
Synthetic strategies for fluorosulfonylated compounds: application to click chemistry reactions
Sebastián
Barata-Vallejo
 *ab,
Damian E.
Yerien
a and
Al
Postigo
*a
*ab,
Damian E.
Yerien
a and
Al
Postigo
*a
aFacultad de Farmacia y Bioquímica, Departamento de Ciencias Químicas, Universidad de Buenos Aires, Junín 954, Buenos Aires, CP 1113, Argentina. E-mail: apostigo@ffyb.uba.ar
bIstituto per la Sintesi Organica e la Fotoreattività ISOF, Consiglio Nazionale delle Ricerche, Via P. Gobetti 101, Bologna, 40129, Italy. E-mail: sebastian.barata@isof.cnr.it
First published on 1st March 2023
Abstract
The syntheses and applications of fluorosulfonylated organic compounds have flourished in the last ten years due to their versatility to participate in click chemistry (SuFEx) reactions. Also, organic architectures that combine the SO2F group and other ancillary functional moieties such as olefins, alkynes, etc. (i.e.: bis-electrophiles) have augmented the applications and diversity of the end compounds. To this effect, the association of an alkyne functionality and the SuFExable group within one structure has been shown to encompass two-in-one click chemistry sequential protocols with the aim of building on the diversity of scaffolds by two consecutive click processes. We next examine the syntheses of (hetero)aromatic-, alkyl-, alkenyl-, and alkynyl-sulfonyl fluorides and β-keto-sulfonyl fluorides and the syntheses of compounds bearing N–SO2F and O–SO2F bonds through diverse catalytic methods, illustrating examples of their SuFEx click chemistry and other ancillary functional group reactivity.
 Sebastián Barata-Vallejo | Sebastián Barata-Vallejo was born in General Villegas (Argentina) and holds degrees in Pharmacy (2007) and Biochemistry (2010). He obtained his Ph.D. degree (2012) from the University of Buenos Aires, studying radical reactions in aqueous and microheterogeneous media under the supervision of Prof. A. Postigo. He has been a research fellow and has held several postdoctoral positions at the Istituto per la Sintesi Organica e la Fotoreattività (ISOF), Consiglio Nazionale delle Ricerche (CNR), Bologna, Italia, under the supervision of Dr. C. Chatgilialoglu, studying biomimetic radical reactions and their mechanisms. He is currently a researcher at the National Council for Scientific and Technical Investigation, CONICET (Argentina), a research associate at ISOF–CNR, Bologna, Italy, and a lecturer at the Department of Chemical Sciences, Faculty of Pharmacy and Biochemistry, University of Buenos Aires. His research activities focus on radical organic chemistry, in particular carbon- and sulfur-centered radical reactivity, fluoroalkylation reactions by radical pathways, and photocatalysis. |
 Damian E. Yerien | Damian E. Yerien was born in Argentina and obtained his degree in Biochemistry from the University of Buenos Aires in 2014. He obtained his Ph.D. degree (2019) from the University of Buenos Aires studying synthetic and mechanistic aspects of radical perfluoroalkylation reactions through photoredox catalysis, under the direction of Prof. Dr. Al Postigo. He is currently a researcher at the National Council for Scientific and Technical Investigation, CONICET (Argentina) and a teaching assistant at the Department of Chemical Sciences, Faculty of Pharmacy and Biochemistry, University of Buenos Aires. |
 Al Postigo | Al Postigo was born in Argentina and obtained his M.Sc. degree from the University of Buenos Aires in 1986. He moved to Canada in 1990 and obtained his Ph.D. from McMaster University in 1994, under the direction of Prof. Dr. W. J. Leigh. After postdoctoral positions in Canada, he returned to Argentina and worked with Prof. Dr. R. Rossi at the University of Córdoba in the area of radical ion reactions. He held assistant and associate professorship positions at the University of Córdoba, the University of Buenos Aires, and the University of Belgrano. He is currently a full professor of Organic Chemistry at the Department of Chemical Sciences, Faculty of Pharmacy and Biochemistry, University of Buenos Aires. His interests are in the areas of radical chemistry and fluorine chemistry. He is devoted to studying radical reactions of these species in water and nonconventional media. |
A. Introduction
The association of sulfur and fluorine atoms has granted organic, biological and medicinal chemistry and materials sciences with most valuable groups, such as –SF5,1–3 –SF4Cl,1,2 –OSO2F,4–6 –SO2CF3,7–10 –S(O)CF3,11,12 –SO2RF, –S(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(N)(R)F,13 and –SO2F,14 among others.
O)(N)(R)F,13 and –SO2F,14 among others.
Sulfonyl fluorides (R(Ar)–SO2F) are chemically stable towards reduction15 and hydrolysis,16 and bear special proton-mediated reactivity.14 The –SO2F group, which is known to be stable under physiological conditions, has relevance in biological chemistry; as such, it encompasses a class of pharmacophores that provide permanent inhibition of target proteins. Furthermore, sulfonyl fluoride biological probes are being used worldwide.17 Aromatic sulfonyl fluorides react chemoselectively with tyrosine in the presence of other nucleophilic amino residues. For instance, phenyl methyl sulfonyl fluoride (PMSF) and 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF, Pefabloc®) are protease inhibitors, and in the case of PMSF, it is widely applied in biochemistry.18,19 In addition, there are above 150 approved drugs in the market that contain S(VI) functionalities.20,21
Sulfonyl fluorides have also widespread applications in the field of organic chemistry, as fluorinating reagents22,23 and synthetic precursors for the preparation of sulfonamides,24 sulfones,25 and sulfonate esters,4 as well as 18F radiolabeling.26,27
Nonetheless, the most relevant application of sulfonyl fluorides is sulfur(VI) fluoride exchange (SuFEx), considered a valuable click reaction as firstly proposed by Sharpless and coworkers in their seminal paper of 2014.14 As definition of click reactions go, “simple synthetic operational processes that work under oxygen, are water tolerant, afford products in high yields and with minimal purification requirements effecting carbon-heteroatom linkages”,14 the sulfonyl fluoride group plays a central role as a precursor of SO2 connectors. Hence, sulfonyl fluorides are of particular interest in drug discovery as click functionalities, which facilitate rapid derivatization of sulfonylated analogs in structure–activity relationships.
A typical approach to introduce SO2F is by fluorination of sulfonyl chlorides, employing KF/18-crown-6 in water28 or KHF2.14 Many reagents have been developed in the SuFEx field. Sulfuryl fluoride (SO2F2),29 a gas reagent, can react with oxygen or nitrogen nucleophiles to afford fluorosulfates (or fluorosulfonates) or fluorosulfonamides, respectively, which in turn can function as click connectors for ulterior nucleophilic substitutions.
Ethene sulfonyl fluoride (ESF) (vide infra, section C.2.2.)20 is considered a bis-electrophile for click chemistry.30a,b Thionyl tetrafluoride (SOF4)30c provides two click sites by reaction with primary amino groups. 1-Bromoethene-1-sulfonyl fluoride (1-Br-ESF), with a bromide moiety connected to the vinyl group adjacent to the fluoride, has also been proposed as a SuFEx reagent.31
Radical sulfur dioxide insertion/fluorination employing DABSO (1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct) as a SO2 surrogate followed by electrophilic fluorination (or by inorganic fluoride sources) is another approach for obtaining sulfonyl fluorides.32 Thus, there is a significant need for alternative methods (one-pot procedures) to synthesize aromatic and aliphatic sulfonyl fluorides that allow the simultaneous incorporation of both SO2 and F groups, without the requirement of previously installed SO2 or F functionalities.
Recent review articles on the applications and reagents,33 properties and reactions34a of sulfonyl fluorides have been advanced, as well as an account on classification34b based on families of organic fluorosulfonylated compounds.33,35 Radical synthetic procedures to obtain fluorosulfonylated aliphatic compounds have very recently been reviewed.36,37 The extraordinary progress in the past few years on the chemistry of sulfonyl fluorides is driven by the SuFEx chemistry which represents one of the most significant click reactions, with relevance in numerous fields and research areas.38
We next examine, from the organic chemistry perspective, the catalytic syntheses of (hetero)aromatic-, alkyl-, alkenyl-, and alkynyl-sulfonyl fluorides and β-keto-sulfonyl fluorides and the syntheses of compounds with N–SO2F and O–SO2F bonds, illustrating examples of their SuFEx chemistry and other ancillary functional group reactivity. A summary of all reactions is presented in Tables 1–4.
| No. | Starting substrate | Product | Reagents & reaction conditions | Ref. |
|---|---|---|---|---|
| 1 | ArSO 2 NHNH 2 | ArSO 2 F | Selectfluor | 43a |
| H2O, 60 °C | ||||
| 2 | Ar–SO 2 Na | ArSO 2 F | Selectfluor | 43a |
| H2O, 60 °C | ||||
| 3 | Ar–SO 2 NH 2 | ArSO 2 F | Pyrylium bromide | 42 |
| MgCl2, KF, MeCN, then H2O | ||||
| 4 |
Ar–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N–SO
2
Me N–SO
2
Me
|
ArSO 2 F |
K
2
S
2
O
5
, NFSI, MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, blue LEDs, r.t., 4 h :H2O, blue LEDs, r.t., 4 h |
55 |
| 5 | Ar–I | ArSO 2 F | 1) Pd(OAc)2DABSO | 4 |
| PAd2Bu, iPrOH 75 °C, 16 h | ||||
| 2) Selectfluor | ||||
| 6 |
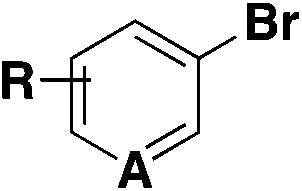
|
(Het)SO 2 F | 1) DABSO, PdCl2(AmPhos)2, Et3N, i-PrOH | 32 |
| 75 °C, 24 h | ||||
| 2) NFSI | ||||
| 7 | (TMS)ArOTf | ArSO 2 F | NHR1R2, SO2F2, KF, 18-crown-6 THF | 44 |
| 8 |
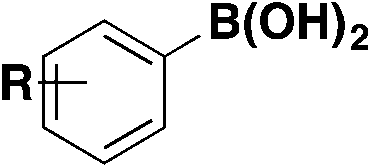
|
ArSO 2 F | 1) NiBr2 (glyme), Tmphen, DABSO, LiOt-Bu | 45 |
| DMI, 100 °C, 16 h | ||||
| 2) NFSI, DIPEA | ||||
| 9 | (Het)aryl-SH or (Het)aryl-SS-aryl(Het) | (Het)aryl-SO 2 F | Batch electrocell, (C/Fe) KF, pyridine, CH3CN/HCl, 20 mA, r.t., 6–48 h | 47 |
| 10 | ArN 2 BF 4 | ArSO 2 F | DABSO, KHF2, CuCl2, 6,6′-dimethyl-2,2′-dipyridyl, MeCN, r.t., 12 h | 49 |
| 11 | ArN 2 BF 4 | ArSO 2 F |
Na
2
S
2
O
5
, NFSI, MeCN/H2O = 20:1 |
50 |
| N2, 60 °C, 6 h | ||||
| 12 | ArN 2 BF 4 | ArSO 2 F | DABSO, 3DPAFIPN | 54 |
| KHF 2 , MeCN, blue LED, r.t., 16 h | ||||
| 13 | ArN 2 BF 4 | ArSO 2 F | K 2 S 2 O 5 , NFSI, MeOH/H2O/AcOH, r.t., 6 h | 51, 52 |
| 14 | ArN 2 BF 4 | ArSO 2 F | Na 2 S 2 O 5 , NFSI, Selectfluor, MeOH, 70 °C, 9 h | 53 |
| 15 | (Het)Ar–MgX | (Het)Ar–SO 2 F | SO 2 F 2 , THF, 23 °C, 1 h | 46 |
| 16 | R–N 3 |
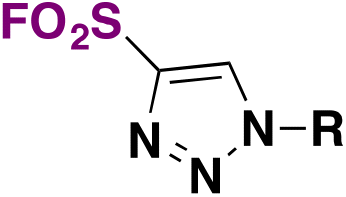
|
DMF, 50 °C, 14 h | 59 |
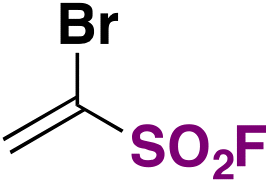
|
||||
| 17 |
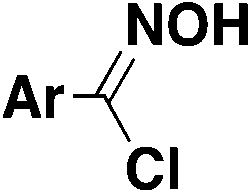
|
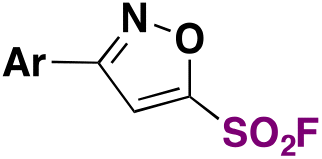
|
N(Pr)3tBuOH, r.t. | 31 |
|
||||
| 18 |
|
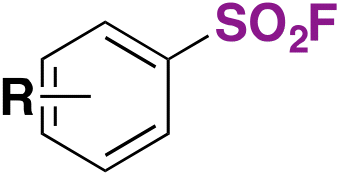
|
DABSO, NFSI, base, [Cu], MeCN, 40 °C, 2 h | 43b |
| Entry | Starting substrate | Product | Reagents & reaction conditions | Ref. |
|---|---|---|---|---|
| 1 | R–Br | R–SO 2 F | 1. Rongalite, DMSO, r.t. | 66 |
| 2. H3PO4 | ||||
| 3. DIPEA, NFSI | ||||
| 2 | R–I |

|

|
60 |
| Mn2(CO)10, HE, DMSO | ||||
| 5 W blue LED, r.t. 24 h, Ar | ||||
| 3 | RCOOH |

|
Na
2
S
2
O
5
, NFSI, Cu, Na2HPO4, MeCN:H2O, r.t. |
70 |
| 4 |
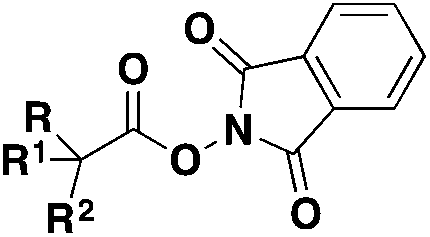
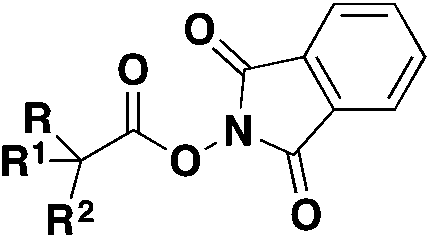
|
|

|
76 |
| Eosin-Y Na2, HE, MeCN | ||||
| Blue LED, Ar, r.t., 12–24 h | ||||
| 5 |
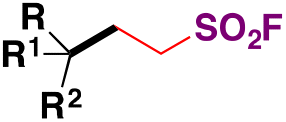
|

|
1. Na2S2O4, Zn, DMPr/H2O, 80 °C, 9 h | 68 |
| 2. NFSI, r.t., 4 h | ||||
| 6 |
|

|
DABSO, NFSI | 65 |
| [Ir(dF(CF3)ppy)2(bpy)]PF6 | ||||
| K3PO4, MeCN:CH2Cl2 |
||||
| 30 W blue LED, r.t., 25 h | ||||
| 7 |
|
|
1. DABSO, HE, base DMA, 16 h, blue LEDs or heat | 74 |
| 2. NFSI, 4 h, r.t | ||||
| 8 |
|
|
|
61 |
| CsF, EtOAc, 50 °C, 12 h | ||||
| 9 |
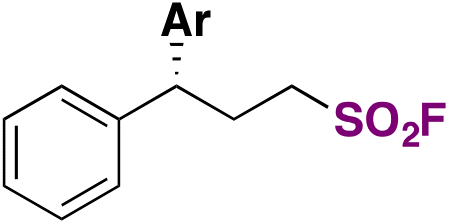
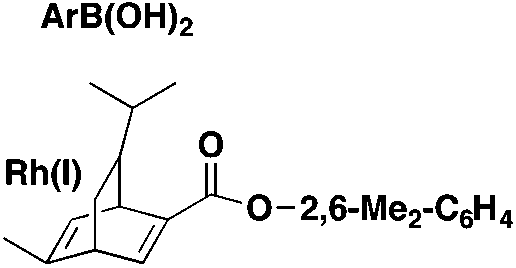
|
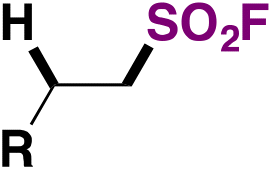
|
|
73 |
| ODA, CHD, 1,4-dioxane | ||||
| Blue LED, r.t., 24 h | ||||
| 10 |
|
|
|
111 |
| ODA, 1,4-dioxane | ||||
| Blue LED, r.t., 24 h | ||||
| 11 |

|

|
Method A | 92 |
| 1. n-BuLi, −78 °C | ||||
| 2. FSO2OSO2F, −78 °C | ||||
| Et2O | ||||
| Method B | ||||
| 1. n-BuLi, −78 °C | ||||
| 2. SO2, −78 °C then to r.t. | ||||
| 3. NFSI | ||||
| 12 |
|
|
|
72 |
| AIBN, EtOAc | ||||
| 85 °C, 24 h |
| Entry | Starting substrate | Product | Reagents & reaction conditions | Ref. |
|---|---|---|---|---|
| 1 |
|
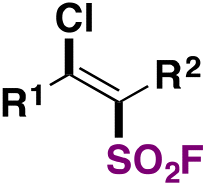
|
FSO 2 Cl, fac-Ir(ppy)3, Et2O/PhCF3 | 79 |
| Blue LEDs, 24–72 h | ||||
| 2 |
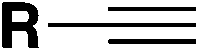
|
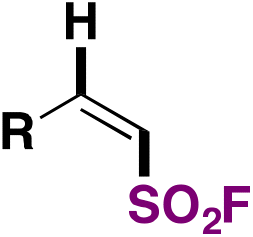
|
(FABI) | 73 |
| ODA, (TMS)3SiH, 1,4-dioxane | ||||
| Blue LED, r.t., 24 h | ||||
| 3 |
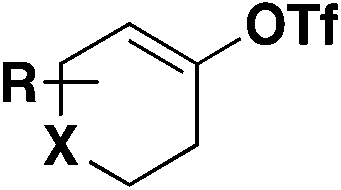
|
|
1) DABSO, PdCl2(AmPhos)2, Et3N, i-PrOH, 75 °C | 80 |
| 2) NFSI | ||||
| EtOAc | ||||
| 4 |
|
|
FABI | 81 |
| fac-Ir(ppy)3, 1,4-dioxane | ||||
| Blue LED, r.t., 12 h | ||||
| 5 |
|
|
IMSF, 4CzIPN, KH2PO4, DME, blue LED | 71 |
| 6 |
|
|
FSO 2 Cl | 78 |
| Ir[dF(CF3)ppy]2(dtbbpy)PF6 | ||||
| Et2O/PhCF3, Ar, r.t. | ||||
| Blue LEDs (460 nm), 12 h | ||||
| Then aq. Na2CO3 | ||||
| 7 |
|
|

|
99 |
| Pd(OAc)2, acetone | ||||
| r.t., 5–15 h | ||||
| 8 |

|
|
|
86 |
| Pd(OAc)2, AgOAc, HFIP | ||||
| 100 °C, 24 h | ||||
| 9 |
|
|

|
88, 100 |
| [Cp*RhCl2]2, AgSbF6 | ||||
| Cu(OAc)2·H2O, Ac2O | ||||
| DCE, 80 °C, 20 h, air | ||||
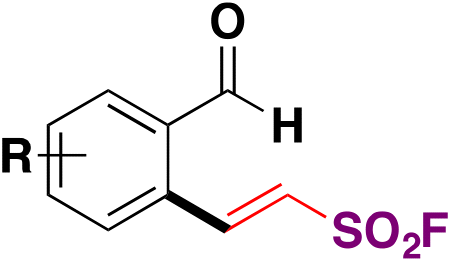
| 10 |
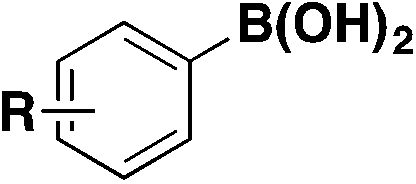
|
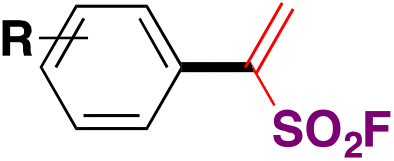
|
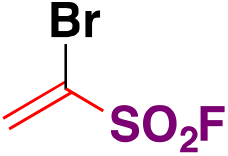
|
90 |
| Pd(dba)3, ligand, K3PO4, toluene, 50 °C, 24 h |
| Entry | Starting substrate | Product | Reagents & reaction conditions | Ref. |
|---|---|---|---|---|
| 1 |
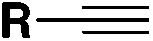
|
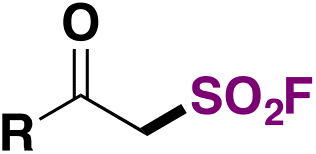
|
FSO 2 Cl | 108 |
| LiClO4, Et2O, Mg(+)/Al(−) | ||||
| U cell = 15 V, O2 (air), r.t., 6 h | ||||
| Undivided cell | ||||
| 2 |
|
|
FSO 2 Cl | 109, 112 |
| Et4NPF6, Et2O, GF(+)/GF(−) | ||||
| U cell = 15 V, O2 (air), r.t., 6 h | ||||
| Undivided cell. r.t., 6 h | ||||
| 3 | RNH 2 |
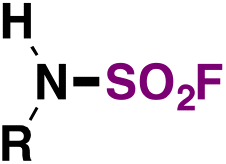
|
 (IMSF)
(IMSF)
|
6 |
| CH3CN, 0 °C to r.t. | ||||
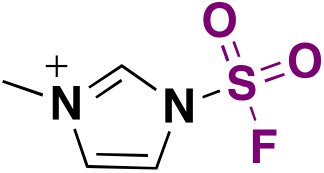
| 4 | ArNH 2 |

|
 (IMSF)
(IMSF)
|
6 |
| CH2Cl2, 0 °C to r.t. | ||||
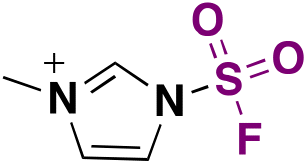
| 5 | R 1 R 2 NH |
|
 (IMSF)
(IMSF)
|
6 |
| CH3CN, r.t. | ||||
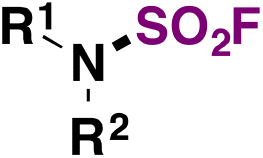
| 6 | RR 1 NH | RR 1 NSO 2 F | AISF, DBU, THF, r.t. 10 min | 5 |
| 7 | Ar–OH | Ar–O–SO 2 F |
 (IMSF)
(IMSF)
|
6 |
| CH3CN, r.t. |
B. Fluorosulfonylation of (hetero)aromatic compounds
Apart from the classical Cl/F exchange in aryl sulfonyl chlorides, the synthesis of aromatic sulfonyl fluorides has been studied for some time. The reaction of disulfides with 6.5 equivalents of Selectfluor in refluxing acetonitrile/water (10:1) provided sulfonyl fluorides in high yields.39–41 Aryl sulfonamides can also be used as starting substrates for the synthesis of aryl sulfonyl fluorides.42
In 2016, an indirect metal-free method (a method employing substrates that already contain S) to obtain aromatic sulfonyl fluorides was reported by Tang, Wang, and collaborators.43a The method required pre-synthesized aryl sulfonyl hydrazides as starting materials but ran without catalysts or additives and was carried out in water under an air atmosphere; the fluorinating agent was Selectfluor. The authors43a also attempted the fluorination in water from sodium arylsulfinates, resulting in good yields of arylfluorosulfonylated products. The scope of the reaction regarding sulfonylhydrazides is depicted in Scheme 1.
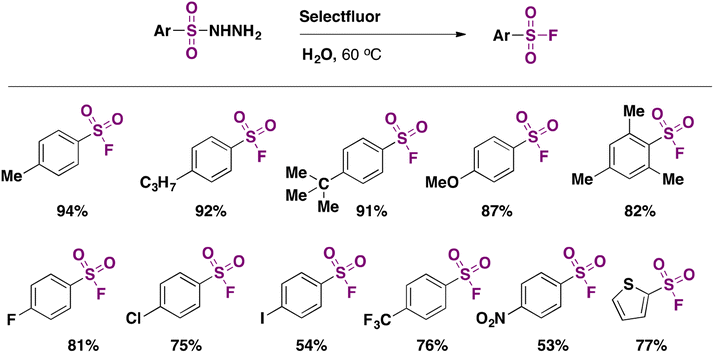 | ||
| Scheme 1 Selected examples of fluorination of arylhydrazides in water. | ||
Substrates bearing both electron withdrawing and releasing groups afforded good yields of fluorosulfonylated products (Scheme 1). The scope regarding sodium arylsulfinates is presented in Scheme 2.
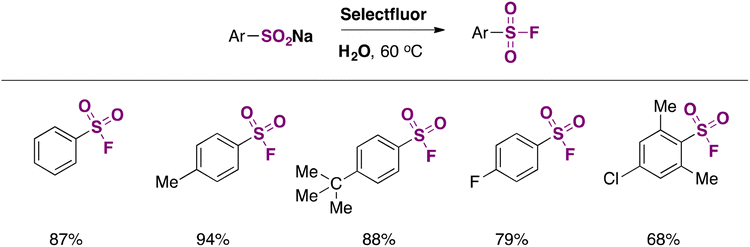 | ||
| Scheme 2 Selected examples for the fluorination of sodium arylsulfinates in water. | ||
The authors studied the reaction mechanism. When radical scavenger TEMPO (2,2,6,6-tetramethyl-1-piperridinyloxy) was introduced in the reaction mixture, the formation of fluorosulfonylated products was suppressed, suggesting a free radical pathway. However, TEMPO had no effect on the reaction of sodium arylsulfinate salts. Consequently, the authors43a proposed a mechanism where the aryl sulfonylhydrazide reacts with Selectfluor to afford a fluorine radical and radical intermediate I, releasing gaseous nitrogen in the presence of water, and an acidic solution is formed (Scheme 3). Resonance forms I and II (Scheme 3) undergo fluorine atom transfer from Selectfluor to yield the sulfonyl fluoride.
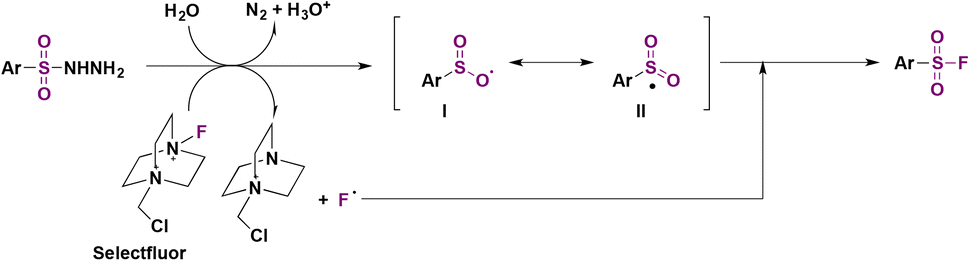 | ||
| Scheme 3 Proposed reaction mechanism. | ||
One advantage of the work of Wang43a is the innocuous reaction medium. However, the use of expensive Selectfluor limits the large-scale application of the protocol.
In 2017, Tribby, Ball and collaborators4 came up with a relatively simple method to convert aryl iodides to aryl sulfonyl fluorides, employing a Pd catalyst, DABSO as a SO2 surrogate, and Selectfluor as the source of fluorine, in isopropyl alcohol as solvent, at 75 °C for 16 h. This protocol is a one-pot procedure and requires only column chromatography for product purification. The scope of the transformation is shown in Scheme 4.
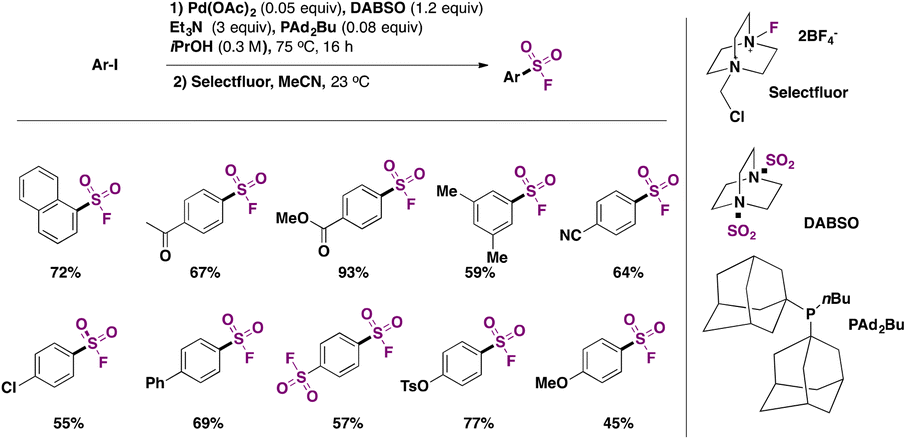 | ||
| Scheme 4 Selected examples for the fluorosulfonylation of aryl iodides. | ||
Aryl iodides with electron releasing and withdrawing groups could be converted to the respective sulfonyl fluorides (Scheme 4). Even 1,4-di-iodobenzene afforded the disubstitution product in good yield. The authors4 utilized the above-described synthesized sulfonyl fluorides in click reactions with nucleophiles, obtaining coupling products with imidazole, phenol, 4-methoxyphenol, and 4-aminophenol, among others.
Later in 2017, Davies, Bagley, Willis and colleagues32 employed (hetero)aryl bromides in Pd-catalyzed fluorosulfonylation in the presence of DABSO as a SO2 surrogate and triethylamine, in isopropanol as solvent, at 75 °C for 24 h. The scope of the transformation is shown in Scheme 5.
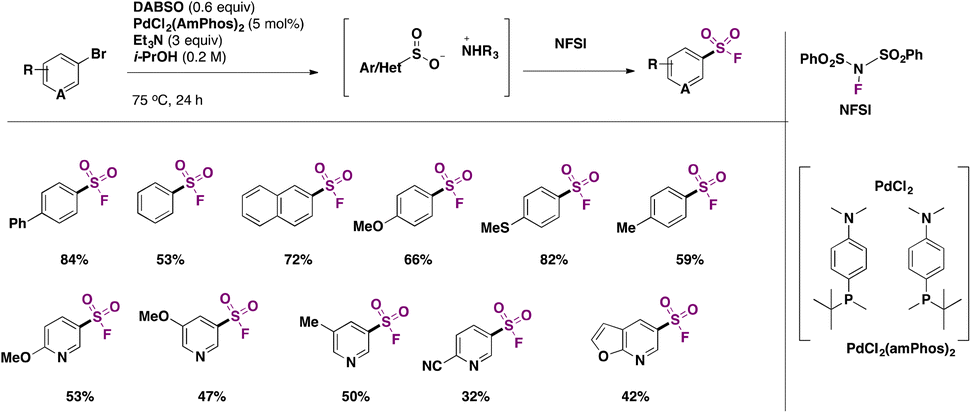 | ||
| Scheme 5 Selected examples for the fluorosulfonylation of (hetero)arylbromides. | ||
Aryl bromides with electron donating and neutral groups afforded good yields of fluorosulfonylated products. Bromopyridines with either electron donating or withdrawing groups afforded good yields of products as well. A comparison with the work of Ball and colleagues4 shows that, in general, bromides give lower overall product yields than iodides, and somewhat harsher reaction conditions with regard to temperature should be applied in the case of bromides as compared to iodides.
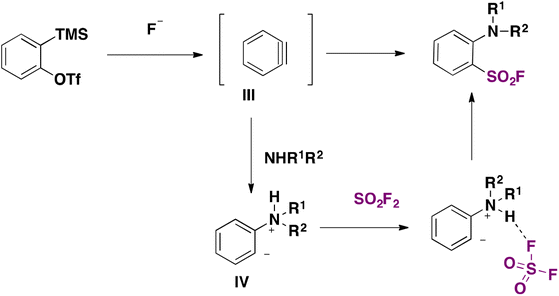
A transition metal-free synthesis of arenesulfonyl fluorides from arynes was developed in 2019 by Kwon and Kim.44 SO2F2 was used as a fluorosulfonylating reagent. The aryne precursor was 2-(trimethylsilyl)phenyl trifluoromethanesulfonate in the presence of an aniline derivative and KF/18-crown-6 in THF as solvent, according to Scheme 6.
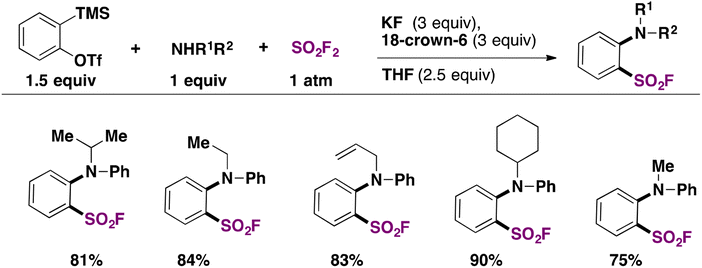 | ||
| Scheme 6 Selected examples for the fluorosulfonylation of arynes. | ||
The authors44 suggested a reaction mechanism such as that presented in Scheme 7. Aryne III is formed in situ by KF attack on 2-(trimethylsilyl)phenyl trifluoromethanesulfonate, giving rise to zwitterion IV, which reacts with SO2F2 to obtain the product.
 | ||
| Scheme 7 Proposed reaction mechanism. | ||
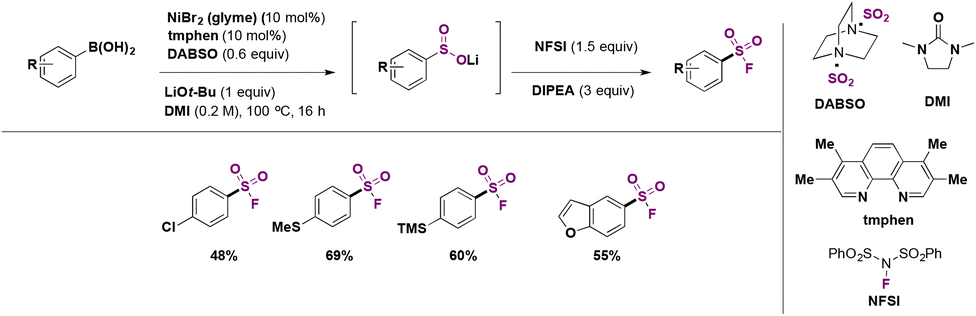
In 2019, Lo, Willis and colleagues45 carried out the syntheses of various (hetero)arylsulfones by Ni catalysis from aryl boronic acids, among which the synthesis of (hetero)aryl fluorosulfones was reported to be quite convenient by this methodology. Selected examples are shown in Scheme 8.
 | ||
| Scheme 8 Selected examples for the fluorosulfonylation of (hetero)aryl boronic acids. | ||
From Scheme 8, it is observed that aryl boronic acids with either electron withdrawing or releasing groups afforded good yields of fluorosulfonylated products.
In 2019, Lee, Ball, and Sammis employed SO2F2 as a fluorosulfonylating reagent of (hetero)aryl and alkyl magnesium salts (see Table 1).46
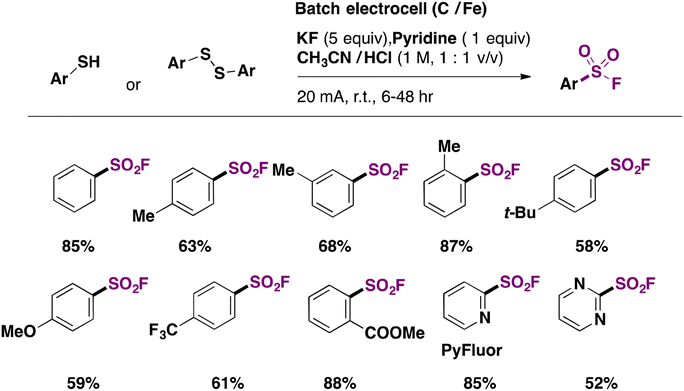
Laudadio, Noël and colleagues47 utilized aromatic sulfides or disulfides in inexpensive graphite/stainless steel electrodes to obtain fluorosulfonylated aromatic compounds. The electrochemical reaction involved the addition of pyridine (1 equiv.) as an electron mediator or phase transfer catalyst, KF as the fluorine source and electrolyte (5 equiv.) in a CH3CN/1 M HCl biphasic reaction mixture. The scope of the transformation is shown in Scheme 9.
 | ||
| Scheme 9 Scope of the electrochemical fluorosulfonylation of thiols. | ||
Electron neutral, donating and withdrawing groups attached to the aryl sulfide provided the respective fluorosulfonylated products in good yields. Also, it was noticed that steric congestion in the ortho positions of the aryl sulfides afforded products in good yields as well. The synthesis of pyridine-2-sulfonyl fluoride (Scheme 9, second row of products), also known as PyFluor (a deoxyfluorination reagent), could be accomplished in excellent yields.22 The authors47 employed aliphatic thiols as well for the electrochemical fluorosulfonylation reaction (not shown) affording excellent yields of fluorosulfonylated aliphatic products.
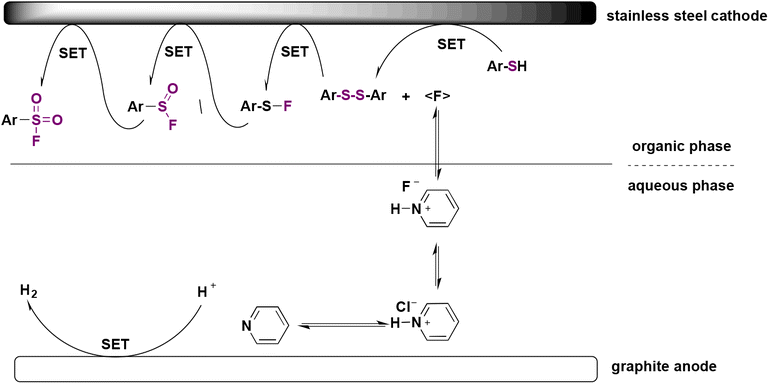
The authors47 investigated the reaction mechanism. Anodic oxidation of aryl sulfide to disulfide was confirmed by kinetic experiments. Oxidation of disulfide results in a radical cation,48 which reacts with fluoride to give sulfenyl fluoride (Ar–SF, nucleophilic fluorination). The addition of TEMPO or butylated hydroxytoluene as radical scavengers lowers the efficacy of the electrochemical event, leading to a radical process. A sulfinyl fluoride (Ar–SOF) intermediate was also detected, but could not be isolated, with sulfonic acid being the major side product. The mechanism is described in Scheme 10.
 | ||
| Scheme 10 Mechanism for the electrochemical fluorosulfonylation of sulfides. | ||
Arene diazonium salts have been employed since 2020 as aryl precursors for the syntheses of fluorosulfonylated arenes. Liu, Chen, Liu, and colleagues49 proposed the use of DABSO as a surrogate of SO2, in the presence of KHF2 and CuCl2 salt and 6,6′-dimethyl-2,2′-dipyridyl as a ligand to accomplish the fluorosulfonylation of tetrafluoroborate arene diazonium salts. Selected examples of the transformation are shown in Scheme 11.
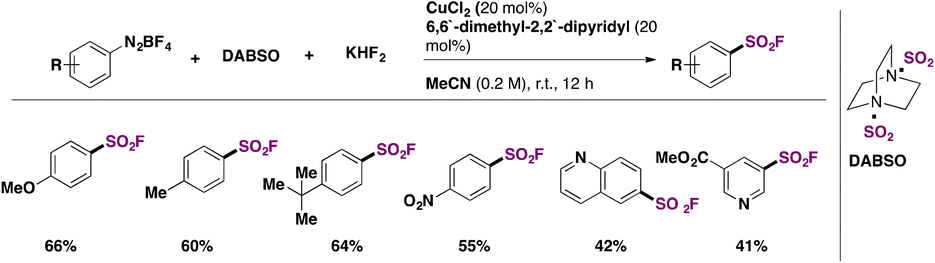 | ||
| Scheme 11 Selected examples for the fluorosulfonylation of aryldiazonium salts employing a Cu catalyst. | ||
Both arenes with electron donating and withdrawing groups afforded the fluorosulfonylated products in good yields. The authors49 investigated the reaction mechanism and concluded that the mechanistic pathways depended on the electronic nature of the arene. They also applied the SuFEx chemistry and obtained coupling products from aryl fluorosulfonates (sulphates, sulfonamides, etc.).
Liu, Qing, and colleagues50 improved on the technique by Liu and co-workers49 doing without the Cu catalyst and the costly DABSO reagent, replacing the latter with Na2S2O5 as the SO2 source. However, the source of F is the more costly NFSI (N-fluorobenzenesulfonimide) rather than the inexpensive KHF2 employed by Liu.49 In Scheme 12, a brief scope of the fluorosulfonylation of arenediazonium salts is presented.
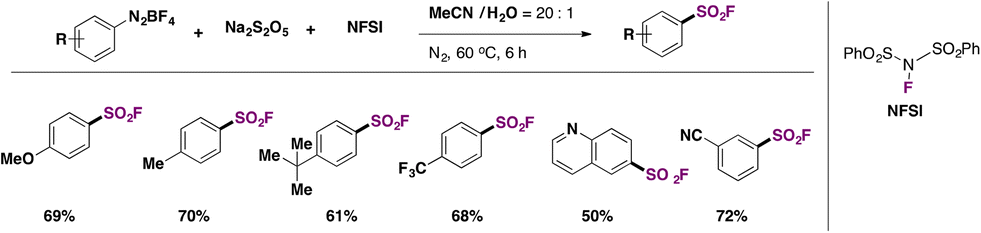 | ||
| Scheme 12 Scope for the fluorosulfonylation of arenediazonium salts with Na2S2O5 as the source of SO2 and NFSI as the F contributor. | ||
Comparing yields from identical products between the two methodologies described in Scheme 11 (Liu and colleagues) and Scheme 12 (Qing and collaborators), similar reaction efficacies are observed from 4-methoxybenzenediazonium, 4-methylbenzenediazonium, 4-tert-butylbenzenediazonium, and 6-(diazenyl)quinoline tetrafluoroborate salts. Qing50 established a radical mechanism to account for product formation. The use of radical scavenger TEMPO inhibited product formation. A postulated mechanism includes the formation of an aryl radical by ET from the benzenediazonium tetrafluoroborate salt. Radical intermediate V (Scheme 13) reacts with Na2S2O5 to generate radical intermediate VI, which, by fluorine atom transfer from NFSI, affords the product.
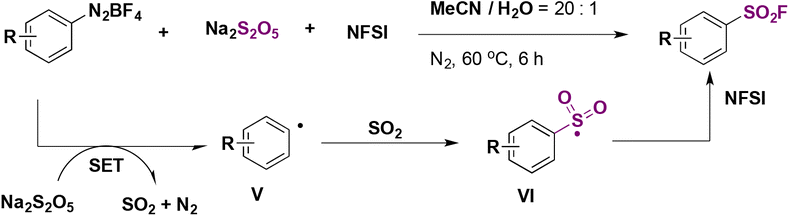 | ||
| Scheme 13 Proposed reaction mechanism. | ||
Confronting the studies of Liu and Qing, the former requires a transition metal catalyst (in sub-stoichiometric quantities) and an expensive SO2 surrogate (DABSO) but a cheap fluorine source (KHF2), while the work of Qing employs an inexpensive source of SO2 (Na2S2O5) but a rather costly source of fluorine (NFSI). From an environmental perspective, the protocol by Qing allows aqueous MeCN mixtures to be employed in the reactions.
At the same time that the report by Lin appeared, Liu and colleagues51 introduced a variant of the work of Qing, this time employing K2S2O5 as the source of SO2 and NFSI as the F atom provider, in MeCN/H2O/HOAc and benzenediazonium tetrafluoroborates as starting substrates. The reactions were carried out at room temperature for 6 h, and the yields of products were comparable to those of the other two methodologies.49,50 After this report, the group of Liu52 revealed a one-pot strategy to obtain fluorosulfonyl arenes directly from anilines.
Zhong, Weng and colleagues,53 also in 2020, reported the fluorosulfonylation of arene tetrafluoroborate salts employing Na2S2O5 as the source of SO2 and Selectfluor as the F atom provider, in methanol as solvent, at 70 °C for 9 h. The yields of the respective fluorosulfonylated arenes are high.
Louvel, Tlili, and collaborators54 reported the photocatalyzed substitution of aryldiazonium salts with the SO2F group employing DABSO as the source of SO2, KHF2 as the source of F, and an organic photocatalyst, 2,4,6-tris(diphenylamino)-5-fluoroisophthalonitrile (3DPAFIPN), in MeCN as solvent irradiated with blue LEDs. A scope of the transformation is shown in Scheme 14.
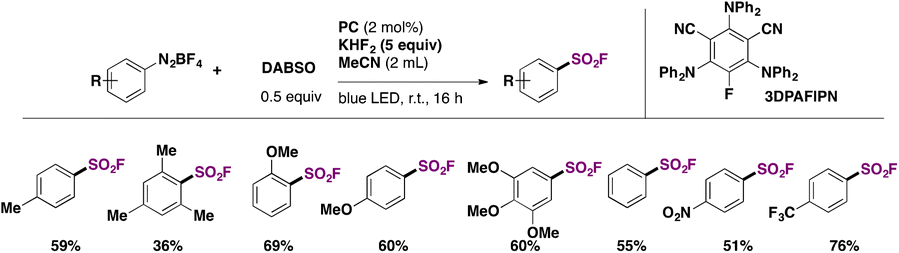 | ||
| Scheme 14 Selected examples for the 3DPAFIPN-photocatalyzed fluorosulfonylation of aryl diazonium salts. | ||
Bui, Tran and Kim55 have very recently developed a visible-light mediated methodology for the synthesis of sulfonyl fluorides starting from aryl azo sulfones. In this report, K2S2O5 and NFSI were used as sulfonyl and fluorine sources, respectively. All the reactions were carried out at room temperature, in aqueous mixtures to obtain the desired products in 60–85% yields.
All the methodologies available to synthesize aromatic fluorosulfonylated products are presented in Table 1.
C. Aliphatic fluorosulfonylation
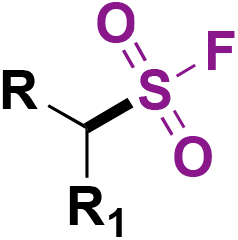
C.1. Syntheses of alkyl sulfonyl fluorides
Alkyl sulfonyl fluorides bear relevance in different fields, such as chemical biology, medicinal chemistry, and synthetic organic chemistry. Aliphatic sulfonyl fluorides have significant relevance as peptide-type inhibitors.56–58Typical methods for the syntheses of aliphatic sulfonyl fluorides involve classical Cl/F exchange from the corresponding chlorides,14 or starting from alkyl halides, thiols or disulfides and performing Cl/F exchange, or the addition of SO2F Michael acceptors such as ESF.59–61 Synthetic methods via radical sulfur dioxide insertion/fluorination also provide aliphatic sulfonyl fluorides.62–65
In 2016, the group of Shavnya66 converted alkyl bromides into alkyl sulfonyl fluorides utilizing rongalite (hydroxymethylsulfinate) and NFSI in reasonably good yields (see Table 2).67
Ma, Ma, Liu and collaborators68 have recently achieved the direct syntheses of aliphatic sulfonyl fluorides, starting from N-hydroxyphthalimide (NHPI) esters (Scheme 15) in the presence of Na2S2O4 and Zn, in a mixture of N,N-dimethylpropionamide (DMPr)/H2O (5:1 v/v) as a reaction medium under an Ar atmosphere at 80 °C for 9 h, and then NFSI at room temperature for an additional 4 h. The scope of the transformation is shown in Scheme 15.
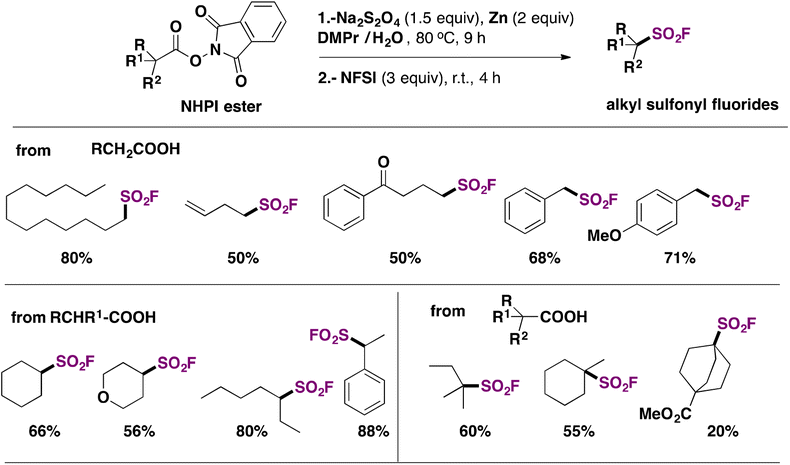 | ||
| Scheme 15 Selected examples for the radical fluorosulfonylation of NHPI esters. | ||
Scheme 15 shows that the current protocol68 can convert primary, secondary and tertiary carboxylic acids into their respective sulfonyl fluorides through the NHPI ester derivatives. The authors also proposed a reaction mechanism to account for product formation, according to Scheme 16.
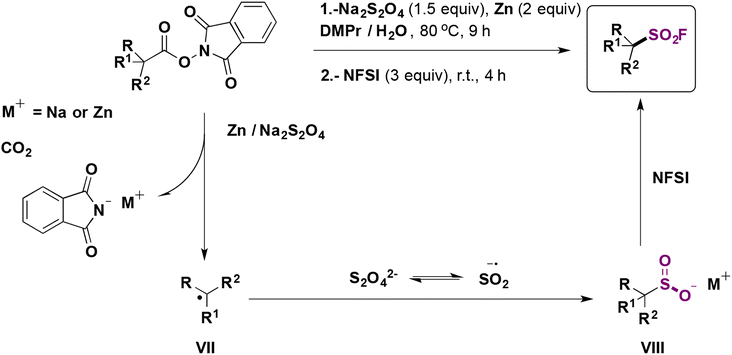 | ||
| Scheme 16 Proposed reaction mechanism. | ||
The authors68 performed some control experiments with radical scavengers (TEMPO) and radical clocks (the NHPI ester derived from 2-cyclopropylacetic acid) which indicated the presence of free radicals. On the basis of experimental results, reductive decarboxylation of the NHPI ester employing Na2S2O4/Zn generates the corresponding alkyl radical intermediate (VII, Scheme 16), which is subsequently trapped by the .SO2 radical anion to form the alkyl sulfinate (VIII). Electrophilic fluorination by NFSI afforded the product.
Nie and collaborators69 reported on a photocatalytic methodology for accessing aliphatic sulfonyl fluorides based on visible light-mediated photocatalyzed decarboxylative fluorosulfonylation of aliphatic NHPI esters. Optimized reaction conditions were achieved when Ir[(dF(CF3)ppy]2(dtbbpy)PF6 ([IrIII]) was employed as a photocatalyst, N-ethyldiisopropylamine (DIPEA) as a sacrificial reductant, and DABSO as a SO2 source, in isopropanol as solvent and under blue light irradiation in an Ar atmosphere. Then NFSI was allowed to react with the sulphonyl radical previously formed, affording the aliphatic sulfonyl fluoride in very good to excellent yields. The methodology proved to work very satisfactorily with a variety of primary, secondary, and tertiary carboxylic acid NHPI esters, including derivatives of pharmacologically active drugs such as oxaprozin and flurbiprofen, among others (Scheme 17). Regarding the reaction mechanism, the authors69 proposed a reductive photocatalytic cycle mediated by the [IrIII] photocatalyst. Upon light excitation, the excited [IrIII]* undergoes a single electron transfer process with DIPEA affording the [IrII] species and DIPEA˙+. Subsequently, the NHPI ester is reduced by [IrII] affording the alkyl radical (R˙) and regenerating the [IrIII] photocatalyst. The reaction of SO2 (released from DABSO) with the R˙ gives rise to a sulphonyl radical which is captured by NFSI, through fluorine atom transfer, affording the aliphatic sulfonyl fluoride product (Scheme 18).
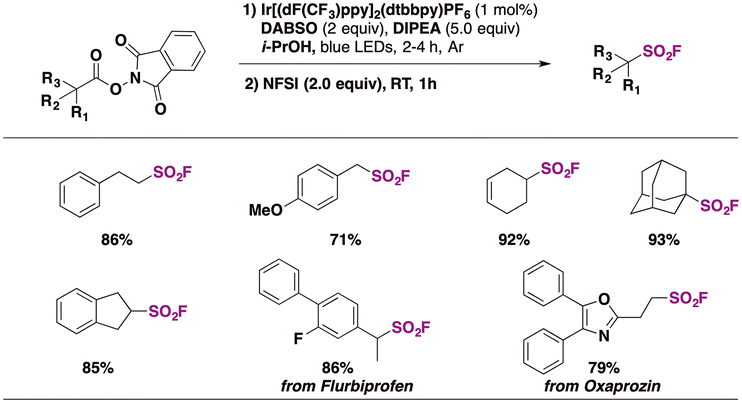 | ||
| Scheme 17 Selected examples for the photocatalytic fluorosulfonylation of aliphatic carboxylic acid NHPI esters. | ||
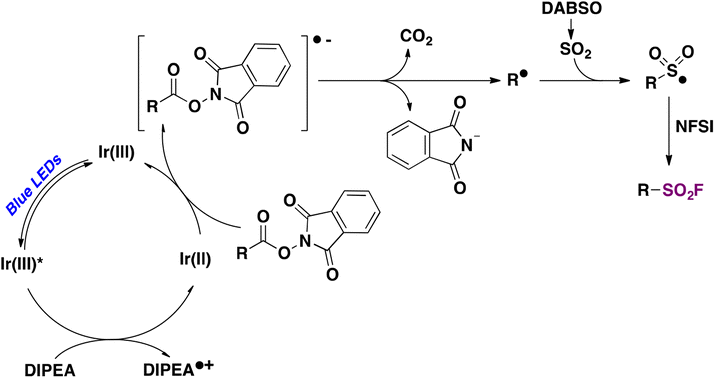 | ||
| Scheme 18 Proposed reaction mechanism. | ||
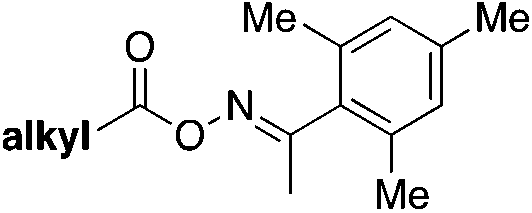
In 2022, Chen, Weng, and colleagues65a achieved the photocatalytic decarboxylative65b direct fluorosulfonylation of oxime esters by energy transfer, in the presence of DABSO and NFSI reagents to yield alkyl-fluorosulfonylated products. The scope of the transformation is presented in Scheme 19.
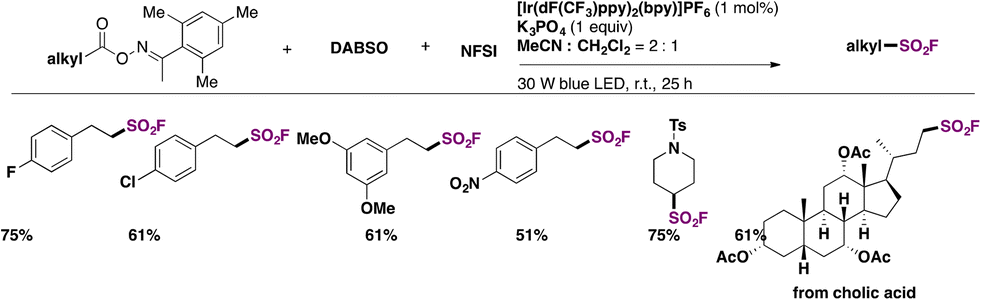 | ||
| Scheme 19 Scope of the Ir-photocatalyzed decarboxylative direct fluorosulfonylation of oxime esters by energy transfer (EnT), in the presence of DABSO and NFSI reagents to yield alkyl-fluorosulfonylated products. | ||
The authors65 investigated the reaction mechanism. They obtained evidence of the N–O bond scission in oxime esters that generated iminyl radicals. To commence with, irradiation from blue LEDs generates the triplet excited state of the photocatalyst (ET = 60.42 kcal mol−1), which transfers its triplet energy to the oxime ester (ET = 49.78 kcal mol−1), undergoing the latter N–O bond homolysis to produce iminyl radicals and alkyl radicals, after releasing CO2. Subsequently, the alkyl radical (IX) is trapped by DABSO to yield the alkylsulfonyl radical (X), which, by F transfer from NFSI, affords the alkylfluorosulfonylation product (Scheme 20).
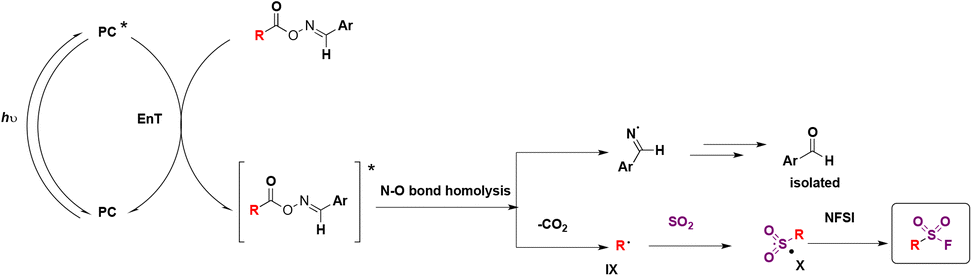 | ||
| Scheme 20 Proposed reaction mechanism. | ||
Weng and collaborators70 developed two different copper-catalyzed direct decarboxylative fluorosulfonylation methodologies of aliphatic carboxylic acids and made the resulting alkyl radicals react with SO2/fluorine sources to afford the fluorosulfonylated alkyl derivative. These strategies are very attractive because they do not require previous derivatization of the carboxylic acid functionality. In the first methodology, limited only to 3-arylpropionic acids, a Cu/N-fluorobenzenesulfonimide (NFSI) catalytic system is employed to perform the decarboxylative fluorosulfonylation, where NFSI is used both as a fluorine source and a hydrogen atom transfer (HAT) agent, assisting in the homolytic cleavage of carboxylic acid O–H bonds.
The second and more appealing strategy, which presents a much wider substrate scope, utilizes 9-mesitylacridine (Acr) as a photocatalyst, DABSO as a SO2 source, and Selectfluor as a fluorine donor, in dichloromethane as solvent and in the presence of Cu and 5,5′-dimethyl-2,2′-bipyridine (L2) under 400 nm LED irradiation and N2 saturation conditions.70 Regarding the substrate scope, the reaction performed well with a range of primary, secondary, and tertiary carboxylic acids, affording the corresponding sulfonyl fluorides in moderate to good yields (Scheme 21). The authors70 proposed a reaction mechanism initiated by hydrogen abstraction of the carboxylic acid O–H bond by the excited photocatalyst (Acr*). The resulting carboxyl radical undergoes CO2 loss affording an alkyl radical (R˙) that reacts with SO2, generated by DABSO decomposition, affording a sulphonyl radical. An L2CuI complex abstracts a fluorine atom from Selectfluor yielding an L2CuIIF adduct and a Selectfluor radical dication. Then, the sulfonyl radical abstracts a fluorine atom from the L2CuIIF adduct giving rise to the aliphatic sulfonyl fluoride product and regenerating the L2CuI complex. On the other hand, the Selectfluor radical dication abstracts a hydrogen atom from AcrH regenerating the photocatalyst and closing the catalytic cycle (Scheme 22).
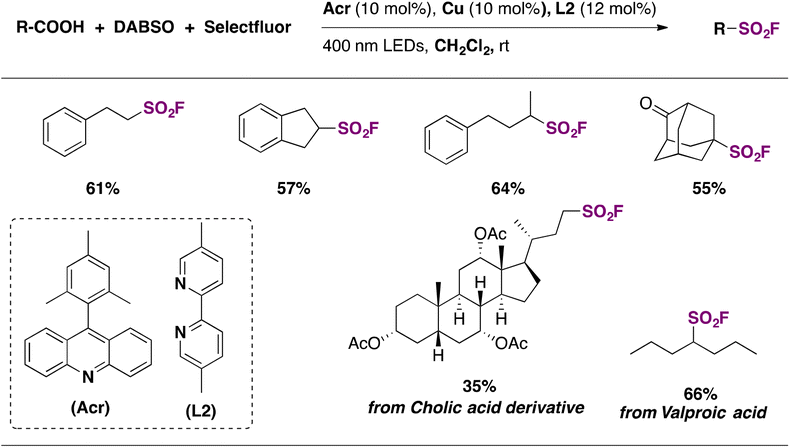 | ||
| Scheme 21 Selected examples for the photocatalytic fluorosulfonylation of aliphatic carboxylic acids. | ||
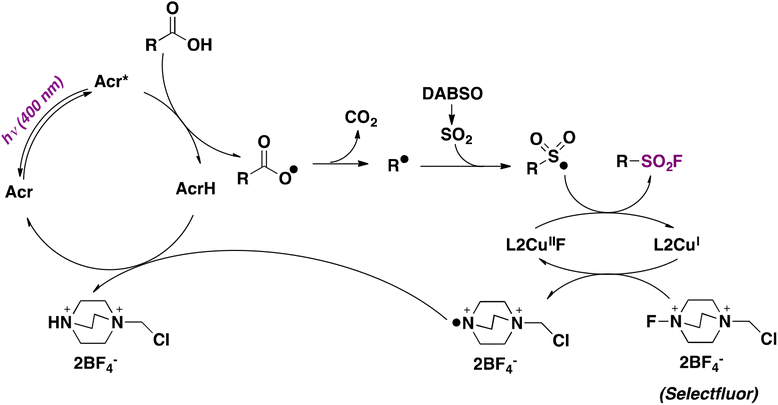 | ||
| Scheme 22 Proposed reaction mechanism. | ||
Wang and colleagues71 employed the redox-active solid imidazolium fluorosulfonyl salt (IMSF) to carry out the late-stage radical hydrofluorosulfonylation of drug molecules and natural products. This reagent reacted through a single electron transfer process and generated the fluorosulfonyl radical under visible light photocatalytic conditions (Scheme 23).
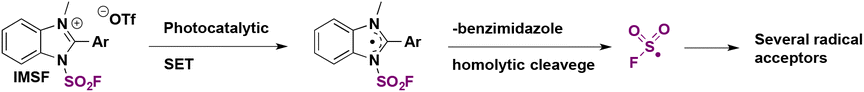 | ||
| Scheme 23 Fluorosulfonyl radical formation with an imidazolium fluorosulfonate salt. | ||
Selected examples for the use of IMSF in relevant substrates are shown in Scheme 24. Using an iridium photocatalyst, 1,4-cyclohexadiene as a hydrogen atom donor and the IMSF salt under visible light irradiation, terminal alkene derivatives of drug molecules such as ibuprofen and 4-methylumbeliferone or natural products (vitamin E) were obtained with good regioselectivity.
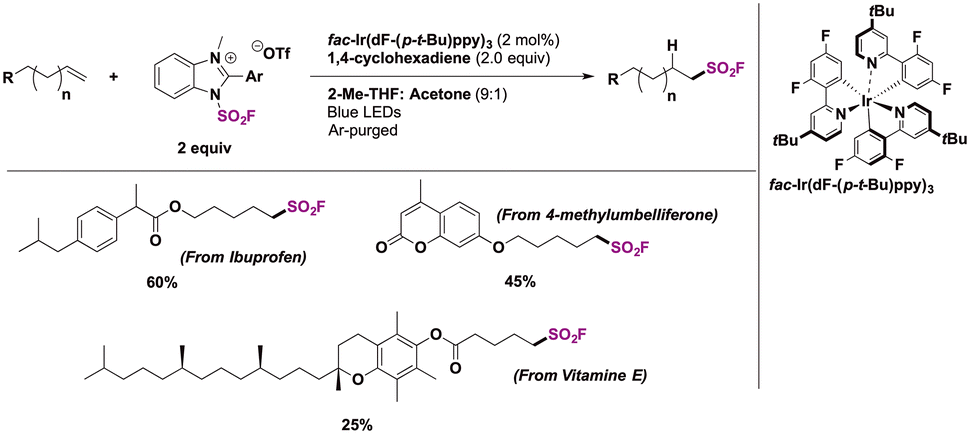 | ||
| Scheme 24 Selected examples of the radical hydrofluorosulfonylation. | ||
An alternative use of the IMSF salt71 was also presented for the difunctionalization of alkenes. Employing unsaturated tertiary alcohols, the reaction proceeded through distal migration induced by the fluorosulfonyl radical. This protocol required an iridium photocatalyst and several aryl groups were well tolerated, as shown in Scheme 25.
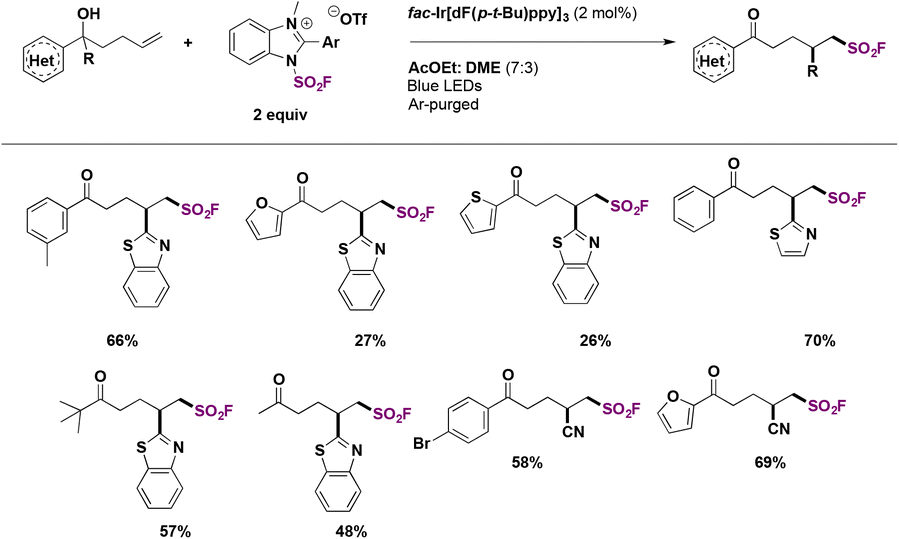 | ||
| Scheme 25 Radical migration fluorosulfonylation: substrate scope. | ||
To explore the mechanistic aspect of the radical fluorosulfonylation protocol, a control experiment was carried out using TEMPO under alkenyl functionalization conditions. The addition of the fluorosulfonyl radical to the unsaturated substrate was completely inhibited, and only the TEMPO-fluorosulfonyl adduct was detected by high resolution mass spectrometry (Scheme 26).
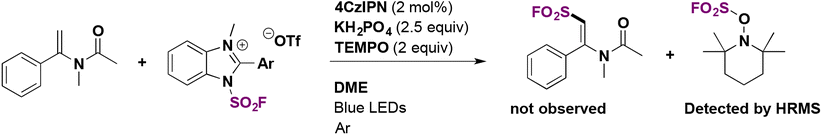 | ||
| Scheme 26 Fluorosulfonylation of alkenes: control experiment with TEMPO. | ||
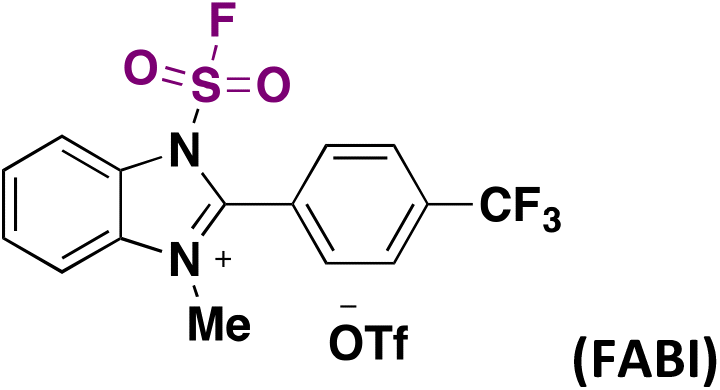
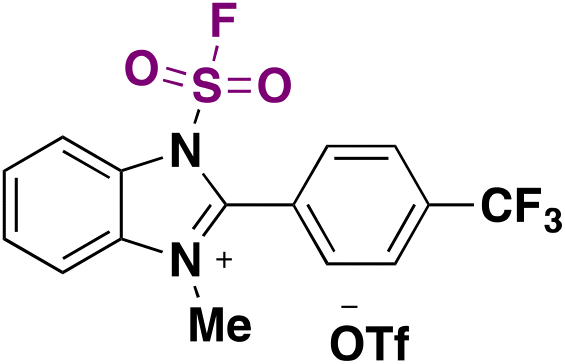
The hydrofluorosulfonylation of alkenes72 was also recently accomplished by Wang, Liao, and colleagues.73 The study was challenged by the fact that when ClSO2F was employed as a SO2F radical precursor, a chloro-fluorosulfonylated product (ATRA product) was obtained from the olefin under photocatalytic conditions in the presence of H atom donors (such as 1,4-CHD), due to the low BDE of the Cl–SO2F bond. The authors73 developed 1-fluorosulfonyl 2-aryl benzoimidazolium triflate (CF3-FABI, vide infra) as a fluorosulfonyl radical precursor. The photocatalyst used was oxygen-doped anthracene (ODA) in 1,4-dioxane as solvent under irradiation with blue LEDs. A brief scope of the transformation is depicted in Scheme 27.
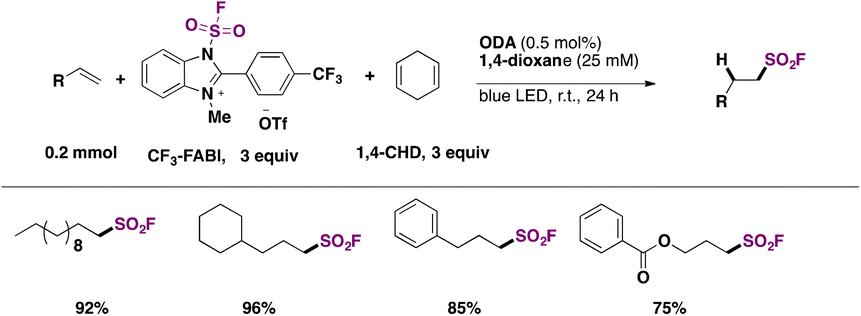 | ||
| Scheme 27 Succinct examples for the oxygen-doped anthracene-photocatalyzed hydrofluorosulfonylation of alkenes with FABI. | ||
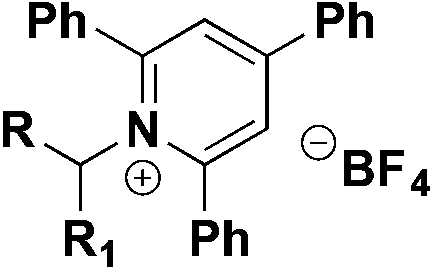
In a 2019 report, Andrews, Willis and colleagues74 presented a protocol for the synthesis of alkyl sulfonyl derivatives from readily available amines via Katritzky pyridinium salt intermediates. This strategy employed a primary or secondary Katritzky salt, DABSO as the SO2 source, an organic base (Et3N, 2,6-lutidine or piperidine) and the Hantzsch ester to prepare a sulfinate salt which could react with several electrophiles to generate alkyl sulfonyl derivatives or fluorine sulfonyl compounds.
The reaction pathway is shown in Scheme 28. The radical species can be generated by photoinduced electron transfer from the electron donor–acceptor complex (EDA) between Katritzky salts and the Hantzsch ester. This very convenient catalyst-free strategy allows generation of the alkyl radical which reacts with the SO2 source (DABSO) and produces the sulfonyl radical. The sulfonyl radical can undergo a hydrogen atom transfer (HAT) reaction from the Hantzsch ester and ulterior deprotonation to afford the sulfinate salt. Subsequently the in situ reaction with several electrophiles provided diverse sulfonyl-derivatives.
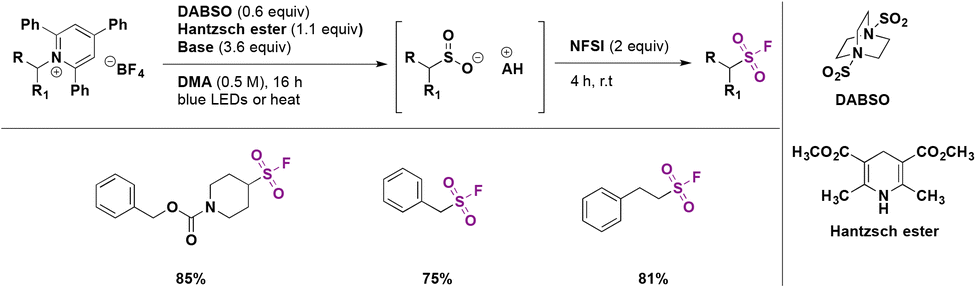 | ||
| Scheme 28 Catalyst-free deaminative synthesis of sulfonyl-derivatives. | ||
This methodology was successfully applied to the synthesis of >60 examples of sulfones (from the corresponding secondary, benzylic and primary amines), sulfonamides and sulfonyl fluorides (Scheme 29). This reaction was carried out by a one-pot procedure and products could be obtained under bulk conditions (10 mmol of substrate) with very good yields.
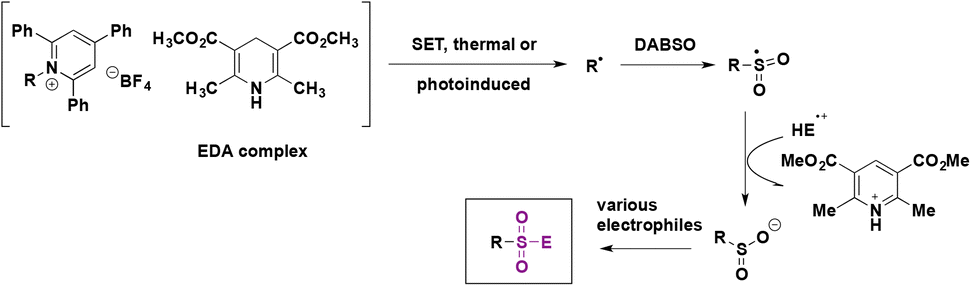 | ||
| Scheme 29 Functionalization of sulfinate salts employing NFSI. | ||
The authors also performed UV-visible experiments from mixtures of Katritzky pyridinium salts and the Hantzsch ester in DMA as solvent, showing a significant bathochromic shift when compared to the isolated components, strongly suggesting the formation of the EDA complex. They also attempted the reaction in the presence of TEMPO, where only the starting materials were observed, supporting the presence of radicals in the reaction mixtures.
In 2021, MacMillan and co-workers reported the development of decatungstate-catalyzed conversion of C(sp3)–H bonds into the corresponding alkyl sulfinic acids.75a The authors75a employed this methodology for the synthesis of several organosulfur compounds. They performed the functionalization of the tricyclic imide A shown in Scheme 30. The sulfinic acid intermediate was converted to the corresponding sulfonyl fluoride using Selectfluor as the fluorine source.
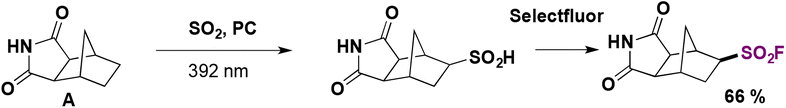 | ||
| Scheme 30 A selected example for the decatungstate-catalyzed conversion of C(sp3)–H bonds into the corresponding alkyl sulfinic acids and ulterior fluorination. | ||
Indirect methods (those making use of fluorosulfonylated hubs such as ESF) to synthesize alkyl fluorosulfonyl compounds have also been developed.
Zhang, Qin, and colleagues60 achieved the syntheses of alkyl sulfonyl fluorides through photocatalysis, employing ESF75b (see section C.2.2.) and an alkyl iodide with Mn2(CO)10 as a photocatalyst and the Hantzsch ester as a reductant in DMSO as solvent, under blue LED illumination. The scope of the reaction is shown in Scheme 31.
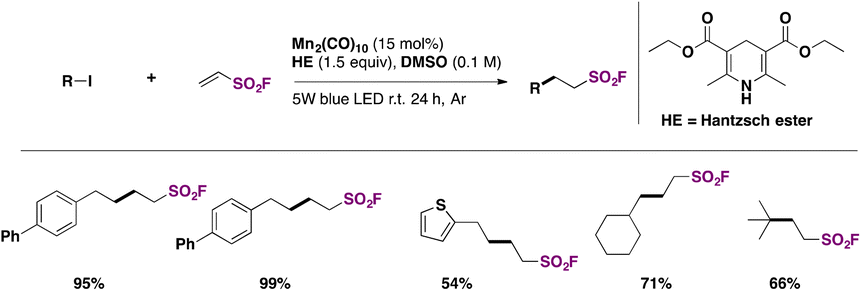 | ||
| Scheme 31 Selected examples for the photocatalyzed syntheses of fluorosulfonylated alkyl compounds from alkyl iodides. | ||
The authors60 expanded the scope of the methodology by synthesizing potentially biologically active alkyl sulfonyl fluorides from steroids, zidovudine, chloramphenicol, thiamphenicol and other pharmacophores, obtaining the respective alkyl-fluorosulfonylated-substituted compounds in good yields.
The authors investigated the reaction mechanism and made a proposal such as that depicted in Scheme 32. The photocatalyst is excited by illumination from the LED source producing Mn–Mn homolytic cleavage affording Mn(CO)5 radicals, which, through I atom transfer from alkyl iodide, produce an alkyl radical intermediate (XI) and Mn(CO)5–I. Alkyl radical intermediate XI adds to ESF to give intermediate XII, which abstracts a H atom from the Hantzsch ester HE to afford the product. In turn, the radical from HE, XIII, is oxidized and rearomatizes to a pyridinium iodide (Scheme 32).
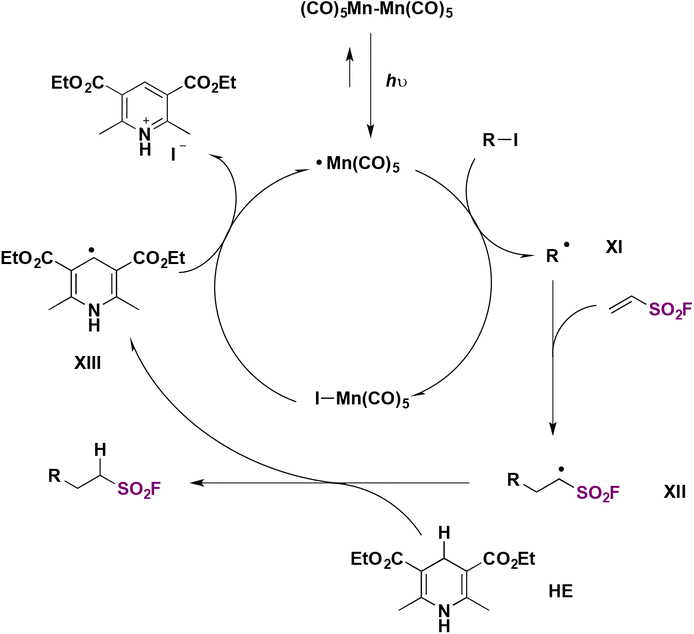 | ||
| Scheme 32 Proposed reaction mechanism. | ||
In 2019, Xu, Liao and colleagues76a carried out the indirect syntheses of alkyl-substituted sulfonyl fluorides by a photocatalyzed decarboxylative reaction of NHPI esters and ESF (section C.2.2.) as the source of sulfonyl fluoride, in the presence of the Hantzsch ester (HE) in MeCN as solvent under irradiation with blue LEDs. A succinct scope of the transformation is presented in Scheme 33.
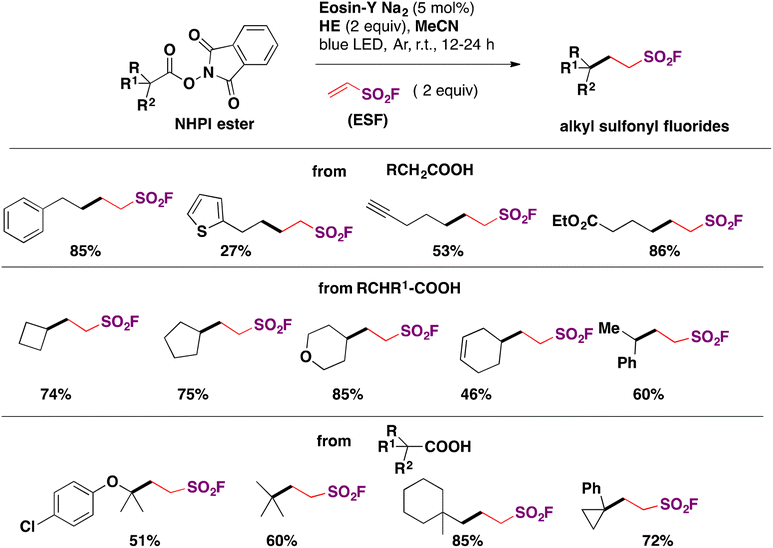 | ||
| Scheme 33 Scope for the photocatalytic preparation of aliphatic sulfonyl fluorides from NHPI esters and ESF. | ||
Primary, secondary and tertiary carboxylic acids afforded good yields of the respective alkyl fluorosulfonylated products. All the methodologies available to synthesize alkyl-substituted fluorosulfonylated products are presented in Table 2.
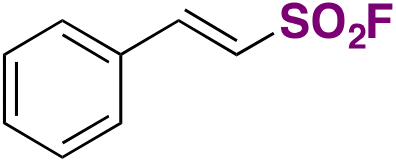
C.2. Syntheses of fluorosulfonylated alkenes
Fluorosulfonylated alkenes76b can be synthesized either through direct fluorosulfonylation strategies (C.2.1.) from olefins or alkynes, or by employing fluorosulfonylating hubs (pre-installed SO2F moiety, C.2.2.), such as ESF and its derivatives or by using SO2 and NFSI.63The difficulty of generation of the FSO2 radical has thwarted advances in the study of this unstable species.77 The group of Liao78 managed to generate the FSO2 radical by photoredox conditions. The precursor of the FSO2 radical was FSO2Cl, employing Ir[dF(CF3)ppy]2(dtbbpy)PF6 as a photocatalyst under blue LED irradiation in an Et2O/PhCF3 mixture of solvents. The scope of the transformation is depicted in Scheme 34.
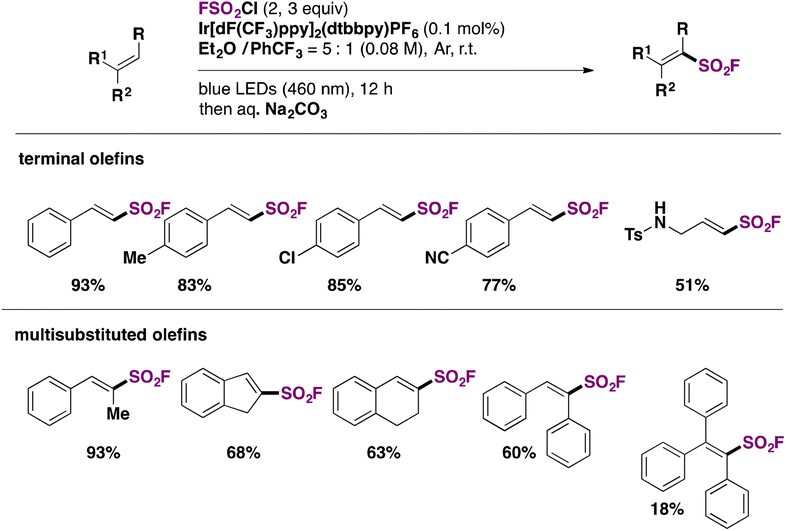 | ||
| Scheme 34 Selected examples for radical fluorosulfonylation of alkenes. | ||
Both electron donating and electron-withdrawing substituted styryl systems afforded the respective fluorosulfonylated products in good yields (Scheme 34). Also, internal 1,2-disubstituted alkenes and trisubstituted alkenes afforded reasonable yields of the fluorosulfonylated olefins. The authors investigated the reaction mechanism78 by performing some radical probe experiments, such as the use of radical scavenger TEMPO. Under these latter conditions, total inhibition of the fluorosulfonylated olefinic product was observed. A radical probe experiment with radical clock 1-phenyl-1-cyclopropyl ethylene afforded the ring opening product shown in Scheme 35.
 | ||
| Scheme 35 Radical clock experiment with 1-phenyl-1-cyclopropyl ethylene. | ||
These latter experiments indicated the presence of free radicals in the reaction. Comparative DFT calculations between the CF3SO2 and FSO2 radicals also showed that the latter has a more planar configuration and a more positive sulfur atom, consistent with the higher electronegativity of F compared to CF3. The authors78 postulated a reaction mechanism such as that shown in Scheme 36. The Ir(III) photocatalyst is excited to its triplet manifold by illumination from the blue LEDs, which by, a SET reaction to FSO2Cl, affords the FSO2 radical and chloride anion. The FSO2 radical readily reacts with the olefin to supply intermediate XIV (Scheme 36), which can either be oxidized by the upper oxidation state of the photocatalyst (i.e.: Ir(IV)) to give XV, which is deprotonated to yield the final product, or undergo a chlorine atom transfer from ClSO2F to give XVI, which loses HCl to give the product (Scheme 36).
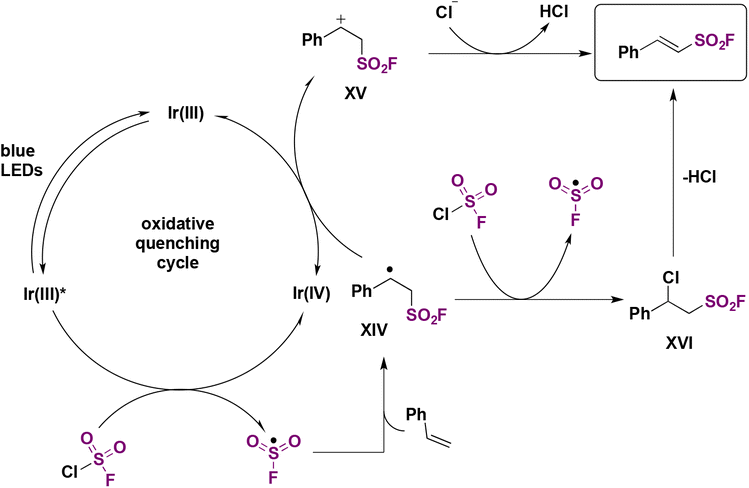 | ||
| Scheme 36 Proposed reaction mechanism. | ||
Nie, Liao, and colleagues,79 utilized FSO2Cl as the chloro-fluorosulfonylating reagent, which, under blue LED (λmax = 460 nm) illumination in the presence of a fac-Ir(ppy)3 photocatalyst and an alkyne substrate in an Et2O/PhCF3 mixture of solvents, afforded β-chloro alkenylsulfonyl fluoride (BCASF) in good yields. The scope of the reaction is shown in Scheme 37.
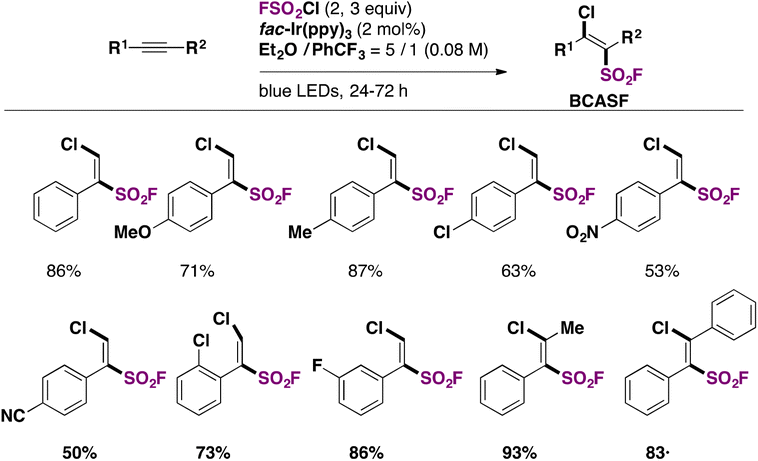 | ||
| Scheme 37 Selected examples for the photocatalyzed syntheses of BCASFs. | ||
Both electron withdrawing and donating groups on the arylacetylenes afforded good yields of the BCASF products. Also, internal alkynes are appropriate substrates for the reaction.
β-Chloro alkenylsulfonyl fluoride (BCASF) hubs are powerful entities for introducing alkyl, alkenyl and alkynyl groups onto the β-position of BCASF through coupling reactions, affording β,β-disubstituted ethylenesulfonyl fluorides and ynenyl-sulfonyl fluorides (vide infra, Scheme 38). The authors79 profited from the chlorine handle of the BCASF substrates to perform coupling reactions, Suzuki coupling with boronic acids and Sonogashira coupling with terminal alkynes. Some of these examples are depicted in Scheme 38.
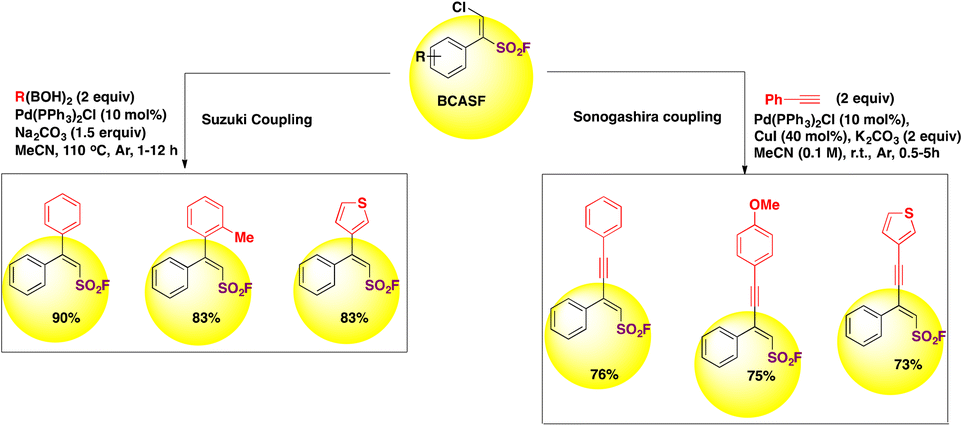 | ||
| Scheme 38 Suzuki- and Sonogashira-type couplings of BCASFs. | ||
The authors79 employed the BCASFs as sulfonyl carriers in amino acids, peptides and drugs. Cysteine and N-terminals of peptides were thus modified by (E)-2-chloro-2-phenylethene-1-sulfonyl fluoride (Ph-BCASF) to afford peptides shown in Scheme 39.
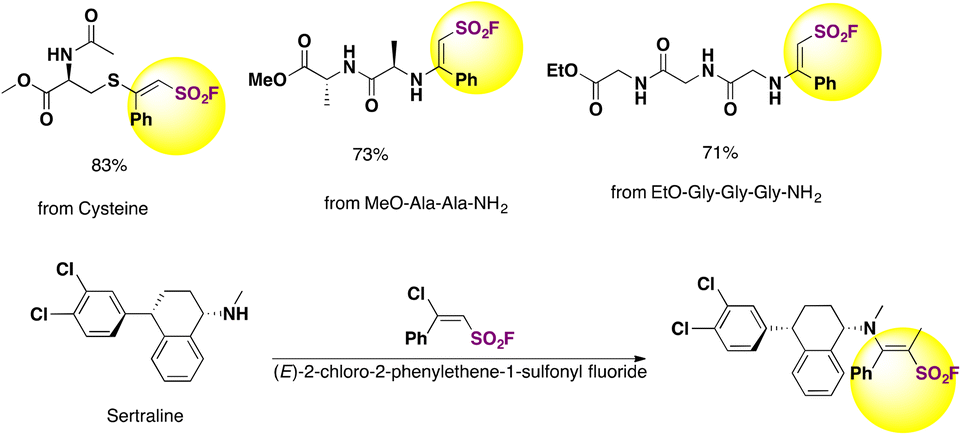 | ||
| Scheme 39 Cysteine and N-terminals of peptides and drugs modified by (E)-2-chloro-2-phenylethene-1-sulfonyl fluoride (Ph-BCASF). | ||
The authors79 also attempted the E/Z isomerization of the BCASFs. N,N-Diisopropylethyleneamine (DIPEA) was found effective for this goal.
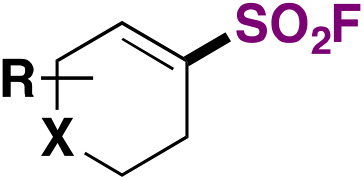
Lou, Willis and collaborators80 achieved the synthesis of fluorosulfonylated cycloalkenes through the use of DABSO, a Pd catalyst and NFSI as the fluorine source. The scope for the syntheses of cyclic alkenyl sulfonyl fluorides is depicted in Scheme 40.
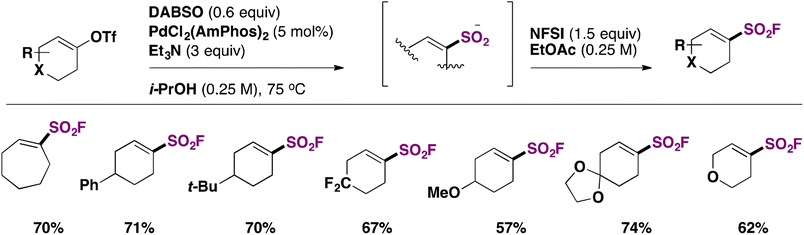 | ||
| Scheme 40 Selected examples for the syntheses of cyclic alkenyl sulfonyl fluorides. | ||
The authors80 investigated derivatization reactions. Substitution of alkenyl sulfonyl fluorides at the sulfur atom was done with 4-methoxyphenol to form sulfonate esters, or with anilines to form sulfonamides (Scheme 41).
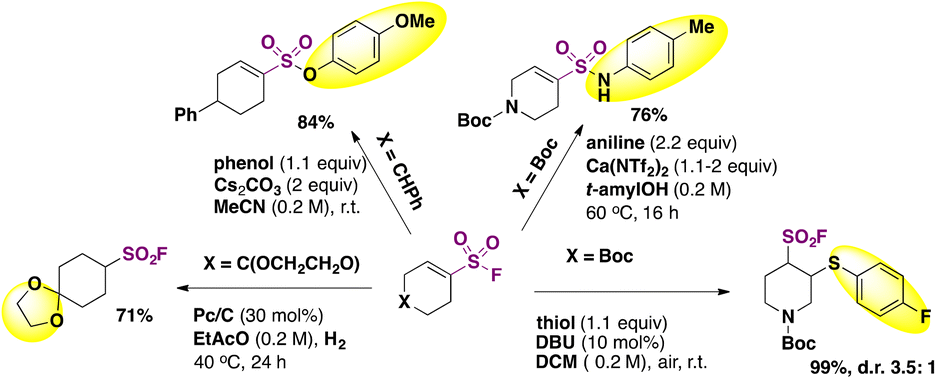 | ||
| Scheme 41 Derivatization of alkenyl sulfonyl fluorides. | ||
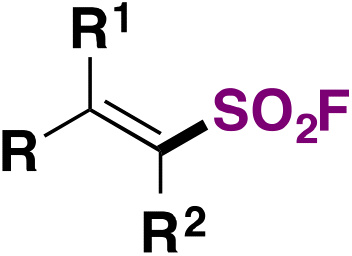
In 2022, the group of Liao81 developed a strategy for the synthesis of fluorosulfonylated alkenes, employing FABI-CF3 (Scheme 42) as a fluorosulfonyl radical precursor. Under fac-Ir(ppy)3 photocatalysis, in 1,4-dioxane as solvent and under blue LED irradiation, fluorosulfonylated alkenes were obtained. Scheme 42 shows some examples of this transformation.
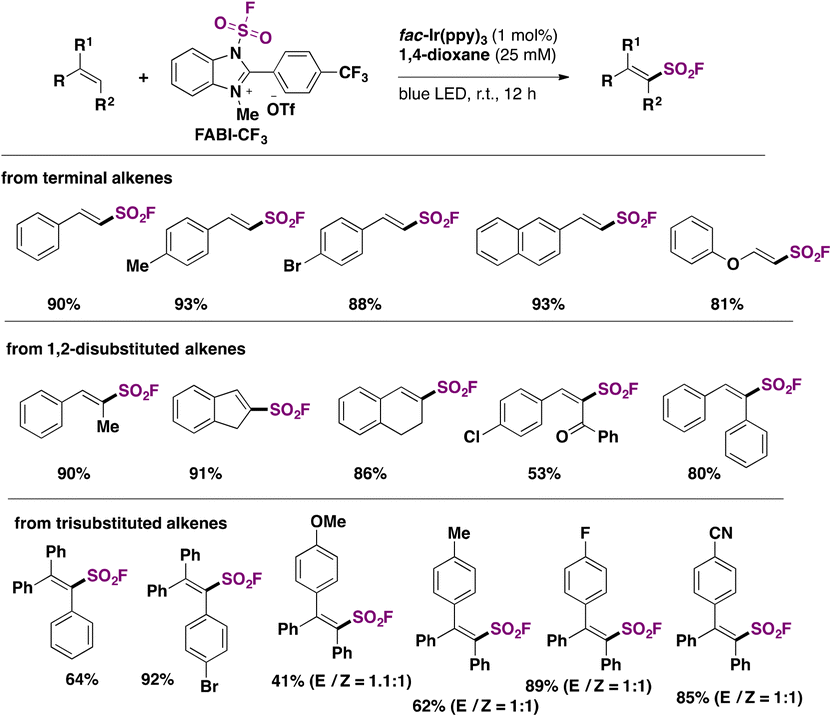 | ||
| Scheme 42 Scope of the photocatalyzed fluorosulfonylation of alkenes. | ||
The authors investigated the reaction mechanism. A mechanistic proposal is depicted in Scheme 43. Irradiation of the photocatalyst Ir(ppy)3 by blue LEDs generates the triplet excited state of Ir(III)* (E1/2 Ir(IV)/Ir(III)* = −1.73 V vs. SCE), undergoing a SET to FABI-CF3 (E1/2 red = −1.07 V vs. SCE), which undergoes homolytic cleavage of the N–S bond to give FSO2 radicals. In turn, the FSO2 radicals add to the styrene affording intermediate XVIb (Scheme 43). XVIb is oxidized by the upper redox state of the photocatalyst affording XVII, which can deprotonate to furnish the fluorosulfonylated alkene (Scheme 43).
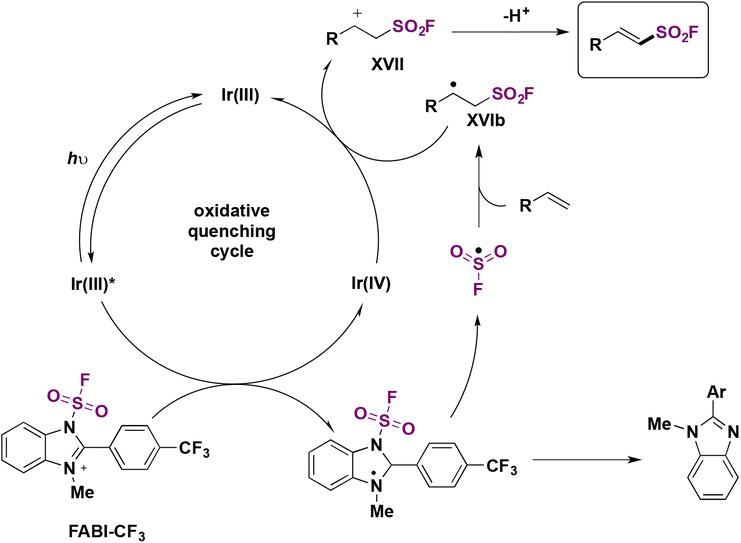 | ||
| Scheme 43 Proposed reaction mechanism. | ||
In a very recent report, Zhang, Wang, and colleagues71 explored the stereoselective radical fluorosulfonylation of several alkenes. The optimized reaction conditions for the synthesis of E-fluorosulfonyl alkenes are shown in Scheme 44. This protocol employed 4CzIPN as a photocatalyst (Scheme 44) and KH2PO4 as a base under visible light irradiation at room temperature. Under these conditions, several examples of unsaturated hydrocarbons were functionalized in good yields with high regio- and stereo-selectivity. They also reported the reaction of two natural derivatized alkenes from cholesterol and estrone and obtained the corresponding products in moderate yields.
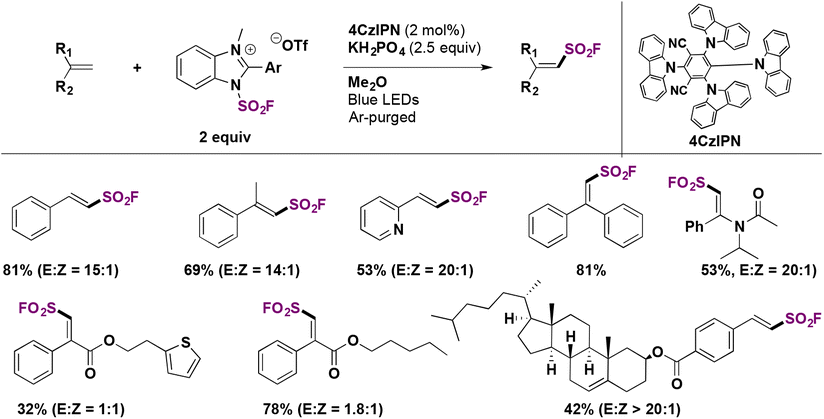 | ||
| Scheme 44 Photocatalytic synthesis of E-fluorosulfonyl alkenes. | ||
The authors71 modified the reaction conditions to obtain the less thermodynamically stable Z-products. With a different solvent mixture and employing a different photocatalyst (PC-1, Scheme 45), the reaction was extended to produce several Z-alkenyl sulfonyl fluorides including derivatives from the bioactive compounds DL-menthol and bexarotene (Scheme 45).
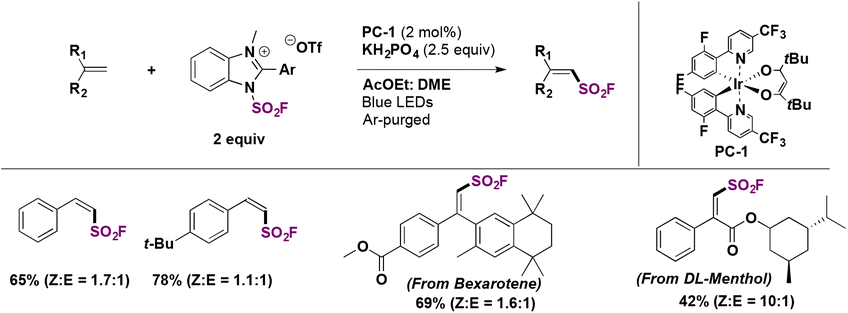 | ||
| Scheme 45 Photocatalytic synthesis of Z-fluorosulfonyl alkenes. | ||
ESF, the primogenial hub, has been synthesized by fluorination of 2-chloroethane-1-sulfonyl chloride with KHF2 and ulterior treatment with MgO.14 It has been employed as a dipolarophile in 1,3-dipolar cycloadditions to give fluorosulfonylated heterocycles.95 When reacted with organic azides, ESF behaves as an acetylene equivalent affording 1,2,3-triazoles with elimination of SO2F.96 However, ESF reacts with diazoalkanes such as diazomalonate, affording fluorosulfonylated heterocycles and cyclopropanes, depending on the reaction conditions.97
ESF has also been employed in conjugate addition reactions,98 palladium-catalyzed Heck-type couplings using aryl iodides,20 diazonium salts,99 and boronic acids,61,84,90 Pd-catalyzed alkenylation,86 and rhodium-catalyzed C–H activation.61,100 Also, the photocatalyzed fluorosulfonylethylation of aryl iodides (vide supra, section B.)60 and the photocatalyzed decarboxylative fluoroethylsulfonylation of N-hydroxyphthalimide esters76 (vide supra, section C.1.) have been recently described with the ESF reagent.
In 2016, Qin, Wu, Sharpless and colleagues99 reported the synthesis of β-arylethenesulfonyl fluorides101 directly from aryldiazonium salts and ESF, via a Heck-type β-C–H arylation pathway through the Heck–Masuda process.102,103 The standard procedure involved the freshly prepared benzenediazonium tetrafluoroborate, treated with ESF14 and Pd(OAc)2 in acetone, at room temperature. The E-stereoisomers from β-phenylethenesulfonyl fluorides were obtained exclusively. The scope of the reaction is represented in Scheme 46.
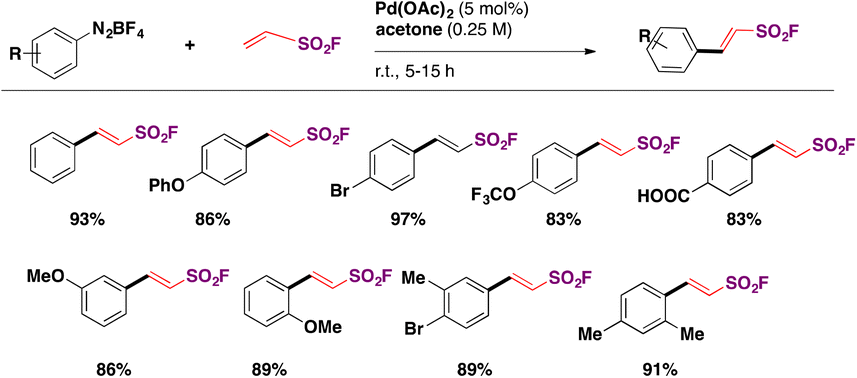 | ||
| Scheme 46 Selected examples for the scope of the synthesis of β-arylethenesulfonyl fluorides from aryldiazonium salts and ESF. | ||
Both electron releasing and withdrawing groups on the benzenediazonium salts afforded the respective β-arylethenesulfonyl fluorides in good yields (Scheme 46). Benzenediazonium salts substituted at the meta and ortho positions, as well as 3,4-, and 2,3-disubstituted-benzenediazonium salts, afforded the respective β-arylethenesulfonyl fluorides in reasonably good yields.
Both the olefin moiety and the sulfonyl fluoride group in β-arylethenesulfonyl fluorides can function as electrophiles. The reaction of β-arylethenesulfonyl fluorides with secondary cyclic amines afforded the Michael addition product as opposed to the substitution at the sulfur atom, whereas the reaction with tert-butyldimethylsilyl (TBS) ether in acetonitrile, using DBU as a base, afforded alkenylsulfonates via SuFEx chemistry (Scheme 47).
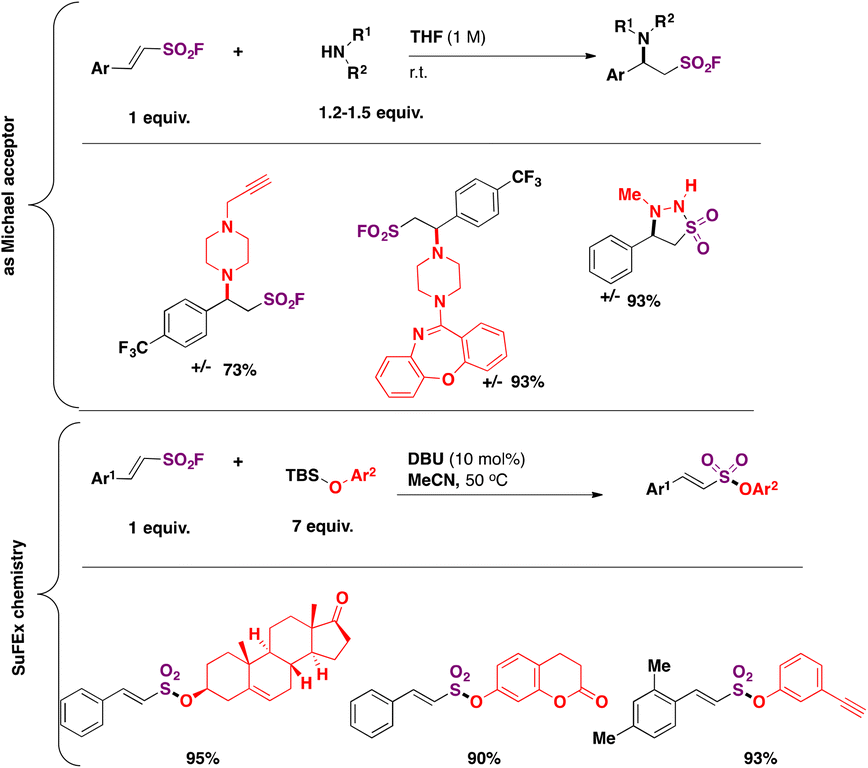 | ||
| Scheme 47 β-Arylethenesulfonyl fluorides as bis-electrophiles: Michael addition and SuFEx chemistry. | ||
The strong preference for the Michael addition86 of secondary amines to ESF over the substitution at S is quite remarkable. However, for β-arylethenesulfonyl fluorides, the reactivity is somewhat diminished, since only electron poor or neutral aromatic moieties are reactive towards the amines (Scheme 47).101
Then Chen, Wang, Yu and colleagues86 informed a variant of the Pd catalysis to achieve homolytic aromatic substitution of arenes with ESF for the synthesis of aryl ethenesulfonyl fluorides. A succinct display of the scope of the reaction is scope in Scheme 48.
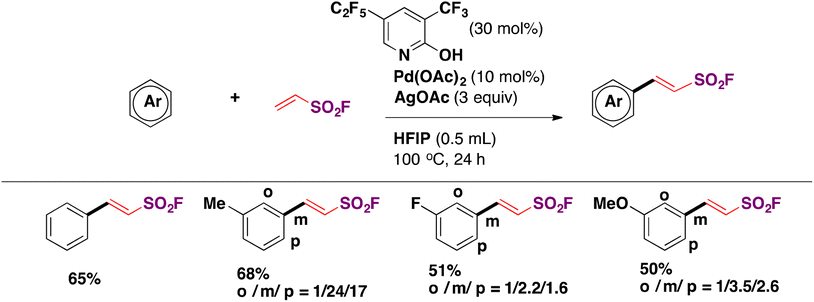 | ||
| Scheme 48 Selected examples for the HAS of arenes with ESF. | ||
The homolytic substitution is poorly regioselective and necessitates harsh reaction conditions.86
In 2018, the group of Qin88,100 reported the Rh-catalyzed ortho-substitution of benzaldehydes and acetophenones with the ESF group to achieve the fluorosulfovinylation of aryl C(sp2)–H bonds from aromatic aldehydes and ketones. A brief scope of the transformation is shown in Scheme 49.
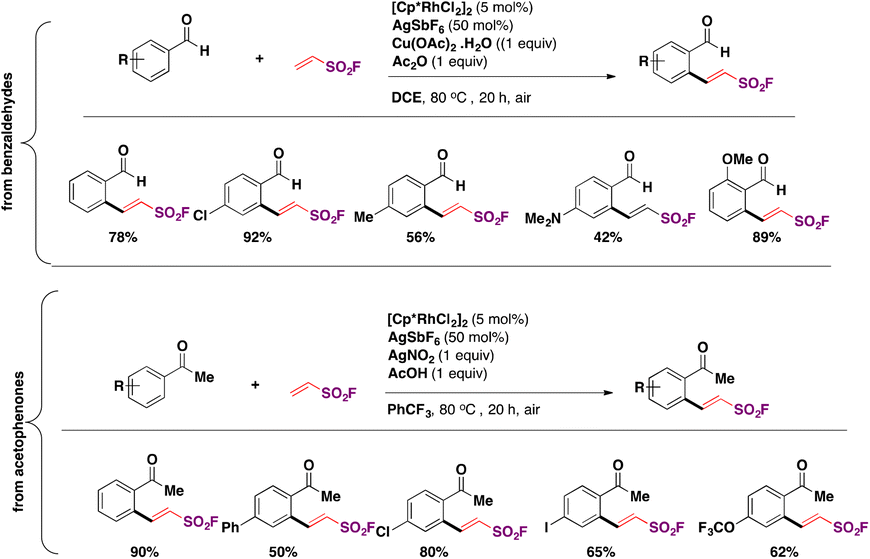 | ||
| Scheme 49 Selected examples for the Rh-catalyzed ortho-substitution of benzaldehydes and acetophenones with ESF. | ||
From Scheme 49, it is observed that both benzaldehydes and acetophenones of diverse electronic nature are good candidates for the Rh-catalyzed incorporation of ESF.
However, the major drawback of ESF and its derivatives is the inherent structural limitation to ethyl or ethylene moieties present in the final products. There are also some concerns with the use of ESF related to its high toxicity (oral LD50 is 50 mg kg−1 for rats) and being a severe lachrymator.35 Its preparation from the toxic 2-chloroethane-1-sulfonyl chloride is also a concern. All the methodologies available to synthesize alkenyl-substituted fluorosulfonylated products are presented in Table 3.
C.3. Syntheses of alkynyl sulfonyl fluorides
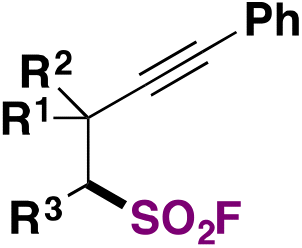
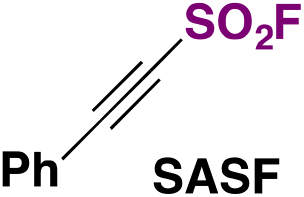
As recently as 2020, Smedley, Sharpless, Moses and colleagues92 synthesized 2-substituted-alkynyl-1-sulfonyl fluorides (SASFs), which bear both an alkyne functionality (ready for a click reaction with azides) and the SO2F SuFExable group. The syntheses of SASFs were readily accomplished by the reaction of terminal alkynes with n-BuLi and FSO2OSO2F (or gaseous SO2 and NFSI afterwards, as the source of F) at −78 °C in Et2O (or THF when SO2 gas is used). The authors uncovered that the method employing the fluorosulfonic acid anhydride (i.e.: FSO2OSO2F) (method A, Scheme 50) worked well with electron-poor substrates, while the method utilizing SO2 and NFSI (method B, Scheme 50) worked satisfactorily with electron-rich substrates. Scheme 50 depicts the syntheses of a series of SASF substrates.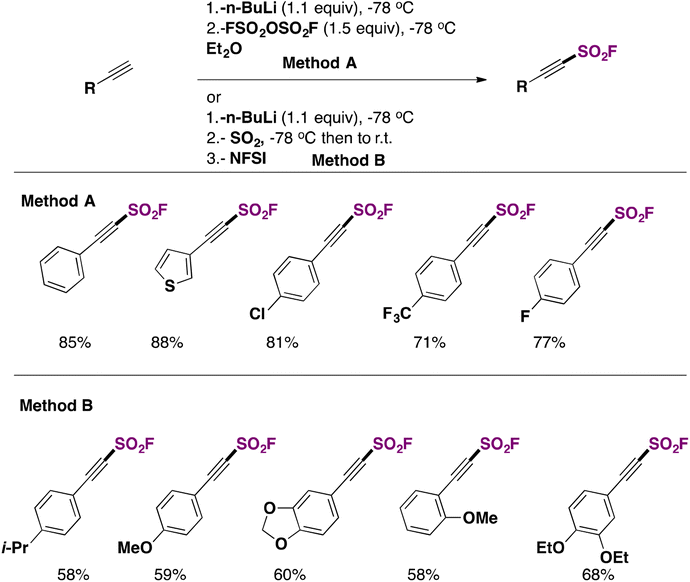 | ||
| Scheme 50 Selected examples for the syntheses of SASFs. | ||
These SASFs were later applied to construct a very interesting diverse set of heterocyclic compounds bearing the SO2F moiety taking advantage of the click chemistry. The authors92 combined the classic click chemistry reaction concept between an alkyne and an azide group104,105 to give triazole-type products with the SuFEx reaction which is associated with the fluorosulfonyl group. This new click associative chemistry was coined diversity oriented clicking (DOC), a methodology which encompasses two-in-one click chemistry sequential protocols with the aim of building on the diversity of scaffolds by click processes using 2-substituted-alkynyl-1-sulfonyl fluorides (SASFs) as starting substrates.
In order to put into practice the DOC methodology, SASFs were made to react with dipoles and cyclic dienes as coupling partners. The 1,3-nitrogen dipoles included nitrile oxides (Scheme 51A), nitrones (Scheme 51B), azides (Scheme 51C), nitrile imines (Scheme 51D), sydnones (Scheme 51E), dienes (Scheme 51F), and pyrroles (Scheme 51G), among others. The reaction click products of these dipoles with SASFs are shown in Scheme 51.
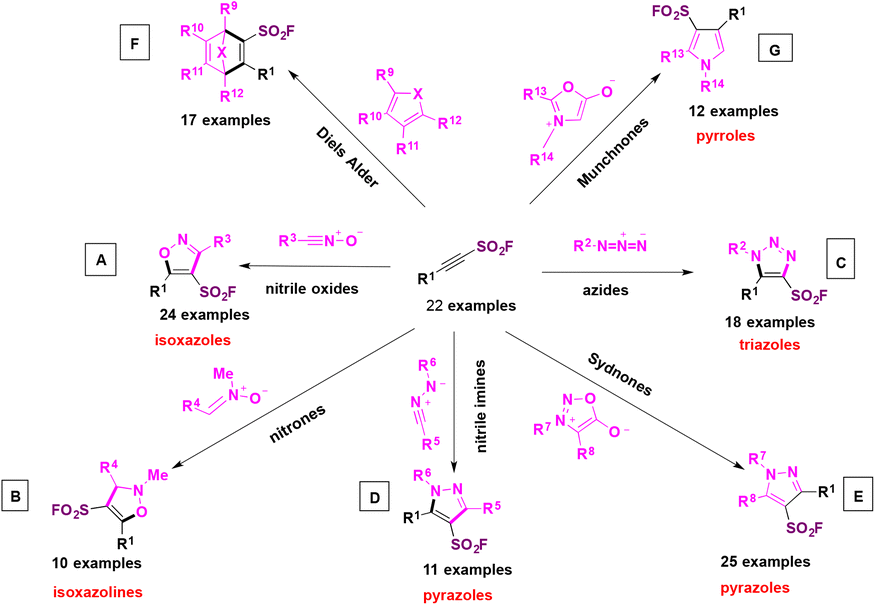 | ||
| Scheme 51 Selected general examples of dipoles reacting with SASFs. A: nitrile oxides. B: nitrones. C: azides. D: nitrile imines. E: sydnones. F: dienes. G: pyrroles. | ||
In this manner, a series of heteroaromatic compounds bearing the fluorosulfonic anchor such as isoxazoles, isoxazolines, pyrazoles, triazoles, and pyrroles could be obtained. It is to be pointed out the high selectivity of the reactions, since single regioisomers in each case were obtained (a recent synthesis of 2,4,5-trisubstituted oxazole sulfonyl fluorides was proposed by H.-L. Qin106 without the employment of SASFs, although through the Rh-catalyzed heterocycloaddition of nitriles with 2-diazo-2-(fluorosulfonyl)acetate).
In order to apply the SuFEx click chemistry on the fluorosulfonyl heterocyclic compounds synthesized (Scheme 51), the authors92 constructed a DOC library. A brief panorama of the scope of the reaction is presented for the syntheses of sulfonates in Scheme 52.
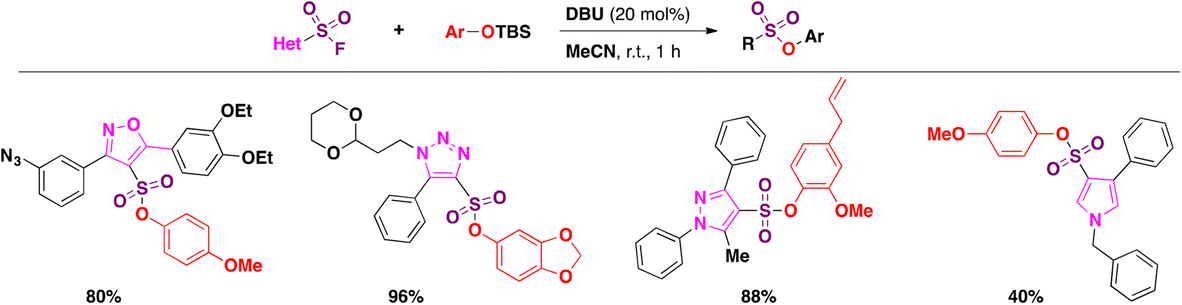 | ||
| Scheme 52 Examples of SuFEx click chemistry reactions from isoxazole, triazole, pyrazole and pyrrole fluorosulfonyl derivatives. | ||
Competition experiments indicated that the reactivity for the SuFEx reaction followed the order triazole > pyrazole > isoxazoline = isoxazole.
Frye, Studer and colleagues72 have employed a SASF reagent (2-substituted-alkynyl-1-sulfonylfluoride) to synthesize a β-alkynyl-fluoro sulfonyl alkane. An excerpt of the reaction is shown in Scheme 53.
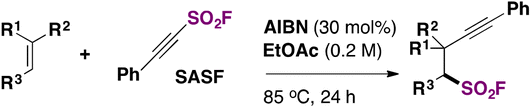 | ||
| Scheme 53 General strategy for the syntheses of β-alkynyl-fluoro sulfonyl alkanes. | ||
C.4. Syntheses of β-keto sulfonyl fluorides
β-Ketosulfonyl fluorides were recognized as analogs to β-ketoesters, in terms of versatile reactivity. The only known method to synthesize β-ketosulfonyl fluorides used the gaseous SF5Cl reagent.107Chen, Huang, Liao and colleagues108 achieved the electrochemical syntheses of β-ketosulfonyl fluorides using a sacrificial anode of magnesium and an aluminum cathode in Et2O as solvent, with LiClO4 as electrolyte. The scope of the transformation is shown in Scheme 54.
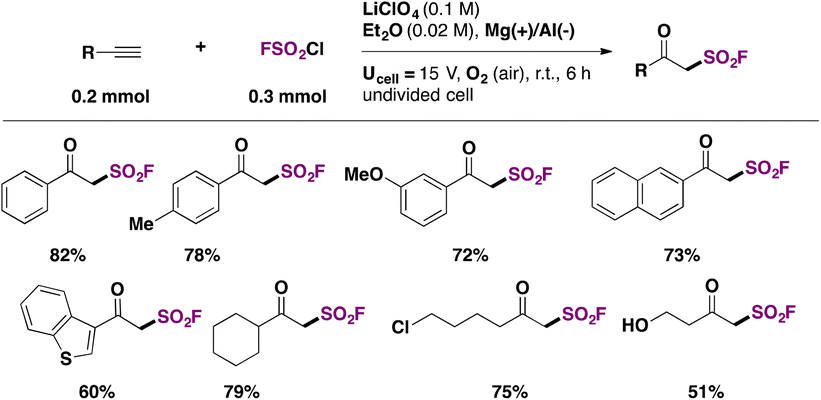 | ||
| Scheme 54 Selected examples for the electrochemical syntheses of β-keto sulfonyl fluorides. | ||
Aryl and heteroaryl acetylenes afforded good yields of the respective β-keto sulfonyl fluorides. Aliphatic acetylenes such as cyclohexylacetylene, 6-chlorohexyne, and 1-hydroxy-3-butyne gave good yields of fluorosulfonylated products as well. Changing the reaction conditions (THF, instead of Et2O, LiClO4 0.2 M, and Ucell = 10 V), the authors108 obtained the α-chloro-β-keto sulfonyl fluorides from phenylacetylenes.
Interestingly, the authors examined the transformation of 2-oxo-2-phenylethane-1-sulfonyl fluoride into derivatives. Scheme 55 depicts some of these relevant transformations.
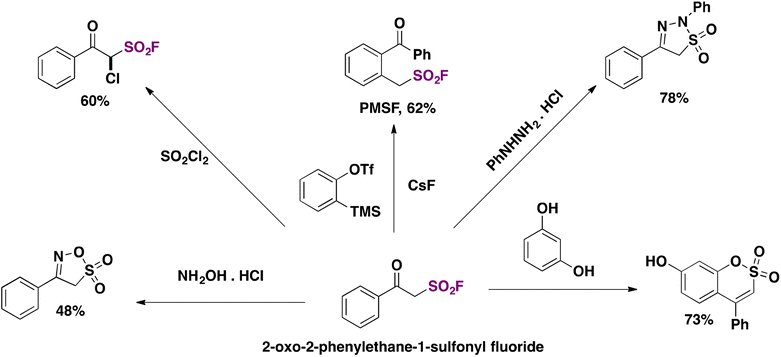 | ||
| Scheme 55 Transformations of 2-oxo-2-phenylethane-1-sulfonyl fluoride into derivatives. | ||
The inhibitor PMSF18,19 (Scheme 55, upper center) is obtained in very good yield. The reaction of 2-oxo-2-phenylethane-1-sulfonyl fluoride with hydroxylamine hydrochloride produced a novel heterocycle “oxathiazole” in reasonably good yield (Scheme 55, lower left side).
Based on radical probe experiments and the necessity for both electrical current and an electrolyte in order to accomplish product formation, the authors postulated a plausible reaction mechanism such as that depicted in Scheme 56. By cathodic reduction, FSO2Cl produced FSO2 radicals and chloride anions. The FSO2 radicals add to the terminal phenylacetylene carbon to produce intermediate XVIII, which in the presence of air generates intermediate XIX. By a Russel mechanism, intermediate XX is formed, which is reduced in situ to intermediate XXI, which upon protonation gives the product in Et2O. In THF, chlorination of the β-keto sulfonyl fluorides by MgCl2 affords the α-chloro-β-keto sulfonyl fluorides (Scheme 56).
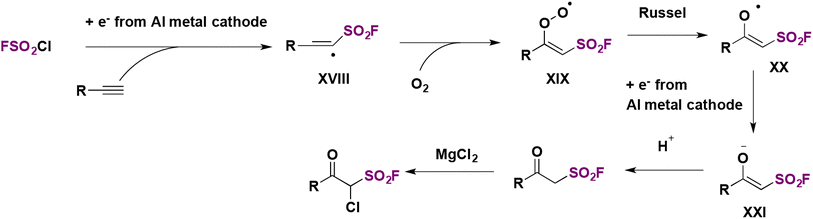 | ||
| Scheme 56 Proposed reaction mechanism. | ||
In 2022, Feng, Huang, and colleagues109 introduced a modification of the electrochemical syntheses of β-keto sulfonyl fluorides through the radical fluorosulfonylation of vinyl triflates. The authors109 used graphite felt (GF) as an electrode to generate FSO2 radicals from FSO2Cl, in diethyl ether as solvent, with Et4NPF6 as electrolyte. The scope of the transformation is shown in Scheme 57.
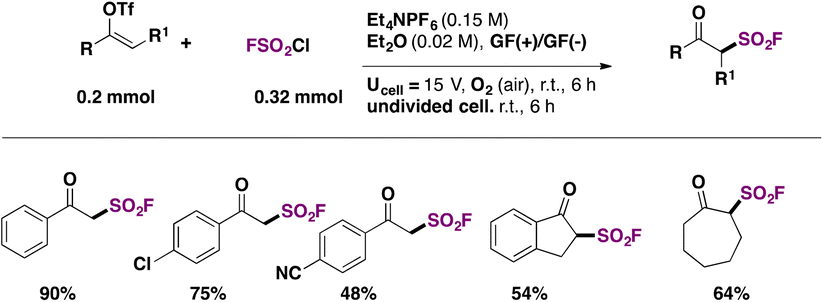 | ||
| Scheme 57 Selected examples for the radical fluorosulfonylation of vinyl triflates. | ||
C.5. Syntheses of fluorosulfonylamines and the fluorosulfonylation of phenols
In 2018, Guo, Sharpless, Dong, and colleagues6 developed an electrophilic fluorosulfonyl donor capable of fluorosulfonylating alcohols, phenols, and primary and secondary amines in excellent yields. The reagent is fluorosulfuryl imidazolium triflate B (Scheme 58), which was observed to be much more reactive and stable than the classic SO2F2 reagent.110B was examined for its ability to fluorosulfonylate a range of phenols and primary and secondary amines, according to Scheme 58.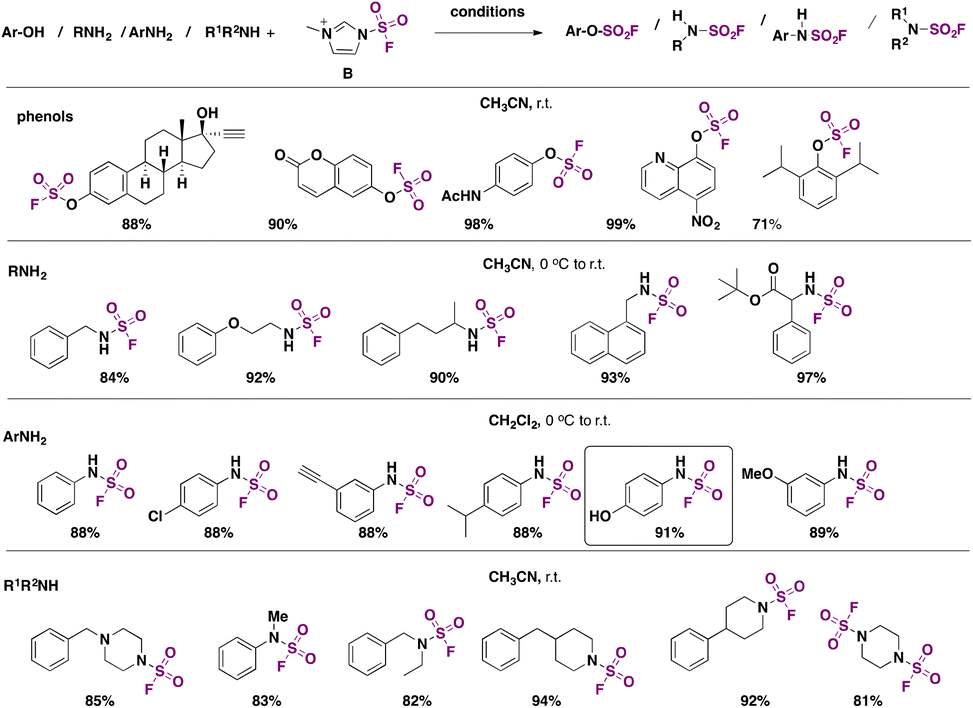 | ||
| Scheme 58 Scope of the fluorosulfonylation of phenols, primary aliphatic amines, primary aromatic amines and secondary amines. | ||
Reagent B was prepared from 2-methylimidazole and SO2F2. Phenols were converted to their respective fluorosulfates in the presence of triethylamine (Scheme 58). Even sterically hindered phenols were transformed into their respective fluorosulfates in very good yields. Unlike SO2F2, B reacted readily with primary and secondary amines, as shown in Scheme 58, affording the corresponding sulfamoyl fluorides without the presence of additives, as opposed to when reagent SO2F2 is used, which necessitates the presence of triethylamine and additives (such as DMAP or DABSO) to afford sulfamoyl fluorides. Both aliphatic and aromatic primary amines reacted under the conditions shown in Scheme 58. Even the less reactive secondary amines provided good yields of products. This represents one of the few protocols to prepare and isolate products bearing the sulfamoyl fluoride group.
It is to be observed that the amine site preferentially reacts in the presence of the phenol function, as depicted in Scheme 58 for aromatic primary amines, in sharp contrast to the SO2F2 reagent, which prefers the phenol reactivity over the amine site.
D. Conclusions
The profound impact attributed to organic compounds bearing the –SO2F functionality has traversed fields, from organic chemistry to biological applications and from drug discovery to materials sciences, most probably driven by the seminal paper by Sharpless in 2014 with the introduction of the SuFEx click chemistry reaction. This constantly growing and expanding area of fluorosulfonylated compounds has demanded a significant need for alternative methods (one-pot procedures) to synthesize aromatic and aliphatic sulfonyl fluorides that allow the simultaneous incorporation of both SO2 and F groups, without the requirement of previously installed SO2 or F functionalities.In this review, we critically discussed, from an organic chemist's perspective, new methodologies for the syntheses and some applications of (hetero)aromatic-, alkyl-, alkenyl-, and alkynyl-sulfonyl fluorides and β-keto-sulfonyl fluorides and the syntheses of compounds with N–SO2F and O–SO2F bonds.
Besides the classical Cl/F exchange from the corresponding chlorides, the syntheses of alkyl-substituted sulfonyl fluorides can be carried out by direct or indirect methods. Synthetic direct methods via radical sulfur dioxide insertion/fluorination provide aliphatic sulfonyl fluorides in good yields or the visible light-photocatalyzed addition of SO2F radicals from newly developed SO2F-reagents affords excellent yields of aliphatic saturated fluorosulfonyl substrates. Indirect methods can resort to the addition of SO2F– Michael acceptors such ESF and derivatives. The drawback with the employment of ESF (and derived hubs) is the use of toxic and hygroscopic starting 2-chloroethane-1-sulfonyl chloride substrate for their syntheses. Also, the synthesis of the precursors for radical fluorosulfonylating reagents derived from benzimidazolium salts necessitates the ClSO2F reagent, which is costly (U$D 10955 per mol).
Fluorosulfonylated alkenes can also be synthesized either through direct fluorosulfonylating radical strategies from olefins or alkynes as substrates, or by employing fluorosulfonylating hubs such as ESF and its derivatives. Direct radical fluorosulfonylating reagents to accomplish the syntheses of fluorosulfonylated alkenes involve ClSO2F and FSO2-substituted benzimidazolium salts precursors, which share limitations concerned with handling and costs.
On the other hand, the syntheses of fluorosulfonylated (hetero)arenes, other than the classical Cl/F exchange in aryl sulfonyl chlorides, can consider the use of disulfides with Selectfluor to obtain the sulfonyl fluoride in high yields. Aryl sulfonamides, sulfonylhydrazides and arylsulfinates can also be used as starting substrates for the synthesis of aryl sulfonyl fluorides. Direct methods, namely those that do not require pre-synthesized/installed sulfur-containing substrates, can employ aryl iodides, aryl bromides, aryl boronic acids, and aryl diazonium salts as starting materials, DABSO and Na2S2O5 as sources of SO2, and a fluorinating reagent (Selectfluor, NFSI, or KHF2). Probably, these latter methods are more friendly in terms of reagent handling and costs. However, to date, no direct radical fluorosulfonylating reagents, such as FSO2-substituted benzoimidazolium salts, have been employed for the incorporation of the SO2F group onto (hetero)aromatic compounds, although radical methods to fluorosulfonylate aromatic substrates are already in place.
Among the catalytic methodologies established for the fluorosulfonylation of aliphatic and aromatic compounds, metal-mediated methods, thermal techniques, and more recently photocatalytic and electrochemical protocols have been reported. However, the application of flow system methodologies has not been reported for fluorosulfonylation reactions, which could be conveniently applied in the photocatalytic protocols established so far. This area will probably witness an expansion in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SBV, DEY and AP are research members of Consejo Nacional de Investigaciones Científicas y Técnicas, CONICET. Financial aid for this project was obtained from Agencia Nacional de Promoción Científica y Técnica-Argentina, Grant PICT2017-2276.Notes and references
- C. R. Pitts, D. Bornemann, P. Liebing, N. Santschi and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 1950–1954 CrossRef CAS PubMed.
- Y. Kraemer, E. N. Bergman, A. Togni and C. R. Pitts, Angew. Chem., Int. Ed. DOI:10.1002/anie.202205088.
- R. Kordnezhadian, B. Li, A. Zogu, J. Demaerel, W. M. De Borggraeve and E. Ismalaj, Chem. – Eur. J. DOI:10.1002/chem.202201491.
- A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294–2299 CrossRef CAS PubMed.
- H. Zhou, P. Mukherjee, R. Liu, E. Evrard, D. Wang, J. M. Humphrey, T. W. Butler, L. R. Hoth, J. B. Sperry, S. K. Sakata, C. J. Helal and C. W. Am Ende, Org. Lett., 2018, 20, 812–815 CrossRef CAS PubMed.
- T. Guo, G. Meng, X. Zhan, Q. Yang, T. Ma, L. Xu, K. B. Sharpless and J. Dong, Angew. Chem., 2018, 130, 2635–2640 CrossRef.
- J. Liao, W. Guo, Z. Zhang, X. Tang, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 1304–1309 CrossRef CAS PubMed.
- K. Zhang, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2015, 80, 7658–7665 CrossRef CAS PubMed.
- Y. Sumii, Y. Sugita, E. Tokunaga and N. Shibata, ChemistryOpen, 2018, 7, 204–211 CrossRef CAS PubMed.
- Y. Liu, S. Bai, Y. Du, X. Qi and H. Gao, Angew. Chem., Int. Ed. DOI:10.1002/anie.202115611.
- L. V. Sokolenko, R. K. Orlova, A. A. Filatov, Y. L. Yagupolskii, E. Magnier, B. Pégot and P. Diter, Molecules, 2019, 24, 1249 CrossRef PubMed.
- S. Xing, Y.-Y. Zhu, W. Liu, Y. Liu, J. Zhang, H. Zhang, Y. Wang, S.-F. Ni and X. Shao, Org. Lett., 2022, 24, 3378–3383 CrossRef CAS PubMed.
- D. Zeng, Y. Ma, W. Deng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2022, 61, 1–9 Search PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. Barry Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- C. Chatgilialoglu, in The Chemistry of Sulphones and Sulphoxides, ed. S. Patai, S. Rappoport and C. Stirling, John Wiley & Sons, Ltd, 1988, pp. 1089–1113 Search PubMed.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS PubMed.
- D. A. Jeffery and M. Bogyo, Curr. Opin. Biotechnol., 2003, 14, 87–95 CrossRef CAS PubMed.
- G. F. Zha, Q. Zheng, J. Leng, P. Wu, H. L. Qin and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 4849–4852 CrossRef CAS PubMed.
- A. J. Brouwer, N. Herrero Álvarez, A. Ciaffoni, H. van de Langemheen and R. M. J. Liskamp, Bioorg. Med. Chem., 2016, 24, 3429–3435 CrossRef CAS PubMed.
- M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed.
- M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS PubMed.
- P. Mukherjee, C. P. Woroch, L. Cleary, M. Rusznak, R. W. Franzese, M. R. Reese, J. W. Tucker, J. M. Humphrey, S. M. Etuk, S. C. Kwan, C. W. Am Ende and N. D. Ball, Org. Lett., 2018, 20, 3943–3947 CrossRef CAS PubMed.
- L. L. Frye, E. L. Sullivan, K. P. Cusack and J. M. Funaro, J. Org. Chem., 1992, 57, 697–701 CrossRef CAS.
- L. Matesic, N. A. Wyatt, B. H. Fraser, M. P. Roberts, T. Q. Pham and I. Greguric, J. Org. Chem., 2013, 78, 11262–11270 CrossRef CAS PubMed.
- J. A. H. Inkster, K. Liu, S. Ait-Mohand, P. Schaffer, B. Guérin, T. J. Ruth and T. Storr, Chem. – Eur. J., 2012, 18, 11079–11087 CrossRef CAS PubMed.
- T. A. Bianchi and L. A. Cate, J. Org. Chem., 1977, 42, 2031–2032 CrossRef CAS.
- J. S. Oakdale, L. Kwisnek and V. V. Fokin, Macromolecules, 2016, 49, 4473–4479 CrossRef CAS.
- (a) C. J. Smedley, J. A. Homer, T. L. Gialelis, A. S. Barrow, R. A. Koelln and J. E. Moses, Angew. Chem., Int. Ed., 2022, e202112375 CAS; (b) M. Wei, D. Liang, X. Cao, W. Luo, G. Ma, Z. Liu and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7397–7404 CrossRef CAS PubMed; (c) S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 2903–2908 CrossRef CAS PubMed.
- J. Leng and H. L. Qin, Chem. Commun., 2018, 54, 4477–4480 RSC.
- A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233–1237 RSC.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- (a) C. Lee, A. J. Cook, J. E. Elisabeth, N. C. Friede, G. M. Sammis and N. D. Ball, ACS Catal., 2021, 11, 6578–6589 CrossRef CAS PubMed; (b) T. Zhong, Z. Chen, J. Yi, G. Lu and J. Weng, Chin. Chem. Lett., 2021, 32, 2736–2750 CrossRef CAS.
- T. S. B. Lou and M. C. Willis, Nat. Rev. Chem., 2022, 6, 146–162 CrossRef CAS.
- F.-S. He, Y. Li and J. Wu, Org. Chem. Front., 2022, 9, 5299–5305 RSC.
- X. Nie and S. Liao, Synlett, 2022, 33, 401–408 CrossRef CAS.
- Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18808–18814 CrossRef CAS PubMed.
- M. Kirihara, S. Naito, Y. Ishizuka, H. Hanai and T. Noguchi, Tetrahedron Lett., 2011, 52, 3086–3089 CrossRef CAS.
- M. Kirihara, S. Naito, Y. Nishimura, Y. Ishizuka, T. Iwai, H. Takeuchi, T. Ogata, H. Hanai, Y. Kinoshita, M. Kishida, K. Yamazaki, T. Noguchi and S. Yamashoji, Tetrahedron, 2014, 70, 2464–2471 CrossRef CAS.
- S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed.
- M. Pérez-Palau and J. Cornella, Eur. J. Org. Chem., 2020, 2020, 2497–2500 CrossRef.
- (a) L. Tang, Y. Yang, L. Wen, X. Yang and Z. Wang, Green Chem., 2016, 18, 1224–1228 RSC; (b) Q. Pan, Y. Liu, W. Pang, J. Wu, X. Ma, X. Hu, Y. Guo, Q.-Y. Chen and C. Liu, Org. Biomol. Chem., 2021, 19, 8999–9003 RSC.
- J. Kwon and B. M. Kim, Org. Lett., 2019, 21, 428–433 CrossRef CAS PubMed.
- P. K. T. Lo, Y. Chen and M. C. Willis, ACS Catal., 2019, 9, 10668–10673 CrossRef CAS.
- C. Lee, N. D. Ball and G. M. Sammis, Chem. Commun., 2019, 55, 14753–14756 RSC.
- G. Laudadio, A. D. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. De Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed.
- K. Lam and W. E. Geiger, J. Org. Chem., 2013, 78, 8020–8027 CrossRef CAS PubMed.
- Y. Liu, D. Yu, Y. Guo, J. C. Xiao, Q. Y. Chen and C. Liu, Org. Lett., 2020, 22, 2281–2286 CrossRef CAS PubMed.
- S. Liu, Y. Huang, X. H. Xu and F. L. Qing, J. Fluorine Chem., 2020, 240, 1–6 CrossRef.
- Q. Lin, Z. Ma, C. Zheng, X. J. Hu, Y. Guo, Q. Y. Chen and C. Liu, Chin. J. Chem., 2020, 38, 1107–1110 CrossRef CAS.
- Z. Ma, L. Shan, X. Ma, X. Hu, Y. Guo, Q. Y. Chen and C. Liu, J. Fluorine Chem., 2022, 254, 109948 CrossRef CAS.
- T. Zhong, M. K. Pang, Z. Da Chen, B. Zhang, J. Weng and G. Lu, Org. Lett., 2020, 22, 3072–3078 CrossRef CAS PubMed.
- D. Louvel, A. Chelagha, J. Rouillon, P. Payard, L. Khrouz, C. Monnereau and A. Tlili, Chem. – Eur. J., 2021, 27, 8704–8708 CrossRef CAS PubMed.
- T. T. Bui, V. H. Tran and H. Kim, Adv. Synth. Catal., 2022, 364, 341–347 CrossRef CAS.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- R. Artschwager, D. J. Ward, S. Gannon, A. J. Brouwer, H. Van De Langemheen, H. Kowalski and R. M. J. Liskamp, J. Med. Chem., 2018, 61, 5395–5411 CrossRef CAS PubMed.
- J. Thomas and V. V. Fokin, Org. Lett., 2018, 20, 3749–3752 CrossRef CAS PubMed.
- X. Zhang, W. Y. Fang, R. Lekkala, W. Tang and H. L. Qin, Adv. Synth. Catal., 2020, 362, 3358–3363 CrossRef CAS.
- (a) B. Moku, W. Y. Fang, J. Leng, L. Li, G. F. Zha, K. P. Rakesh and H. L. Qin, iScience, 2019, 21, 695–705 CrossRef CAS PubMed; (b) H.-R. Chen, Z.-Y. Hu, H.-L. Qin and H. Tang, Org. Chem. Front., 2021, 8, 1185–1189 RSC.
- Y. Liu, H. Wu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Angew. Chem., 2017, 129, 15634–15637 CrossRef.
- Q. Lin, Y. Liu, Z. Xiao, L. Zheng, X. Zhou, Y. Guo, Q. Y. Chen, C. Liu and C. Zheng, Org. Chem. Front., 2019, 6, 447–450 RSC.
- Y. Liu, Q. Lin, Z. Xiao, C. Zheng, Y. Guo, Q. Y. Chen and C. Liu, Chem. – Eur. J., 2019, 25, 1824–1828 CrossRef CAS PubMed.
- (a) Z. Da Chen, X. Zhou, J. T. Yi, H. J. Diao, Q. L. Chen, G. Lu and J. Weng, Org. Lett., 2022, 24, 2474–2478 CrossRef PubMed; (b) T. Zhong, J.-T. Yi, Z.-D. Chen, Q.-C. Zhuang, Y.-Z. Li, G. Lu and J. Weng, Chem. Sci., 2021, 12, 9359–9365 RSC.
- A. Shavnya, S. B. Coffey, K. D. Hesp, S. C. Ross and A. S. Tsai, Org. Lett., 2016, 18, 5848–5851 CrossRef CAS PubMed.
- A. Shavnya, K. D. Hesp and A. S. Tsai, Adv. Synth. Catal., 2018, 360, 1768–1774 CrossRef CAS.
- Z. Ma, Y. Liu, X. Ma, X. Hu, Y. Guo, Q. Y. Chen and C. Liu, Org. Chem. Front., 2022, 9, 1115–1120 RSC.
- H. Zhang, S. Li, H.-L. Zheng, G. Zhu, S. Liao and X. Nie, Org. Chem. Front., 2022, 9, 4854–4860 RSC.
- J.-T. Yi, X. Zhou, Q.-L. Chen, Z.-D. Chen, G. Lu and J. Weng, Chem. Commun., 2022, 58, 9409–9412 RSC.
- W. Zhang, H. Li, X. Li, Z. Zou, M. Huang, J. Liu, X. Wang, S. Ni, Y. Pan and Y. Wang, Nat. Commun., 2022, 13, 3515 CrossRef CAS PubMed.
- N. L. Frye, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2022, 61, 1–6 Search PubMed.
- P. Wang, H. Zhang, M. Zhao, S. Ji, L. Lin, N. Yang, X. Nie, J. Song and S. Liao, Angew. Chem., Int. Ed., 2022, 61, 1–9 Search PubMed.
- J. A. Andrews, L. R. E. Pantaine, C. F. Palmer, D. L. Poole and M. C. Willis, Org. Lett., 2021, 23, 8488–8493 CrossRef CAS PubMed.
- (a) P. J. Sarver, N. B. Bissonnette and D. W. C. Macmillan, J. Am. Chem. Soc., 2021, 143, 9737–9743 CrossRef CAS PubMed; (b) J. Chen, D.-y. Zhu, X.-j. Zhang and M. Yan, J. Org. Chem., 2021, 86, 3041–3048 CrossRef CAS PubMed.
- (a) R. Xu, T. Xu, M. Yang, T. Cao and S. Liao, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed; (b) Y. Liu, H. Wu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Angew. Chem., 2017, 129, 15634–15637 CrossRef.
- X. Zeng, H. Beckers and H. Willner, J. Am. Chem. Soc., 2013, 135, 2096–2099 CrossRef CAS PubMed.
- X. Nie, T. Xu, J. Song, A. Devaraj, B. Zhang, Y. Chen and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 3956–3960 CrossRef CAS PubMed.
- X. Nie, T. Xu, Y. Hong, H. Zhang, C. Mao and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 22035–22042 CrossRef CAS PubMed.
- T. S. B. Lou, S. W. Bagley and M. C. Willis, Angew. Chem., Int. Ed., 2019, 58, 18859–18863 CrossRef CAS PubMed.
- P. Wang, H. Zhang, X. Nie, T. Xu and S. Liao, Nat. Commun., 2022, 13, 3370 CrossRef CAS PubMed.
- Q. Chen, P. Mayer and H. Mayr, Angew. Chem., Int. Ed., 2016, 55, 12664–12667 CrossRef CAS PubMed.
- H.-L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, Angew. Chem., 2016, 128, 14361–14364 CrossRef.
- P. K. Chinthakindi, K. B. Govender, A. S. Kumar, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, Org. Lett., 2017, 19, 480–483 CrossRef CAS PubMed.
- T. S. Lou, S. W. Bagley and M. C. Willis, Angew. Chem., 2019, 131, 19035–19039 CrossRef.
- X.-Y. Chen, Y. Wu, J. Zhou, P. Wang and J.-Q. Yu, Org. Lett., 2019, 21, 1426–1429 CrossRef CAS PubMed.
- J. J. Krutak, R. D. Burpitt, W. H. Moore and J. A. Hyatt, J. Org. Chem., 1979, 44, 3847–3858 CrossRef CAS.
- C. Li, S. M. Wang and H. L. Qin, Org. Lett., 2018, 20, 4699–4703 CrossRef CAS PubMed.
- C. J. Smedley, M. C. Giel, A. Molino, A. S. Barrow, D. J. D. Wilson and J. E. Moses, Chem. Commun., 2018, 54, 6020–6023 RSC.
- J. Leng, N. S. Alharbi and H.-L. Qin, Eur. J. Org. Chem., 2019, 2019, 6101–6105 CrossRef CAS.
- C. Li, Y. Zheng, K. P. Rakesh and H. L. Qin, Chem. Commun., 2020, 56, 8075–8078 RSC.
- C. J. Smedley, G. Li, A. S. Barrow, T. L. Gialelis, M. C. Giel, A. Ottonello, Y. Cheng, S. Kitamura, D. W. Wolan, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2020, 59, 12460–12469 CrossRef CAS PubMed.
- X. Zhang, B. Moku, J. Leng, K. P. Rakesh and H. L. Qin, Eur. J. Org. Chem., 2019, 2019, 1763–1769 CrossRef CAS.
- S. Xu and S. Cui, Org. Lett., 2021, 23, 5197–5202 CrossRef CAS PubMed.
- V. L. Mykhalchuk, V. S. Yarmolchuk, R. O. Doroschuk, A. A. Tolmachev and O. O. Grygorenko, Eur. J. Org. Chem., 2018, 2018, 2870–2876 CrossRef CAS.
- M. C. Giel, C. J. Smedley, E. R. R. Mackie, T. Guo, J. Dong, T. P. Soares da Costa and J. E. Moses, Angew. Chem., Int. Ed., 2020, 59, 1181–1186 CrossRef CAS PubMed.
- L. Li, P. Mayer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem. DOI:10.1002/ejoc.202200865.
- J. Chen, B. Q. Huang, Z. Q. Wang, X. J. Zhang and M. Yan, Org. Lett., 2019, 21, 9742–9746 CrossRef CAS PubMed.
- H. L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2016, 55, 14155–14158 CrossRef CAS PubMed.
- S. M. Wang, C. Li, J. Leng, S. N. A. Bukhari and H. L. Qin, Org. Chem. Front., 2018, 5, 1411–1415 RSC.
- P. Wipf, D. C. Aslan, E. C. Southwick and J. S. Lazo, Bioorg. Med. Chem. Lett., 2001, 11, 313–317 CrossRef CAS PubMed.
- K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 6, 159–162 CrossRef.
- N. Oger, M. Dhalluin, E. Le Grognec and F. X. Felpin, Org. Process Res. Dev., 2014, 18, 1786–1801 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- W.-Y. Fang, S.-M. Wang, Z.-W. Zhang and H.-L. Qin, Org. Lett., 2020, 22, 8904–8909 CrossRef CAS PubMed.
- T. Henkel, T. Krügerke and K. Seppelt, Angew. Chem., Int. Ed. Engl., 1990, 29, 1128–1129 CrossRef.
- D. Chen, X. Nie, Q. Feng, Y. Zhang, Y. Wang, Q. Wang, L. Huang, S. Huang and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 27271–27276 CrossRef CAS PubMed.
- Q. Feng, Y. Fu, Y. Zheng, S. Liao and S. Huang, Org. Lett., 2022, 24, 3702–3706 CrossRef CAS PubMed.
- S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS PubMed.
- H. Zhang, N. Yang, J. Li, P. Wang, S. Li, L. Xie and S. Liao, Org. Lett., 2022, 24, 8170–8175 CrossRef CAS PubMed.
- P. Wang, S.-J. Li, H. Zhang, N. Yang and S. Liao, Synlett, 2023, 34(05), 471–476 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |