
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A data-driven high-throughput workflow applied to promoted In-oxide catalysts for CO2 hydrogenation to methanol†
Mohammad
Khatamirad‡
 *a,
Edvin
Fako‡
b,
Chiara
Boscagli
c,
Matthias
Müller
c,
Fabian
Ebert
a,
Raoul
Naumann d'Alnoncourt
*a,
Ansgar
Schaefer
b,
Stephan Andreas
Schunk
bcd,
Ivana
Jevtovikj
c,
Frank
Rosowski
ab and
Sandip
De
*b
*a,
Edvin
Fako‡
b,
Chiara
Boscagli
c,
Matthias
Müller
c,
Fabian
Ebert
a,
Raoul
Naumann d'Alnoncourt
*a,
Ansgar
Schaefer
b,
Stephan Andreas
Schunk
bcd,
Ivana
Jevtovikj
c,
Frank
Rosowski
ab and
Sandip
De
*b
aBasCat – UniCat BASF JointLab, Technische Universität Berlin, Berlin, Germany. E-mail: r.naumann@bascat.tu-berlin.de
bBASF SE, Group Research, Ludwigshafen, Germany. E-mail: sandip.de@basf.com
chte GmbH, Heidelberg, Germany
dInstitute of Chemical Technology, Universität Leipzig, Leipzig, Germany
First published on 27th March 2023
Abstract
We propose a novel high-throughput workflow, combining DFT-derived atomic scale interaction parameters with experimental data to identify key performance-related descriptors in a CO2 to methanol reaction, for In-based catalysts. Utilizing advanced machine learning algorithms suitable for small datasets, secondary descriptors with high predictive power for catalytic activity were constructed. These descriptors, which highlight the crucial role of hydroxyl sites, can be applied to designing new materials and to bringing them to the test with high-throughput screening, paving the path for accelerated catalyst design.
The environmental impact of CO2 emissions calls for urgent action towards converting this environmentally detrimental waste product into carbon feedstock and recycling it to valuable products.1,2 Achieving this objective requires catalysts that are both inexpensive to produce and highly efficient in given process scenarios. The high thermodynamic stability of CO2 presents a significant hurdle in its utilization, which can only be overcome by lowering the high reaction barrier through the use of suitable catalyst materials.3 While alternative CO2 activation methods do exist,4,5 conventional, thermal heterogeneous catalysis is a preferred method in industrial applications. However, the process of designing new catalysts is traditionally decelerated by the circular dependency between catalyst testing, comprehensive characterization, and computational modelling. Experimentally observed performance trends from experimental data can provide insight into catalytic activity and allow for the traditional design of experiments based on simple composition changes and major reaction parameters. But when it comes to designing new catalyst classes, data based on experiments alone may have certain limitations.6,7
On the other hand, in-depth characterization of individual catalysts requires a significant investment of resources, and may under certain conditions be limiting. The advent of quantum-mechanics (QM) methods such as density functional theory (DFT) in the late 1990s has helped to increase the speed of discovering new materials via revealing structure–activity relationships.8 However, one of the main challenges in employing QM methods in screening processes for new catalysts is the high computational cost.9 Traditional simulation protocols involve a systematic exploration of reaction mechanisms, which can be tedious and require experienced modellers' careful interventions, making them challenging for high-throughput screening applications. Widely studied reactions with community-accepted mechanisms, such as the one being discussed here, in principle, allow for identification of crucial rate-limiting steps referring to earlier work. However, it would be desirable to replace the need for very expensive, accurately pre-determined reaction steps with a high-throughput simulation-friendly, data-driven protocol. Such a protocol should consist of programmatically generated, chemically relevant reaction intermediate adsorption energies on the desired surface-active site. These “generic” descriptors combined with relevant experiment parameters can then be correlated with target response parameters to identify the design rules with key important parameters.10,11 These design rules hold the promise of reducing the number of configurations that need to be considered, and in turn, reducing the computational cost and increasing the speed of prediction.
In this study, we propose a workflow that combines high-throughput computation and experimentation, along with machine learning algorithms (as shown in Fig. 1). With this workflow, we identify descriptors and rules that have the desired predictive power for the potential development of new catalytic materials. It is important to note that this protocol does not require prior knowledge of curated reaction steps; only a general knowledge of possible intermediates is sufficient. This makes the protocol suitable for new catalyst compositions where the reaction mechanisms have not been experimentally proven, and data-driven identification of important intermediates that can provide more insight quickly on the hypothesized mechanism. The desired target product in the study discussed here is methanol, which can be used as an alternative clean fuel and intermediate for the production of a variety of chemical commodities.12,13
 | ||
| Fig. 1 Overall workflow of the study, showing the link between material selection, DFT descriptor computation, performance correlation, and design rule identification via machine learning methods. This workflow combines high-throughput computation and experimentation, allowing the identification of descriptors and rules that have the best predictive power for the potential development of new catalytic materials. It is important to note that this protocol does not require prior knowledge of curated reaction steps; only a general knowledge of possible intermediates is sufficient. | ||
We focus our attention on In2O3/ZrO2 as a benchmark system for CO2 hydrogenation. In2O3/ZrO2 catalysts have gained attention in recent years due to the enhanced stability under CO2-rich conditions compared to extensively studied Cu-based catalysts,14,15 making In2O3/ZrO2 a promising candidate for industrial application. To diversify the chemical space sampled in this study, we employ a simple promotion strategy in which 13 different promoters from transition and post-transition metals were selected to synthesize In-modified catalysts via co-impregnation (see Fig. 1). A molar ratio of promoter![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In = 1:3 was used. To synthesize the catalysts, ZrO2 was initially crushed and sieved to reach a target fraction of 250–315 μm. For impregnation of In, as well as co-impregnation of each of the 13 co-promoters, the crushed and sieved ZrO2 was added to an aqueous nitrate solution of In and the respective co-promoter. Subsequently, the solvent was evaporated and the catalyst was calcined at 300 °C.
:In = 1:3 was used. To synthesize the catalysts, ZrO2 was initially crushed and sieved to reach a target fraction of 250–315 μm. For impregnation of In, as well as co-impregnation of each of the 13 co-promoters, the crushed and sieved ZrO2 was added to an aqueous nitrate solution of In and the respective co-promoter. Subsequently, the solvent was evaporated and the catalyst was calcined at 300 °C.
The catalytic test was carried out in the gas phase in a 16-fold parallel reactor system at hte GmbH (Heidelberg, Germany). The reactors were filled with 0.5 ml of calcined catalysts, with a pre-/post-bed of corundum (Fig. 1). The catalysts in each reactor channel were reduced under H2:N2:Ar = 30:60:10, and afterwards tested for CO2 hydrogenation to methanol. The reaction was conducted under a pressure = 80 bar, total flow = 48 L h−1, CO:CO2:H2 = 1.9:17.1:76 and three different temperatures: 225, 250 and 275 °C. The yields were measured using in-line TCD-GC. Only data in the kinetic regime outside of the thermodynamic equilibrium limits were considered, and therefore higher temperatures above 275 °C were not included in the screening.
To develop a descriptive model for catalytic performance, one should consider the behaviour of the activated catalyst, which is formed in situ and under reaction conditions. Obtaining a model under such conditions via the classical approach on the one hand requires extensive operando characterization and on the other hand, relies on several multi-level theoretical concepts.16–18 Therefore, obtaining a functional relation between performance and computed descriptors stemming from a simplistic yet representative DFT model is extremely valuable. To this extent, we construct an oxygen vacancy on the (110) surface of cubic In2O3 and introduce the chemical impact of the promoter species by replacing one of the In atoms at the O vacancy by the promoter (Fig. 1 and S1†). This site is probed by all intermediates, along two competing pathways for CO and methanol formation15 (Fig. S2†). In total, 1350 intermediate relaxations were performed on the DFT level (Fig. S3–S6†). Thereby, adsorption energies of 90 unique adsorbates, as well as formation energies of oxygen vacancies on two different adjacent sites with respect to the promoter site, were computed for each of the 14 catalysts, resulting in a total of 92 DFT-obtained descriptors. The energies of many expected intermediates along the investigated CO or methanol formation pathways are correlated to each other (Fig. S7–S11†). A bivariate correlation matrix was generated to detect descriptor pairs whose absolute value of the Pearson coefficient was above 0.9. Based on this correlation analysis, a reduced subset of 27 DFT descriptors (from the total of 92), which are not correlated with each other, were selected for the model generation.
Many of the DFT-derived descriptors are expected to have an impact on the catalyst's performance, some more than others. These correlations may be linear, but are not limited to that. To identify the key descriptors and account for non-linear correlations, we use the SISSO (sure independence screening and sparsifying operator) algorithm as introduced by Ouyang et al.19 to develop a predictive model for the methanol yield. The SISSO algorithm creates a mathematical expression for the target feature (Ymethanol), as a function of a non-linear combination of input primary features.20–23 The employed primary features for our study consist of 27 DFT-obtained descriptors and the reaction temperature T. We use the term secondary descriptor to refer to any non-linear combination of primary features, generated by SIS (sure independence screening).
The model complexity is determined by two parameters: the number of non-zero coefficients or dimension, and the number of allowed mathematical operations to create secondary descriptors. For the sake of brevity, we use D and O to refer to the dimension and number of allowed operations, respectively. Considering the high sensitivity of ML models to data scaling, prior to determining the optimum model complexity, various methods for pre-processing of the primary features were employed to obtain the most suitable scaling method in terms of accuracy. We report four different methods, which were used to generate SISSO models with a reference complexity of 3D,3O, and use the root mean square error (RMSE) for each method to assess their performance (see Table 1). For numerical stability, all methods employed a common step of applying a shift of Emin − E0 to all DFT-computed values, where Emin is the lowest adsorption energy among calculated values, and E0 is a small positive value (0.1 eV for all DFT-computed descriptors) to ensure that there are no zero values in the dataset. Afterwards, the following methods were employed one at a time to scale all primary features, i.e. DFT-obtained and T: (i) scale values for each primary feature between 0 and 1, (ii) divide values for each primary feature by the smallest value, (iii) replace T with log(T), (iv) remove T from primary features, and divide all DFT-obtained values by T/T0, where T0 is the lowest reaction temperature, 225 °C.
| Employed method | i | ii | iii | iv |
|---|---|---|---|---|
| RMSE-test | 2.66 | 3.81 | 3.90 | 3.85 |
| RMSE-train | 2.17 | 2.23 | 2.35 | 2.23 |
As shown in Table 1, the first pre-processing method yields the lowest RMSE for both test and train sets, and is therefore selected for creating the SISSO model. After scaling the primary features, the optimum model complexity should be determined. The accuracy of the trained model is expected to increase with complexity. However, overfitting by highly complex models must be avoided to facilitate generalization of the model to validation data, i.e. data not used in training. To establish the optimum trade-off between model accuracy and generalization ability, we adapt the leave-one-group-out cross validation procedure.24 In our work, we consider 16 levels of model complexity; from 1D,1O to 4D,4O. For each complexity level, a leave-one-group-out cross validation25 is carried out, where each group is represented by one of the 14 tested catalysts. This means that for each cross validation, a model is trained with data from 13 catalysts, and tested with the unseen data from the 14th catalyst. Thereby, the predictive power of each trained model is tested with unseen data from a catalyst with a completely different composition than that of the training set. The overall cross validation performance is evaluated by averaging the regression coefficient R2 across the trained models, which is a widely accepted method for evaluating the accuracy of ML models with small datasets.26,27
The cross-validation results are shown in Fig. 2a. As expected, the highest complexity (4D,4O) gives the highest average R2 of 0.95. But the respective RMSE values (Fig. S12†) suggest that a model with D = 3 and O = 1 is optimal for avoiding overfitting and yields the lowest average RMSE for the test set, which is selected for generation of the SISSO model in this study, which is shown in eqn (1):
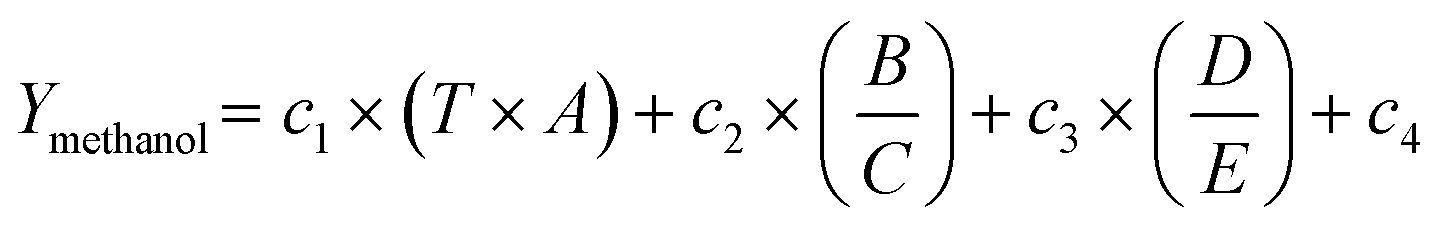 | (1) |
| Symbol in eqn (1) | Value |
|---|---|
| A | E ads of linear CO2 [config. Fig. 2d, A] |
| B | E ads of linear CHO2 [config. Fig. 2d, B] |
| C | E ads of linear CO [config. Fig. 2d, C] |
| D | E ads of linear CO2 [config. Fig. 2d, D] |
| E | E ads of linear CO + OH [config. Fig. 2d, E] |
| c 1 | 49.48 |
| c 2 | −29.04 |
| c 3 | 7.06 |
| c 4 | −18.40 |
 | ||
| Fig. 2 a) Evaluation of trained models after cross validation for different model complexities. b) Model accuracy for a selected complexity of 3D and 1O. c) The hatched lines show the detected constraints on SIS-constructed descriptors for outstanding subgroups. The color bar on the right shows the respective values for Ymethanol. d) Relaxed structures of intermediates identified by SISSO. A Pt promoted system is shown as an example. Carbon, oxygen and hydrogen atoms are shown as gray, red and white spheres respectively. Letters A–E are used to refer to each relaxed configuration (see Table 2). | ||
The scaling of primary features between 0 and 1 allows for linking the values of fitting constants c1–c3 to the importance of the respective SIS-constructed descriptors. The first term in eqn (1) which includes the temperature, has the highest weight (c1 = 49.48), meaning that the employed SISSO algorithm follows the general expectation of higher yield at elevated temperature. This highlights the relevance of test conditions to performance optimization. Despite having a significant impact on the performance, the reaction temperature should not be vastly varied to enhance the performance, because elevated temperatures, apart from reaching thermodynamic limits, may also increase the risk of structural changes that degrade the catalyst.28 A valid indicator of a potentially promising catalyst candidate may be the ability to perform well under industrially relevant conditions close to conventional methanol process conditions.29,30 In this study, the registered performance data were chosen in a way that the data used were not recorded under thermodynamic equilibrium conditions and test parameters are chosen accordingly.
From an atomistic perspective, eqn (1) shows us the desirable properties of a high-performing catalytic site. For the DFT-derived transformed and scaled descriptors, low values imply strong adsorption and vice versa. We notice that strongly bound but not activated linear CO2 (Fig. 2d A) negatively impacts performance. The other contributing factor is the optimal balance between the propensity of the site to bind an OH group, next to a CO (CO2) molecule, and a solely bound CO adsorbed at the active site. The key to improving performance is improving the affinity towards the substrate with an OH group in close proximity (Fig. 2d B and D) in relation to the substrate bound alone (Fig. 2d C and E). This can be interpreted as a leaving group from the substrate or H atom to be transferred to the substrate backbone.
The primary and secondary features from eqn (1) are used as potential descriptive parameters to establish design criteria for increased catalytic performance. To accomplish this, we used the subgroup-discovery (SGD) algorithm6,31,32 which is designed to identify distinct subgroups in the data and describe the most exceptional subgroup through the most relevant descriptive parameters from the provided set of parameters. The algorithm detects several competing subgroups, which are evaluated based on a quality function (see ESI† Methods-2 for more information).
The SGD results reconfirm that the reaction temperature and the SIS-constructed descriptors B/C and D/E are the key features for describing outstanding performance. Fig. 2c depicts the detected constraints for B/C ≤ 1.04 and D/E > 1.58 with hatched lines. The color map reflects the obtained values from eqn (1) for T = 250 °C and A = AIn, i.e. the respective value for the In reference catalyst. The variation ranges for B/C and D/E cover the minimum and maximum for each descriptor across the entire dataset. The colour of each scatter point in this figure reflects the measured value for the respective catalyst at the same temperature. The prediction and measurement show good agreement, which is particularly relevant in the area detected by SGD (Fig. 2c). The catalysts that form the subgroup and are located in this area are In–Pt, In–Pd, and In–Rh, while the catalyst that is furthest away from the detected subgroup area is In–Sn, which in fact shows the lowest measured yield among tested catalysts. The overlay of SISSO-generated values (shown by the colour map) and SGD-detected area (shown by hatched lines) highlights the ability of the employed workflow in autonomous prediction of exceptional performance. The developed design criteria in Fig. 2c can be used to explore new catalytic materials by computing the values for SIS-constructed descriptors and identifying materials that meet the constraints depicted. SGD provides numerical targets for the atomistic interpretation, allowing us to flag materials as good quality candidates during high-throughput computational screening.
Conclusions
We propose a workflow that utilizes atomic scale interaction parameters derived from high-throughput computational simulations to supplement the information available from high-throughput experimental data. This allows for a more comprehensive understanding of the catalyst optimization process going beyond traditional design of experiment methods. Our machine learning models trained with this enriched input descriptor space have the potential to predict the performance of new catalysts, such as the unseen Pt–In system, based on the training on other promoted systems (e.g. Rh, Pd, Nb, etc.) alone. Additionally, the workflow also opens prospects to further reduce future computational cost by identifying the key descriptors that have the highest predictive power for a given catalytic performance, and simultaneously construct design criteria for new catalysts. By applying this workflow to extensively studied indium based catalysts, we show that the design rules obtained align well with our understanding of chemical principles and highlight the influence of hydroxyl sites on the catalytic activity. The machine learning algorithms (SISSO and SGD) used in this study also offer the advantage of explainability, making them useful in catalyst applications, where experimental data are often limited. The results of this study demonstrate the potential of the workflow to identify descriptors and rules with sufficiently high predictive power for the potential development of new catalyst materials, making it a valuable tool for addressing the needs of a more modern and agile approach to industrial catalyst and process development.Data availability
This dataset is to be referred to as ‘InOX-14 dataset’, and the code will be available upon acceptance.Author contributions
The authors' contributions are described according to the CRediT (Contributor Roles Taxonomy from CASRAI). Conceptualization: S. D., E. F., M. K. S. A. S., and F. R.; data curation: M. K., E. F., and S. D.; formal analysis: M. K., E. F., S. D., and I. J.; funding acquisition: S. A. S. and F. R.; supervision: S. A. S., F. R., and S. D.; investigation: M. K., E. F., and S. D; methodology: S. D., E. F., M. K., C. B., I. J., M. M., and F. E.; project administration: C. B. and S. A. S.; resources: F. R. and S. D.; validation: M. K., E. F., and S. D.; visualization: M. K. and E. F.; writing – original draft: M. K., E. F., and S. D.; writing – review and editing: M. K., E. F., S. D., S. A. S., F. R., A. S., and R. N. A.Conflicts of interest
There are no conflicts to declare.References
- C. F. Shih, T. Zhang, J. Li and C. Bai, Powering the Future with Liquid Sunshine Choon, Joule, 2018, 2, 1925–1949 CrossRef CAS
.
- X.-Y. Meng, C. Peng, J. Jia, P. Liu, Y.-L. Men and Y.-X. Pan, Recent progress and understanding on In2O3-based composite catalysts for boosting CO2 hydrogenation, J. CO2 Util., 2022, 55, 101844 CrossRef CAS
- A. Adamu, F. Russo-Abegão and K. Boodhoo, Process intensification technologies for CO2 capture and conversion – a review, BMC Chem. Eng., 2020, 2, 1–18 CrossRef
- S. Liu, L. R. Winter and J. G. Chen, Review of Plasma-Assisted Catalysis for Selective Generation of Oxygenates from CO2 and CH4, ACS Catal., 2020, 10, 2855–2871 CrossRef CAS
- R. Snoeckx and A. Bogaerts, Plasma technology-a novel solution for CO2 conversion?, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC
- L. Foppa,
et al., Learning Design Rules for Selective Oxidation Catalysts from High-Throughput Experimentation and Artificial Intelligence, ACS Catal., 2022, 12, 2223–2232 CrossRef CAS PubMed
- F. Göltl, Three Grand Challenges for the Computational Design of Heterogeneous Catalysts, J. Phys. Chem. C, 2022, 126, 3305–3313 CrossRef
- B. R. Goldsmith, J. Esterhuizen, J. X. Liu, C. J. Bartel and C. Sutton, Machine learning for heterogeneous catalyst design and discovery, AIChE J., 2018, 64, 2311–2323 CrossRef CAS
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed
- S. Pablo-García,
et al., Generalizing Performance Equations in Heterogeneous Catalysis from Hybrid Data and Statistical Learning, ACS Catal., 2022, 12, 1581–1594 CrossRef
- A. J. Saadun,
et al., Performance of Metal-Catalyzed Hydrodebromination of Dibromomethane Analyzed by Descriptors Derived from Statistical Learning, ACS Catal., 2020, 10, 6129–6143 CrossRef CAS
- O. Martin and J. Pérez-Ramírez, New and revisited insights into the promotion of methanol synthesis catalysts by CO2, Catal. Sci. Technol., 2013, 3, 3343–3352 RSC
- P. Sharma, J. Sebastian, S. Ghosh, D. Creaser and L. Olsson, Recent advances in hydrogenation of CO2into hydrocarbonsviamethanol intermediate over heterogeneous catalysts, Catal. Sci. Technol., 2021, 11, 1665–1697 RSC
- O. Martin,
et al., Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed
- S. Dang,
et al., Rationally designed indium oxide catalysts for CO2 hydrogenation to methanol with high activity and selectivity, Sci. Adv., 2020, 6, eaaz2060 CrossRef CAS PubMed
- A. Trunschke, Prospects and challenges for autonomous catalyst discovery viewed from an experimental perspective, Catal. Sci. Technol., 2022, 12, 3650–3669 RSC
- B. M. Weckhuysen and J. Yu, Recent advances in zeolite chemistry and catalysis, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC
- A. Urakawa, Trends and advances in Operando methodology, Curr. Opin. Chem. Eng., 2016, 12, 31–36 CrossRef
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler and L. M. Ghiringhelli, SISSO: A compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates, Phys. Rev. Mater., 2018, 2, 1–12 Search PubMed
- L. M. Ghiringhelli,
et al., Learning physical descriptors for materials science by compressed sensing, New J. Phys., 2017, 19, 023017 CrossRef
- L. M. Ghiringhelli, J. Vybiral, S. V. Levchenko, C. Draxl and M. Scheffler, Big data of materials science: Critical role of the descriptor, Phys. Rev. Lett., 2015, 114, 1–5 CrossRef PubMed
-
J. Cutler and M. Dickenson, Introduction to Machine Learning with Python, 2020, DOI:10.1007/978-3-030-36826-5_10
- T. Aonishi, K. Mimura, M. Okada and Y. Yamamoto, L0 regularization-based compressed sensing with quantum-classical hybrid approach, Quantum Sci. Technol., 2022, 7, 3 Search PubMed
- Y. Jung and J. Hu, A K-fold averaging cross-validation procedure, J. Nonparametric Stat., 2015, 27, 167–179 CrossRef PubMed
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel and B. Thirion, Scikit-learn: Machin Learning in Python, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- Y. Zhang,
et al., Phase prediction in high entropy alloys with a rational selection of materials descriptors and machine learning models, Acta Mater., 2020, 185, 528–539 CrossRef CAS
- C. Zou,
et al., Integrating data mining and machine learning to discover high-strength ductile titanium alloys, Acta Mater., 2021, 202, 211–221 CrossRef CAS
-
J. U. Bauer, Kinetic and Mechanistic Studies for the Direct Conversion of Syngas to Ethanol, PhD thesis, Technische Universität Berlin, 2020 Search PubMed
- M. Khatamirad,
et al., A Systematic Approach to Study Complex Ternary Co-Promoter Interactions: Addition of Ir, Li, and Ti to RhMn/SiO2 for Syngas Conversion to Ethanol, Catalysts, 2022, 12, 1321 CrossRef CAS
- M. Khatamirad,
et al., Silica-supported Catalyst System Rh-Mn-Ir-Li-Ti in Syngas to Ethanol Reaction: Reactivity Trends and Performance Optimization, ChemCatChem, 2023, 15, e202201104 CrossRef CAS
- M. Atzmueller, Subgroup discovery, Wiley Interdiscip. Rev.: Data Min. Knowl. Discov., 2015, 5, 35–49 Search PubMed
- J. H. Friedman and N. I. Fisher, Statistics and Computing, Volume 9, Number 2 - SpringerLink, Stat. Comput., 1999, 9, 123–143 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00148b |
| ‡ These authors have an equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |