
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-responsive materials: the (photo)chemistry and applications of donor–acceptor Stenhouse adducts in polymer science
Michèle
Clerc
 abcd,
Sara
Sandlass
e,
Omar
Rifaie-Graham
f,
Julie A.
Peterson
g,
Nico
Bruns
*cdh,
Javier
Read de Alaniz
*g and
Luciano F.
Boesel
*ad
abcd,
Sara
Sandlass
e,
Omar
Rifaie-Graham
f,
Julie A.
Peterson
g,
Nico
Bruns
*cdh,
Javier
Read de Alaniz
*g and
Luciano F.
Boesel
*ad
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biomimetic Membranes and Textiles, 9014 St. Gallen, Switzerland. E-mail: luciano.boesel@empa.ch
bUniversity of Fribourg, Department of Chemistry, 1700 Fribourg, Switzerland
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK
dSwiss National Center of Competence in Research Bio-Inspired Materials, Switzerland
eDepartment of Chemical Engineering, University of California, Santa Barbara, California 93106, USA
fDepartment of Chemistry, School of Physical and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK
gDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: javier@chem.ucsb.edu
hDepartment of Chemistry, Technical University of Darmstadt, 64287 Darmstadt, Germany. E-mail: nico.bruns@tu-darmstadt.de
First published on 31st October 2023
Abstract
Donor–acceptor Stenhouse adduct (DASA) photoswitches have gained a lot of attention since their discovery in 2014. Their negative photochromism, visible light absorbance, synthetic tunability, and the large property changes between their photoisomers make them attractive candidates over other commonly used photoswitches for use in materials with responsive or adaptive properties. The development of such materials and their translation into advanced technologies continues to widely impact forefront materials research, and DASAs have thus attracted considerable interest in the field of visible-light responsive molecular switches and dynamic materials. Despite this interest, there have been challenges in understanding their complex behavior in the context of both small molecule studies and materials. Moreover, incorporation of DASAs into polymers can be challenging due to their incompatibility with the conditions for most common polymerization techniques. In this review, therefore, we examine and critically discuss the recent developments and challenges in the field of DASA-containing polymers, aiming at providing a better understanding of the interplay between the properties of both constituents (matrix and photoswitch). The first part summarizes current understanding of DASA design and switching properties. The second section discusses strategies of incorporation of DASAs into polymers, properties of DASA-containing materials, and methods for studying switching of DASAs in materials. We also discuss emerging applications for DASA photoswitches in polymeric materials, ranging from light-responsive drug delivery systems, to photothermal actuators, sensors and photoswitchable surfaces. Last, we summarize the current challenges in the field and venture on the steps required to explore novel systems and expand both the functional properties and the application opportunities of DASA-containing polymers.
 Michèle Clerc | Michèle Clerc obtained her BSc and MSc degree from the University of Zurich and then joined Luciano Boesel's group at Empa, Swiss Federal Laboratories for Materials Science and Technology, for her doctoral studies. In 2023, she received her PhD degree in chemistry from the University of Fribourg under the joint supervision of Luciano Boesel, Prof. Stefan Salentinig (University of Fribourg) and Prof. Nico Bruns (University of Strathclyde). Her thesis work focuses on the control of polymer materials with light using donor–acceptor Stenhouse adduct photoswitches. |
 Sara Sandlass | Sara Sandlass is a doctoral student working under Professor Michael J. Gordon at the University of California, Santa Barbara. Her research is in the design of photo-responsive materials and structures for applications in light powered soft robotics. As part of this work, she is interested in studying the isomerization kinetics of DASA in polymers to better leverage these materials for applications in photoactuation. |
 Omar Rifaie-Graham | Omar Rifaie-Graham is an Assistant Professor in Chemistry at Queen Mary University of London since 2023. He received his PhD in Chemistry from the Adolphe Merkle Institute, University of Fribourg in 2018. He then became a Swiss National Science Foundation Early Postdoc Mobility Fellow and a Marie Skłodowska-Curie Actions Individual Fellow at Imperial College London. His research interests span stimuli-responsive polymers and polymer nanotechnology. In particular, his research focuses on the emergence of cell behavior from ensembles of polymers and enzymes to understand fundamentals of biological phenomena and to generate micro- and nanomachines for application in biomedicine and biotechnology. |
 Julie A. Peterson | Julie Peterson joined Bowling Green State University as an Assistant Professor in 2023. She received her PhD in Organic Chemistry at Iowa State University under the supervision of Dr Arthur Winter in 2020. She then moved to University of California, Santa Barbara to work with Dr Javier Read de Alaniz as an Elings Postdoctoral fellow. Her current research focuses on designing light responsive small molecules for use in materials and photobiochemical applications. |
 Nico Bruns | Nico Bruns is Professor of Sustainable Functional Polymers at the Technical University of Darmstadt, Germany. He received his PhD in Macromolecular Chemistry from the University of Freiburg, Germany, was a postdoc at the University of California, Berkeley, and then moved to the University of Basel, Switzerland, for his Habilitation. From 2013 to 2018 he was Associate Professor of Macromolecular Chemistry at the Adolphe-Merkle-Institute of the University of Fribourg, Switzerland, and between 2018 and 2021 Professor of Macromolecular Chemistry at the University of Strathclyde, Glasgow, UK. His research interests are biocatalysis in polymer chemistry, stimuli-responsive polymersomes, and bio-inspired materials, including DASA–polymer conjugates. |
 Javier Read de Alaniz | Javier Read de Alaniz is the co-Director of the NSF BioPACIFIC Materials Innovation Platform and Professor of Chemistry at UC Santa Barbara (UCSB). He obtained his PhD under the supervision of Professor Tomislav Rovis at Colorado State University in 2006. He then moved as a UC President's Postdoctoral Fellow to UC Irvine to work with Professor Larry Overman, where he worked in the area of total synthesis. In 2009 he started his independent career at UCSB and in 2015 became a full Professor. His research interest includes the development of stimuli-responsive molecules, as well as the synthesis of functional polymers and biomaterials. |
 Luciano F. Boesel | Luciano F. Boesel is Group Leader and Deputy Head of the Laboratory for Biomimetic Membranes and Textiles at Empa, Swiss Federal Laboratories for Materials Science and Technology. He received a PhD in Materials Science and Technology (Biomaterials) from the University of Minho (Portugal). Before joining Empa in 2010, he was a Marie Skłodowska-Curie Research Fellow at the Institute of Science and Technology of Polymers (CSIC, Spain) and an Alexander von Humboldt Fellow at the Max-Planck Institute for Polymer Research (Germany). His research interests are in the development of wearable biophysical and biochemical sensors, stimuli-responsive materials, and wearable solar energy harvesting. |
1. Introduction
Molecular photoswitches are small organic molecules that can be reversibly interconverted between different (meta)stable isomers in response to light.1 The reversible structural changes induced by photoisomerization render photoswitches attractive as tools for optical manipulation of materials.2–5 Photochromic materials can be composed of (bio-)macromolecules functionalized with photoswitches, whereby photoisomerization events at the molecular level cause changes in the surrounding polymer chains. This can lead to interesting effects across length scales, from light-induced nanoscale morphology changes up to macroscopic motion, enabling a broad spectrum of applications in material science and biology.2–5 A selection of relevant fields of application of photoswitches and specific material classes have been recently reviewed, targeting applications in medicine,6 catalysis,7 optoelectronics,8 fluorescence bioimaging,9 chemosensing,10 photoinduced solid-to-liquid transitions,11 photoresponsive hydrogels,12,13 photo-triggered polymeric nanomedicines,14 photoswitchable molecular amphiphiles,15 peptides16 or nanomaterials in general.17The majority of photoswitch classes require energy-rich ultraviolet (UV) light to undergo photoisomerization in at least one direction.1 This is clearly a limitation for applications in the life sciences, since UV light causes photodamage in biological samples and also shows reduced tissue penetration.18 Therefore, visible light and near-infrared (NIR) addressable photoswitches (wavelength, λ, above 400 nm of the electromagnetic spectrum) offer a clear advantage. As a result, different strategies have emerged to extend activation windows to longer wavelengths in well-established classes of photoswitches, such as azobenzenes,19 spiropyrans/spirooxazines,20 and diarylethenes.21 These strategies include structural modifications of the photoswitch (increase of π-conjugation, push–pull substitution patterns, etc.), multi-photon excitation, or indirect photoexcitation via energy or electron transfer.22,23 However, red-shifting often compromises other properties of the system, such as a reduction in isomerization quantum yield, photoconversion efficiency, fatigue resistance, or solubility, a shortened thermal half-live of the metastable isomer, or decreased synthetic accessibility.22,23 The discovery of photoswitches with completely novel core structures that allow direct activation by visible/NIR light23 is therefore crucial for expanding the applicability of smart photochromic materials,24 especially for controlling new and more complex systems and for biomedical applications. In the past decade, different visible/NIR light addressable photoswitch classes were (re-)discovered, including dihydropyrenes,25 (hemi-)indigoids,26–28 phenoxyl-imidazolyl radical complexes29 and donor–acceptor Stenhouse adducts (DASAs).30,31
Among visible/NIR photoswitches, DASAs stand out owing to their synthetic accessibility and large structural changes during photoisomerization (Fig. 1). In practice, DASAs offer high molar absorptivity and present both optical and photoswitching tunability through synthetically easy to implement structural modifications.32–37 In addition, DASA photoisomerization involves a transition from a stable colored isomer to a thermally metastable colorless state (classified as negative photochromism).38 For applications requiring high absorber concentrations in materials, this feature is advantageous since controlling the spatial distribution of colored and colorless isomers allows for tunability in the depth of light penetration.4 Hence, it is not surprising that DASAs have attracted tremendous interest in the field, leading to a steep increase in publications over the past nine years since their discovery. Alongside elucidation of photoswitching mechanisms39–43 and establishing structure–property relationships,32–37,44 a number of interesting and diverse applications in functional systems and materials have been reported. While a solid basis for the fundamental understanding of design principles on the molecular level has been established in recent years, DASA behavior in more complex environments is still not well understood. However, understanding the interplay between the influence of a (polymeric) support on the properties of DASAs and vice versa is essential to realize their full potential in applications. Due to their novelty, previous review45–47 and book chapters48,49 on DASA photoswitches primarily focused on their structure, synthesis, switching mechanism and properties at the small molecule level. In this review, we focus on DASAs in macromolecular environments and discuss in detail their implementation, performance and applications in smart polymeric materials.
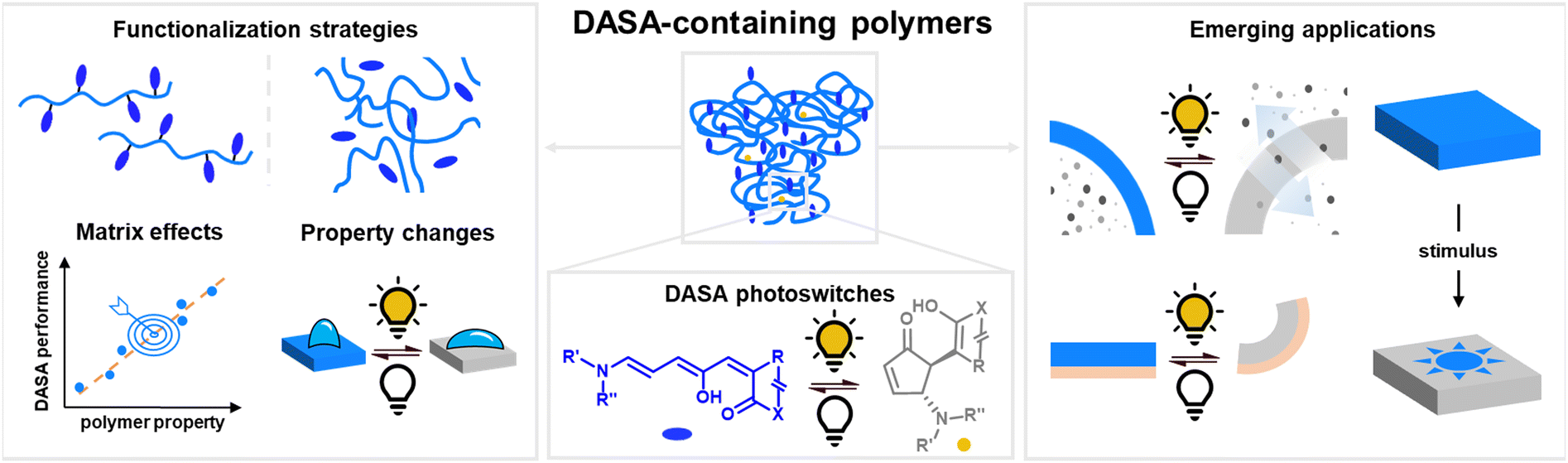 | ||
| Fig. 1 Visible light-responsive polymeric materials based on donor–acceptor Stenhouse Adducts (DASAs). This article focuses on DASA polymer conjugation and functionalization strategies, polymer matrix effects on DASA photoswitching, (photo)induced effects on polymer properties, and application examples of DASA-containing polymers. | ||
In the first introductory part of this article, we summarize the current state of knowledge on DASA (photo)chemistry with an emphasis on aspects that are relevant for applications in materials, such as the influence of environmental factors on the physical and chemical properties of DASAs. In the second part, we discuss DASA polymer conjugation and functionalization strategies, (photo)induced effects on polymer properties, photoswitching and matrix effects, DASA stability and fatigue, as well as previously applied methods and models for DASA photoswitching analysis in materials. The third part covers a comprehensive overview on application examples of DASA-containing polymers. The majority of literature reports on DASA-functionalized materials involve synthetic polymers, which is the main focus here. The few examples of DASA in combination with biopolymers/-oligomers are briefly discussed in the third part. Examples of the integration of DASAs into functional materials other than polymers lie outside the scope of this review, including inorganic and metal nanoparticles,50–53 graphene,54 metal–organic frameworks (MOFs),55,56 polyoxometalates,57 supramolecular complexes with metal–ligand/organic molecular vessels,58,59 or with hydrogen bonding receptors,60 and self-assembled structures of small molecule DASA-based amphiphiles.61,62
1.1 Donor–acceptor Stenhouse adducts
In 2014, the photochromism of DASAs was discovered by Read de Alaniz and co-workers.30,31 They termed the compounds in reference to the structurally closely related Stenhouse salts.63,64 These first discovered DASAs were referred to as first-generation DASAs and were later expanded by second32,33 and third34 generation derivatives in order to overcome a number of drawbacks associated with the first generation (Fig. 2 and Table 1). The structure of the brightly colored open (“linear”) isomer consists of a donor–acceptor substituted triene (“polymethine”) with a hydroxy-functional group in C2 position. The hydroxy functional group distinguishes DASAs from non-photochromic merocyanine dyes.65 The donors are aliphatic (first-generation) or aromatic (second-generation) secondary amines. The acceptors consist of cyclic β-carbonyl carbon acids, such as Meldrum's and barbituric acid (first and second generation) or other heterocycles, such as pyrazolone or isoxazolone (third-generation).30,32,34 The donor–acceptor substituted triene is synthetically derived from furfural (2-furaldehyde) with the furan oxygen providing the hydroxy group in the structure of the open isomer. DASAs can be synthesized via a two-step route.66 First, furfural is activated by condensation with carbon acid moieties through a Knoevenagel condensation (Fig. 2a). The resulting furan adducts are then ring-opened by reaction with a secondary amine nucleophile to yield the open DASA isomer. Synthetic attempts to replace the furyl group with pyrrole or thiophene or to extend the triene chain were unsuccessful.67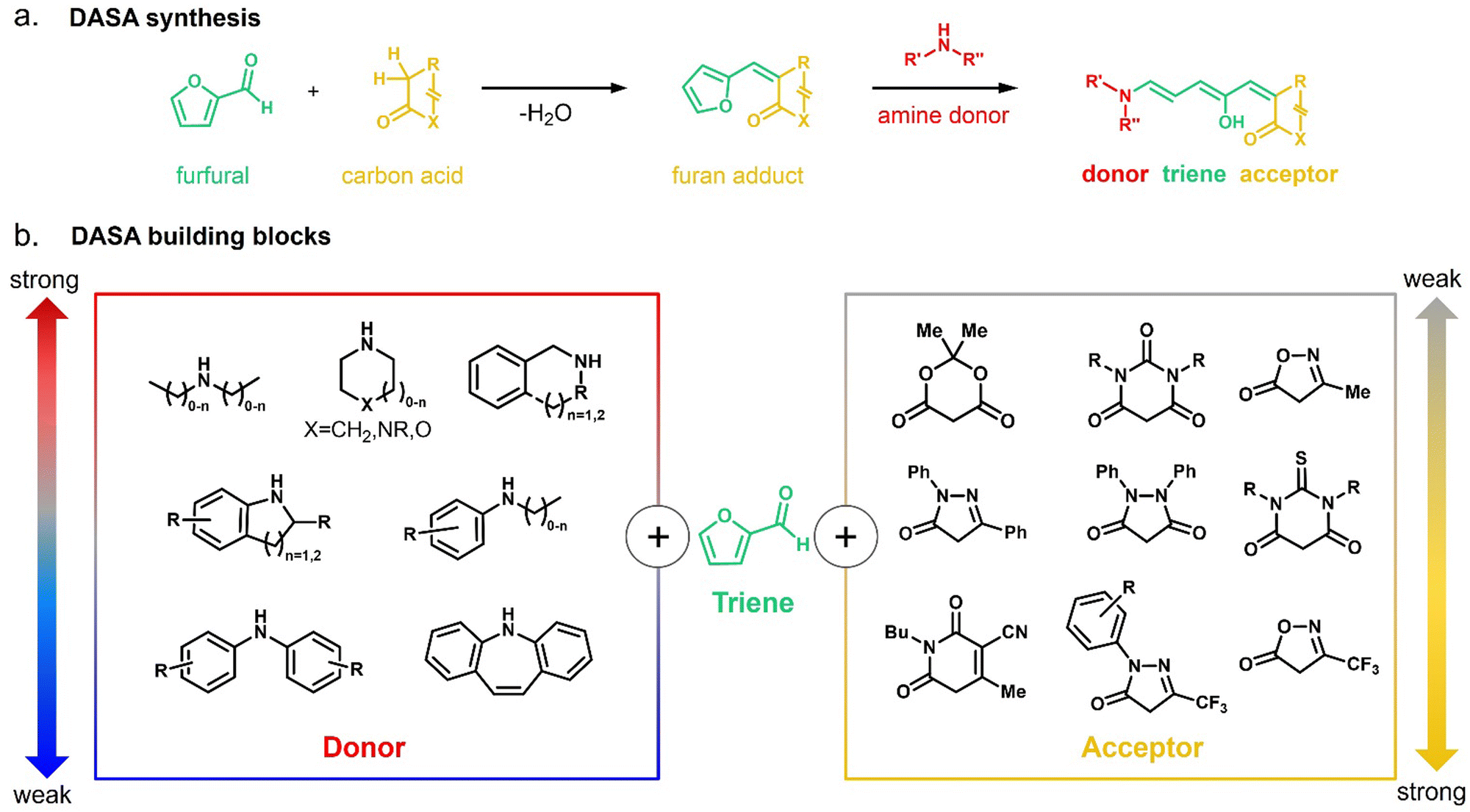 | ||
| Fig. 2 DASA synthesis and building blocks. (a) DASA synthesis is modular and based on the Knoevenagel condensation of furfural with a cyclic β-carbonyl carbon acid, followed by a ring-opening reaction of the resulting furan adduct with a secondary amine. (b) Chemical structures of different secondary amine donors and carbon acid acceptors previously utilized for DASA synthesis. Donors and acceptors are ranked tentatively according to their approximate strength (as indicated by reported or predicated pKa(H)). The properties of DASAs are largely dictated by the relative strength of their donor and acceptor groups. The triene bridge of the open isomer is derived from furfural. | ||
|
|
|||
|---|---|---|---|
| First generation | Second generation | Third generation | |
| Strong donor + weak acceptor | Weak donor + weak acceptor | Various donors and (strong) acceptors | |
| Absorption (toluene) | λ max ∼ 550 nm | λ max ∼ 550–700 nm | λ max ∼ 550–700 nm |
| Dark equilibrium | Open isomer preferred | Closed isomer preferred | Varied |
| Solvent compatibility | Limited to apolar solvents with exceptions | Various apolar/polar aprotic solvents | Various apolar/polar aprotic solvents |
| Half-life closed isomer | Seconds–minutes | Minutes–hours | Varied |
| Nature of open isomer | Zwitterionic | Hybrid | Varied |
| Preferred closed form tautomer | Zwitterionic | Keto | Varied |
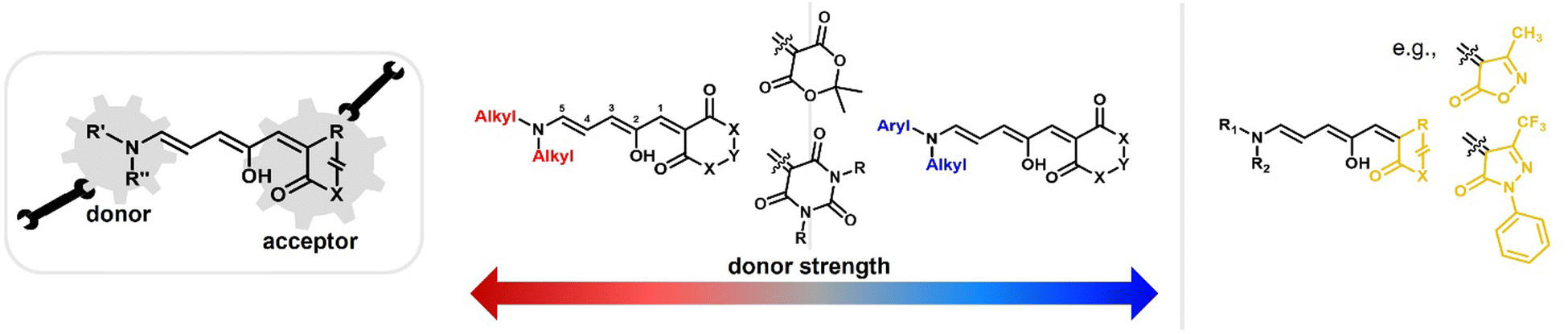
Depending on the donor and acceptor group, the absorption of the open DASA isomer is tunable between 450–750 nm (Fig. 3a; see also Section 1.1.1.2).34,68 Upon irradiation of the colored triene isomer, a conversion to a colorless 4,5-disubstituted cyclopentenone closed isomer can be observed (Fig. 1). This closed isomer is metastable in many organic aprotic solvents at room temperature and thermally reverts back in the dark to the open isomer on a timescale of minutes to hours.34 The closed isomer does not show considerable absorption above ∼300–350 nm and is not addressable by light.45 Such a transition from a stable colored isomer to a thermally metastable colorless state is classified as negative T-type photochromism (whereby “T-type” stands for a thermal recovery pathway, as opposed to a photochemical recovery pathway in the case of “P-type”).38 In materials containing negative T-type photochromes, photoswitching is not localized to a surface but proceeds through the bulk of the material as a propagating bleaching front and photoswitching at greater depth of the sample becomes easier (Fig. 3b). First-generation DASAs typically form a zwitterionic closed form, for which the strongly basic amine donor deprotonates the acceptor and forms an enolate (Fig. 3c, 1). However, depending on the DASA derivative, solvent and DASA concentration, the formation of a neutral ketone or enol isomer instead of the zwitterionic enolate can be observed.33,44,60,69 In addition, the strength of the push–pull ability dictates the degree of charge separation of the conjugated π-system of the open isomer in the ground state, which is important for the optical properties and reactivity of the DASA in relation on its surroundings.44 As the strength of the donor and acceptor groups increases, the polarization of the triene increases (Fig. 3d). The two border cases are that of a neutral form with alternating bond lengths along the triene and a charge-separated (zwitterionic) form with reverse bond length alterations.70 When these two resonance forms contribute equally to the ground state structure, a fully delocalized (“hybrid”) DASA is present (with equivalent bond lengths). The absorption properties of a DASA are sensitive to the polarity of the solvent, and this is more pronounced for DASAs with a more dipolar nature (solvatochromism, cf. Section 1.1.1.2). Therefore, the measurement of the solvatochromic shift of the open form absorption by UV-Vis spectroscopy in solvents of different polarity allows to establish an empirical scale for the degree of zwitterionic character of different DASA derivatives (e.g., solvatochromic slope in Fig. 3d).44 The first generation of DASAs that is based on strong dialkylamine donors and relatively weak electron-withdrawing acceptor groups shows a high bond length alternation (BLA) suggesting a zwitterionic open form, which is supported by solvatochromic shift analysis, theoretical calculations, and experimental crystals structures.33,44,71,72 In the second generation, the dialkylamine donor is replaced by weakly donating aromatic amines, which leads to less BLA and a more hybrid triene structure. Combining a weak aryl amine donor with a stronger electron-withdrawing carbon acid acceptor than Meldrum's or barbituric acid in some third-generation derivatives in turn results in a more zwitterionic electronic ground state.44 Interestingly, a non-photochromic DASA derivative based on indandione (4, Fig. 3d) has a neutral ground state.44 In addition, an increase in solvent polarity generally leads to a stabilization of the zwitterionic resonance form for all derivatives.
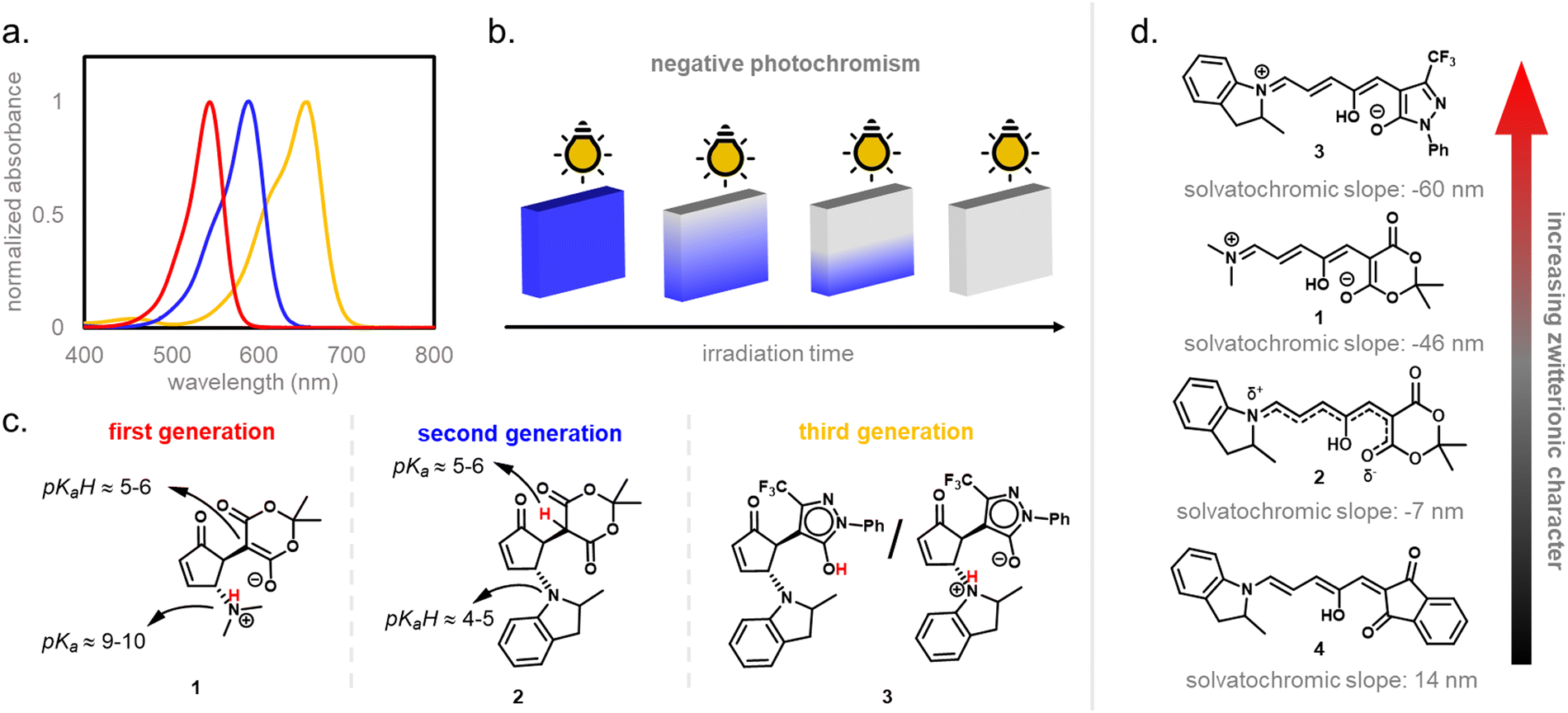 | ||
| Fig. 3 DASA properties and nature of closed and open isomers. (a) Representative absorbance spectra of open isomers. Red: first-generation; blue: second-generation; orange: third-generation. (b) Schematics showing the negative photochromism of DASAs in a material in the solid-state. The transition from the stable colored isomer to the thermally metastable colorless closed form is classified as negative T-type photochromism. Here, photoswitching is not localized to the surface but proceeds through the bulk of the material as a propagating bleaching front and photoswitching at greater depth of the sample becomes easier. (c) Preferred closed form tautomer for representative DASAs of different generations. The tautomeric state mainly depends on the donor and acceptor groups and the pKa value difference of the protonated amine and the carbon acid moiety. (d) Nature and degree of charge separation of the conjugated π-system in the open isomer and slope from solvatochromic shift analysis in solvents of differing polarity. As the strengths of the donor and acceptor groups increases, the polarization of the triene bridge increases (resonance forms from bottom to top: neutral triene form, fully delocalized hybrid form and zwitterionic triene form). In the neutral and hybrid ground state structures, the push–pull donor–acceptor conjugation is weakened as compared to the highly dipolar (i.e., charge-separated) zwitterionic form. Increasing solvent polarity leads to a stabilization of the zwitterionic resonance form. | ||
Changing the structure of the donor moiety or carbon acid acceptor also influences other properties of the DASA, such as the position of the dark equilibrium between the isomers, the solvent compatibility and switching kinetics.34,44 These effects will be discussed in more detail in the Section 1.1.1. Beside electronic effects, the influence of steric effects from substituents on the donor and acceptor have also been explored.35,37 Moreover, substitutions along the triene also induce substantial effects on the photoswitching kinetics, absorption wavelengths, dark equilibrium and stability of the formed DASAs, but research in this direction is still in its early stages.36,73,74
1.1.1.1 Mechanism. The mechanism of DASA photoswitching is considered to follow a complex multistage pathway involving a combination of light-triggered (actinic) and thermal isomerization steps (Fig. 4).39 In the proposed mechanism, a photoisomerization along the C2–C3 double bond of the thermodynamically most stable open isomer (EEZZ) is followed by a thermal bond rotation along C3–C4. A thermally allowed, conrotatory 4π-electrocyclization with concomitant proton-transfer reaction is then responsible for the formation of the five-membered-ring.39,40 The metastable intermediate B obtained after the initial actinic step is generated within picoseconds, whereas the thermal steps occur on a longer time scale (>nanoseconds).39,40 In addition, interconversion of the closed form C and other tautomeric closed forms (C′ to C′′′′) was observed, which strongly depends on the DASA type (cf. Section 1.1) and environmental influences.33,44,60,69
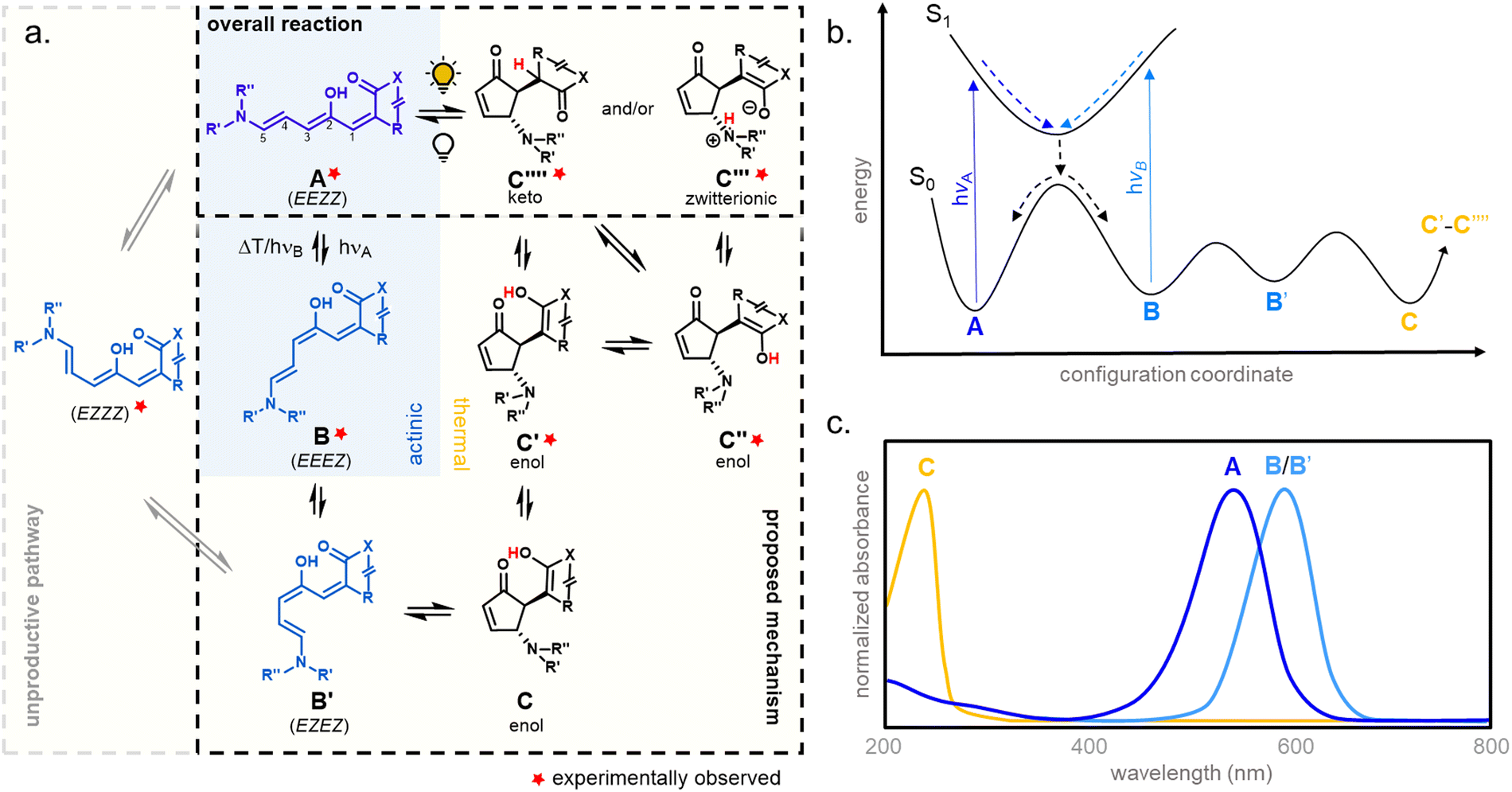 | ||
| Fig. 4 DASA photoswitching mechanism. (a) Proposed photoswitching mechanism of DASAs showing the complex multistage pathway involving a combination of light-triggered (actinic, blue background) and thermal isomerization steps (yellow background). Note: the isomers C and C′ are interconverted into each other by a nitrogen inversion. E/Z refer to the stereochemical configuration of the double bonds along the triene bridge. (b) Schematic representation of potential energy surfaces involved in DASA photoisomerization. The photochemical reaction involves the electronically excited state of the thermodynamically most stable open isomer A, which is reached by excitation of the π–π* transition that lies in the visible/NIR spectral range. (c) Representative schematics of normalized UV-Vis absorbance spectra of the different DASA isomers. | ||
The mechanistic proposal of DASA photoswitching is based on experimental observations of key intermediates, the stereochemistry of the closed isomer, and experiments at different temperatures (ultrafast UV-Vis spectroscopy,39,40,65,75 ultrafast and rapid-scan FTIR spectroscopy,41,65,75 time-resolved UV-Vis steady state spectroscopy,37,39,65,75 NMR spectroscopy,37,39,41,65 and X-ray structure analysis30,31,33,35). It was also supported in theoretical approaches by computations of the (excited) energy surfaces (TD-DFT calculations,37,39,40,65,76 CASSCF calculations,37,76,77 ADC(2) and CC2 calculations,78ab initio molecular dynamics42,43,79).
The B/B′ intermediate stage is typically short-lived, with the reaction either progressing to the closed form C or reverting to the most stable open isomer A. A sufficiently high thermal barrier between A and B/B′ is therefore crucial for the closed isomer to be formed.41,60 Importantly, the metastable isomers B and B′ exhibit a red-shifted absorption relative to A, which in some cases can be observed in steady state UV-Vis spectroscopy as a transient ∼50 nm red-shifted shoulder to the main absorption peak during illumination.39 The back-reaction from the closed isomer to the open isomer happens thermally. However, transient absorption experiments below −40 °C revealed that irradiation of the shoulder in the UV-Vis spectra arising from B/B′ facilitates the reformation of A, demonstrating that the actinic step for DASAs is both T- and P-type reversible.40 Recently, a DASA derivative with longer-lived B/B′ intermediates at room temperature could be accessed through introduction of sterically demanding groups into both the acceptor and donor moieties.37 The B/B′ intermediate stage in this system was addressable by a second wavelength of light, interfering with the formation of the closed form and enabling a three-stage switch. In polymer films, addressing B/B′ with a second wavelength of light had a reduced effect on the rate of isomerization from the open to closed forms (vide infra, Section 2.7).
1.1.1.2 Optical properties. The activation with visible light occurs due to a strong absorption band of the DASA open isomer corresponding to a π–π* transition (cf.Fig. 3a and 4c). Only the lowest excited singlet state is involved in the spectroscopic signature.80 This absorption band is around λmax = 550 nm for dialkylamines with Meldrum's acid acceptors (first-generation DASAs) and can be shifted to the red spectral region by extending conjugation either through the donor or acceptor group.32,34,80,81 The open DASA isomers also have very high molar absorptivity (∼100
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in solution).82–84 Moreover, second-order nonlinear optical properties and two-photon absorption properties of DASAs have been predicted.84–87
000 M−1 cm−1 in solution).82–84 Moreover, second-order nonlinear optical properties and two-photon absorption properties of DASAs have been predicted.84–87
Alongside other chromophores or photoswitches with push–pull groups that introduce charge-transfer character, the photophysical properties of DASAs are highly dependent on the polarity of the environment. DASA open isomers typically show strong negative solvatochromism,44i.e. for solvents with higher polarity, a blue-shift of the absorption band is observed (the electronic ground state structure is more stabilized in polar solvents as compared to the electronic excited state, meaning that the ground state is more polar than the excited state).88 These effects are less pronounced for second-generation DASAs with a weakened push–pull ability that exhibit a less dipolar nature.44
There is usually a band asymmetry (broadening) observed towards absorption at shorter wavelengths. This higher energy part of the spectra could be computationally well reproduced by accounting for vibrational contributions with a DFT level description, which are most likely caused by the vibronic progression corresponding to the S1 ← S0 electronic transition.71,80 Spectral signs for DASA aggregation in the solid state (pure) or in materials in the form of band broadening and enhancement of the vibronic features at shorter wavelengths were also observed (vide infra, Section 2.5.3). As mentioned before, in solvents with low polarity, a more hybrid electronic ground state structure of the open isomer is favored with a higher delocalization of the electron density and low BLA. As a result, excitation in these solvents leads to minimal changes in bond order and only weak vibronic interactions within the excited state, in accordance with the Frank–Condon principle.72 DASA absorption bands typically become narrower and their intensities increase in nonpolar environments when compared to polar solvents, such as alcohols and water.65,72
Fluorescence from DASAs is not as well studied as their absorption properties. The open DASA isomers do not fluoresce strongly in organic solvents or water, despite their high molar absorptivites.72,89 A major reason for this is that radiative relaxation directly competes with photoswitching by rotational relaxation of the excited state. Fluorescence measurements of DASAs have also been recorded in different polymers (cf. Sections 3.1.1 and 3.1.2).90–92
1.1.1.3 Methods and models for DASA photoswitching analysis. Despite the complexity of the proposed DASA photoswitching mechanism (cf.Fig. 4 and Section 1.1.1.1), most DASA kinetic studies model the interconversion of A (colored) to C (colorless) as a two-step reversible process that proceeds through a lumped B/B′ intermediate state (“three-state” model, Fig. 5a).35,39,41,93 Representative absorbance profiles of A, B/B′ and C are illustrated in Fig. 4c. Single step kinetic models have also been used to describe the photoswitching of A and thermal recovery from C as a simple first order reversible isomerization (“two-state” model, Fig. 5b).32,44,84,94–96 In several studies first-order exponential functions were used to fit kinetic data and to extract values for the apparent half-lives of the open or closed isomers under specific conditions (noting that DASA photoswitching is not a first order process). While single step models provide little insight into individual steps of the mechanism, they can be helpful for experimentally observing qualitative trends in the interconversion rates of A and C with changes to the DASA architecture,36,84 concentration,94 and environment.44 In addition, interconversions between the different isomers are categorized according to their thermal and photochemical character. For actinic steps, rate constants are often measured as a function of light intensity to differentiate between thermal and photochemical contributions to the observed isomerization rate.35,39,40,93,97,98 Usually, the thermal and photochemical rates are assumed to be independent, but this is only true if negligible photothermal heating occurs during illumination. Importantly, the rates of the photon-driven steps depend on the spectrum and intensity of the light source that is used.
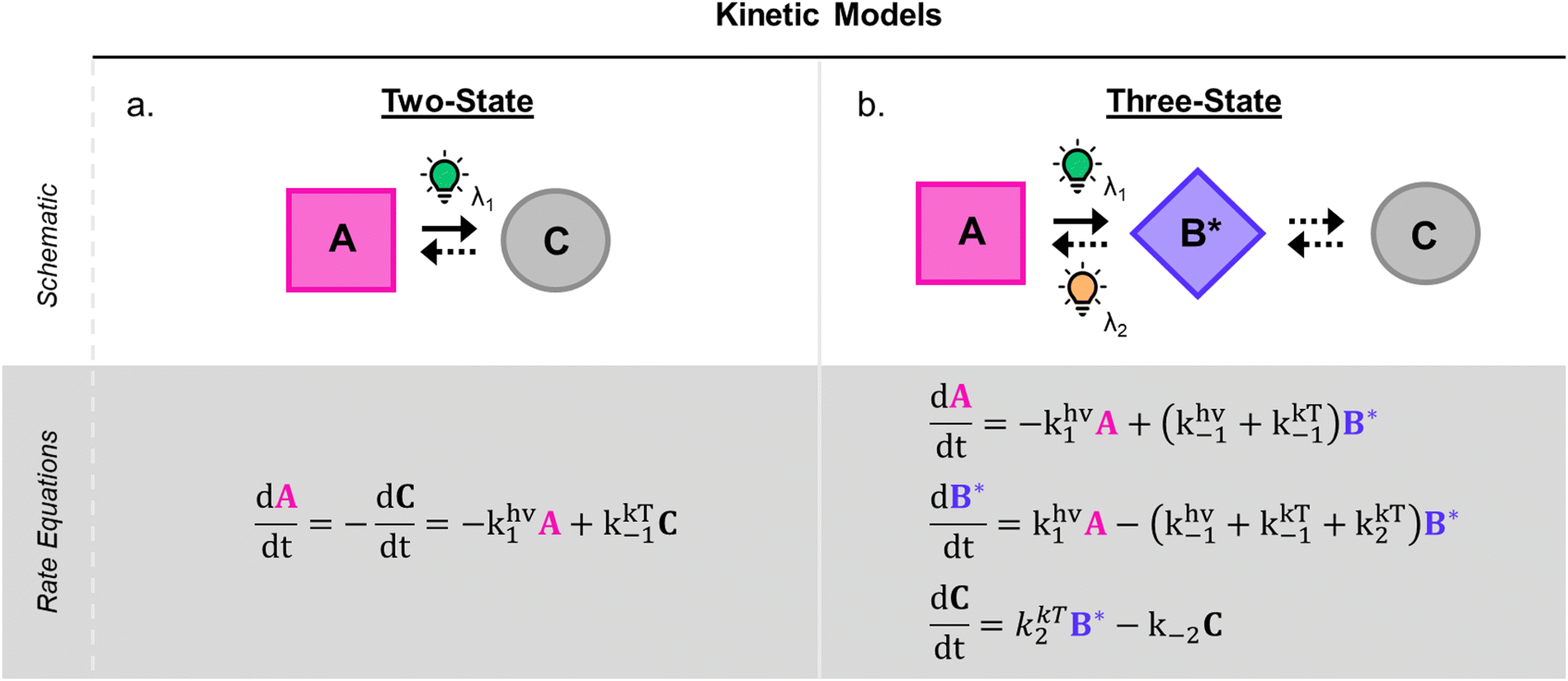 | ||
| Fig. 5 DASA photokinetic models. (a) Schematic illustrations and differential rate equations of two-state model. (b) Schematic illustrations and differential rate equations of three-state model. Steps known to be photochemical are indicated pictorially with a lightbulb and mathematically with a superscripted “hν” on the corresponding rate constant. Thermally accessible isomerizations are shown with dashed arrow and denoted by superscripted “kT” on the corresponding rate constants. A, B*, and C indicate spatially averaged isomer concentrations in A, B/B′ and C, respectively. Structures for these isomers are provided in Fig. 4 along with an energy diagram. First order photochemical rate equations are only valid in low absorbance limit of the relevant photoactive species. A general derivation outside this limit is provided by Stranius and Börjesson in ref. 99. | ||
Another important consideration when modelling photochemical reaction rates is the lack of a well-defined order with respect to the concentrations of the photoactive species. In all of the single and two-step DASA kinetic models applied in published works, the actinic interconversions of A and B/B′ are assumed to obey first order kinetics in their respective concentrations with characteristic rate constants that reflect the reaction quantum efficiencies.35,94 However, the rate of actinic isomerization is actually proportional to the rate of photon absorption, which scales logarithmically with the concentration of the absorbing species according to Beer's law. As a result, traditional first order kinetics can only be assumed in the low absorbance limit, where the scaling is approximately linear.99 In the opposite limit of total absorbance, the photochemical rate law approaches zeroth order in the absorber concentration.99 For absorbances below 0.04 a.u. and above 1.3 a.u., these convenient approximations introduce less than 5% error in the model validity.98 In addition to complicating the kinetic modelling of photochemical processes, this phenomenon may result in the appearance of false trends when normalizing photoswitching datasets with different starting absorbance for a qualitative comparison. As such, care must be taken when modelling the photochemical reaction rates.
Regardless of the kinetic model used, however, isomerization is modelled to proceed until a state of dynamic equilibrium is attained, the equilibrium between the photoreaction(s) and the thermal back reaction under visible light irradiation, i.e. the photothermal stationary state (PTSS), or the dark equilibrium state. Comparing the equilibrium distribution of open and closed form isomers often provides additional insight into the chemical and mechanical factors driving or impeding DASA photoswitching,32–37,81,100 especially when the kinetics of photoswitching are too fast to be reliably measured in real time. However, it should be noted that the distribution between open and closed isomers at equilibrium is also sensitive to solvent quality and the presence of trace amounts of impurities or additives (cf. Section 2.6), concentration, as well as potentially unknown other factors. Therefore, comparison across papers should always be done with care.
Experimental techniques for tracking the photochemical and thermal isomerization of DASAs in solution include pump probe UV-Vis spectroscopy (∼0.1–1 s resolution), rapid scan FTIR spectroscopy (96 ms resolution),41 ultrafast pulse probe UV-Vis spectroscopy (∼1–10 ps resolution),39,401H NMR spectroscopy,33,35,95 and mass spectrometry.97 The primary differences between these approaches are their temporal resolution, their ability to resolve the thermodynamically distinct isomers produced during photoswitching, and their capability for passive observation of photoactive species. In addition, the sensitivity and precision of the measurement device must be considered when operating in the low or high absorbance regimes.
1.1.1.4 Cyclization efficiency under illumination. Under illumination with visible light, the DASAs rapidly form the photoisomer B in the actinic step (cf.Fig. 4), which is then followed by a slow thermal equilibration between the different metastable open and closed isomers along the multistep reaction pathway. For efficient formation of the closed isomer under irradiation, both the actinic step and thermal steps in forward direction need to be fast and outcompete the thermal back reaction. The photo stationary state (PSS, relative concentrations of thermodynamically stable and metastable isomers under irradiation) for DASAs is governed in part by the thermal back reaction and not by a photochemical reaction from the B/B' intermediates back to A (it is therefore often referred to as photothermal stationary state (PTSS)99).
The electronics of both the donor and acceptor groups influence the photochemical reaction and the lifetime of the excited state.40 The quantum yield for the formation of intermediate B (actinic step) was measured (≈10–20% for first- and second-generation DASAs), but it is not directly indicative for the DASA cyclization efficiency.40,75 To understand DASA photoisomerization as a whole, the thermal steps for the forward isomerization and thermal recovery must also be considered.33,35,41
The energy levels and barriers of intermediates along the energy landscape of the thermal steps are also heavily influenced by electronic effects of the donor and acceptor group in the different DASA derivatives and also by external factors (e.g., solvent polarity, Fig. 6a).35,37,40 In some examples, effective quantum yields for the full transition from the open to closed isomer were reported based on assuming a simplified two state system (see Section 1.1.1.3).84,94,96
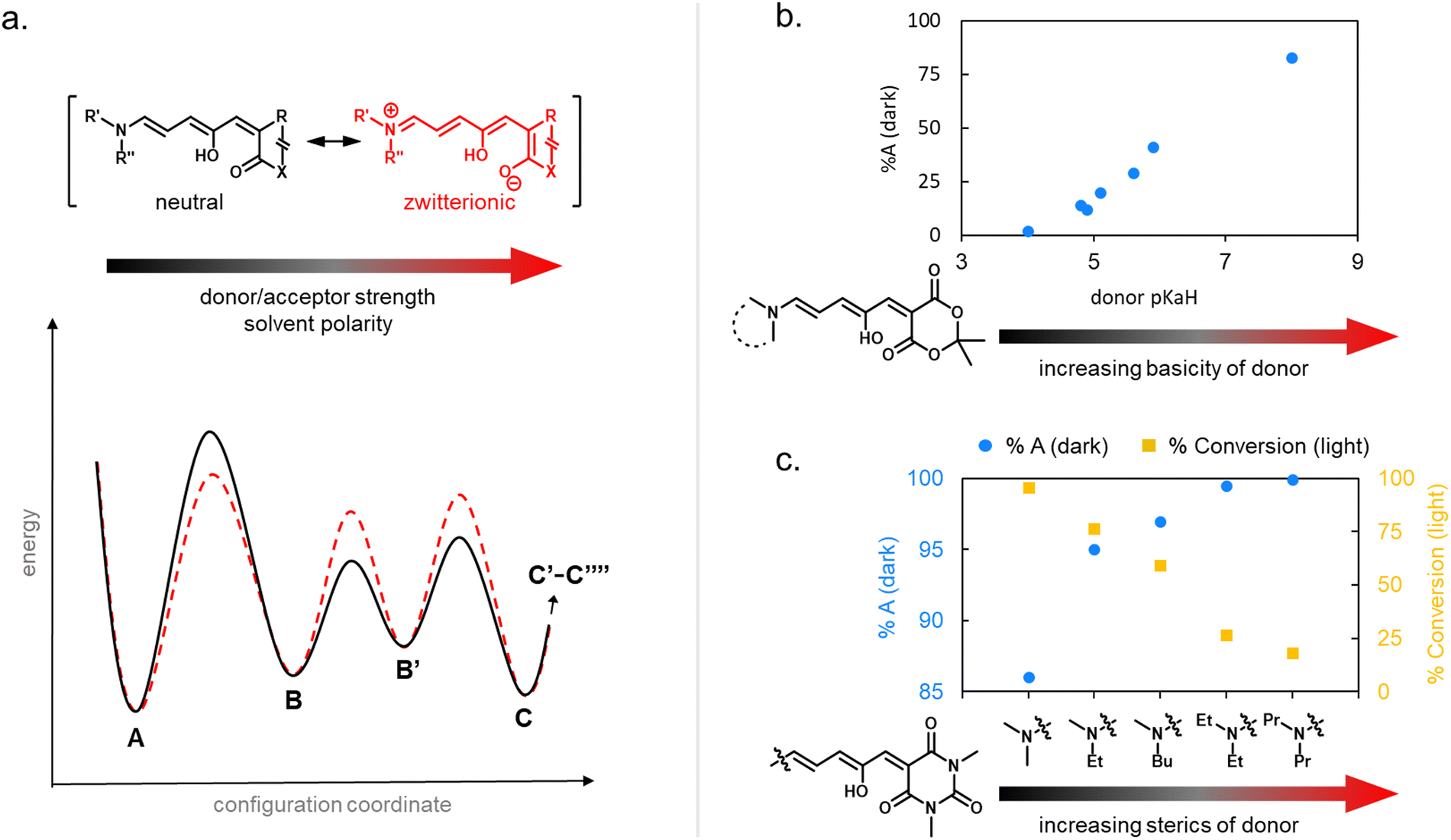 | ||
| Fig. 6 Cyclization efficiency under illumination or in the dark in relation to DASA structure. (a) The influence of the zwitterionic nature of DASA photoswitches on their energy landscape. As the strength of the donor and acceptor groups of a DASA increases, the polarization of the triene increases. A stronger push–pull ability favors the open isomer in the DASA energy landscape. (b) The impact of basicity of the donor on the thermodynamic equilibrium (“dark equilibrium”) between open and closed isomers of a Meldrum's acid derived DASA (equilibrium measured by NMR integration in chloroform). For DASAs with less basic (i.e., weaker) donors and weakened push–pull character, the amount of open isomer at equilibrium decreases. This is often accompanied with longer half-lives for the closed isomer and higher amounts of closed isomer under irradiation. Note that steric effects can also play a role (e.g., sterically demanding donors, such as diaryl derivatives, are not shown here). Data taken from ref. 32 and 33. (c) The impact of sterics of the donor on the dark equilibrium and the conversion to the closed form under irradiation of a series of 1,3-dimethylbarbituric acid derived first-generation DASA. Increasing the bulkiness of both amine substituents favors the open isomer in the dark equilibrium, but at the same time leads to faster thermal recovery rates and less closed form formation under irradiation due to more steric hindrance in the thermal steps. All data in chloroform under light irradiation with a 567 nm LED until no further changes observed and dark equilibrium measured by NMR integration in deuterated chloroform. Data taken from ref. 35. | ||
Generally, a stronger push–pull ability favors the open isomer in the DASA energy landscape (Fig. 6b).49 First-generation DASAs with strong donors and third-generation DASAs with strong acceptors typically present short-lived closed isomers and lower amounts of closed isomer at the PTSS under irradiation. A weakening of the push–pull character leads to longer half-lives for the closed isomer and more closed isomer under irradiation, as observed in second-generation DASAs.32
Electronic factors most likely also influence the accessibility of possible side pathways in the DASA switching mechanism, including competing photochemically or thermally induced double bond isomerizations (cf. Section 1.1.1.1), which reduce the cyclization efficiency to the closed isomer.79 However, this topic requires further study and experimental validation.
Notably, besides electronic factors, sterics of the acceptor and donor groups also play a role.35,37 For example, first generation DASAs based on dimethylamine undergo more efficient cyclization under light in comparison to diethylamine derivatives (Fig. 6c) owing to reduced steric hindrance in the thermal steps (i.e., bond rotation along C3–C4 and the 4π electrocyclization ring-closure step).35,41
1.1.1.5 Dark equilibria and cyclization under exclusion of light. The thermodynamic equilibrium (“dark equilibrium”) between the isomers of a photoswitch is an important parameter, since it determines the proportion of compound that is available for undergoing the photochemical reaction. For DASAs, it is strongly dependent on their individual donor and acceptor groups as well as their solvent environment.34 The open isomer is stabilized in DASAs that present stronger push–pull ability (first- and third-generation).49 Thus, increasing amine donor basicity increases the concentration of the open isomer in the thermodynamic equilibrium (Fig. 6b).32 Similar effects can be achieved by introducing stronger acceptors in third-generation DASA to counteract the electronic effects of weak donors.34 Moreover, increasing the bulkiness of both amine substituents in first-generation DASAs favors the open isomer in the thermodynamic equilibrium, but at the same time this leads to faster thermal recovery and a decrease of closed form formation under irradiation (Fig. 6c).35,49 Substitutions along the triene structure can also affect the DASA thermodynamic equilibrium by either increasing the energy of the closed form relative to the open form,36 or vice versa,73,74 depending on the substituent size and position on the backbone.
The stabilization of different tautomeric forms of the closed isomer in different media determine the dark equilibrium distribution.49 A purely thermal isomerization from the open to the closed isomer is typically observed in polar protic solvents, whereby the overall equilibrium is strongly shifted to the more hydrophilic closed isomer. This is most likely due to stabilization effects of the closed isomer through hydrogen bonding interactions. Thus, DASAs are considered to be “hydrochromic” molecules (i.e., molecules that change color when exposed to water or moisture).101,102 Recently, it was reported that the dark equilibrium is also sensitive to acid/base chemistry (acidochromism).103 For example, protonation of first-generation or second-generation DASAs in halogenated organic solvents led to cyclization of the open isomer to a cationic closed form (an amine-protonated keto tautomer as opposed to the typically observed zwitterion shown in Fig. 3), and could be accelerated by light irradiation and reversed by addition of base.104,105 In an aqueous environment, however, decreasing the pH has been shown to increase the ability of DASA to recover to the open form.106 Under some acidic conditions, conversion of DASA to Stenhouse salts has also been observed.107 Importantly, understanding these effects as they relate to the structure of DASA in both the open and closed forms is still in its early stages.
1.1.1.6 Effects of the medium. While solvent effects play a huge role in DASA photoswitching, the kinetics of the actinic step are only slightly affected.75 This large influence in photoswitching stems from differences in energy levels and barriers involved in the thermally induced steps after the photochemical reaction in the different media (cf.Fig. 6a). In general, the barrier from B to A decreases with increasing solvent polarity, while the barriers from B to B′ and B′ to C increase (this is associated to the variation in the double bond character of the different bonds of the triene bridge, which affects the activation energy to bond rotation).108 Since different DASAs present variable starting energy landscapes (e.g., strength of push–pull ability), equal environment polarity induced effects can lead to a different photoswitching performance.108 While most first-generation DASAs only showed efficient photo-induced formation of the closed isomer and thermal reversibility in apolar solvents such as toluene, newer generations can also reversibly switch in a range of more polar aprotic solvents, including chlorinated solvents and tetrahydrofuran.34 First-generation DASAs with sterically less demanding substituents on the amine donor, such as two methyl groups, can also switch in a wider range of solvents.35 Moreover, the addition of polar ionic liquids to organic solvents influence the DASA photoswitching ability in that solvent.44 The effect of such polarity changes was smaller for DASAs that have open and closed isomers with lower dipole moments (e.g., second-generation DASAs).
Reversible photoswitching of DASAs in a spectrum of polar protic solvents, such as pure water, has not been possible to date. Contributing factors include the lowered energy barrier between B and A in these solvents,108 favoring fast thermal back reaction from B to A instead of isomerization to B′ and C. The increased spectral overlap between A and B/B′ in these solvents (negative solvatochromism, cf. Section 1.1.1.2) may also affect the photostationary state of the actinic step and lead to an enhanced photochemical reaction from B to A.75
Another factor influencing DASA photoswitching is DASA concentration. While unimolecular photoisomerization reactions are assumed to be relatively insensitive to photoswitch concentration up to millimolar concentrations, pronounced effects occur on the rates of both the forward photoisomerization and the thermal recovery for DASAs in the concentration range between 10−4–10−1 M.94 A third-generation DASA built from a CF3 pyrazolone-based acceptor and a 2-methyl indoline donor (3, cf.Fig. 3) showed faster thermal recovery and reduced open-to-closed conversion under irradiation with increasing DASA concentration with rate constants changing by factors of 20 or more.94 The reason for this is not entirely clear, but no experimental evidence for aggregation or excited state quenching was observed. It is likely that these effects are caused by long-range dipolar interactions between the DASAs that interfere with the thermal steps of the photoisomerization, as both open and closed isomer can present substantial ionic character (cf. Section 1.1).94 This was further supported by the demonstration that second-generation DASAs with a hybrid open isomer and a neutral closed isomer are less susceptible to concentration effects.44 While solvated DASA small molecules are often analyzed in dilute concentrations (micromolar range), application in materials often results in an increase in local DASA concentration or generally requires higher concentrations. Understanding the effects of concentration in materials as they relate to photothermal effects and the corresponding contribution to thermal reversibility remains to be investigated.
1.1.1.7 Summary. As described above, a number of studies have provided a solid foundation to better understand and predict DASA behaviour in solution. This understanding may carry over to their behaviour in materials, and the combination of DASA with a given matrix should be carefully considered. DASA derivatives with different levels of zwitterionic character will result in different properties and should be chosen to fit the desired application. Additionally, the environmental sensitivity of DASA derivatives, particularly to water, should be taken into consideration when choosing applications. While many of the structure–property relationships established for small molecules in solution are critical to applications in materials (as detailed below), future studies are still needed to better understand how the polymeric matrix itself influences the properties of DASAs and vice versa.
2. DASA-containing polymers
2.1 Functionalization through covalent conjugation
When integrating photoswitches into polymers, it is often required that the photoswitch is covalently conjugated (Fig. 7). This can prevent leaching, phase separation, and aggregation, provide higher stability, or be necessary to transmit and couple the molecular isomerization events to the polymer chains to cause changes and movements at the nano- or even macroscale. While examples of mixing and dissolving DASAs directly into polymer matrices without covalent attachment have been showcased (see Section 2.2), many of the DASA-materials with applications beyond simple color change rely on covalent attachment. To this end, various strategies have been developed and are described in detail in the following section.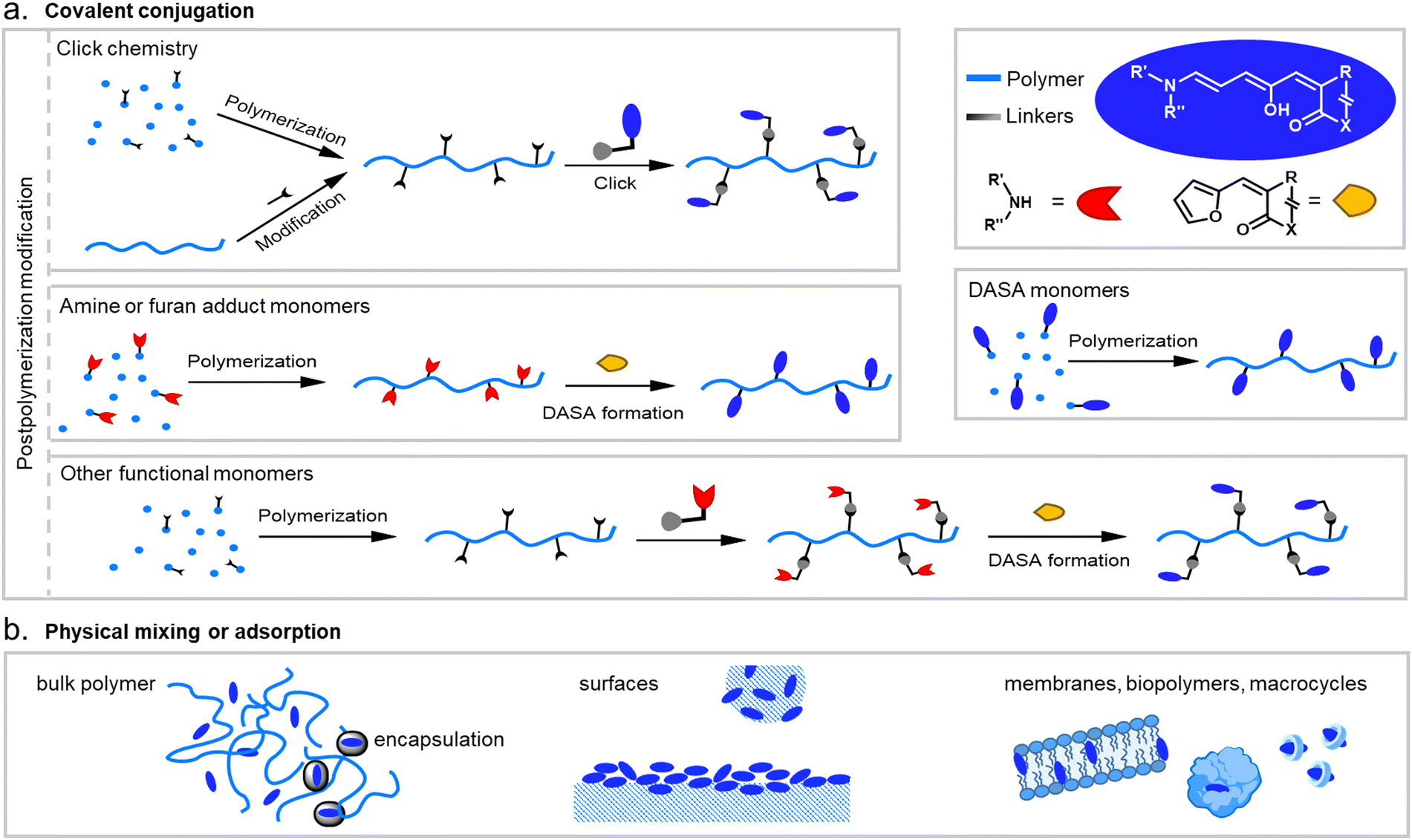 | ||
| Fig. 7 DASA integration into polymeric materials via different strategies. These can be divided into methods based on (a) covalent conjugation, or (b) physical mixing or adsorption. | ||
While other photoswitches, such as spiropyrans, can be modified to yield monomers to be incorporated into polymers trough common polymerization methods, such as free and controlled radical polymerizations, polycondensations or ring-opening metathesis polymerization (ROMP),109 DASAs typically show limited stability in the presence of radicals and strong nucleophiles.45 Besides degradation of the open isomer, it is also known that the closed isomer can undergo (reversible) addition of nucleophiles, such as thiols and amines, into the Michael acceptor of the cyclopentenone structure.103,110–112 Hence, radical polymerization conditions compatible with DASA monomers have not yet been reported and current conjugation strategies are largely based on post-polymerization methods.
Click chemistry providing high reaction specificity has been employed to conjugate DASAs to pre-formed polymers in one-step (Section 2.1.1). The most commonly used route for functionalization of polymers with DASAs involves the conjugation of the acceptor (Section 2.1.2) or the donor (Section 2.1.3) to a polymer chain followed by the DASA-forming reaction as the final functionalization step (“two-step” approach). Therefore, most examples rely on the incorporation of the amine donor into the polymer instead of the furan adduct (acceptor). This is due to the reactive nature of furan adducts, which makes it more difficult to find orthogonal conjugation chemistries. For example, besides nucleophilic furan ring opening, it was recently demonstrated that furan adducts can undergo addition reactions at the exocyclic double bond, including cycloadditions.113
Two-step DASA conjugation strategies and one-step click chemistry methods are both modular, allowing access to different DASA–polymers from the same precursor polymer.96,114,115 In the first approach, this can even be achieved by simply replacing the furan adduct or amine component during the final functionalization step. On the other hand, click chemistry circumvents issues with slow reaction rates and incomplete functionalization that may be encountered when relying on the DASA-forming reaction as the final functionalization step. Alternatively, promotion of the DASA formation on the polymer using special reaction conditions can also help to achieve higher conversions and to shorten the time required for DASA formation on materials (Section 2.1.5).
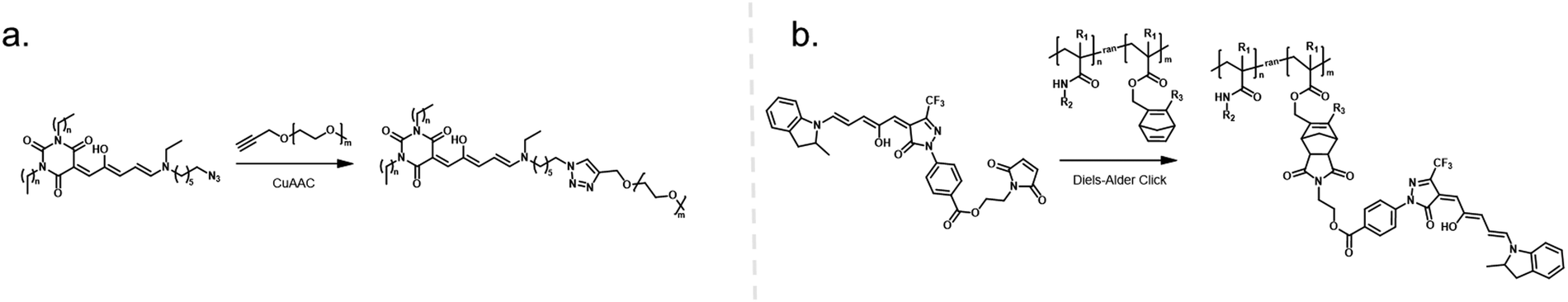 | ||
| Fig. 8 One-step click chemistry for DASA conjugation to polymers and the structures of resulting DASA–polymer conjugates. (a) Copper-catalyzed azide–alkyne cycloaddition (CuAAC) Click reaction. (b) Diels–Alder click approach based on reaction between a masked cyclopentadiene and a maleimide moiety. The reactive cyclopentadiene is generated in situ (“unmasked”) from a stable norbornadiene precursor via a retro-Diels Alder reaction with equimolar amounts of tetrazine. | ||
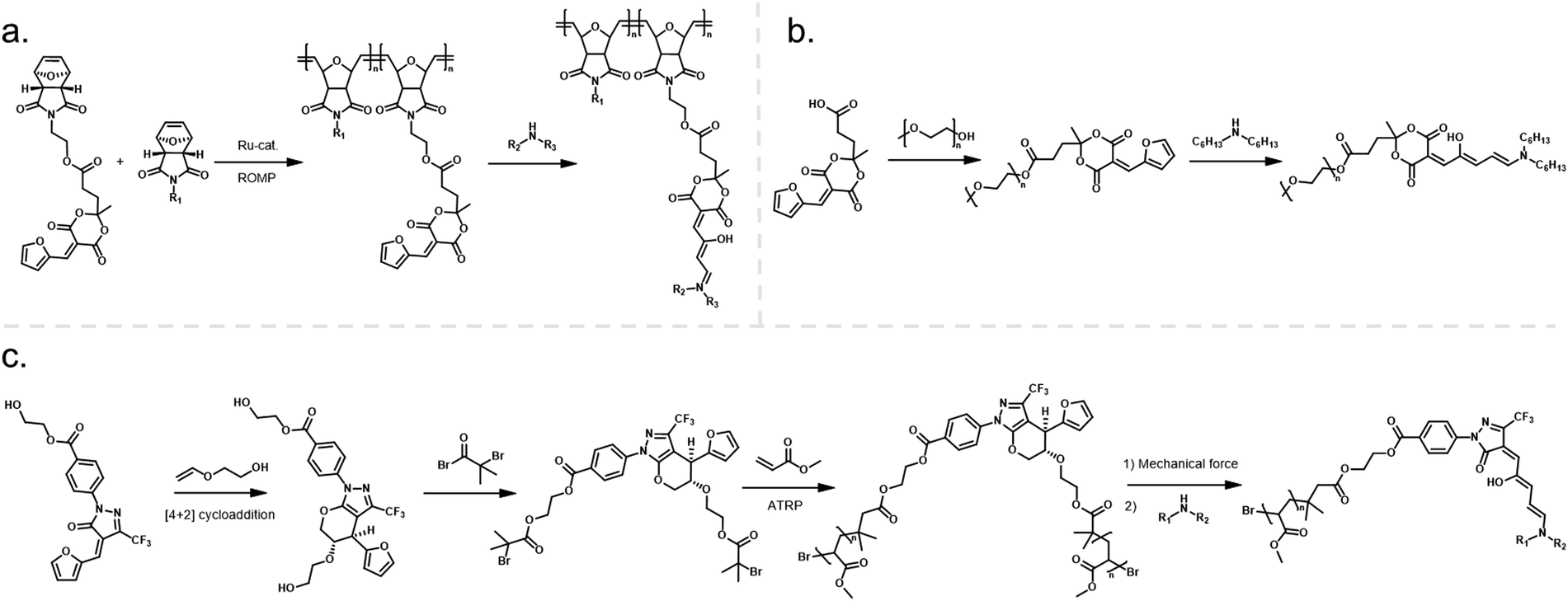 | ||
| Fig. 9 Different strategies for DASA conjugation to polymers via the acceptor moiety. (a) Furan adduct conjugation to oxa-norbornene monomers and subsequent ring-opening metathesis polymerization (ROMP) and DASA formation. (b) Conjugation of a carboxylic acid-functional Meldrum's acid derived furan adduct to PEG via Steglich esterification and subsequent DASA formation. (c) Synthesis of linear poly(methyl acrylate) (PMA) polymers via ATRP) containing a masked furan adduct at the chain midpoint. A mechanically triggered retro-[4+2] cycloaddition reaction then unmasks the furan adduct on the polymer, which reacts with a secondary amine to form a DASA. | ||
In an early example, inherently present amine functionalities in branched polyethylenimine (PEI) were used to synthesize DASAs on polycarbonate (PC) surfaces by reaction with Meldrum's acid furan adduct (Fig. 10a).120 Later PEI was also used to prepare DASA-functionalized nanoparticles in the same way.121 Similarly, the amines present in polydopamine were used to functionalize silica micro- and nanoparticles with DASAs.122 However, both strategies suffer from the limitation that primary amines are present in these materials. Branched PEI contains primary, secondary, and tertiary amines, whilst polydopamine features a primary amine instead of a secondary amine. Typically, the reaction of primary amines with the furan adducts results in unstable adducts.31 In addition, the presence of unreacted amines on the polymer can also cause decomposition of DASAs, or affect DASA photoswitching properties through changes in medium polarity or basicity.45
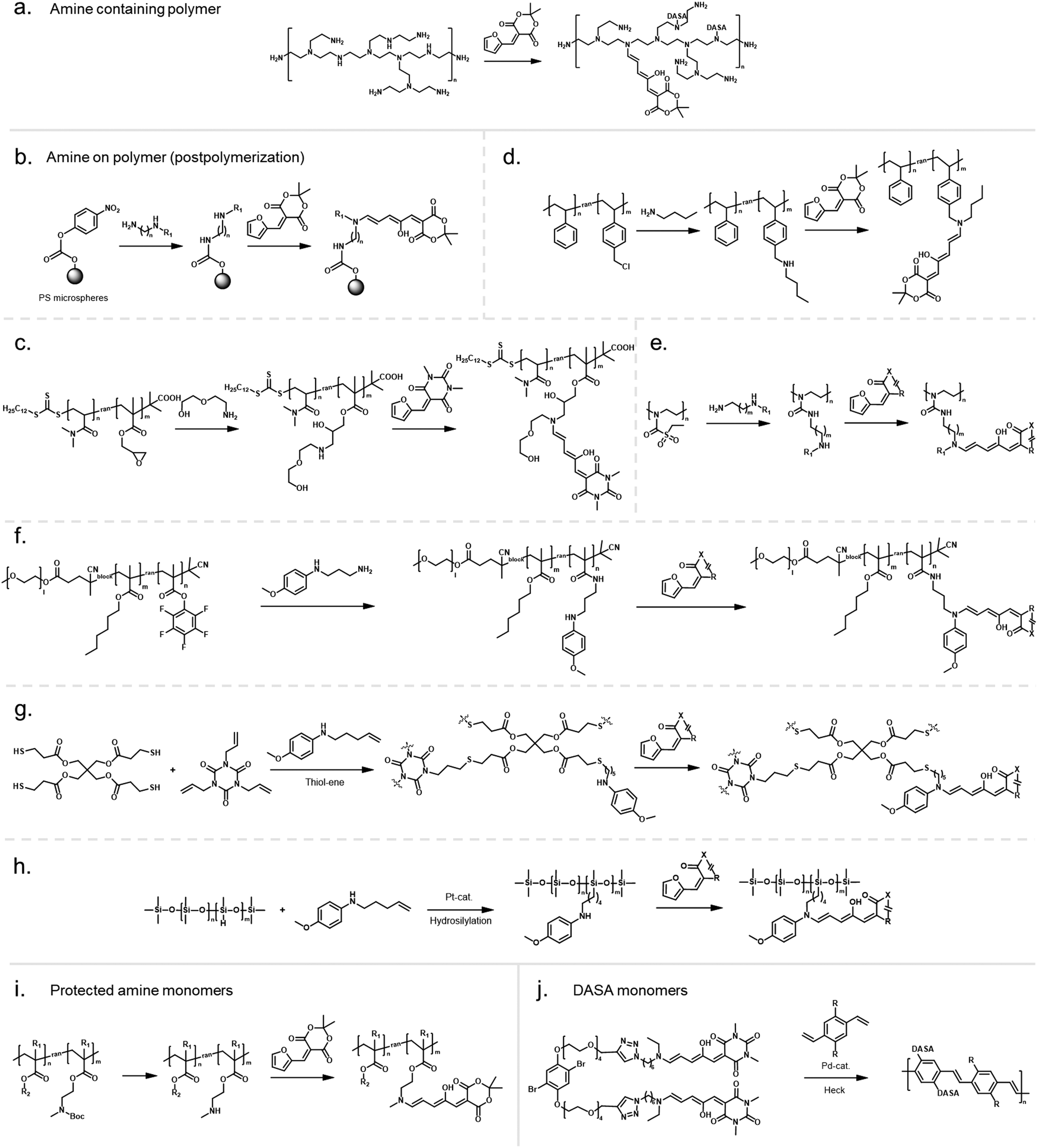 | ||
| Fig. 10 Different strategies for DASA conjugation to polymers via the donor moiety and polymerization of DASA monomers. (a) Utilization of amines inherently present in a polymer backbone, e.g., PEI, for DASA formation. In most examples, amine conjugation was performed via postpoylmerizaiton followed by DASA formation by reaction with furan adducts. Different functional monomers or functional groups were used to introduce secondary amines postpolymerization, such as (b) carbonates, e.g., p-nitrophenyl carbonate (c) epoxide groups, e.g., glycidyl methacrylate, (d) alkyl halides, e.g., 4-vinylbenzyl chloride (e) sulfonyl groups, or (f) activated esters, e.g., pentafluorophenyl(meth)acrylate. (g) Amine functionalization of thiol–ene resins during the network formation step by using alkene-functional amines. (h) Amine functionalization of PDMS via hydrosilylation with alkene-functional amines. (i) Copolymerization of protected alkylamine (meth)acrylate derivatives to introduce the amine functionality during the polymerization step. (j) Only reported example of polymerization of DASA monomers involving Heck coupling of dibromobenzene DASA-functionalized monomers with a divinylbenzene derivative. | ||
Another method for functionalization of PC or poly(styrene) (PS) surfaces involved the reaction of surface carbonate groups or polymer-bound p-nitrophenyl carbonate reacted with various diamines being subsequently converted into DASAs by reaction with a Meldrum's acid furan adduct (Fig. 10b).123 Epoxide functionalities were also used to install 2-(2-aminoethoxy)ethanol as a DASA precursor through epoxide ring opening. To this end, glycidyl methacrylate was applied as comonomer in reversible addition–fragmentation chain-transfer (RAFT) polymerization to synthesize poly(glycidyl methacrylate-co-dimethylacrylamide) (Fig. 10c).124 The amine-functionalized polymer was then converted to a first-generation DASA-bearing polymer by reaction with a barbituric acid derived furan adduct. Similarly, the same epoxide comonomer and amine linker were used to functionalize the surface of poly(N-vinylcaprolactam)-based microgels with DASAs.125
Besides carbonate aminolysis and epoxide ring opening, nucleophilic substitution reactions on alkyl halides or sulfonyl groups have been used for post-polymerization amine conjugation to create DASA-bearing materials. In further detail, reaction of chloromethyl units in a styrene-vinylbenzyl chloride copolymer with n-butylamine was used to synthesize amine-functional PS. Subsequent reaction with a Meldrum's acid furan adduct yielded DASA-functionalized PS (Fig. 10d).126 Analogously, N-phenylpropane-1,3-diamine was used to modify cross-linked poly(styrene-co-vinylbenzyl chloride) microspheres, which enabled the formation of third-generation DASAs.127 Sulfonyl groups in poly(2-ethylsulfonyl-2-oxazoline) gels were substituted with N-ethyl-1,3-propanediamine or N-phenylethylenediamine to obtain secondary alkyl and aryl amines on the polymer (Fig. 10e).128 The poly(2-ethylsulfonyl-2-oxazoline) was synthesized by cationic ring-opening polymerization of 2-ethylthio-2-oxazoline and subsequent oxidation of the thiocarbamate group to become activated for substitution reactions. The amine-functionalized polymers were then reacted with different furan adducts to synthesize first- and second-generation DASAs.128
Our groups used activated ester co-monomers of pentafluorophenyl(meth)acrylate in various (meth)acrylate copolymers including block copolymers (BCPs) made by RAFT polymerization that were reactive to amines (Fig. 10f).114,129 The activated ester-functionalized motifs were subjected to aminolysis with aliphatic primary amines carrying an indoline or N-methylaniline functionality for the synthesis of the DASAs. This approach enabled the introduction of second- and third-generation DASAs,114,129 and has the advantage that pentafluorophenyl(meth)acrylates are hydrophobic, compatible with free radical and RAFT polymerization, and also substantially more stable towards hydrolysis in comparison to other groups used to install amines on polymers. More recently, other groups have relied on this functionalization strategy130,131 or expanded it by using N-methyl-1,3-propanediamine as linker to couple first-generation DASAs.132 Moreover, polymer gels functionalized with pendant DASAs were synthesized from networks made of pentafluorophenylacrylate and hexamethylene diacrylate as a crosslinker.119
In another approach, an alkene-functional methoxy-N-methyl aniline was integrated into the network structure of cross-linked thiol–ene photopolymerizable resins directly during the network formation step (Fig. 10g).91 Subsequently, these networks were reacted with different furan adducts through swelling in a solvent to synthesize DASAs from the amines attached to the network structure. Similarly, alkene-functional secondary aryl amines were attached via hydrosilylation chemistry to different linear and cross-linked polydimethylsiloxanes (PDMS) containing hydrosiloxane groups.118 The process enabled to covalently attach second and third-generation DASAs to PDMS networks or chain- and sidechains of linear PDMS (Fig. 10h).118 However, the same approach was not applicable to a one step-conjugation of an alkene-functional DASA due to side reactions of the hydrosilylation catalyst with the triene-enol bridge of the DASA.118
Stenzel and co-workers copolymerized protected alkylamine (meth)acrylate derivatives via RAFT polymerization, which were subsequently unmasked and subjected to reaction with a Meldrum's acid furan adduct (Fig. 10i).100,133 This procedure enabled the synthesis of DASA-functionalized homopolymers and BCPs, with the latter presenting one block that entirely consisted of DASA units.
However, first-generation DASAs formed from basic alkylamines are not compatible with these reaction conditions since they form hydrogen-bond complexes with HFIP. Instead, the addition of triethylamine as an auxiliary base was reported to promote the formation of first-generation DASAs on the side chain of (meth)acrylate copolymers with pendant N-methyl-alkyl amines,100 or in the synthesis of several second-generation derivatives.33,56 The addition of bases increased isolated yields in the synthesis of small molecule first generation DASAs, but also produced mixtures of open and closed isomers (cf. acid/base properties of DASAs, Section 1.1.1.6).31 The addition of basic amines to different DASAs in organic solvents post-synthesis also led to the formation of the closed isomer.119
Depending on the synthesis conditions, different ratios of the open and closed isomer are produced. The conditions are therefore important because thermal equilibration in a new medium after isolation may be slow, incomplete, or completely prevented, especially in solid materials.
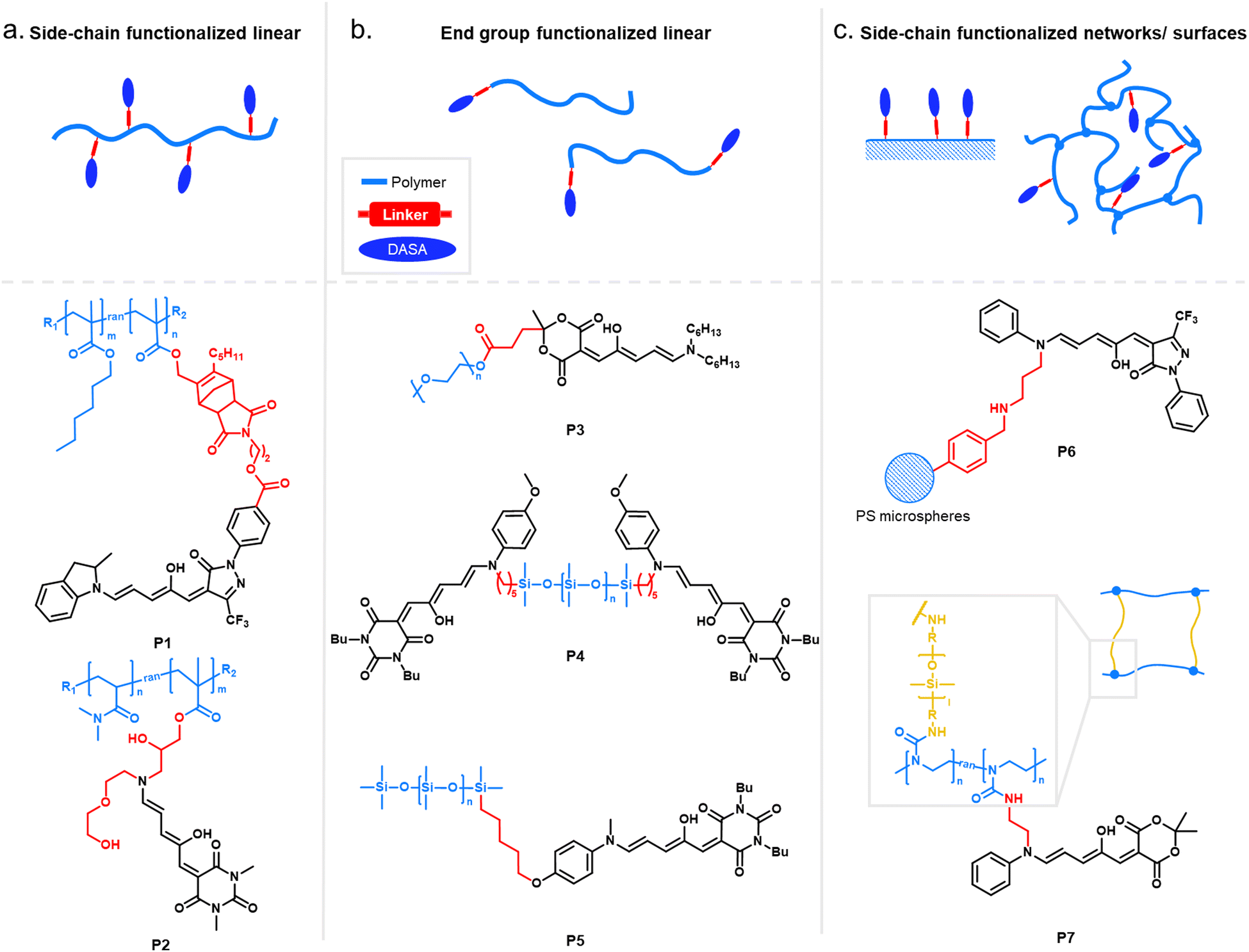 | ||
| Fig. 11 Schematics and structures of examples of linkers and macromolecular architectures of DASA-functionalized polymers. Linker attachment was either via the acceptor or donor site of the DASA. (a) Side-chain functionalized linear (block) copolymers. (b) End-group functionalized polymers including mono-functional and telechelic species. (c) DASA functionalization of networks or surfaces (side-chain functionalization). | ||
As outlined in the sections above, most DASAs are conjugated to polymers via the donor moiety. However, direct substitutions on the amine linker attachment can strongly influence the photoswitching properties through both electronic and steric effects, as discussed before for small molecule DASAs (Section 1.1). For example, DASAs that were coupled to PDMS via different aniline linkers displayed stabilization of the open isomer for the derivative bearing bulky N-alkyl groups (see Fig. 10, P4 and P5 for structural formulae of the linkers).118 This dependency could be reduced by attaching linkers at distal sites of the photoswitch, such as via click chemistry allowing DASA–polymer conjugation via the acceptor part.83,115
For other photoswitch families it is well established that linker length or rigidity can affect photoisomerization in materials.109 Longer and more flexible linkers often allow for more conformational flexibility or encourage solvation by the solvent molecules for compounds immobilized on supports or surfaces. Interestingly, a linker containing both an aniline and a benzylic amine attached to cross-linked PS microspheres (P6, Fig. 11) primarily reacted with furan adducts via the less nucleophilic aniline moiety during synthesis.127 This is likely due to increased steric hindrance for units in closer proximity to the main styryl network. In addition, linker chemistry and rigidity can also dictate thermal or mechanical properties of a polymer. The effects of linker type have not yet been systematically studied for DASA materials. However, a higher increase in Tg as a result of DASA conjugation relative to the unmodified parent polymer was found for methacrylate copolymers with DASAs connected through a Diels–Alder motif (e.g., P1, Fig. 11)83,96 when compared to acrylate copolymers attached via a less bulky linker (cf. Section 2.3.2).114
Moreover, photosurfactants based on two types of DASA-end-functionalized PEG, either connected through the Diels–Alder motif (P1, Fig. 11), or an esterification reaction (P3, Fig. 11) led to substantial difference in interfacial tension of surfactant-stabilized toluene droplets in water and also in the DASA photoswitching kinetics (see also Section 3.2).115 This example underlines the importance of linker design for the functional performance of DASA polymers. The effects of dissimilar linkers must also be considered when comparing different systems.
2.2 Functionalization through physical mixing or adsorption
DASAs have also been embedded inside bulk polymers without covalent attachment. The investigated materials differ in terms of their preparation process, polymer matrix properties (chemical structure, polarity, glass transition temperature (Tg), crystallinity etc.), as well as the type and concentration of DASAs in the polymer. In most cases, mixing has been achieved by co-dissolving the polymer and the DASA and fabricating polymer films or thin coatings by different methods, such as drop-casting,32,94,135 spin coating44,136 doctor blading,93 or dip coating,118 followed by a drying process to remove the solvent from the films. To date, glass substrates have been mainly employed to support the formation of DASA polymer films. Importantly, the glass cleaning procedure can affect the interactions of the glass with the DASAs at the surface of the polymer matrices.135 For spectrokinetic analysis of DASA photoswitching in polymer films (Section 2.7), film deposition techniques that deliver consistent film thickness over a large area, such as spin coating, may be preferred over simple drop-casting. Besides variation in thickness, the preparation process can also affect DASA distribution in the films (increase in local concentration and possibility of self-aggregation/stacking, cf. Section 2.5.4). Overall, the investigated DASA concentrations in the solid polymer matrices were rather low in the range of 0.5–8 wt%,32,44,93,135 but sometimes higher, up to 17 wt%.136The incorporation of DASAs into polymer films by physical mixing has proven to be versatile, being incorporated into matrices of poly(methylmethacrylate) (PMMA)32,44,94,136,137 and other (meth)acrylate copolymers,93 polystyrene (PS),44,89,94,135,136 poly(vinyl chloride) (PVC),93,136 poly(ethylene glycol) (PEG),136 poly(vinyl acetate) (PVA),93 poly(vinyl pyrrolidone) (PVP),136 polycaprolactone (PCL)136 and polyacrylonitrile.44 Recently, DASAs were also studied in PVP films containing various small molecule additives, such as hexyl benzoate, benzophenone, or terphenyl, to study their effects on DASA photoswitching.136 In terms of polymer composition, key requirements that have to be taken into account are compatibility of the DASA and the polymer (in terms of their chemical structure and polarity), for example, to avoid phase separation during film fabrication. The polarity of the matrix is also relevant for DASA photoswitching in these materials (refer to Sections 1.1.1.6 and 2.5.2). Another crucial handle to tune photochemical performance of DASAs in bulk polymer matrices is rigidity (i.e. Tg, refer to Section 2.5.1.1).93 Moreover, it was recently reported that commonly found impurities in polymer matrices can accelerate DASA (photo)degradation and therefore affect DASA stability and fatigue (Section 2.6).138
Besides direct dissolution into the polymer matrix, DASAs have been nanoencapsulated inside MOFs prior to immobilization inside PDMS networks.55 During the network immobilization step, the DASAs were added to the resin before curing. While free DASAs lost their color in the PDMS cross-linking step, MOF-encapsulated DASAs remained in their open form.55 A different approach to DASA “nanoencapsulation” consisted of covalently attaching PDMS chains to DASAs enabling to modulate the flexibility of the DASA microenvironment when mixed with rigid methacrylates.118 Moreover, a first-generation DASA presenting a dioctylamine donor was embedded in a urethane-cross-linked polybutadiene network structure by directly addition to the resin prior to the curing process.90 However, this approach delivered a colorless material with the DASAs residing in their closed isomer (while the open isomer could be recovered upon heating). Other examples showcase neat DASA film fabrication on different polymer surfaces such as paper, i.e. cellulose, or PEG.102,136,139 Cellulose nanoparticles have also been physically mixed with different DASAs (2–40 wt% relative to the particle mass) in isopropanol.140 Finally, non-covalent association of DASAs with other biopolymers72,106,141 or nanoparticles142 in solution or suspension has also been reported (see Sections 3.5 and 3.3.2).
2.3 Effects of DASA incorporation on parent polymer properties
Maximizing the effect of photoswitches in driving material property changes by light is usually an important prerequisite for successful applications of DASA–polymer materials (Section 2.4). On the other hand, it is often desirable that the integration of photoswitches into a material only minimally affects the material properties in the absence of the stimulus. This can involve scenarios where a material must meet certain mechanical requirements, maintain the self-assembly behavior for a given morphology after DASA functionalization, or preserve advantageous properties of the parent polymer, such as biocompatibility. Therefore, understanding the effects of DASA incorporation on the physical, chemical, or mechanical properties of a polymer material, even prior to photoisomerization, is critical for optimizing system design and material selection.000 M−1 cm−1 in solution),82–84 DASA-containing polymers are usually deep purple to blue in color, even at very low levels of functionalization. The intensity of the coloration is, however, dependent on the thermodynamic equilibrium between the open and closed DASA isomers in the material (see Section 2.5.3). The polymer environment, in turn, can also affect the optical properties of the switch, e.g., shift or broaden the absorption band due to polarity effects (solvatochromism), aggregation or polymer–DASA interactions (see Sections 1.1.1.2 and 2.5.3).
DASA conjugation can have substantial effects on polymer solubility in different solvents. Some applications rely on this effect, e.g., DASA conjugation at the chain end of water-soluble PEG yields amphiphilic polymers owing to the more hydrophobic DASA end-group, allowing the formation of micelles in aqueous media (cf. Section 3.3.1).116 PS films doped with DASAs (8 wt%) showed a slight increase in water contact angle relative to pristine PS at pH = 7 (87.4 ± 1° vs. 85.6 ± 0.7°), while at pH = 2 the trend was found to be reversed (84.0 ± 0.3° vs. 86.0 ± 0.6°).135 The hydrodynamic volume of polymer chains or their aggregation behavior in solution can also change as a result of DASA conjugation, which may be observed with gel permeation chromatography (GPC). For example, previous reports on DASA-functionalized acrylate or methacrylate homopolymers with 100 mol% DASA functionalization degree showed well defined and monomodal peaks for the protected-amine precursor polymers, but after DASA conjugation broadening towards higher molecular weights as well as the formation of bimodal or even trimodal distributions was observed.100 Similar observations were made with DASA-modified PEI121 or poly(dimethylacrylamide) (DMA).124 The dipolar character of both the open and closed DASA isomers (cf.Fig. 3) on the polymer chains may contribute to increased intermolecular interactions, interaction with the stationary phase in GPC, or aggregation.
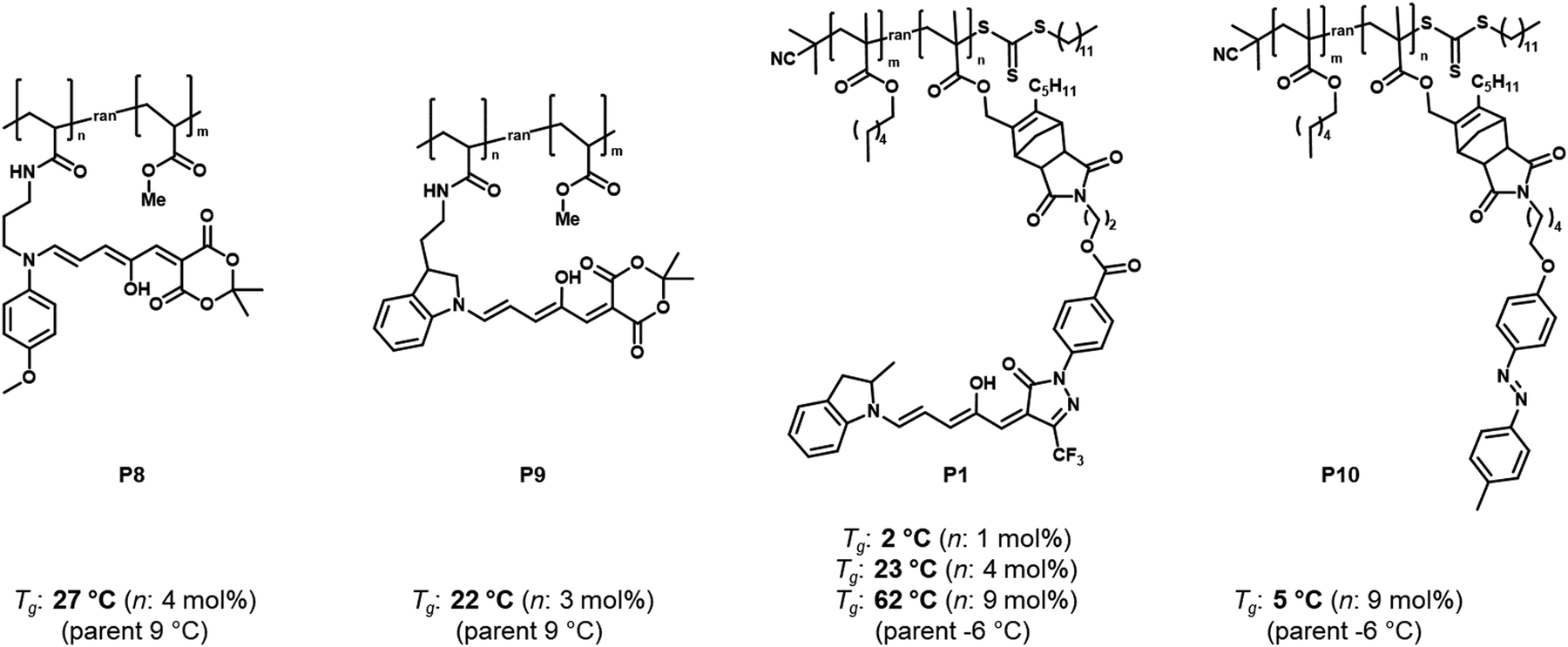 | ||
| Fig. 12 Structures of different DASA or azobenzene polymer conjugates and influence of photoswitch integration on the Tg (determined by differential scanning calorimetry, DSC) before photoisomerization. The “parent” Tg refers to the Tg of the polymers carrying no functional units. Data taken from ref. 96 and 114. | ||
2.4 Photo-induced material property changes
The attainable photo-induced macroscopic property changes of a material are of primary importance for DASA applications (Fig. 13). In most cases, these changes are driven by the transient non-equilibrium formation of the closed isomer upon photoisomerization and the substantial physicochemical differences between open and closed DASA isomers, such as electronic ground state structure, hydrophilicity and molecular shape and volume (see Section 1.1). The molecular level changes upon DASA photoisomerization can also affect the surrounding polymer chains and result in a range of optical and chemical material property changes (Section 2.4.1) as well as influencing the polymer's thermal or mechanical properties (Section 2.4.2). Therefore, a key requirement for achieving (reversible) photoinduced changes at the macroscale is efficient DASA photoswitching within the environment of the material (see Section 2.5). However, it is not yet clear whether and to what extent other metastable intermediates in the complex switching pathway of DASAs may play a role in changing material properties. In addition, due to the high molar absorptivity of the open DASA isomer (∼100000 M−1 cm−1 in solution),82–84 light absorption can also lead to local heating of the material caused by the non-radiative, thermal relaxation of the photo-excited DASAs (photothermal effect). Through these heating effects, light irradiation can also indirectly cause physicochemical variations, such as thermal expansion of solid polymer materials (Section 2.4.2). Macroscopic effects that can arise from one or a combination of the above-mentioned photo-induced changes are also revisited and discussed in more detail in the applications section (Section 3).
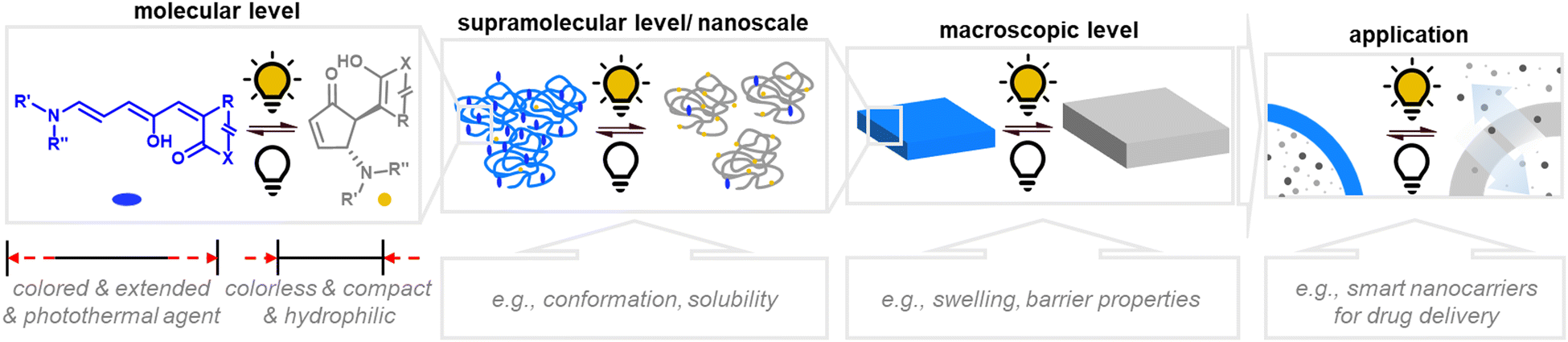 | ||
| Fig. 13 Photoinduced material property changes as a result of DASA photoswitching on different levels: molecular, supramolecular, macroscopic level and systems/devices for applications. | ||
Apart from optical effects, material changes related to hydrophilicity and chemical properties have been studied in a number of DASA-functionalized polymer systems, including surface and interface wettability and adhesive properties,115,135,140 (Section 3.2) or the size and structure of polymer nanoparticles (Section 3.3).92,125,133 Previous studies on the small molecule level showed that that the closed DASA isomer is substantially more water-soluble (hydrophilic) than the open isomer (cf. Section 1.1.1.5),31,48,68 which can be exploited to drive hydrophilicity-related changes in materials. More specifically, this can include photo-induced hydrophilicity increase of polymer film surfaces122,140 or an increase in polymer water-solubility, which can then induce swelling,125 changes in polymer self-assembly behavior or conformation in water,116 as well as changes in solute permeability of polymer membranes.129
However, it should be reiterated that the open DASA isomer, which has often been considered highly apolar, can exhibit a substantial dipolar character depending on the DASA derivative and the polarity of the environment (cf.Fig. 3d).44 The effective dipole moment changes between the two isomers can in fact be rather small (calculated ground state dipole moment change of ∼1 D for open/closed first-generation DASA71vs. 3 D for cis/trans azobenzenes144). Nevertheless, hydrophilicity is not only governed by the magnitude of a compound's dipole moment (polarity), but also by its ability to act as a water proton acceptor or to form hydrogen bonds with water molecules and water soluble molecules.145 “In addition, different DASAs can adopt different tautomeric forms in their closed state (cf.Fig. 3c). For example, the preferred population of closed isomer formed upon irradiation is not in all cases a charged species,44 which likely influences the overall extent of hydrophilicity change a DASA can undergo from cyclization.
Moreover, light-induced heating of materials resulting from photothemal effects due to the high molar absorptivity of the open form DASAs can also drive thermal, mechanical and other material property changes (e.g., photothermal actuation, Section 3.4).
2.5 Photoswitching and matrix effects
For many applications, important requirements for a “well” performing photoswitch are rapid switching in forward (or both) directions, large changes in isomer distribution upon switching (i.e., high amounts of closed DASA isomer at the PTSS), high proportions of open isomer at dark equilibrium, as well as high reversibility and fatigue resistance. However, the photoswitching behavior of DASAs can change quite dramatically when transitioning from dilute solution to macromolecular systems, while the nature and extent of these changes also depend on the DASA derivative that is used. Establishing general design principles to transfer a beneficial photoswitching performance from solution to environments of increasing complexity is therefore critical for the development of DASA–polymer materials. Conversely, material integration also offers potential for DASAs to have their photoswitching properties tuned by the local environment of the polymer, for example, with the goal of improving photoswitching properties in solvents where the isolated switch performs poorly (e.g., DASAs in water, see Section 1.1.1.6).Both mechanical effects and chemical properties of the macromolecular environment can affect the DASA photoswitching kinetics and thermodynamics (Fig. 14), e.g., by altering thermal barriers to isomerization or affecting the actinic step. Mechanical effects (Section 2.5.1) become relevant in materials where the DASAs are confined to microenvironments with larger viscosities as compared to organic solvents, which are typically Newtonian fluids with low viscosities of similar magnitude. This particularly applies to DASAs in or on polymers in the solid state but can also be of relevance for DASAs in/on dispersed polymer nanoparticles or otherwise confined DASAs (encapsulated, surface attached etc.146). In addition, the chemical environment given by the presence of polymer chains (Section 2.5.2) or the proximity to other DASAs (Section 2.5.3), changes in dark equilibrium as a result of polymer integration (Section 2.5.4), as well as fatigue resistance (Section 2.6), are other crucial factors that determine the overall photoswitching performance of DASAs in a polymer material.
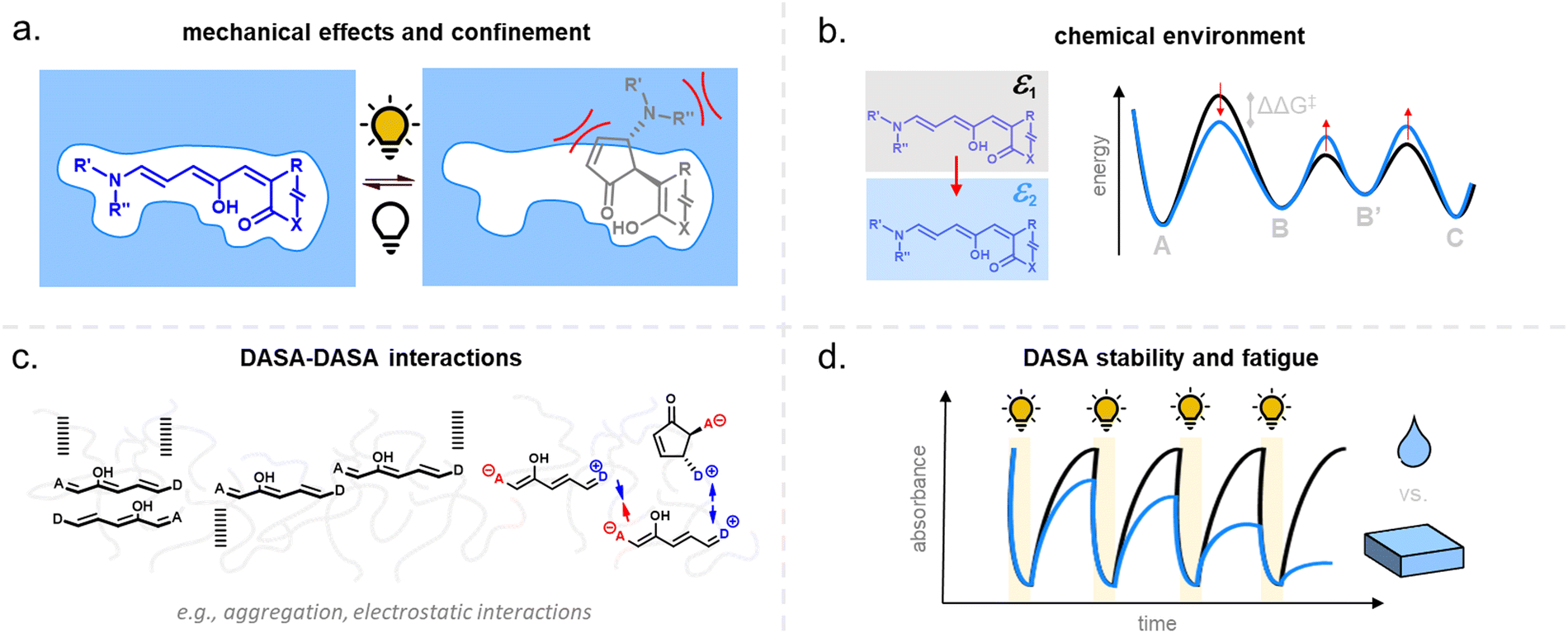 | ||
| Fig. 14 Matrix effects for DASA photoswitching in polymers. (a) Polymer matrix mechanics. (b) Chemical properties of the polymer (e.g., polarity, hydrogen bonding interactions). (c) Increasing DASA–DASA interactions as a result of incorporation into a polymer (e.g., aggregation, electrostatic interactions), due to, for example, increase in local DASA concentration and/or reduced diffusivity. (d) Effects of host environment on DASA stability and fatigue. | ||
Moreover, light must be able to penetrate into a material in order to induce the photoisomerization process. Sufficient light penetration in materials can constitute an additional challenge due to absorption of light by the polymer matrix, which can lower the overall photoresponse. Scattering and reflection effects can further complicate the picture and contribute to light defocusing. Finally, the current lack of understanding of DASA photoswitching in more complex environments is due in part to the limited applicability of many analytical techniques used for solutions, as well as the challenges imposed by the kinetic modelling of their multistep photoswitching mechanism (refer to Sections 1.1.1.3 and 2.7).
2.5.1.1 Bulk polymers. Mechanical effects can be relevant when the DASAs are confined by being covalently coupled or embedded into a polymer matrix, whereby photoswitching is typically slower when compared to solvents of similar polarity. The magnitude of this effect largely depends on the rigidity of the matrix polymer, as quantified by its Tg. For example, the photoswitching kinetics of second-generation DASAs conjugated to the side chains of different (meth)acrylate copolymers clearly correlated with the rigidity of the polymer in the solid state.114 The discoloration kinetics under illumination become slower with increasing Tg of the matrix, while thermal reversion is practically shut down at temperatures below Tg. When the materials are held at a temperature above Tg, increased recovery is observed (Fig. 15a). Most likely, glassy polymers prohibit the molecular mobility that is required for efficient photoisomerization. Importantly, DASAs can substantially increase the parent Tg of a polymer (Section 2.3.2), which in turn may hinder photoswitching in the solid state when the materials become too stiff after functionalization (Fig. 15b).96 It is not yet fully understood which individual step(s) along the multi-stage DASA switching paths (cf.Fig. 4) are influenced by the mechanical effects (see also Section 2.7).
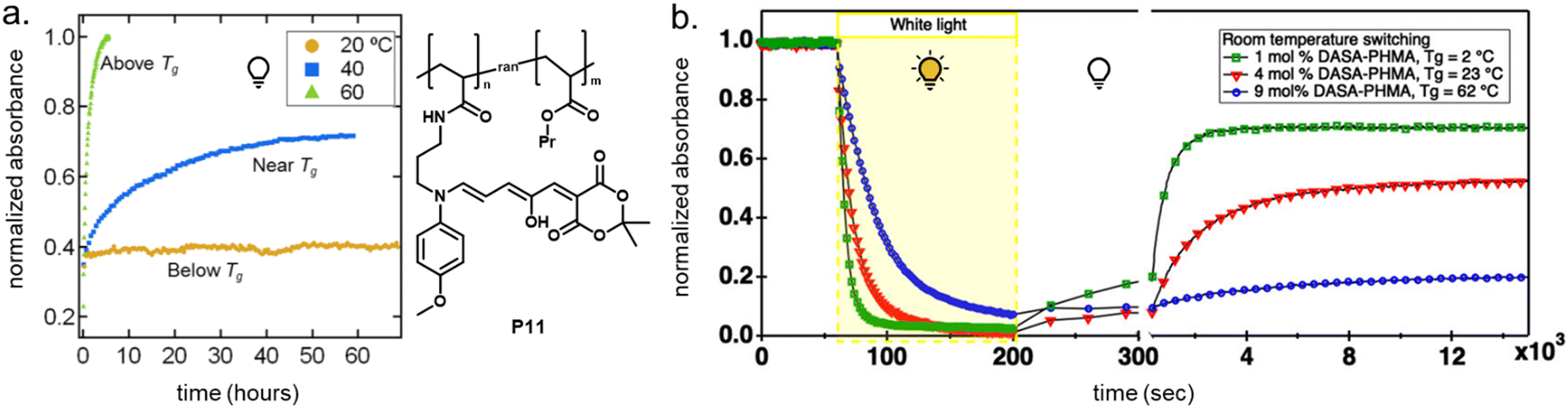 | ||
| Fig. 15 Mechanical effects on DASA photoswitching in solid polymer films. Kinetics were followed by time-dependent UV-Vis spectroscopy measured at the absorption wavelengths of the open isomer. (a) Thermal recovery kinetics of a second-generation DASA-functionalized propyl acrylate copolymer, P11, at 60 °C, 40 °C, and 20 °C, which are above, near, and below the Tg for the parent polymer (35 °C). Functionalization degree, n, was 3–4 mol%. Figure adapted with permission from ref. 114 Copyright 2017 American Chemical Society. (b) Forward isomerization under light and thermal recovery in the dark of third-generation DASA-functionalized PHMA (see P1, Fig. 10) with different Tg as a result of varying DASA concentration. A stiffening of the matrix polymer after covalent attachment of DASAs can lead to hindered photoisomerization and largely limited thermal recovery. Figure adapted with permission from ref. 96 Copyright 2022 American Chemical Society. | ||
DASA-functionalized networks synthesized from thiol–ene photoresins showed faster photoswitching responses when they were less cross-linked and softer.91 Polymer films containing non-conjugated embedded DASAs presented comparable dependence of the photoswitching on matrix Tg.93,136 For example, photoswitching rates of a library of second- and third-generation DASAs were studied as a function of contributions from Tg and Hansen solubility parameters (accounting for polar and hydrogen bonding interactions).93 DASA photoswitching was found to be primarily dependent on matrix rigidity. Moreover, the morphology of the solid polymer may also be relevant, since higher crystallinity leads to more steric obstruction. Photoswitches can also crystallize inside the polymer, as previously shown for, e.g., azobenzenes.147 Aggregation effects for DASAs are discussed in more detail in Section 2.5.3.
Overall, polymers that are both soft and that provide a suitable physicochemical environment for a DASA (see Section 2.5.2) allow for photoswitching kinetics that resemble solvated DASAs. For example, second-generation DASAs that are covalently attached to soft and hydrophobic PDMS elastomer led to efficient photoisomerization under illumination (fast and low amounts of open isomer at PTSS) and full thermal reversion in the solid state.118 However, soft polymers are not always suitable for a certain applications. Therefore, several strategies have been developed to circumvent unfavorable matrix mechanics and/or increase the free volume for isomerization in solids.148 Some of these methods have also been explored for DASAs, including MOF nanocages,55 structural modifications of the DASA with bulky, flexible groups (e.g., long alkyl chains),54 and other nanoencapsulation strategies.118
2.5.1.2 Polymers in solution and suspension. While polymer rigidity is the dominant factor for DASA photoswitching in bulk applications, it is less relevant for DASA-functionalized polymers in solutions where the DASAs are partially or fully solubilized. For example, the effect of polymer chain length (20 vs. 100 monomer units) and polymer backbone in a series of first-generation DASA-functionalized acrylate or methacrylate homopolymers was investigated in anhydrous dimethyl sulfoxide (DMSO) solutions (Fig. 16).100 Both, chain length and polymer type led to differences in the switching ability of the polymer-bound DASAs in solution, but the more rigid methacrylate polymer outperformed the acrylate polymer in terms of photoswitching performance and stability. Higher conversions to the closed isomer under irradiation were reached for the polymers with higher molecular weights, but the time to reach the PTSS was the fastest for the short methacrylate polymer. All the samples had similar thermal recovery rates, but only the DASAs conjugated to the short methacrylate polymer showed good reversibility with high recovery percentage of the open isomer in the dark. The dark equilibrium was also affected by polymer conjugation (refer to Section 2.5.4). The authors observed that some of these differences between the acrylate and methacrylate polymers, specifically recovery percentage, may be due to the limited hydrolytic stability for the acrylates.100 On the other hand, the reason for the limited recovery of the DASAs on the long chain poly(methacrylate) in solution is not clear. It could be related to differences in polymer conformation/aggregation state in solution with increased confinement of the DASAs when compared to the short chain analogue, or possibly be a result from increased DASA–DASA interactions (see also Section 2.5.3) or from fatigue.
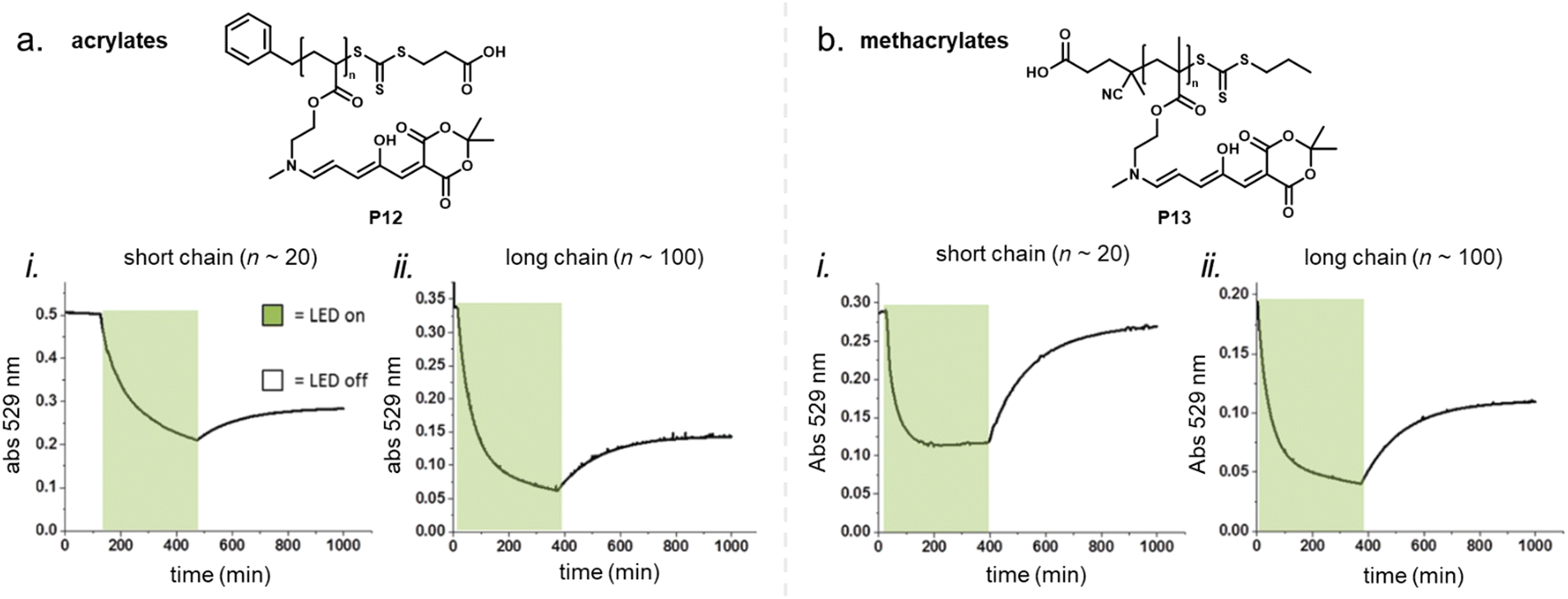 | ||
| Fig. 16 Photoswitching of DASA-functionalized polymers in solution. (a) DASA acrylate homopolymer (P12). (b) DASA methacrylate homopolymer (P13). Absorbance changes of the open isomer for short (i) and long (ii) chain poylmers in anhydrous DMSO solution overtime are shown. The samples were irradiated for 6 h with a green LED (530 nm) and allowed to thermally recover overnight. Figure adapted with permission from ref. 100 Copyright 2019 Royal Society of Chemistry. | ||
Glassy PS-based cross-linked microparticles decorated with DASAs exhibited incomplete bleaching under irradiation and low thermal recovery when dispersed and swollen in toluene.127 This is likely due to mechanical effects on DASAs within the cross-linked rigid network, whereby the cross-linking inhibits local segmental motion even when the material is swollen in a solvent. In this case, the glassy structure is not affected by increases in temperature (i.e. no Tg was observed by DSC) and the thermal recovery to the open DASA isomer after irradiation was not promoted at elevated temperatures.127 This is in contrast to observations on DASA-modified polymers in the bulk that were not highly crosslinked (Section 2.5.1.1).
2.5.2.1 Polarity. Solvent effects, especially solvent polarity, play a major role in DASA photoswitching as influence important barriers in the thermal steps of the isomerization mechanism (see Section 1.1.1.6 and Fig. 6). The importance of polar interactions also extends to macromolecular environments, including polymers in the solid state (Fig. 14b). For example, in solid polymer films, the ground state properties of the open isomers are affected by the polarity of the surrounding polymer matrix exhibiting an increasing degree of charge separation in more polar matrices, as shown by solvatochromic absorption shift analysis.44
Solvent compatibility of different DASA derivatives and their susceptibility to polarity changes of the medium observed in solution (refer to Fig. 2), also affect DASA photoswitching behavior in polymer materials, although sometimes these effects are overshadowed or convoluted by additional material-specific effects (e.g., matrix mechanics). To separate polarity effects in materials form other matrix effects, it can be helpful to investigate the photoswitching of the freely diffusing DASA in solution, ideally in a solvent of comparable polarity. For example, for most first-generation DASAs, efficient cyclization under irradiation and reversibility is limited to highly apolar solvents, such as toluene (cf. Section 1.1.1.6).30 These switches also show absent or slow cyclization under irradiation and/or irreversible recovery in numerous polymer materials, particularly in the solid state (e.g., also in moderately polar, soft matrices, such PMA).114 An exception are first-generation DASAs with sterically less demanding methyl substituents on the amine donor undergoing cyclization more efficiently in a wider range of solvents35 and conjugated to polymers (Section 1.1.1.4).100,132,133
For many second-generation and third-generation DASAs a “good” photochromic performance (cf. introduction to Section 2.5) is observed over a wider range of solvent polarities, while the kinetics are still solvent dependent and can vary considerably from solvent to solvent.32–34 These DASAs tend to also be better switches in various macromolecular environments. Particularly, DASAs with a more hybrid open isomer (mainly second generation, see Section 1.1.1) are the least sensitive to variations in solvent polarity and remain good switches in polymes of varying polarity. This also includes solid materials, such as soft acrylate and methacrylate copolymers,93,114 PDMS,118 cross-linked thiol–ene photoresins,91 or PCL.136 Importantly, the solubility of open and closed isomer, as well as the thermodynamic equilibrium between the isomers, are also affected by the polarity and protic nature of the polymer (Section 2.5.4).
2.5.2.2 Hydrogen bonding. Apart from polar intermolecular interactions, specific interactions between the DASAs and polymer functional groups (e.g., hydrogen bonding) could influence their photoswitching ability. For example, it has been computationally predicted that a modulation of the strength of the intramolecular hydrogen bond (from the acceptor carbonyl to the hydroxy-functional group on C2) is an important “turning knob” for altering different stages in the DASA photoswitching pathway.42,43,65 In particular, this hydrogen bond is involved in the proton-transfer that accompanies the ring-closing step (for switching mechanism cf.Fig. 4).41 The bond length and bond energy of this internal hydrogen bond may be susceptible to the local environment, for example, polarity or the presence of competitive hydrogen bond donors or acceptors in the solvent81,87 or surrounding polymer material. In addition, it was also suggested that the enol form of the closed isomer can form hydrogen-bonded pairs in solution.35
Recently, a promotion effect on the DASA discoloration rates under illumination was explored in ester-containing polymers (i.e. PCL, PMMA) and by addition of ester-containing small molecules to other polymers in the solid state.136 The authors hypothesized that the better cyclization efficiencies observed are related to a strengthened (shortened) internal hydrogen bond of a second-generation DASA in these environments. DFT calculations suggested that the intramolecular hydrogen bond of the open isomer B′ is shortened by nearby ester-containing repeating units, which could be a reason for facilitated proton transfer during ring closure.136 In addition, Lewis and co-workers analyzed contributions from hydrogen bonding of different solvents on DASA photoswitching, reporting acceleration on ring opening for solvents with higher hydrogen bonding parameters, δH (Hansen solubility parameter).93 Further research is needed to fully understand the origin and magnitude of these effects. In addition, the ability to form hydrogen bonds may be one of the mediating factors for inclusion of DASAs within host macrocycles, such as cyclodextrins (refer to Section 3.5).
Increasing the functionalization degree of DASAs in covalently functionalized polymers generally leads to a substantial increase in their Tg (Section 2.3.2), which in turn increases resistance to photoswitchability (Section 2.5.1.1). A series of methacrylate copolymers with varying DASA functionalization degrees and varying comonomers was analyzed for comparing similar materials with differing DASA content but similar Tg in order to decouple the effects of DASA concentration and Tg on material performance.96 Solid films with 1 mol% to 9 mol% DASA and a similar Tg around 60 °C showed similar rates of forward isomerization and thermal recovery regardless of DASA content.96 This finding indicates that in the investigated concentration range, the DASA concentration effects from DASA interactions are negligible in this system.
Besides long-range dipolar interactions between the DASAs described above, other interactions should be considered at high concentrations, such as aggregation of the open DASA isomer. For example, aggregation as a result of dipole–dipole interactions (alongside π–π stacking) is a well-known problem of the planar open-ring isomer of spiropyrans, which readily forms H- and J-aggregates (“face-to-face” and “head-to-tail” arrangement of the transition dipoles, Fig. 17a) in hydrophobic solvents.109 Aggregation of DASAs leads to steric and molecular mobility restrictions and is, therefore, likely to be counterproductive for efficient switching. This is one reason for the limited photochromism observed in pure DASAs in the solid state (crystalline and amorphous).54,94,136 Aggregation could also contribute to reduced fatigue resistance (cf. Section 2.6). However, even in solution at high concentration of 10−3 M in chloroform or toluene, no sign of absorption band broadening or shifting, which would indicate aggregation or electronic coupling could be found for a third-generation DASA derived from 2-methylindoline and CF3-pyrazolone.94 Similarly, for the same DASA dispersed in PMMA, only at a very high concentration of 10−1 M a very slight band broadening was observed. In contrast, films composed of PS produced a broadening of the DASA band and an enhancement of the 0–1 vibronic feature was found at high DASA concentrations (≥10−2 M), which are indicative of the presence of H-type aggregates in these films (Fig. 17b).94 A band broadening with increasing DASA incorporation and/or Tg was also found for DASAs conjugated to a series of methacrylate copolymers with different alkyl chain lengths (hexyl, butyl, or propyl) in solid films, but not for the polymers in solution.96 However, PHMA retained an amorphous microstructure, as shown by X-ray diffraction (XRD) analysis and DSC, even at relatively high DASA functionalization degree of 10 mol%.83
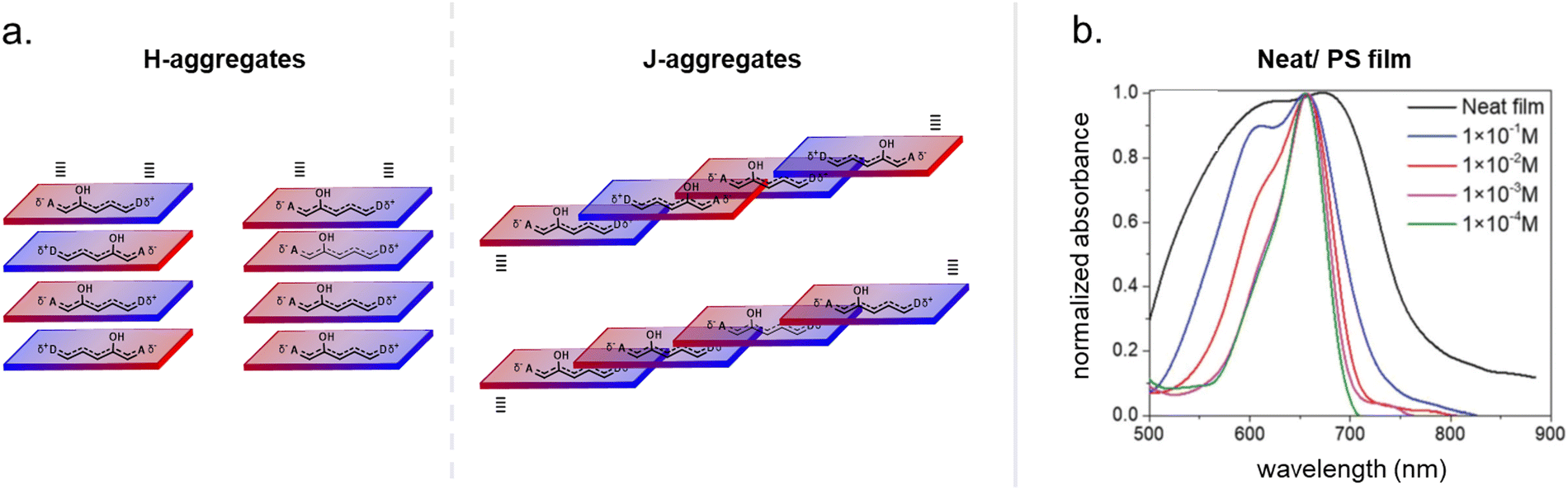 | ||
| Fig. 17 Concentration and aggregation effects. (a) Aggregation of open DASA isomer as a result of dipole–dipole interactions alongside π–π stacking as H- and J-aggregates (“face-to-face“ and “head-to-tail“ arrangement of the transition dipole). A and D stand for acceptor and donor, respectively. (b) Absorption spectra of DASA 3 (cf.Fig. 3) as neat films or dispersed in PS at different concentrations. A broadening of the DASA band and an enhancement of the 0–1 vibronic feature was found at high DASA concentrations, which are indicative for the presence of DASA aggregates in these films. No such effects were found for the same DASA in solution up to the solubility limit. Figure adapted with permission from ref. 94 Copyright 2019 Royal Society of Chemistry. | ||
In general, aggregation is mitigated since most open DASA isomers are not entirely planar, which hinders strong intermolecular π–π and efficient stacking. For example, the use of 2-methylindoline instead of indoline for third-generation DASAs actively hinders stacking interactions and markedly improves DASA solubility in organic solvents. By tailoring the type of DASA (dipole moment and planarity of the open isomer) and DASA integration into a macromolecular system (covalent conjugation, isolation from other entities by encapsulation, etc.), the possibility of DASA–DASA interactions could be further reduced, if needed.
Excluding assumptions based on open form absorption, the evaluation of the DASA equilibrium distribution in more complex systems generally poses analytical challenges, because analytical methods, such as liquid-state NMR spectroscopy are not suitable for solid state investigations or because measurements are complicated by signals of the macromolecule. Because of this, systems were analyzed mostly in the solvated state. For example, (bi)end-functionalized and pendant DASA–PDMS conjugates dissolved in chloroform did not display changes in the DASA dark equilibrium when compared to small molecule models.118 However, in a series of first-generation acrylate and methacrylate homopolymers with pendant DASA groups, it was found that the isomer ratio at the thermodynamic equilibrium in DMSO was shifted toward higher concentration of closed isomer after polymer conjugation (78–99% vs. 62%).100 This effect was more pronounced for the methacrylate backbone, but not substantially influenced by the chain length. In contrast, conjugation to the shorter methacrylate homopolymer led to a stabilization of the open isomer in water enabling reversible switching, which is not observed for the small molecule analogue.100 The proportion of open DASA isomer at equilibrium in DMSO was lower for a short methacrylate homopolymer in comparison to the same polymer attached to poly(ethyleneglycol) methyl ether methacrylate (poly(PEGMEMA)) (< 10% for the homopolymer vs. 43% for the diblock copolymer).100
Polymer conjugation or non-covalent encapsulation (“cages”) can lead to stabilization of the open isomer in solvents where usually cyclization to the closed isomer would be observed under exclusion of light, such as aqueous environments. This is usually a result of compartmentalization, whereby the (typically hydrophobic) polymer environment prevents access of solvent molecules, such as, for example, in supramolecular assemblies of amphiphilic polymers (cf. Section 3.3).129,133 In the case of “tight” encapsulation and confinement, effects that retard or prevent cyclization through limited rotational freedom and steric restrictions may also come into play.72 However, in this case, photoinduced cyclization is also often impaired. In addition, encapsulation or polymer conjugation can also enhance solubility of a DASA in certain media, such as water (see for example cyclodextrins, Section 3.5). In solid materials, DASA-functionalized hydrophobic polymer networks prevented water from diffusing into the gels, leading a loss of the hydrochromic behavior of the DASAs (see Section 1.1.1.5).119
2.6 DASA stability and fatigue resistance
Although photoisomerization of DASAs, in principle, is a nondestructive process, side reactions can lead to by-products resulting in gradual degradation with increasing number of switching cycles (“fatigue”, Fig. 14d). While small molecule DASAs in dilute solutions can show remarkable fatigue resistance (e.g., decrease of only 50% in 610 switching cycles for one derivative),35 DASA stability in materials is often compromised. This is interesting, since spiropyran photoswitches often display the opposite behavior. Spiropyran immobilization is associated with substantially reduced photodegradation, mostly by suppressing bimolecular (with respect to spiropyran) degradation pathways and through the restriction of H- and J-aggregate formation.109The recovery percentage for DASAs in solid polymer films and other DASA materials is often <100% already in the first cycle and does not reach 100% when the polymer is held at temperatures above the Tg (although often higher recovery and faster kinetics can be achieved when heated above Tg, see Section 2.5.1.1).93,96,138 Upon repetitive cycling, this incomplete thermal recovery typically compounds with each subsequent cycle. It is not yet understood whether this is due to the DASAs reaching a new thermodynamic equilibrium or due to degradation. It is also well established that the interaction of a triplet excited state of a chromophore with oxygen can generate singlet oxygen (1O2) and other reactive oxygen species (ROS), which can then lead to photooxidation.149 Removal of oxygen led to an improved fatigue performance of a small molecule DASA in solution (41% decomposition after 100 cycles under air to just 25% under argon),35 which indicates that photodecomposition via triplet excited states is important (Fig. 18a).
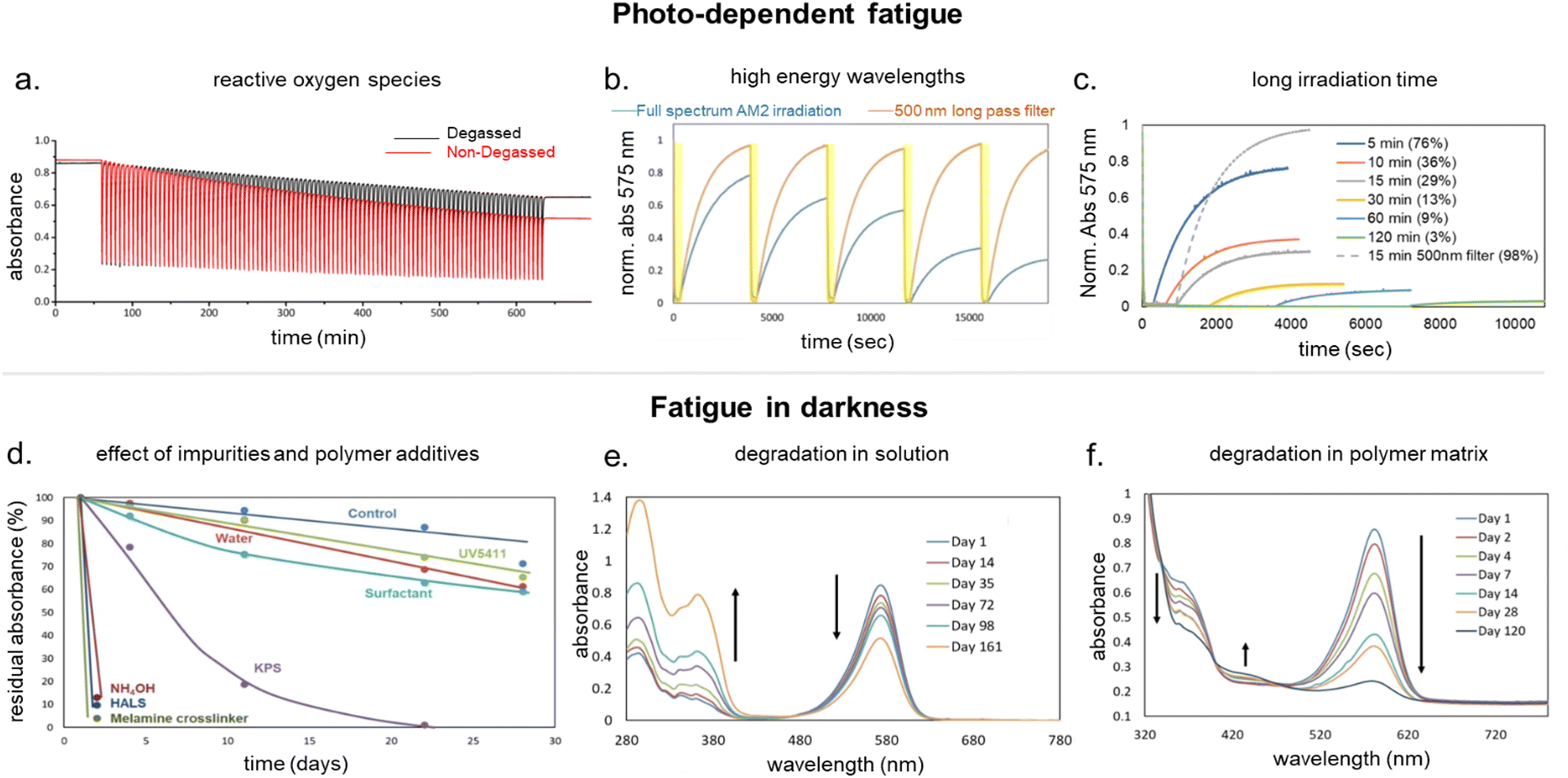 | ||
| Fig. 18 Mechanisms of DASA fatigue in presence of light irradiation and in darkness. (a) Fatigue measurements of a barbituric acid-based first-generation DASA measured by UV-Vis spectroscopy at 565 nm under atmospheric conditions (red) and degassed with argon (black). After 100 cycles of 45 sec irradiation (567 nm) followed by 5 min equilibration in the dark, the degassed sample decomposed by 25%, while the non-degassed sample decomposed by 41%. Figure adapted with permission from ref. 35 Copyright Royal Society of Chemistry 2018. (b) Repetitive photo-cycling of a second-generation DASA based on N-methyl aniline and 1,3-dimethylbarbituirc acid in a solid polymer matrix (PBA-co-PMMA; 2 wt%) by irradiation with an AM2 solar simulated light (blue) and in presence of a 500 nm long pass filter (orange). (c) Photoswitching of a second-generation DASA based on N-methyl aniline and 1,3-dimethylbarbituirc acid in a polymer matrix (poly(tert-butyl methacrylate)-co-PBA; 2 wt%) after different irradiation times with an AM2 solar simulated light directly and in presence of a 500 nm long pass filter. The light exposure time was found to decrease the thermal recovery to the DASA open isomer (recovery percentage is given in brackets). (d) Residual open isomer absorbance of a second-generation DASA based on N-methyl aniline and 1,3-dimethylbarbituirc acid at dark equilibrium overtime in acetonitrile (0.15 mM) in presence of different impurities at 0.2 wt% (UV5411: common UV absorber based on benzotriazole; KPS: potassium persulfate; HALS: hindered amine light stabilizer). (e) Decomposition of a second-generation DASA based on N-methyl aniline and 1,3-dimethylbarbituirc acid in acetonitrile (0.15 mM) stored in the dark over time. A decrease in the open isomer absorbance band at 575 nm and an increase in absorbance in the UV region (370 nm) could be observed. The latter changes are likely caused by the retro formation of the furan adduct. (f) Decomposition of a second-generation DASA based on N-methyl aniline and 1,3-dimethylbarbituirc acid within a solid polymer matrix (PBA-co-PMMA; 2 wt%) over time stored in darkness. An increase of absorbance band at 430 nm, and decrease at 370 nm and 585 nm is observed. Figures adapted with permission from ref. 138 Copyright 2023 Elsevier. | ||
Recently, it was discovered that blocking the shorter wavelengths (<500 nm) when irradiating a second-generation DASA in solution or dispersed in solid polymer films with a broadband white light source (AM2 solar simulator) led to greatly improved fatigue resistance (Fig. 18b).138 This was achieved by using a long pass filter or by addition of common UV absorbers. Increasing the exposure time to light was also found to decrease the thermal recovery (Fig. 18c).138 This suggests the existence of a photo-dependent fatigue mechanism for DASAs from higher-energy light. NMR analysis of the DASA in acetonitrile solution additionally revealed the formation of an aldehyde photodegradation product upon prolonged irradiation without the filter, which was not observed upon irradiation with filter.138 The degradation product could stem from oxidative addition and cleavage reaction with the open DASA isomer.138 Degradation of first- and second-generation DASAs as a result of UV light irradiation (365 nm) has also been reported elsewhere.150
In some cases, DASA stability may be limited in certain polymer systems even under exclusion of light. For example, acrylate polymers (as opposed to the methacrylate polymers) with aliphatic secondary amine side groups, including first generation DASA-functionalized acrylate polymers, have limited stability in water and can hydrolyze over time.100 In addition, DASAs were found to be susceptible to degradation by different polymer additives or impurities, e.g., residual redox initiator, polymerization catalysts, common cross-linkers, and bases (Fig. 18d).138 The presence of only trace amounts of stabilizers or additives in common solvents (e.g., ethanol in chloroform at 0.75 vol%) was also found to strongly influence DASA photoswitching properties in solution.108 Lewis and co-workers investigated structure–property relationships associated to the fatigue in DASAs embedded in polymer matrices and in organic solution.138 Even in darkness, substantial levels of irreversible DASA discoloration both in solution and in polymers were observed over time. These changes were accompanied by an increase in absorption in the UV region (Fig. 18e and f) that was associated to the retro-formation of the activated furan adduct. However, this was only evidenced by NMR spectroscopy in the fatigued DASA solutions and not in the polymer environments directly.138 The degradation was faster in polymers than in acetonitrile or ethanol solution, but the differences in time dependency and changes in absorbance bands suggest different or additional degradation mechanisms occurring in the polymer, possibly due to the presence of additives. In acetonitrile and in polymer matrices, the tendency of a DASA to degrade increased with the strength of the acceptor and increasing push–pull ability. Moreover, precipitation in the polymer matrices was observed for DASAs with greater push–pull ability (most likely due to formation of an ionic closed isomer), which may have resulted in a change in the dark equilibrium distribution toward the closed isomer.138 Covalent immobilization in matrices has been proposed to improve fatigue resistance compared to embedding free DASAs in polymer films,114 however, experimental evidence is still needed to confirm this.
While photo-dependent fatigue can be mitigated by using suitable light sources, addition of UV absorbers, or radical and ROS inhibitors,138 it is still unclear how the observed fatigue in the dark involving reversion to the furan adduct can be prevented or to what extent this degradation pathway occurs with different DASA derivatives and DASA-materials. Limited DASA stability from retro-formation of the starting materials has also recently been observed or predicted in studies of various small molecule DASAs in solution.36,73,79,107 It was reported that a DASA composed of an indoline donor and a Meldrum's acid-based acceptor with methyl substitution at position 5 of the triene bridge led to release of the amine and recovery of the furan adduct when in solution overtime.36 This reaction was shown to be faster in more polar solvents such as methanol. DASA stability and their tendency to undergo the retro-reaction as a function of different substitution patterns on the triene bridge have also been studied by DFT calculations.73 In addition, acid triggered in situ formation of Stenhouse salts from DASAs was observed, which most likely proceeded via a retro-reaction to the furan adduct with subsequent release of furfural.107 A mechanistic characterization of this dissociative side reaction has not yet been reported. However, a computational study recently suggested a possible pathway via an alternative thermally-induced double bond isomerization of the open form that leads to the formation of the EZZZ isomer (cf.Fig. 4).79 In this potential pathway, the EZZZ isomer undergoes cyclization and proton transfer followed by dissociation into a secondary amine and furan adduct.
The potential instability of DASAs and degradation to their starting materials needs further investigation, as this could be detrimental to emerging biomedical applications of DASA-functionalized polymers (e.g., phototherapy, drug delivery, see Section 3.3), since furan adducts are known to be cytotoxic151 and could present carcinogenic effects.152 In addition, exposure to released amine donor groups such as aniline could lead to various anomalies, e.g., hypoxemia due to binding to hemoglobin.153
2.7 Kinetic analysis of DASA switching in polymer materials
Kinetic models previously applied to the analysis of DASA photoswitching are discussed in detail in Section 1.1.1.3. The various spectroscopic techniques introduced in Section 1.1.1.3 differ in their suitability for studying DASA-photoswitching in complex environments and solid samples, including polymer films. Most macromolecules are IR and NMR active, which complicates kinetic analysis by FTIR and NMR spectroscopy as the environment accounts for most of the signal relative to the comparatively dilute DASA units. Therefore, UV-Vis spectroscopy was used in almost all examples to characterize DASA switching kinetics in polymers.93,94,96 Few studies have instead monitored the weak fluorescence signal of the open isomer.90,91Although interference from other absorbing species in the visible spectral range is rarely a problem, analysis of DASA photoswitching in polymers by UV-Vis spectroscopy can be challenging for other reasons. For example, low transmittance samples, such as polymer films with high DASA content, can result in large noise levels and low measurement accuracy. Another difficulty often encountered with solid samples or suspensions of colloids is light scattering, which can complicate UV-Vis absorption measurements. An integrating sphere was used to monitor DASAs absorption overtime in diffuse reflectance mode, overcoming issues arising from the inherent scattering of a polymer microsphere suspension.127 As described in Section 2.2, the polymer film preparation method (e.g., drop-casting, spin coating, drawing, etc.) may introduce inhomogeneities in the film thickness and surface quality, resulting in non-uniform absorption of the pumping and probing beams that may need to be taken into account. Additionally, anisotropic confinement of the DASA moieties, e.g., in a liquid crystal network, can theoretically result in absorption cross sections and photoswitching kinetics that depend on the polarization and incidence angle of light.99 Aside from complications arising from their optical properties, polymers (in the solid state) tend to drastically slow or completely inhibit the equilibration of A and C isomers in the dark (Section 2.5.1.1).114 This makes it nearly impossible to estimate the molar absorptivities of A and B/B′ in polymer films. In solution, these values are typically obtained by comparing the isomer distribution measured by 1H NMR with the sample absorbance measured at equilibrium in the dark.35
Additionally, the limited diffusion of DASAs in polymer films often affects the kinetics of photochemical isomerizations in strongly absorbing films. This is because light intensity decreases with increasing penetration depth and photoisomerization generates a concentration gradient over the thickness of the film, causing the reaction to slow as it propagates deeper into the material.99 Consequently, in conditions where the thermal back reaction rate from C to A is competitive with the forward photoisomerization rate, the resulting PTSS is expected to be enriched in the open form isomer at increased depths from the irradiated surface. This effect, while never experimentally observed, is expected to be amplified in systems where the initial absorbance is large relative to the optical pumping power used to promote photoswitching. In rubbery polymers, like PDMS, thermal open form recovery occurs readily over the course of several minutes at room temperature in the dark.118 As a result, spatial dependencies in the rates of photoswitching and the thermal back reaction must be taken into account when inferring local DASA isomerization kinetics from the spatially averaged kinetics probed experimentally.37 On the other hand, in glassy polymeric environments, such as PMMA and PS, where the molecular confinement forces are much larger (cf. Section 2.5.1.1), the back reaction does not proceed at a measurable rate over several hours at room temperature. When this is the case, photoswitching can often be considered an irreversible process to generate a visible light inactive state and spatial variations in the rate of photoswitching do not need to be taken into account in the kinetic analysis.
The above-described challenges related to quantitatively measuring the DASA photoswitching kinetics in polymers have generally slowed progress towards understanding the effects of polymeric microenvironments on the step-wise switching kinetics of DASA relative to the strides made in characterizing DASA kinetics in solution. Recently, Lewis and co-workers measured the photoswitching and thermal recovery kinetics of a variety of second-generation and third-generation DASAs dispersed in different polymer matrices.93 By employing the three state kinetic model described in Section 1.1.1.3, they uncovered a negative and roughly linear correlation between the polymer glass transition temperature and the kinetics of isomerization from A to B/B′ and from B/B′ to A. However, the transient population of B/B′ established under irradiation could not be experimentally observed in this work because the absorbance of this state was too low to accurately measure. As a result, it was difficult to validate the kinetic model. Without a better fundamental understanding of how polymer matrix effects shape the energetic landscape of DASA photoswitching, specifically regarding the stability of intermediates, phenomena that require leveraging these intermediate states, including the photokinetic switching control demonstrated by Stricker et al. under irradiation of B/B′ in solution (Section 1.1.1.1),37 are largely unexplored in DASA-containing materials. With greater insight into the effects of polymeric microenvironments on the kinetics and thermodynamics of individual steps of DASA photoswitching, one could rationally design solid materials that promote more desirable photoswitching properties for a variety of applications (cf. Section 3).
To address this lack of suitable spectroscopic tools for measuring DASA kinetics in solid polymers, Gordon, Read de Alaniz and co-workers developed a new method for performing UV-Vis absorption spectroscopy using frequency modulation to “fingerprint” each absorbance state. This method was introduced with the explicit aim of better characterizing the thermal and photochemical isomerizations of A and B/B′ in polymers and solvents.98,108 The technique, termed frequency modulated pump probe spectroscopy, or FMPPS, as illustrated schematically in Fig. 19a, relied on periodic optical modulation to encode the spectral information of two low intensity probe beams. Wavelength indiscriminate phase locked detection was then used to isolate the transient absorbance of the two distinct isomeric states based on the known spectral properties of each probe beam. By compromising the wavelength resolution of traditional spectrometers, their detection scheme could provide accurate absorbance measurements down to 0.002 a.u. while exposing the sample to anywhere between several watts and a few nanowatts of light to tune the passivity of the measurement. In addition, the temporal resolution of the measurement could be scaled from a few milliseconds to several seconds, allowing thermal and photochemical processes occurring on vastly different time scales to be measured in real time.
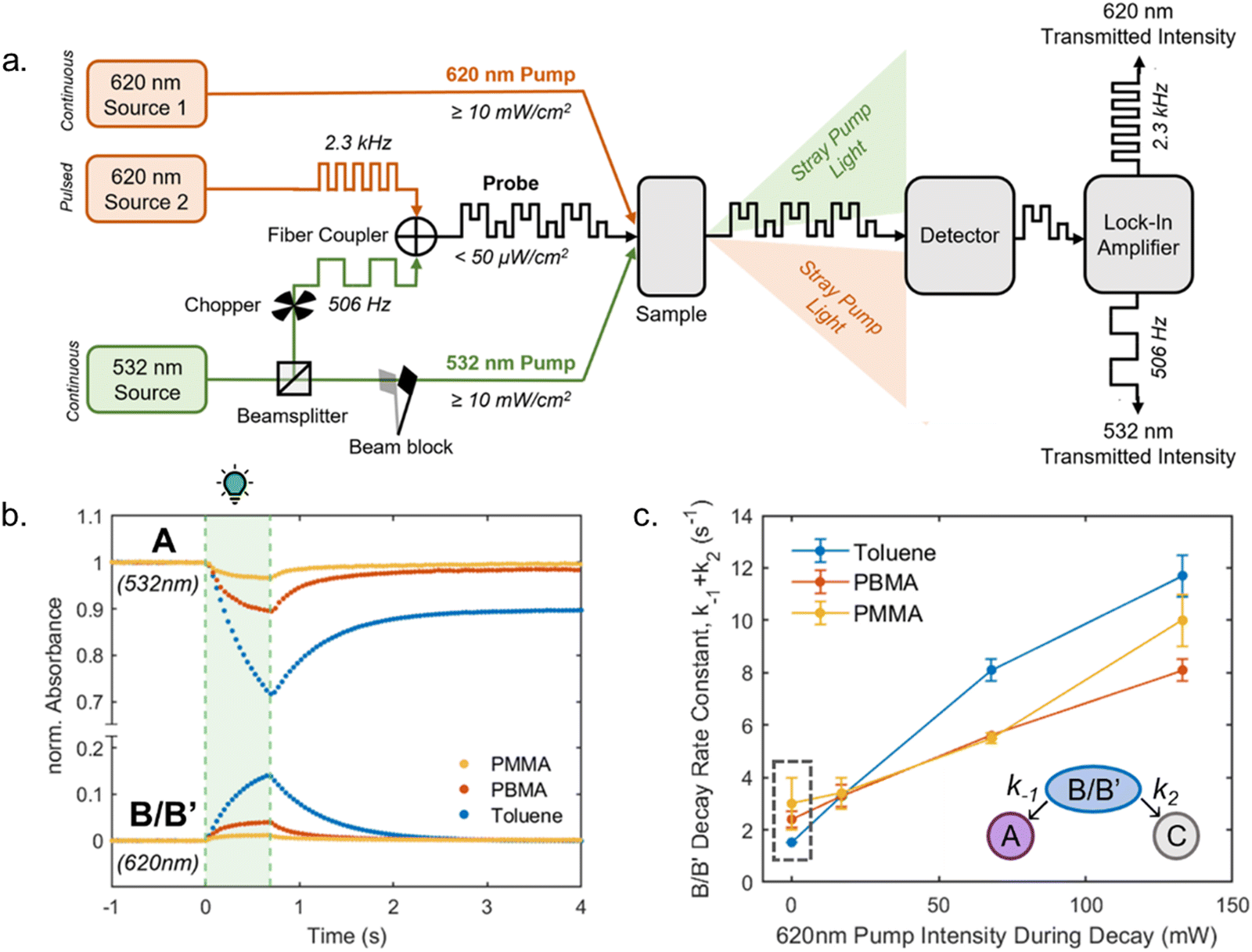 | ||
| Fig. 19 Frequency modulated pump probe spectroscopy (FMPPS) for studying DASA kinetics in bulk polymers. (a) Schematic illustration of the FMPPS technique using a 532 nm laser to excite and probe A and 620 nm LED to excite and probe B/B′. (b) Using FMPPS to observe the approach to A/B pseudoequilibrium in the first 600 ms of A irradiation with 532 nm light (24 mW cm−2) in toluene, PBMA, and PMMA. The absorbances of A (top) and B/B′ (bottom) were measured with the 532 nm and 620 nm probe beams illustrated in (a), respectively. (c) Comparing thermal and photochemically accelerated decay kinetics of B/B′ in PBMA, PMMA, and toluene measured by FMPPS. Varying intensities of 620 nm light were used to excite B and B′ during decay and promote photochemical reversion to A. The active pumping area was approximately 1.7 cm2. The strictly thermal decay rate constants are boxed and were measured under the sub-microwatt intensities of 620 nm light needed to probe the B/B′ state. Figures adapted with permission from ref. 98 Copyright 2023 Elsevier. | ||
Recently, it was shown how FMPPS could be used to provide mechanistic insight as to why the same DASA architecture supported a stable and photochemically accessible B/B′ intermediate state in toluene, but exhibited increasingly binary photo-responsive properties when mixed in poly(butyl methacrylate) (PBMA) and PMMA.98 By characterizing the thermal and photochemical rates of B/B′ decay, it was found that while the photochemical isomerization from A to B was inhibited in PBMA and PMMA relative to toluene (Fig. 19b), the thermal decay of B/B′ showed no significant dependence on the nature of the host environment (Fig. 19c). As a result, the pseudo-equilibrium populations of A and B established under irradiation of A alone favored lower concentrations of B and B′ in the polymeric hosts. Furthermore, it was hypothesized that under simultaneous irradiation of A and B/B′ in PBMA and PMMA, the rate of photochemical isomerization from B to A, which scaled exponentially with the concentration of B, could not compete with the rate of thermal decay at low B/B′ populations, which scaled linearly with the concentration of B. This made it prohibitively inefficient to photochemically address the intermediate B/B′ state in polymers, and the unique three state nature of DASA was functionally lost.
3. Emerging applications of DASA-containing polymers
In this section, applications of DASA–polymers are presented. The different examples are broadly classified into materials for modulation of optical properties, changes in surface chemistry/interfacial properties, photoactuation, and polymeric carriers (Fig. 20). At the end of the section, applications of DASAs in biopolymers are also briefly outlined. | ||
| Fig. 20 Overview of different categories of applications of DASA-containing polymers. | ||
3.1 Changes in optical properties
In another example, a bi-photochromic first-generation DASA containing an azobenzene moiety attached to its amine donor was dispersed in PS films.89 This DASA switched faster and reversibly in the polymer films as opposed to the previous examples on first-generation derivatives, which is likely due to the weaker benzylamine donor moiety used for the conjugation of the DASA to azobenzenes rendering it more hybrid in nature with a greater tolerance to higher polarity media (cf.Fig. 3 and Section 1.1.1.6). This enabled more efficient photopatterning and reversion of the patterns through heating of the polymer films. This material was also used to obtain a photonic logic device from the four isomeric states the DASA-azobenzene conjugate can reside in, which are addressable with different wavelengths of light. The compatibility of DASA with orthogonal photoswitches such as azobenzenes,68,89 diarylethenes,150 or other phototriggers155 is of great interest for the development of multiresponsive and multifunctional materials, whereby each photoactive unit can be addressed independently and in parallel with light of different wavelengths. For example, a drop-cast film made from a mixture of PMMA and a second-generation DASA in combination with a diarylethene photochrome could be switched between four different distinct colors by using three different wavelengths of light and heat.150 The multiresponsiveness of DASAs to stimuli other than light, such as heat or water, has been used for the development of secret inks and polymer-based data encryption systems (cf. Section 3.1.4), as well as sensor platforms (cf. Section 3.1.5).
Second-generation DASAs with more hybrid DASA open forms in comparison to the first generation (cf.Fig. 3 and Section 1.1.1.6) could be photoswitched more efficiently in polymer films and the open isomer could be recovered when the films were held at temperatures above their Tg (Fig. 21a).114,150 For example, ternary photopatterning was demonstrated by blending two acrylate copolymers conjugated to two different DASAs that could be selectively addressed with two different wavelengths of light.114 Although not specifically analyzed in the above examples, the photopatterned regions in DASA–polymer films may also present differences in other properties besides color changes (e.g., polymer morphology, mechanical, thermal or adhesive properties, or surface chemistry), which could offer a range of interesting opportunities in diagnostics, sensing, micro- and nanoactuation, biotechnology or bioseparation. However, this will require further investigation in the future.
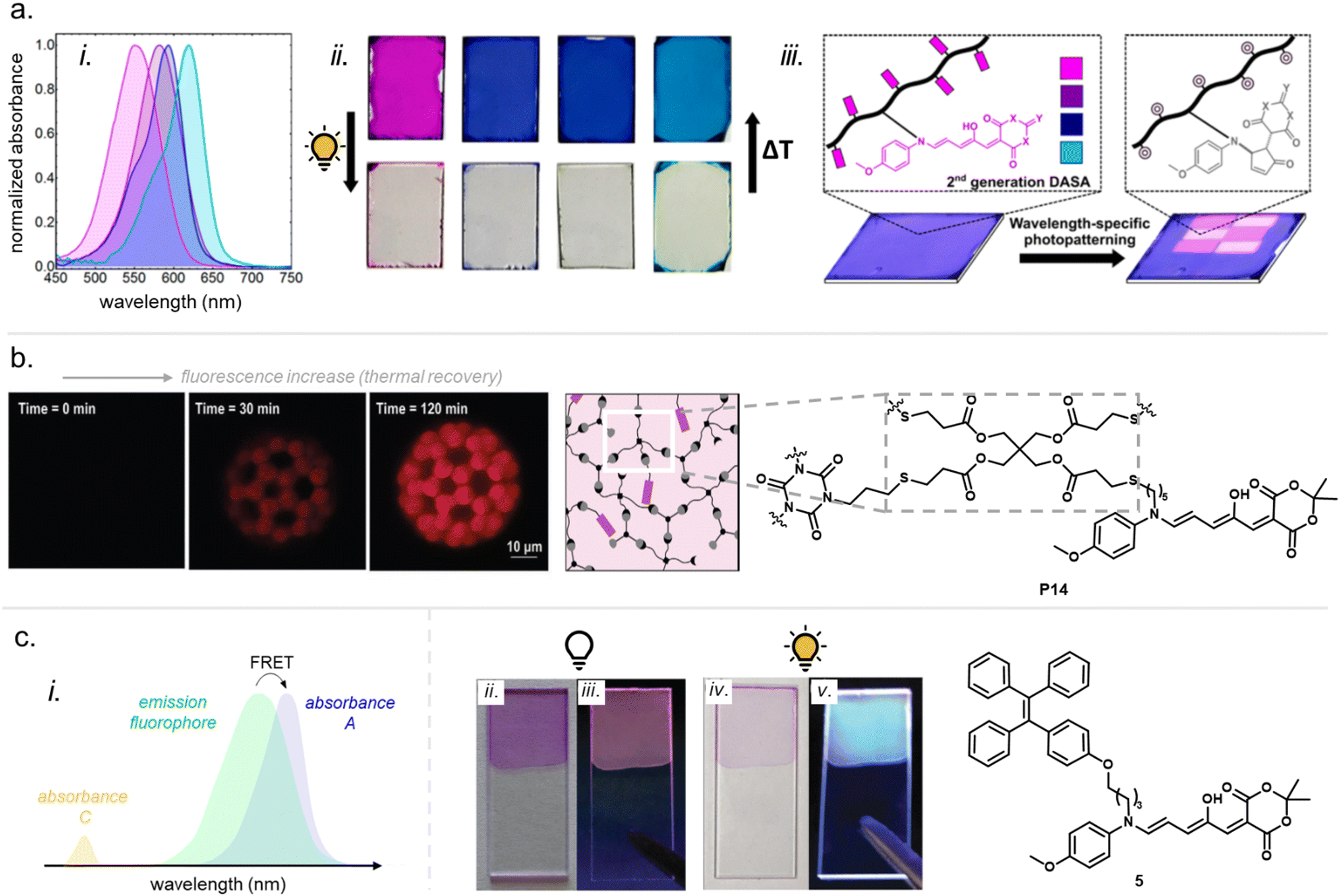 | ||
| Fig. 21 Applications of DASA–polymer materials based on changes in optical material properties due to DASA photochromism. (a) Photochromic and photopatternable polymer films made from PMA conjugates with different second-generation DASAs. (i) Normalized UV-Vis absorbance spectra of spin-coated thin films of four different DASA-PMA conjugates derived from Meldrum's acid or 1,3-dibutylbarbituric acid in combination with indoline and aniline donors (cf.P8 and P9, Fig. 11). (ii) Photographic images of the polymer films before and after white light illumination or thermal equilibration, respectively. (iii) Selective switching of a blend of two different DASA–polymer conjugates shown in (ii). A ternary pattern was achieved by using two different irradiation wavelengths (white light and orange light, 617 nm, at 40 °C) and different photomasks. The pattern can be erased through heating the sample above 70 °C. Figures adapted with permission from ref. 114 Copyright 2017, American Chemical Society. (b) Microscopic images of photochromic icosahedron-shaped microstructure obtained from nanoscribe 3D-printing via two-photon polymerization of a thiol–ene resin and subsequent DASA conjugation to the pre-formed network (P14). The microstructures showed reversible color changes upon photoswitching and the thermal recovery kinetics could be monitored by fluorescence confocal microscopy following the weak fluorescence of the open DASA isomer. Images of the microstructures at different time points during thermal recovery are shown. Figures adapted with permission from ref. 91 Copyright 2021 by the authors. (c) Switchable fluorescence from DASA photoswitching. (i) Representative schematics of UV-Vis absorbance spectra of the open DASA isomer A and the closed isomer C and the emission spectrum of a fluorophore that overlaps with the open isomer absorbance band. Förster Resonance energy transfer (FRET) from the fluorophore to the open DASA isomer is possible due to spectral overlap. An on/off fluorescence response as a result of DASA photoisomerization could be achieved by transferring energy selectively to one of the DASA isomers. (ii)–(v) Films made from spin-coating mixtures of PMMA and DASA 5, which is conjugated to a tetraphenylethylene fluorophore. Fluorescence quenching of the fluorophore by FRET was observed when the DASAs resided in the open form. Photographic images of a film before (ii) and (iii) and after (iv) and (v) visible light irradiation (532 nm, 15 min) taken under ambient light (ii) and (iv) or under 365 nm UV light (iii) and (v). The appearance of blue fluorescence (λmax = 455 nm) was observed after DASA switching (fluorescence quantum yields in the films after light ∼6%, vs. before light <0.1%). The fluorescence could be reversibly modulated by alternating irradiation and heating to 55 °C. Figure adapted with permission from ref. 137 Copyright 2018 Royal Society of Chemistry. | ||
Apart from two-dimensional surfaces, complex DASA-based photochromic micro objects were recently fabricated by laser writing via two-photon polymerization of a thiol–ene resin and a subsequent post-polymerization modification process of the networks for functionalization with DASAs.91 These three-dimensional microstructures employing second or third-generation DASAs showed reversible color changes upon photoswitching, which could be monitored by fluorescence confocal microscopy following the weak fluorescence of the open isomer (Fig. 21b). DASAs are promising candidates for the development of novel photodynamic networks.156
Photopatterning of polymer films in combination with fluorescence has also been reported for DASAs covalently conjugated to different fluorophores. For example, a third-generation DASA was conjugated to a fluorophore unit with aggregation-induced emission properties by using an indoline-tri- or tetraphenylethylene as the donor.143 These DASAs were dispersed in solid PMMA films, where they showed irreversible photoisomerization to the closed form under visible light irradiation. Interestingly, the open-to-closed transition was accompanied by a strong fluorescence increase of the fluorophore moiety when excited at 365 nm. The authors attributed this behavior to an intramolecular charge transfer between the fluorophore and the open DASA isomer that quenches its fluorescence.143 This allowed photoprinting on the polymer films with a readout by both disappearance of the DASA color and by increased fluorescence of the fluorophore moiety. Recently, PMMA films with comparable properties based on a barbituric acid-derived DASA conjugated to fluorophores containing tetraphenylethylene, phenanthroimidazole, and triphenylamine moieties, were presented.105 In addition, a Meldrum's acid-based DASA bearing a tetraphenylethylene fluorophore on its aniline donor showed reversible photoswitching in PMMA.137 In contrast to the above examples, these films could be repeatedly switched from pink/non-fluorescent to colorless/blue fluorescent upon alternating visible light irradiation and heating to 55 °C (Fig. 21d). The fluorescence quenching mechanism was attributed to FRET from the tetraphenylethylene fluorophore to the open DASA isomer.137 Surface relief gratings fabricated from this material also showed reversible on/off fluorescence as a result of DASA photoisomerization, however, no change in polymer morphology could be observed.137
In a different approach owing to the high extinction coefficient of DASAs, Robb and co-workers demonstrated the mechanically mediated synthesis of DASA–polymer conjugates both in solution under sonication and in solid state bulk polymer materials under compression and tensile forces (Fig. 22).113 This was achieved by generating a masked mechanically active hetero-Diels–Alder adduct from an activated furan that acts as mechanophore, which was located in the chain center of PMA (cf. Section 2.1.2) for experiments in solution or as a cross-linker in bulk PDMS. Under mechanical stress, a retro-[4+2] cycloaddition reaction revealed the furan adduct that could readily react with donor amines present in the medium to yield DASAs. Importantly, the approach enabled spatiotemporally controlled multicolored responses by exposing the unmasked activated furan adducts to a variety of donor amines that acted as developers by DASA formation.
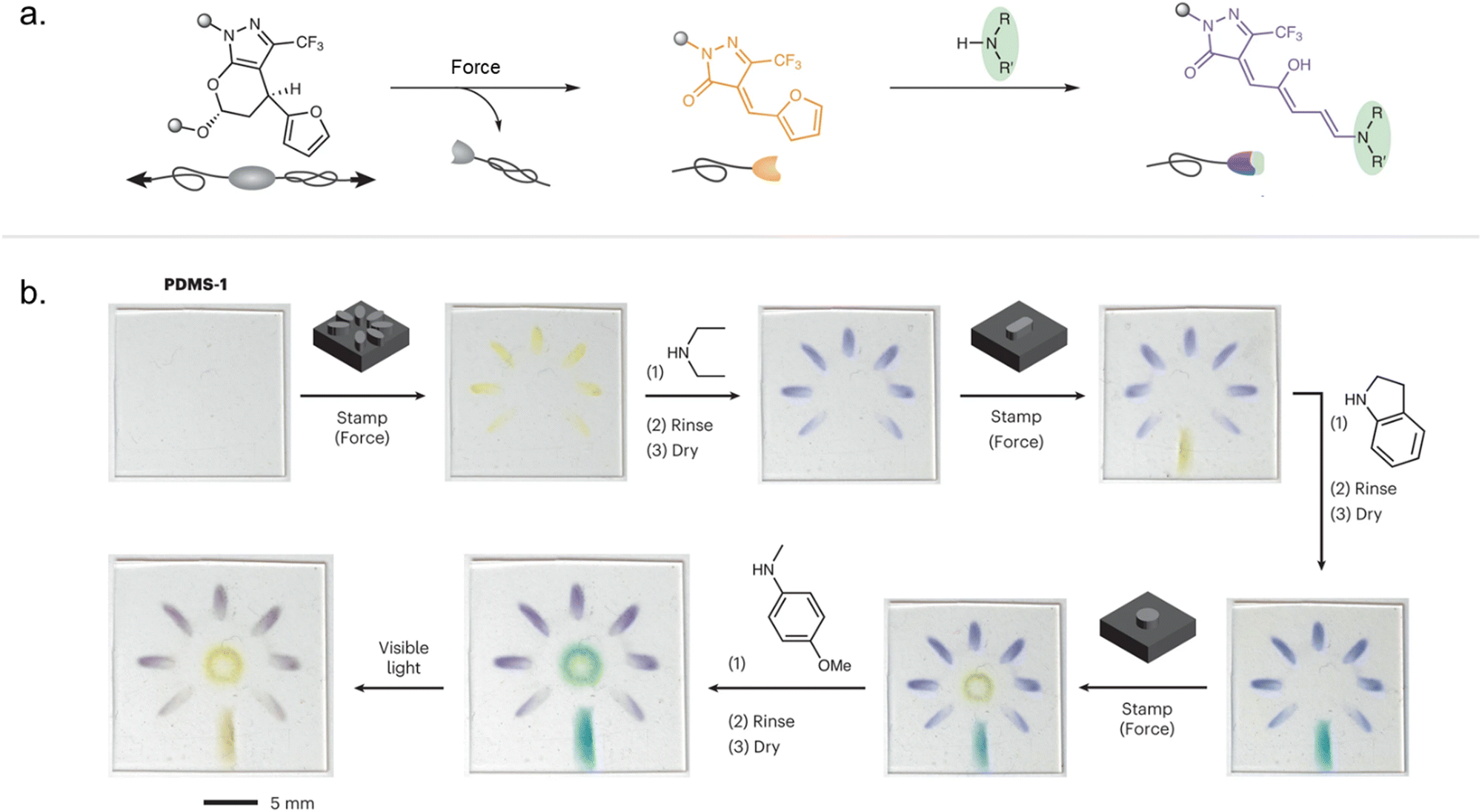 | ||
| Fig. 22 Mechanochomism based on a masked furan adduct formed from a mechanically active hetero-Diels–Alder adduct and DASA formation by reaction with amines. (a) Mechanism for mechanochromism. After a mechanically-induced retro-[4+2] cycloaddition, the resulting activated furan adduct can react with secondary amines to yield DASAs. (b) Photographic images of PDMS films modified with the masked furan adduct after force application with a stamp and exposure to amines. By selectively applying a library of secondary amines after force stimulation, multicolored mechanochemical soft lithography can be achieved. Figures adapted with permission from ref. 113. Copyright 2023 Springer Nature. | ||
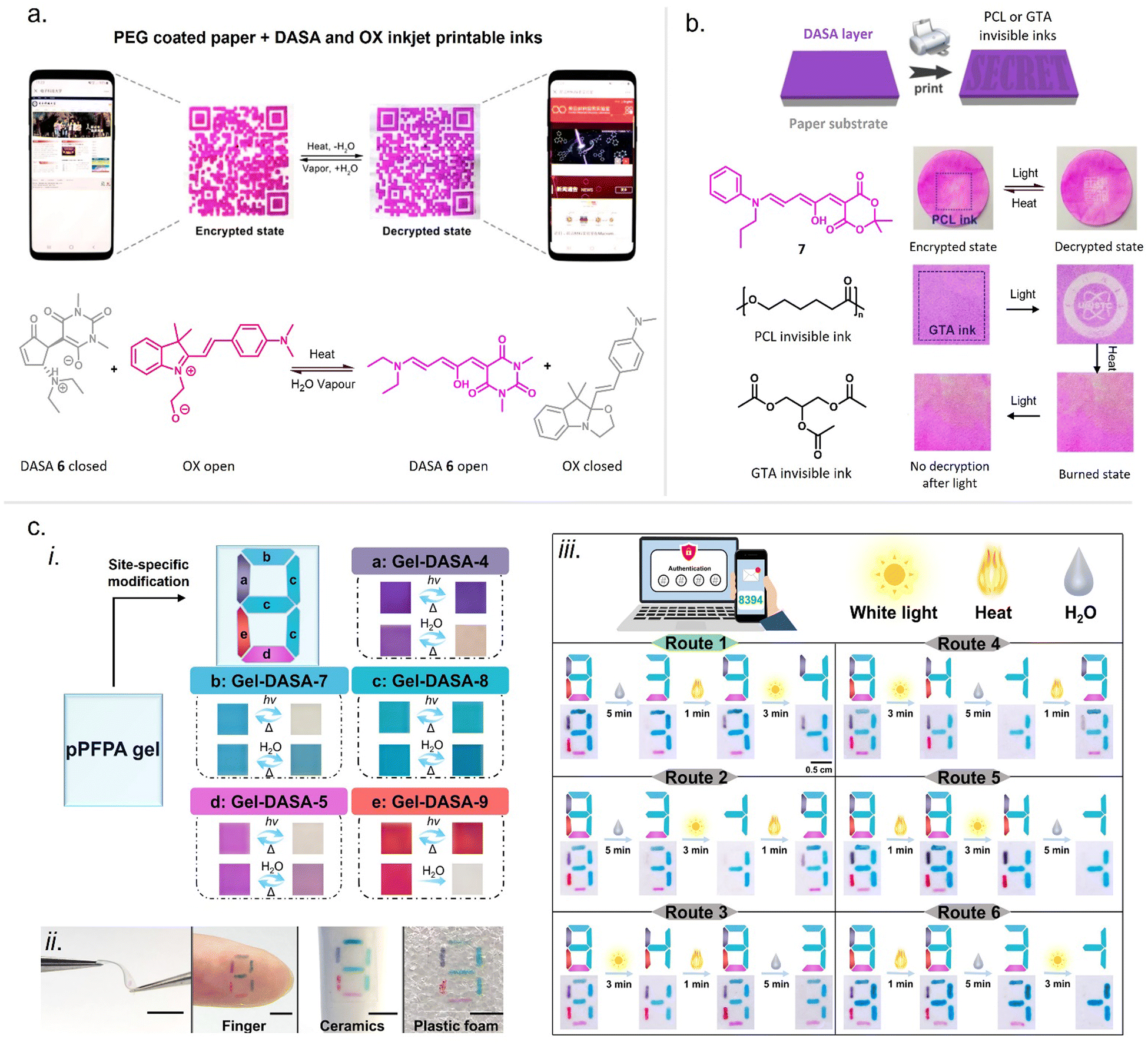 | ||
| Fig. 23 Applications of DASAs for information encryption. (a) Photographic images of the switching of printed QR codes with heat and vapor. The QR code on the left was printed with an ink made with hydrochromic oxazolidine (OX) in its open colorful state, and the QR code on the right was printed with an ink made from DASA 6 in its colorless closed form isomer. Double-encryption of the message was enabled by the opposite on/off color response of the two compounds to heat or water. Figure adapted with permission from ref. 102 Copyright 2020 American Chemical Society. (b) Secrecy system based on open form DASA coatings on paper or glass surfaces. Invisible inks made from ester-containing polymers or small molecules promote photo-induced DASA decoloration when applied to the neat DASA coatings, which reveals the encrypted message under light. Polycaprolactone (PCL) as the invisible ink provided a reversible encryption/decryption system with response to light/heat, while glyceryl triacetate (GTA) provided a “burn after reading” secrecy system due to the removal of GTA from the surface during the heating process. Figures adapted with permission from ref. 136 Copyright 2022 Elsevier. (c) Sequential logic encryption system based on pPFPA gels covalently functionalized with DASAs. (i) Schematic illustration of site-specific modification on the pPFPA gel with different DASAs to form a number 8 for the purpose of digital number encryption. (ii) Photographic images of the flexible gels with encrypted data under bending or attached to different surfaces (scale bars are 1 cm). (iii) The outputs of the data-encryption system based on the input of three different stimuli (white light, heating to 80 °C, exposure to deionized water at 25°) when applied in different order. Only the correct sequence of inputs leads to data decryption. Figures adapted with permission from ref. 119 Copyright 2022 the authors. | ||
In a more recent study, another DASA-based encryption system was reported relying on a different mechanism.136 It was found that the photoinduced discoloration of DASAs was accelerated in the presence of compounds with ester-functional groups (compare Section 2.5.2.2), which was exploited by using ester-containing polymers, such as PCL, as invisible inks on DASA coated paper surfaces. Upon irradiation with light, the DASAs in the areas in contact with PCL isomerize faster and lose their color making the encrypted message visible (Fig. 23b). In addition, using a small ester-containing molecule, glyceryl triacetate (GTA), as the invisible ink led to the same effect, but this molecule could be removed from the surface upon heating. After thermal recovery of the DASAs to the open isomer, the encrypted message was therefore “burned” and could not be revealed anymore after another cycle of illumination. Interestingly, simple scotch tape with the main component being poly(acrylic ester) also helped to promote the forward isomerization of neat DASA films under illumination on various surfaces (i.e. paper, glass, metal, wood).136
A polymer gel-based data encryption system was reported using a combination of different DASAs providing sequential logic encryption, where not only the presence but also the order of the input stimuli played a role in revealing the encrypted information (Fig. 23c).119 This was achieved by site-specific covalent modification of the gels with different DASAs that responded differently to different stimuli, i.e., light, water, and heat (see also poly(2-ethylsulfonyl-2-oxazoline) gel sensing system in Section 3.1.5). Numbers or QR codes could be “written” on the gels by exposing only specific areas of a pentafluorophenyl acrylate precursor network to the amine reagent and furan adduct for the formation of DASAs. By carefully selecting DASAs with different photochromic and hydrochromic behavior in the gels, as well as different degrees of thermal recovery after light irradiation or exposure to water, only a defined sequence of visible light illumination, immersion into water, and heat treatment at 80 °C led to the decryption of the message.119 On the other hand, a faulty sequence of input stimuli generated a false output with no reversal, which led to a loss of the encoded information. Moreover, the DASA-gels were transparent, highly flexible and could conform and adhere to a variety of substrates, including glass, ceramics and plastic, making them attractive for use as smart labels.
| Type of sensors | Material/reference | Probing mechanism | Sensitivity/limit of detection (response time, conditions) |
|---|---|---|---|
| a 3-Dimensional red-green-blue color space value. b Color distance metric values. c Diethyl cyanophosphate. | |||
| Amines | Poly(oxa-norbornene)s with grafted Meldrum's acid furan adduct117 | Turn-on coloration as a result of DASA formation | Diethylamine: 20 ppm |
| n-Butylamine: 10 ppm | |||
| Indoline: 20 ppm | |||
| p-Methoxyaniline: 100 ppm (3 min at 75 °C in aqueous solutions; λmax ≥ 0.05 a.u.) | |||
| Electrospun PCL fibers loaded with furan adducts151 | Turn-on coloration as a result of DASA formation | Diethylamine: 10 ppb (for a CF3-pyrazolone furan adduct; gas phase, exposure time of 30 min; ΔRGBa = 74); dynamic range for different furan adducts: CF3-pyrazolone 10 ppb – 0.1 ppm; Meldrum's acid furan adduct 40 ppb – 1 ppm; N-methyl barbituric acid furan adduct 1 ppm – 10 ppm | |
| Meldrum's acid furan adduct mixed into poly(2-ethylsulfonyl-2-oxazoline) gels128 | Turn-on coloration as a result of DASA formation | Dimethylamine: 245 ppm (gas phase, exposure time of 3 h, ΔE*b = 50) |
|
| Temperature | Poly(oxa-norbornene)s with grafted second-gen. DASA117 | Turn-on coloration from closed-to-open DASA isomerization | ΔT ≈ 5 °C around Tg of the polymer (heating thin films from 50–100 °C; ΔEb = 20) |
| Urethane-cross-linked polybutadiene with mixed in first-gen. DASA90 | Turn-on coloration from closed-to-open DASA isomerization | n.d. | |
| Nerve agent mimics | First-gen. DASA bound to poly(dimethylacrylamide)124 | Turn-off coloration as a result of reaction with open isomer | DCNPc: 1.0 mM (in dioxane solution) |
| Fe3+/Cu2+ ions | PEI polymer dot nanoparticles with covalently attached first-gen. DASAs121 | Turn-off fluorescence signal of polymer dot | Fe3+: 10.1 nM (in water) |
| Cu2+: 1.3 nM (in water) | |||
| Humidity | First- and second-gen. DASA bound to poly(2-ethylsulfonyl-2-oxazoline) gels128 | Turn-off coloration due to water vapor induced DASA cyclization | n.d. |
| Light | First- and second-gen. DASA bound to poly(2-ethylsulfonyl-2-oxazoline) gels128 | Turn-off coloration due to DASA photochromism | n.d. |
Among the latter, poly(oxa-norbornene)s with Meldrum's acid furan adducts grafted to their sidechains were reported as both amine and thermal sensors.117 Unlike low molecular weight furan adduct sensors, this platform allowed the detection of amines in water because the DASAs are protected by the hydrophobic polymer matrix and remain in their colored form. For the amine sensing, the polymer films were dipped for 3 min into aqueous amine solutions at 75 °C (see Table 1 for the limit of detection (LOD) of different amines).117 For the temperature sensing, the furan adducts on the polymer were converted to second-generation DASAs. Spin coated thin films of the DASA–polymer were then photoswitched with light to convert the DASAs to their closed isomers. Due to the high Tg of the polymers (≥65 °C), the DASAs stayed in their closed state when the films were kept at low temperatures (0 °C). Increasing the temperature then led to thermal recovery of the colored, open isomer (cf. Section 2.5.1.1), which can be used as a sensor response for temperature monitoring. The films were heated from 50 to 100 °C, whereby the required temperature to induce visually detectable coloration largely depended on the Tg of the polymer. For example, polymers with Tg values of 90 °C or 65 °C required temperatures of 85 °C and 70 °C, respectively, to reach a color response (color distance metric values ΔE* = 20 were used as a benchmark). Thus, relatively small differences in temperature were distinguishable by the naked eye (≥5 °C).
The incorporation of furan adducts into electrospun polymer meshes made from PCL was reported for the detection of volatile amines.151 The dissolution of furan adducts without conjugation into PCL was studied, as well as meshes made from blends of PCL and the aforementioned poly(oxa-norbornene) with pendant Meldrum's acid furan adduct. For the later approach, cytocompatibility in both indirect and direct contact with the meshes could be achieved by preventing leaching of the furan adducts through covalent attachment.151 Additionally, by changing the type of furan adduct from Meldrum's acid to a highly reactive CF3-substituted pyrazolone a LOD as low as 10 ppb for diethylamine in the gas phase could be achieved in the meshes based on non-covalent association.151 As the other amine sensor described above, this sensor is also nonreversible and only allows for single-use or cumulative detection.
Another thermally activated “turn-on” sensor was developed by embedding a first-generation DASA in a urethane-cross-linked polybutadiene to detect micron-scale localized temperature increases after impact or rapid heating.90 The obtained elastomer contained the DASAs in their closed colorless form, but temperature increases led to thermal reversion to the colored open isomer. The application of this sensor platform was demonstrated by determining the peak temperatures reached during bullet perforation of the material utilizing the strong coloration and weak fluorescence of the open DASA isomer for detection and quantification.
Recently, a DASA-based colorimetric sensing platform for amine detection, humidity, and light irradiation based on poly(2-ethylsulfonyl-2-oxazoline) gels was presented.128 For this approach, DASAs were covalently attached to the polymer, while for the amine sensing Meldrum's acid furan adduct was mixed into the gels. By choosing either a first, second or a non-photochromic (derived from diethylamine and indandione34) DASA, it was shown that orthogonal responses can be achieved to light irradiation, water, and heat. For example, due to the relative insensitivity of second-generation DASAs to their environment (Section 1.1.1.6), this DASA responds to light irradiation, but not to water when immobilized inside the gels, while the opposite behavior was observed for the non-photochromic derivative. In the gels the thermal recovery to the open isomer was also limited, which allows the sensors to maintain their responses after removal of the stimuli.
On the other hand, the gels containing the furan adducts were used to detect accumulated levels of fish degradation through increasing levels of coloration as a result of reaction with volatile amines that are released upon spoilage.128 In calibration measurements with known amine concentrations, color distance metric (ΔE*) values around 50 could be achieved with dimethylamine at concentrations as low as 245 ppm, which are already well distinguishable by the naked eye.128
Chemosensing for compounds other than amines was also demonstrated in two examples. A first-generation DASA bound to poly(dimethylacrylamide) was used for the “turn-off” colorimetric detection of nerve agent mimics (diethyl cyanophosphate, DCNP) in both solution and in the vapor phase using polymer films.124 The DASA thereby changed from colored to colorless upon reaction with DCNP, possibly by a DCNP-promoted intramolecular N-alkylation of the donor in the open isomer.124 However, a turn-off response may limit the selectivity and therefore the practical use of such a sensing platform, since DASAs also become colorless as a result of isomerization, either by photochromism or as a result of external factors, such as humidity. In another example, DASA-functionalized PEI-based fluorescent polymer dot nanoparticles were reported to respond to the presence of metal ions and pH changes and were used as fluorescence-based chemosensor for the detection of Fe3+ and Cu2+ ions in aqueous solutions.121 In these solutions the DASAs were present in their closed state in this system and the origin and variation of the fluorescence signal were likely associated with aggregation changes of the fluorescent polymer dot particles.121 However, in ethanol the particles underwent a substantial size decrease as a result of both light irradiation and changes in pH probably as a result from dissociation of the PEI aggregates that form the particles.121 Although the authors stressed the application of this system for chemosensing, the observed size changes could also be relevant for stimuli-induced substance release (Section 3.3).
3.2 Changes in surface chemistry and interfacial properties
The wettability switching of different surfaces through DASA photoisomerization has also been reported. For example, a third-generation DASA was mixed into PS films to control the adhesion of the films to glass surfaces with light (Fig. 24a).135 It was found that after light exposure, the shear and pull-off adhesion of the polymer to the glass is weakened leading to a large change in the detachment rate of the films from glass in aqueous solutions. This phenomenon was harnessed to enable the photocontrolled release of molecules from a closed glass container held together by the DASA-PS films into the surrounding water. The authors suggested that the physical origin of these effects is likely due to molecular-level noncovalent interactions between the open DASA and the silanol groups on the glass surface, rather than a change in morphology or mechanics of the polymer host.135 The importance of surface-specific chemical interactions is suggested, for example, by the sensitivity of the surface water contact angle to pH (see also Section 2.3.1), as well as the sensitivity of the photoinduced deadhesion to the glass cleaning procedure. In addition, the DASA PS mixture was photoswitched in solution and then cast into films. It showed the same rapid detachment in water as the irradiated solid films. Using DASAs to modulate solid–solid adhesion through changes in noncovalent forces between surfaces is an innovative approach that could find other applications in, for example, solvent-free methods for removal of protective coatings and smart adhesives. However, this first example showcased that samples at the required DASA concentrations leading to measurable changes in adhesion (>1 wt%), required prolonged light exposure (10–15 h) for complete disappearance of the open isomer. In addition, the process was not reversible (cf. matrix mechanics in Section 2.5.1.1 and concentration effects in Section 2.5.3).135 These disadvantages currently render the concept less viable and requires optimization in the future.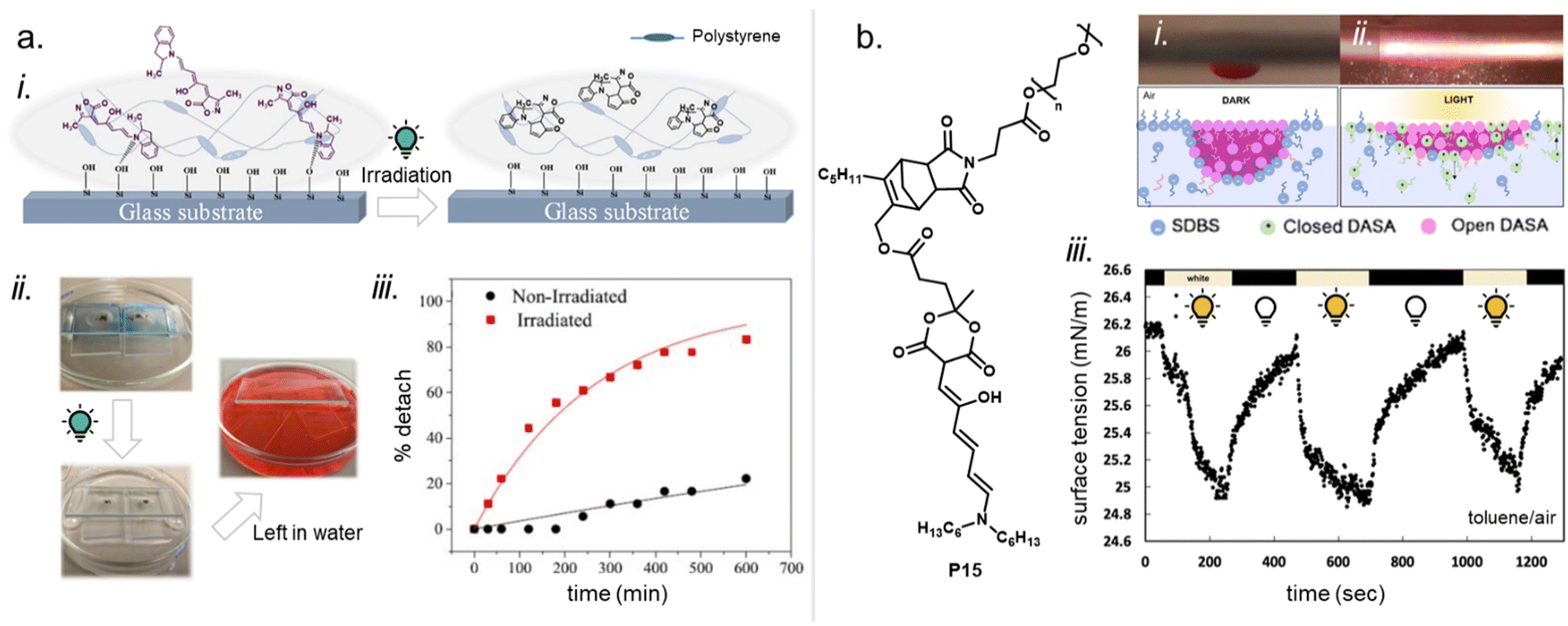 | ||
| Fig. 24 DASAs applied for photomodulation of surface chemistry and interfacial properties. (a) Light induced deadhesion of a polystyrene (PS) film doped with third-generation DASAs from a glass surface. (i) Illustration of the proposed mechanism for the deadhesion. Photoisomerization leads to ring-closure and molecular contraction that interferes with the amine–silanol interaction of the open isomer and the glass surface. (ii) Release of a dye powder (Allura Red) encapsulated between two glass slides held together by the DASA-modified PS film (8 wt% DASA). Photographs of the sample before and after irradiation (532 nm, 6 mW cm−2) and after deadhesion and the release of the dye into the surrounding water. Under irradiation, the assembly separated after 5 h, while in the dark it remained intact >24 h. (iii) The fraction of drop-cast PS films (1 wt% DASA) detached from glass overtime when placed in water before or after irradiation for 10–15 h (532 nm). After irradiation, more than 50% of films detached after 3 h, whereas no detachment occurred for the films with no irradiation during this time. Figures adapted with permission from ref. 135 Copyright 2019 American Chemical Society. (b) PEG-derived DASA photosurfactants led to photoinduced changes in wetting and interfacial properties at toluene–water–air interfaces of macroscopic droplets in the presence of an ionic co-surfactant, such as sodium dodecyl benzenesulfonate (SDBS). Photographic images and schematic of the droplet wetting experiment (10 mM P15 in toluene wetting to the water−SDBS interface) before (i) and after (ii) irradiation (214 mW cm−2 white light source for 5 seconds). (iii) Reversible surface tension change of 20 mM photosurfactant P3 (cf.Fig. 11) in toluene measured against air with cycled irradiation (white light source). Figures adapted with permission from ref. 115 Copyright 2021 American Chemical Society. | ||
The wetting behavior of cellulose-derived nanoparticle surface coatings could be controlled with light through functionalization of the nanoparticles with first-generation DASAs.140 The DASAs were coated onto hydroxypropyl cellulose stearoyl ester nanoparticles by hydrophobic non-covalent interactions and the particles were spray-coated onto a porous paper substrate. This yielded a surface that is initially superhydrophobic (water contact angle of >150°) and becomes hydrophilic when switched with visible light causing complete penetration of aqueous droplets into the porous substrate (water contact angle of 0°). This can be interesting for controlling water flow on surfaces and paper-based fluid timers,140 for example, in diagnostic applications or lab-on-a-chip devices. However, no reversibility to the open isomer was observed after light irradiation.
In another example, silica micro- and nanoparticles were coated with polydopamine, which was then reacted with a barbituric acid-based furan adduct.122 The particles were then immobilized onto glass surfaces with the help of a thin layer of cross-linked PDMS. However, polydopamine presents primary amines instead of secondary amine functional groups, which usually do not allow for the synthesis of stable DASAs.31 Nonetheless, a large green-light initiated change toward hydrophilic contact angles was reported (from ∼140° towards ∼100°).122 Heating also switched the surface back to more hydrophobic contact angles. However, low absorbance maxima values at λmax ≈ 550 nm (<0.5) in the UV Vis absorption spectra of the samples, as well as their brown coloration (before irradiation),122 indicated the presence of only small amounts of an open DASA isomer on the surface. Therefore, further studies would be helpful to better understand these materials, for example, on the new dopamine-based DASA derivative and its properties.
Cross-linked PS-based microparticles decorated with third-generation DASAs exhibited swelling behavior after white light irradiation both in toluene and aqueous suspensions.127 The swelling behavior in toluene was associated with the molecular contraction of the DASA closed isomer, offering more free volume for solvent penetration into the polymer matrix. In contrast, the swelling behavior in water was associated to the higher hydrophilicity of the closed isomer compared to the open isomer. The microparticles displayed higher dispensability in water after light irradiation. This property was further exploited by suspending the microparticles in heterogeneous mixtures of chloroform and water showing that after pre-irradiation in chloroform followed by vigorous mixing, stable transitioning to the aqueous phase was favored.127 This property could find applications in heterogenous polymer supports for catalysis.
In addition, two different PEG-derived first-generation DASA photosurfactants (P15, Fig. 24b and P3, Fig. 11) were recently analyzed for their influence on changes in wetting and interfacial properties at toluene–water–air interfaces of macroscopic droplets.115 Specifically, the photoisomerization of DASA from its hydrophobic open form to its zwitterionic hydrophilic closed form resulted in the loss of surfactant amphiphilicity, lowering the interfacial surface tension at the toluene–water interface and inducing droplet wetting. At concentrations of DASA ranging from 1 to 20 mM, these droplets spread with wetted diameter increasing by ∼150–200% upon irradiation. The response was reversible and repeatable with the removal of light, leading to an immediate dewetting and return to the original diameter. In addition, the use of an anionic non-switchable co-surfactant (sodium dodecyl benzenesulfonate, SDBS) was found to amplify the photo-induced wetting effect, increasing the change in droplet spreading by up to 3 times the change observed in the absence of SDBS.115 Such an efficient remote control of the properties and motion of fluid interfaces represents many potential applications in material science and chemical engineering, allowing control over droplet or particle motions on solid and fluid surfaces, as well as emulsion and micelle aggregation or phase separation.
3.3 Release from polymeric carriers and nanoreactor applications
DASAs are particularly promising as tools for biomedical research because they absorb visible light that is harmless to organisms. As result of the large structural and chemical differences between the open and closed isomer, DASAs have proved to be useful for the light-mediated control of substance release from polymeric carries in the context of drug delivery applications. Various micro-and nanoparticular systems have been developed for this purpose, including self-assembled micelles and vesicles from polymer-derived surfactants or amphiphilic BCPs, solid core particles, micro- and nanogels, as well as polymer coatings on nanoparticle substrates (Table 3). Besides carriers for targeted release, applications as light-controlled polymeric nanoreactors containing enzymes were also reported. These systems will be discussed in the following sections.| Morphology, reference | Material | Size [nm],a Zeta potential (ζ) [mV] | DASA gen. | Cargo | Cargo loading content [wt%], encapsulation efficiency [wt%] | Photoinduced release/uptake mechanism | Reversible switching in water |
|---|---|---|---|---|---|---|---|
| a Hydrodynamic diameter determined by dynamic light scattering (DLS). b Poly(ethylene glycol-methyl ether methacrylate). c Upconversion nanoparticles. d Doxorubicin. e Poly(hexyl acrylate). f Poly(butyl acrylate). g 1,2-Dilauroyl-sn-glycero-3-phosphocholine. h Camptothecin. i The authors stressed the application of these particles for chemosensing (see Section 3.1.3). j Measured in ethanol. k Suggested for use as heterogeneous polymer supports for catalysis (cf. Section 3.2). l Determined by scanning electron microscopy (SEM). m Polyphenylmethacrylate. | |||||||
| Micelles116 | PEG | 22, n.d. | 1st | Nile red, paclitaxel | 0.17, 99 | Micelle disassembly | No |
| Micelles133 | BCP of poly(PEGMEMAb) and DASA block | 60, −14.7 | 1st | Ellipticine | 4.8/7.4, 92 | Micelle disassembly | Yes |
| Micelles loaded with UCNPs134c |
PEG | 138, n.d. | 1st | DOXd, nitroglycerin | 1.25 (DOXd), n.d. | Micelle disassembly | No |
| Micelles loaded with UCNPs132c |
PEG-b-PHAe | 160–185, n.d. | 1st | Nile red | n.d. | Slight swelling, permeability change | n.d. |
| Polymersomes129 | PEG-b-PHMA | 122 (for 3rd gen.), n.d. | 2nd/3rd | Sodium fluorescein, enzymes | n.d., 0.2–7.3 (enzymes) | Permeability change | Yes |
| Polymersomes131 | PEG-b-PBAf | 207, n.d. | 2nd | Enzymes | n.d, 7.5 | Permeability change | Yes |
| Liposomes142 | DLPCg + lauric acid + Pluronic F127 | 145, n.d. | 1st | Fluorescein isothiocyanate–dextran | n.d., 20 | Liposome-to-cubosome phase transition | Yes |
| Solid-core NPs92 | Poly(p-phenylene vinylene) | 80, −7.8 | 1st | DOXd, CPTh, Cy5 | n.d., 27.3 (CPTh)/34.2 (DOXd)/35.6 (Cy5) | Swelling, opening of structures | No |
| Solid-core NPs121i |
PEI | 200 (water)/342 (EtOH), n.d. | 1st | n.a. | n.a. | Aggregate dissociationj | No |
| Micro-/nanogels125 | Poly(N-vinylcaprolactam) | 360–680, n.d. | 1st | Fluorescein isothiocyanate | n.d. | Swelling, opening of structures | No |
| Cross-linked microparticles127k | PS | 4000–5000,l n.d. | 3rd | n.a. | n.a. | Swelling and increased dispersibility | No |
| Polymer layer on silica or enzyme covered UCNPs130c |
PEG-b-PPhMAm | 200–280, −10 | 2nd | D-Luciferin, SYTO™ nucleic acid stain | n.d. | Permeability change | Yes |
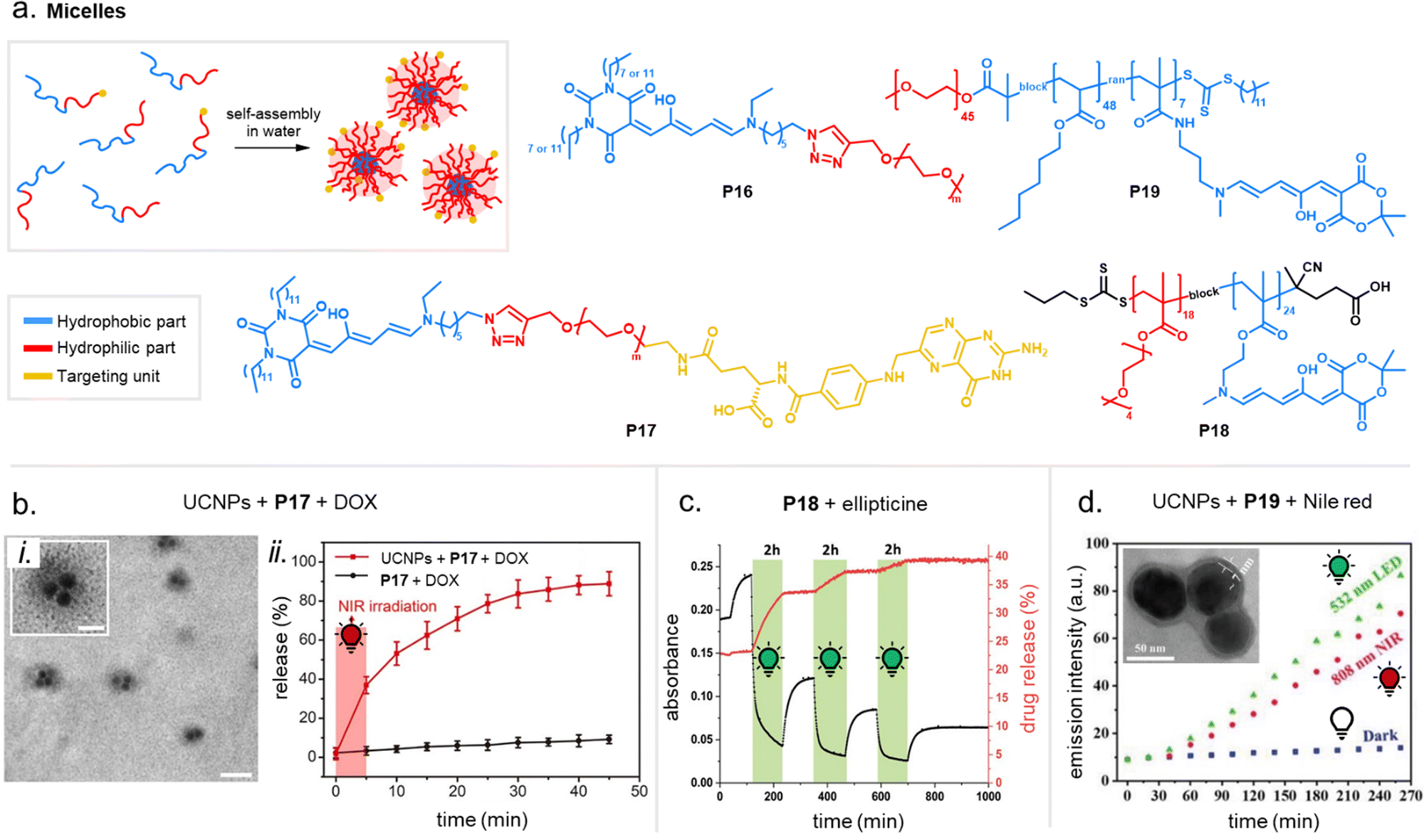 | ||
| Fig. 25 Light-responsive micelles containing DASAs. (a) Schematic illustration of micellization process from amphiphilic polymeric compounds and structures of DASA-modified polymers used for micelle formation. (b) Micelles of P17 assembled with upconversion nanoparticles (UCNPs) that are responsive to NIR light. (i) Transmission electron microscopy (TEM) image of micelles loaded with an average of 2-4 UCNPs with the scale bar of 150 nm (inset: 75 nm). (ii) Release of doxorubicin (DOX) from micelles with and without UCNPs after 5 min NIR irradiation (808 nm laser, 2 W cm−2). Release was quantified by monitoring the fluorescence of DOX in the supernatants overtime. Figures adapted with permission from ref. 134 Copyright 2021 Elsevier. (c) Ellipticine release from micelles made from P18 in water at 37 °C. UV-Vis absorption of DASA-modified micelles (518 nm, black trace) and ellipticine drug release (red trace) for 3 irradiation cycles with a 530 nm LED (2.15 W intensity; 2 h ON, 2 h OFF). The release from the micelles was quantified by monitoring the polarity-sensitive fluorescence of ellipticine in situ. Figure adapted with permission from ref. 133 Copyright 2019 Royal Society of Chemistry. (d) Nile red release from micelles of P19 assembled with UCNPs that are responsive to NIR light. The release was monitored via a dialysis method and the graph displays the increase in Nile red fluorescence intensity (at 640 nm, λex = 550 nm) in the receiver compartment over time for micelles under light exposure (0.2 W 808 nm laser, 6.37 W cm−2, or 532 nm LED, 12.8 mW cm−2) and in the dark. The inset shows a TEM image of a micelle after NIR irradiation (three UCNPs covered by the polymer layer). Figures adapted with permission from ref. 132 Copyright 2021 John Wiley and Sons. | ||
Upon irradiation, the formation of the closed and less hydrophobic DASAs lead to micelle dissociation and a dissolution of the PEG in water, which is accompanied by the release of encapsulated hydrophobic compounds (Nile red dye, paclitaxel). Moreover, the chemotherapeutic agent paclitaxel was also successfully delivered to MCF-7 human breast cancer cells.116 In this system, the photoswitching of the DASAs was not reversible and the open form could not be recovered after light irradiation. This is due to the direct exposure of the DASAs to water after the micelle disassembly, which trapped them as the closed isomer.75 As such, this system did not make use of the reversibility of DASA photoswitching and the irreversible manipulation was more similar in behavior and outcome to photolabile protecting groups (uncaging).165
Micelles were also prepared from similar PEG-DASA conjugate P17 in combination with upconversion nanoparticles (UCNPs) and a targeting unit (folic acid) at the other end of the PEG chain (5000 g mol−1).134 This system presents the advantage that it can be activated by NIR light at 808 nm, which leads to visible-light emission from the Er3+/Yb3+ codoped UCNPs. The UCNPs were loaded inside the micellar cores (micelles of ∼120 nm diameter with 2–4 UCNPs). The emitted light could then trigger photoswitching of the DASAs and lead to a fast micelle disassembly and drug release. The NIR-triggered release of the anticancer drug doxorubicin (DOX) and the generation of nitric oxide from encapsulated nitroglycerin was studied in vitro, intracellularly and in a mouse model. Only five minutes of NIR irradiation (808 nm laser, 2 W cm−2) led to almost complete DOX release (84%) within 30 minutes (Fig. 25b).134 Two other PEG-derived DASA photosurfactants have been recently reported for the modulation of toluene–water–air interfaces of macroscopic droplets (refer to Section 3.2).
DASA-functionalized BCPs were synthesized by RAFT polymerization employing a poly(PEGMEMA) hydrophilic block and a block made from methacrylate-derived monomers with first-generation DASAs pendant groups (P18, Fig. 25a).133 In contrast to aforementioned polymers with terminal substitution, this system consisted of a polymer block with 100% DASA sidechains. Besides this, the DASA derivative used here bears a methyl instead of ethyl substituent on the amine donor, which was previously shown to strongly influence the photoswitching properties and solvent compatibility as a result of reduced steric hindrance (see Section 2.5.2.1).35,41 The self-assembled micelles could be loaded with the hydrophobic anticancer drug ellipticine, which could then be released upon visible light irradiation (LED, 530 nm). Interestingly, this system showed reversibility of the DASA photoswitching, although the recovery to the open form was rather limited already after the first irradiation cycle.133 Upon repeated cycles of irradiation and darkness, the release could be turned on and off, but most of the encapsulated drug was released within the first cycle of the irradiation (Fig. 25c). The release also ceased immediately after removal of the light stimulus. After three on/off cycles (3 times 2 h of light irradiation) at 37 °C in water or PBS the micelles lost their structural integrity. However, in biological media, the micelles were also not particularly stable even in the non-irradiated state. As a result, in cell viability studies on MCF-7 breast cancer cells the difference in measured IC50 values between the micelles in the dark and under irradiation was not statistically significant.133 In contrast to the previously described examples, it is nevertheless interesting to note that some degree of reversibility of DASA switching was observed in this system, suggesting that the structural integrity of the particles is at least partially preserved after stimulation with light. It is likely that this is a result of the higher kinetic and thermodynamic stability of BCP self-assemblies vs. assemblies from other photosurfactants. Moreover, in an earlier detailed study by the same group, micelles made from a very similar amphiphilic BCP with slightly different balance of the hydrophilic block and the DASA block showed negligible changes in size or structure after the same irradiation time.100 However, no release experiments were reported.
Micelles made from a different DASA-functionalized amphiphilic BCP, P19, were also combined with Er3+/Yb3+ codoped UCNPs allowing activation with NIR light.132 Similar to the aforementioned example involving UCNP composites,134 808 nm NIR light could be used to trigger DASA photoswitching by leveraging absorption–emission spectral matching of the DASA and the UCNP emission. The nanoparticles here are not “standard micelles”, instead they consist of non-spherical aggregates of an average of three UCNPs (diameter ∼50 nm) covered with a thin layer of BCP (∼5 nm).132 The BCP design consisted of a PEG hydrophilic block and a hydrophobic block made from a random copolymer of methacrylate-derived DASA monomers (13 mol%) and hexyl acrylate. Here, a first-generation DASA with a methyl substituent was used, as described above.133 In this case, the nanoparticles could release a hydrophobic model drug (Nile red) with low-power 808 nm excitation (0.2 W laser, 6.37 W cm−2) (Fig. 25d). The photothermal effect caused by the 0.2 W 808 nm irradiation to the aqueous particle dispersions was found to be negligible. Interestingly, no dissociation of the structures, but only a slight swelling after irradiation was observed in this case (∼11 nm increase in hydrodynamic diameter for a particle with an initial diameter of ∼185 nm). Unfortunately, these micelles also showed fast and considerable open-to-closed isomerization overtime in the dark, which might be due to instability of the micelle and to water penetration into the structures.
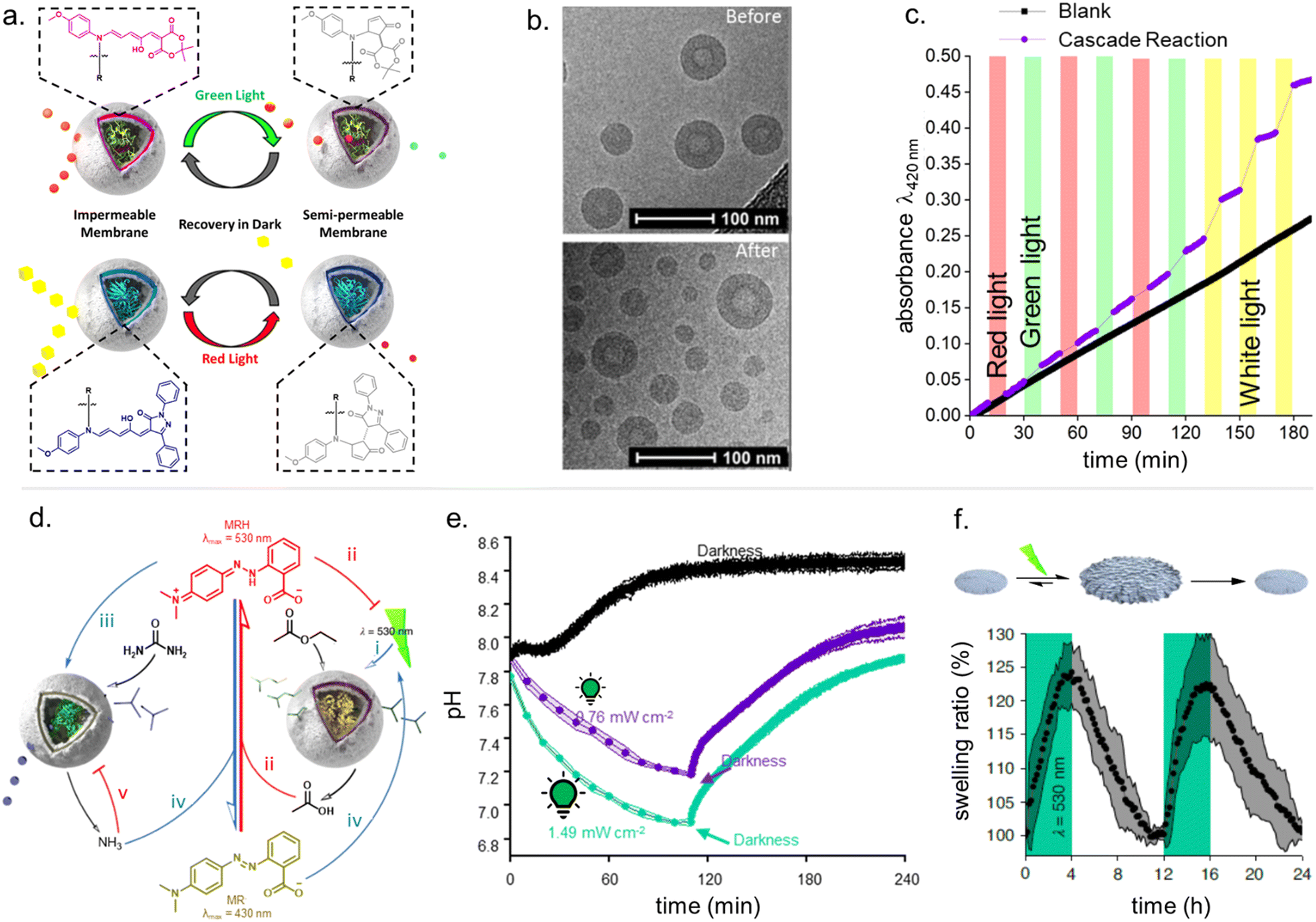 | ||
| Fig. 26 Light-responsive enzyme nanoreactors based on DASA-functionalized polymersomes. (a) Schematics of photo-induced polymersome permeabilization and structural formulae of the employed DASA. In darkness, the DASA equilibrium is shifted to the open isomer while in presence of green (MELD, top) or red (PYRA, bottom) light the equilibrium is shifted toward the closed isomer. This process generates a change in polymersome membrane permeability that enables the diffusion of water-soluble compounds through the membrane. In darkness, the process is reverted. The polymersomes were filled with enzymes to obtain nanoreactors: MELD was filled with horseradish peroxidase (HRP) and PYRA was filled with glucose oxidase (GOx). (b) Cryo-TEM images of polymersomes before and after light irradiation showing no change in morphology. (c) Catalysis of a cascade reaction mediated by alternating green and red LED light. Red light stimulates PYRA-GOx to produce hydrogen peroxide from glucose, while green light stimulates MELD-HRP to transform hydrogen peroxide and pyrogallol into the colored purpurogallin (absorbance at λ420nm). (d) Schematics of green light-induced oscillation of pH mediated by communicating binary populations of DASA-esterase polymersome nanoreactors and light-insensitive permeable polymersome nanoreactors filled with urease. (i) Green light induces the permeation of ethyl acetate into DASA-esterase nanoreactors and allows for the production of acetic acid. (ii) The accumulation of acid protonates the pH pigment methyl red (MRH) that acts as a photomask for the absorption of green light by DASA-esterase polymersomes inhibiting further acetic acid synthesis in presence of light through a negative feedback loop. (iii) the accumulation of acidic MRH activates the urease-filled permeable nanoreactor to produce ammonia from urea. (iv) the basic environment deprotonates MRH to produce MR− which simultaneously permits the penetration of further green light. (v) the accumulation of ammonia generates a basic environment that inhibits further catalysis by the urease-filled nanoreactor. (e) Monitoring of pH evolution over time of a mixture of DASA-esterase and urease nanoreactors at different light intensities and after withdrawal of light irradiation in the presence of ethyl acetate and urea. (f) Reversible swelling and deswelling of an acid responsive hydrogel in presence of DASA-esterase and urease polymersome nanoreactors and enzyme substrates after light irradiation followed by darkness. Figures adapted with permission from ref. 129 and 131 Copyrights 2018 American Chemical Society and 2023 Springer Nature. | ||
This concept was recently expanded by coupling the photoresponsiveness of DASA polymersomes to fluctuations of pH (Fig. 26d).131 The polymersomes were composed of PEG-block-poly(butyl acrylate) with randomly distributed residues of a Meldrum's acid-based second-generation DASA in the hydrophobic block. By encapsulation of an esterase enzyme, catalytic formation of acetic acid from ethyl acetate was possible in presence of green light (530 nm). This chemistry was exploited to generate fluctuations of pH inspired by circadian rhythm processes that generate controlled non-equilibrium fluctuations of metabolites in response to different light intensities. Thus, the formation of acid by esterase-loaded DASA polymersomes were employed to activate a second population of light-insensitive permeable polymersomes that contained the antagonistic enzyme urease. The urease polymersomes generated ammonia in presence of urea in acidic conditions. Moreover, the formation of acid induced the protonation of a pH pigment, methyl red, that overlapped in absorbance (λmax = 530 nm) with the DASA. The accumulation of the protonated pigment acted as photomask competing for the light absorption with the DASA while decreasing its ability to photoisomerize to the closed form and change permeability of the polymersome membrane (Fig. 26e). This process gradually interrupted the catalytic formation of acid and was only restored when the photomask was deprotonated by the base-forming urease nanoreactors, enabling the open DASA isomer to absorb green light again.131 Overall, the system imitated biological communication processes by encompassing three delayed oscillatory processes: (i) oscillation of closed DASA formation on the polymersome membranes, (ii) oscillation of medium pH, (iii) oscillation of the swelling ratio of a pH-sensitive hydrogel that was present in the reaction medium (Fig. 26f).
Although not strictly derived from polymers, Boyd and co-workers recently investigated vesicles made from lipids and other self-assembled lipid-based liquid crystalline systems functionalized with DASA photoswitches.142,167 These liposomes were made from a mixture of lipids (1,2-dilauroyl-sn-glycero-3-phosphocholine, DLPC, and lauric acid) doped with small molecule DASA (first-generation), and the BCP “Pluronic F127“ as stabilizer. The DASAs contained octyl-substituents on the aliphatic amine donor, likely to promote solubilization and anchoring in the hydrophobic membrane. This is the only example where the DASA were physically mixed and not covalently bonded to the nanocarrier. Unlike most other materials used to fabricate DASA-functionalized nanocarriers, DLPC and pluronic polymers are well-established compounds for use in pharmaceutical products. It was shown via time-resolved small-angle X-ray scattering (SAXS) that these nanocarries undergo a rapid and fully reversible order–order phase transition from liposomes to cubosomes under green light irradiation, with the impact on the phase behavior being dependent on the concentration of DASAs.142 The thermal reversion of the DASAs in this system was rather limited. Nevertheless, “on/off” control (three cycles) over the release kinetics of the encapsulated model macromolecular cargo (fluorescein isothiocyanate–dextran, 3–5 kDa) was achieved, while also a three-to-four-fold amount of cargo release was observed under irradiation for one hour as compared to dark conditions. DLS analysis of the structures before and after irradiation showed a small change in hydrodynamic diameter (with 145 nm prior and 153 nm after irradiation). As for the mechanism, the authors suggested that the DASA photoswitching from an extended linear to compact closed form could induce more negative curvature on the host-lipid packing and thus trigger a phase transition.142 However, the reverse phase transition upon removal of the light stimulus was complete and very rapid despite the much slower thermal recovery of the DASAs. This suggests that the observed effects may be partially associated with an increase in temperature, as also acknowledged by the authors.142 Photothermal effects arising from the high extinction coefficients of the open DASA isomer in the visible range were recently also described in the context of photothermal polymer actuators (Section 3.4).
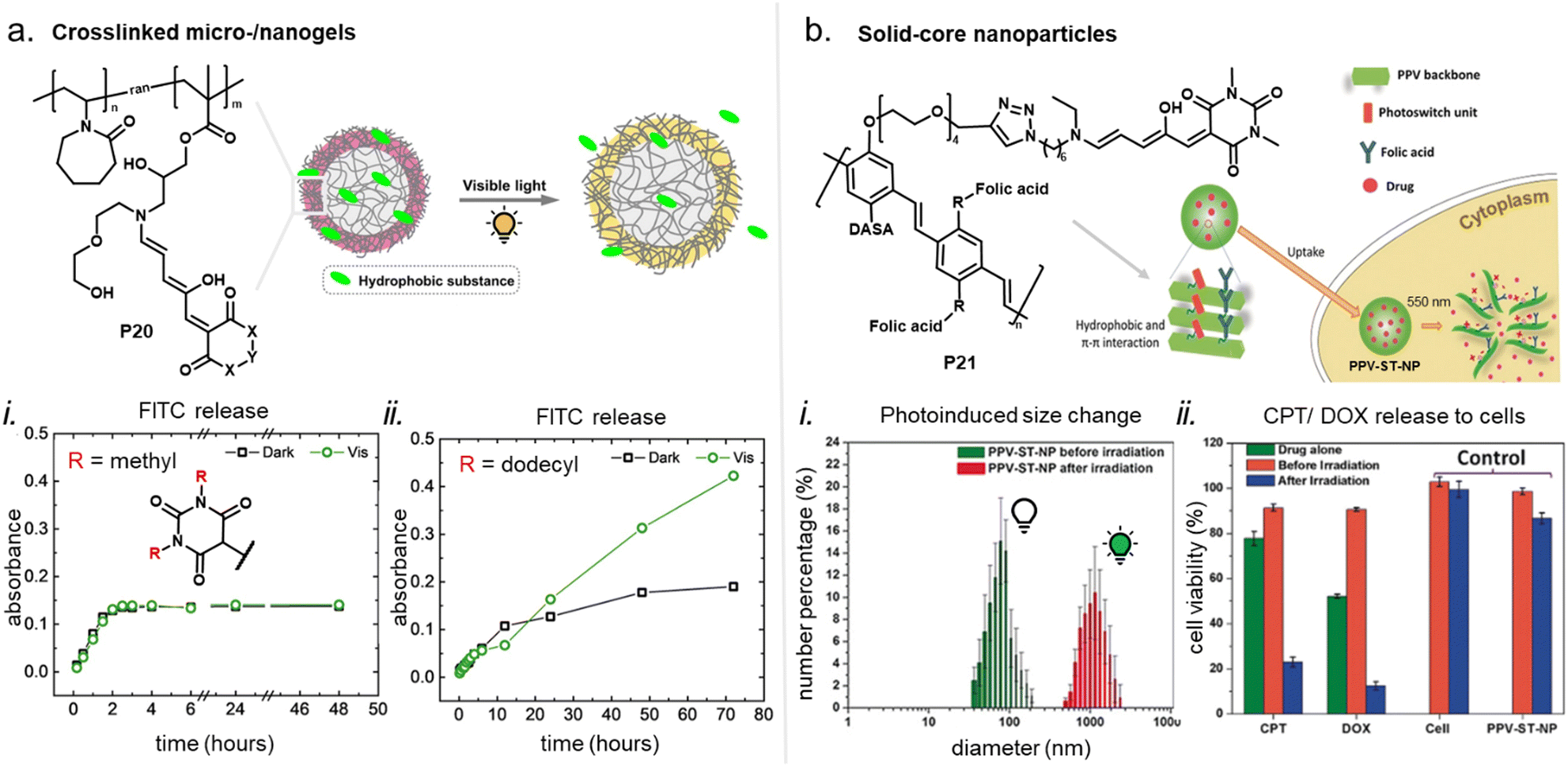 | ||
| Fig. 27 Light-responsive gel and solid-core NPs containing DASAs. (a) Mico-/nanogels from temperature-responsive cross-linked poly(N-vinylcaprolactam) with first-generation DASA covalently attached to the outer layer (P20). Schematic illustration of the light-triggered uptake and release of hydrophobic compounds, such as fluorescein isothiocyanate (FITC). (i) and (ii) Release experiments of FITC from the DASA-modified gels in the dark and under visible light irradiation (cold light source, 100 W halogen lamp) using a dialysis method. Data from gels containing DASAs derived from N-methyl (i) and N-dodecyl (ii) barbituric acid are shown. Light-accelerated release of FITC could be achieved only for the latter sample, due to the slower light-independent cyclization in water as a result of the increased hydrophobicity of the DASA. Figures adapted with permission from ref. 125 Copyrights 2022 American Chemical Society. (b) Conjugated polymer nanoparticles made by nanoprecipitation of a DASA-functionalized hydrophobic poly(p-phenylene vinylene) (PPV, P21) in water. Schematic representation of the drug-loaded NPs, uptake in cells facilitated by folic acid targeting units, and structural change upon irradiation (550 nm, 30 W metal halide lamp with cut-off filter) leading to intracellular drug release. (i) Size distribution histograms from dynamic light scattering (DLS) analysis of the NPs before and after irradiation for 1 h with visible light. (ii) Viabilities of HeLa cells incubated with drug-loaded NPs (8 μM; camptothecin, CPT, or doxorubicin, DOX) before or after irradiation at 550 nm for 1 h. Before irradiation the cells display good viability (>90%) in presence of the encapsulated drugs, whereas after illumination viabilities decrease substantially after illumination (<25%). Figures adapted with permission from ref. 92 Copyrights 2018 John Wiley and Sons. | ||
In another example, nanoparticles were made by nanoprecipitation of hydrophobic poly(p-phenylene vinylene), which was covalently functionalized with first-generation DASAs and folic acid units (Fig. 27b).92 The nanoparticles were held together by hydrophobic and π–π interactions. It was demonstrated that DASA photoswitching led to a swelling and opening of the nanoparticles to release anticancer drugs from within the structures. The average size of the particles dramatically increased under irradiation form 80 nm to up to 1 mm, which was also accompanied by a major change in surface charge (zeta potential change from −7.8 mV to surface-charge polarization in both positive and negative charges of +60 and −60 mV).92 This was also demonstrated in cell assays, where cell uptake into cancer cell lines was facilitated by the folic acid targeting units on the nanoparticles. Cells exposed to particles loaded with hydrophilic DOX or hydrophobic camptothecin (CPT) showed good cell viability in the dark, whereas cell viability decreased markedly after illumination with visible light, indicating successful photoinduced release of the drugs.92 The particles also display switchable fluorescence as a result of DASA photoisomerization (see Section 3.1.2). However, in this example no reversible photoswitching of the DASA was observed.
Zhang, Liu and co-workers reported a layer-by-layer approach to coat UCNPs with different functional materials including a polymer layer made from a DASA-functionalized BCP to gate permeation of substances into and out of the nanoparticles.130 The design of the DASA-functionalized BCP is similar to the enzyme nanoreactors previously reported for DASA-based nanoreactors129 consisting of a PEG-block and a hydrophobic block made from a random copolymer of methacrylate-derived DASA monomers (10 mol%, second-generation) and phenyl methacrylate.130 Two different types of particles were shown: For the first one, the UCNPs were coated with a layer of enzymes (glucose, lactate or urate oxidases, approximately 10 nm thick) and then with a polymer layer (approximately 20–60 nm thick layer for particles with a hydrodynamic diameter of ∼200–280 nm). For the second one, the UCNPs were coated with a mesoporous layer of silica, loaded with cargo (D-luciferin or SYTO− nucleic acid stain), and subsequently coated with the polymer layer. The system was mainly presented as enzyme nanoreactor platform (first type of particles), but for the second type of particles, visible and NIR light-induced cargo release in a luciferase-transfected CT26 tumor-bearing mouse model or in cell assays was also demonstrated.130 However, no information on the morphology or ordering of the DASA-functionalized BCP gating membrane was provided, neither before nor after photoswitching.
3.4 Photothermal actuation
The joint photochromic and photothermal nature of DASAs (Section 2.4) was exploited in solution to generate convective flows in fluid cells.82 In polymers, the offset in thermal expansion coefficient between a DASA functionalized PHMA active layer (P1, Fig. 12) and a poly(imide) (PI) substrate was leveraged to build a simple crawler propelled by photothermal bilayer bending actuation (Fig. 28).83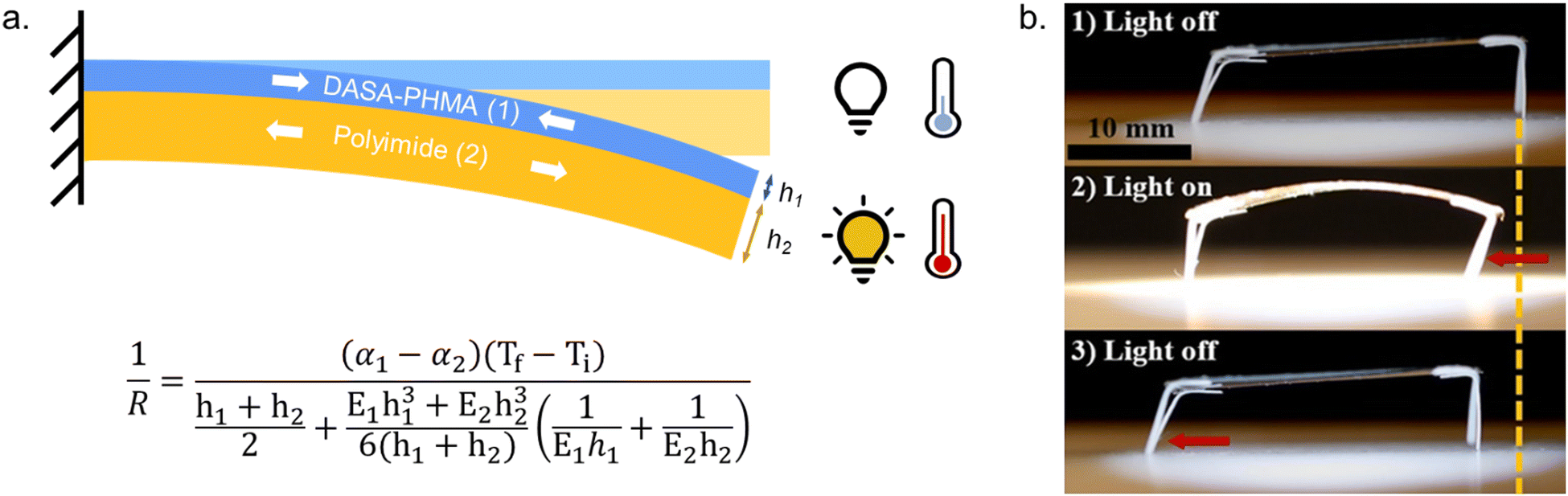 | ||
| Fig. 28 Photothermal DASA–polymer film actuators. (a) Schematic illustration of photothermal bilayer actuation (top) with an active layer based on P1 (cf.Fig. 10). The radius of curvature (R) achieved upon photothermal heating from temperature Ti to Tf is defined according to the thermal bilayer beam bending equation (bottom) in terms of the elastic modulus (E), thickness (h), and thermal expansion coefficient (α) of each layer. Converging and diverging white arrows indicate the layers in compressive and tensile stress, respectively. Beam deflection occurs towards the layer in tensile stress, in this case, polyimide (α2 < α1). (b) Demonstration of a DASA-powered photothermal crawler based on the bilayer bending principle in (a). Figure adapted with permission from ref. 83 Copyright 2020, American Chemical Society. | ||
The work output of such a DASA bilayer photoactuator depends on a variety of factors, including the thickness and elasticity of the comprising layers, the DASA loading and isomerization kinetics, the intensity and spectral properties of the photoexcitation source, and the optical history of the film.83,96 For example, bilayers of DASA conjugated methacrylate copolymer on PI had greater work output when the Tg of the active layer was increased from 23 °C to 62 °C,96 resulting in an increase in the film elastic modulus. This observation is supported on a strictly mechanical basis by beam theory (cf. equation in Fig. 28).168 In addition, a slight increase in the photothermal conversion efficiency was found for stiffer active layers, suggesting that the mechanical properties of the active layer have also affected the photoswitching energetics of the DASA.96 A less efficient switching to the closed isomer in the stiffer materials thereby reduces the loss of photothermal agent (i.e., the open isomer) under illumination (see Section 2.5.1.1). Longer pre-exposure times of DASA-PHMA/PI bilayer cantilevers also led to a depletion of the open form population, resulting in reduced photothermal heating and less bending actuation.83
3.5 Biopolymers
A few reports have been published on DASAs in combination with biopolymers, such as polypeptides,141 cellulose102,139,140 and cyclic oligosaccharides.72,106 DASAs in combination with cellulose have been described in the form of coatings on paper substrates102,139 or cellulose nanoparticles140 for applications in rewritable surfaces or wettability switchable surfaces (refer to previous sections). In addition, Ran and co-workers recently reported on the non-covalent association of a small molecule third-generation DASA to the hydrophobic pockets of β-amyloid peptides (Aβ40, Fig. 29a).141 It was demonstrated that this binding event slightly slows down light-independent DASA cyclization in water and also affects the tertiary structure of the peptide leading to reduced protein aggregation.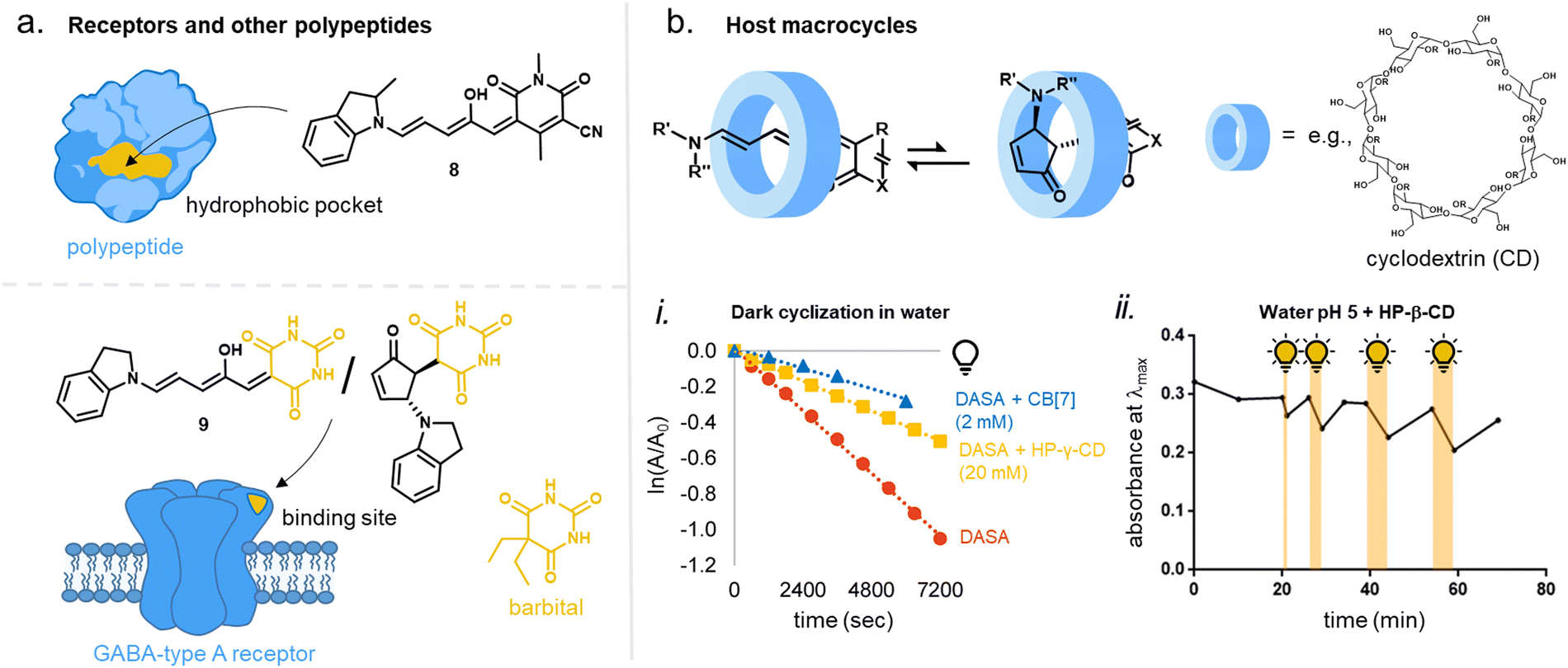 | ||
| Fig. 29 Non-covalent association/interaction of DASAs with biopolymers or –oligomers in aqueous media. (a) Interaction of DASAs with polypeptides. Top: Binding to and stabilization of an open DASA isomer in the hydrophobic pocket of a β-amyloid peptide (Aβ40). Bottom: A barbituric acid derived second-generation DASA is active as an antagonist in γ-aminobutyric acid (GABA)-type A receptors. Barbiturate drugs, such as barbital, that are structurally related to the acceptor in 9 bind to GABA-type A receptors and thereby enhance GABA-mediated chloride currents. (b) First and second-generation DASA undergo host–guest complexation with hydroxypropyl β- or γ-cyclodextrins (HP-CD) or other host macrocycles, such as cucurbit[7]uril (CB[7]). (i) Dark switching of a Meldrum's acid derived first-generation DASA in water with no host or after inclusion in different host macrocycles. Semi-log plot of absorption changes of open isomer over time showing stabilization of the open isomer in the presence of the macrocycles. Figure adapted with permission from ref. 72 Copyright 2020 by the authors. (ii) Reversible photoswitching and stabilization of the open isomer of 9 in water at acidic pH when encapsulated in HP-β-CD (500 μM DASA in solution containing 10 wt% CD). Time course at the absorption peak (605 nm) with illumination times of 1, 3, and 5 min (broad spectrum halogen lamp, 150 W, orange bars). Figure adapted with permission from ref. 106 Copyright 2022 American Chemical Society. | ||
Given by the structural similarity of the acceptor in barbituric-acid derived DASAs and common barbiturate drugs, Gorostiza and co-workers showed that a second-generation DASA (9, Fig. 29a) is active as an antagonist in γ-aminobutyric acid (GABA)-type A receptors (GABAARs) and alters the firing rate of non-transfected neurons in a physiological medium at neutral pH.106 The observed pharmacological activity is interesting and may raise prospects for future applications in photopharmacology.169 However, differences in receptor binding between open and closed isomers or visible light-induced effects on neuronal activity have not yet been investigated.
β- and γ-cyclodextrins (CD) and other host macrocycles were used to encapsulate DASAs in their hydrophobic interior (Fig. 29b). Wagner and co-workers were the first to establish the protective nature of this supramolecular complexation and that it reduces the cyclization of first-generation DASAs in aqueous environments.72 More recently, Gorostiza and co-workers reported on the use of hydroxypropyl-β-CD complexes to encapsulate DASA 9 and could show that under acidic pH these inclusion complexes showed reversible photoswitching behavior in water, although with rather limited degrees of forward isomerization.106 The solubility of the DASA in pure water could also be increased reaching millimolar concentrations with the help of 25 wt% CD.106 However, CD was not included in the neuronal activity assays of this research group described above. Nevertheless, the utility of CD for compartmentalization of the DASAs in biologically relevant media is promising, particularly since it is an approved drug excipient.
4. Current challenges and next steps
Complexity of the DASA switching mechanism and environmental sensitivity: opportunity or limitation?
While different behavior in different environments can be beneficial by allowing for complex responses and tunability, this can only be leveraged if it is properly understood. Even though substantial progress has been made in elucidating the photoswitching mechanism and establishing structure–property relationships at the small molecule level, many aspects of DASA behavior in more complex environments are still not fully understood. For applications in macromolecular systems, the strong environmental sensitivity of the photophysical and photochemical properties of DASAs is currently more of a limitation than a useful “turning knob”.The photoswitching mechanism of DASA (cf.Fig. 4) is far more complex than for other classes of photoswitches, such as azobenzenes, interconnecting both photodynamic and thermodynamic equilibria. While the initial actinic step determines the immediate photoresponse, the thermal steps that occur on longer time scales are more important for understanding and controlling DASA photoswitching. Therefore, DASAs result in complex materials with more than two (meta)stable states. The main challenge in understanding such materials is that a variety of parameters can impact the various steps along the complex energy landscape of the multistep (photo)isomerization pathway in a non-trivial way. These parameters can include, for example, mechanical effects, polar and hydrogen bonding interactions, aromatic interactions, temperature, or acid–base properties. Already on small molecule models, it was shown that only small changes in the local environment, such as slight polarity differences, can have a substantial influence on the overall photoswitching performance (Section 1.1.1.6).44,108 In addition, the nature and extent of these influences also depend on the DASA type.
The role of unproductive or alternative (photo)isomerization pathways (see Section 1.1.1.1) of DASAs when going from dilute solutions to different macromolecular systems is also almost unexplored. The same applies to the dark equilibrium distribution and the final tautomerization of the closed isomer (Fig. 3), which is important for their overall behavior, but is not well studied in materials, particularly in the solid state. This is also due to the limited applicability of many analytical techniques used for studying photoswitching in solutions. Overall, more work on establishing general design principles for transferring the advantageous photoswitching performance of a DASA in solution to macromolecular systems is needed, also with respect to the DASA structure. This is important to improve current system designs in a rational way, which is ultimately required to realize the full potential of DASA-functionalized polymeric materials in applications. However, given the rapid increase in the fundamental understanding of the DASA photoswitching mechanism and structure–property relationship in solution, progress in this area is certainly on a solid foundation.
Concentration effects and fatigue in materials
Owing to the high sensitivity to the presence of polar neighboring units, the limited efficiency for cyclization under irradiation of DASAs at higher DASA concentration is another, but closely related challenge (refer to Section 2.5.3). This is a serious problem, especially for applications in materials, where in some cases high concentrations are required to drive property changes and it is desirable to achieve close to quantitative conversion to the metastable isomer under irradiation to maximize differences in material properties. Another difficulty is that conjugation of DASAs to polymers, even at a low percentage, can substantially affect the properties of the parent polymer, including thermal and mechanical properties. The increase in rigidity of a polymer as a result of the incorporation of DASA can negatively impact the photoswitching performance. This factor must be considered in the system design to meet the material requirements for a particular application.Other highly relevant open questions concern short and long-term DASA stability, as well as innovative solutions for DASA fatigue in materials (Section 2.6). Recent findings raise the concern that DASA stability even in the dark, is limited for some DASAs under certain conditions, and that, for example, a reversion to its starting materials can occur.36,73,107,138 It still needs to be investigated in more detail how general this problem is and when exactly it occurs. Apart from this, further studies are required focusing on elucidating system-dependent degradation pathways both in the dark and under light irradiation. It is important to emphasize that to meet the requirements of real-world applications and to compete with more established photoswitches, it is necessary for the DASAs to become a lot more robust when integrated in materials. Guided by the creative power of synthetic chemistry and material science, there is certainly a great opportunity to meet these challenges.
Understanding the origin of photoinduced material property changes
A useful feature of DASAs is the vastly different physicochemical properties of the open and closed isomers. However, for complex reactions in materials, it is often difficult to identify specifically which photoinduced effect(s) at the molecular level are responsible for changes at larger length scales, up to macroscopic effects. For example, the role of Tg drop and increasing fluidity of DASA-functionalized polymers upon photoisomerization that was recently reported,96 has not yet been considered or investigated in the context of release or permeabilization applications with polymeric nanocarriers. This is also largely applicable to potential effects as a result form local heating (photothermal effects). Moreover, the role of other intermediates along the multistage DASA switching pathway in driving or affecting material property changes also remains unclear. The sensitivity of DASAs to other environmental factors in materials such as hydrochromism, acidochromism, thermochromism, and possibly others, must also be considered. In application- and device-oriented studies, the origin of observed macroscopic changes is often explained by a particular isomeric state or property of DASA without experimental evidence or adequate characterization of the material itself. A better understanding of the underlying mechanisms that induce a functional output in a dynamic material can help to further improve it, but this also requires proper characterization of the system. An interdisciplinary and multi-scale approach spanning from molecular photochemistry to modeling and fabrication of polymeric systems can provide notable advances in this rapidly growing area of research.Photoswitching in water, red-shifting and P-type systems
Achieving full control over DASA photoswitching kinetics and DASA dark equilibrium under physiological conditions remains an important goal. As water is the universal solvent of life, advances that enable reversible DASA photoswitching in an aqueous media will most likely open new opportunities in photopharmacology, biomedical research and biomedical applications (drug delivery, super-resolution imaging, dynamic cell culture materials, reversible activation/deactivation of biomolecules etc.). From the polymer field, particularly photoresponsive polymer hydrogels have received much attention in medicine, due to their unique mechanical properties and biocompatibility.12While encapsulation and compartmentalization strategies in water, e.g., nanoparticles, such as polymersomes, or DASA conjugation to polymers (Sections 3.3 and 3.5), could partially circumvent this problem by providing a protected, suitable microenvironment, the direct use of DASAs in polar protic media (including hydrophilic protic polymer matrices) presents a number of difficulties. Firstly, the overall dark equilibrium is strongly shifted to the more hydrophilic closed isomer with current DASA derivatives (Section 1.1.1.5). Secondly, reversible and efficient photoswitching in highly polar solvents is more difficult (contributing factors to this are the lowered energy barrier between B and A, which favors fast thermal back reaction from B to A instead of isomerization to B′ and C, refer to Section 1.1.1.6). Thirdly, solubility and stability of DASAs in water and the development of biocompatible switches are also critical for direct application in aqueous media.
Addressing these issues through synthetic structural changes of DASAs is challenging. For example, modifying stability and/or solubility of the open or closed isomers independently from modifying the degree of charge separation of the triene bridge (which plays a major role in defining the photoswitching behavior with regards to polarity effects) is difficult to achieve. Promising investigations on substitutions along the DASA triene have revealed that the dark equilibrium and degree of charge separation of the open isomer can be modulated independently of donor and acceptor strengths,36,73,74 providing an additional “turning knob” for future designs to address these issues. In addition, photoswitches operating in the 400–600 nm window still have limited bio-applications due to insufficient tissue penetration depth of light of these wavelengths and NIR photoswitches are clearly favorable for applications in photopharmacology or use in vivo. NIR light-induced photoswitching of DASAs inside materials without requiring photon upconversion strategies is still lacking.
Another important goal yet to be achieved in the field of molecular photoswitches is the development of compounds that can be driven with red light/NIR light in both directions. A P-type DASA that can be photocontrolled in both directions from the open to the closed isomer is currently not possible, mainly since the closed isomer that absorbs in the UV spectral region cannot be ring-opened with light. However, DASAs with responsiveness to two or more different wavelengths of light, such as by conjugation to or combination with other classes of photoswitches,68,89,155 or by modulating the PSS of the A to B/B′ intermediate stage of the actinic step in the DASA photoswitching pathway,37 were demonstrated. The latter example has drawn attention to the photothermal equilibrium between A and B, which is essentially a P-/T-type E/Z switch coupled with a (multistep) intramolecular thermal reaction and provides inspiration on how to tune the lifetime of B/B′ in future studies.
Potential applications in new areas
The development of DASA-compatible polymerization and/or cross-linking strategies would be particularly interesting for accessing complex macromolecular architectures that have not yet been investigated, such as polymers incorporating disubstituted DASAs as a part of the polymer backbone. Such materials could be interesting for applications in photochemical/photomechanical actuation or force sensing (mechanochromism), both areas largely unexplored in the DASA field. The ability to control and execute actuation with white light is uniquely possible with negatively photochromic absorbers, like DASA, and could potentially enable self-modulating photoactuation under continuous broadband irradiation, such as by sunlight. While unreported to date, DASA-based polymer actuators relying on photochemical/photomechanical mechanisms, like those demonstrated primarily using azobenzene, have the potential to support greater localized strains that are possible by photothermal actuation and would be well suited to applications in shape programming materials or soft robotics. DASA integration into polymers that provide increasing order between the photoswitches for enabling more cooperative action upon isomerization (e.g., as photoswitchable mesogen in liquid crystal polymer systems170), or for obtaining materials that are optically anisotropic, also remain to be shown.In addition, the versatility of spiropyran photoswitches being responsive to other stimuli than light, including thermo-, acido-, mechano-, solvato-, and electrochromic responsivity, was one of the reasons for their popularity and use in a variety of dynamic materials. Multi stimuli-responsiveness of DASAs was also explored, including metal ions,69,103,112,171–173 water,101 acids/bases,104,105 or electrochemistry,174 which could present interesting opportunities also in polymer and material science in the future. Moreover, the large drop in Tg and elastic modulus of polymers upon photoswitching of covalently attached DASAs could offer many opportunities for polymer phase transition control,96 for example, for self-healing materials, smart adhesives or photorheological fluids.
A major field of application for photoswitches are drug delivery systems and biomaterials, including photodynamic therapy and light-triggerable release systems. While first steps have been taken into this direction with DASA–polymer materials, there is still a lot of room for research into DASA materials for biomedical applications. One important point that needs to be elucidated in this context is the level of biocompatibility and if there are any cytotoxic effects of DASAs, their different isomers, and their degradation products. Finally, to move to increasingly complex systems, as found in biology, we must introduce complexity on a molecular level and break the binary on/off performance of traditional photoswitches. DASA photoswitches exhibit more than two independently addressable stages, which can expand beyond the currently possible material responses and allow not only for complex switching phenomena but also for advanced control of species population in a material. The first step towards this was recently demonstrated by the development of a multi-stage DASA-based photoswitch.37 Because the kinetics and thermodynamics are affected by the matrix environment, new approaches will need to be developed in the solid-state to stabilize key intermediates along the photoswitching pathway, further accelerating discoveries in this growing area.
5. Conclusion
Judging from the large physicochemical changes between open and closed isomers and their responsiveness to visible light, DASAs are ideally suited for creating stimuli-responsive materials. To date, a solid foundation has been set to establish DASAs as complementary photoswitches to more established classes, such as azobenzenes or spiropyrans, for a range of material science applications. In summary, several strategies have been developed for the covalent conjugation of DASAs to a variety of different polymer types and macromolecular architectures, including various post-polymerization modification methods and click chemistry. These efforts delivered a wide range of visible light-responsive polymeric materials for applications in solution, suspension, at surfaces, in bulk configuration, as well as (supramolecular) (self-) assemblies and nano- and microstructures. Application examples show that DASAs have the potential to enable visible light modulation of a wide range of polymer properties and functions, from optical, thermal and mechanical properties, to surface or interface wettability and morphology of nanoparticles.Several strategies available today to (partially) circumvent current limitations of DASAs under physiological conditions (cyclization under exclusion of light and lack of reversible photoswitching in water, hydrolytic stability, limited solubility of open isomer) came from the polymer field, including compartmentalization or encapsulation, where the polymer environment prevents access of solvent molecules to the DASAs. Sophisticated and tailored polymer constructs have shown great potential for enabling visible/NIR light-mediated control over species release and transport for drug delivery applications in aqueous environment. Composites of DASA–polymers and UCNPs thereby enabled addressability with light in the “near-infrared phototherapeutic window”, which could serve as a powerful future tool for biomedical applications and research. Moreover, wavelength-selective activation of multiple different DASA derivatives or DASAs in combination with other phototriggers was demonstrated both at the small molecule level and in polymeric materials, which opens the door to more advanced multiresponsive materials that offer photoorthognal control over their properties and functions. Taken together, it can easily be envisioned that DASAs and DASA-containing polymers will enable breakthroughs in soft robotics, smart materials and photopharmacology.
Abbreviations
| ATRP | Atom transfer radical polymerization |
| BCP(s) | Block copolymer(s) |
| CB[7] | Cucurbit[7]uril |
| CD | Cyclodextrin |
| CMC | Critical micelle concentration |
| CPT | Camptothecin |
| CuAAC | Copper-catalyzed azide–alkyne cycloaddition |
| DASA(s) | Donor–acceptor Stenhouse adduct(s) |
| DCNP | Diethyl cyanophosphate |
| DFT | Density functional theory |
| DMA | Dynamic mechanical anaylsis |
| DMSO | Dimethyl sulfoxide |
| DLPC | 1,2-Dilauroyl-sn-glycero-3-phosphocholine |
| DLS | Dynamic light scattering |
| DOX | Doxorubicin |
| DSC | Differential scanning calorimetry |
| FITC | Fluorescein isothiocyanate |
| FMPPS | Frequency modulated pump probe spectroscopy |
| FRET | Förster resonance energy transfer |
| FTIR | Fourier transform infrared |
| GABA | γ-Aminobutyric acid |
| HALS | Hindered amine light stabilizers |
| HFIP | 1,1,1,3,3,3-Hexafluoro-2-propanol |
| IR | Infrared |
| KPS | Potassium persulfate |
| LED | Light emitting diode |
| LOD | Limit of detection |
| MCF-7 | Michigan Cancer Foundation-7 |
| MOF | Metal organic framework |
| MR | Methyl red |
| n.a. | Not applicable |
| n.d. | Not determined |
| NIR | Near-infrared |
| NMR | Nuclear magnetic resonance |
| NP(s) | Nanoparticle(s) |
| PBA | Poly(butyl acrylate) |
| PBMA | Poly(butyl methactrylate) |
| PC | Polycarbonate |
| PCL | Polycarpolacton |
| PDMS | Polydimethylsiloxane |
| PEG | Poly(ethylene glycol) |
| PEI | Poly(ethylene imine) |
| PHA | Poly(hexyl acrylate) |
| PHMA | Poly(hexyl methacrylate) |
| PI | Polyimide |
| PMA | Poly(methyl acrylate) |
| PMMA | Poly(methyl methacrylate) |
| PEGMEMA | Poly(ethylene glycol)-methyl ether methacrylate |
| PPhMA | Poly(phenyl methacrylate) |
| PS | Polystyrene |
| PSS | Photostationary state |
| PTSS | Photothermal stationary state |
| PVA | Poly(vinyl acetate) |
| PVC | Poly(vinyl chloride) |
| PVP | Poly(vinyl pyrrolidone) |
| RAFT | Reversible addition–fragmentation chain transfer |
| ROMP | Ring-opening metathesis polymerization |
| ROS | Reactive oxygen species |
| SAXS | Small angle X-ray scattering |
| SDBS | Sodium dodecyl benzenesulfonate |
| SEM | Scanning electron microscopy |
| TD-DFT | Time-dependent density functional theory |
| TEM | Transmission electron microscopy |
| T g | Glass transition temperature |
| THF | Tetrahydrofuran |
| UCNP(s) | Upconversion nanoparticle(s) |
| UV | Ultraviolet light |
| Vis | Visible light |
| XRD | X-Ray diffraction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation through Grant No. 200021_172609 and through the National Center of Competence in Research (NCCR) Bio-Inspired Materials (Grants No 51NF40-182881 and 51NF40-205603). We also acknowledge the support from the Novartis Research Foundation (project “Lightloop”). This work was also partially supported by the office of Naval Research through the MURI on Photomechanical Materials Systems (N00014-18-1-2624) and the National Science Foundation (NSF) under Award No. EFMA 193527. J. A. P. acknowledges the support of an Elings Prize Fellowship from the CNSI at UCSB.References
- Molecular Photoswitches, ed. Z. L. Pianowski, Wiley-VCH, Weinheim, 2022 Search PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- Z. L. Pianowski, Chem. – Eur. J., 2019, 25, 5128–5144 CrossRef CAS PubMed.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 7, 1900404 CrossRef.
- H. Nie, J. L. Self, A. S. Kuenstler, R. C. Hayward and J. Read de Alaniz, Adv. Opt. Mater., 2019, 7, 1900224 CrossRef.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymanski, Chem. Sci., 2020, 11, 11672–11691 RSC.
- R. Dorel and B. L. Feringa, Chem. Commun., 2019, 55, 6477–6486 RSC.
- Y. Zhuang, X. Ren, X. Che, S. Liu, W. Huang and Q. Zhao, Adv. Photonics, 2020, 3, 014001 Search PubMed.
- M. Olesinska-Monch and C. Deo, Chem. Commun., 2023, 59, 660–669 RSC.
- A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS.
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- P. F. Luo, S. L. Xiang, C. Li and M. Q. Zhu, J. Polym. Sci., 2021, 59, 2246–2264 CrossRef CAS.
- L. Beaute, N. McClenaghan and S. Lecommandoux, Adv. Drug Delivery Rev., 2019, 138, 148–166 CrossRef CAS PubMed.
- S. Chen, R. Costil, F. K. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed.
- L. Albert and O. Vazquez, Chem. Commun., 2019, 55, 10192–10213 RSC.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- N. R. B. Boase, Macromol. Rapid Commun., 2020, 41, 2000305 CrossRef CAS PubMed.
- M. Gao, D. Kwaria, Y. Norikane and Y. Yue, Nat. Sci., 2023, 3, e220020 CrossRef CAS.
- J.-X. Wang, C. Li and H. Tian, Coord. Chem. Rev., 2021, 427, 213579 CrossRef CAS.
- Z. Li, C. He, Z. Lu, P. Li and Y.-P. Zhu, Dyes Pigm., 2020, 182, 108623 CrossRef CAS.
- D. Bleger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS PubMed.
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202205758 CrossRef CAS PubMed.
- A. L. Leistner and Z. L. Pianowski, Eur. J. Org. Chem., 2022, e202101271 CrossRef CAS.
- K. Klaue, Y. Garmshausen and S. Hecht, Angew. Chem., Int. Ed., 2018, 57, 1414–1417 CrossRef CAS PubMed.
- M. W. H. Hoorens, M. Medved, A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. Jan Buma and W. Szymanski, Nat. Commun., 2019, 10, 2390 CrossRef PubMed.
- D. Roke, M. Sen, W. Danowski, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2019, 141, 7622–7627 CrossRef CAS PubMed.
- V. Josef, F. Hampel and H. Dube, Angew. Chem., Int. Ed., 2022, 61, 202210855 CrossRef PubMed.
- K. Mutoh, N. Miyashita, K. Arai and J. Abe, J. Am. Chem. Soc., 2019, 141, 5650–5654 CrossRef CAS PubMed.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Chem. Commun., 2016, 52, 13576–13579 RSC.
- J. R. Hemmer, Z. A. Page, K. D. Clark, F. Stricker, N. D. Dolinski, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2018, 140, 10425–10429 CrossRef CAS PubMed.
- N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242–8252 RSC.
- J. A. Peterson, F. Stricker and J. Read de Alaniz, Chem. Commun., 2022, 58, 2303–2306 RSC.
- F. Stricker, D. M. Sanchez, U. Raucci, N. D. Dolinski, M. S. Zayas, J. Meisner, C. J. Hawker, T. J. Martinez and J. Read de Alaniz, Nat. Chem., 2022, 14, 942–948 CrossRef CAS PubMed.
- S. Aiken, R. J. L. Edgar, C. D. Gabbutt, B. M. Heron and P. A. Hobson, Dyes Pigm., 2018, 149, 92–121 CrossRef CAS.
- M. M. Lerch, S. J. Wezenberg, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 6344–6347 CrossRef CAS PubMed.
- M. Di Donato, M. M. Lerch, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, S. P. Ihrig, M. Medved, D. Jacquemin, W. Szymanski, W. J. Buma, P. Foggi and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 15596–15599 CrossRef CAS PubMed.
- H. Zulfikri, M. A. J. Koenis, M. M. Lerch, M. Di Donato, W. Szymanski, C. Filippi, B. L. Feringa and W. J. Buma, J. Am. Chem. Soc., 2019, 141, 7376–7384 CrossRef CAS PubMed.
- D. M. Sanchez, U. Raucci, K. N. Ferreras and T. J. Martinez, J. Phys. Chem. Lett., 2020, 11, 7901–7907 CrossRef CAS PubMed.
- D. M. Sanchez, U. Raucci and T. J. Martinez, J. Am. Chem. Soc., 2021, 143, 20015–20021 CrossRef CAS PubMed.
- M. Sroda, F. Stricker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Chem. – Eur. J., 2021, 27, 4183–4190 CrossRef CAS PubMed.
- M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- Y. Duan, H. Zhao, C. Xiong, L. Mao, D. Wang and Y. Zheng, Chin. J. Chem., 2021, 39, 985–998 CrossRef CAS.
- Y. Huang, Y. Du, L. Yuan, Z. Chu and L. He, J. Macromol. Sci., Part A: Pure Appl. Chem., 2021, 58, 717–724 CrossRef CAS.
- S. Helmy and J. Read de Alaniz, Photochromic and Thermochromic Heterocycles, Elsevier Inc., 2015, vol. 117, pp. 131–177 Search PubMed.
- F. Stricker, S. Seshadri and J. Read de Alaniz, in Molecular Photoswitches, ed. Z. L. Pianowski, Wiley-VCH, Weinheim, 2022, vol. 1, pp. 304–324 Search PubMed.
- D. E. Nanasi, A. Kunfi, A. Abraham, P. J. Mayer, J. Mihaly, G. F. Samu, E. Kiss, M. Mohai and G. London, Langmuir, 2021, 37, 3057–3066 CrossRef CAS PubMed.
- B. Bończak and M. Fiałkowski, J. Phys. Chem. C, 2022, 126, 7096–7106 CrossRef.
- G. Sobczak, I. Misztalewska-Turkowicz and V. Sashuk, J. Phys. Chem. C, 2021, 125, 5306–5314 CrossRef CAS.
- J. Ahrens, T. Bian, T. Vexler and R. Klajn, ChemPhotoChem, 2017, 1, 230–236 CrossRef CAS.
- Y. Duan, M. Song, F. Sun, Y. Xu, F. Shi, H. Wang, Y. Zheng, C. He, X. Liu, C. Wei, X. Deng, L. Chen, F. Liu and D. Wang, Adv. Sci., 2023, 2207443, DOI:10.1002/advs.202207443.
- F. Sun, X. Xiong, A. Gao, Y. Duan, L. Mao, L. Gu, Z. Wang, C. He, X. Deng, Y. Zheng and D. Wang, Chem. Eng. J., 2022, 427, 132037 CrossRef CAS.
- N. D. Shepherd, T. Wang, B. Ding, J. E. Beves and D. M. D'Alessandro, Inorg. Chem., 2021, 60, 11706–11710 CrossRef CAS PubMed.
- A. Boulmier, M. Haouas, S. Tomane, L. Michely, A. Dolbecq, A. Vallee, V. Brezova, D. L. Versace, P. Mialane and O. Oms, Chemistry, 2019, 25, 14349–14357 CrossRef CAS PubMed.
- R. Saha, A. Devaraj, S. Bhattacharyya, S. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2019, 141, 8638–8645 CrossRef CAS PubMed.
- S. Mukhopadhyay, A. Sarkar, S. Ghoshal, P. Sarkar, K. Dhara and P. Chattopadhyay, J. Phys. Chem. B, 2021, 125, 7222–7230 CrossRef CAS PubMed.
- N. Mallo, A. Tron, J. Andréasson, J. B. Harper, L. S. D. Jacob, N. D. McClenaghan, G. Jonusauskas and J. E. Beves, ChemPhotoChem, 2020, 4, 407–412 CrossRef CAS.
- A. K.-H. Chau, L.-H. Cheung and F. K.-C. Leung, Dyes Pigm., 2022, 208, 110807 CrossRef CAS.
- L. H. Cheung, T. Kajitani and F. K. Leung, J. Colloid Interface Sci., 2022, 628, 984–993 CrossRef CAS PubMed.
- J. Stenhouse, Justus Liebigs Ann. Chem., 1850, 74, 278–297 CrossRef.
- R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, Chem. – Eur. J., 2018, 24, 9170–9186 CrossRef CAS PubMed.
- M. M. Lerch, M. Medved, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, W. Szymanski, W. J. Buma, P. Foggi, M. Di Donato and B. L. Feringa, J. Phys. Chem. A, 2018, 122, 955–964 CrossRef CAS PubMed.
- F. Stricker, J. Peterson and J. Read de Alaniz, Org. Synth., 2022, 99, 79–91 CrossRef CAS.
- G. Noirbent, Y. Xu, A. H. Bonardi, S. Duval, D. Gigmes, J. Lalevee and F. Dumur, Molecules, 2020, 25, 2317 CrossRef CAS PubMed.
- M. M. Lerch, M. J. Hansen, W. A. Velema, W. Szymanski and B. L. Feringa, Nat. Commun., 2016, 7, 12054 CrossRef CAS PubMed.
- Y. Duan, H. Zhao, G. Xue, F. Sun, F. Stricker, Z. Wang, L. Mao, C. He, J. Read de Alaniz, Y. Zheng and D. Wang, J. Phys. Chem. B, 2022, 126, 3347–3354 CrossRef CAS PubMed.
- S. R. Marder, B. Kippelen, A. K.-Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS.
- A. D. Laurent, M. Medved and D. Jacquemin, Chem. Phys. Chem., 2016, 17, 1846–1851 CrossRef CAS PubMed.
- L. Payne, J. D. Josephson, R. S. Murphy and B. D. Wagner, Molecules, 2020, 25, 4928 CrossRef CAS PubMed.
- B. Peñín, N. Sanosa, D. Sampedro and I. Funes-Ardoiz, ACS Omega, 2022, 7, 22811–22817 CrossRef PubMed.
- D. Martinez-Lopez, E. Santamaria-Aranda, M. Marazzi, C. Garcia-Iriepa and D. Sampedro, Chemistry, 2021, 27, 4420–4429 CrossRef CAS PubMed.
- M. M. Lerch, M. Di Donato, A. D. Laurent, M. Medved, A. Iagatti, L. Bussotti, A. Lapini, W. J. Buma, P. Foggi, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 8063–8068 CrossRef CAS PubMed.
- C. García-Iriepa, M. Marazzi and D. Sampedro, ChemPhotoChem, 2019, 3, 866–873 CrossRef.
- Y. Li, C. Zhu, F. Gu and F. Liu, Phys. Chem. Chem. Phys., 2023, 25, 7417–7422 RSC.
- M. Ugandi and M. Roemelt, J. Phys. Chem. A, 2020, 124, 7756–7767 CrossRef CAS PubMed.
- U. Raucci, D. M. Sanchez, T. J. Martinez and M. Parrinello, J. Am. Chem. Soc., 2022, 144, 19265–19271 CrossRef CAS PubMed.
- R. Berraud-Pache, E. Santamaría-Aranda, B. de Souza, G. Bistoni, F. Neese, D. Sampedro and R. Izsák, Chem. Sci., 2021, 12, 2916–2924 RSC.
- M. Clerc, F. Stricker, S. Ulrich, M. Sroda, N. Bruns, L. F. Boesel and J. Read de Alaniz, Angew. Chem., Int. Ed., 2021, 60, 10219–10227 CrossRef CAS PubMed.
- S. Seshadri, L. F. Gockowski, J. Lee, M. Sroda, M. E. Helgeson, J. Read de Alaniz and M. T. Valentine, Nat. Commun., 2020, 11, 2599 CrossRef CAS PubMed.
- J. Lee, M. M. Sroda, Y. Kwon, S. El-Arid, S. Seshadri, L. F. Gockowski, E. W. Hawkes, M. T. Valentine and J. Read de Alaniz, ACS Appl. Mater. Interfaces, 2020, 12, 54075 CrossRef CAS PubMed.
- S. Dubuis, A. Dellai, C. Courdurie, J. Owona, A. Kalafatis, L. Vellutini, E. Genin, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2023, 145, 10861–10871 CrossRef CAS PubMed.
- C. Tonnele, B. Champagne, L. Muccioli and F. Castet, Phys. Chem. Chem. Phys., 2018, 20, 27658–27667 RSC.
- Y. Yao, H. L. Xu and Z. M. Su, Dyes Pigm., 2021, 193, 109502 CrossRef CAS.
- C. Garcia-Iriepa and M. Marazzi, Materials, 2017, 10, 1025–1038 CrossRef PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- F.-Y. Tang, J.-N. Hou, K.-X. Liang, Y. Liu, L. Deng and Y.-N. Liu, New J. Chem., 2017, 41, 6071–6075 RSC.
- B. P. Mason, M. Whittaker, J. Hemmer, S. Arora, A. Harper, S. Alnemrat, A. McEachen, S. Helmy, J. Read de Alaniz and J. P. Hooper, Appl. Phys. Lett., 2016, 108, 041906 CrossRef.
- S. Ulrich, X. Wang, M. Rottmar, R. M. Rossi, B. J. Nelson, N. Bruns, R. Muller, K. Maniura-Weber, X. H. Qin and L. F. Boesel, Small, 2021, 17, 2101337 CrossRef CAS PubMed.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114–13119 CrossRef CAS PubMed.
- R. McDonough, N. Rudgley, O. Majewski, M. V. Perkins, R. A. Evans and D. A. Lewis, ChemPhotoChem, 2022, 6, e202200076 CrossRef CAS.
- B. F. Lui, N. T. Tierce, F. Tong, M. M. Sroda, H. Lu, J. Read de Alaniz and C. J. Bardeen, Photochem. Photobiol. Sci., 2019, 18, 1587–1595 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, F. Eisenreich, J. Niu, S. Hecht, J. Read de Alaniz and C. J. Hawker, ChemPhotoChem, 2017, 1, 125–131 CrossRef CAS.
- M. M. Sroda, J. Lee, Y. Kwon, F. Stricker, M. Park, M. T. Valentine and J. Read de Alaniz, ACS Appl. Polym. Mater., 2022, 4, 141–149 CrossRef CAS.
- J. N. Bull, E. Carrascosa, N. Mallo, M. S. Scholz, G. da Silva, J. E. Beves and E. J. Bieske, J. Phys. Chem. Lett., 2018, 9, 665–671 CrossRef CAS PubMed.
- S. Sandlass, F. Stricker, D. Fragoso, J. Read de Alaniz and M. J. Gordon, J. Photochem. Photobiol., A, 2023, 444, 114964 CrossRef CAS.
- K. Stranius and K. Borjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- J. E. Yap, N. Mallo, D. S. Thomas, J. E. Beves and M. H. Stenzel, Polym. Chem., 2019, 10, 6515–6522 RSC.
- D. Wang, L. Zhao, H. Zhao, J. Wu, M. Wagner, W. Sun, X. Liu, M.-S. Miao and Y. Zheng, Commun. Chem., 2019, 2, 118 CrossRef.
- H. Zhao, X. Qin, L. Zhao, S. Dong, L. Gu, W. Sun, D. Wang and Y. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 8952–8960 CrossRef CAS PubMed.
- Y. D. Cai, T. Y. Chen, X. Q. Chen and X. Bao, Org. Lett., 2019, 21, 7445–7449 CrossRef CAS PubMed.
- A. Fiorentino, B. Sachini, S. Corra, A. Credi, C. Femoni, A. Fraix and S. Silvi, Chem. Commun., 2022, 58, 11236–11239 RSC.
- C. Wang, Q. Yan, S. Wang and D. Cao, Dyes Pigm., 2022, 207, 110773 CrossRef CAS.
- R. Castagna, G. Maleeva, D. Pirovano, C. Matera and P. Gorostiza, J. Am. Chem. Soc., 2022, 144, 15595–15602 CrossRef CAS PubMed.
- Y. Shpinov, A. Schlichter, P. Pelupessy, T. Le Saux, L. Jullien and B. Adelizzi, Chem. – Eur. J., 2022, 28, e202200497 CrossRef CAS PubMed.
- F. Stricker, J. Peterson, S. K. Sandlass, A. D. Tagyos, M. Sroda, S. Seshadri, M. Gordon and J. Read de Alaniz, Chem, 2023, 9, 1994–2005 CAS.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- J. Alves, S. Wiedbrauk, D. Grafe, S. L. Walden, J. P. Blinco and C. Barner-Kowollik, Chem. – Eur. J., 2020, 26, 809–813 CrossRef CAS PubMed.
- J. Alves, S. Wiedbrauk, C. Barner-Kowollik and J. P. Blinco, ChemPhotoChem, 2021, 5, 711–715 CrossRef CAS.
- T. Y. Chen, Y. D. Cai, S. Q. Jiang, W. Cai, M. L. Tong and X. Bao, ChemPhotoChem, 2021, 5, 559–564 CrossRef CAS.
- A. C. Overholts, W. Granados Razo and M. J. Robb, Nat. Chem., 2023, 15, 332–338 CrossRef CAS PubMed.
- S. Ulrich, J. R. Hemmer, Z. A. Page, N. D. Dolinski, O. Rifaie-Graham, N. Bruns, C. J. Hawker, L. F. Boesel and J. Read de Alaniz, ACS Macro Lett., 2017, 6, 738–742 CrossRef CAS PubMed.
- S. Seshadri, S. J. Bailey, L. Zhao, J. Fisher, M. Sroda, M. Chiu, F. Stricker, M. T. Valentine, J. Read de Alaniz and M. E. Helgeson, Langmuir, 2021, 37, 9939–9951 CrossRef CAS PubMed.
- S. O. Poelma, S. S. Oh, S. Helmy, A. S. Knight, G. L. Burnett, H. T. Soh, C. J. Hawker and J. Read de Alaniz, Chem. Commun., 2016, 52, 10525–10528 RSC.
- Q. Chen, Y. J. Diaz, M. C. Hawker, M. R. Martinez, Z. A. Page, S. Xiao-An Zhang, C. J. Hawker and J. Read de Alaniz, Macromolecules, 2019, 52, 4370–4375 CrossRef CAS.
- M. Clerc, C. Tekin, S. Ulrich, R. V. M. Freire, S. Salentinig, N. Bruns and L. F. Boesel, Macromol. Rapid Commun., 2022, 43, 2200120 CrossRef CAS PubMed.
- Y. Dong, Y. Ling, D. Wang, Y. Liu, X. Chen, S. Zheng, X. Wu, J. Shen, S. Feng, J. Zhang and W. Huang, Sci. Adv., 2022, 8, eadd1980 CrossRef CAS PubMed.
- S. Singh, K. Friedel, M. Himmerlich, Y. Lei, G. Schlingloff and A. Schober, ACS Macro Lett., 2015, 4, 1273–1277 CrossRef CAS PubMed.
- D. Zhong, Z. Cao, B. Wu, Q. Zhang and G. Wang, Sens. Actuators, B, 2018, 254, 385–392 CrossRef CAS.
- H. Zhao, D. Wang, Y. Fan, M. Ren, S. Dong and Y. Zheng, Langmuir, 2018, 34, 15537–15543 CrossRef CAS PubMed.
- S. Singh, P. Mai, J. Borowiec, Y. Zhang, Y. Lei and A. Schober, R. Soc. Open Sci., 2018, 5, 180207 CrossRef PubMed.
- A. Balamurugan and H.-i Lee, Macromolecules, 2016, 49, 2568–2574 CrossRef CAS.
- C. Hu, Y. Sun, G. van Wissen, Y. Peng and A. Pich, Chem. Mater., 2022, 34, 4774–4784 CrossRef CAS.
- G. Sinawang, B. Wu, J. Wang, S. Li and Y. He, Macromol. Chem. Phys., 2016, 217, 2409–2414 CrossRef CAS.
- J. P. Wesseler, G. M. Cameron, P. A. G. Cormack and N. Bruns, Polym. Chem., 2023, 14, 1456–1468 RSC.
- Y. Ling, Y. Dong, W. Huang, J. Liu, S. Feng and W. Huang, ACS Appl. Polym. Mater., 2022, 4, 6505–6513 CrossRef CAS.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS PubMed.
- M.-D. Liu, Q.-X. Huang, C.-J. Liu and X.-Z. Zhang, Sci. China Mater., 2022, 66, 375–384 CrossRef.
- O. Rifaie-Graham, J. Yeow, A. Najer, R. Wang, R. Sun, K. Zhou, T. N. Dell, C. Adrianus, C. Thanapongpibul, M. Chami, S. Mann, J. Read de Alaniz and M. M. Stevens, Nat. Chem., 2023, 15, 110–118 CrossRef CAS PubMed.
- J. Lin, H. Ma, Z. Wang, S. Zhou, B. Yan, F. Shi, Q. Yan, J. Wang, H. Fan and J. Xiang, Macromol. Rapid Commun., 2021, 42, 2100318 CrossRef CAS PubMed.
- J. E. Yap, L. Zhang, J. T. Lovegrove, J. E. Beves and M. H. Stenzel, Macromol. Rapid Commun., 2020, 41, 2000236 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, W. Chen, Y. He, Y. Liu and H. Ju, J. Controlled Release, 2021, 336, 469–479 CrossRef CAS PubMed.
- S. H. Mostafavi, W. Li, K. D. Clark, F. Stricker, J. Read de Alaniz and C. J. Bardeen, Macromolecules, 2019, 52, 6311–6317 CrossRef CAS.
- X. Xiong, F. Sun, A. Gao, Z. Wang, Y. Duan, Z. Yao, C. He, R. Han, X. Deng, Y. Zheng and D. Wang, J. Chem. Eng., 2022, 450, 138090 CrossRef CAS.
- B. Wu, T. Xue, W. Wang, S. Li, J. Shen and Y. He, J. Mater. Chem. C, 2018, 6, 8538–8545 RSC.
- R. McDonough, N. Rudgley, O. Majewski, M. V. Perkins, R. A. Evans and D. A. Lewis, ChemPhotoChem, 2023, 7, e20220024 CrossRef.
- L. Mao, Z. Wang, Y. Duan, C. Xiong, C. He, X. Deng, Y. Zheng and D. Wang, ACS Nano, 2021, 15, 10384–10392 CrossRef CAS PubMed.
- M. Nau, D. Seelinger and M. Biesalski, Adv. Mater. Interfaces, 2019, 6, 1900378 CrossRef.
- C. Zheng, Y. Yu, S. Kuang, B. Zhu, H. Zhou, S. Q. Zhang, J. Yang, L. Shi and C. Ran, Anal. Chem., 2021, 93, 9887–9896 CrossRef CAS PubMed.
- S. Jia, A. Tan, A. Hawley, B. Graham and B. J. Boyd, J. Colloid Interface Sci., 2019, 548, 151–159 CrossRef CAS PubMed.
- Q. Yan, C. Li, S. Wang, Z. Lin, Q. Yan and D. Cao, Dyes Pigm., 2020, 178, 108352 CrossRef CAS.
- X. Tong, G. Wang, A. Soldera and Y. Zhao, J. Phys. Chem. B, 2005, 109, 20281–20287 CrossRef CAS PubMed.
- N. Sagawa and T. Shikata, Phys. Chem. Chem. Phys., 2014, 16, 13262–13270 RSC.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- C. Pakula, C. Hanisch, V. Zaporojtchenko, T. Strunskus, C. Bornholdt, D. Zargarani, R. Herges and F. Faupel, J. Mater. Sci., 2010, 46, 2488–2494 CrossRef.
- F. Sun and D. Wang, J. Mater. Chem. C, 2022, 10, 13700–13716 RSC.
- I. Pibiri, S. Buscemi, A. Palumbo Piccionello and A. Pace, ChemPhotoChem, 2018, 2, 535–547 CrossRef CAS.
- J. Zhang, J. Zhang, X. Teng, X. Liu, X. Jiao, Y. Li, X. Xie, Q. Yan, X. Wang and B. Tang, J. Phys. Chem. Lett., 2022, 13, 3611–3620 CrossRef CAS PubMed.
- S. Ulrich, S. O. Moura, Y. Diaz, M. Clerc, A. G. Guex, J. Read de Alaniz, A. Martins, N. M. Neves, M. Rottmar, R. M. Rossi, G. Fortunato and L. F. Boesel, Sens. Actuators, B, 2020, 322, 128570 CrossRef CAS.
- H. K. Knutsen, J. Alexander, L. Barregard, M. Bignami, B. Bruschweiler, S. Ceccatelli, B. Cottrill, M. Dinovi, L. Edler, B. Grasl-Kraupp, C. Hogstrand, L. R. Hoogenboom, C. S. Nebbia, I. P. Oswald, A. Petersen, M. Rose, A. C. Roudot, T. Schwerdtle, C. Vleminckx, G. Vollmer, K. Chipman, B. De Meulenaer, M. Dinovi, W. Mennes, J. Schlatter, D. Schrenk, K. Baert, B. Dujardin and H. Wallace, EFSA J., 2017, 15, e05005 Search PubMed.
- A. Hartwig and M. Commission, The MAK-Collection for Occupational Health and Safety, Wiley-VCH Weinheim, 2019, vol. 4, pp. 1–19 Search PubMed.
- A. Bianco, S. Perissinotto, M. Garbugli, G. Lanzani and C. Bertarelli, Laser Photonics Rev., 2011, 5, 711–736 CrossRef CAS.
- M. Li, S. Yang, W. Liang, X. Zhang and D. Qu, Dyes Pigm., 2019, 166, 239–244 CrossRef CAS.
- V. X. Truong, K. Ehrmann, M. Seifermann, P. A. Levkin and C. Barner-Kowollik, Chem. – Eur. J., 2022, 28, e202104466 CrossRef CAS PubMed.
- D. Hu, Z. Tian, W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 15279–15281 CrossRef CAS PubMed.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- O. Rifaie-Graham, E. A. Apebende, L. K. Bast and N. Bruns, Adv. Mater., 2018, 30, 1705483 CrossRef PubMed.
- M. H. Barbee, T. Kouznetsova, S. L. Barrett, G. R. Gossweiler, Y. Lin, S. K. Rastogi, W. J. Brittain and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 12746–12750 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- S. Cho and Y. Kim, Sens. Actuators, B, 2022, 355, 131274 CrossRef CAS.
- L. Zeußel, P. Mai, S. Sharma, A. Schober, S. Ren and S. Singh, ChemistrySelect, 2021, 6, 6834–6840 CrossRef.
- Y. J. Diaz, Z. A. Page, A. S. Knight, N. J. Treat, J. R. Hemmer, C. J. Hawker and J. Read de Alaniz, Chem. – Eur. J., 2017, 23, 3562–3566 CrossRef CAS PubMed.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- S. Jia, J. D. Du, A. Hawley, W. K. Fong, B. Graham and B. J. Boyd, Langmuir, 2017, 33, 2215–2221 CrossRef CAS PubMed.
- S. Timoshenko, J. Opt. Soc. Am., 1925, 11, 233–255 CrossRef CAS.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- K. Mehta, A. R. Peeketi, L. Liu, D. Broer, P. Onck and R. K. Annabattula, Appl. Phys. Rev., 2020, 7, 041306 CAS.
- Z. Lin, S. Wang, Q. Yan, Q. Yan and D. Cao, Dyes Pigm., 2021, 191, 109384 CrossRef CAS.
- S. Yang, J. Liu, Z. Cao, M. Li, Q. Luo and D. Qu, Dyes Pigm., 2018, 148, 341–347 CrossRef CAS.
- P. Fu, Q. Yan, S. Wang, H. Wu and D. Cao, New J. Chem., 2022, 46, 12600–12608 RSC.
- N. D. Shepherd, H. S. Moore, J. E. Beves and D. M. D’Alessandro, Chemistry, 2021, 3, 728–733 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |