
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulated self-assembly of three flexible Cr(III) PCPs for SO2 adsorption and detection†
Valeria B.
López-Cervantes‡
 a,
Dominic
Bara‡
b,
Ana
Yañez-Aulestia‡
a,
Eva
Martínez-Ahumada
a,
Alfredo
López-Olvera
a,
Yoarhy A.
Amador-Sánchez
a,
Diego
Solis-Ibarra
*a,
Elí
Sánchez-González
a,
Ilich A.
Ibarra
*a and
Ross S.
Forgan
*b
a,
Dominic
Bara‡
b,
Ana
Yañez-Aulestia‡
a,
Eva
Martínez-Ahumada
a,
Alfredo
López-Olvera
a,
Yoarhy A.
Amador-Sánchez
a,
Diego
Solis-Ibarra
*a,
Elí
Sánchez-González
a,
Ilich A.
Ibarra
*a and
Ross S.
Forgan
*b
aLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacan, 04510, Ciudad de Mexico, Mexico. E-mail: argel@unam.mx; Fax: +52-55-5622-4595
bWestCHEM School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: ross.forgan@glasgow.ac.uk
First published on 1st June 2023
Abstract
Modulated self-assembly protocols are used to develop facile, HF-free syntheses of the archetypal flexible PCP, MIL-53(Cr), and novel isoreticular analogues MIL-53(Cr)-Br and MIL-53(Cr)-NO2. All three PCPs show good SO2 uptake (298 K, 1 bar) and high chemical stabilities against dry and wet SO2. Solid-state photoluminescence spectroscopy indicates all three PCPs exhibit turn-off sensing of SO2, in particular MIL-53(Cr)-Br, which shows a 2.7-fold decrease in emission on exposure to SO2 at room temperature, indicating potential sensing applications.
Porous coordination polymers (PCPs), a class of hybrid materials comprised of extended metal–ligand coordination networks, have attracted attention due to their high porosity and tuneable and adaptable structures.1 Many “flexible” PCPs undergo reversible crystal-to-crystal structural transformations upon guest inclusion and removal,2 combining a highly ordered network with structural transformability2a and/or local molecular motion, often with relatively large movements of >5–10 Å.3 Phase changes can occur in response to external stimuli, e.g., light, temperature, pressure, electric and/or magnetic fields and guest adsorption/desorption.4 The most common dynamic mode is “breathing”,2b,4c often characterised by a phase transition between two or more distinct states causing a unit cell volume change.3 “Gate-opening”4c generally results in the pore opening in the presence of adsorbate molecules, offering the attractive possibility of pressure-swing processes for selective gas adsorption and separation.5
The MIL-53 isoreticular series comprises one of the most investigated families of flexible PCPs.6 One-dimensional chains of octahedral CrO4(OH)2 units containing axially bridging μ2-OH moieties and four equatorial carboxylate oxygen atoms from four benzene-1,4-dicarboxylate (BDC) ligands are connected into a diamondoid net with sra topology. First isolated as the Cr congener,6a,6b examples with Sc, V, Fe, Al, In, and Ga, as well as derivatives with functionalised BDC ligands, have since been synthesised.6c Introduction of –Br and –NO2 substituents to MIL-53(Al) and MIL-53(In) tuned their flexibilities and gas uptakes; locating functional groups within the diamondoid channel stiffens the pore due to steric hindrance.7 There are relatively few reports, however, of functionalised MIL-53(Cr) derivatives.8
The functional diversity and structural adjustability of PCPs mean they can achieve efficient and specific host–guest recognition, while the presence of many π and n electrons can induce fluorescence, making PCPs attractive candidates as sensors.9 It has been suggested that flexible PCPs could be more suited to luminescent detection compared to rigid materials,10 for example, [Zn2(BDC)2(dpNDI)] has a doubly-interpenetrated dynamic structure and exhibits strong fluorescence only if there is movement between its two nets; if it remains rigid, no luminescence is observed.11 However, research on flexible PCPs that can be used as luminescent sensors is limited. Herein we report the modulated self-assembly of three PCPs of the flexible MIL-53(Cr) family, avoiding the use of HF in their syntheses, and detail the SO2 uptake and detection behaviours of these PCPs.
MIL-53(Cr) and its novel –Br and –NO2 substituted analogues were obtained by a modification of the original literature protocol (ESI,† Section S2),6a where we applied the principles of modulated self-assembly12 that were previously successful for the synthesis of MIL-53(Fe).13 Equimolar amounts of the linker and CrCl3·6H2O, with concentrated HCl as modulator, were reacted hydrothermally at 220 °C in a Teflon-lined autoclave for 72 h to yield as-synthesised (as) PCPs after washing. Samples were heated in DMF for 16 h at 220 °C to isolate the PCPs as DMF solvates (DMF),14 removing any residual bound linker, and then refluxed in MeOH for 16 h before drying under vacuum (MeOH). Samples were activated (act) for adsorption experiments by degassing on a turbomolecular vacuum pump at 150 °C for 20 h. Comparison of powder X-ray diffractograms (Fig. 1 and ESI,† Section S3) showed all three DMF solvates were structurally similar to the MIL-53(Fe) DMF solvate reported previously,13 while washing with MeOH allowed the samples to close. MIL-53(Cr) was isolated as its hydrated narrow pore form, MIL-53_lt (Fig. 1a).6a,6b Comparison with the predicted diffractograms from the crystal structures of the In analogues7b confirmed the formation of MIL-53(Cr)-Br (Fig. 1b) and MIL-53(Cr)-NO2 (Fig. 1c). However, upon drying, they were isolated in a more open form than that of both the Al and In analogues,7 indicating that the nature of the metal ion influences the behaviour of these two new PCPs.
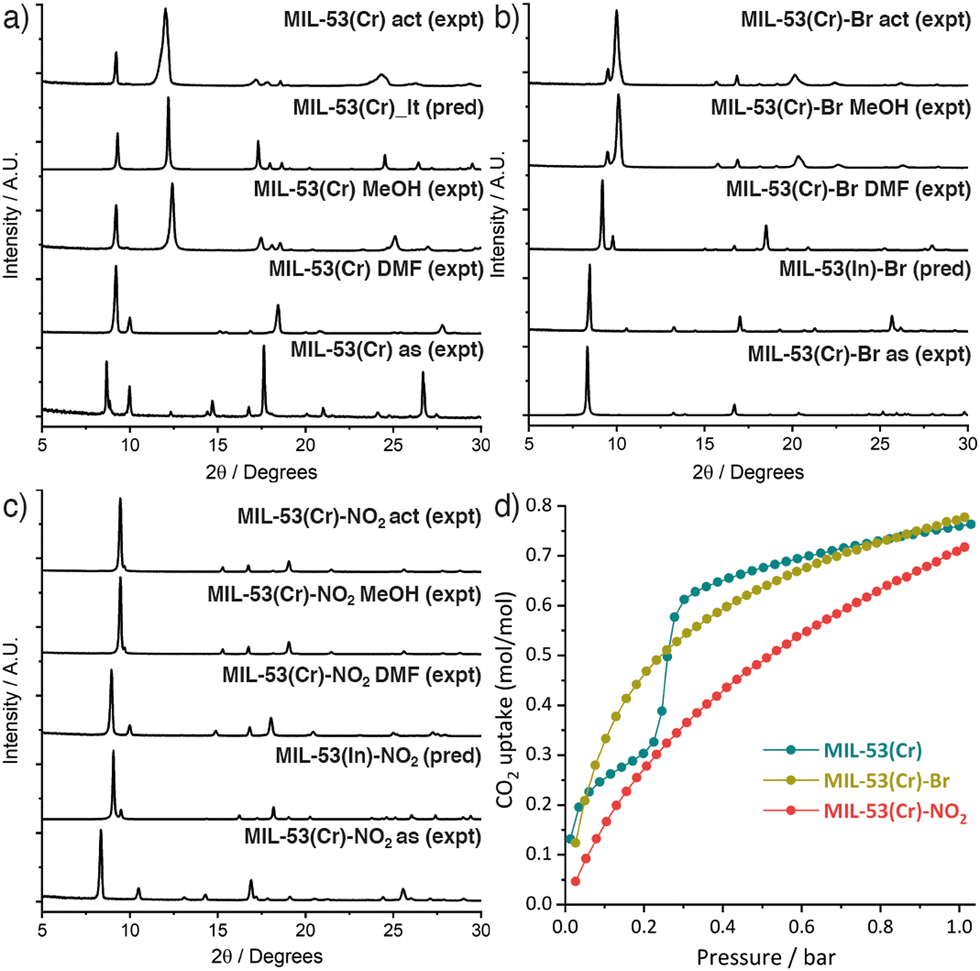 | ||
| Fig. 1 Stacked powder X-ray diffractograms of (a) MIL-53(Cr), (b) MIL-53(Cr)-Br, and (c) MIL-53(Cr)-NO2 across their respective synthesis and activation procedures (as = as-synthesised, act = activated). (d) CO2 adsorption isotherms (273 K) of the three MOFs. | ||
Only MIL-53(Cr) adsorbed N2 at 77 K (BET area = 1411 m2 g−1) while MIL-53(Cr)-Br and MIL-53(Cr)-NO2 again behaved differently from the Al and In analogues in adsorbing negligible amounts of N2. In contrast, all three Cr MOFs showed good CO2 uptake at 273 K (Fig. 1d) and 298 K (Fig. S20, ESI†) confirming their porosity, with MIL-53(Cr) exhibiting a characteristic stepped adsorption isotherm consistent with a structural transition to an open pore phase.15 The CO2 uptakes of the functionalised MOFs are comparable to the Al analogues,7a as are their isosteric enthalpies of CO2 adsorption (∼20–40 kJ mol−1, Fig. S22, ESI†). Using HCl as a modulator in hydrothermal syntheses avoids the highly toxic HF, which has only been achieved previously through mechanochemical approaches,16 and is clearly applicable to producing a range of MIL-53(Cr) derivatives.
Adsorption–desorption SO2 isotherms (298 K, 0–1 bar) were collected on samples that had been further activated at 473 K under vacuum for 2 h (Fig. 2). The SO2 adsorption-desorption isotherm for MIL-53(Cr) again shows a stepwise profile, indicative of guest-induced structural changes in the material. After a sharp initial SO2 uptake at low pressure (ca. 0.66 mol mol−1), gate-opening is observed at around 0.4 bar, with SO2 uptake reaching 1.86 mol mol−1 at 0.6 bar and gradually increasing to reach the total SO2 capture value of 1.87 mol mol−1 at 1 bar. Significant hysteresis is observed on the desorption branch, with the onset of desorption and concomitant gate-closing at less than 0.4 bar.
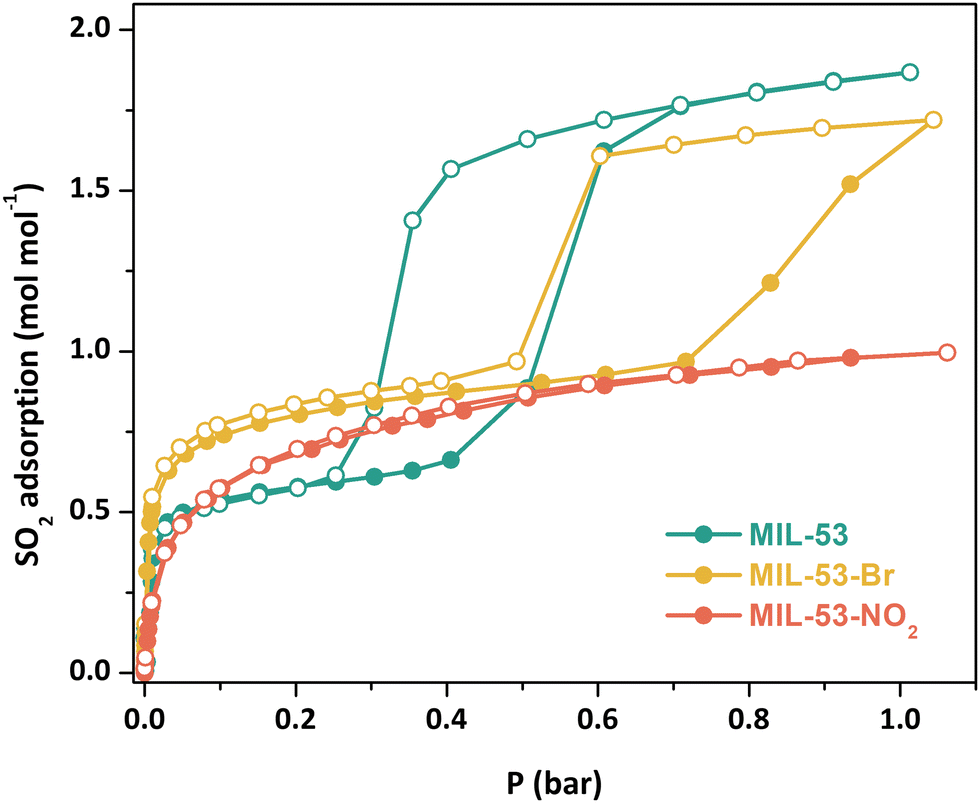 | ||
| Fig. 2 Experimental SO2 adsorption–desorption isotherms for fully activated MIL-53(Cr) (green), MIL-53(Cr)-Br (yellow), and MIL-53(Cr)-NO2 (red) samples at 298 K and up to 1 bar. Filled circles = adsorption; open circles = desorption. | ||
MIL-53(Cr)-Br shows a similar stepwise profile, with a sharp increase in SO2 adsorption at low pressure reaching 0.97 mol mol−1 at 0.72 bar, where gate-opening occurs, with a second increase in SO2 capture reaching a total uptake of 1.72 mol mol−1 at 1 bar. Hysteresis in the desorption branch is again present, with the onset of gate-closing occurring around 0.6 bar. MIL-53(Cr)-Br requires a higher SO2 pressure to achieve the gate opening effect than the unsubstituted MOF, likely due to the steric bulk of the Br functional group modifying the inclination of the phenyl ring, making ligand rotation more difficult.7,17 It is also notable that the initial SO2 uptake for MIL-53(Cr)-Br is higher than MIL-53(Cr), likely due to it being held in a more open pore state because of the Br group, whilst at 1 bar SO2 uptake is slightly lower than MIL-53(Cr), as would be expected given the latter would have a larger pore volume.
In contrast, MIL-53(Cr)-NO2 shows a typical type-I adsorption-desorption isotherm, reaching a total SO2 uptake of 0.99 mol mol−1 at 1 bar. The NO2 groups are larger than the Br groups, making ligand rotation even more difficult due to the higher steric hindrance, thus the gating phenomenon presumably occurs at higher pressure.17a The flexibilities of functionalised MIL-53(Al) derivatives also depend on the chemical nature of the functional groups,17b in particular hydrogen bonding between the functional groups and the OH ligands; strong interactions between the NO2 group and the μ2-OH may hinder the easy opening of the pore.
Powder X-ray diffractograms of the three materials collected after SO2 adsorption and desorption (ESI,† Section S7) revealed that they were stable to SO2 exposure, and the three PCPs returned to their pre-activated structures. Similarly, infrared spectra of the PCPs showed no changes after SO2 exposure.
In our previous examination of SO2 adsorption by MIL-53(Al), Monte Carlo simulations showed that strong interactions exist between SO2 molecules via their O-atoms, and the H-atom of the μ2-OH group in the inorganic secondary building unit; there are also interactions between SO2 and the organic linker.18 Thus, we propose a similar SO2 adsorption mechanism for the MIL-53(Cr) material in this work. In addition to this binding motif, electronegative substituents on PCPs, such as fluoro units, have been observed to enhance SO2 uptake through electrostatic attraction between Sδ+ and Fδ− units in DFT calculations.19 Thus, we propose a similar SO2 adsorption mechanism for the substituted PCPs, in which the main adsorption interaction is between the Sδ+ of SO2 and Brδ− for MIL-53(Cr)-Br, and Oδ− atoms of the highly polar –NO2 groups20 of MIL-53(Cr)-NO2, respectively.
We also evaluated the SO2 selectivity over CO2 for these materials by comparison of single component adsorption isotherms (298 K, 1 bar, Fig. S31, ESI†). For all three samples, the SO2 adsorbed amount is much higher than the CO2 over the whole pressure range. The pyIAST python package21 was used to apply the ideal adsorbed solution theory (IAST) model to the single component adsorption isotherms to calculate the SO2/CO2 selectivities at 1 bar for different molar SO2/CO2 compositions. These results are summarized in Table S1 (ESI†).
The SO2 uptake of MIL-53(Cr) and its derivatives compare reasonably well to other systems, but are lower than MIL-53(Al) and some larger pore MOFs.18,19 Thus, we sought to explore the possibility of using these materials as efficient fluorescent SO2 detectors. Under UV light irradiation at λex = 300 nm (chosen based on solid-state UV-vis spectroscopy experiments, Fig. S15, ESI†), MIL-53(Cr) displays a broad photoluminescence peak centred at λmax = 415 nm (Fig. 3a), whose origin can be attributed to the organic linker.9 The emission of MIL-53(Cr) is similar to that of the free ligand (Fig. S35 (ESI†), λmax = 383 nm) with a slight red shift, indicating metal-to-ligand charge transfer that perturbs the ligand energy levels.9 After exposure to SO2 in our homemade in situ adsorption system, the MIL-53(Cr) sample was packed in a quartz holder for solids to measure its photoluminescence properties (ESI,† Section S11), and a shift in emission to λmax = 420 nm was observed, with emission intensity decreasing 1.4-fold (quenching). For MIL-53(Cr)-Br (λex = 360 nm), the maximum of the photoluminescence for the activated sample is located at λmax = 450 nm and changes to λmax = 436 nm after the SO2 exposure (Fig. 3b). Remarkably, the emission intensity is quenched 2.7-fold, suggesting potential application as a turn-off sensor. Finally, MIL-53(Cr)-NO2 (λex = 350 nm) exhibits broad photoluminescence centred at λmax = 507 nm and, although small, a 1.2-fold decrease in intensity is also observed upon SO2 exposure (Fig. 3c). The quenching observed in all three materials can be attributed to the electronic effect that SO2 exerts on their structures after exposure.9,22 Further, the fact that the most significant quenching effect is observed for MIL-53(Cr)-Br is consistent with previous observations of halogenated PCPs showing stronger affinities for SO2.19 When these three materials were exposed to H2O and CO2 (Fig. S36, ESI†), or left for some time after activation (Fig. S37, ESI†) no significant changes in the shape or intensity of the emission were observed with respect to the spectrum of the activated samples. These facts support that the change in fluorescence is due to the adsorption of SO2 and not to other factors, and that there is some selectivity in the response.
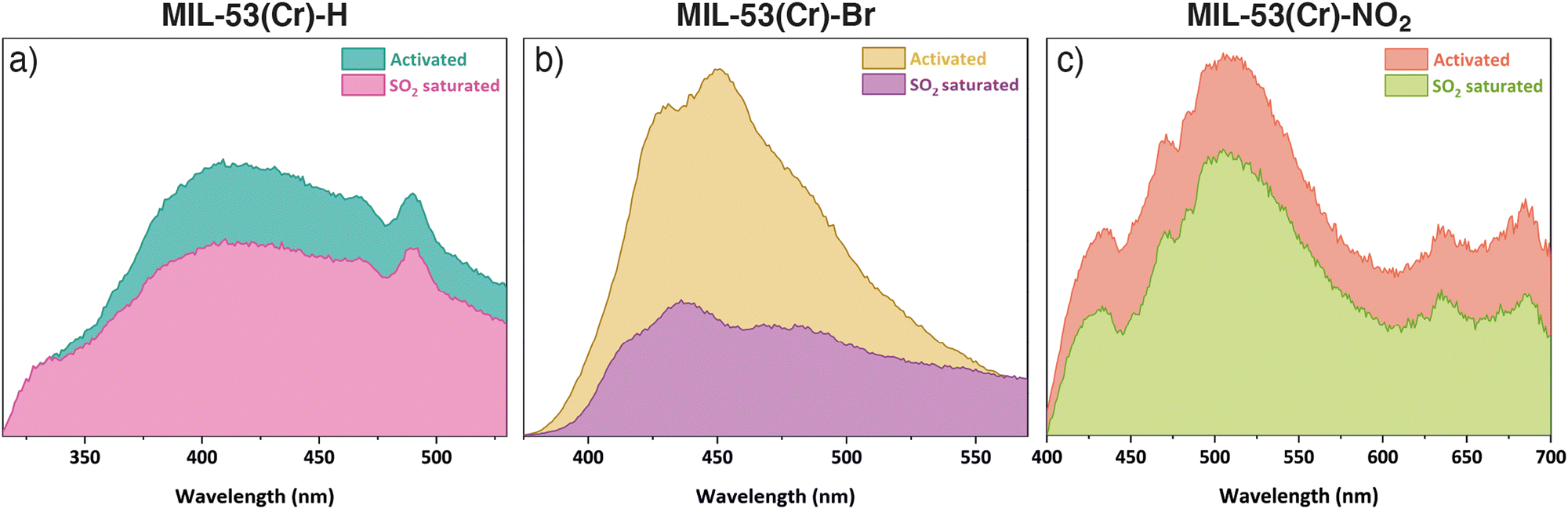 | ||
| Fig. 3 Solid-state emission spectra of activated and SO2 saturated samples of (a) MIL-53(Cr), (b) MIL-53(Cr)-Br, and (c) MIL-53(Cr)-NO2. | ||
In this work, HCl-modulated hydrothermal syntheses of MIL-53(Cr), MIL-53(Cr)-Br and MIL-53(Cr)-NO2 were developed, which are comparable to existing green mechanochemical routes to MIL-53(Cr)16 but with enhanced substrate scope offering a new route into this valuable isoreticular series. All three materials exhibit reversible SO2 adsorption–desorption with high chemical stability to dry and wet SO2. Changes in the emission spectra of the MOFs after the SO2 adsorption, particularly the large quenching observed for MIL-53(Cr)-Br, suggest this material has potential as a sensor for SO2.
This project received financial support in part from the European Research Council (ERC) under the European Union's Horizon 2020 Programme for Research and Innovation (grant agreement no. 677289, SCoTMOF, ERC-2015-STG). I.A.I. thanks PAPIIT UNAM (IN201123), México, for financial support. We thank Mohammad Reza Alizadeh Kiapi (University of Cambridge) for assistance with BETSI analysis23 of gas adsorption data, and Matthew Liddle (University of Glasgow) for thermogravimetric analysis. The data which underpin this submission are available at http://dx.doi.org/10.5525/gla.researchdata.1446.
Conflicts of interest
There are no conflicts to declare.References
- (a) J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS; (b) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- (a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed; (b) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC; (c) S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed; (d) S. K. Ghosh, J. P. Zhang and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 7965–7968 CrossRef CAS PubMed; (e) A. Hazra, D. P. van Heerden, S. Sanyal, P. Lama, C. Esterhuysen and L. J. Barbour, Chem. Sci., 2019, 10, 10018–10024 RSC.
- P. Zhao, S. C. E. Tsang and D. Fairen-Jimenez, Cell Rep. Phys. Sci., 2021, 2, 100544 CrossRef CAS.
- (a) A. Clearfield, Dalton Trans., 2016, 45, 4100–4112 RSC; (b) M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem. – Eur. J., 2019, 25, 5105–5112 CrossRef CAS PubMed; (c) S. Rahman, A. Arami-Niya, X. Yang, G. Xiao, G. Li and E. F. May, Commun. Chem., 2020, 3, 186 CrossRef CAS PubMed.
- (a) L. Li, R. Krishna, Y. Wang, J. Yang, X. Wang and J. Li, J. Mater. Chem. A, 2016, 4, 751–755 RSC; (b) X. Wang, R. Krishna, L. Li, B. Wang, T. He, Y. Z. Zhang, J. R. Li and J. Li, Chem. Eng. J., 2018, 346, 489–496 CrossRef CAS; (c) N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus, T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201–15204 CrossRef CAS PubMed.
- (a) F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822–823 RSC; (b) C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed; (c) F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58, 1019–1035 CrossRef CAS.
- (a) S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed; (b) L. Wu, G. Chaplais, M. Xue, S. Qiu, J. Patarin, A. Simon-Masseron and H. Chen, RSC Adv., 2019, 9, 1918–1928 RSC.
- P. G. Yot, K. Yang, V. Guillerm, F. Ragon, V. Dmitriev, P. Parisiades, E. Elkaïm, T. Devic, P. Horcajada, C. Serre, N. Stock, J. P. S. Mowat, P. A. Wright, G. Férey and G. Maurin, Eur. J. Inorg. Chem., 2016, 4424–4429 CrossRef CAS.
- (a) T. Wu, X.-J. Gao, F. Ge and H.-G. Zheng, CrystEngComm, 2022, 24, 7881–7901 RSC; (b) R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li, J.-P. Zhang, R.-B. Lin, S.-Y. Liu, J.-W. Ye, X.-Y. Li and J.-P. Zhang, Adv. Sci., 2016, 3, 1500434 CrossRef PubMed; (c) H. Y. Li, S. N. Zhao, S. Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC; (d) W.-T. Koo, J.-S. Jang and I.-D. Kim, Chemistry, 2019, 5, 1938–1963 CrossRef CAS.
- X. L. Lv, L. H. Xie, B. Wang, M. Zhao, Y. Cui and J. R. Li, J. Mater. Chem. C, 2018, 6, 10628–10639 RSC.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- D. Bara, E. G. Meekel, I. Pakamorė, C. Wilson, S. Ling and R. S. Forgan, Mater. Horiz., 2021, 8, 3377–3386 RSC.
- S. Bourelly, B. Moulin, A. Rivera, G. Maurin, S. Devautour-Vinot, C. Serre, T. Devic, P. Horcajada, A. Vimont, G. Clet, M. Daturi, J.-C. Lavalley, S. Loera-Serna, R. Denoyel, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2010, 132, 9488–9498 CrossRef PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Férey, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS.
- (a) L. Han, J. Zhang, Y. Mao, W. Zhou, W. Xu and Y. Sun, Ind. Eng. Chem. Res., 2019, 58, 15489 CrossRef CAS; (b) W. L. Teo, S. K. B. Ariff, W. Zhou, D. Jana, S. Z. F. Phua and Y. Zhao, ChemNanoMat, 2020, 6, 204–207 CrossRef CAS.
- (a) T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J. M. Grenéche, B. le Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J. C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS PubMed; (b) A. S. Munn, R. S. Pillai, S. Biswas, N. Stock, G. Maurin and R. I. Walton, Dalton Trans., 2016, 45, 4162–4168 RSC.
- A. López-Olvera, J. A. Zárate, E. Martínez-Ahumada, D. Fan, M. L. Díaz-Ramírez, P. A. Sáenz-Cavazos, V. Martis, D. R. Williams, E. Sánchez-González, G. Maurin and I. A. Ibarra, ACS Appl. Mater. Interfaces, 2021, 13, 39363–39370 CrossRef PubMed.
- (a) M. R. Tchalala, P. M. Bhatt, K. N. Chappanda, S. R. Tavares, K. Adil, Y. Belmabkhout, A. Shkurenko, A. Cadiau, N. Heymans, G. de Weireld, G. Maurin, K. N. Salama and M. Eddaoudi, Nat. Commun., 2019, 10, 1328 CrossRef CAS PubMed; (b) X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29, 1606929 CrossRef PubMed; (c) E. Martínez-Ahumada, A. López-Olvera, V. Jancik, J. E. Sánchez-Bautista, E. González-Zamora, V. Martis, D. R. Williams and I. A. Ibarra, Organometallics, 2020, 39, 883–915 CrossRef.
- (a) S. S. Iremonger, R. Vaidhyanathan, R. K. Mah and G. K. H. Shimizu, Inorg. Chem., 2013, 52, 4124–4126 CrossRef CAS PubMed; (b) D. K. Maity, A. Halder, B. Bhattacharya, A. Das and D. Ghoshal, Cryst. Growth Des., 2016, 16, 1162–1167 CrossRef CAS.
- C. M. Simon, B. Smit and M. Haranczyk, Comput. Phys. Commun., 2016, 200, 364–380 CrossRef CAS.
- J. X. Wang, J. Yin, O. Shekhah, O. M. Bakr, M. Eddaoudi and O. F. Mohammed, ACS Appl. Mater. Interfaces, 2022, 14, 9970–9986 CrossRef CAS PubMed.
- J. W. M. Osterrieth, et al. , Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01685d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |