
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of an organometallic yttrium bismuth cluster and elucidation of its electronic structure†
Elizabeth R.
Pugliese
 ,
Florian
Benner
and
Selvan
Demir
*
,
Florian
Benner
and
Selvan
Demir
*
Department of Chemistry, Michigan State University, 578 South Shaw Lane, East Lansing, Michigan 48824, USA. E-mail: sdemir@chemistry.msu.edu
First published on 15th November 2023
Abstract
A rare organometallic yttrium bismuth cluster complex with a heterometallocubane structure at the core was isolated and characterised by single-crystal X-ray diffraction analysis and UV-Vis spectroscopy. The anionic Bi66− core is best described as a Zintl ion. Computational exploration of its electronic structure reveals polarised Y–Bi bonds alongside delocalisation of the Bi–Bi bonds.
The inclusion of bismuth into rare earth (RE) chemistry is exceedingly rare, with only four known metal complexes in which bismuth is immediately bound to a RE element.1–4 Bismuth is slightly radioactive,5 non-toxic, and possesses a large nuclear charge which evokes relativistic effects and large spin–orbit coupling.6 These attributes lead to striking physical properties rendering bismuth compounds relevant for catalysis7 and photovoltaic perovskite materials.8 Our group discovered successful applications of bismuth for the design of single-molecule magnets with real magnetic memory.2,3 Compounds containing bismuth–metal bonds are underexplored relative to other p-block elements, possibly due to enhanced reactivity. Bismuth can form higher nuclearity entities ushering in cluster formation,9 which are known with differing topology and metal count where some follow the Zintl nomenclature. Bismuth-metal clusters bearing six bismuth atoms are particularly rare,2,9–11 and we published the first two molecular lanthanide (Ln) bismuth cluster compounds of the formula [K(THF)4]2[Cp*2Ln2Bi6], where Cp* = pentamethylcyclopentadienyl, and Ln = Tb, Dy. Here, the core is a heterometallocubane with a Bi66− anion bridging the two Ln(III) ions. Attaining a thorough understanding of their electronic structure and bonding situation has been impossible owing to the involved paramagnetic Ln ions and their intrinsic multiconfigurational nature. This prompted the question whether an analogous compound with yttrium ions is synthetically feasible as the slightly smaller ionic radius of Y can cause differing products, especially under reductive synthetic conditions. Here, we report the synthesis and isolation of the first organometallic yttrium bismuth cluster compound [K(THF)4]2[Cp*2Y2Bi6] (1), featuring Bi in a chair conformation with a −6 charge. This molecule constitutes simultaneously the first dinuclear transition metal (TM) complex containing a Bi66− core. Yttrium is innate to a stable isotope (89Y) with a natural abundance of 100% and a nuclear spin of 1/2 allowing a deep insight into the electronic structure of Y compounds. Thus, 1 was characterised via single-crystal X-ray diffraction and density functional theory (DFT) computations. Strikingly, 1 is the only organometallic TM complex containing a metallocubane moiety.
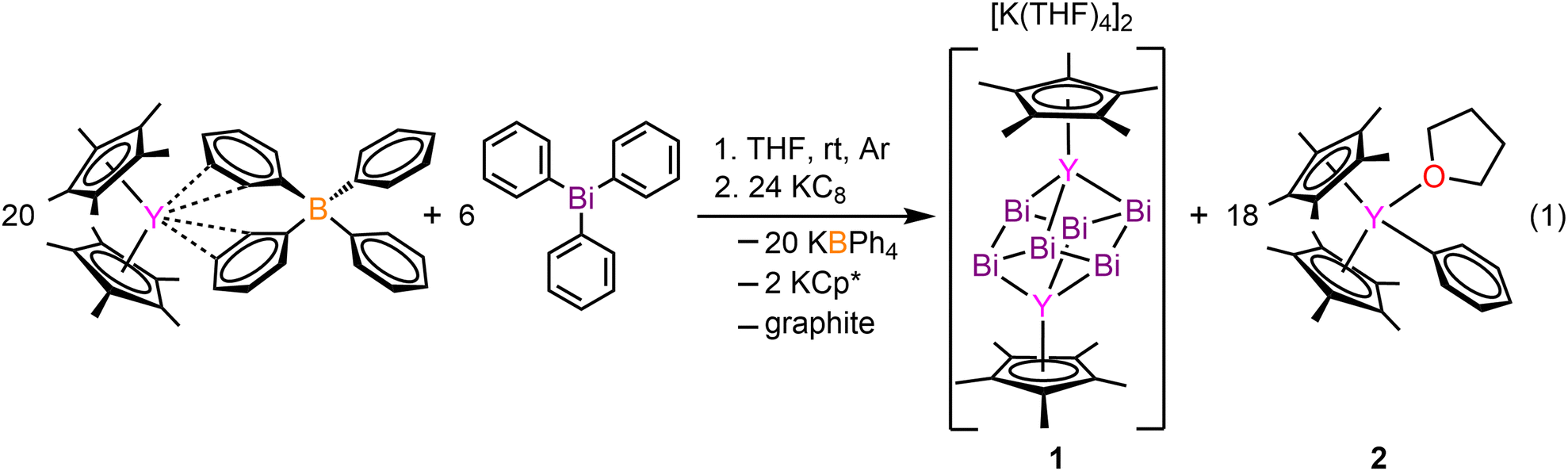
[K(THF)4]2[Cp*2Y2Bi6] (1) was synthesised from the treatment of Cp*2Y(BPh4) with triphenylbismuth (BiPh3) in THF at room temperature followed by addition of the strong reductant potassium graphite, eqn (1). This synthetic route required development of differing reaction conditions to generate 1 relative to the procedure devised for the terbium and dysprosium analogs.2 The addition of KC8 allows for the reduction of the BiIII present in BiPh3 to BiI− in the Bi66− cluster core. The formed byproducts are the insoluble KBPh4 and graphite, alongside Cp*2YPh(THF) (2) which is readily hexane soluble, all of which can be easily separated from 1 through filtration. Black block-shaped crystals of 1 were grown from a diffusion of diethyl ether into a concentrated THF solution at −35 °C. This crystallisation method is notable as Zintl ions are typically synthesised from the mixture of alkali/alkaline earth metals and main group elements, and oftentimes necessitate the use of highly polar solvents such as ethylenediamine (en).12 Such crystallisation conditions are not compatible with organometallic RE chemistry. Thus, we note that 1 is readily synthesised from BiPh3 as the Bi source which is soluble in THF. 1 crystallises in the P21/n space group and features a diyttrium complex that resides on a crystallographic inversion centre, such the two yttrium ions are equivalent by symmetry, Fig. 1. Each Y site is tripositive, six-coordinate, and ligated to one Cp*− ligand. The Y(III) centres are bridged by six Bi(I) ions arranged to a Bi66− core which adopts a chair conformation reminiscent to cyclohexane. Thus, 1 constitutes the first complex of a molecular yttrium bismuth heterometallocubane. The Y–Bi distances in 1 of 3.04(2), 3.09(2), and 3.20(2) Å are in excellent agreement with intermetallic solid state compounds such as Y5Bi3 (3.051–3.395 Å),13 and are similar to that of the Tb and Dy complexes.2 The distances are shorter than 3.2335(2) Å and 3.1879(6) Å in dinuclear Y complexes containing a Bi22− unit, (Cp*2Y)2(μ-η2:η2-Bi2) and a Bi23− radical, [K(crypt-222)][(Cp*2Y)2(μ-η2:η2-Bi2˙)]·2THF,3 respectively. This discrepancy may be attributed to the decreased steric bulk of the ancillary half-metallocene moiety.3 The Bi–Bi distances in 1 are 3.022(9), 3.046(10), and 3.060(8) Å, suggestive of a Bi–Bi single bond (3.02 Å).2,141 constitutes the only compound comprising a 4d metal that forms a cube or a true cyclohexane conformer for Bi6. The closest TM complex serves the icosahedral complex [K(18-c-6)][Nix@{(CO)8Ni6Bi6}], containing a Bi6 unit albeit with an interstitial Ni atom and a −4 charge.11 The average Y–Cnt distance is 2.228(22) Å, where Cnt = Cp* ring centroid, is shorter than that of the Tb (2.338(1) Å) and Dy (2.365(1) Å) congeners owing to slight differences in ionic radii.15 Relative to other halfmetallocene complexes such as Cp*Y(CH2C6H4NMe2-o)2 the Y–Cnt bond in 1 is considerably shorter (2.390(1) Å),16 likely due to reduced steric repulsion between the Cp* methyl groups and the Bi ions compared to the bulkier organic ligands. 1 is isostructural with the Tb and Dy homologs.2
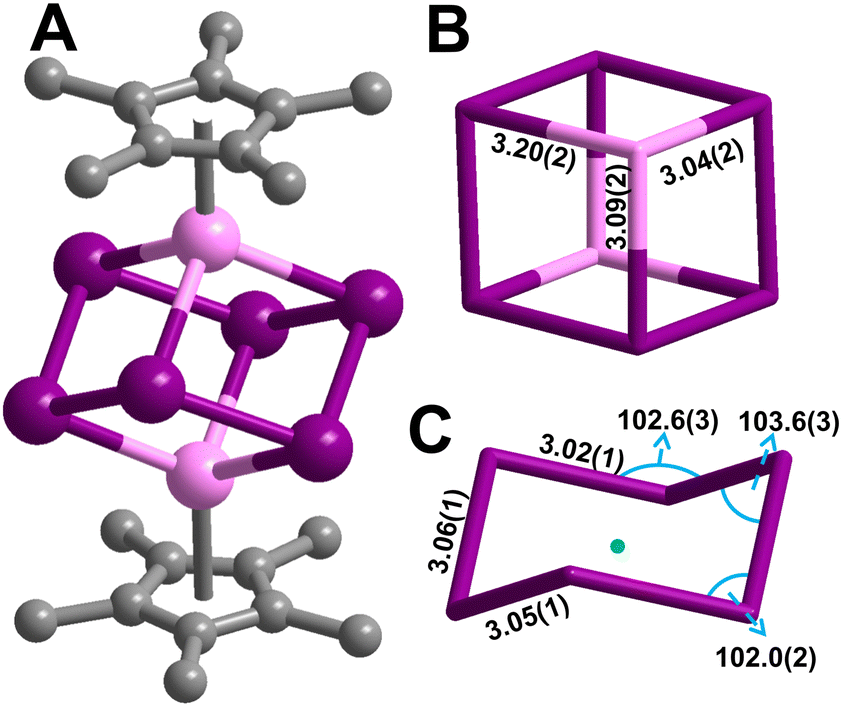 | ||
| Fig. 1 (A) Structure of [K(THF)4]2[Cp*2Y2Bi6] (1). Pink, purple, and grey spheres represent Y, Bi, and C atoms, respectively. H atoms and both [K(THF)4]+ cations have been omitted for clarity. (B) Neutral {Y2Bi6} cube enlarged with Y–Bi distances labelled. (C) Bi66− chair enlarged with distances and angles labelled. The green mark represents the inversion centre. All distances (Å) and angles (°) are rounded up for clarity and full values are in the main text. | ||
UV-Vis spectra of 1 were recorded at concentrations of 20–100 μmol L−1 in THF at room temperature (Fig. S10, ESI†). In all cases, absorption was observed over the entire range of the visible region, never reaching absorption levels as low as a blank THF sample. Several distinguishable peaks in the visible region between 677 and 410 nm could be identified, which primarily result from transitions of occupied Bi66−-centred orbitals to Bi66−-centred virtual orbitals as confirmed via TDDFT calculations (Table S7, ESI†). Notably, excitations into the two near-degenerate lowest-lying unoccupied π* orbitals are of negligible importance for transitions above 420 nm.
Excitingly, 1 allows for the first-time a thorough analysis of the electronic structure of bismuth clusters in the vicinity of organometallic RE scaffolds, due to the diamagnetic Y ions. In addition, their most stable +III oxidation state precludes ambiguities regarding the Bi oxidation state, which often cannot be assigned in Zintl clusters based on TM.9 To this end, we performed quantum chemical calculations via DFT methods, and the obtained electron density was analysed via natural localised molecular orbitals (NLMOs), nucleus independent chemical shift (NICS) and Bader's quantum theory of atoms in molecules (QTAIM).
The calculated frontier orbitals reveal that the highest occupied orbitals (HOMO−2–HOMO) involve Y and Bi66− contributions (Fig. 2), while the low-lying unoccupied orbitals (HOMO+1–HOMO+3) are primarily Bi66− centred (Fig. 2). Noticeably, the HOMO features a π-bonding symmetry, a feature indicative of metal ϕ- or σ-aromatic systems.9,10
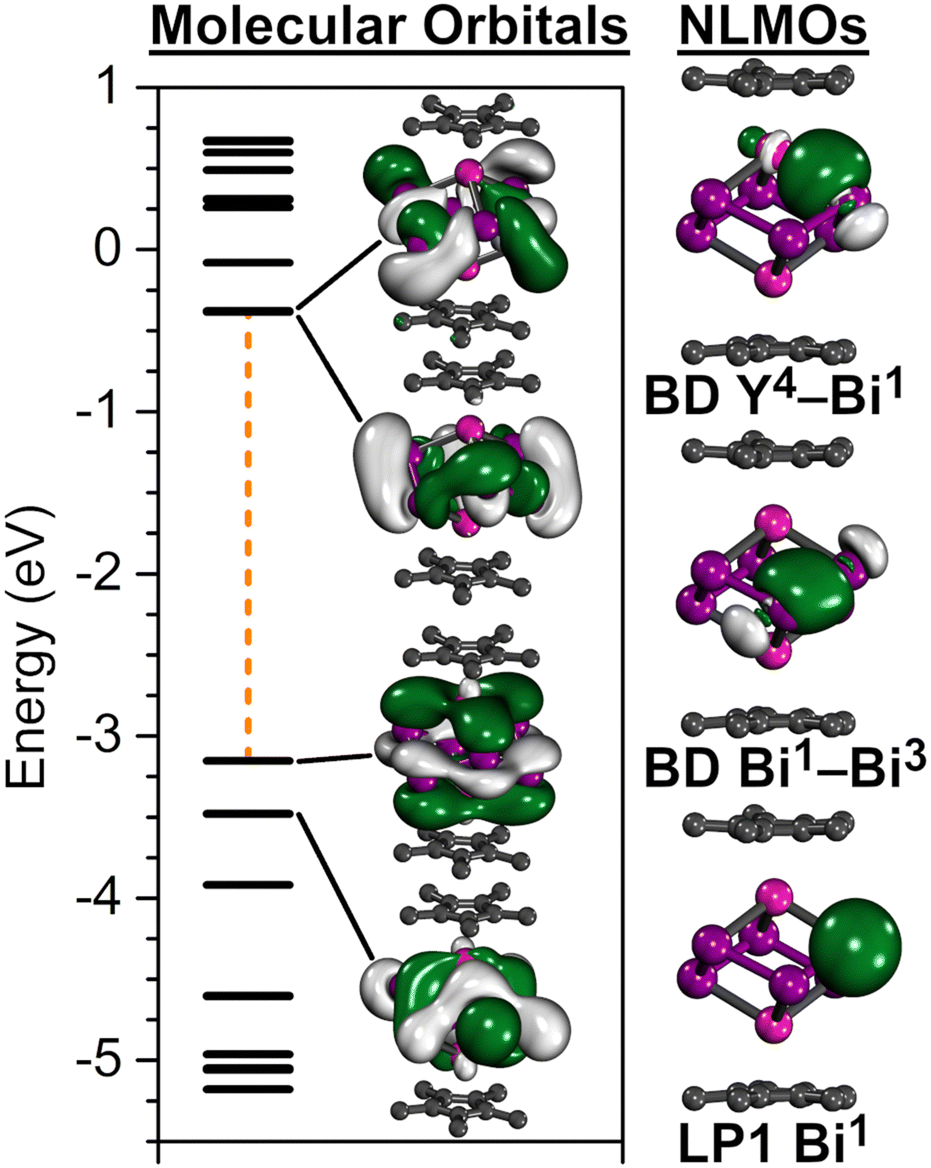 | ||
| Fig. 2 Left: Energy diagram of the 10 highest occupied and 10 lowest unoccupied orbitals with graphic depiction of the respective two highest occupied and lowest unoccupied frontier orbitals. The orange dashed line highlights the HOMO–LUMO gap (3.14 eV). Right: Depiction of selected representative natural localised molecular orbitals (NLMO) illustrating a strongly polarised Y–Bi interaction (top), a Bi–Bi interaction through 6p orbitals (middle), and a Bi lone pair (bottom). | ||
NLMO localisation of the canonical orbitals provide further insight into the bonding situation. For every Bi atom two bonding interactions to neighbouring Bi atoms were observed, which are primarily of 6p character with minor contributions from the Y atoms (∼6%) (Fig. 2). Each Bi atom partakes in one Y–Bi bond, which is strongly polarised towards the Bi atoms (∼78% Bi and ∼18% Y contribution) with ∼16% s-/∼83% p-hybridisation in Bi and ∼37% s-/∼62% d-hybridisation in Y. In addition, each Bi atom features one lone pair (LP) of primary s-character (Fig. 2). Although all bonds appear to be localised within the cluster, a small degree of electron delocalisation is indicated via the occupation difference between NBO and NLMO, where the LP NBOs exhibit slightly decreased occupancies of 1.93. This is also reflected within the second order perturbation analysis, where the Bi LPs are donating into vacant Y d-orbitals as well as antibonding Y–Bi and Bi–Bi bonds (Table S5, ESI†).
The electron localisation function (ELF) serves as a visualisation tool for the probability to localise an electron pair within a given spatial extent. ELF values range between 0 and 1, where 1 represents ideal localisation, 0.5 corresponds to electron-gas like probability, and <0.5 indicates delocalisation in low-density regions.17 The ELF for 1 shows interacting electrons between neighbouring Bi atoms are delocalised, while the LPs are localised and visible as outward-facing clouds (Fig. 3 and Fig. S7, ESI†). This can be correlated to the cluster interacting weakly with two [K(THF)4]+ cations in the solid state where K⋯Bi = 3.9 (2) Å (Fig S4, ESI†). Lastly, the highly ionic nature of the Y–Bi interactions are visible as gaps between the Bi and Y atoms.
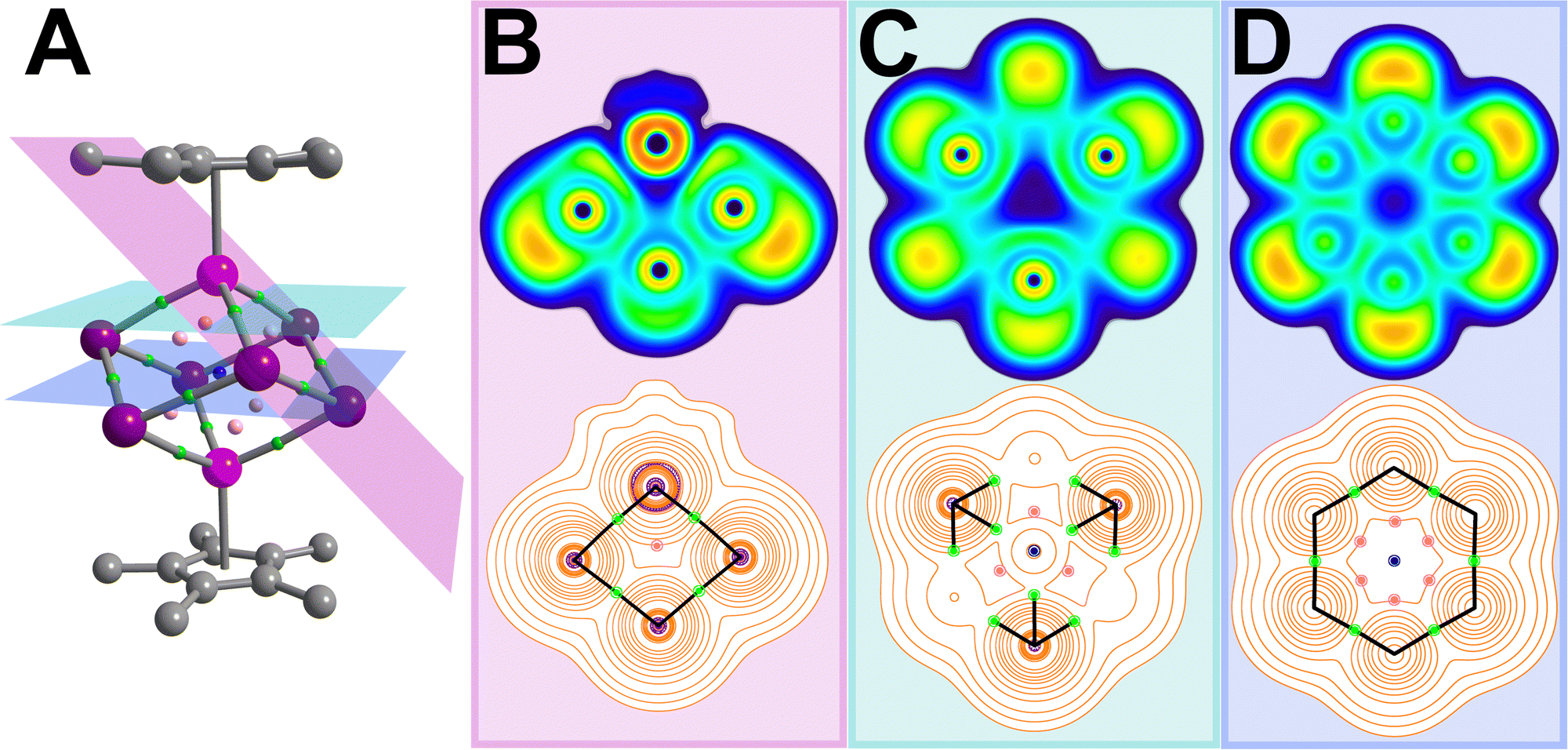 | ||
| Fig. 3 Results of the QTAIM analysis of 1, where large pink, purple, grey spheres represent Y, Bi and C atoms, and small pale pink, green and blue spheres represent bond critical (BCP), ring critical (RCP) and cage critical points (CCP), respectively (A). The electron localization function (ELF, top) and Laplacian of the electron density (bottom) are plotted along the pink, green and blue planes through the inner-cluster BCPs (B–D). ELF values are defined between 0 (dark blue) and 1 (red) to map electron pair probability within a spatial region, where a value of 1 represents ideal localisation and 0.5 (green) correspond to electron-gas like probability, while values <0.5 indicate delocalisation in low-density regions. In the Laplacian plots, orange and purple contour lines correlate with charge depletion and accumulation, and black lines represent bond paths connecting neighbouring Bi and Y atoms, respectively. In (B) contributions from the Cp* ligand were omitted for clarity. Real space values at all critical points are listed in Table S6 (ESI†). | ||
Bi atoms in close proximity, i.e. positioned in a prismatic array, may feature substantial ϕ aromaticity.9 The Bi6 assembly in 1 is peerless and potentially aromatic. To probe this, the powerful method NICS was chosen as a phenomenological descriptor for the aromatic shielding/deshielding. NICS values were calculated along the axial (through Cnt–Y bonds) and equatorial (through opposing Bi atoms) axis (Fig. S8, ESI†). The obtained values were plotted against the distance in Å from the cluster core (Fig. S8, ESI†). The NICS0 value (centre of the cluster) is negative (δNICS0 = −7.77 ppm), which at first glance suggests a diatropic ring current and aromaticity. Relative to other metal aromatic systems, this value is much smaller; for example [K(crypt-222)][{CpRu}3Bi6] exhibits δNICS0 = −21.3 ppm,9 and [Re2(CO)8(μ-BiPh)2] δNICS0 = −37.8/−41.78 ppm.18 The equatorial NICS profile reveals progressively negative NICS when approaching the Bi atoms. Conversely, traversing the axial direction yields positive NICS values between ±1.25 and 2.25 Å near the Y atoms, before adopting negative values close to the aromatic Cp rings again. A planar, organic, and aromatic compound exhibits a gradual NICS profile,18,19 which is only seen close to the cluster centre (∼±1 Å) before contributions from either Y or Bi dominate the NICS values. Hence, the slight deshielding observed at the NICS0 position likely arises from the proximity of the Bi− ligands and their large ionic radii rather than a true ring current and associated metal aromaticity (see space filing model in Fig. S2, ESI†). The Bi66− chair was also calculated without the {Cp*Y}2+ fragments for comparison (Fig. S9, ESI†). Here, the NICS0 and values perpendicular to the chair are positive, indicative of antiaromaticity. Hence, the highly Lewis acidic halfmetallocene moieties affect the electronic structure of the Bi66− chair, due to the vicinity of the cluster core and the Y centre (2.146 Å).
The electronic structure analysis of 1via QTAIM visualises first the Laplacian of the electron density (∇2ρ) which proves the highly ionic bonding suggested via NLMO investigation (Fig. 3). The real space values at bond critical points (bcp) show that all bonding interactions within the [Y2Bi6]2− cluster fall within the range of intermediate bonding regime between ideal covalent and ionic bonding, best exemplified by the slightly positive ∇2ρ and slightly negative H(r) values and negative H(r)/ρ ratios. The ellipticity of the electron density ε(r) is near 0 for each bond, further hinting at single bonds in the cluster moiety. The lack of electron delocalisation is reflected in the curvature of electron density perpendicular to the ring critical points (RCPs, λπ3) of the cubane faces, which is two orders of magnitude smaller than the value found for benzene.20,21 The associated Mayer bond indices (MBOs) clarify the bond strengths, where values ∼1 are calculated for each Bi–Bi bond and ∼0.5 for each Y–Bi bond.
In conclusion, the first heterometallocubane complex, [K(THF)4]2[Cp*2Y2Bi6], 1, containing a Bi66− cluster and a transition metal was isolated, wherein the bismuth core bridges two {Cp*Y}2+ fragments and the hexabismuth moiety features a chair-like conformation akin to cyclohexane. Computational bonding analysis of 1 revealed polarised Y–Bi bonds and delocalised electrons within the Bi–Bi bonds. NICS calculations uncovered potential diatropic ring current, which however is ascribed to the proximity of the large Bi ions, rather than reflecting an authentic ring current. Notably, analysis of the bismuth chair alone reveals a paratropic ring current, which implies that the YIII centres alter the electronic structure of the cluster core significantly. These foregoing results are crucial for a thorough understanding of currently underexplored RE-Bi complexes, which are relevant for SMM design and potentially other future applications such as in small molecule activation.
E. R. P. prepared and characterised all compounds. F. B. performed DFT calculations and interpreted results with help of E. R. P. S. D. assisted with data analysis, formulated, and directed the research, and wrote the manuscript with input from all authors.
S. D. is grateful to the Department of Chemistry at Michigan State University (MSU) for start-up funds and the Institute for Cyber-Enabled Research for support. Funding for a single-crystal X-ray diffractometer was provided by the National Science Foundation under grand No. CHE-1919565.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 1589–1591 Search PubMed.
- P. Zhang, F. Benner, N. F. Chilton and S. Demir, Chem, 2022, 8, 717–730 CAS.
- P. Zhang, R. Nabi, J. K. Staab, N. F. Chilton and S. Demir, J. Am. Chem. Soc., 2023, 145, 9152–9163 CrossRef CAS PubMed.
- E. R. Pugliese, F. Benner and S. Demir, Chem. – Eur. J., 2023 DOI:10.1002/chem.202302687.
- P. De Marcillac, N. Coron, G. Dambier, J. Leblanc and J. P. Moalic, Nature, 2003, 422, 876–878 CrossRef CAS PubMed.
- P. Pyykko, Chem. Rev., 1988, 88, 563–594 CrossRef CAS.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- L. Zhang, K. Wang and B. Zou, ChemSusChem, 2019, 12, 1612–1630 CrossRef CAS PubMed.
- B. Peerless, A. Schmidt, Y. J. Franzke and S. Dehnen, Nat. Chem., 2023, 15, 347–356 CrossRef CAS PubMed.
- L. Qiao, D. Chen, J. Zhu, A. Muñoz-Castro and Z. M. Sun, Chem. Commun., 2021, 57, 3656–3659 RSC.
- J. M. Goicoechea, M. W. Hull and S. C. Sevov, J. Am. Chem. Soc., 2007, 129, 7885–7893 CrossRef CAS PubMed.
- K. Beuthert, B. Peerless and S. Dehnen, Commun. Chem., 2023, 6, 109 CrossRef CAS PubMed.
- B. C. Wang, E. J. Gabe, L. D. Calvert and J. B. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1440–1445 CrossRef.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, 3rd edn, 1960 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- T. Shima, M. Nishiura and Z. Hou, Organometallics, 2011, 30, 2513–2524 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- E. Schott, X. Zárate and R. Arratia-Pérez, Polyhedron, 2011, 30, 846–850 CrossRef CAS.
- E. R. Pugliese, F. Benner, E. Castellanos, F. Delano and S. Demir, Inorg. Chem., 2022, 61, 2444–2454 CrossRef CAS PubMed.
- S. T. Howard and T. M. Krygowski, Can. J. Chem., 1997, 75, 1174–1181 CrossRef CAS.
- N. A. Da Silva, R. E. Bruns and R. L. A. Haiduke, J. Phys. Org. Chem., 2021, 34, 1–12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2292178 and 2292179. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04520j |
| This journal is © The Royal Society of Chemistry 2023 |