
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of penta- and hexa(3,4-thienylene): size-dependent structural properties of cyclic oligothiophenes†
Mai
Nagase
 ab,
Sachiko
Nakano
a and
Yasutomo
Segawa
*ab
ab,
Sachiko
Nakano
a and
Yasutomo
Segawa
*ab
aInstitute for Molecular Science, Myodaiji, Okazaki, 444-8787, Japan. E-mail: segawa@ims.ac.jp
bThe Graduate University for Advanced Studies, SOKENDAI, Myodaiji, Okazaki, 444-8787, Japan
First published on 23rd August 2023
Abstract
Penta- and hexa(3,4-thienylene)s were synthesized as a potential precursor for thiophene-containing polyarenes, and the structures were determined via X-ray crystallography. The interconversion of thiophene rings is fast in penta(3,4-thienylene), and slow in hexa(3,4-thienylene) reflecting the activation energy for enantiomerization. Size-dependent bathochromic shifts were observed in UV-vis absorption spectra.
Cyclic oligoarylenes have long received considerable attention due to their structural, electronic, and optical properties.1 The most representative arylenes are phenylenes, in which the phenyl groups can be connected to each other in three ways, i.e., at the o-, m-, or p-positions. Of these, cyclo-o-phenylenes are of little interest and are not very well explored. Even so, biphenylene, triphenylene, tetraphenylene, hexaphenylene, and octaphenylene derivatives have already been synthesized (Fig. 1a), and their reactivity, structural properties, and applications have been investigated.2 Replacing the benzene rings with thiophene rings allows modifying the electronic and structural properties to induce, for example, weaker aromaticity, lower steric hindrance, and intermolecular S⋯S interactions in the solid state.3 Structurally, cyclo-o-phenylenes correspond approximately to cyclic oligo(3,4-thienylene)s in as much that the thiophene rings are linked at the β,β′-positions (Fig. 1b). To date, cyclic oligo(3,4-thienylene)s with two to four thiophene rings4 have been explored. Given that all their α-positions are reactive sites, tri- and tetra(3,4-thienylene)s (3T and 4T) can be transformed into a variety of electron-rich π-conjugated molecules, such as heterosumanenes5 and hetero[8]circulenes,6 as well as tetra(3,4-thienylene)s can be employed as e.g., 3D building blocks7 or solvent-responsive molecular assemblies.8 In addition, tri(3,4-thienylene)s have been studied as charge-transfer complexes9 or planar [6]radialenes.10 However, cyclic oligo(3,4-thienylene)s with five or more thiophene rings have not yet been synthesized.11 To create larger β,β′-linked cyclic oligo(3,4-thienylene)s, building blocks that contain functional groups at the β-positions are a prerequisite. In 2017, a practical synthetic method for a β-substituted unit, i.e., a 3,4-diborylated thiophene derivative, was developed by Osuka, Tanaka, and co-workers, which widened the range of synthetic approaches for cyclic compounds that contain 3,4-thienylenes and other heteroarenes.12
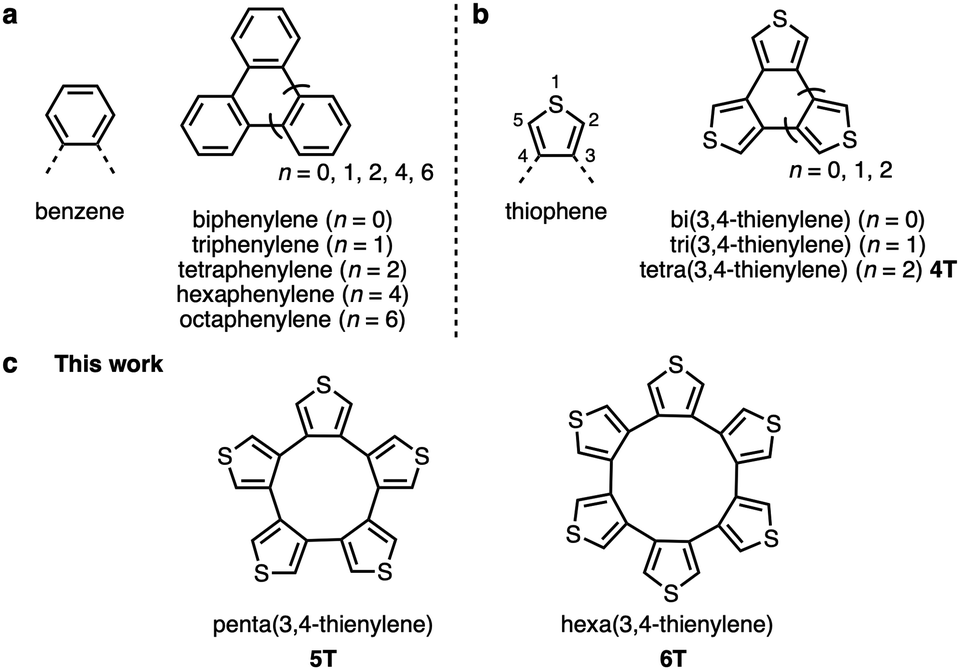 | ||
| Fig. 1 Representative cyclic oligoarylenes. (a) Cyclo-o-phenylenes. (b) Cyclic oligo(3,4-thienylene)s. (c) Penta- and hexa(3,4-thienylene)s (5T and 6T). | ||
Herein, we report the synthesis of penta- and hexa(3,4-thienylene)s (5T and 6T) via Pd-catalyzed cross-coupling and Ni-mediated homocoupling reactions using 3,4-diborylthiophene 1 or the newly synthesized 4,4′-diboryl-3,3′-bithiophene 2 (Fig. 1c and 2a). The structures and electronic properties of 5T and 6T were determined using NMR and X-ray crystallography as well as UV-vis absorption spectroscopy. The structural characteristics and optoelectronic properties of 5T and 6T were compared to those of 4T, and examined using density functional theory (DFT) calculations.
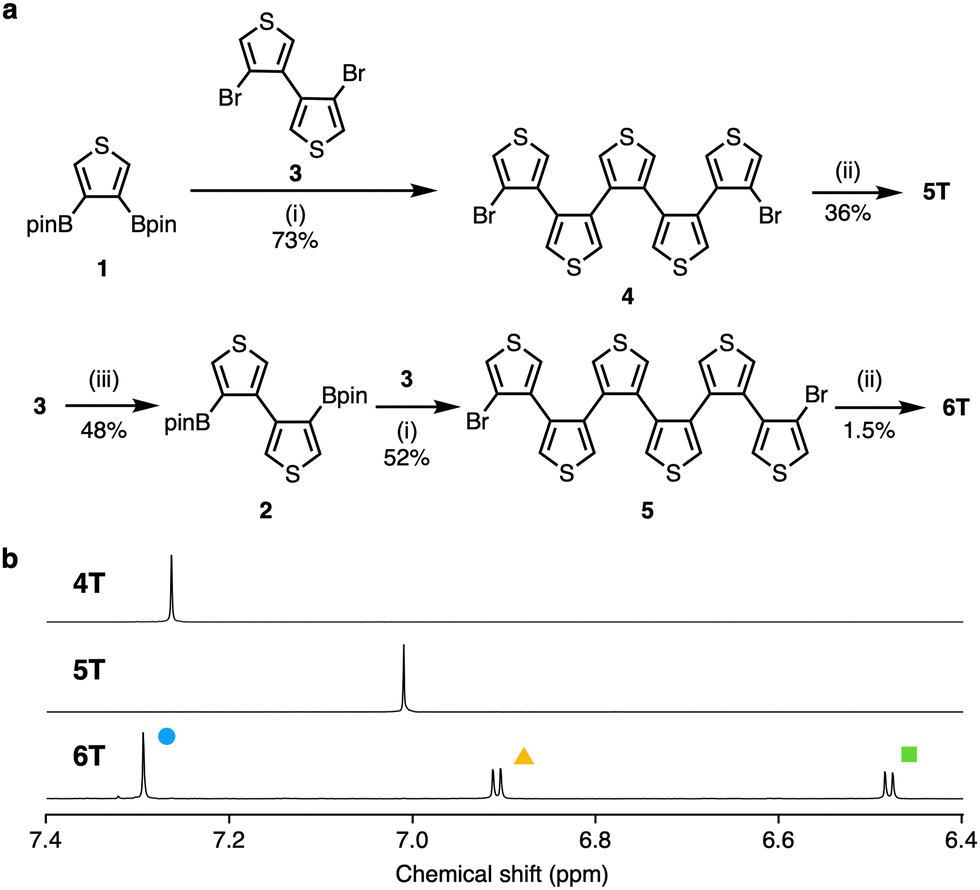 | ||
| Fig. 2 (a) Synthesis of 5T and 6T; (i) Pd2(dba)3, XPhos, K3PO4, THF/H2O, 40 °C, 22–23 h; (ii) Ni(cod)2, 2,2′-bipyridine, THF, reflux, 2.5 h; (iii) HBpin, Zn powder, Et3N, NiCl2(dppp), DPPF, toluene, reflux, 60 h. (b) 1H NMR spectra of 4T–6T in CD2Cl2. The signals of 6T are indicated by colored marks. Abbreviations: dba = dibenzylideneacetone; XPhos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl; cod = 1,5-cyclooctadiene; Bpin = 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl; dppp = 1,3-bis(diphenylphosphino)propane; DPPF = 1,1′-bis(diphenylphosphino)ferrocene. | ||
Penta- and hexa(3,4-thienylene)s (5T and 6T) were synthesized via sequential Suzuki–Miyaura cross-coupling and Ni(0)-mediated intramolecular homocoupling reactions (Fig. 2a). For that purpose, diborylthiophene 1 and 4,4′-dibromo-3,3′-bithiophene (3) were prepared initially according to reported methods.12,13 The Suzuki–Miyaura cross-coupling reaction of 1 with an excess of 3 (8.0 equiv.) was carried out using Pd2(dba)3 (2.5 mol%), XPhos (10 mol%), and K3PO4 (4.0 equiv.) to produce dibromoquinquethiophene 4 in 73% yield. Dibromobithiophene 3 was used in excess in order to suppress the generation of polymers. The resulting 4 was treated with Ni(cod)2 (2.2 equiv.) and 2,2′-bipyridine (2.2 equiv.) for 3 h in refluxing tetrahydrofuran (THF) (2 mM), which allowed isolating the desired cyclic oligothiophene 5T in 36% yield. Increasing the substrate concentration provided 5T in low yield in this reaction step. Next, a cyclic oligothiophene containing an additional thiophene ring, 6T, was synthesized by applying analogous reaction conditions. A borylation reaction similar to that used to synthesize 112,14 was performed to obtain 2 in 48% yield. The reaction of 2 with an excess of 3 under Pd catalysis furnished dibromosexithiophene 5 in 52% yield. The obtained 5 was subsequently treated with Ni(cod)2 (3.3 equiv.) and 2,2-bipyridine (3.3 equiv.) in THF. In contrast to 5T, 6T was obtained in low yield (1.5%), presumably due to the formation of polymeric compounds. Both 5T and 6T were slightly soluble in common organic solvents (see Table S1 in ESI† for details).
To identify the thus-obtained compounds 5T and 6T, and to investigate their size-dependent structural characteristics, NMR spectra were recorded (Fig. 2b). In the room-temperature 1H NMR spectrum of 5T in dichloromethane-d2, the α-protons of the thiophene rings appear as a singlet at 7.01 ppm. Even at −95 °C, the singlet remains almost unchanged. In the 13C NMR spectrum in chloroform-d1, two signals were observed at 124.6 and 137.6 ppm. These results indicate that the dynamic interconversion of the thiophene rings is fast on the NMR timescale. Unlike that of 5T, the 1H NMR spectrum of 6T in dichloromethane-d2 at room temperature displays three signals, i.e., doublets at 6.5 and 6.9 ppm as well as a singlet at 7.3 ppm. Only very slight broadening of the signals was observed even at 140 °C in tetrachloroethane-d2, indicating that 6T has three diastereotopic protons and those are not exchanged in the NMR timescale. The 1H NMR spectra of 4T–6T presented in Fig. 2b clearly demonstrate the size dependence of the symmetry in solution. The NMR assignments were supported by chemical shift calculations using the GIAO method (see Fig. S2 in ESI† for details).
The solid-state structures of 5T and 6T were determined via X-ray diffraction analysis of single crystals obtained from a hexane/chloroform and a pentane/THF solution, respectively (Fig. 3a and b). Compound 5T adopts a C2-symmetric structure in which five thiophene rings are fused at the 3,4-positions to form a 10-membered ring. Compound 6T adopts a screw (or helical) structure with D2 symmetry, similar to hexaphenylene2b,e–h and hexa(2,3-thienylene)s.11b,f The distance between the centroids of the central thiophene rings (av. 3.4 Å) is typical for intramolecular π–π interactions. In both crystals, the enantiomers are aligned alternatingly reflecting the centric space group (I2/a). The average dihedral angles (θ) of the α–β–β–α and β–β–β–β positions of the 3,3′-bithiophene moieties are shown in Fig. 3a and b. The dihedral angles in 5T and 6T are 46.0° (θa), 90.8° (θb), and 87.8° (θc), and 41.8° (θd), 46.0° (θe), and 124.5° (θf), respectively (see Fig. S3 in ESI† for details). Considering the dihedral angle in 4T (54.3°), determined using X-ray crystallography,4e the smallest dihedral angle in 4T, 5T, and 6T decreases with increasing number of thiophene rings.
 | ||
| Fig. 3 (a) and (b) Molecular structures of 5T (a) and 6T (b) with thermal ellipsoids at 50% probability; gray = carbon; yellow = sulfur; white = hydrogen. The C2 axis of 5T and one of the C2 axes of 6T are noted with dashed lines. The hydrogen atoms indicated by the colored marks correspond to the 1H NMR signals shown in Fig. 2b. | ||
In order to gain insight into the size-dependent structural characteristics, DFT calculations were conducted. The structures of 5T and 6T, optimized at the B3LYP-D3/6-31G(d) level nicely reproduced the structures observed by X-ray crystallography, including the dihedral angles (see Fig. S3 in ESI† for details). Compound 6T has two possible stable conformations, i.e., a screw and a crown conformation.‡ The Gibbs free energy of 6T with the screw conformation is 3.5 kcal mol−1 lower than that of the crown conformation, which was not observed by NMR spectroscopy in this study (see Fig. S4 in ESI† for details). Strain energies of 5T and 6T were calculated to be 19.9 and 1.1 kcal mol−1, respectively, using hypothetical homodesmotic reactions.15 This result indicates that 5T has a strained structure, and the decrease in energy of 6T might be due to the stabilization by intramolecular π–π interaction (see Fig. S5 in ESI† for details). The enantiomerization between C2-symmetric 5T and 5T′ as well as between D2-symmetric 6T and 6T′ is illustrated in Fig. 4a and b. Our calculations suggest Cs symmetry for the transition states (TSs) of 5T and 6T, and activation energies for the interconversion of 5T and 6T of 5.0 and 26.5 kcal mol−1, respectively. The highlighted thiophene ring resides initially on the C2 axis in 5T; after interconversion via the enantiomerization pathway, this thiophene ring does not reside anymore on the C2 axis in 5T′. By repeating these enantiomerization steps, all α-protons become magnetically equivalent. This dynamic motion, together with the low activation energy, leads to a rapid exchange of the α-protons even at low temperatures. In the enantiomerization process of 6T, the highlighted thiophene rings on the C2 axis become the central thiophene rings, and four thiophene rings are symmetrically identical. Although this process leads to the magnetic equivalence of the α-protons, the reaction rate should be slow because of the high activation energy (26.5 kcal mol−1). Taken together, the NMR spectrum of 4T shows a singlet because of its highly symmetric structure (D2d). Although 5T has low symmetry (C2), the fast dynamic motion of the molecule enables the α-protons of the thiophene rings to reach magnetic equivalence. Compound 6T also has low symmetry (D2), albeit that its unsymmetrical NMR signals indicate a slow exchange of the diastereotopic α-protons on the NMR timescale. Thus, the size-dependent structural characteristics of 4T, 5T, and 6T were determined using a combination of NMR measurements and DFT calculations.
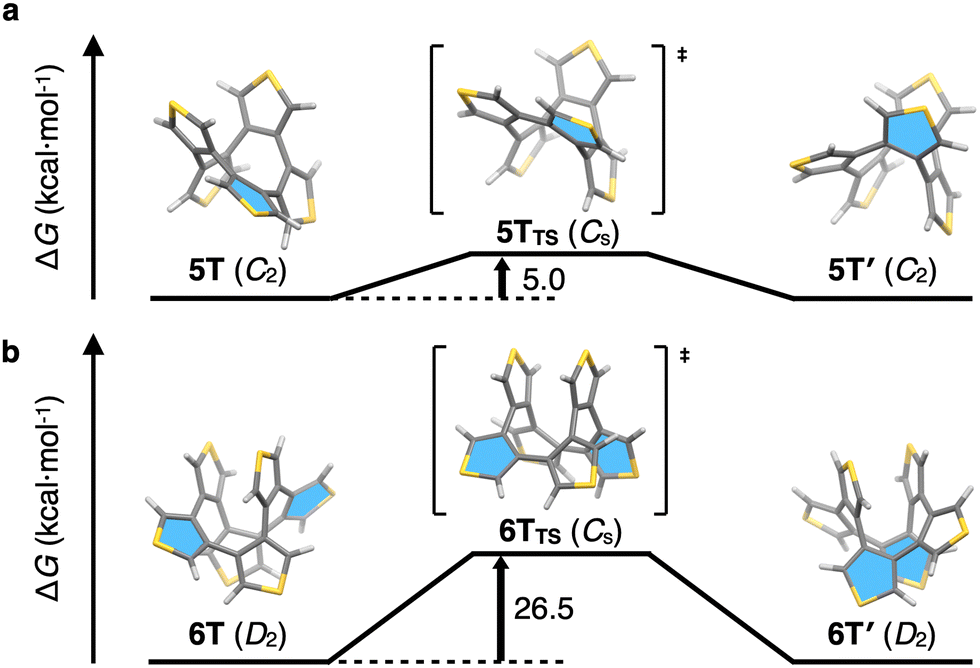 | ||
| Fig. 4 Energy diagrams for the enantiomerization of 5T (a) and 6T (b). Selected thiophene rings are highlighted in blue to illustrate the enantiomerization. | ||
To further examine the size-dependent characteristics of 5T, 6T, and reference compound 4T, their UV-vis absorption spectra were recorded (Fig. 5a). The maximum absorption for all three compounds was observed at ∼230 nm, and the longest-wavelength absorption maxima were observed at 263, 273, and 276 nm for 4T, 5T, and 6T, respectively. Fluorescence of 5T and 6T was negligible in dichloromethane solution. The frontier molecular orbitals related to the longest absorption bands and their energy levels are depicted in Fig. 5b, taking into account the vertical excitation energies, oscillator strengths, and transition contributions obtained from time-dependent (TD) DFT calculations (see Fig. S6 in ESI† for details). Because 4T adopts a highly symmetrical structure, the electronic wavefunctions of the HOMO and LUMO are distributed over the entire molecule, and the HOMO and the HOMO−1 are degenerate. In contrast, the HOMO and LUMO of 5T and 6T are relatively localized, presumably because of their lower-symmetry structures. The shoulder peaks in the range of 260–280 nm can be attributed to a combination of the HOMO−1 → LUMO and HOMO → LUMO transitions for 4T, and the HOMO → LUMO transitions for 5T and 6T. With increasing number of thiophene rings, the HOMO level rises gradually from −6.09 eV to −5.76 eV, while the LUMO energy decreases from 4T to 5T and is similar for 5T and 6T. Consequently, it can be concluded that the HOMO–LUMO energy gap decreases from 4T to 6T, which is consistent with the bathochromic shift observed for the longest-wavelength absorption maxima.
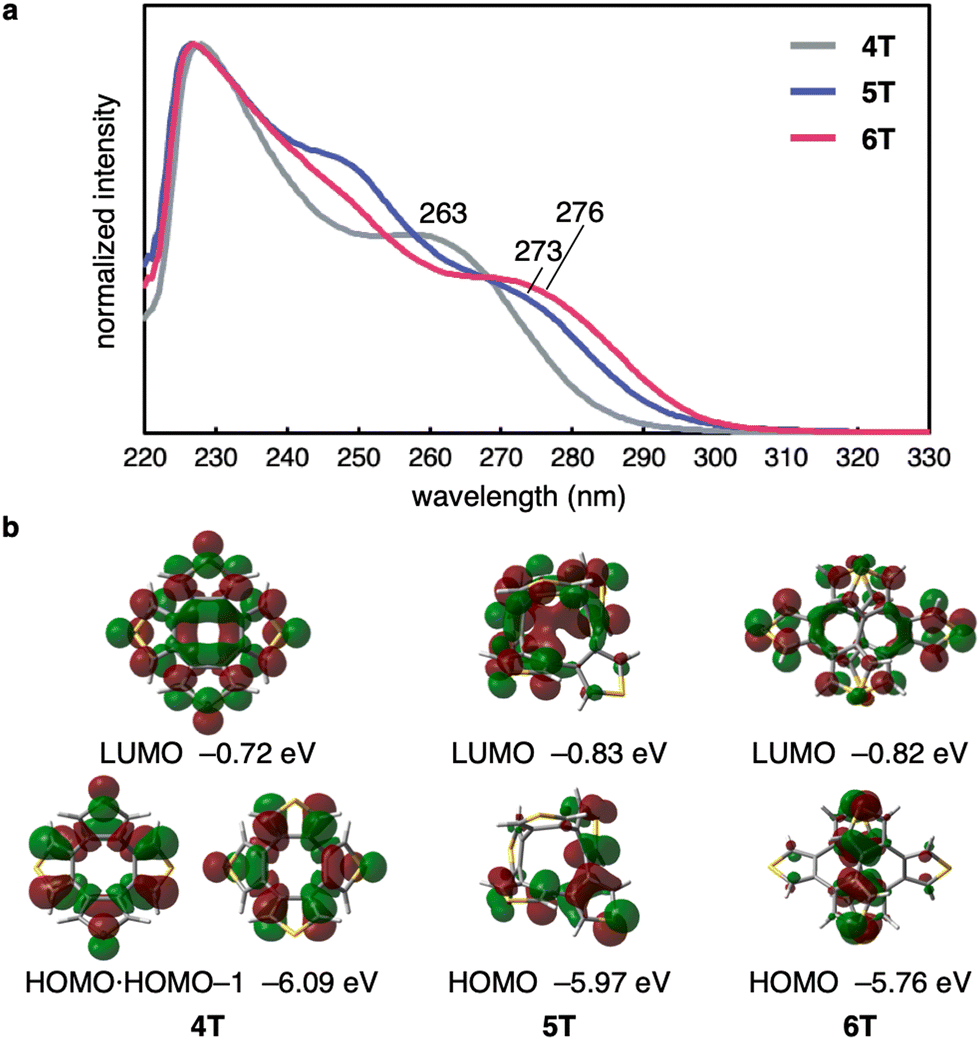 | ||
| Fig. 5 Optoelectronic properties of 4T, 5T, and 6T. (a) UV-vis absorption spectra of 4T, 5T, and 6T in CH2Cl2. (b) Frontier molecular orbitals and the energy levels of 4T, 5T, and 6T calculated at the B3LYP-D3/6-31G(d) level (isovalue = 0.03). | ||
In summary, we have developed synthetic routes to 5T and 6Tvia Suzuki–Miyaura cross-coupling and a Ni-mediated homocoupling reaction that involved the synthesis of a new β,β′-substituted moiety 2. Using an excess of 3 effectively suppressed the formation of polymers. While only a single singlet was observed in the NMR spectra of 4T and 5T, three different signals were observed for 6T. The solid-state structures of 5T and 6T were determined using X-ray crystallography; a corresponding analysis of the metric parameters revealed that the smallest dihedral angle of the 3,3′-bithiophene moiety decreases with increasing number of thiophene rings. The size-dependence observed in the NMR spectra can be rationally interpreted in terms of the activation energies for the interconversion of the thiophene rings. The size-dependence manifests in the optoelectronic measurements, in which the longest-wavelength absorption maxima are by 13 nm bathochromically shifted with increasing size of the molecules. The results of our DFT calculations suggest that gradually increasing the number of thiophene rings gradually increases the HOMO energy and decreases the LUMO energy. With synthetic routes to 5T and 6T established, it will now be possible to explore the synthetic chemistry of a variety of unprecedented thiophene-containing arenes.
This work was supported by FOREST program (JPMJFR211R to Y. S.) from JST, JSPS KAKENHI (JP22K19038 and JP22H02068 to Y. S.), UBE Foundation, Mitsubishi Foundation, and Asahi Glass Foundation. We thank Tetsuro Kusamoto, Ryota Matsuoka (IMS) and Takayuki Tanaka (Kyoto Univ.) for their support of experiments and fruitful advices. M. N. is a recipient of JSPS Research Fellowship for Young Scientists (DC2) and IMS SRA fellowship. This work was conducted in IMS supported by ARIM (JPMXP1223MS5012). Calculations were performed using the resources of the Research Center for Computational Science, Okazaki, Japan (23-IMS-C202).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS PubMed; (b) M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522 CrossRef CAS PubMed; (c) M. Stȩpień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288 CrossRef PubMed; (d) J.-i Setsune, Chem. Rev., 2017, 117, 3044 CrossRef CAS PubMed; (e) M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 15743 CrossRef CAS PubMed.
- (a) W. C. Lothrop, J. Am. Chem. Soc., 1941, 63, 1187 CrossRef CAS; (b) G. Wittig and G. Lehmann, Liebigs Ann. Chem., 1957, 90, 875 CAS; (c) G. Wittig and F. Bickelhaupt, Chem. Ber., 1958, 91, 883 CrossRef CAS; (d) C. M. Buess and D. D. Lawson, Chem. Rev., 1960, 60, 313 CrossRef CAS; (e) G. Wittig and K.-D. Rümpler, Liebigs Ann. Chem., 1971, 751, 1 CrossRef CAS; (f) H. Irngartinger, Isr. J. Chem., 1972, 10, 635 CrossRef CAS; (g) L. Ernst, A. Mannschreck and K.-D. Rümpler, Org. Magn. Reson., 1973, 5, 125 CrossRef CAS; (h) H. Irngartinger, Acta Crystallogr., 1973, B29, 894 CrossRef; (i) H. J. S. Winkler and G. Wittig, J. Org. Chem., 1963, 28, 1733 CrossRef CAS; (j) J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892 CrossRef CAS; (k) D. Sonet and B. Bibal, Tetrahedron Lett., 2019, 60, 872 CrossRef CAS; (l) H. Takano, T. Ito, K. S. Kanyiva and T. Shibata, Eur. J. Org. Chem., 2019, 2871 CrossRef CAS; (m) N. Contreras-Pereda, S. Pané, J. Puigmartí-Luis and D. Ruiz-Molina, Coord. Chem. Rev., 2022, 460, 214459 CrossRef CAS; (n) H.-R. Ma, X.-S. Peng, J.-F. Cui and H. N. C. Wong, Tetrahedron Lett., 2023, 119, 154429 CrossRef CAS.
- (a) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 4347 CrossRef CAS; (b) Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC; (c) T. Okamoto, C. P. Yu, C. Mitsui, M. Yamagishi, H. Ishii and J. Takeya, J. Am. Chem. Soc., 2020, 142, 9083 CrossRef CAS PubMed; (d) Q. Shen, C. He, S. Li, L. Zuo, M. Shi and H. Chen, Acc. Mater. Res., 2022, 3, 644 CrossRef CAS.
- (a) T. Kauffmann, B. Greving, J. König, A. Mitschker and A. Woltermann, Angew. Chem., Int. Ed. Engl., 1975, 14, 713 CrossRef; (b) H. Hart and M. Sasaoka, J. Am. Chem. Soc., 1978, 100, 4326 CrossRef CAS; (c) H. Irngartinger, U. Huber-Patz and H. Rodewald, Acta Crystallogr., 1985, C41, 1088 CAS; (d) M. K. Shepherd, J. Chem. Soc., Chem. Commun., 1985, 880 RSC; (e) T. Fujimoto, R. Suizu, H. Yoshikawa and K. Awaga, Chem. – Eur. J., 2008, 14, 6053 CrossRef CAS PubMed.
- K. Imamura, K. Takimiya, Y. Aso and T. Otsubo, Chem. Commun., 1999, 1859 RSC.
- (a) K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367 CrossRef CAS PubMed; (b) A. Dadvand, F. Cicoira, K. Y. Chernichenko, E. S. Balenkova, R. M. Osuna, F. Rosei, V. G. Nenajdenko and D. F. Perepichka, Chem. Commun., 2008, 5354 RSC; (c) S. Kato, Y. Serizawa, D. Sakamaki, S. Seki, Y. Miyake and H. Shinokubo, Chem. Commun., 2015, 51, 16944 RSC; (d) Y. Serizawa, S. Akahori, S. Kato, H. Sakai, T. Hasobe, Y. Miyake and H. Shinokubo, Chem. – Eur. J., 2017, 23, 6948 CrossRef CAS PubMed; (e) S. Akahori, H. Sakai, T. Hasobe, H. Shinokubo and Y. Miyake, Org. Lett., 2018, 20, 304 CrossRef CAS PubMed; (f) L. Li, S. Zhao, B. Li, L. Xu, C. Li, J. Shi and H. Wang, Org. Lett., 2018, 20, 2181 CrossRef CAS PubMed; (g) S. Kato, S. Akahori, Y. Serizawa, X. Lin, M. Yamauchi, S. Yagai, T. Sakurai, W. Matsuda, S. Seki, H. Shinokubo and Y. Miyake, J. Org. Chem., 2020, 85, 62 CrossRef CAS PubMed; (h) Y. Miyake and H. Shinokubo, Chem. Commun., 2020, 56, 15605 RSC; (i) S. Akahori, T. Sasamori, H. Shinokubo and Y. Miyake, Chem. – Eur. J., 2021, 27, 8178 CrossRef CAS PubMed; (j) S. Akahori, T. Fujihara, Y. Tsuji, H. Shinokubo and Y. Miyake, Synthesis, 2021, 2995 CAS.
- (a) S. Zhang, X. Liu, C. Li, L. Li, J. Song, J. Shi, M. Morton, S. Rajca, A. Rajca and H. Wang, J. Am. Chem. Soc., 2016, 138, 10002 CrossRef CAS PubMed; (b) C. Zhao, X. Cai, Z. Ma, J. Shi, L. Xu and H. Wang, J. Photochem. Photobiol., A, 2018, 355, 318 CrossRef CAS; (c) A. Rajca, C. Shu, H. Zhang, S. Zhang, H. Wang and S. Rajca, Photochem. Photobiol., 2021, 97, 1376 CrossRef CAS PubMed.
- T. Takeda, M. Ozawa and T. Akutagawa, Cryst. Growth Des., 2019, 19, 4784 CrossRef CAS.
- T. Sugano, T. Hashida, A. Kobayashi, H. Kobayashi and M. Kinoshita, Bull. Chem. Soc. Jpn., 1988, 61, 2303 CrossRef CAS.
- A. Patra, Y. H. Wijsboom, L. J. W. Shimon and M. Bendikov, Angew. Chem., Int. Ed., 2007, 46, 8814 CrossRef CAS PubMed.
- Examples of cyclic structures including β,β′-linkages: (a) M. J. Marsella, K. Yoon and F. S. Tham, Org. Lett., 2001, 3, 2129 CrossRef CAS PubMed; (b) Y. Wang, Z. Wang, D. Zhao, Z. Wang, Y. Cheng and H. Wang, Synlett, 2007, 2390 CAS; (c) F. Sannicolò, P. R. Mussini, T. Benincori, R. Cirilli, S. Abbate, S. Arnaboldi, S. Casolo, E. Castiglioni, G. Longhi, R. Martinazzo, M. Panigati, M. Pappini, E. Q. Procopio and S. Rizzo, Chem. – Eur. J., 2014, 20, 15298 CrossRef PubMed; (d) K. Asai, A. Fukazawa and S. Yamaguchi, Chem. Commun., 2015, 51, 6096 RSC; (e) C. Zhao, L. Xu, Y. Wang, C. Li and H. Wang, Chin. J. Chem., 2015, 33, 71 CrossRef CAS; (f) M. Ueda, K. Jorner, Y. M. Sung, T. Mori, Q. Xiao, D. Kim, H. Ottosson, T. Aida and Y. Itoh, Nat. Commun., 2017, 8, 346 CrossRef PubMed; (g) E. Quartapelle Procopio, T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, C. Carbonera, R. Cirilli, A. Cominetti, L. Longo, R. Martinazzo, M. Panigati and R. Pò, New J. Chem., 2017, 41, 10009 RSC.
- K. Kise, F. Chen, K. Kato, T. Tanaka and A. Osuka, Chem. Lett., 2017, 46, 1319 CrossRef CAS.
- A. Rajca, M. Miyasaka, M. Pink, H. Wang and S. Rajca, J. Am. Chem. Soc., 2004, 126, 15211 CrossRef CAS PubMed.
- P. Leowanawat, A.-M. Resmerita, C. Moldoveanu, C. Liu, N. Zhang, D. A. Wilson, L. M. Hoang, B. M. Rosen and V. Percec, J. Org. Chem., 2010, 75, 7822 CrossRef CAS PubMed.
- V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2281496 (2), 2281497 (5T) and 2281498 (6T). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03508e |
| ‡ Crown conformer is observed in hexaphenylene2b,e–g and hexa(2,3-thienylene)s.4a,11a |
| This journal is © The Royal Society of Chemistry 2023 |