
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional silicone oils and elastomers: new routes lead to new properties
Michael A.
Brook

Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, ON L8S 4M1, Canada. E-mail: mabrook@mcmaster.ca
First published on 5th October 2023
Abstract
Silicones are mostly utilized for their stability to a range of vigorous environmental conditions, which arises, in part, from the lack of functionality in finished products. The commonly used functional groups in silicones, e.g., SiH, SiCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, are mostly consumed during final product synthesis. Organic functional groups may also be found in silicone products, including organic alcohols, amines, polyethers, etc., that deliver functionality not achieved by traditional organic polymers (e.g., aminosilicones, softening of fabrics; silicone polyethers, superwetting agricultural adjuvants). However, relatively little organic chemistry is practiced in commercial silicones, limiting the types of desirable functionality that can be attained. We report the utilization of a series of simple-to-practice organic reactions that take place efficiently on silicone oils to allow the preparation of a wide variety of functional silicones. The silicone oil starting materials typically act as both solvent and educt to allow many of the newer reactions, such as Click processes, to be used to tune the properties of both silicone oil and elastomer products. The review considers the concept of ‘functionality’ to include: the reactive groups used to enable synthesis of more complicated structures; and separately, the functional properties of the product silicones. One such property that is considered throughout is degradability at end-of-life, which is related to the sustainability of silicones.
CH2, are mostly consumed during final product synthesis. Organic functional groups may also be found in silicone products, including organic alcohols, amines, polyethers, etc., that deliver functionality not achieved by traditional organic polymers (e.g., aminosilicones, softening of fabrics; silicone polyethers, superwetting agricultural adjuvants). However, relatively little organic chemistry is practiced in commercial silicones, limiting the types of desirable functionality that can be attained. We report the utilization of a series of simple-to-practice organic reactions that take place efficiently on silicone oils to allow the preparation of a wide variety of functional silicones. The silicone oil starting materials typically act as both solvent and educt to allow many of the newer reactions, such as Click processes, to be used to tune the properties of both silicone oil and elastomer products. The review considers the concept of ‘functionality’ to include: the reactive groups used to enable synthesis of more complicated structures; and separately, the functional properties of the product silicones. One such property that is considered throughout is degradability at end-of-life, which is related to the sustainability of silicones.
 Michael A. Brook | Mike Brook, PhD McGill (1994); PDF ETH-Zürich (1985) is a Distinguished University Professor, Chair of Sustainable Silicone Polymers in the Chemistry and Chemical Biology Department at McMaster University. He won the F. S. Kipping Award in Silicon Chemistry (ACS, 2016), the Canadian Institute for Chemistry Award in Green Chemistry (CIC, 2023), Macromolecular Science & Engineering Award (CIC, 2017), and was a Canadian Killam Research Fellow. Brook's research is focussed on improving sustainability of useful silicones by: creating them while avoiding catalysts (click reactions) and developing methods to incorporate natural materials into silicones to add new properties, including facile environmental degradation. |
1. Introduction
Synthetic chemists spend much of their time attempting to coerce functional groups to form bonds leading to desired small molecules, polymers, resins and, more broadly, to create molecular structures that possess desired properties. Therefore, when considering ‘functional silicones’ there are two aspects to consider: the functionality (reactive groups) that will enable specific bonds to form and the resulting, hopefully beneficial, structures that result with specific functionality – interesting properties. This review is structured to present both types of meaning: normal text refers to functional reactivity, while italics refers to properties of the molecules (including applications) produced to reflect these two complementary objectives.A brief overview of traditional silicone chemistry is provided as a standard against which new approaches can be measured. The introduction of ‘traditional’ functional groups in the world of silicones (traditional = commercial) – both oils and elastomers – is then presented. The bulk of the review involves non-traditional functional groups introduced first with, and second without, the need for catalysts. An attempt was made to consider functional oils first then elastomers, and finally silicones at interfaces. However, in some cases it was more logical to consider processes that lead to the oils and elastomers at the same time. To help the reader “Elastomers” are called out in headings. In each of the cases, the functionality – utility – of the silicone systems will be reported. It is hoped that the reader sees this review a challenge: why not add even more functionality to silicones to make them yet more valuable?
We note this review is written with a bias related to sustainability. Particular attention is focused on both products and processes that better fit the principles of Green Chemistry,1 including use of catalysts instead of stoichiometric reagents, or no catalyst at all, Click reactions without byproducts, alternatives to covalent bonding to create elastomers, etc. The products themselves should be endowed with interesting functionality that, when possible, should first include the possibility of reuse and, second, degradation in the environment to give safe compounds at end of life. The review concludes with some thoughts about the benefits of functionality in the context of sustainability.
2. What is so great about silicones? What they don’t do!
2.1 Resistance to thermal, oxidative and irradiative stress
Silicones are mostly comprised of Me2SiO fragments that are very stable to oxidation (it is hard to oxidize methyl groups).2,3 The C–Si bonds are prone to oxidation on thermodynamic grounds (Si–C vs. much stronger Si–O) but are kinetically quite stable. Si–O bonds are very strong; virtually all the silicon compounds found in nature are silica or silicates with oxygen forming all bonds to silicon atoms. While thermodynamically stable, the Si–O bonds in silicones can be kinetically reactive when 1 oxygen ligand is exchanged for another, particularly at pH conditions away from neutrality (especially pH < 5 and pH > 9) or with good nucleophiles in non-aqueous conditions (Fig. 1A). For the purposes of this paper, the traditional silicone moieties Me2SiO (D) and Me3SiO (M) will be considered ‘non-functional’, by which we mean they are much, much less reactive than the other functional groups present in reaction mixtures (note: phenylsilicones that also contain MePhSiO and Ph2SiO monomers are a subset of commercial silicones that are prone to protiodesilylation to give benzene and trifunctional silanes, but only under highly acidic conditions. We will not consider such extreme conditions and phenylsilicones will also be considered ‘non-functional’).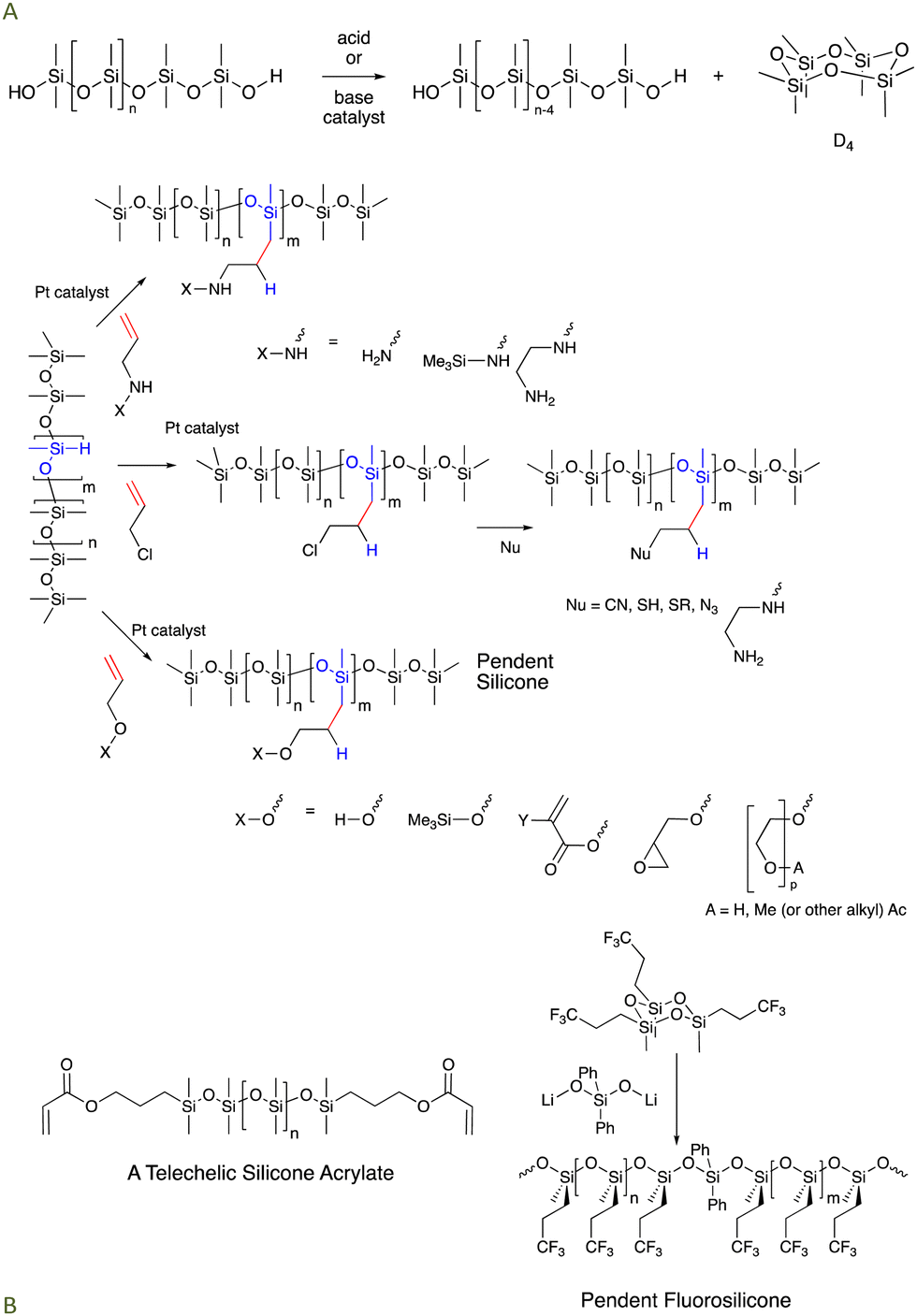 | ||
| Fig. 1 (A) Silicone equilibration under acidic or basic conditions. (B) Hydrosilylation of γ-functional allyl groups to prepare amino-, halo-, and alcohol/ether modified silicones. Examples of telechelic and pendent silicones are provided, including a pendent fluorosilicone.15 | ||
Silicone oils only start to thermally degrade – in air, or the presence of oxygen – near 325 °C; phenylsilicones have even higher thermal stability. Silicones are used, for example, as domes for LEDs or in the O-rings for UV lamps because of their much higher resistance to thermal and photodegradation, including oxidation, than traditional organic polymers.
2.2 Mobility, water repellency and gas permeability
The surface energy of silicones is very low – between alkanes and fluorocarbons – so they are much less soluble in water than non-polar organic compounds. They are also oleophobic and can form dispersions rather than solutions with organic oils.4 Unlike organic polymers, linear dimethylsilicones are oils regardless of molar mass; even a polymer of molar mass of 1 × 106 g mol−1 is a highly viscous oil, not a solid. These oils also have an exceptionally low glass transition temperature ∼−120 °C, so they remain processable at temperatures far below those for organic polymers. Finally, silicones love air interfaces. Their mobility and the thermodynamic advantage in creating interfacial area with air drives many of the interesting physical properties, for example, as foaming agents, defoamers and film formers (the origins of these effects have been extensively reviewed5,6).The very large free volume of silicone polymers leads to very high gas permeability5 which, in turn, leads to utilization in applications like contact lenses that must allow oxygen to pass to the cornea through the lens.7
2.3 Silicone oil as a solvent
Traditional synthetic organic chemistry frequently makes use of hydrophobic media for bond forming processes and hydrophilic media to both quench reactions and facilitate organic product separation from spent reagents: oil vs. water. The physical properties of silicone oils suggest that we could consider silicone oils as very non-polar solvents. In the discussion below, silicone polymers are often used as both the educt for reaction and the solvent/medium in which the reactions take place. This is a gift to silicone chemists that is not generally available to other (polymer) chemists. To the degree that the proposal holds, it means the search for new functionality, based on organic chemistry, can start with a basic undergraduate textbook (i.e., where it says solvent, think silicone oils).3. Traditional functional silicones: SiH, OH, ∼CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH2
CH2
Silicone polymers originate from sand converted to silicon metal and then, via the Direct Process, to chlorosilanes (e.g., Me2SiCl2, Me3SiCl, MeHSiCl2). The details of these industrial processes are beyond the scope of this paper but have been extensively reviewed.8–10 Although the monomers used to prepare silicone polymers are highly functional, there are only a few functional groups that remain in commercial polymer products: SiH, Si-Vinyl (Si–CHCH2), and SiOH. Silanols (HOSiR3) are typically found at chain ends of PDMS polymers (PDMS = polydimethylsiloxane (∼Me2SiO)n∼), where they mostly serve as nucleophiles that react with small silanes for moisture cure elastomers (RTV – room temperature vulcanization, see the section on Curing Processes for Traditional Elastomers). The most useful functional group for synthetic chemists is the SiH group; monomers containing this group (Me2HSiCl, MeHSiCl2) are produced as by-products in the Direct Process in excess to needs of the silicone industry. Si–H bonds are much weaker than C–H bonds. Furthermore, unlike alkanes silanes are hydrides, Siδ+–Hδ−, and the silicon atom is sterically very accessible, so both kinetics and thermodynamics favor reactions, particularly under (aqueous) base or in the presence of silaphilic nucleophiles that convert Si–H to much stronger Si–O bonds. The reaction with aqueous base can be violent, and leads to production of flammable hydrogen gas (note that, under controlled conditions, this process is used for creating elastomeric silicone foams).11
3.1 Alkoxysilane reactivity
Alkoxysilanes, for example, R′′Si(OR′)3 (R′′ = Me, Ph, OR′) are small molecules frequently used to crosslink silicones in the presence of moisture. The reaction of alkoxy groups with silanols or water to first produce SiOH groups (plus the alcohol by-product – R′OH), and then a disiloxane R′′(RO′)2SiOSi(OR′)2R′′ is thermodynamically favored12 and always occurs even in the absence of catalysts, for example in reagent bottles once opened to air, albeit slowly. The rates of hydrolysis and condensation are significantly accelerated by catalysts, particularly those based on tin (see RTV Elastomers below).Within the silicone business, “Si–O–C” linkages are always understood to be temporary, which is not always the case. Phenols are more acidic than alcohols and, on that basis, should be better leaving groups under basic conditions. However, just as phenyl esters can be slower to hydrolyze than alkyl esters,13 phenoxysilanes are very stable to hydrolysis under both basic and acidic conditions. The formation of phenoxysilanes using the Piers-Rubinsztajn (PR) reaction14 is described in several places in this review because the products have surprising stability towards hydrolysis.
3.2 Organic functional groups on silicones
Imagine if synthetic organic chemists were told they were limited to 3 or 4 functional groups when developing new products? How could one design and create complex molecules with such limitations? While new processes and new functional groups bring complexity and, of course, some bad outcomes, much more importantly they provide an opportunity to leverage different synthetic protocols to create compounds with new interesting properties, in this case silicones. The silicone industry has introduced a limited number of organofunctional silanes and silicones for this purpose. While completely speculative on the part of the author, the limited presence of organofunctional silicones in industry arises from the need to curate product lines to focus on a narrow range of higher volume products. In other words, once the functional materials described below can be shown to be commercially viable, they will be developed into products.3.3 Further modification of organofunctional groups in traditional silicones
Anyone schooled in organic synthesis will recognize that the organic functional groups (Fig. 1B) are entry points for much broader synthesis. Using them, it should be possible to modify and tune silicones to target specific desired properties; both silicone reactivity and physical properties like solubility/melting point/dispersibility in water can be encoded. For example, simple organic chemistry has been used to turn these basic functional silicones into silicones bearing γ-propyl-phosphates, quaternary ammonium salts, and other functional groups. It should be noted that most silicone oils have functional groups either at both ends of a linear chain (telechelic) or randomly spread along the chain (pendent, Fig. 1B).3.4 Desirable properties of traditional, functional silicone oils
Most of the applications of functional silicone oils arise from their interfacial properties. 5 The materials readily migrate across solid surfaces in air and also to water/silicone or oil/silicone interfaces.Aminoalkylsilicones spread across cotton and other fibers and convey ‘softness’ to fabrics. 18 The use of high density cationic charge is a common strategy for killing bacteria on surfaces. 19–21 Silicone quaternary ammonium surfactants are often used to size fabrics to convey this antibacterial activity. 22 Polyether-modified trisiloxanes are widely used as adjuvants in agriculture where they facilitate spreading of pesticides/herbicides on leaf surfaces. 23 Silicone polyethers aid in controlling bubble density and size 24 to structure polyurethane foams, 25 such as those used in furniture. Ironically, silicones are also excellent defoamers and, because of their safety with humans, are used in a myriad of industries, including the food industry, pulp and paper, etc.
4. Traditional silicone elastomers: curing processes
Curing processes consume most of the functional groups in silicone oils (pre-elastomers) to form crosslinks in commercial silicone elastomers. Three different curing processes dominate the formation of commercial silicone elastomers: radical cure; platinum-catalyzed hydrosilylation; and moisture cure (nucleophilic substitution, room temperature vulcanization RTV. Note: some platinum cured materials are also described in the literature as RTV, Fig. 2).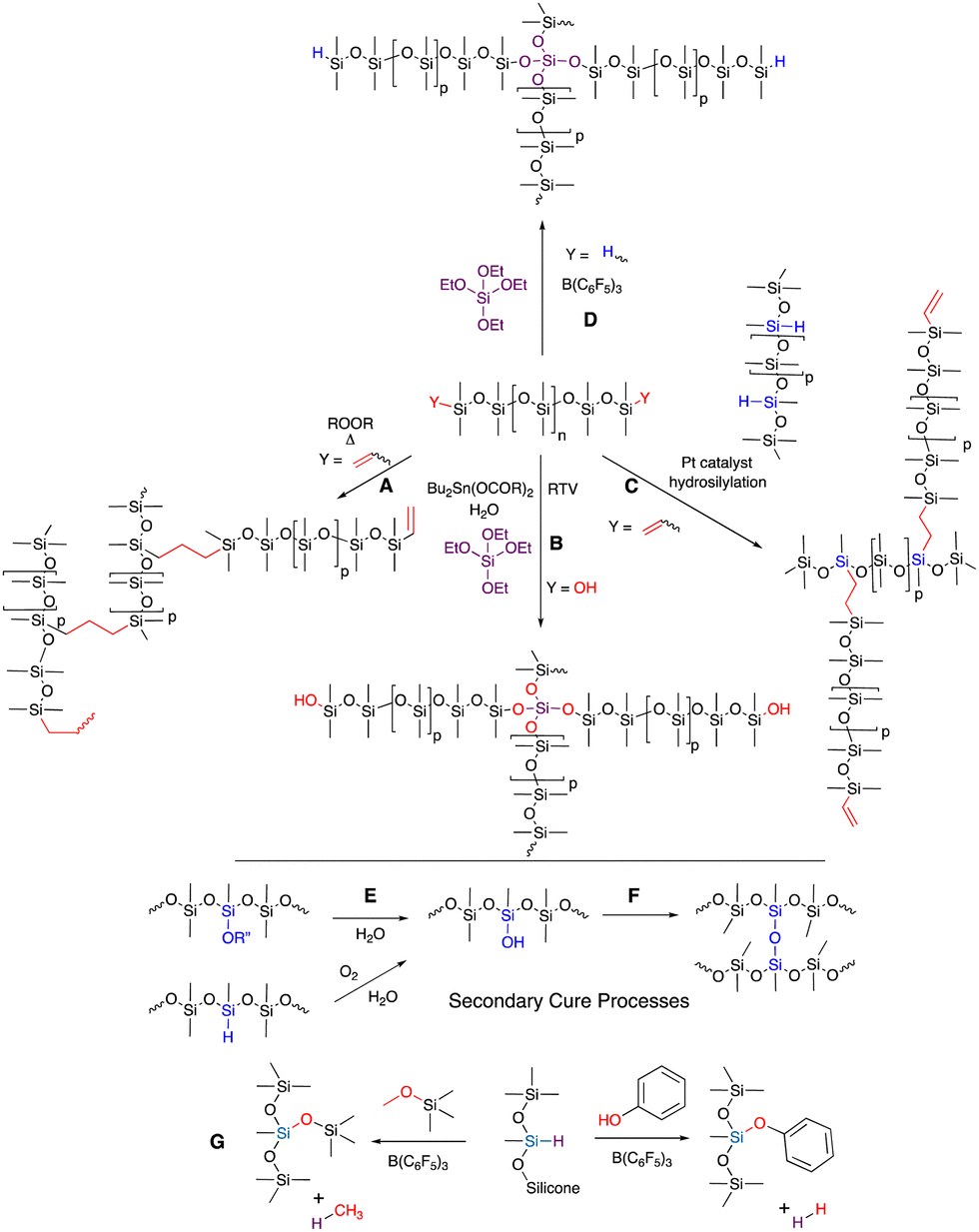 | ||
| Fig. 2 (A) Radical cure with vinylsilicones. (B) RTV moisture cure. (C) Hydrosilylation cure. (D) Piers–Rubinsztajn (PR) cure. (E) Secondary oxidation/hydrolysis processes that generate free silanols that, over time, (F) lead to further cure. (G) The PR reaction with alkoxysilanes and phenols. | ||
4.1 Organic radical cure
Radical cure on silicones usually involves use of a radical initiator to randomly generate SiCH2˙ radicals that then recombine to give a 2-carbon interchain link, or a more specific reaction (often a lower temperatures) with vinylsilicones that react with a randomly generated SiCH2˙ to form a 3-carbon link (Fig. 2A). While somewhat better network control is obtained when vinylsilicones are used, in neither case does the control match that of either RTV or hydrosilylation (Fig. 2B and C). Radical catalysts and residues will typically remain within the silicone rubber after cure and, for some applications, must be extracted out before use.4.2 Hydrosilylation
Hydrosilylation is widely utilized to make silicone elastomers because of its high efficiency, and the ability to control the reaction temperature for cure, from room temperature to nearly 200 °C. The strength of the catalyst/inhibitor complex determines the offset temperature for cure that accompanies inhibitor decomplexation from Pt (Fig. 2C). This allows the user to work with the material at lower temperatures with a reasonable ‘pot life’, but to lock in cure when desired by heating. Hydrosilylation cure is used in applications ranging from in vivo cure of biomedical implants to injection molded bread loaf pans. Very little platinum is required to catalyze the process – a few ppm – but one wonders at the logic of using an endangered and expensive element in a material that, at end of life, will be placed in the environment without the ability recover the platinum. Note that other catalysts may also work effectively, including those based on Ru, Ir, Co, Pd, Fe etc., but they may be more expensive or need special conditions (e.g., water/air free).26 The workhorse catalysts are based on platinum, including Speier's catalyst (H2PtCl6) in the early days of hydrosilylation cure and Karstedt's catalyst,16,17 currently.4.3 Room temperature vulcanization
RTV is a process that requires moisture as a co-reagent; water may be explicitly provided in a formulation or be present in moisture in the air (Fig. 2B). The rates of cure, based on the SN2 reaction of silanols with functional silanes, are controlled by use of different leaving groups, with alkoxy groups slower, acetoxy groups faster and oximes somewhere in the middle. The rates of nucleophilic substitution at a tri- or tetrafunctional silane gradually slow as each leaving group is displaced by a silanol terminated silicone chain. Complete cure can take a long time.The commonly used catalyst for these processes is dialkyltin dicarboxylate (e.g., dibutyltin dilaurate) or related tin compounds, particularly if there is moisture present; titanium catalysts operate under drier conditions. Alkyltin compounds are not benign. They are known to be toxic to many organisms and relatively high catalyst loadings of 0.1–1 wt% or more are required.
4.4 Beneficial properties of silicone elastomers
These curing processes will consume most, but not all the functional groups in the pre-elastomers. Hydrosilylation cure products are usually formulated to leave a known excess of vinyl or SiH groups to ensure complete loss of one of the functional groups and allow reproducible control of crosslink density. In RTV, silanols on silicone chains are normally matched to leaving group concentration on the crosslinker. However, residual groups can be converted to silanols that then couple to give very slow secondary cure (Fig. 2E and F). In spite of this comment, a strong benefit of any of these silicone elastomers is that they possess very, very little residual reactivity, which accounts for their utility at high/low temperatures; in the presence of water or other reagents (except acids/bases); high current/voltage or high degree of irradiation with light.One very special silicone elastomer is derived from trifluoropropyl groups (Fig. 1B). These rubbers have even lower surface energy than traditional silicones. Unlike fluoropolymers based on carbon, the silicones are much more mobile and are both hydrophobic and lipophobic (hence their use as gaskets in the presence of petroleum products).
4.5 A new reaction to prepare traditional silicones: Piers Rubinsztajn (PR)
The Piers Rubinsztajn reaction (PR) efficiently converts hydrosilanes/hydrosilicones to disiloxanes upon reaction with alkoxysilanes, silanols, water or phenols to co-generate alkanes or hydrogen by-products. Piers reported this reaction as a undesired side reaction of carbonyl reduction and Rubinsztajn noted this was a new way to make silicones.27,28 PR reaction conditions convert phenols and alkyl aryl ethers, including Vitamin E,29 to silyl aryl ethers (Fig. 2G) and the process can be used to create hydrolytically stable foams and elastomers (Fig. 2D).5. Using traditional processes to develop new functionality
5.1 Solubilization of triarylamines using the PR reaction
Triarylamines comprise a class of organic semiconductors useful in organic photovoltaics, for example, as a hole transport material. These compounds can be very difficult to process, particularly because of their low solubility in most solvents. The PR reaction could be used to convert suitably functional arylamines bearing an alkylether group into aryloxysilicones that were much more processable. For example, they are soluble even in non-polar solvents because of the silicone chains (Fig. 3A).30,31 Amines are excellent Lewis bases that would normally complex with and thus inhibit the boron catalyst. However, the extensive conjugation of the nitrogen lone pair with the aryl groups precludes catalyst inhibition.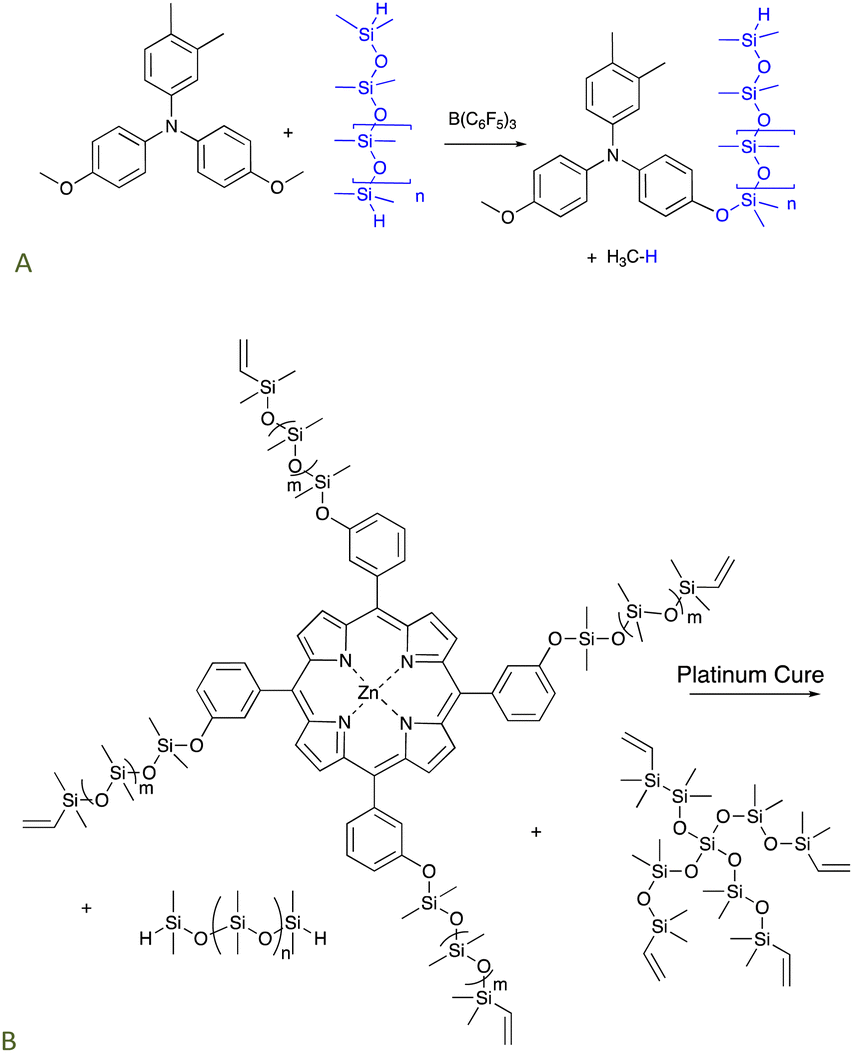 | ||
| Fig. 3 The PR process used to prepare (A) processable silicone-modified triarylamines,30,31 and, (B) silicone-modified porphyrins that cure (via hydrosilylation) to give elastomers that release 1O2 upon irradiation.32 | ||
5.2 Silicones that deliver reactive oxygen species: photodynamic oxidation
The use of platinum-catalyzed hydrosilylation can be extended to new organic functionalities. The relative stability of silicones to oxidation makes them a perfect vehicle to deliver reactive oxygen species (ROS). Singlet oxygen 1O2 – a powerful ROS – can be used to perform a variety of tasks including removing odors, killing bacteria, or in photodynamic therapy.33–35 A zinc porphyrin known to generate 1O2 was first modified using a PR process to introduce vinylsilicone side chains. The resulting product bearing 4 vinyl groups was crosslinked using hydrosilylation with telechelic HSi-silicones and, optionally, additional pure silicone crosslinkers depending on the desired crosslink density and porphyrin content. Photoirradiation of the elastomers released singlet oxygen from the elastomer surface in concentrations sufficient to kill bacteria on the silicone surface but which did not change the properties of the silicone elastomer (Fig. 3B).325.3 Antioxidants by hydrosilylation of eugenol
The allyl group found in the natural oil from cloves – eugenol – readily reacts with HSi-functional silicones to give silicone oils terminated with phenols 1 (Fig. 4A and B). The phenol groups in these polymers were shown to be equally effective antioxidants as the parent eugenol. The eugenol silicones are less soluble in water than eugenol and readily spread to form antioxidant films on surfaces.29 The resulting phenolic oils have been used to create polycarbonate copolymers (Siltech Corp.). An analogous process allows the preparation of vitamin A bound to silicones.36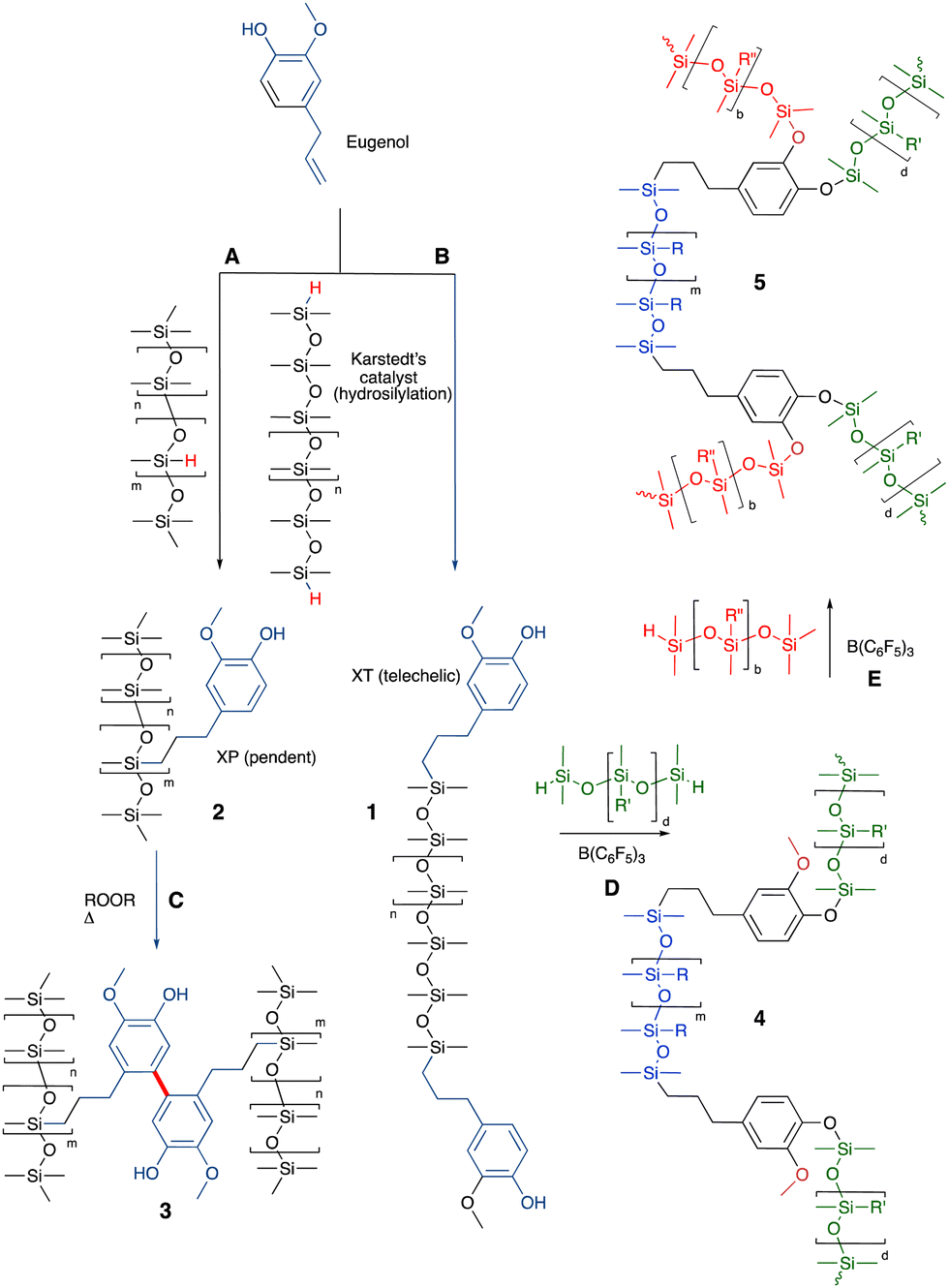 | ||
| Fig. 4 (A) and (B) Hydrosilylation to create silicone/eugenol oils.29 (C) Crosslinking using oxidative phenolic coupling.37 (D) and (E) Sequential crosslinking with up to 3 different types of silicones to generate silicone foams (note: performing the PR reactions first and hydrosilylation last leads to elastomers rather than foams).38 | ||
6. Alternative reactions to introduce organic functionality into silicone oils; with catalysts
6.1 Click reactions with catalysts
The utilization of the moniker “Click Reaction” keeps broadening in the literature. Here, a click reaction is defined as one that has no by-products and may or may not need catalysis.6.1.1.1 Elastomers – 3D printing. The thiol–ene process occurs very rapidly when photoinduced; the thiol + vinylsilicone materials cure completely within about 10 seconds. These compounds can be used as functional, curable inks in a UV-driven 3D printing process.39 The modulus of the elastomeric object could be varied simply by changing, on the fly, the relative amount of chain extender added. The speed of cure permits printing of complex shapes without the need for sacrificial material, including ‘voids’, overhangs and hollow shapes (Fig. 5A and B).
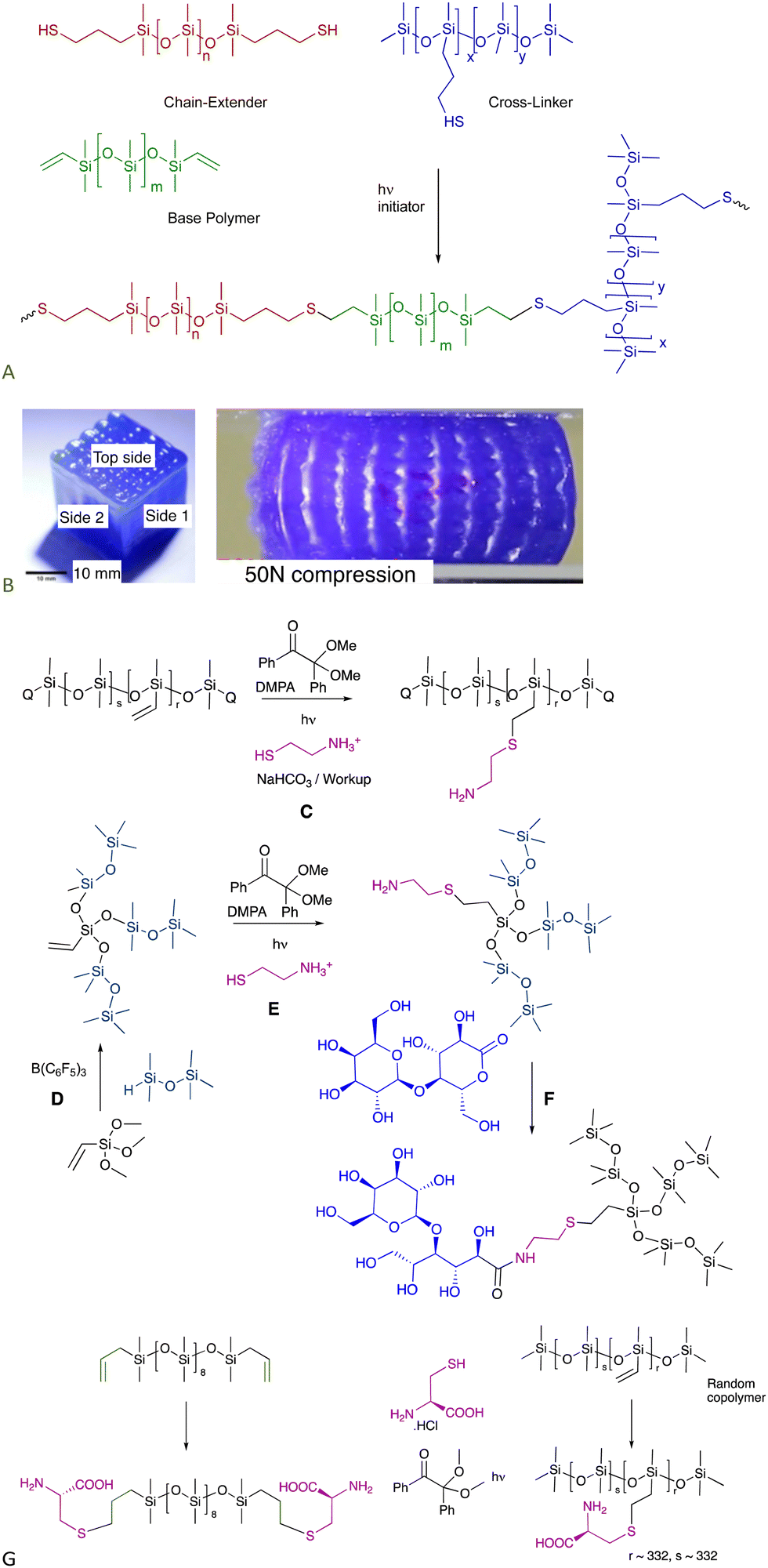 | ||
| Fig. 5 (A) Functional thiol–ene inks for 3D printing.39 (B) Voids in 3D printed cube as printed and under compression. Left photo reproduced from ref. 39 with permission from Elsevier. (C) Thiol–ene reaction with cysteamine to provide aminosilicones. (D) PR then (E) thiol–ene to generate highly branched aminosilicones.42 (F) Catalyst-free lactone opening to make sugar silicone surfactants (see also Fig. 12A).40 (G) Cysteine-modified silicones using the thiol–ene reaction.43 | ||
6.1.1.2 Incorporation of natural amines, and other thiol-containing natural materials silicones. Aminosilicones are, arguably, the workhouse organofunctional silicones available in commerce. They are typically prepared by hydrosilylation (Fig. 1B), which normally involves protection/deprotection sequences that contravene Green Principle 8 – reduce derivatives.1 Without protection, the amine inhibits the Pt catalyst. The thiol–ene reaction with commercial vinylsilicones using either thermal (e.g., AIBN) or photochemical (e.g., PhI(OAc)2) initiators is extremely efficient and leads to the creation of pendent aminosilicones or branched aminosilicones (Fig. 5C and E) without the need for protection sequences.40–42 The amine utilized here is the natural material cysteamine, but can also be the amino acid cysteine (Fig. 5G).43 Thus, the thiol–ene reaction provides a protection free way to introduce thiol-containing natural materials in synthetic silicone polymers.
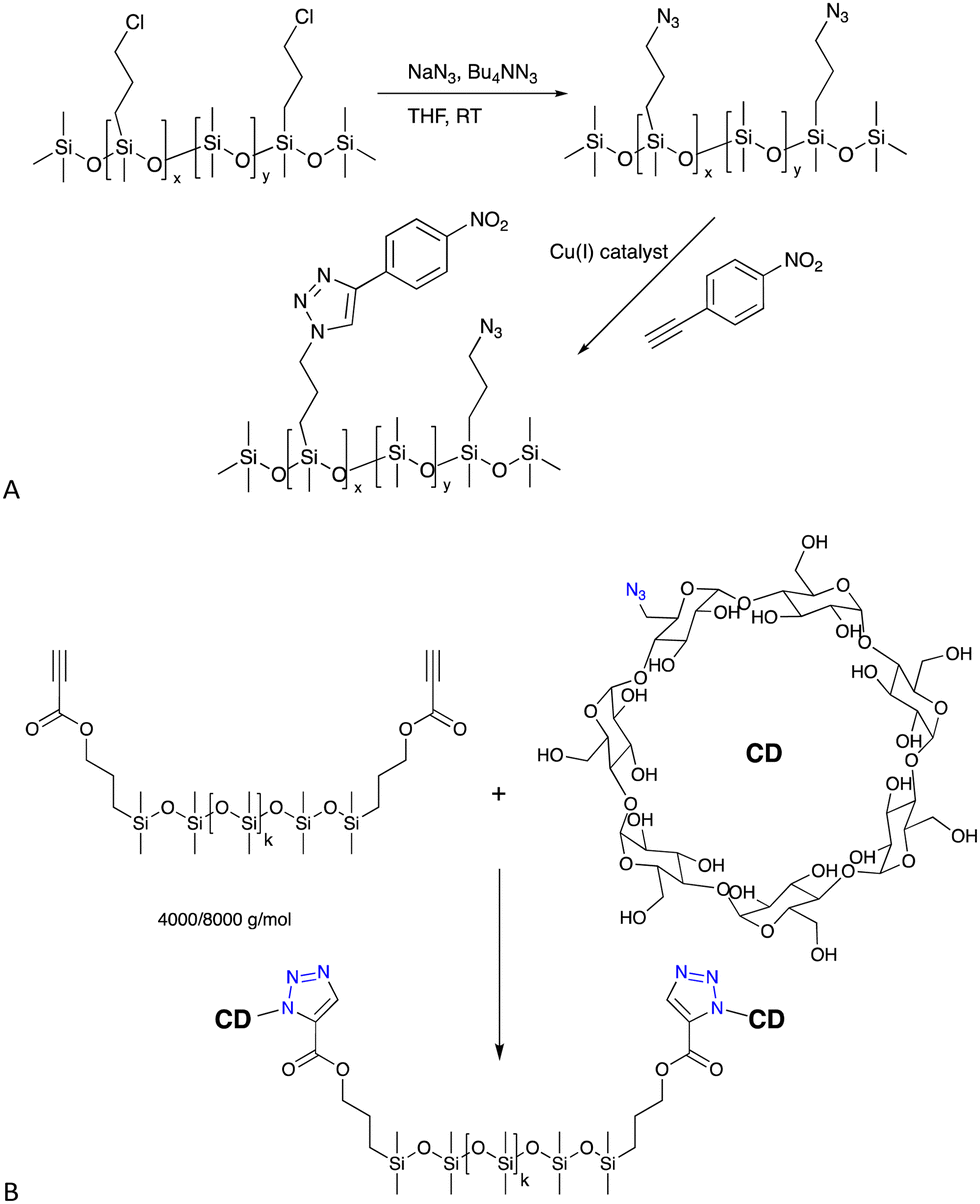 | ||
| Fig. 6 (A) Formation of azidoalkylsilicones and then the CuAAC reaction with an alkyne based on nitrobenzene.47 (B) Cyclodextrin silicone surfactants/elastomers formed by a CuAAC reaction.48,49 | ||
6.1.2.1 Surfactants. Azido-modified silicones will react with propargyl-modified sugars to create new sugar silicone surfactants48 or, alternatively, the silicone can be modified with alkynes – propionates are particularly reactive – to permit CuAAC reactions with azido-modified cyclodextrin (Fig. 6B), also leading to unusual surfactant molecules that self-assemble (see also H-bonding Crosslinks, below).48,49
6.2 Click chemistry without catalysts: Woodward–Hoffmann allowed reactions
6.2.1.1 Surfactants, hydrogels and elastomers. The combination of azide-modified silicones prepared from commercial chloropropylsilicones (Fig. 6A), with propionate-terminated polyethers provides an alternative route to polyether surfactants (Fig. 7A)53,54 or, with polyether dipropionates, hydrogels that exhibit very interesting dynamic behavior with water at an air interface (Fig. 7B).55,56 If a silicone dipropionate is instead used as a chain extender, the process is similarly effective at creating silicone elastomers without the need for catalysts (Fig. 7C).57 The potential fragility of the ester linkages may be of interest for recycling or end of life degradation (see End of Life, last section of the paper).
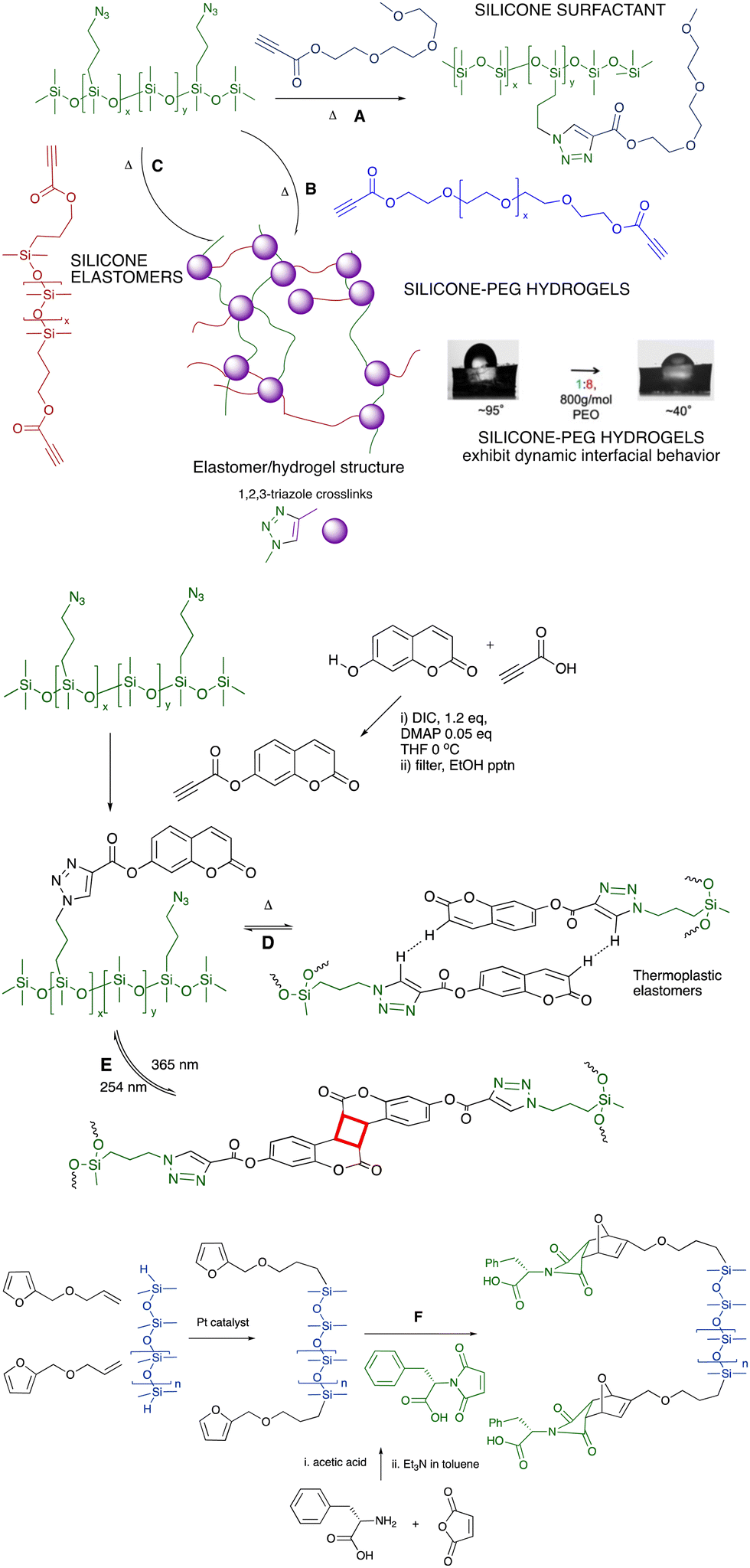 | ||
| Fig. 7 Azidoalkylsilicones participate in Huisgen reactions with (A) PEG-propynoates to give surfactants,53,54 (B) PEG-dipropynoates to produce hydrogels,55,56 and (C) with silicone-propynoates to create silicone elastomers.57 Contact angle photographs reproduced from ref. 56 with permission from the American Chemical Society. (D) Coumarin silicones self-associate to give thermoplastic elastomers,58 or (E) photodimerize to give thermosets.59 (F) Diels–Alder reactions to form silicones modified with phenylalanine.60 | ||
6.2.1.2 Elastomers: aromatic self-assembly of coumarin for crosslinking. The impact of aromatic association within a silicone fluid can be enormous. As silicone oils were increasingly modified with the aromatic group coumarin using a Click process, the polymers changed from low to high viscosity fluids or elastomers; increases in viscosity of up to 5 orders of magnitude were observed. The elastomers were thermoplastic (Fig. 7D).58
6.3 Other click reactions without catalysts
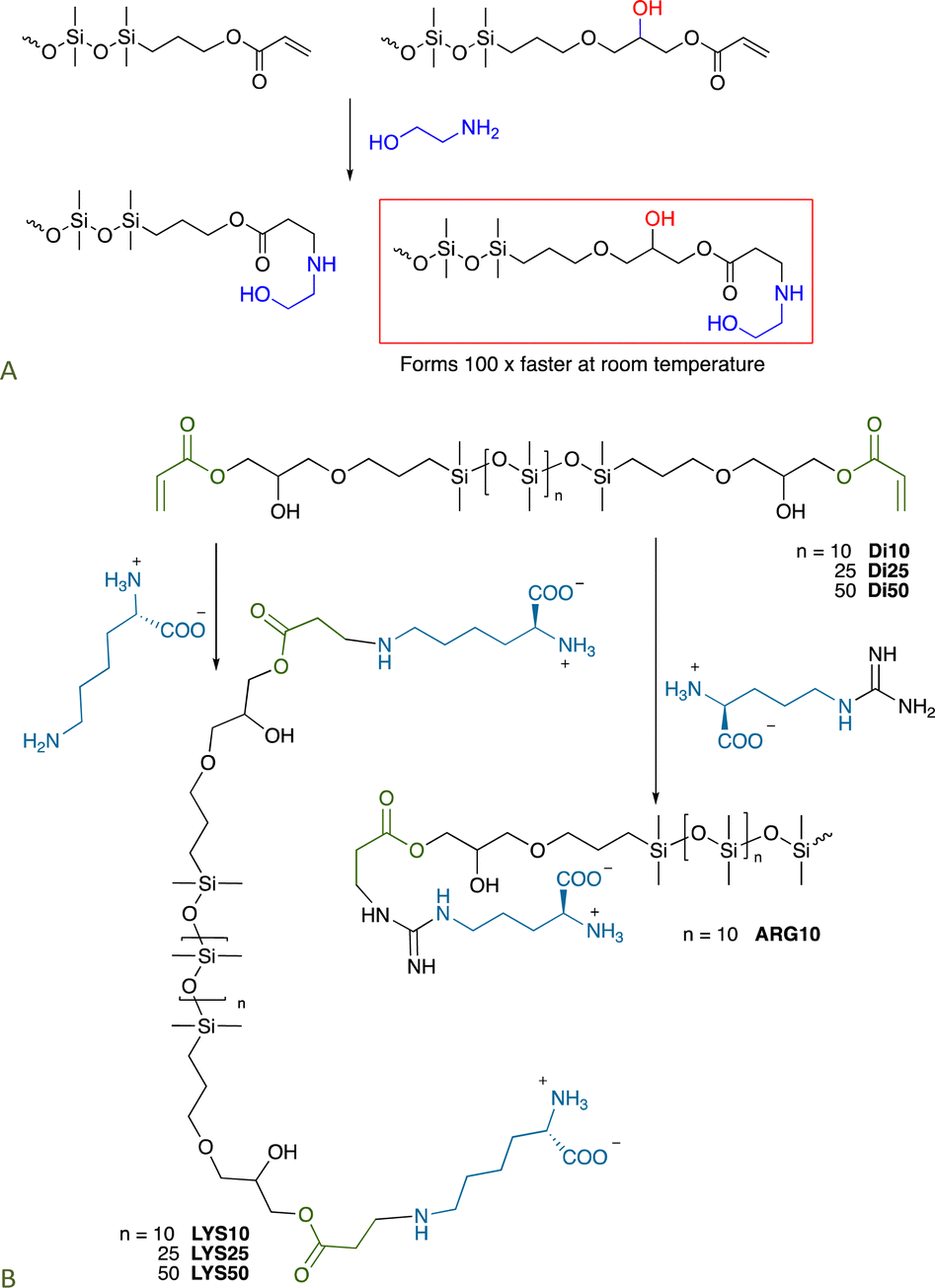
7. Making silicone elastomers: better catalysts or none at all
As noted above, the most common way to introduce organic functionality onto a silane or silicone backbone is hydrosilylation. Although very elegant syntheses can be done with Ru, Ir, Co, Pd, etc., the workhorse catalysts are based on platinum: Speier's catalyst in the early days and Karstedt's catalyst currently.16,17,26 The very high efficiency (only a few ppm is required) is hard to beat. When the product is an oil the use of platinum catalysts make sense, as the expensive spent metal can be recovered and purified. That is not the case with elastomers. The ultimate destination of the platinum used to catalyze crosslinking in elastomers is the environment per se, including in landfills. That is, one loses an expensive and difficult to replenish metal. The crosslinking in moisture cure RTV silicones may be catalyzed by tin compounds, which have a poor toxicity profile. Thus, in both cases better alternatives to these catalysts must exist to create silicone rubber, including the use of organic crosslinks.7.1 Organic crosslinks in silicone elastomers
7.1.1.1 Thioacetals. Acetals are widely used protecting groups for aldehydes/ketones when strong base/nucleophiles are to be used.13 Thioacetals are also created from aldehydes, but are more resilient protecting groups. Rather than simple acid treatment for acetal deprotection, thioacetals require strong electrophiles, reducing agents, or sulfur-complexing metals (e.g., Hg). They can also be deprotected oxidatively.
Commercial thiopropylsilicones were covalently crosslinked using simple aromatic aldehydes, like benzaldehyde. The ability of aromatic groups to self-associate to give secondary crosslinks within the elastomer was demonstrated by an increase in modulus as a benzene group was replaced with the larger fluorenyl group (Fig. 9A).67
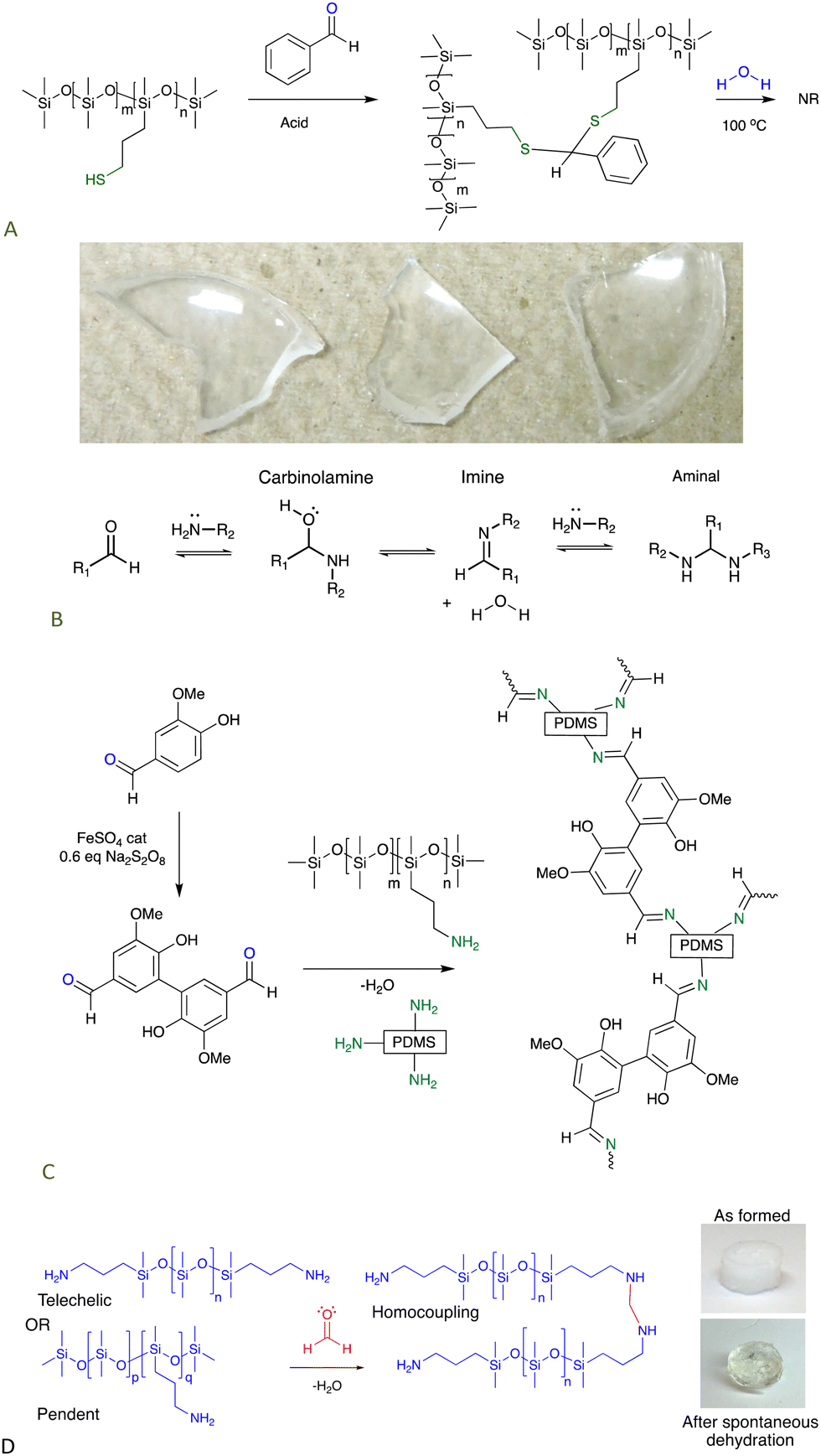 | ||
| Fig. 9 (A) Crosslinking thiopropylsilicones with aldehydes.67 Photo from ref. 67 reproduced with permission from the Royal Society of Chemistry. (B) Reactions between aldehydes and amines. (C) Schiff base crosslinks,69 and (D) aminal-based crosslinks from aminosilicones.71 | ||
7.1.1.2 Imines. Amines form strong, but reversible, bonds with aldehydes to give imines (Fig. 9B). Aliphatic imines are easy to hydrolyze by pushing the equilibrium towards the aldehyde, but imines derived from aromatic aldehydes – Schiff bases – are much more stable. It was straightforward to turn commercial aminosilicones into aldehydes simply by adding a difunctional aromatic aldehyde, e.g., terephthaldehyde. High quality dynamic elastomers resulted that, at about 80 °C, became thermoplastic. Complete degradation of the elastomers by adding monofunctional amines was straightforward.68 A greener analogue based on divanillin exhibited higher strength because,69 it is inferred, the phenolic groups phase separate from the silicone providing additional physical and H-bonding crosslinks (Fig. 9C); in some cases, the latter bonding motifs are sufficient to crosslink silicones.70
7.1.1.3 Aminals. When the extremely reactive (and toxic) aldehyde formaldehyde was reacted with aminosilicones, it was shown not to stop at the imine stage but reacted further to give aminal crosslinks. The process was both rapid and efficient; it cleanly occurred underwater to give robust elastomers in less than 10 seconds and acted as effective sealant (Fig. 9B and D).71
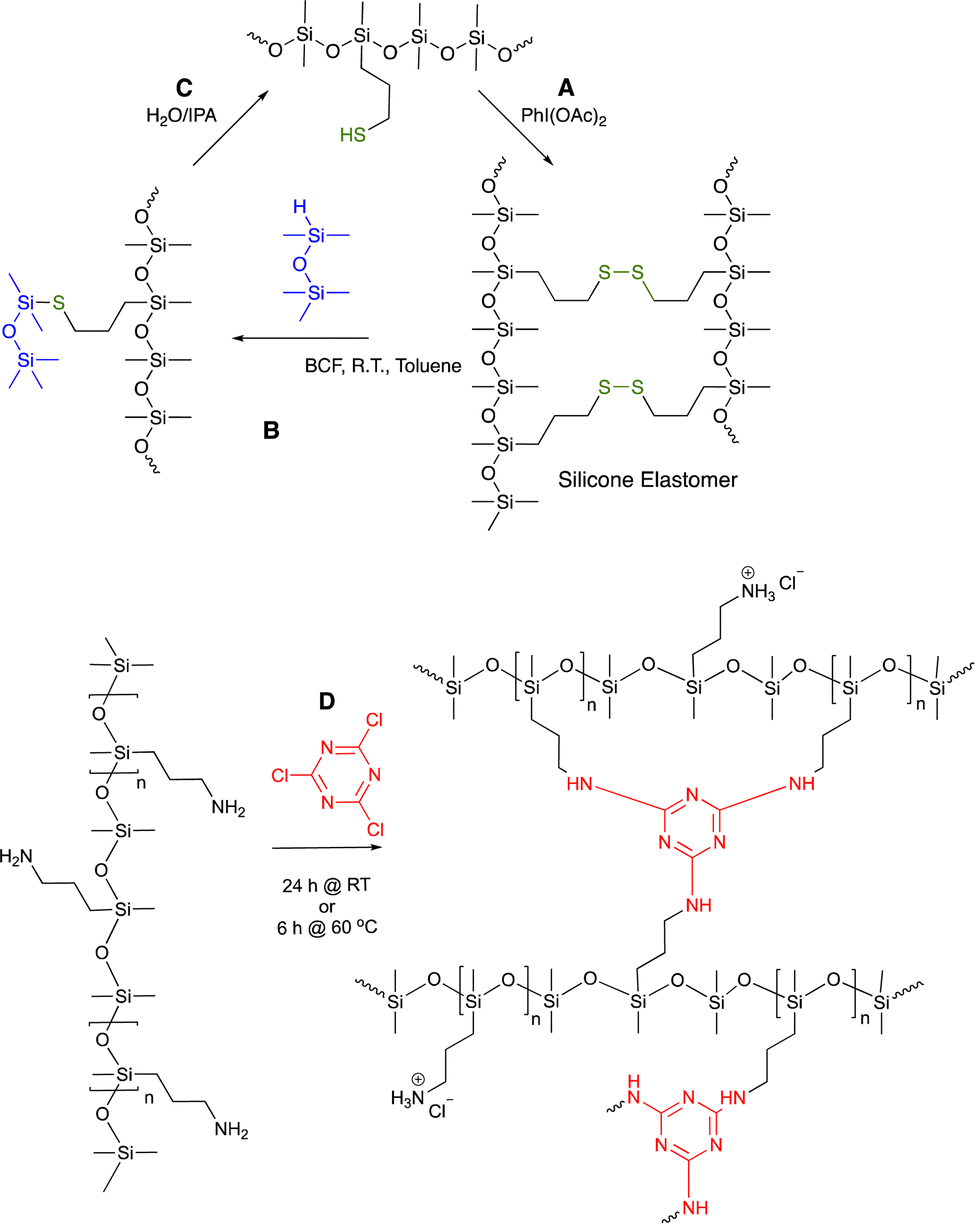 | ||
| Fig. 10 (A) Disulfide crosslinks in silicone elastomers formed by thiol oxidation are, (B) reduced using hydrosilanes under Piers’ conditions, and, (C) converted back to starting materials by hydrolysis/alcoholysis of the silyl thio ether.73 (D) Triazine crosslinks from simple nucleophilic addition of aminosilicones.75 | ||
8. Silicone elastomers without covalent bond crosslinks
Chemists love covalent bonds, and why not, with their high strength and well understood kinetic reactivity. However, there can be advantages that accrue when molecules are held together by weaker bonds, particularly with crosslinked materials. Silicones are very low surface energy materials. As a consequence, the effective strength of highly polar or ionic bonds is effectively higher within a silicone than in other media. In addition to individual bonds, there is a tendency for polar/ionic groups to self-assemble, leading to a secondary type of crosslinking, as was seen with the vanillin imine and catechol-based elastomers (Fig. 9C).69,70 Very resilient bonding can result from the combination of any bonds of lower energies/bond, including aromatic association (that can include π–π bonding, Fig. 7D), ionic bonds and H-bonding – e.g., DNA. When desired, for example for recycling, it is often easier to cleave (assemblies) of weaker bonds than a few stronger covalent bonds.8.1 Ionic bonding
8.1.1.1 Self-healing elastomers. Ionic bonding between silicone carboxylates and aminosilicones has been shown to create self-healing elastomers.76 If an analogous dicarboxylate, the product of a thia-Michael reaction with maleate (Fig. 11A), is simply mixed with commercial ethylenediaminosilicones, 2+2 (Gemini) ionic crosslinks form that are more effective than twice as many crosslinks formed from mono COOH + mono amine. The product elastomers are thermoplastic, facilitating reuse and recycling.77
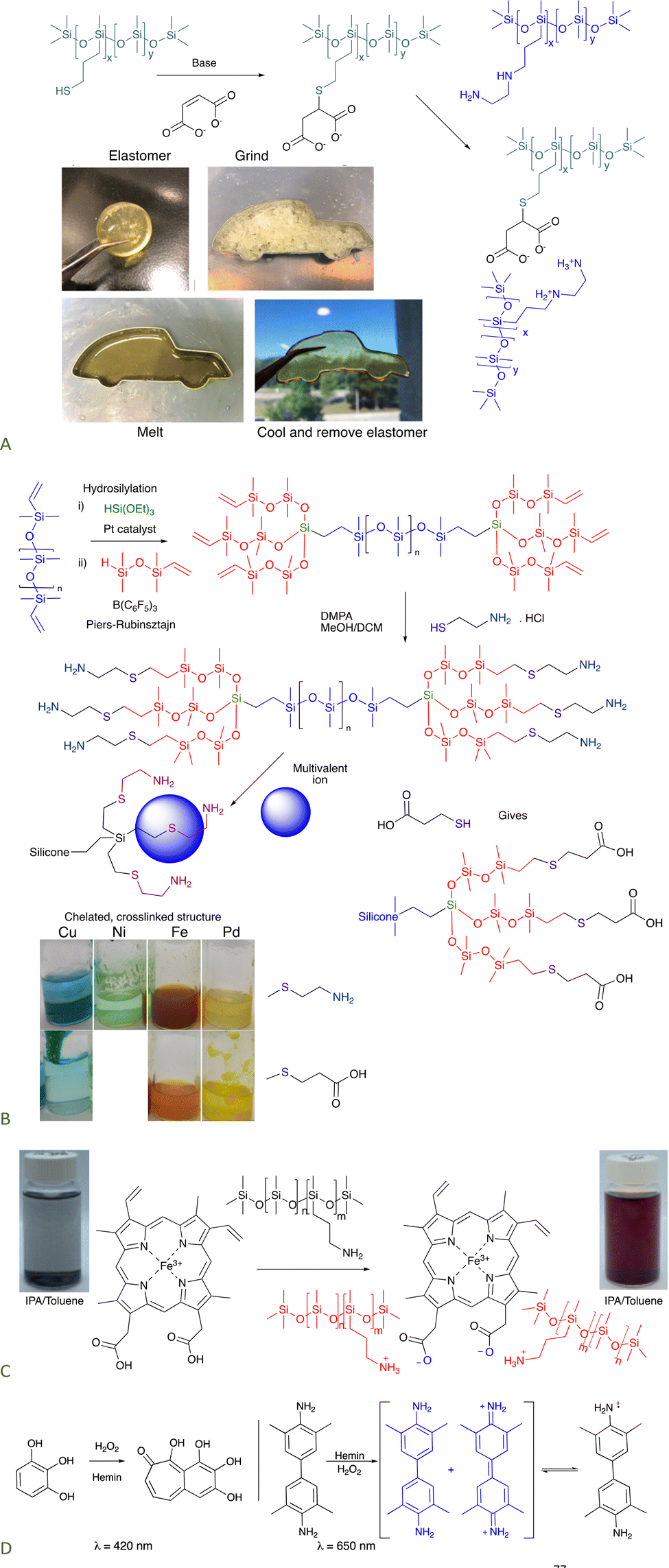 | ||
| Fig. 11 (A) Gemini ionic crosslinking to give a thermoplastic silicone.77 Photographs from ref. 77 reproduced with permission from the Royal Society of Chemistry. (B) Tri- or hexadentate ligands formed on a silicone backbone from cysteamine or thiopropionic acid.42 (C) Hemin can crosslink silicones to give peroxidase mimics that (D) can oxidize organic molecules.78 Photographs Photo from ref. 78 reproduced with permission from John Wiley & Sons. | ||
8.1.1.2 Hemin-containing silicones. Hemin is a mimic of peroxidase enzymes that is comprised of a porphyrin structure bearing 2 carboxylic acids. Isopropanol (IPA) is an effective solvent for both silicones and hemin. Mixing hemin with a pendent aminosilicone in IPA leads to complete dissolution of the hemin, which is otherwise insoluble. With an appropriate match of silicone amines/COOH on hemin, elastomers result after solvent removal. The elastomers behave like an immobilized version of hemin itself in that they are able to oxidize organic materials in the presence of light, as shown with pyrogallol or tetramethylbenzidine (Fig. 11C and D).78
8.2 H-Bonding crosslinks
To examine the role that H-bonds could play as sites of assembly, a series of telechelic silicones was prepared from gluconic acid–amide formation from the lactone is an uncatalyzed click reaction.41 Even the silicone with the lowest sugar content, <0.1 wt% (n = 671, Fig. 12A, for disaccharide surfactants analogously produced, see Fig. 5F), exhibited very unusual properties. While at rest the silicone was a viscous fluid, application of force led to shear thickening behavior as – under stress – the sugar units formed crosslinking, H-bonded clusters; after about 15 minutes, the transient networks dissipated. It was possible to select the desired elasticity of the fluids simply by controlling the density of sugar groups: at high sugar content, they were glasses; with dilution became hard then soft elastomers (Fig. 12A–C); and finally viscous fluids. Except for the glasses, all became more rigid/viscous/crosslinked by applying force.82 The elastomeric properties could be eliminated simply by providing competing hydrogen bonding partners, for example, by adding small quantities of IPA.83 Analogous sugar silicones formed from cyclodextrin were robust elastomers crosslinked by separate sugar domains (Fig. 6B). These H-bonded structures could only be broken apart by chaotropic agents.49
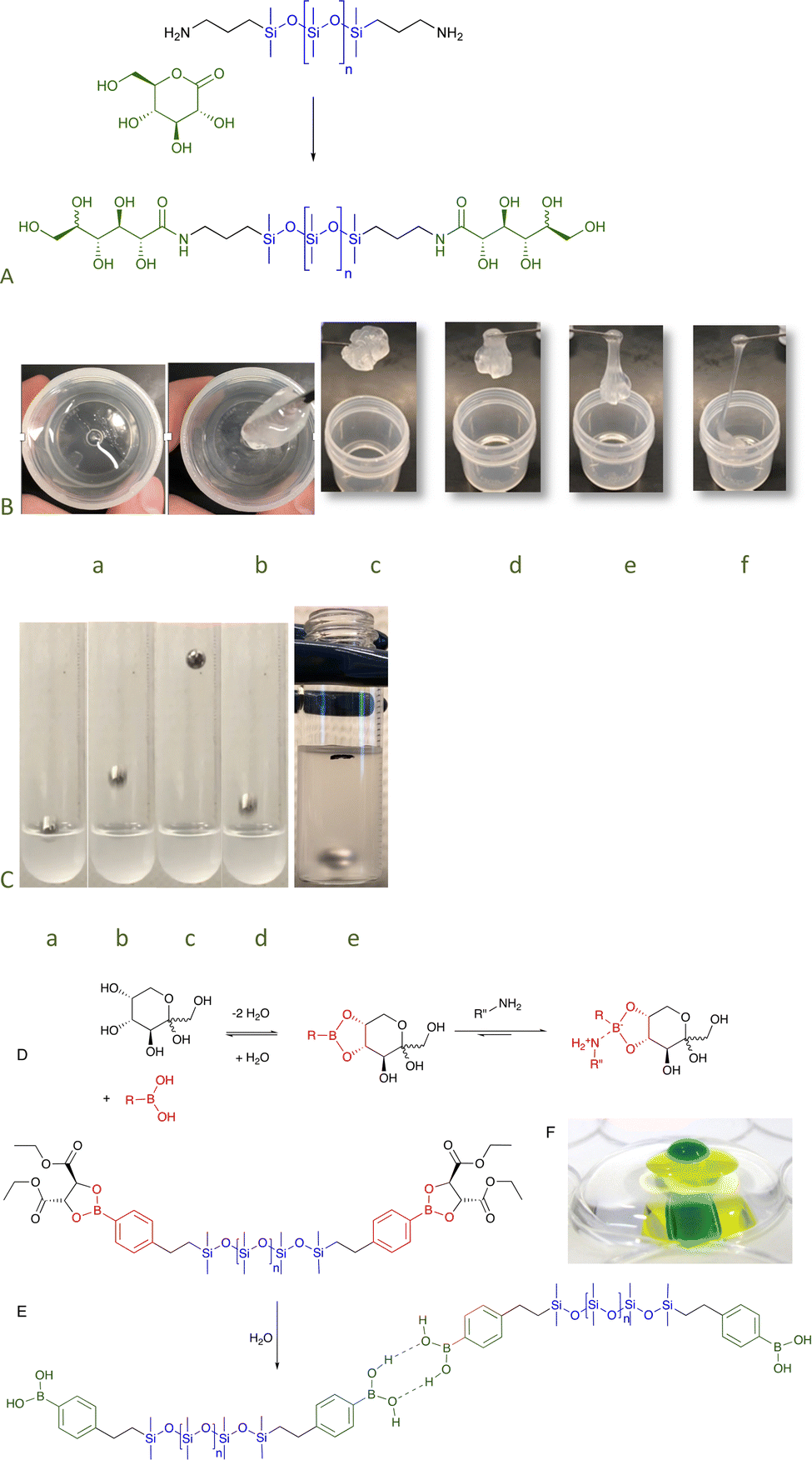 | ||
| Fig. 12 (A) Preparation of sugar amides by gluconic acid ring opening.82 (B) Stirring the sugar silicones leads to thickening and then elastomer formation a → b that, over about 15 minutes, returns to its fluid state c → f. (C) Sequence showing the ball bouncing on 30% solids sugar silicones (n = 61) in (Me2SiO)5 a → d and the ball sinking over about ½ hour e. (D) Equilibrium between boronic acids and sugars. (E) Hydrolysis of the tartrate protecting group leads to H-bonding elastomers (that can be disrupted by sugars and other binding agents). Before hydrolysis, the compound spreads across interfaces. (F) A picture of water (green) on water (yellow) on water (clear), each layer is isolated by a silicone boronate elastomer.86 Photo from ref. 86 reproduced with permission from the Royal Society of Chemistry. | ||
8.3 Combination of ionic and hydrogen bonding
The pKa of ammonium salts and phenols is very similar. As a consequence, it is possible to create elastomers by using a combination of H- and ionic bonding;70,87,88 both small molecules like catechol and large molecules like tannic acid participate as crosslinkers with telechelic aminosilicones. The product elastomers are excellent antioxidants. Depending on crosslink density they may be thermoplastic; all can be completely degraded by exposure to small amines. Some readers will not appreciate the intense colors that accompany these and many other silicones containing natural products (Fig. 13A). However, the simplicity of the synthesis, ability to reprocess or degrade the elastomer at will, and the use of natural materials as elastomer constituents should overcome these limitations in many applications. These elastomers retain the antioxidant activity of the contained phenols.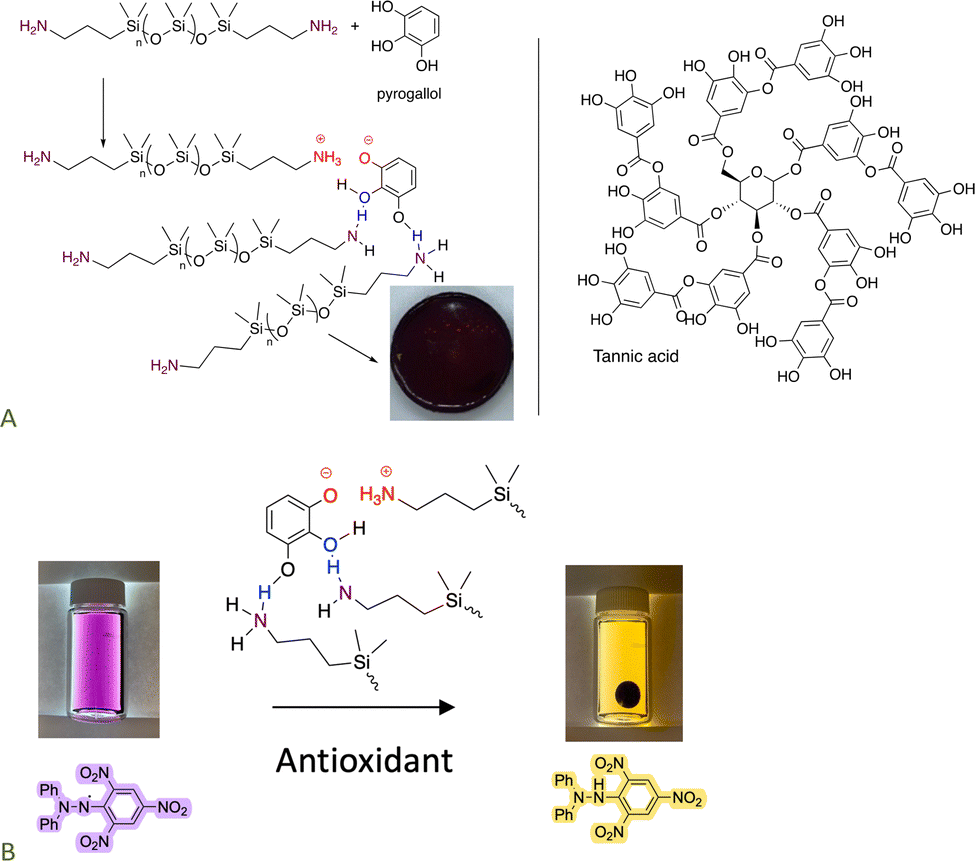 | ||
| Fig. 13 (A) Formation of elastomers simply by mixing aminosilicones with phenols. (B) Antioxidant activity of the elastomer formed.70 Photographs from ref. 70 reproduced with permission from the American Chemical Society. | ||
9. Controlling interfaces
There are many applications that require interfacial control ranging from painted steel surfaces that prevent oxidation of the underlying substrate to sunscreen to protect the skin from UV light. Biological environments are particularly challenging for the interfaces of synthetic materials, including silicones. While small biological molecules may adsorb without problems, larger molecules can undergo conformational changes, for example leading to protein denaturation, once in contact with the synthetic surface; such changes at the interface can elicit very negative biological responses. Silicone polymers are widely used in both topical and implanted biomedical devices. They are also frequently found in coatings, sealants, and adhesives. For all such applications, organic modification of the silicone to facilitate stabilization at/on/of the substrate interface is normally required.9.1 Proteins
The denaturation of proteins is, in many cases, facilitated by exposure to hydrophobic interfaces, such as solvents and polymer surfaces,89 including silicones.90,91 However, the inclusion of just a few hydrophilic groups – unreactive polyethers or reactive alkoxysilanes – can lead to complexes in which selected proteins are stabilized against denaturation.92–94 The resilience of the protein/silicone fluid interface was demonstrated by the ability to deliver from the same water-in-silicone oil dispersion both enzymes and bleach (which normally degrades enzymes) (Fig. 14A).95–97 Alternatively, the protein/silicone materials, particularly when filled with starch particles, led to model oral/nasal vaccines.98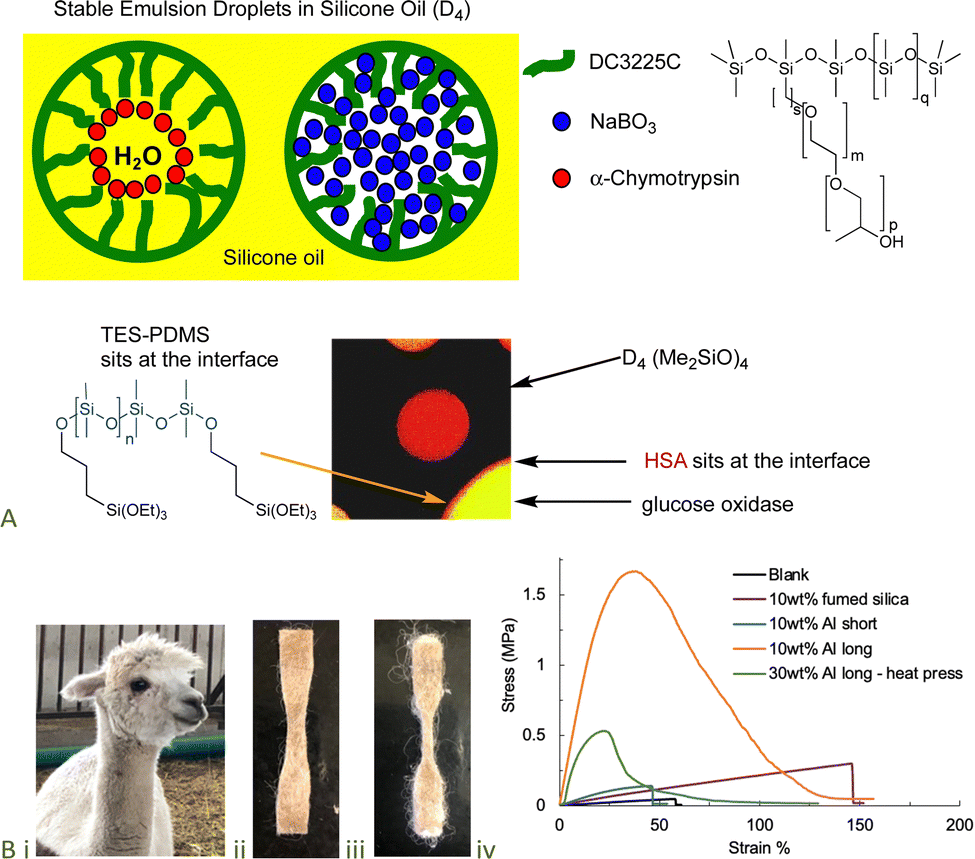 | ||
| Fig. 14 (A) Enzyme-containing water-in-silicone oil emulsions.97,108 Scheme and photo from ref. 97 and 108 reproduced with permission from the American Chemical Society. (B) (i) Alpaca, silicone elastomer containing (ii) 10 wt% short and (iii) 10 wt% long alpaca fibers, (iv) enormous difference in materials properties based on fiber length at the same mass loading.100 Photographs from ref. 100 reproduced with permission from the Royal Society of Chemistry. | ||
A related, but nearly water-free, process involves solutions of proteins dissolved in the biocompatible, natural solvent glycerol. Dispersions of glycerol droplets in silicone pre-elastomers form highly viscous glycerol-in-oil emulsions. The emulsions do not easily undergo gross phase separation and it is straightforward to cure the silicone component of the emulsions. The interconnectedness of glycerol droplets was demonstrated by release of enzymes in active form from the glycerol/silicone elastomer.99
9.2 Biomaterials interfaces: esters and amides
Silicones are somewhat distal from the locus of functional group manipulation in the next two applications. We,101,102 and many others,103–105 have utilized polyethers as a type of insulation to prevent direct contact of biological media – including in vivo – with a silicone elastomer surface. The low surface energy of silicones can lead to protein denaturation but the hydrophilic, spacing PEG layer can reduce or completely avoid denaturation.Non-functional silicone elastomers can be etched under acidic conditions in the presence of HSi-containing silicone oils to generate SiH elastomer surfaces. This process takes advantage of the fragility of silicone polymers at low pH. Once the SiH group is present on the surface, it is possible to use hydrosilylation to graft organofunctional moieties on the surface using a PEG (poly(ethylene glycol)) linker that separates the silicone surface from the organic functionalization. A tosylate-containing surface was converted to an amine-grafted surface by SN2 reaction with amines, that, in turn, permitted hyaluronic acid (HA) to be grafted to the surface (Fig. 15A).106 HA is widely used as a degradable biological filler and on interfaces because it is often recognized by biological entities as a ‘normal’ material. An analogous process was used to crosslink silicone elastomers using an SN2 reaction to form quaternary ammonium (quat) crosslinks. Quat salts are recognized for their antibacterial activity.107
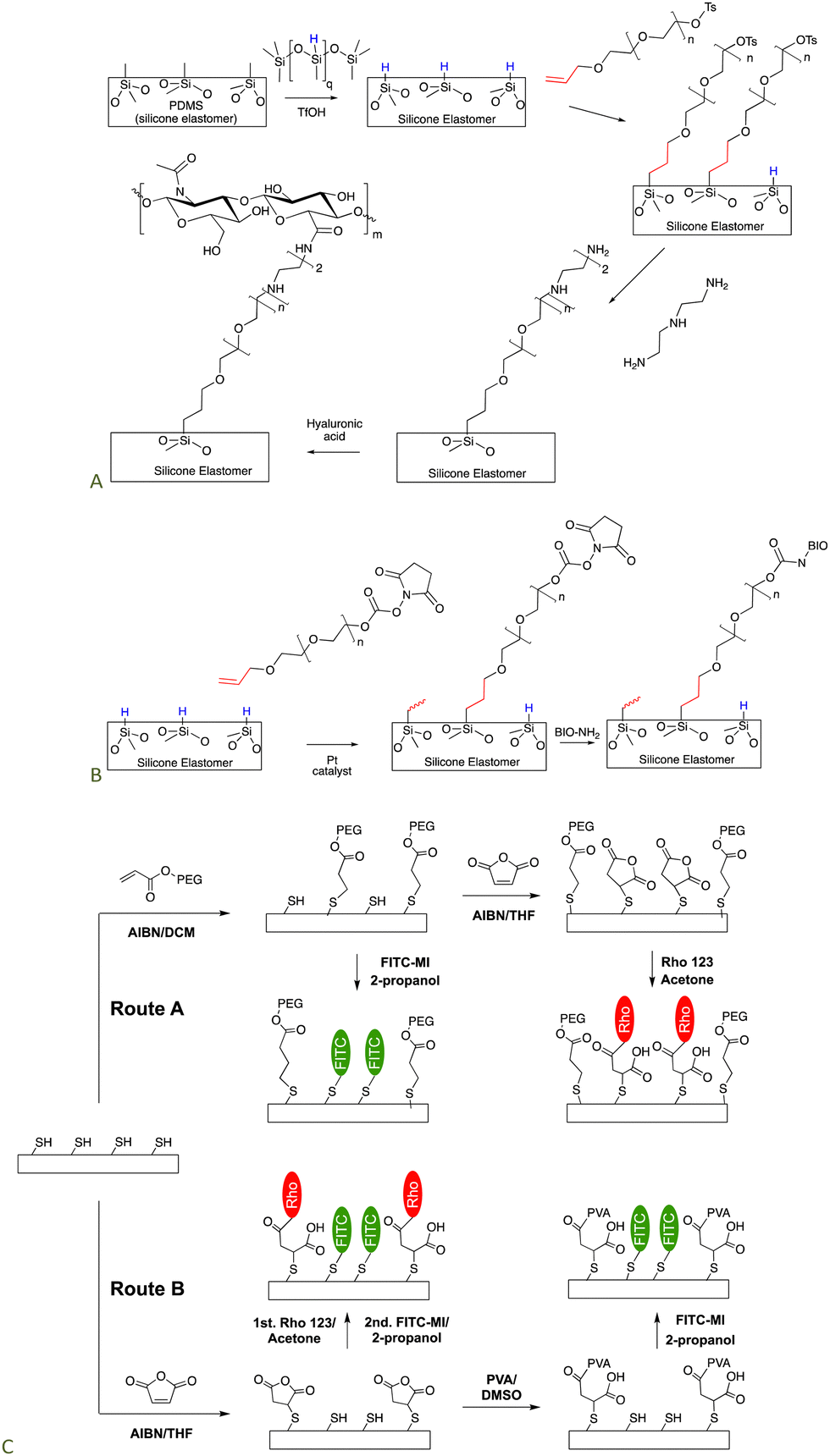 | ||
| Fig. 15 Strategy to prepare silicone biointerfaces by grafting a polyether to the surface and then using (A) an SN2,106 or (B) esterification processes to tether biological molecules to the surface (BIO are biological molecules including polysaccharides, peptides and proteins).102 (C) Silicone surfaces can be rendered functional with multiple organic groups, including with photopatterning, using combinations of thia–Michael, and esterification reactions.74 | ||
The HSi-silicone surfaces could alternatively be modified with an activated ester-terminated PEG chain (Fig. 15B). A wide variety of biological molecules containing H2N-groups readily reacted to produce surface-grafted biological molecules (BIO) including polysaccharides like heparin, the adhesion peptides RGD and YIGSR, and a range of proteins, such as mucin, heparin, lysozyme, and albumin. The biological molecules maintained their biological activity once tethered to the silicone surface, consistent with an effective insulating layer provided by the PEG.101
It is possible to introduce esters and other functional groups onto thiol-modified silicone surfaces using a radical thia-Michael reaction (note that this reaction operates in analogy with the thia-Michael reaction (Fig. 11A) but under radical rather than ionic conditions). One enormous advantage of the radical process is that it can be induced by light, so that patterns of (bio)molecules can be introduced onto the surface with spatial precision. For example, it was possible to pattern two different types of fluorescent markers on silicone elastomer surfaces using a combination of thia–Michael and esterification reactions (Fig. 15C).74
9.3 Functional fillers for silicone elastomers
The physical properties of unfilled silicone elastomers are relatively poor, such that commercial samples are typically filled with silica; fumed silica with very small particle diameters (aggregates of silica are typically ∼50 nm in diameter) is particularly useful. The improvements in tear resistance and extension at break of the elastomers are attributed to favorable interactions between the silica surface (OH groups) and oxygen atoms on the silicone polymers. The fumed silica is prepared from a waste product, SiCl4, from the Direct Process, which is a nice example of Green Chemistry (one process’ waste product is another's feedstock). However, the silica is expensive. Any displacement of silicone, with its high energy content, with natural materials better fits the principles of Green Chemistry, but of course the products filled with natural materials must have comparable or different (better) properties to those filled with silica (see also, Wool fillers, Section 9.1.1, Fig. 14B).9.4 Saccharide and edible oil esters
Anhydrides react directly at elevated temperatures with alcohols. This process can be used to modify polysaccharide surfaces, including starch and cellulose, without catalysts. In the first case, an anhydride derived from maleic acid was hydrosilylated with a telechelic silicone and then reacted to provide starch/silicone composites containing up to 70% starch (Fig. 16A). Alternatively, maleic anhydride was first introduced on a cellulose surface, the generated carboxylic acid opened the epoxide of allyl glycidyl ether without the need for catalyst, and then silicones were introduced using hydrosilylation (Fig. 16C, see also Fig. 15C).109 A resilient hydrophobic coating of the wood surface results.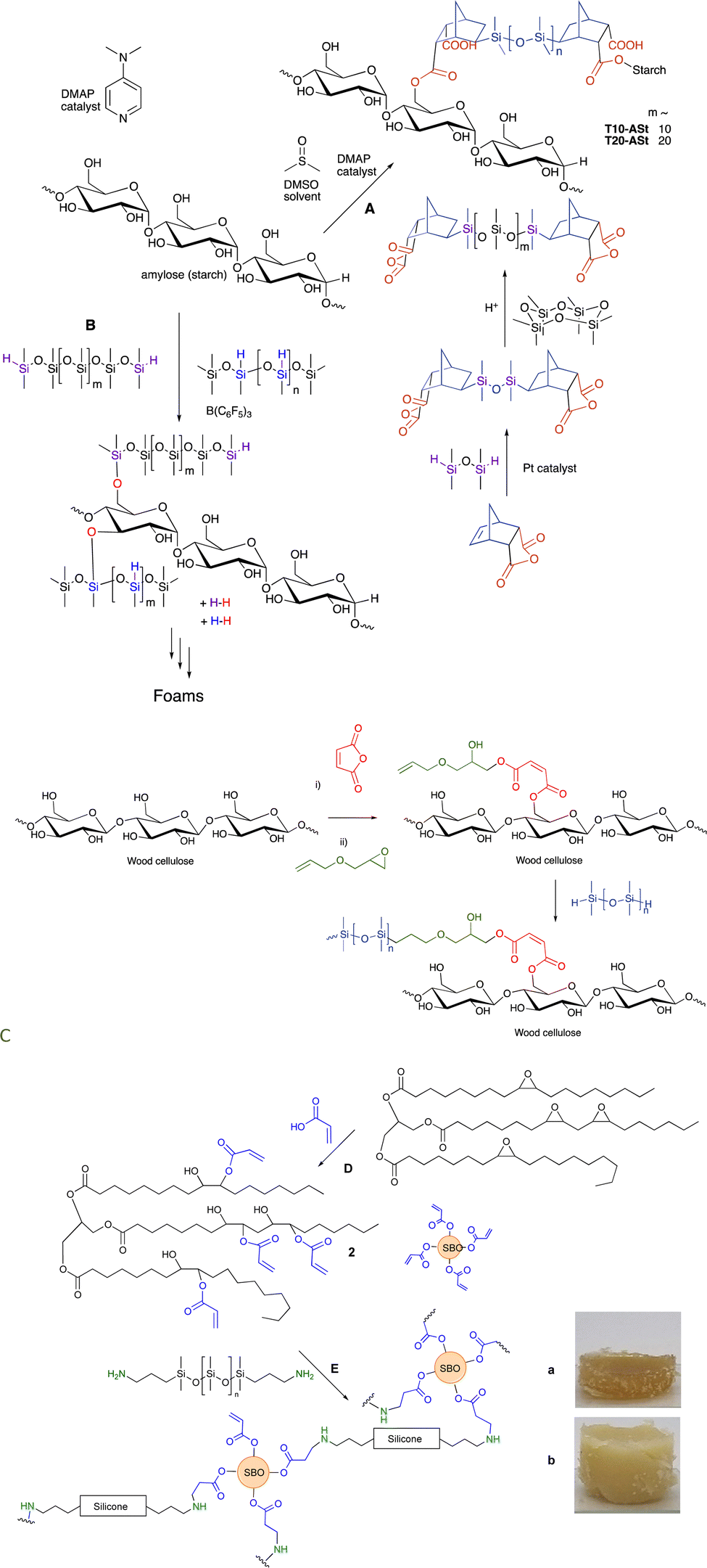 | ||
| Fig. 16 Silicone/starch conjugates formed using either (A) ester or (B) silyl ether linkages.61 (C) Silylation of wood surfaces using esters.109 (D) Creation of soy acrylates, and, (E) crosslinking with aminosilicones (a and b have both 25 wt% silicone but were made from telechelic silicones with (a) 12 and (b) 59 monomers in the backbone).110 Photographs from ref. 110 reproduced with permission from the Royal Society of Chemistry. | ||
An analogous process allows one to incorporate triglycerides into silicones. Epoxidized triglycerides are readily prepared by oxidation of oils, for example, soybean oil; increasingly, these epoxidized compounds are commercially available. The catalyst free reaction with acrylic acid gave acrylated oils. The degree of acrylation is controlled by the stoichiometry of the reaction.
This key material now serves as a (partly) natural crosslinker. The aza-Michael reaction (see Fig. 8) with aminosilicones leads to silicone elastomers that can contain between 5–90 wt% soy oil (Fig. 16D and E).110 The products readily undergo degradation via ester hydrolysis.
9.5 Starch foams based on alkoxysilanes
As noted above, alkoxysilanes normally form disiloxanes with hydrosilanes under PR conditions (Fig. 2G). They are also susceptible to hydrolysis accelerated by acid or base, but even under neutral conditions (e.g., moisture) slow hydrolysis is normal. Starch, one of the most abundant biopolymers on the planet, is highly polar and immiscible with silicone oils. However, if one takes powdered starch in the presence of various silicone polymers containing SiH groups and B(C6F5)3 (in analogy to the work on lignin, see below Fig. 17), rapid foaming occurs to give low density starch foams containing up to 50 wt% starch (Fig. 16B).61 The products are surprisingly resistant to water; little change was seen after boiling in water for 3 days.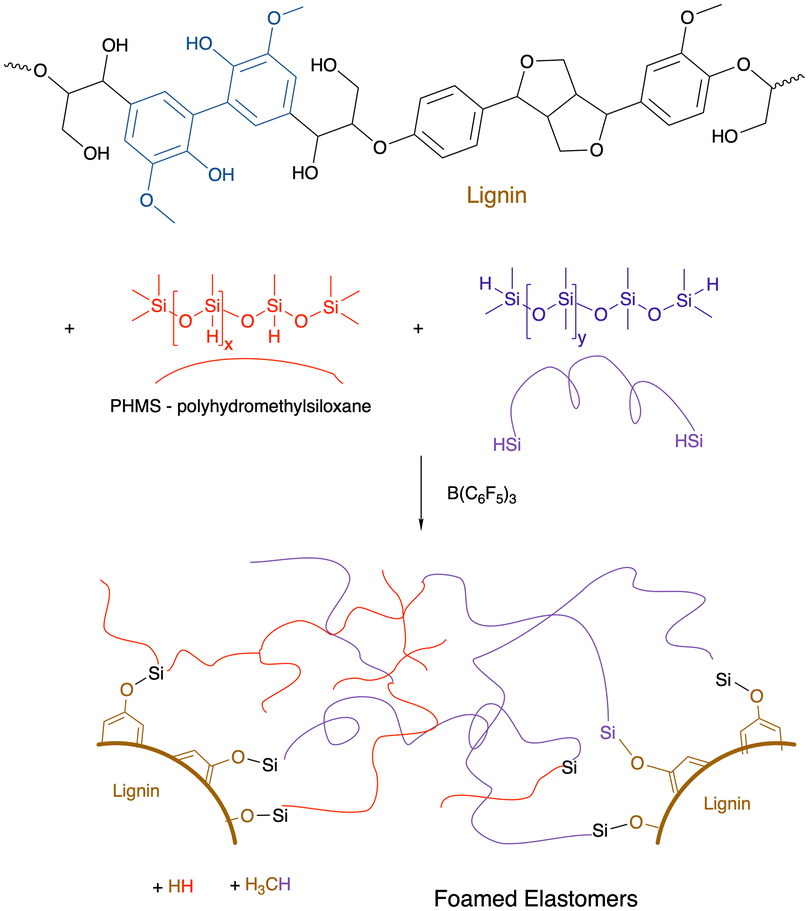 | ||
| Fig. 17 Formation of lignin/silicone elastomer foams using the PR process.111 | ||
9.6 Lignin
The Piers–Rubinsztajn reaction14 allows new siloxane bonds to be formed between hydrosilanes and alkoxysilanes (Fig. 2G). Lignin is an increasingly available feedstock that is a by-product of the pulp and paper industry. It is a highly aromatic polymer derived from phenolic monomers (Fig. 17).111 The direct reaction between the phenolic groups on lignin with various mixtures of hydrosilanes leads – on demand – to either elastomers (if one pulls a vacuum during synthesis) or foams.112The resulting elastomers/foams have lost their ability to be antioxidants because the phenolic groups are blocked by silicone groups. However, a surprising type of functionalization of the lignin/silicone foams is resistance to combustion.112 While the external surfaces of both pure silicone foams and lignin/silicone foam are converted to a thermally insulating silica layer upon combustion, the lignin containing foams resisted flames better, it is hypothesized, because absorption of thermal energy by lignin without combustion provides additional time for the protective silica layer to form. Up to about 40% of the silicone in these materials is displaced by lignin.
10. End of life – degrading silicones
At the beginning of this article, it was noted that silicones are mostly valued for what they don’t do; they exhibit a lack of reactivity under even vigorous conditions. However, this works against the need for silicones to degrade smoothly in the environment at end of life. It will be recognized by many of the readers that the functionality encoded in the product silicones described above will render the silicones to be much more reactive than pure silicones and, therefore, less stable under some conditions. For many applications, that sensitivity will not matter in normal use but should facilitate degradation once placed in the environment. For example, esters will eventually hydrolyze, but will need enzymes, acids, bases or other catalysts to facilitate the reaction, conditions that will arise in the environment, but not in normal use. One can, therefore, design silicones whose stability/reactivity balance better meets the needs of function while increasing their circularity and, therefore, their sustainability.11. Looking forward for functional silicones
I was once told (in the late 1980s) by a senior chemist in a global polymer company that, “There is no need for new monomers.” On the one hand he was right, in that the path from new monomer to new polymer with new function to commercial reality is long and fraught. However, the changes that have occurred in polymer science and products since that time show the limitations of the statement. Traditional silicone chemistry works really well and, within the industry, there is some reticence to moving beyond to robust chemistry that exists. However, there are increasing technical needs that will not be met by traditional silicone oils and elastomers, nor by organic polymers. New monomers and new polymers with new reactive functionality and new properties will be needed.Many of the examples provided in this paper are designed to be enabling. They show that the very unusual properties of silicones can be modified with both traditional and more recently developed organic chemical processes or encoded in natural and synthetic polymer systems. Thus, the practitioner can select from a broad range of readily available chemistry to create silicones in new ways – functionality in the traditional sense – to generate new materials with new properties, that is, to be more broadly functional.
While the desired properties from any silicone are seen from the viewpoint of the synthetic chemist, one property of silicones that is currently overlooked is their sensitivity to acid and base and catalysts found in soil. Silicone oils undergo relatively rapid degradation under environmental conditions.113,114 Could this be a strategy to induce a weak spot – built in obsolesce – in silicone and other polymers to facilitate degradation at end of life, to improve their sustainability. That type of functionality is the focus of our ongoing research.
12. Conclusions
Silicone polymers are deeply integrated in the lives of many of us, both in our personal contact with them and in the many products that benefit from their use. This review has attempted to show that some simple organic chemistry on a silicone backbone provides new routes to current classes of silicones but, more importantly, opens up opportunities to make new functional silicone oils and elastomers with tailored, beneficial properties, including an augmented degree of sustainability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to express my deep appreciation for the long-standing funding provided to my research program by the Natural Sciences and Engineering Research Council of Canada, and to my group members over the years who have invested their hard work and creativity in pursuit of interesting silicone polymers and routes to prepare them.Notes and references
- P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, OUP, 2000 Search PubMed.
- M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, ch. 9, pp. 256–308 Search PubMed.
- S. J. Clarson and J. A. Semlyen, Siloxane Polymers, Prentice Hall, Englewood Cliffs, NJ, 1993 Search PubMed.
- A. J. O'Lenick Jr. and J. K. Parkinson, Cosmet. Toilet, 1996, 111, 37–44 Search PubMed.
- M. J. Owen, in Silicon-Based Polymer Science: A Comprehensive Resource, ed. J. M. Zeigler and F. W. G. Fearon, American Chemical Society, Washington, DC, 1990, ch. 40, pp. 705–739 Search PubMed.
- M. J. Owen, in Siloxane Polymers, ed. S. J. Clarson and J. A. Semlyen, Prentice Hall, Englewood Cliffs, 1993, ch. 7, p. 309 Search PubMed.
- B. J. Tighe, Eye Contact Lens Clin. Pract., 2013, 39, 4–12 CrossRef PubMed.
- L. N. Lewis, Silicones and Silicone-Modified Materials, American Chemical Society, 2000, vol. 729, ch. 2, pp. 11–19 Search PubMed.
- M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, ch. 12, pp. 381–458 Search PubMed.
- A. J. O’Lenick, Basic Silicone Chemistry – A Review, https://web.archive.org/web/20110516234405/https://www.siliconespectator.com/articles/Silicone_Spectator_January_2009.pdf, (accessed Sept. 11, 2023) Search PubMed.
- M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, ch. 7, pp. 171–188 Search PubMed.
- A. F. Schneider and M. A. Brook, Chem. – Eur. J., 2019, 25, 15367–15374 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley-Interscience, New Jersey, 4th edn, 2006 Search PubMed.
- M. A. Brook, Chem. – Eur. J., 2018, 24, 8458–8469 CrossRef CAS PubMed.
- J. C. Saam, J. Inorg. Organomet. Polym., 1999, 9, 3–16 CrossRef CAS.
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platinum Metal. Rev., 1997, 41, 66–75 CAS.
- P. B. Hitchcock, M. F. Lappert and N. J. W. Warhurst, Angew. Chem., Int. Ed. Engl., 1991, 30, 438–440 CrossRef.
- P. Habereder and A. Bereck, Rev. Prog. Color. Relat. Top., 2002, 32, 125–137 CAS.
- T. Wright, D. Karis, S. C. Millik, T. Tomkovic, S. G. Hatzikiriakos, A. Nelson and M. O. Wolf, ACS Appl. Mater. Interfaces, 2021, 13, 22195–22203 CrossRef CAS PubMed.
- D. D. Iarikov, M. Kargar, A. Sahari, L. Russel, K. T. Gause, B. Behkam and W. A. Ducker, Biomacromolecules, 2014, 15, 169–176 CrossRef CAS PubMed.
- L. A. T. W. Asri, M. Crismaru, S. Roest, Y. Chen, O. Ivashenko, P. Rudolf, J. C. Tiller, H. C. van der Mei, T. J. A. Loontjens and H. J. Busscher, Adv. Funct. Mater., 2014, 24, 346–355 CrossRef CAS.
- N. A. Falk, J. Surf. Deter., 2019, 22, 1119–1127 CrossRef CAS PubMed.
- P. J. G. Stevens, Pesticide Sci., 1993, 38, 103–122 CrossRef.
- R. M. Hill, Silicone Surfactants, Dekker, New York, 1999 Search PubMed.
- S. A. Snow, U. C. Pernisz and R. J. Braun, Silicon Chem., 2006, 3, 1–10 CrossRef CAS.
- B. Marciniec, J. Gulinski, W. Urbaniak and Z. W. Kornetka, Comprehensive Handbook on Hydrosilylation Chemistry, Pergamon, Oxford, 1992 Search PubMed.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- S. Rubinsztajn and J. Cella, Polym. Prepr., 2004, 45(1), 635–636 CAS.
- M. A. Brook, A. Yepremyan, G. Lu, M. Melendez-Zamudio, D. J. Hrabowyj and C. B. Gale, Green Chem., 2022, 24, 8751–8759 RSC.
- B. A. Kamino and T. P. Bender, Chem. Soc. Rev., 2013, 42, 5119–5130 RSC.
- B. A. Kamino, J. B. Grande, M. A. Brook and T. P. Bender, Org. Lett., 2011, 13, 154–157 CrossRef CAS PubMed.
- C. B. Gale, Z. B. Yan, M. Fefer, G. R. Goward and M. A. Brook, Macromolecules, 2021, 54, 4333–4341 CrossRef CAS.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- P.-S. Lai and C.-Y. Hsu, WO2014189796A1, 2014 Search PubMed.
- G. Lu, A. Yepremyen, K. Tamim, Y. Chen and M. A. Brook, Polymers, 2022, 14, 5040 CrossRef CAS PubMed.
- A. Y. Li, M. Melendez-Zamudio, A. Yepremyan and M. A. Brook, Green Chem., 2023, 25, 5267–5275 RSC.
- S. E. Laengert, A. F. Schneider, E. Lovinger, Y. Chen and M. A. Brook, Chem. – Asian J., 2017, 12, 1208–1212 CrossRef CAS PubMed.
- S. Zheng, M. Zlatin, P. R. Selvaganapathy and M. A. Brook, Addit. Manuf., 2018, 24, 86–92 CAS.
- A. Lusterio, M. Melendez-Zamudio and M. A. Brook, Ind. Eng. Chem. Res., 2021, 60, 3830–3838 CrossRef CAS.
- A. Lusterio and M. A. Brook, Molecules, 2021, 26, 4802 CrossRef CAS PubMed.
- M. Melendez-Zamudio, K. Chavda and M. A. Brook, Molecules, 2022, 27, 1869 CrossRef CAS PubMed.
- A. Camacho Ramirez, M. Melendez-Zamudio, A. Guerra Contreras and M. A. Brook, Sustain. Chem., 2023, 4, 197–208 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS PubMed.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polymer, 2014, 55, 6212–6219 CrossRef CAS.
- S. Halila, M. Manguian, S. Fort, S. Cottaz, T. Hamaide, E. Fleury and H. Driguez, Macromol. Chem. Phys., 2008, 209, 1282–1290 CrossRef CAS.
- T. Rambarran, A. Bertrand, F. Gonzaga, F. Boisson, J. Bernard, E. Fleury, F. Ganachaud and M. A. Brook, Chem. Commun., 2016, 52, 6681–6684 RSC.
- D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- C. D. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- T. Rambarran, F. Gonzaga, M. A. Brook, F. Lasowski and H. Sheardown, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1082–1093 CrossRef CAS.
- T. Rambarran, F. Gonzaga and M. A. Brook, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 855–864 CrossRef CAS.
- F. Lasowski, T. Rambarran, M. Brook and H. Sheardown, Compatability of PEG-containing siloxane materials by metal-free click-chemistry, Front. Bioeng. Biotechnol., Conference Abstract: 10th World Biomaterials Congress, 2016 DOI:10.3389/conf.FBIOE.2016.01.00120.
- T. Rambarran, F. Gonzaga and M. A. Brook, Macromolecules, 2012, 45, 2276–2285 CrossRef CAS.
- F. Gonzaga, G. Yu and M. A. Brook, Macromolecules, 2009, 42, 9220–9224 CrossRef CAS.
- A. S. Fawcett and M. A. Brook, Macromolecules, 2014, 47, 1656–1663 CrossRef CAS.
- A. S. Fawcett, T. C. Hughes, L. Zepeda-Velazquez and M. A. Brook, Macromolecules, 2015, 48, 6499–6507 CrossRef CAS.
- F. J. LaRonde, M. A. Brook and G. Hu, Silicon Chem., 2002, 1, 215–222 CrossRef CAS.
- Y. Chen, D. A. Valentini and M. A. Brook, Sustainability, 2023, 15, 9941 CrossRef CAS.
- P. Azadi Namin, P. Booth, J. Treviño Silva, L. J. Voigt and P. M. Zelisko, Macromolecules, 2023, 56, 2038–2051 CrossRef CAS.
- A. Genest, D. Portinha, E. Fleury and F. Ganachaud, Prog. Polym. Sci., 2017, 72, 61–110 CrossRef CAS.
- A. Genest, S. Binauld, E. Pouget, F. Ganachaud, E. Fleury and D. Portinha, Polym. Chem., 2017, 8, 624–630 RSC.
- A. Genest, D. Portinha, E. Pouget, K. Lamnawar, F. Ganachaud and E. Fleury, Macromol. Rapid Commun., 2021, 42, 2000372 CrossRef CAS PubMed.
- G. Lu, A. Yepremyan, S. Godfrey, C. Mohr, M. Herrlein and M. A. Brook, J. Polym. Sci., 2021, 59, 1935–1941 CrossRef CAS.
- A. Fatona, J. Moran-Mirabal and M. A. Brook, Polym. Chem., 2019, 10, 219–227 RSC.
- R. Bui and M. A. Brook, Polymer, 2019, 160, 282–290 CrossRef CAS.
- R. Bui and M. A. Brook, Green Chem., 2021, 23, 5600–5608 RSC.
- K. Tamim, C. B. Gale, K. E. C. Silverthorne, G. Lu, C. H. Iao and M. A. Brook, ACS Sustainable Chem. Eng., 2023, 11(18), 7062–7071 CrossRef CAS PubMed.
- R. Bui and M. A. Brook, Adv. Funct. Mater., 2020, 30, 2000737 CrossRef CAS.
- S. Zheng and M. A. Brook, ACS Appl. Polym. Mater., 2021, 3, 2045–2053 CrossRef CAS.
- S. Zheng and M. A. Brook, Macromol. Rapid Commun., 2021, 42, 2000375 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and M. A. Brook, Langmuir, 2013, 29, 12432–12442 CrossRef CAS PubMed.
- A. Fatona, A. Osamudiamen, J. Moran-Mirabal and M. A. Brook, J. Polym. Sci., 2020, 58, 1949–1959 CrossRef CAS.
- F. B. Madsen, L. Yu and A. L. Skov, ACS Macro Lett., 2016, 5, 1196–1200 CrossRef CAS PubMed.
- S. Zheng, Y. Chen and M. A. Brook, Polym. Chem., 2020, 11, 7382–7392 RSC.
- C. B. Gale and M. A. Brook, Adv. Funct. Mater., 2021, 31, 2105453 CrossRef CAS.
- M. A. Brook and R.-S. Himmeldirk, US Pat. 6,566,322, 2003 Search PubMed.
- P. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef CAS PubMed.
- L. J. Voigt, K. E. Tucker and P. M. Zelisko, Biomacromolecules, 2023, 24, 3463–3471 CrossRef CAS PubMed.
- K. Faiczak, M. A. Brook and A. Feinle, Macromol. Rapid Commun., 2020, 41, 2000161 CrossRef CAS PubMed.
- A. Yepremyan, A. Osamudiamen, M. A. Brook and A. Feinle, Chem. Commun., 2020, 56, 13555–13558 RSC.
- M. A. Brook, L. Dodge, Y. Chen, F. Gonzaga and H. Amarne, Chem. Commun., 2013, 49, 1392–1394 RSC.
- L. Dodge, Y. Chen and M. A. Brook, Chem. – Eur. J., 2014, 20, 9349–9356 CrossRef CAS PubMed.
- L. Zepeda-Velazquez, B. Macphail and M. A. Brook, Polym. Chem., 2016, 7, 4458–4466 RSC.
- S. Kong, R. Wang, S. Feng and D. Wang, Front. Chem., 2021, 9, 778896 CrossRef CAS PubMed.
- M. Kraszni, A. Marosi and C. K. Larive, Anal. Bioanal. Chem., 2013, 405, 5807–5816 CrossRef CAS PubMed.
- C. Tanford, Adv. Protein Chem., 1968, 23, 121–282 CrossRef CAS PubMed.
- L. M. Nayef, M. F. Khan and M. A. Brook, Colloids Surf., B, 2015, 128, 586–593 CrossRef CAS PubMed.
- L. Nayef, M. F. Khan and M. A. Brook, Heliyon, 2017, 3, e00264 CrossRef PubMed.
- V. Bartzoka, M. R. McDermott and M. A. Brook, Adv. Mater., 1999, 11(3), 257–259 CrossRef CAS.
- V. Bartzoka, M. A. Brook and M. R. McDermott, Langmuir, 1998, 14, 1887–1891 CrossRef CAS.
- V. Bartzoka, M. A. Brook and M. R. McDermott, Langmuir, 1998, 14, 1892–1898 CrossRef CAS.
- P. M. Zelisko and M. A. Brook, Langmuir, 2002, 18, 8982–8987 CrossRef CAS.
- P. M. Zelisko, A. M. Ragheb, M. Hrynyk and M. A. Brook, in Science and Technology of Silicones and Silicone-Modified Materials, ed. S. J. Clarson, J. J. Fitzgerald, M. J. Owen, S. D. Smith and M. E. VanDyke, 2007, vol. 964, pp. 256–266 Search PubMed.
- P. M. Zelisko, A. L. Aguilar and M. A. Brook, Langmuir, 2007, 23, 3620–3625 CrossRef CAS PubMed.
- M. R. McDermott, P. L. Heritage, V. Bartzoka and M. A. Brook, Immunol. Cell Biol., 1998, 76, 256–262 CrossRef CAS PubMed.
- P. Mazurek, P. M. Zelisko, A. L. Skov and M. A. Brook, ACS Appl. Polym. Mater., 2020, 2, 1203–1212 CrossRef CAS.
- S. Zheng, A. D'Angelo, U. Zell, Y. Chen, K. E. C. Silverthorne and M. A. Brook, Green Chem., 2021, 23, 7692–7700 RSC.
- H. Chen, M. A. Brook, H. D. Sheardown, Y. Chen and B. Klenkler, Bioconjugate Chem., 2006, 17, 21–28 CrossRef CAS PubMed.
- H. Chen, Y. Chen, H. Sheardown and M. A. Brook, Biomaterials, 2005, 26, 7418–7424 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- P. Kingshott and H. J. Griesser, Curr. Opin. Solid State Mater. Sci., 1999, 4, 403–412 CrossRef CAS.
- J. M. Harris, Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Springer, 1992 Search PubMed.
- J. G. Alauzun, J. N. Fortuna, H. Sheardown, F. Gonzaga and M. A. Brook, J. Mater. Chem., 2009, 19, 5033–5038 RSC.
- S. S. Filippova, K. V. Deriabin, I. Perevyazko, O. V. Shamova, D. S. Orlov and R. M. Islamova, ACS Appl. Polym. Mater., 2023, 5(7), 5286–5296 CrossRef CAS.
- P. M. Zelisko, K. K. Flora, J. D. Brennan and M. A. Brook, Biomacromolecules, 2008, 9, 2153–2161 CrossRef CAS PubMed.
- G. Sèbe and M. A. Brook, Wood Sci. Technol., 2001, 35, 269–282 CrossRef.
- M. Melendez-Zamudio, E. Donahue-Boyle, Y. Chen and M. A. Brook, Green Chem., 2023, 25, 280–287 RSC.
- J. Zhang and M. A. Brook, Mobilizing Chemistry Expertise To Solve Humanitarian Problems vol. 2, American Chemical Society, 2017, vol. 1268, ch. 6, pp. 91–116 Search PubMed.
- J. Zhang, E. Fleury, Y. Chen and M. A. Brook, RSC Adv., 2015, 5, 103907–103914 RSC.
- R. G. Lehmann, S. Varaprath, R. B. Annelin and J. L. Arndt, Envir. Tox. Chem., 1995, 14, 1299–1305 CrossRef CAS.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Envir. Tox. Chem., 1994, 13, 1061–1064 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |