocsis
 a,
Kornélia
Baán
b,
Sándor B.
Ötvös
c,
Ákos
Kukovecz
b,
Zoltán
Kónya
b,
Pál
Sipos‡
d,
István
Pálinkó
a and
Gábor
Varga§
*e
a,
Kornélia
Baán
b,
Sándor B.
Ötvös
c,
Ákos
Kukovecz
b,
Zoltán
Kónya
b,
Pál
Sipos‡
d,
István
Pálinkó
a and
Gábor
Varga§
*e
aDepartment of Organic Chemistry and Materials and Solution Structure Research Group, University of Szeged, Dóm tér 8, Szeged, H-6720 Hungary
bDepartment of Applied and Environmental Chemistry and Interdisciplinary Excellence Centre, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged, H-6720 Hungary
cInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, Graz, A-8010 Austria
dDepartment of Inorganic and Analytical Chemistry and Materials and Solution Structure Research Group, University of Szeged, Dóm tér 7, Szeged, H-6720 Hungary
eDepartment of Physical Chemistry and Materials Science and Materials and Solution Structure Research Group, University of Szeged, Rerrich Béla tér 1, Szeged, H-6720 Hungary. E-mail: gabor.varga5@chem.u-szeged.hu
First published on 20th April 2023
Abstract
Oxidative dehydrogenative coupling (ODC) is a useful tool for the formation of new C–C, C–O and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in an atom-economic and sustainable manner. Although this strategy is versatile, it is much less introduced to prepare N-heterocycles. Herein, we report the oxidative dehydrogenative coupling of anilines and 2-aminoanilies with vicinal diols to selectively produce azobenzenes (34–95% yield), quinolines (65–100% yield) and quinoxalines (78–100% yield) utilizing an environmentally friendly synthesis strategy. The reactions were catalysed by cooperative bifunctional catalysts based on transition metal oxides/Bi(III) which allow solvent-, additive-, oxidant- and base-free cyclisations/couplings. In addition, the catalysts were proven to be highly robust and reusable.
N bonds in an atom-economic and sustainable manner. Although this strategy is versatile, it is much less introduced to prepare N-heterocycles. Herein, we report the oxidative dehydrogenative coupling of anilines and 2-aminoanilies with vicinal diols to selectively produce azobenzenes (34–95% yield), quinolines (65–100% yield) and quinoxalines (78–100% yield) utilizing an environmentally friendly synthesis strategy. The reactions were catalysed by cooperative bifunctional catalysts based on transition metal oxides/Bi(III) which allow solvent-, additive-, oxidant- and base-free cyclisations/couplings. In addition, the catalysts were proven to be highly robust and reusable.
1. Introduction
Nitrogen-containing heterocycles, such as quinolines and quinoxalines, are extremely important organic intermediates that have been used in many industrial processes for the synthesis of polymers,1–4 dyes,5–8 pharmaceuticals9–15 and pesticides.16–18 For the industrial-scale synthesis of quinoxaline,19 the vapor phase double condensation of ethylenediamine derivatives with 1,2-dicarbonyl compounds (Scheme 1A), and the Lewis acid mediated Skraup synthesis (Scheme 1B) for quinoline are the most widely applied procedures.20–23 However, these processes exhibit well-known drawbacks, namely the necessity of environmentally non-reliable reagents and solvents, harsh reaction conditions and limited substrate scope. On manufacturing scales, despite all their weaknesses, these cyclisations remain the most popular methods because they are profitable and technically feasible.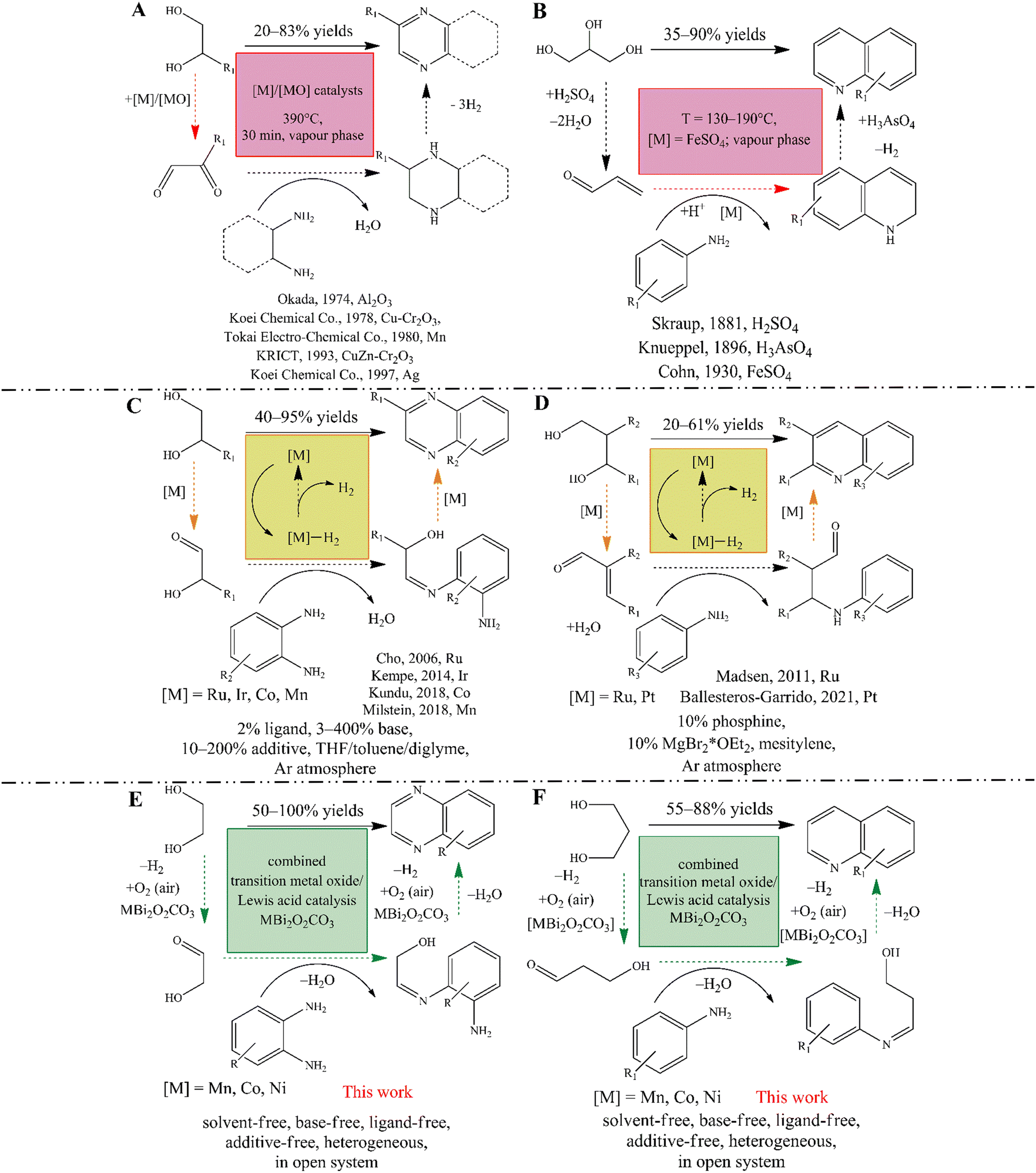 | ||
| Scheme 1 Synthesis of N-heterocycles via industrial oxidative traps (A and B); acceptorless dehydrogenative couplings (C and D) and our envisioned oxidative dehydrogenative couplings of diols and amines (E and F). | ||
Leaving aside classical multistep reactions,24 acceptorless dehydrogenative couplings (ADC) of readily available25–27 biomass-derived alcohols, amines and nitroarenes are nowadays the most advanced methods for accessing N-heterocycles in academia.28–31 Numerous studies focusing on the modified Friedländer synthesis of quinolines described that both mono dehydrogenative coupling of 2-aminobenzyl alcohols and ketones32–34 and the double dehydrogenative coupling of secondary alcohols with 2-aminobenzyl alcohols35 can be realized according to the ADC strategy. In parallel, quinoxaline synthesis based on ADC of non-symmetric 1,2-diols and 2-nitroanilines36,37 or o-phenylenediamines (Scheme 1C) was also successfully initiated.37–40 Similar to quinoxalines,36–39 indoles39 and pyrroles,41 quinoline can also be produced when diols are used as reactants if an aniline derivative is present in the system (Scheme 1D).42,43 Nevertheless, the usefulness this synthetic strategy has proven to be limited. This is probably due to lower product yields associated with the non-efficient Michael addition step of the tandem process.42 It should be noted however, that these acceptorless dehydrogenation strategies allow the selective conversion of diols without producing an overoxidized or C–C bond-cleaved product, making them more advantageous for the syntheses of these N-heterocycles in contrast to the oxidative dehydrogenation processes (see below).
The popularity of these strategies is due to their high selectivity and also to the fact that one can avoid the use of halogenated reactants and the pre-functionalization of the starting materials.30 On the other hand, catalysts for ADCs are usually based on expensive and chemically sensitive 4d and 5d (noble) metal complexes, mainly Ir and Ru.28–31 Although remarkable progress has been made in the last two decades using noble metal catalysis, including key contributions by Milstein,30,41,44–47 Kempe,38,48–50 Beller51,52 and Sun,53 a long-term goal is to develop an ADC strategy based on the most abundant, biocompatible, non-toxic and cost-effective transition metals, even under phosphine-free conditions. By developing pincer-type transition metal catalysts and their phosphine-free alternatives, these goals can be achieved more or less readily, which has made the production of quinolines and quinoxalines more efficient and sustainable than before.54–72 However, these ADCs have numerous limitations that are very similar to those of their noble metal-based counterparts. When using these complex systems, the only way to obtain the desired product is to use special ligands, additives and/or stoichiometric amounts of bases, inert atmosphere in most of the cases, and usually in non-green solvents (e.g., toluene). These conditions are not acceptable for larger scale production strongly limiting practical usefulness of this process. Moreover, all ADC conversions seem to be a strongly monopolized for homogeneous catalysis.
In contrast, oxidative reaction pathways have long been known to allow the desired products under mild reaction conditions using simple transition metal salts as the actual catalysts, even in heterogeneous phase. These are very popular and easy-to-perform reactions, such as the oxidation trap of α-hydroxycarbonyl compounds with 1,2-diamines73–75 or the bismuth-mediated oxidative coupling of epoxides with ene-1,2-diamines.76 However, these methods have major drawbacks compared to ADCs, both because they require pre-functionalization of the starting materials, which excludes the use of renewable reactants, and also because of the fact that their versatility and selectivity are lower. Moreover, it is not precedented to use green solvents in these methods. Therefore, it has become a challenge to find heterogeneous catalytic methods that provide an alternative, environmentally friendly reaction pathway with a simple experimental procedure for ADC involving readily available reactants. Corma et al. reported an elegant, oxidative dehydrogenative, one-pot two-step synthesis of quinoxalines that relied on the aerobic oxidation of biomass-derived vicinal diols leading to the corresponding dicarbonyls, followed by their cyclic condensation with 2-aminoaniline and its derivatives.77 This reaction combines the simplicity of oxidative conversions and the efficiency of ADCs, but uses noble metal-based catalysts, particularly Au-containing ones. They found that the rate-limiting step is the oxidation of the corresponding diol, which can also determine the selectivity of the reactions. Indeed, Au/CeO2 catalysts are able to promote oxidative C–C bond cleavage as well, which leads to the appearance of the corresponding benzimidazoles and reduces product yields. A similar aerobic oxidative dehydrogenative strategy exists for quinoline synthesis using primary alcohols instead of diols as starting materials. However, this was also strongly limited by the noble metal catalyst (Pd(OAc)2) together with various additives, in the absence of which the alcohol oxidation step is found to be incomplete.78
These examples also showed that the selective oxidation of diols/alcohols plays a key role in these oxidative or acceptorless dehydrogenative couplings. Both platinum metals and other noble metals have the ability to catalyse the aerobic oxidative dehydrogenation of alcohols, but with the possibility of over-oxidation or C–C cleavage of the alcohols.79,80 It is also known that catalytic properties can be fine-tuned by using bismuth or lead promoters.79 Some reports have shown that pure, heterogeneous copper oxides also have the ability to promote these oxidation reactions with good yields.81,82 Other catalysts based on transition metal oxides (TMO) or transition metal ion-containing specimens, even cooperating with Lewis acidic promoters, were able to play the main role in the oxidative dehydrogenative transformations and showed promising catalytic abilities.82–85 In this context, it is noteworthy that no oxidative dehydrogenative coupling (ODC) of diols and anilines has yet been carried out for the synthesis of N-heterocycles using a TMO-based catalyst, although this reaction strategy has already proven to be an indispensable tool for the construction of new C–C86 or azo-NN87–89 bonds via aniline coupling.
In view of the above findings, our hypothesis is that a bifunctional composite catalyst based on Co, Mn or Ni oxides combined with Lewis acidic sites should be capable of promoting oxidative dehydrogenative cross-coupling reactions of the corresponding diols and anilines/o-phenylenediamines, even producing quinoxalines or quinolines. Considering the activity and selectivity of the bismuth catalyst in the oxidative coupling reaction for quinoxalines,76,90 there is a great opportunity to build a supported tandem catalyst system.91 However, these active sites should work cooperatively and mimic the operation of Milstein complexes.47 For this reason, a constant well-dispersed cation distribution should be ensured. This can be achieved if transition metal oxides are immobilized on the surface of a bismuth-containing host material (Scheme 1E and F).
We have recently shown that as-prepared bismutite-supported (Bi2O2CO3) TMO cooperative catalysts92,93 (referred to as MBi2O2CO3; M = Co(II), Mn(II), Ni(II)) can enable efficient aerobic oxidative annulations to yield imines and benzimidazoles.94 These inorganic composites practically meet the requirements of promising catalyst candidates to promote the proposed oxidative dehydrogenative cross-coupling reaction of (amino)anilines and diols. However, in order to test our hypothesis in relation to the performance of these composites as actual ODC catalysts, simple oxidative dehydrogenative cross-couplings, especially homo- and heterocouplings of anilines, were selected as test reactions.87,88
In this report, we show that heterogeneous catalytic oxidative dehydrogenative coupling reactions of (amino)anilines and diols as well as oxidative dehydrogenative coupling reactions of anilines are possible under mild reaction conditions using ambient air as oxidant without any additives, bases and special ligands or an inert atmosphere or stochiometric wastes other than water thereby offering a sustainable methodology (Scheme 1E and F). To achieve our goal, cooperative TMO/Bi2O2CO3 catalysts were employed, which were found to be active, selective and recyclable catalysts for 1) homo- and hetero-coupling of anilines, 2) synthesis of quinolines and 3) quinoxalines via an oxidative dehydrogenative reaction pathway.
2. Experimental part
2.1. General information
All chemicals used were analytical grade Sigma-Aldrich products and were applied without further purification: manganese(II)nitrate-hydrate (Mn(NO3)2 × H2O); cobalt(II)nitrate-hexahydrate (Co(NO3)2 × 6H2O); nickel(II)nitrate-hexahydrate (Ni(NO3)2 × H2O); bismuth(III)nitrate-pentahydrate (Bi(NO3)3 × 5H2O); ammonia solution (25%; NH4OH); sodium carbonate anhydrous (Na2CO3); concentrated nitric acid (cc. HNO3); toluene (C6H5CH3); 5-methyloxolan-2-one (C5H8O2, γ-valerolactone); dimethyl sulfoxide (C2H6OS; DMSO); acetonitrile (C2H3N); methanol (CH3OH); ethyl acetate (CH3COOCH2CH3; EtOAc); sodium sulfate anhydrous (Na2SO4); aniline (C6H5NH2), o-anisidine (CH3OC6H4NH2); p-anisidine (4-(CH3O)C6H4NH2); 3-nitroaniline (O2NC6H4NH2); 4-bromoaniline (BrC6H4NH2); 4-cloroaniline (ClC6H4NH2); o-toluidine (CH3C6H4NH2); p-toluidine (4-(CH3)C6H4NH2); 4-aminobenzonitrile (H2NC6H4CN); 1,3-propanediol (HO(CH2)3OH); 2-aminoaniline (C6H4(NH2)2); ethan-1,2-diol (HOCH2CH2OH, ethylene-glycol); 4-bromo-1,2-diaminobenzene (C6H3(NH2)2Br); 4-cloro-1,2-diaminobenzene (C6H3(NH2)2Cl); 4-nitro-1,2-diaminobenzene (C6H3(NH2)2NO3); 3,4-diaminobenzoic acid ((H2N)2C6H3CO2H); 3,4-diaminotoluene (CH3C6H3(NH2)2); 2-aminobenzylamine (H2NC6H4CH2NH2); 1,2,3,4-tetrahydroquinoline (C9H11N); cobalt(II) oxide (CoO); 1,2-propanediol (HOCH2CH(CH3)OH); 2-hydroxypropanal (C3H6O2).2.2. Synthesis of transition metal-containing bismutites via modified co-precipitation method
A modified co-precipitation method was used to prepare bismutite-based composite catalysts with strongly bound transition metal oxides. Bi(NO3)3 × 5H2O of 3.75 mmol in the presence of the corresponding transition metal salts (3.75 mmol) were dissolved in 25 mL of 5 wt% aqueous citric acid solution. Then 40–40 mL of 0.4–0.4 M ammonia and sodium carbonate solutions were added dropwise to the metal salt solution and stirred at 100 °C for 24 hours. The crude product obtained was filtered, washed several times and dried at 60 °C overnight. The solidified product was labelled as MBi2O2CO3 (M: Ni, Co or Mn). For comparison, transition metal-free bismutite (Bi2O2CO3) was also produced in the same way without loading the corresponding transition metal salt.2.3. Materials characterization
X-ray diffraction (XRD) patterns were recorded with a Rigaku XRD-MiniFlex II instrument using CuKα radiation (λ = 0.15418 nm), 40 kV accelerating voltage at 30 mA. The JCPDS-ICDD (Joint Committee of Powder Diffraction Standards-International Centre for Diffraction Data) database was used for the identification of the characteristic reflections of the synthesized solid inorganics obtained.The morphology of the prepared specimens was examined by scanning electron microscopy (SEM). The SEM images were taken by an S-4700 scanning electron microscope (SEM, Hitachi, Japan) with an accelerating voltage of 10–18 kV. The elemental imaging of the freshly prepared samples was studied by scanning electron microscopy coupled to energy dispersive X-ray spectrometer (SEM-EDX).
The thermal behaviour of the composites was investigated by thermogravimetry (TG) and differential thermogravimetry (DTG). The bismutite-based samples were analysed using a Setaram Labsys derivatograph operating in air at a heating rate of 5 °C min−1. 20–30 mg of the samples were used for the measurements. The metal ion ratio of the composites in the initial state and the possible leaching during the catalytic processes were measured with ICP-MS on an Agilent 7700× instrument. Prior to the measurements, a few micrograms of the samples measured with analytical accuracy were dissolved in 1 mL of concentrated nitric acid and then diluted to 50 mL with distilled water and filtered.
Specific surface areas were determined by using the Brunauer–Emmett–Teller method (BET) (adsorption of N2 at 77 K). A NOVA3000 instrument was used for these measurements (Quantachrome, USA). The samples were rinsed with N2 at 150 °C for 5 hours to clean the surface of any adsorbents.
The basic sites were also characterised by using CO2-temperature programmed desorption (TPD) technique. TPD measurements were carried out on a Hewlett-Packard 5890 GC system equipped with a TCD detector. Prior to the measurements, a quartz tube was loaded with a portion of the sample (100 mg) followed by an initial purge in a He stream (50 mL min−1) at room temperature for 10 min to remove impurities. The temperature was then raised to 650 K using the ramp rate of 10 K min−1 and then held for 1 hour to remove water and other impurities. The temperature was then lowered to 373 K. Finally, the gas was changed to CO2 in He (30 mL min−1 CO2, 50 mL min−1 He) and circulated over the sample for 1 h. The gas was then removed from the sample by systematically increasing the temperature.
The surface acidity of the catalysts was characterised by NH3-TPD. The samples were pre-treated as above to remove impurities and water. Ammonia was then adsorbed for 30 minutes. Finally, the sample was heated from room temperature to 823 K at a rate of 10 K min−1 and the desorbed gases were analysed with a TC detector.
2.4. Optimized procedure for the catalytic reactions
In an optimised aniline heterocoupling reaction, a mixture of aniline (0.5 mL), the appropriate catalysts (10 mol% metal ion loading) and the appropriate substituted aniline (0.5 M) was placed in a 2.5 mL round bottom flask with a condenser. The reaction mixture was heated at 150 °C with stirring (500 rpm) for the appropriate time in an open system. The progress of the reaction was monitored by thin layer chromatography (TLC) using mixtures of ethyl acetate and hexane as eluent. After the reaction, the mixture was cooled and the catalyst removed by centrifugation. The resulting liquid was extracted with distilled water (2 × 15 mL), brine (1 × 15 mL) and ethyl acetate (10 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The homocoupling reactions were carried out in exactly the same way without using substituted anilines.During the optimized procedure for the quinoline synthesis, aniline derivatives (0.25 M), 1,3-propanediol (2 mL) and the appropriate catalyst of 10 mol% were added to a round bottom flask of 2.5 mL with a condenser. The flask was then immersed in the preheated oil bath (150 °C) and heated for the appropriate time. The progress of the coupling reaction was monitored by TLC using mixtures of ethyl acetate and hexane as eluent. The oil bath was then cooled to room temperature followed by the removal of the catalyst by filtration. Then the reaction mixture was diluted with 6 mL water and the organic part was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure.
During the optimized procedure for the quinoxaline synthesis, the corresponding catalyst of 2.5–10 mol%, o-phenylenediamine derivatives (0.25 M) and ethylene-glycol of 2 mL were placed into a 2.5 mL round bottom flask. After vigorous stirring (500 rpm) the mixture of 5 minutes, the mixture was heated up to 90–110 °C and it was maintained at that temperature for 24 h stirring continuously. The reaction mixture was then cooled in an ice bath and the catalyst was removed by filtration. The organic part was extracted with ethyl acetate (3 × 8 mL) and the solvent was evaporated under reduced pressure after drying over anhydrous Na2SO4.
The conversion and selectivity were determined after each reaction by GC-MS and 1H NMR spectroscopy. For the GC-MS measurements, a Thermo Scientific Trace 1310 gas chromatograph coupled with a Thermo Scientific ISQ QD Single Quadrupole Mass Spectrometer using a Thermo Scientific TG-SQC column (15 m × 0.25 mm ID × 0.25 μm film) was applied. The measurement parameters were as follows: column oven temperature: from 50 to 300 °C at 15 °C min−1; injection temperature: 240 °C; ion source temperature: 200 °C; electrospray ionisation: 70 eV; carrier gas: He at 1.5 mL min−1; injection volume: 2 μL; split ratio: 1 to 33.3; and mass range: 25–500 m/z. A Bruker DRX500 500 MHz NMR spectrometer was used for NMR spectroscopy (see section S4†). All samples were dissolved in 0.55 ml DMSO-d6 and 1H NMR spectra were recorded at room temperature. The spectra were internally fixed to the remaining resonance of the DMSO-d6 at 2500 ppm. For both analyses, the starting materials, products and by-products were identified using reference samples.
2.5. Control experiments
In the catalytic ethylene glycol oxidation (Scheme 2a and b), a CoO/CoBi2O2CO3/Bi2O2CO3 catalyst of ∼65 mg was added to 2.5 mL of aqueous ethylene glycol solution (0.3 M). This mixture was stirred for 72 h at reflux temperature in an open system and also in a system purged with argon. After removal of the catalyst, the liquid products were analyzed.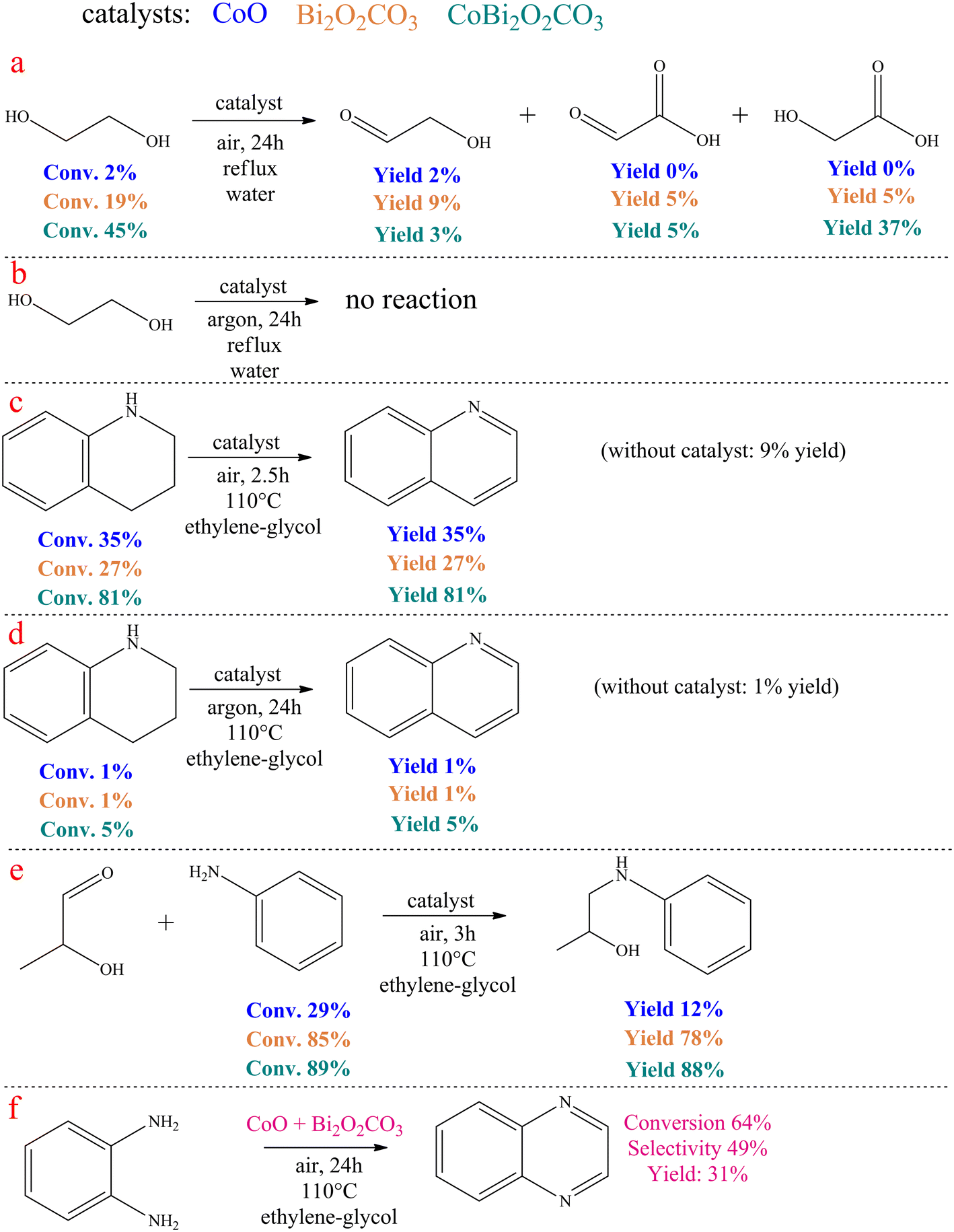 | ||
| Scheme 2 Control experiments. | ||
In the catalytic dehydrogenation of 1,2,3,4-tetrahydroquinoline (Scheme 2c and d), 2.5 mL of aniline and the appropriate amount (∼65 mg) of catalyst were added to one portion of the tetrahydroquinoline to give a solution with a tetrahydroquinoline concentration of 0.25 M. The reaction mixture was then stirred either for 2.5 hours in an open system or for 24 hours in an argon-purged system at reflux temperature. After removal of the catalyst, the liquid products were analyzed.
In the catalytic coupling reaction of 2-hydroxypropanal and aniline (Scheme 2e), an appropriate amount of 2-hydroxypropanal (0.25 M) was mixed with aniline (0.26 M) and 2.5 mL of ethylene glycol, followed by the addition of the catalyst. The resulting mixture was then stirred for 3 hours at 110 °C in an open system. After removal of the catalyst, the liquid products were analyzed.
The conversion and selectivity were determined after each reaction by GC-MS. The instrument and the protocol used were the same as detailed in the Optimized procedure for the catalytic reactions section.
3. Results and discussion
CoBi2O2CO3, NiBi2O2CO3 and MnBi2O2CO3 were prepared and activated according to our procedure published previously.94Fig. 1 shows the XRD patterns of the as-prepared and activated samples. The identified Bragg reflections are due to phase-pure, highly crystalline rhombohedral polycrystals whose crystal parameters are analogous to that observed for previously described surface modified bismutite (Bi2O2CO3; PDF#41-1488) structures. All collected samples exhibited a cauliflower-like morphology and their dimensions are almost in the same order of magnitude between 400 and 800 nm. As shown in Table S1,† the results of the elemental analysis are also in good agreement with those reported for the bismutite analogues, indicating the successful preparation of the catalyst candidates.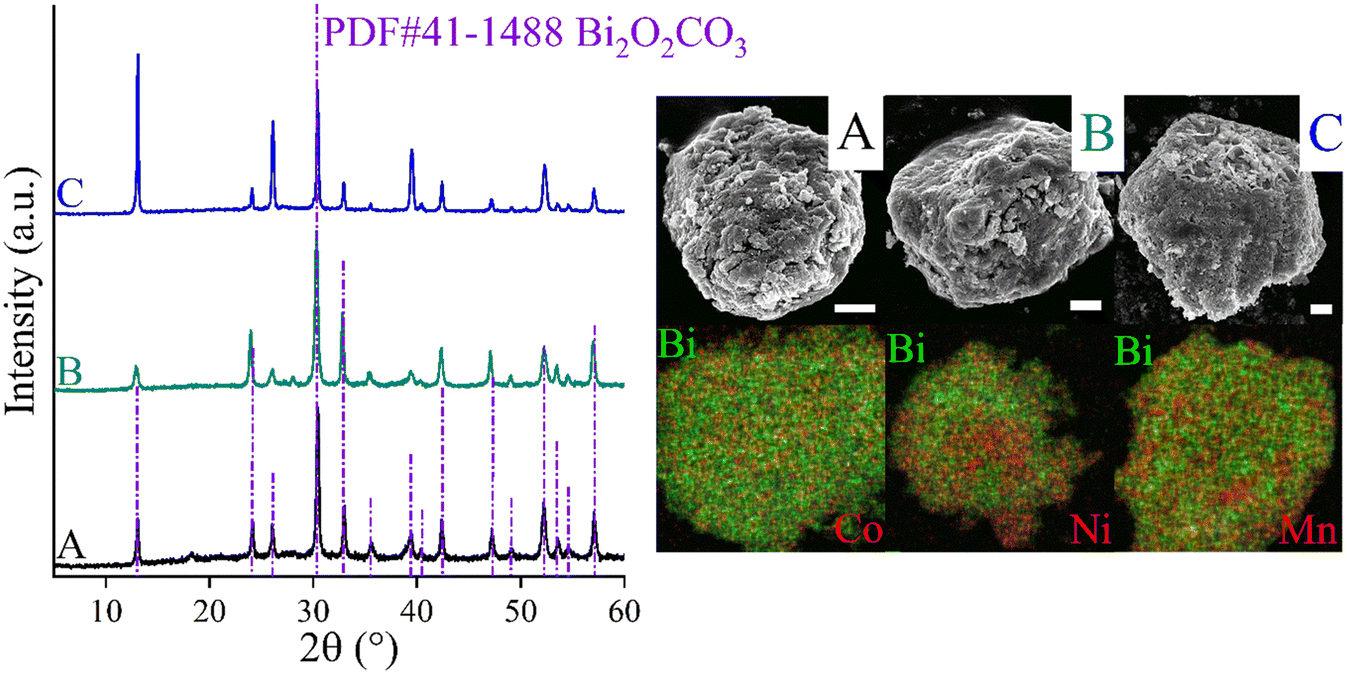 | ||
| Fig. 1 XRD patterns, SEM images and SEM-EDX elemental maps of CoBi2O2CO3 (A); NiBi2O2CO3 (B) and MnBi2O2CO3 (C). The scale bars represent 400 nm. The purple dashed-dot lines represent the positions and intensities of the characteristic reflections of a pure bismuth subcarbonate sample (bismutite, Bi2O2CO3). | ||
In order to verify the usefulness of the as-prepared catalysts in oxidative dehydrogenative reactions, homo- and hetero-couplings of anilines have been carried out as test reactions using bismutite analogues as the actual catalysts (Table 1; for all identified products, NMR data can be found in the ESI† in section S2). For these reactions, the oxidative dehydrogenative reaction route is also well-known.87,88 All the investigated model systems afforded moderate yields of azobenzene without the occurrence of significant amount of by-product after stirring in an open flask at 150 °C for 72 hours in DMSO as solvent (Table S2†). It should be noted that no reaction took place in the absence of the catalysts or under argon atmosphere. Detailed optimization procedure to maximize azobenzene yields were performed with the most promising manganese-based catalyst only. When solvents other than DMSO were used, there were no significant changes in the yield, while solvent-free conditions were shown to be promising and led to good yields of azobenzene (71%; Table S2,† entry 13). Systematic alteration of the reaction parameters led to a significant decrease in conversions in all cases (Table S2,† entries 6–12 and 15–19). It should be pointed out that ambient air was found sufficient as oxidant eliminating the need for a pure oxygen atmosphere. With the optimized reaction conditions in hand, the catalytic capabilities of the bismutites were investigated (Table 1). The reactions conducted by means of cobalt–bismuth and nickel–bismuth catalysis showed that the catalytic abilities of the bismutite analogues differ significantly. When using the nickel analogue, only small amounts of product 3 was formed (23%). In contrast, an almost quantitative reaction (95% yield) took place when the cobalt-containing analogue was used. There is no better indication for the efficiency of CoBi2O2CO3 than the fact that a comparably good yield of azobenzene (51%) could be obtained in only 24 hours.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| MnBi2O2CO3 | CoBi2O2CO3 | NiBi2O2CO3 | |||||
| Conversion of 1 (mol%) | Yielda of 3 (mol%) | Conversion of 1 (mol%) | Yielda of 3 (mol%) | Conversion of 1 (mol%) | Yielda of 3 (mol%) | ||
| 2.5 mol% CoBi2O2CO3 ; t = 24 h.a NMR yield. | |||||||
| 1 | R = H | 71 | 71 | 95/51 | 95/51 | 23 | 23 |
| 2 | R = o-OMe | 78 | 44 | 100/93 | 88/80 | 14 | 13 |
| 3 | R = p-OMe | 100 | 52 | 100/100 | 60/56 | 100 | 72 |
| 4 | R = m-nitro | 43 | 4 | 100/56 | 75/34 | 3 | 2 |
| 5 | R = p-Br | 52 | 52 | 100/66 | 99/65 | 21 | 21 |
| 6 | R = p-Cl | 24 | 21 | 51/10 | 48/10 | 4 | 4 |
| 7 | R = o-Me | 29 | 22 | 100/75 | 84/57 | 22 | 17 |
| 8 | R = p-Me | 51 | 47 | 100/98 | 94/81 | 84 | 2 |
| 9 | R = p-CN | 12 | 9 | 35/5 | 34/4 | 5 | 1 |
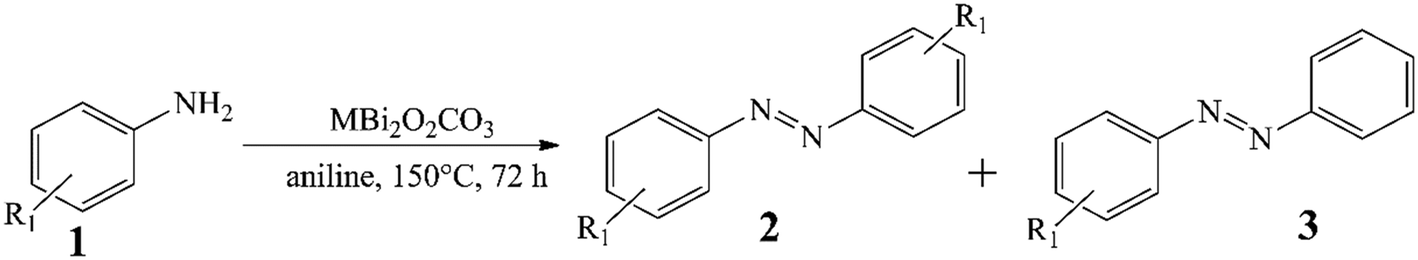
The differences between the performance of the catalysts become even more spectacular upon introducing heterocoupling reactions of anilines (Table 1, entries 2–9). The CoBi2O2CO3 catalyst appeared not only the most active (34–88% yields), but also the most selective towards the unsymmetrical azo compounds. This pronounced activity allowed the reduction of the catalyst loading to 2.5 mol% generally with moderate to excellent yields (56–80%) of the heterocoupled products. Noticeably, all three bismutite-based catalysts exhibited unique selectivity toward the unsymmetrical product, which was mainly independent of the substituents of the aromatic ring. Indeed, this can readily be explained by the significant excess of aniline employed. Taking advantage of the robustness of the CoBi2O2CO3 catalyst, it was next aimed to detach the effect of the aniline excess and re-investigate the selectivity results (Table S3†). On repeating three different asymmetric heterocoupling reactions in DMSO and toluene as well, almost identical selectivities could be achieved in addition to altered conversions despite the lower aniline excess. This should be regarded as direct evidence for the selectivity of the CoBi2O2CO3 catalyst toward unsymmetrical products. It should be noted here that aniline homocoupling took place in all cases because of the aniline excess.
These test reactions for oxidative dehydrogenative aniline couplings have shown that the bismutite analogues have considerable activity and good selectivity that are comparable to the heterogeneous and homogeneous oxidative dehydrogenative benchmark catalysts known from the literature (Table S4†).87,88 In other words, our hypothesis that these composites are suitable for promoting oxidative dehydrogenative reactions is correct. This is especially true for the cobalt-based bismutite analogue. However, there are also some interesting aspects that should be mentioned. In contrast to the benchmark catalysts, which offered excellent azobenzene yields exclusively in non-green solvents, especially toluene, our composites can work in pure aniline in such a way that ambient air is involved as an oxidant (Table S4†). Furthermore, aniline is chemoselectively coupled with its substituted counterparts in the presence of the bismutites to form the corresponding asymmetric azobenzenes. This procedure proves to be much more selective than the reactions carried out with benchmark catalysis (Table S4†). Furthermore, the bismutites were generally able to function with much lower catalyst loadings (2.5 mol%) compared with the benchmark heterogeneous catalyst (32 mol%). Finally, similar to the Mn2O3 heterogeneous benchmark catalyst, no additional bases or additives had to be used to perform these couplings.
To prove the applicability of our hypothesis for effective oxidative dehydrogenative couplings for the synthesis of N-heterocycles, various aniline derivatives were investigated in their reactions with propane-1,3-diol to produce the corresponding quinolines (Table 2). Using the same reaction conditions optimized earlier for homo- and heterocouplings of aniline derivatives and propane-1,3-diol as reaction partner, moderate to excellent quinoline yields were obtained in almost all cases. These were completely unexpected results, because quinoline could previously only be produced – in the presence of diols – by double ADC combined with a Michael addition step of aniline derivatives with unsymmetrical vicinal diols. In this case, the unsymmetrical property of the diols and the resulting presence of a secondary alcohol group are crucial for the selective dehydrogenation of the diols, which enables the coupling. Moreover, only low to moderate yields of quinolines (18–61%; Table S5†) were obtained with this strategy using only ruthenium- or platinum catalysis in the presence of various additives.42,43 This result might be due to the soft Lewis acidic feature95 of the as-prepared catalysts allowing a more attractive N-alkylation–cyclization tandem96,97 sequence instead of the Michael addition-cyclization mechanism.42 This N-heterocyclisation mechanism is known in quinoline synthesis, but has so far only been described in the presence of hydrogen acceptors.98,99 On the other hand, the electronic nature or steric effect of the substituent of the aniline derivative had no significant influence on the dehydrogenative coupling, except for the nitro group in meta position. The yield of the reaction between 3-nitroanline and propane-1,3-diol was low, except for the appearance of 3-hydroxypropanal as a by-product, which could indicate oxidative dehydrogenation of the diol. In general, bismutite-based catalysts were found to be highly selective toward quinoline. Homo-coupled azo compounds of aniline derivatives could also be detected, which were formed as a by-product to a small extent. It should also be emphasized that the CoBi2O2CO3 catalyst showed the highest activity so far and gave the quinoline products in excellent yields (isolated yields: 55–92%). Due to its high catalytic activity, it was possible to reduce reaction time to 48 h, even when introducing only 2.5 mol% of catalyst loading. The performance of CoBi2O2CO3 appears to be comparable to transition metal or noble metal catalysed acceptorless dehydrogenative strategies and base metal catalysed borrowing hydrogen strategies (Table S5†), which usually suffer from the use of additional inorganic bases and non-green solvents. In contrast, the reactions promoted by CoBi2O2CO3 were carried out in propane-1,3-diol (one of the reaction partners), without the need for any additives, ligands or bases, and without the use of an inert atmosphere, allowing more applicable and environmentally friendly processes than before.
|
|
|||||
|---|---|---|---|---|---|
| Desired product (dp) | Aniline conversion (mol%) | dp selectivity (%) | dp yielda (%) | Maximal isolated dp yieldb (%) | |
| 2.5 mol% CoBi2O2CO3.a NMR yield.b On using 10 mol% CoBi2O2CO3. | |||||
| 1 |
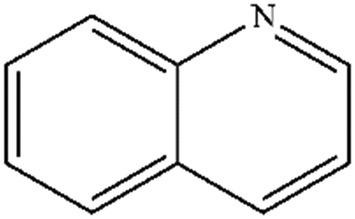
|
Mn: 100 | Mn: 94 | Mn: 94 | 92 |
| Co: 100 | Co: 92 | Co: 92 | |||
| Co: 100 | Co: 100 | Co: 100 | |||
| Ni: 82 | Ni: 93 | Ni: 76 | |||
| 2 |
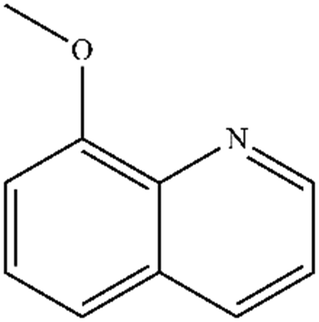
|
Mn: 67 | Mn: 100 | Mn: 67 | 79 |
| Co: 95 | Co: 81 | Co: 77 | |||
| Co: 100 | Co: 91 | Co: 91 | |||
| Ni: 85 | Ni: 83 | Ni: 71 | |||
| 3 |
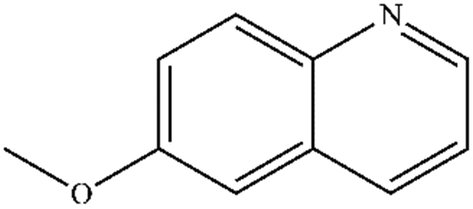
|
Mn: 66 | Mn: 76 | Mn: 50 | 77 |
| Co: 68 | Co: 63 | Co: 43 | |||
| Co: 95 | Co: 96 | Co: 91 | |||
| Ni: 79 | Ni: 46 | Ni: 36 | |||
| 4 |
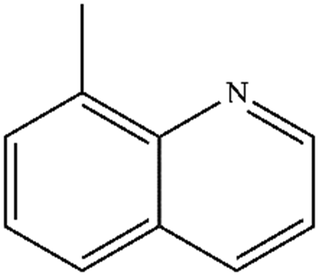
|
Mn: 98 | Mn: 96 | Mn: 94 | 75 |
| Co: 51 | Co: 92 | Co: 47 | |||
| Co: 93 | Co: 100 | Co: 93 | |||
| Ni: 52 | Ni: 100 | Ni: 52 | |||
| 5 |
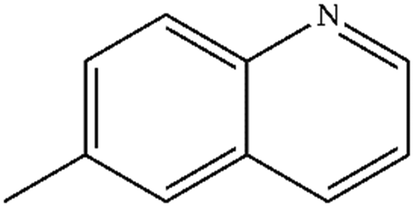
|
Mn: 89 | Mn: 82 | Mn: 73 | 91 |
| Co: 85 | Co: 82 | Co: 70 | |||
| Co: 100 | Co: 100 | Co: 100 | |||
| Ni: 84 | Ni: 87 | Ni: 73 | |||
| 6 |
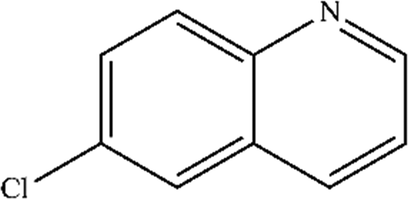
|
Mn: 52 | Mn: 58 | Mn: 30 | 55 |
| Co: 61 | Co: 41 | Co: 25 | |||
| Co: 100 | Co: 65 | Co: 65 | |||
| Ni: 33 | Ni: 100 | Ni: 33 | |||
| 7 |
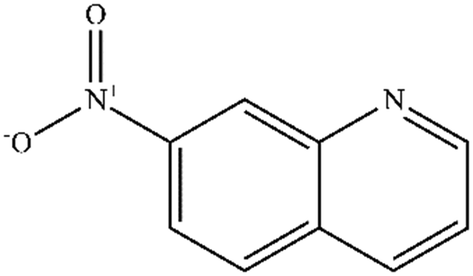
|
Mn: 0 | Mn: 0 | Mn: 0 | 7 |
| Co: 6 | Co: 100 | Co: 6 | |||
| Co: 11 | Co: 88 | Co: 10 | |||
| Ni: 4 | Ni: 100 | Ni: 4 | |||
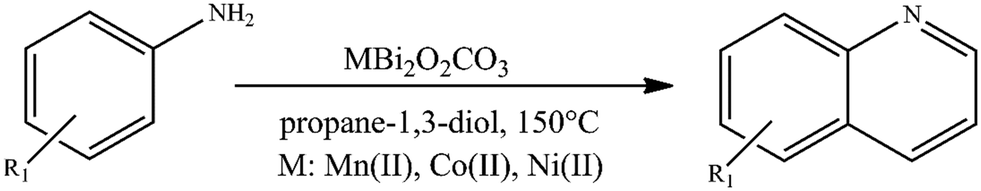
To further investigate the catalytic ability of bismutites, attempts were made to synthesize quinoxaline derivatives using a procedure that is almost identical to the one described above (Fig. 2, Table S6†). Therefore, cross-coupling reactions of 2-aminoaniline and its derivatives with electron donating and electron withdrawing substituents as well as ethylene glycol were initiated. In contrast to quinoline synthesis, these reactions could be achieved in shorter reaction times (8–24 h) and at lower reaction temperatures (90–110 °C) while achieving 34–100% yields of the desired quinoxaline derivatives by using either Ni- or Co-based bismutites. This means that the use of unsymmetrical diols containing secondary alcohol groups, which are essential for ADR reactions, was not required to ensure high yields. Interestingly, MnBi2O2CO3 was less selective for the coupling product than other bismutite analogues. When using Ni- or Co-based catalysts, the occurrence of azobenzene derivatives as a by-product would be possible. MnBi2O2CO3 promoted the formation of the by-product phenazine-2,3-diamine, which is a self-coupling product of 2-aminoaniline (Fig. 2). In addition, CoBi2O2CO3 without any auxiliary substances excelled in catalytic activity over other homogeneous or heterogeneous catalysts, including benchmark catalysts (Table S7†). The Co-bismutite-based catalytic system is thus suitable for producing quinoxalines with excellent conversions selectively via base-free oxidative hydrogenative cross-coupling of 2-aminoaniline derivatives and ethylene glycol under ambient conditions, using ethylene glycol as solvent. The observed differences in the yields obtained are probably due to the catalytic activity of the composites in alcohol oxidation reactions. This is assumed given that all the catalysts have been shown to be active and reusable catalysts for the synthesis of various N-heterocycles, as shown previously,92,94 so their abilities to promote alcohol oxidations must necessarily differ. As shown by the determined isolated yield of quinoxaline derivatives (63–85%) and the calculated E-factor of about 2.1–5.6, the developed process can be considered a green chemical process compared to the best available technologies in the fine chemical industry (Table S8†).100
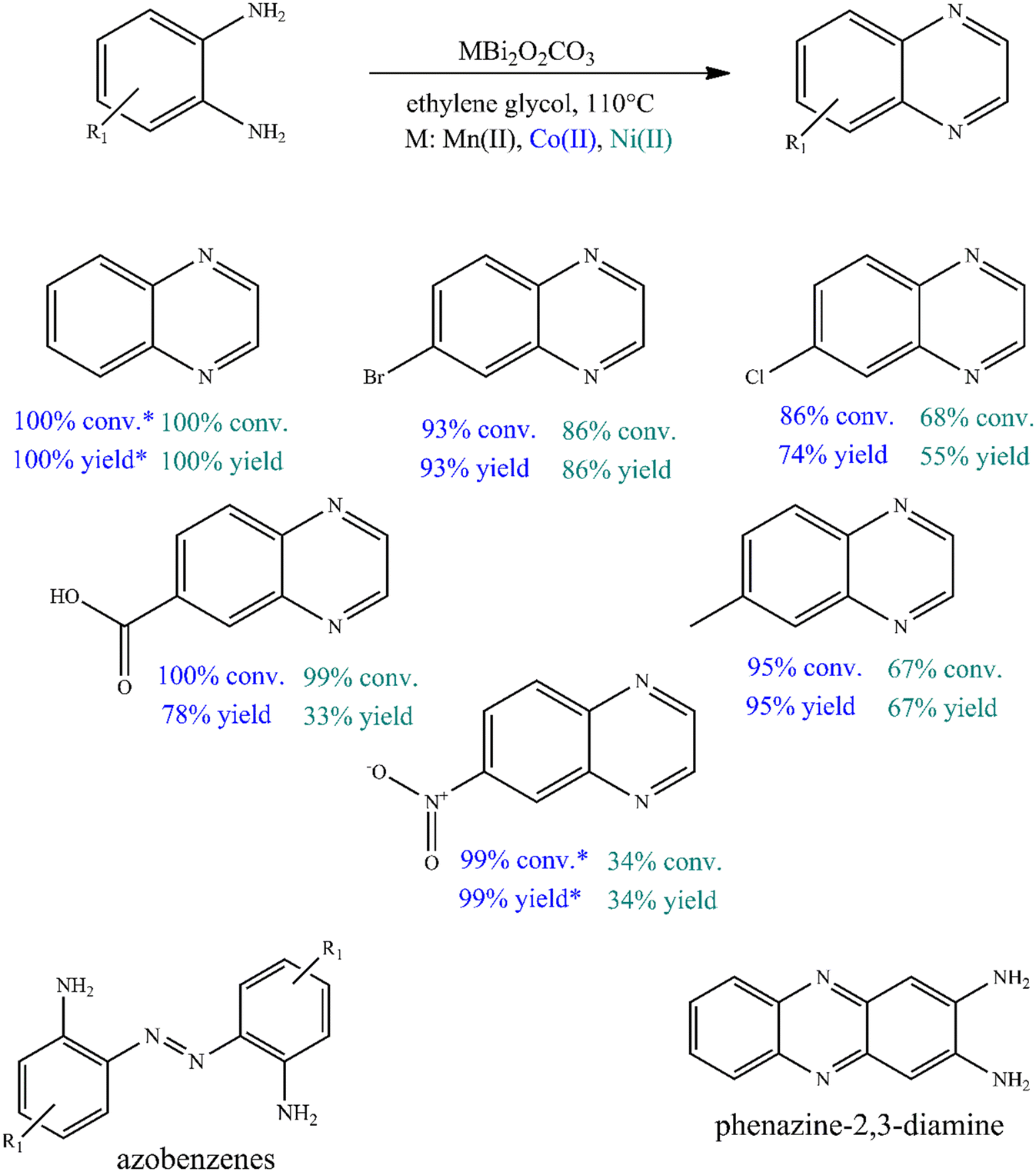 | ||
| Fig. 2 Oxidative dehydrogenative heterocoupling of o-phenylenediamines and ethylene glycol. Reaction conditions: 1 equiv. (0.25 M) o-phenylenediamine or its substituted derivative, 2 mL ethylene glycol, 110 °C for 24 h and 10 mol% catalyst. (GC yields were shown, *2.5 mol% CoBi2O2CO3). | ||
However, as a self-supported catalyst, CoBi2O2CO3 must be physically and chemically stable so that it can be reused. To investigate the stability of the catalyst, five recycling runs were performed (Fig. 3A). To avoid masking the presence of catalyst deactivation, 4 hours-long cycles were used in the recycling tests. It was found that both the catalytic activity and the selectivity of the actual catalyst remained almost unchanged in five cycles, with no leaching, as shown in the results from ICP-MS. When examining the changes in the catalytic markers over time, it was also possible to show that the selected catalyst functions in the same way in the fifth run as it did in the first run, which proves the stability of the catalyst (Fig. S1†). After the fifth run, the structural integrity of the reused catalyst was found to be unchanged, which was confirmed by XRD measurements (Fig. S2†). However, these results alone are not sufficient as evidence of the heterogeneous nature of the Co-based bismutite-promoted reaction. To confirm the heterogeneous catalytic nature, the solid was filtered out from the reaction slurry by centrifugation at low 2-aminoaniline conversion and the resulting clear solution was heated further under unchanged reaction conditions (Fig. 3B). The constant conversion and product yield indicated that the active components did not dissolve in the reaction mixture and the cross-coupling was catalysed heterogeneously. So far, only a few heterogeneous catalytic pathways have been designed and developed for N-heterocycles based on noble metals.
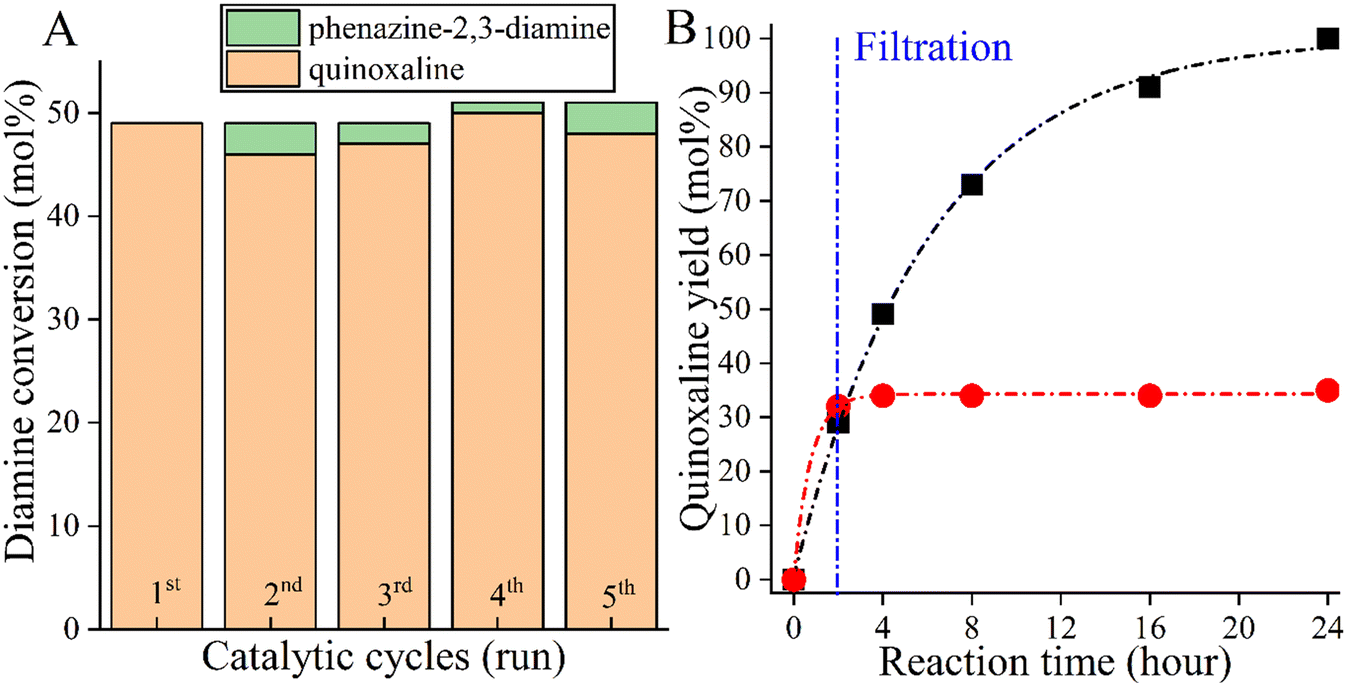 | ||
| Fig. 3 Five run recycling test (A) and the hot filtration test (B) of the CoBi2O2CO3 catalyst. Reaction conditions: 1 equiv. (0.25 M) o-phenylenediamine, 2 mL ethylene glycol, 110 °C for 24 h (4 h in the case of recycling) and 2.5 mol% catalyst. | ||
An attempt was made to explain the outstanding performance of the Co-containing bismutite. Both the beneficial effects of the interfaces demonstrated in our earlier work,94 and the effects of the surface dimensions, which were shown to be very similar for all catalysts prepared (Table S1†), could be excluded. Furthermore, there are no significant differences in the acidity of the catalysts as determined by the NH3-TPD technique (Fig. 4B). However, CO2-TPD (Fig. 4A) measurements indicated significant differences in the basicity of the catalysts in the order of CoBi2O2CO3 > NiBi2O2CO3 > MnBi2O2CO3 > Bi2O2CO3. This is essential, because basic properties could enhance catalytic activity, facilitating dehydrogenation of vicinal diols or adsorption of alcohol on the active sites.81
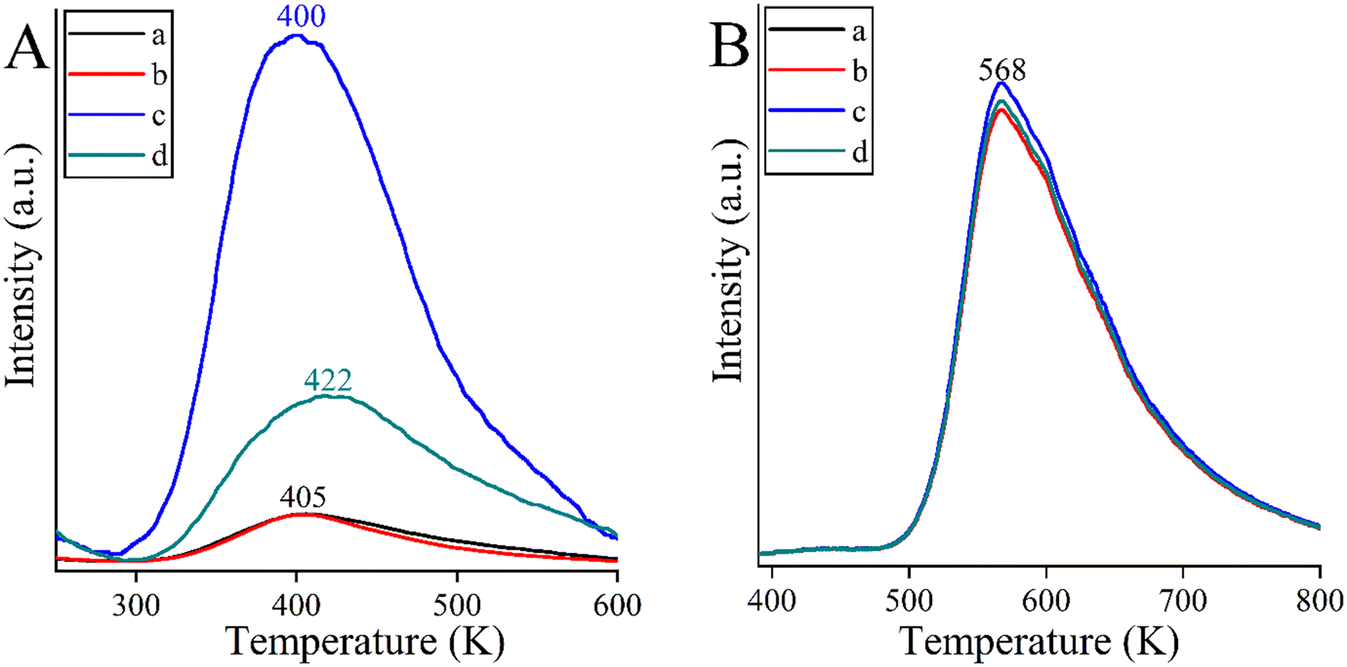 | ||
| Fig. 4 CO2-TPD curves (A) and NH3-TPD curves (B) of the bismutite-based composites: Bi2O2CO3 (a), MnBi2O2CO3 (b), CoBi2O2CO3 (c) and NiBi2O2CO3 (d). | ||
Some control experiments were carried out to explore the reaction mechanism of quinoxaline formation catalysed by CoBi2O2CO3 (Scheme 2). For comparison, the control experiments were also carried out by replacing CoBi2O2CO3 with pure CoO or Bi2O2CO3 as the catalyst.
Surprisingly, no dicarbonyl product was detected when pure ethylene glycol was treated in the presence of the catalysts in water at reflux temperature for 24 hours under air atmosphere (Scheme 2a). This fact refutes that the catalytic reaction occurred in the same way as reported for Au/CeO2 catalysed ODC for quinoxalines.77 Both the performance of pure CoO and pure bismutite were negligible compared to CoBi2O2CO3. In this latter case, a remarkable ethylene glycol conversion was observed. This reaction yielded glyoxylic acid as the main product, while glycolic acid and glycolaldehyde were also identified, which is in good agreement with the widely accepted mechanism of ethylene glycol oxidation in water.101 It is striking that the conversion of ethylene glycol is far from the stochiometric value, indicating that the dehydrogenation equilibrium of vicinal diols is probably shifted in the presence of anilines during the actual coupling reactions. Moreover, there was no reaction when these experiments were repeated under an argon atmosphere (Scheme 2b). This confirms that CoBi2O2CO3 is able to promote efficient oxidative dehydrogenation of diols with molecular oxygen as oxidant; however, this catalyst cannot catalyse acceptorless dehydrogenation.
In control experiments performed under the optimized reaction conditions, using 1,2,3,4-terahydroquinoline and the corresponding catalysts, it was found that aerobic oxidative dehydrogenation of the N-containing heterocycle was possible in the presence of CoBi2O2CO3 (Scheme 2c and d). In contrast, catalyst building blocks CoO and Bi2O2CO3 alone were not able to effectively promote this reaction, further strengthening the suggestion that Co-bismutite operates cooperatively. In another control experiment performed under the same conditions, the trapping of 2-hydroxipropanal on aniline took place (Scheme 2e) indicating that the condensation step probably occurred with the participation of the bismuth centres as active catalytic sites.
Finally, the envisioned ODC of ethylene glycol and o-phenylenediamine was repeated in the presence of the physical mixture of CoO and Bi2O2CO3 (Scheme 2f). It was found that the reaction could also be carried out in this mode, but with significantly reduced selectivity and conversion. This shows that randomly distributed multiple active sites are less effective than those with ordered structures, highlighting the need for proper catalyst fabrication process.91
Considering the results of the control experiments, it can be assumed that the oxidative dehydrogenations are cooperatively catalysed by the CoO and bismutite components of the catalyst. However, the role of Bi2O2CO3 also includes the promotion of the C–N coupling, which can also take place at the Bi3+ Lewis acidic centres, similarly as reported by Yadav and co-workers.90
In accordance with our observations and considering the earlier proposals of Milstein,57 Corma,77 Yadav90 and Riisager,81 a reasonable mechanism for the CoBi2O2CO3 catalysed cross-coupling reaction of o-phenylenediamine and ethylene glycol is presented in Scheme 3. In the first step, ethylene glycol adsorbs on the basic/Lewis acidic surface of the catalyst, probably at the Bi(III) centers,102 forming a metal alkoxide intermediate. Subsequently, the alcoholic hydroxy proton is abstracted by a surface oxygen – which is, in principle, a surface oxygen of the CoO specimen – followed by β-H elimination catalysed by Co(II) centres. As a result, a Co(II)-hydride coupled with a protonated surface oxygen and a Bi(III)-coordinated α-hydroxycarbonyl compound is generated. After then, Bi(III)-mediated condensation of the amino group of o-phenylenediamine with the carbonyl specimen – in parallel with Bi(III)-decomplexation – affords an intermediate that undergoes further conversion in two rapid steps, a sequential proton shift and an enol-oxo tautomerization. The next step is the condensation of the second amino group generating a 1,2-dihydroxiquinoxaline intermediate, which rapidly undergoes dehydrogenation in a manner similar to the first step. This leads to the formation of the final product quinoxaline – which subsequently desorbs – as well as another Co(II)-hydride coupled with a protonated surface oxygen. In the final step, molecular oxygen reacts with the Co(II)-hydride to form a peroxide anion, which immediately reacts with a second Co(II)-hydride to generate two molecules of hydroxides. These hydroxides then undergo a rapid acid–base reaction with the protonated surface oxygen that regenerates the original catalyst surface and also produces two molecules of water as the only by-product.
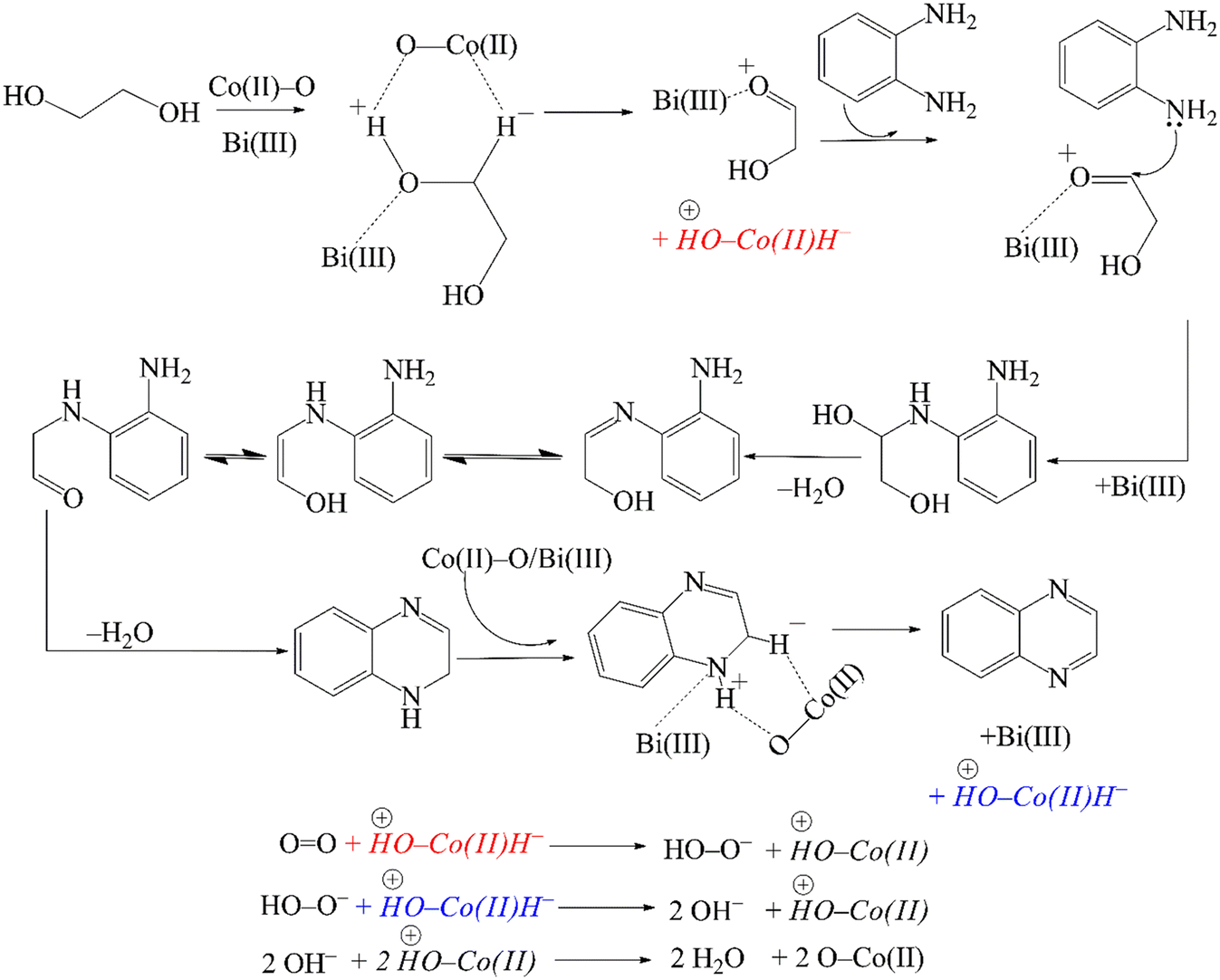 | ||
| Scheme 3 Proposed mechanism of the oxidative dehydrogenative coupling reaction of o-phenylenediamine and ethylene glycol catalysed by CoBi2O2CO3, providing quinoxaline. | ||
4. Conclusions
A noble metal-free, aerobic, oxidative dehydrogenative, heterogeneous catalytic strategy for the synthesis of N-heterocycles was reported using self-supported, reusable, cooperative transition metal oxide/Bi2O2CO3 catalysts. With this strategy, azobenzene, unsymmetrical azobenzenes, quinoline and quinoxaline and their substituted derivatives were selectively produced in good to excellent yields, with only H2O as stoichiometric waste. Moreover, these reactions could simply be carried out in green solvents without the use of any additives or bases using only atmospheric air as terminal oxidant. The active bismutites can be easily recovered and reused without leaching or structural collapse of the catalyst. The catalytic performance of the CoBi2O2CO3 system was found exceptionally high compared to other as-prepared catalysts, which could be due to its relatively high inherent basicity. This cobalt-based catalyst showed an excellent substituent tolerance and remarkable robustness, providing good to excellent yields of substituted quinolines and quinoxalines as well as unsymmetrical azobenzenes under useful reaction conditions. Control experiments verified CoO centres as the active component for the dehydrogenative transformations, while Bi(III) centres were able to promote the condensation reactions and mediate the dehydrogenations.The reported catalytic procedures can be regarded as significant improvement in terms of process sustainability as compared with earlier approaches due i) the cheap, robust and reusable heterogeneous catalysts employed, ii) the highly chemoselective nature of the transformations, iii) the lack of any auxiliary substances or additives and iv) the generation of only minimal amounts of waste.
Author contributions
M. Kocsis: investigation, data curation, writing – original draft. K. Baán: investigation, data curation. S. B. Ötvös: conceptualization, data curation, writing – original draft. Á. Kukovecz: resources, data curation. Z. Kónya: resources, data curation. P. Sipos: funding acquisition, resources, writing – original draft. I. Pálinkó: funding acquisition, resources. G. Varga: conceptualization, supervision, investigation, methodology, data curation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Szeged Open Access Fund (Grant number: 6190).Notes and references
- L. Xu, T. Zhou, M. Liao, R. Hu and B. Z. Tang, ACS Macro Lett., 2019, 8, 101–106 CrossRef CAS PubMed.
- H. Deng, T. Han, E. Zhao, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2016, 49, 5475–5483 CrossRef CAS.
- J. Yang, P. Cong, L. Chen, X. Wang, J. Li, A. Tang, B. Zhang, Y. Geng and E. Zhou, ACS Macro Lett., 2019, 8, 743–748 CrossRef CAS PubMed.
- J. Yang, M. A. Uddin, Y. Tang, Y. Wang, Y. Wang, H. Su, R. Gao, Z. K. Chen, J. Dai, H. Y. Woo and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 23235–23246 CrossRef CAS PubMed.
- H. Jiang, Y. Wu, A. Islam, M. Wu, W. Zhang, C. Shen, H. Zhang, E. Li, H. Tian and W. H. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 13635–13644 CrossRef CAS PubMed.
- X. Li, L. P. Zhang, L. Kang and Y. Zhao, Chem. Commun., 2019, 55, 11390–11393 RSC.
- P. Tarnow, C. Zordick, A. Bottke, B. Fischer, F. Kühne, T. Tralau and A. Luch, Chem. Res. Toxicol., 2020, 33, 742–750 Search PubMed.
- J. V. Jun, E. J. Petersson and D. M. Chenoweth, J. Am. Chem. Soc., 2018, 140, 9486–9493 CrossRef CAS PubMed.
- K. Murugan, C. Panneerselvam, J. Subramaniam, M. Paulpandi, R. Rajaganesh, M. Vasanthakumaran, J. Madhavan, S. S. Shafi, M. Roni, J. S. Portilla-Pulido, S. C. Mendez, J. E. Duque, L. Wang, A. T. Aziz, B. Chandramohan, D. Dinesh, S. Piramanayagam and J. S. Hwang, Sci. Rep., 2022, 12, 1–11 CrossRef PubMed.
- S. Zhang, Z. Xu, C. Gao, Q. C. Ren, L. Chang, Z. S. Lv and L. S. Feng, Eur. J. Med. Chem., 2017, 138, 501–513 CrossRef CAS PubMed.
- Y. Q. Hu, S. Zhang, F. Zhao, C. Gao, L. S. Feng, Z. S. Lv, Z. Xu and X. Wu, Eur. J. Med. Chem., 2017, 133, 255–267 CrossRef CAS PubMed.
- O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS PubMed.
- L. N. Rusere, A. N. Matthew, G. J. Lockbaum, M. Jahangir, A. Newton, C. J. Petropoulos, W. Huang, N. Kurt Yilmaz, C. A. Schiffer and A. Ali, ACS Med. Chem. Lett., 2018, 9, 691–696 CrossRef CAS PubMed.
- G. Tangherlini, D. V. Kalinin, D. Schepmann, T. Che, N. Mykicki, S. Ständer, K. Loser and B. Wünsch, J. Med. Chem., 2019, 62, 893–907 CrossRef CAS PubMed.
- D. Nageswara Rao, J. Zephyr, M. Henes, E. T. Chan, A. N. Matthew, A. K. Hedger, H. L. Conway, M. Saeed, A. Newton, C. J. Petropoulos, W. Huang, N. Kurt Yilmaz, C. A. Schiffer and A. Ali, J. Med. Chem., 2021, 64, 11972–11989 CrossRef CAS PubMed.
- X. H. Liu, W. Yu, L. J. Min, D. E. Wedge, C. X. Tan, J. Q. Weng, H. K. Wu, C. L. Cantrell, J. Bajsa-Hirschel, X. W. Hua and S. O. Duke, J. Agric. Food Chem., 2020, 68, 7324–7332 CrossRef CAS PubMed.
- Z. Sun, C. Wei, S. Wu, W. Zhang, R. Song and D. Hu, J. Agric. Food Chem., 2022, 70, 7029–7038 CrossRef CAS PubMed.
- W. Wang, S. Zhang, J. Wang, F. Wu, T. Wang and G. Xu, J. Agric. Food Chem., 2021, 69, 491–500 CrossRef CAS PubMed.
- Y. S. Higasio and T. Shoji, Appl. Catal., A, 2001, 221, 197–207 CrossRef CAS.
- Z. H. Skraup, Monatsh. Chem., 1881, 2, 139–170 CrossRef.
- C. A. Knueppel, Ber. Dtsch. Chem. Ges., 1896, 29, 703–709 CrossRef CAS.
- E. W. Cohn, J. Am. Chem. Soc., 1930, 52, 3685–3688 CrossRef CAS.
- O. Doebner and W. V. Miller, Ber. Dtsch. Chem. Ges., 1884, 17, 1712–1721 CrossRef.
- J. J. Li, Name Reactions in Heterocyclic Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004 Search PubMed.
- R. A. Urban and B. R. Bakshi, Ind. Eng. Chem. Res., 2009, 48, 8068–8082 CrossRef CAS.
- B. Winter, R. Meys and A. Bardow, J. Cleaner Prod., 2021, 290, 125818 CrossRef CAS.
- W. H. Faveere, S. Van Praet, B. Vermeeren, K. N. R. Dumoleijn, K. Moonen, E. Taarning and B. F. Sels, Angew. Chem., 2021, 60, 12204–12223 CrossRef CAS PubMed.
- N. Hofmann and K. C. Hultzsch, Eur. J. Org. Chem., 2021, 2021, 6206–6223 CrossRef CAS.
- G. Chelucci, Coord. Chem. Rev., 2017, 331, 37–53 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2006, 312, 257–261 CrossRef PubMed.
- K. Sun, H. Shan, G. Lu, C. Cai and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 25188–25202 CrossRef CAS PubMed.
- M. Subramanian, S. Sundar and R. Rengan, Appl. Organomet. Chem., 2018, 32, e4582 CrossRef.
- D. Wei, V. Dorcet, C. Darcel and J. B. Sortais, ChemSusChem, 2019, 12, 3078–3082 CrossRef CAS PubMed.
- A. Maji, A. Singh, N. Singh and K. Ghosh, ChemCatChem, 2020, 12, 3108–3125 CrossRef CAS.
- H. Vander Mierde, P. Van Der Voort, D. De Vos and F. Verpoort, Eur. J. Org. Chem., 2008, 2008, 1625–1631 CrossRef.
- F. Xie, M. Zhang, H. Jiang, M. Chen, W. Lv, A. Zheng and X. Jian, Green Chem., 2015, 17, 279–284 RSC.
- K. Chakrabarti, M. Maji and S. Kundu, Green Chem., 2019, 21, 1999–2004 RSC.
- T. Hille, T. Irrgang and R. Kempe, Chem. – Eur. J., 2014, 20, 5569–5572 CrossRef CAS PubMed.
- M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2019, 21, 1116–1120 CrossRef CAS PubMed.
- C. S. Cho and S. G. Oh, Tetrahedron Lett., 2006, 47, 5633–5636 CrossRef CAS.
- P. Daw, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 11931–11934 CrossRef CAS PubMed.
- R. N. Monrad and R. Madsen, Org. Biomol. Chem., 2011, 9, 610–615 RSC.
- D. Bellezza, R. J. Zaragozá, M. José Aurell, R. Ballesteros and R. Ballesteros-Garrido, Org. Biomol. Chem., 2021, 19, 677–683 RSC.
- B. Gnanaprakasam, E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., 2011, 123, 12448–12452 CrossRef.
- D. Srimani, Y. Ben-David and D. Milstein, Chem. Commun., 2013, 49, 6632–6634 RSC.
- D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., 2013, 125, 4104–4107 CrossRef.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140–144 CrossRef CAS PubMed.
- S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326–6329 CrossRef CAS PubMed.
- N. Deibl, K. Ament and R. Kempe, J. Am. Chem. Soc., 2015, 137, 12804–12807 CrossRef CAS PubMed.
- M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384–11388 CrossRef CAS PubMed.
- M. Zhang, H. Neumann and M. Beller, Angew. Chem., 2013, 125, 625–629 CrossRef.
- B. Pan, B. Liu, E. Yue, Q. Liu, X. Yang, Z. Wang and W. H. Sun, ACS Catal., 2016, 6, 1247–1253 CrossRef CAS.
- D. Panja, B. Paul, B. Balasubramaniam, R. K. Gupta and S. Kundu, Catal. Commun., 2020, 137, 105927 CrossRef CAS.
- S. Shee, D. Panja and S. Kundu, J. Org. Chem., 2020, 85, 2775–2784 CrossRef CAS PubMed.
- K. Das, A. Mondal and D. Srimani, Chem. Commun., 2018, 54, 10582–10585 RSC.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734–7741 CrossRef CAS PubMed.
- M. Maji, D. Panja, I. Borthakur and S. Kundu, Org. Chem. Front., 2021, 8, 2673–2709 RSC.
- A. Maji, S. Gupta, M. Maji and S. Kundu, J. Org. Chem., 2022, 87, 8351–8367 CrossRef CAS PubMed.
- A. K. Bains, V. Singh and D. Adhikari, J. Org. Chem., 2020, 85, 14971–14979 CrossRef CAS PubMed.
- S. Shee, K. Ganguli, K. Jana and S. Kundu, Chem. Commun., 2018, 54, 6883–6886 RSC.
- S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2017, 54, 90–93 RSC.
- S. Das, S. Sinha, D. Samanta, R. Mondal, G. Chakraborty, P. Brandaõ and N. D. Paul, J. Org. Chem., 2019, 84, 10160–10171 CrossRef CAS PubMed.
- G. Chakraborty, R. Sikari, S. Das, R. Mondal, S. Sinha, S. Banerjee and N. D. Paul, J. Org. Chem., 2019, 84, 2626–2641 CrossRef CAS PubMed.
- S. Das, D. Maiti and S. De Sarkar, J. Org. Chem., 2018, 83, 2309–2316 CrossRef CAS PubMed.
- S. Parua, R. Sikari, S. Sinha, S. Das, G. Chakraborty and N. D. Paul, Org. Biomol. Chem., 2018, 16, 274–284 RSC.
- C. Zhang, B. Hu, D. Chen and H. Xia, Organometallics, 2019, 38, 3218–3226 CrossRef CAS.
- M. K. Barman, A. Jana and B. Maji, Adv. Synth. Catal., 2018, 360, 3233–3238 CrossRef CAS.
- S. Elangovan, J. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS PubMed.
- M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543–15546 CrossRef CAS PubMed.
- A. Mondal, M. K. Sahoo, M. Subaramanian and E. Balaraman, J. Org. Chem., 2020, 85, 7181–7191 CrossRef CAS PubMed.
- C. S. Cho and S. G. Oh, J. Mol. Catal. A: Chem., 2007, 276, 205–210 CrossRef CAS.
- S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Org. Biomol. Chem., 2004, 2, 788 RSC.
- S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Chem. Commun., 2003, 2286–2287 RSC.
- S. Antoniotti and E. Duñach, Tetrahedron Lett., 2002, 43, 3971–3973 CrossRef CAS.
- M. J. Climent, A. Corma, J. C. Hernández, A. B. Hungría, S. Iborra and S. Martínez-Silvestre, J. Catal., 2012, 292, 118–129 CrossRef CAS.
- J. Li, J. Zhang, H. Yang and G. Jiang, J. Org. Chem., 2017, 82, 3284–3290 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283 CrossRef CAS.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- R. Poreddy, C. Engelbrekt and A. Riisager, Catal. Sci. Technol., 2015, 5, 2467–2477 RSC.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- P. Chandra, T. Ghosh, N. Choudhary, A. Mohammad and S. M. Mobin, Coord. Chem. Rev., 2020, 411, 213241 CrossRef CAS.
- A. Mondal, D. Mukherjee, B. Adhikary and M. A. Ahmed, J. Nanopart. Res., 2016, 18, 342–347 CrossRef.
- T. K. Slot, D. Eisenberg, D. van Noordenne, P. Jungbacker and G. Rothenberg, Chem. – Eur. J., 2016, 22, 12307–12311 CrossRef CAS PubMed.
- A. M. Faisca Phillips and A. J. L. Pombeiro, ChemCatChem, 2018, 10, 3354–3383 CrossRef CAS.
- B. Dutta, S. Biswas, V. Sharma, N. O. Savage, S. P. Alpay and S. L. Suib, Angew. Chem., 2016, 128, 2211–2215 CrossRef.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- Á. Georgiádes, S. B. Ötvös and F. Fülöp, ACS Sustainable Chem. Eng., 2015, 3, 3388–3397 CrossRef.
- J. S. Yadav, B. V. Subba Reddy, K. Premalatha and K. S. Shankar, Synthesis, 2008, 23, 3787–3792 CrossRef.
- D. E. Fogg and E. N. Dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- M. Kocsis, S. B. Ötvös, G. F. Samu, Z. Fogarassy, B. Pécz, Á. Kukovecz, Z. Kónya, P. Sipos, I. Pálinkó and G. Varga, ACS Appl. Mater. Interfaces, 2021, 13, 42650–42661 CrossRef CAS PubMed.
- R. Mészáros, A. Márton, M. Szabados, G. Varga, Z. Kónya, Á. Kukovecz, F. Fülöp, I. Pálinkó and S. B. Ötvös, Green Chem., 2021, 23, 4685–4696 RSC.
- M. Kocsis, M. Szabados, S. B. Ötvös, G. F. Samu, Z. Fogarassy, B. Pécz, Á. Kukovecz, Z. Kónya, P. Sipos, I. Pálinkó and G. Varga, J. Catal., 2022, 414, 163–178 CrossRef CAS.
- J. Ramler and C. Lichtenberg, Chem. – Eur. J., 2020, 26, 10250–10258 CrossRef CAS PubMed.
- M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- Y. Tsuji, K. T. Huh and Y. Watanabe, J. Org. Chem., 1987, 52, 1673–1680 CrossRef CAS.
- Y. Tsuji, H. Nishimura, K.-T. Huh and Y. Watanabe, J. Organomet. Chem., 1985, 286, c44–c46 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- H. Yan, S. Yao, J. Wang, S. Zhao, Y. Sun, M. Liu, X. Zhou, G. Zhang, X. Jin, X. Feng, Y. Liu, X. Chen, D. Chen and C. Yang, Appl. Catal., B, 2021, 284, 119803 CrossRef CAS.
- I. J. Casely, J. W. Ziller, B. J. Mincher and W. J. Evans, Inorg. Chem., 2011, 50, 1513–1520 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Quantitative analysis of the as-prepared catalysts, optimization procedure for the synthesis of azobenzene, selectivity tests of the CoBi2O2CO3, supporting tests for the catalytic quinoxaline synthesis, comparative tables with the catalytic performance of benchmark catalysts, XRD patterns of the CoBi2O2CO3 used, NMR data of the products. See DOI: https://doi.org/10.1039/d3cy00327b |
| ‡ Passed away. |
| § Present address: Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. |
| This journal is © The Royal Society of Chemistry 2023 |