
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmidation by reactive extrusion for the synthesis of active pharmaceutical ingredients teriflunomide and moclobemide†
Matthieu
Lavayssiere
 and
Frédéric
Lamaty
*
and
Frédéric
Lamaty
*
IBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: frederic.lamaty@umontpellier.fr
First published on 22nd February 2023
Abstract
The solventless synthesis of an amide was performed in a twin-screw extruder in the presence of a coupling agent, providing a high yielding and productive method. The reaction conditions were optimized to prepare APIs, teriflunomide and moclobemide.
The amide bond is ubiquitous in nature, structuring biomacromolecules such as proteins and playing a major role in their biological activities. It is involved in numerous interactions (e.g., hydrogen bonding or electrostatic interactions) related to biological processes. Amide bonds are also present in a large number of manufactured molecules such as materials and pharmacologically active compounds. This includes marketed drugs: 25% of their active pharmaceutical ingredients (APIs) possess an amide bond,1 as depicted in Fig. 1, with examples of major drugs.
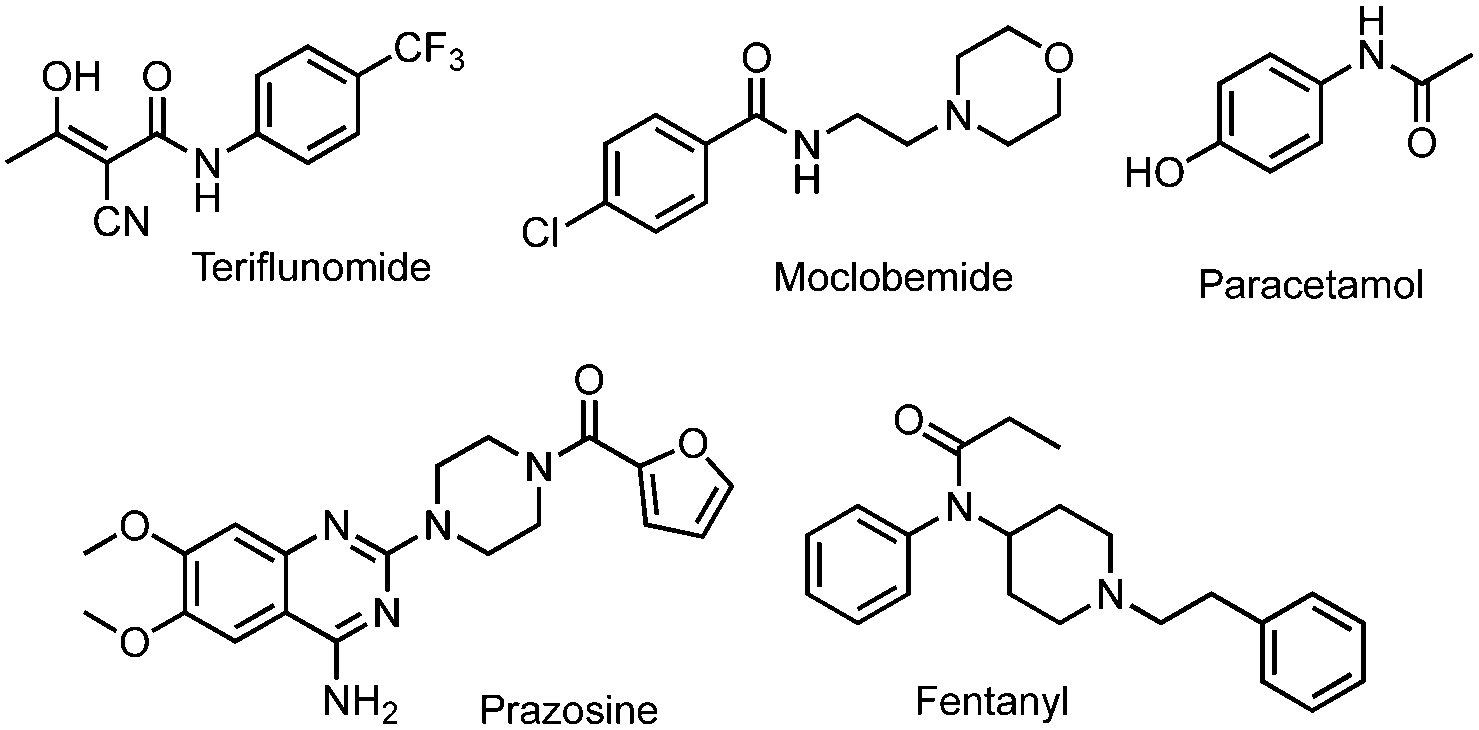 | ||
| Fig. 1 Selected examples of amides as APIs. | ||
While alternative methods to prepare amides are under development,2 the amide bond is typically synthesized from a carboxylic acid and an amine.3 However, the synthesis of these compounds, including at the industrial level,4 often requires reaction conditions involving hazardous and toxic reagents (coupling agents, bases…) in stoichiometric quantities, and sources of pollution, including the use of large amounts of solvent to solubilize the reaction medium and purify the synthesized product. Research teams have developed a mechanochemistry approach, namely solvent-free or solventless methods by ball-milling for the preparation of organic molecules including APIs.5 In 2012, our team reported the first example of solvent-free amide bond synthesis starting from a carboxylic acid and an amine, in a ball-mill, with CDI‡,6 as a coupling agent.7
This method was applied to the organic-solvent free synthesis of the API teriflunomide.7,8 Nearly concomitantly, the group of Štrukil and Margetić reported amidation reaction including EDC.HCl‡ as an efficient coupling agent.9 Recently, further developments were reported with the use of various coupling agents in a ball-mill to prepare an amide.10 Furthermore, the mechanochemical approach to the synthesis of peptides has pioneered and complemented the evaluation of coupling systems in the absence of solvent.5f,11
Nevertheless, the ball-milling approach corresponds to a discontinuous batch process and the scaling-up of the corresponding reaction with larger mills, currently employed in the mineral industry, raises problems of safety for organic synthesis. While flow chemistry is currently revolutionizing the processing of chemicals, this approach necessitates a large amount of solvent and is not well-adapted to the presence of solids or of concentrated mixtures. Recently, it has been demonstrated that extruders, another mechanochemistry tool, could provide a continuous approach for solvent-free or solventless organic reactions, involving solids or viscous mixtures,12 with few reports regarding the preparation of APIs.13 This approach was also recognized by IUPAC as one of the ten most important chemical innovations that could change our world,14 including at the industrial level. In order to fill in the gap of the number of reactions developed by reactive extrusion, further developments are needed to provide chemists with practical tools to synthesize efficiently organic molecules, including widespread amidation. We now disclose a first report on the continuous synthesis of amides by twin-screw extrusion (TSE) including the preparation of two important APIs, teriflunomide and moclobemide.
Capitalizing on previous results obtained in the synthesis of peptides,15 the amidation reaction was studied and optimized using a corotating twin-screw extruder (Pharma Melt Extruder (PME) Xplore). This extruder can be used in a continuous mode when the reaction time is short, or with a recirculation to increase the residence time in the barrel, corresponding to a longer length of the screw. The previously reported model reaction, used for studying the reaction conditions in a ball-mill,7 namely the coupling of 3-phenylpropionic acid 1 and benzyl amine 2 was investigated in various conditions including coupling agents and bases/additives if needed. Two important extruder parameters that could be optimized are the temperature of the barrel and the rotation speed of the screws. Many coupling agents and conditions are available and reported in the literature to carry out efficiently an amidation reaction. Among them, a limited number has been effectively used in the pharmaceutical industry for large scale applications.4 Selection criteria for choosing a coupling system (including coupling agent, base, additives, solvent) are efficiency, availability, practical handling and ease of purification of the final product, as well as safety, toxicity and environmental impact. We evaluated the toolbox for amidation using an extruder focusing on three coupling agents: CDI, EDC.HCl and COMU.‡
Firstly CDI, one of the most employed coupling agents, efficient, cheap and considered non-toxic, was explored. CDI generates carbon dioxide and imidazole as by-products, and the latter can serve as a base when the coupled amine is provided as a salt. The reaction was carried out in two steps, first an activation step of carboxylic acid 1, for a given mixing time t1 with CDI, followed by the addition of benzyl amine 2 (Table 1). The reaction mixture was mixed for a further time (t2). The loading of the experiment was fixed at about 2 g, filling completely the available volume of the barrel equipped with screws (2 mL). A slight excess of acid compared to the amine was used. The temperature of the barrel was set to 30 °C and the rotation speed to 200 rpm. Premixing15a or not the starting materials, with a spatula in a beaker, before introduction in the barrel did not change the final result. At the end of the transformation, the output valve was adjusted to recover the crude mixture.
|
|
||||
|---|---|---|---|---|
| Entry | η (mL g−1) of EtOAC | Activation step t1 (min) | Reaction time t2 (min) | Conv.a (%) |
| a Determined by HPLC (see ESI). b In this case, recovery of the extrudate was incomplete. c 4% of urea was also obtained. d Isolated yield. | ||||
| 1 | 0 | 5 | 10 | 0 |
| 2 | 0.6 | 5 | 10 | >99 |
| 3b | 0.15 | 5 | 10 | >99 |
| 4 | 0.3 | 5 | 10 | >99 |
| 5 | 0.3 | 0 | 5 | 94c |
| 6 | 0.3 | 1 | 1 | 98 (81)d |
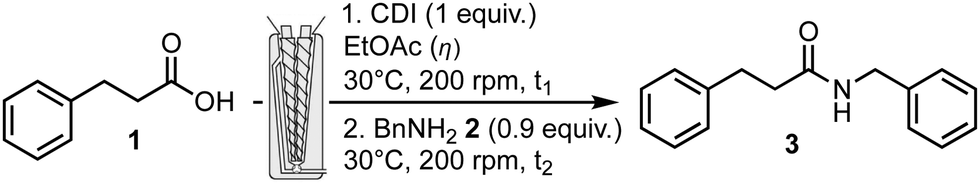
In the first experiment (entry 1), when reactants were mixed neat, recirculation did not happen and no conversion was observed. In the next experiment (entry 2), EtOAc was used in a small amount (1.2 mL, η = 0.6 mL g−1)§ as a liquid additive (LA) to facilitate the fluidity and the homogeneity of the reaction mixture. The conversion sharply increased to completion towards the expected product. Optimization of the quantities of LA and reaction time was realized. Decreasing the amount of EtOAc (η = 0.15 mL g−1), conversion was still excellent but the introduction of reagents was not facilitated and recovery of the extrudate was difficult (entry 3). Increasing to η = 0.3 mL g−1 solved this problem (entry 4). When the three reaction components and LA were mixed together (entry 5) avoiding a pre-activation time, high conversion was obtained albeit with the formation of about 4% of urea arising from the reaction of CDI with benzyl amine 2 only. Gratifyingly, reducing both the activation and reaction time to 1 minute each (entry 6) afforded almost complete conversion. In this case, the extrudate was solubilized in EtOAc, followed by acidic then basic aqueous washings to afford pure expected N-benzyl-3-phenylpropanamide 3 in 81% yield. The efficient use of CDI required a two-step procedure since mixing all reagents together provided urea as a side product. To develop a potentially more efficient process, other coupling agents were considered. EDC.HCl is another much employed coupling agent and does not require the use of an additional base unless the amine is used as its salt form. EDC.HCl may be preferred over other carbodiimide type coupling agents because the urea by product thus obtained can be removed by a simple aqueous work up. To improve the kinetics of the amidation and reduce the potential epimerization of a chiral center present in the starting material, additives have been used to enhance the reactivity of the coupling agent. One of the most used additives is HOBt,‡ but its sensitivity to explosion represents some concern. Recently, other additives were explored more specifically for peptide synthesis, including oximes. One of the mostly used is ethyl oxyma,‡ which was investigated in this study.16 Finally, carbodiimides being sensitizing molecules, a carbodiimide-free oxyma-based coupling agent, COMU was also tested.17
EDC.HCl, widely used together with DMAP as a base, in a catalytic amount, in solution or in solvent-free conditions,4,9,10a,11e,f was investigated. 10 minutes of mixing 1 and 2 in the extruder together with EDC.HCl and DMAP yielded an incomplete conversion towards the expected product (Table 2, entry 1). Adding EtOAc (η = 0.6 mL g−1) did not improve the conversion (entry 2). In the absence of base, conversion was improved but not complete (entry 3). Less toxic K2CO3 (entry 4) was more effective when the amount of EtOAc was increased to η = 0.75 mL g−1 and the temperature was raised to 50 °C. Diisopropylethyl amine (DIPEA) also gave excellent conversion (entry 5). To avoid the need for recirculation in the extruder, the possibility to reduce further the reaction time was explored, by adding oxyma. Surprisingly, in the first experiment (entry 6), the conversion decreased, but switching to CH3CN as a liquid additive radically improved the outcome of the reaction, most probably because it provides a more homogeneous reaction mixture in the presence of oxyma and facilitates the recovery of the extrudate (entry 7). The reaction time could be decreased to 1 min (entry 8), still providing excellent conversion with an isolated yield of 86% with the same recovery procedure, as described above. Noteworthily, in this case, recirculation was not needed and molecule 3 was produced in a continuous mode. Oxyma-based coupling agent COMU was tested in the optimal conditions for amidation in a ball-mill, reported in the literature,10c with K2HPO4 as a base (entry 9) but this resulted in an average conversion. COMU in the presence of DIPEA and EtOAc (entry 10) yielded full conversion in 10 minutes. Switching from DIPEA to less toxic K2CO3 did not impact the conversion (entry 11). CH3CN, for the reasons mentioned above, was also used (entry 12) to give a similar result with an isolated yield of 80%. Attempts to reduce the reaction time to 1 min (entry 13) were not satisfactory. Scaling up the synthesis by running the same reaction three times successively in the extruder (entry 14) yielded 99% conversion and an excellent isolated yield of 93% to provide 1.71 g of 3. Finally, reaction with COMU, K2CO3 and CH3CN (entry 15) was not the best combination regarding conversion but the process could be carried out in a continuous mode on a larger scale, providing 1.29 g of 3 (77% isolated yield, 85% conversion).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Coupling agent | Base (equiv.) | Liquid additive (η)/additive (equiv.) | t (min) | Conv.a (%) |
| a Determined by HPLC (see ESI). b Extruder barrel temperature set at 50 °C. c Isolated yield. | |||||
| 1 | EDC.HCl | DMAP (0.3) | — | 10 | 78 |
| 2 | EDC.HCl | DMAP (0.3) | EtOAc (0.6) | 10 | 79 |
| 3 | EDC.HCl | — | EtOAc (0.6) | 10 | 85 |
| 4b | EDC.HCl | K2CO3 (0.15) | EtOAc (0.75) | 10 | 97 |
| 5 | EDC.HCl | DIPEA (0.3) | EtOAc (0.6) | 10 | 98 |
| 6 | EDC.HCl | DIPEA (0.3) | EtOAc (0.75)/oxyma (1) | 10 | 81 |
| 7 | EDC.HCl | DIPEA (0.3) | CH3CN (0.6)/oxyma (1) | 10 | >99 |
| 8 | EDC.HCl | DIPEA (0.3) | CH3CN (0.3)/oxyma (1) | 1 | 96 (86)c |
| 9 | COMU | K2HPO4 (1) | EtOAc (0.6) | 1 | 52 |
| 10 | COMU | DIPEA (1) | EtOAc (0.75) | 10 | >99 |
| 11 | COMU | K2CO3 (0.5) | EtOAc (0.75) | 10 | >99 (75)c |
| 12 | COMU | DIPEA (1) | CH3CN (0.6) | 10 | >99 (80)c |
| 13 | COMU | DIPEA (1) | CH3CN (0.6) | 1 | 94 |
| 14 | COMU | DIPEA (1) | CH3CN (0.6) | 3 × 10 | >99 (93)c |
| 15 | COMU | K2CO3 (0.5) | CH3CN (0.6) | 3 × 1 | 85(77)c |

These methods were applied to the preparation of APIs owning an amide bond. Two molecules were chosen, according to their therapeutic and economical value in the pharmaceutical industry, teriflunomide 4 and moclobemide 5 (Fig. 1 and Table 3). Teriflunomide is an oral disease-modifying therapy for relapsing forms of multiple sclerosis.18 Moclobemide is used to treat depression and social anxiety disorder.19 In the case of teriflunomide, a similar scheme as the one developed in our original work7 was considered, with the first step (amidation) performed herein in the extruder. Then opening of the isoxazole ring in solution in acidic conditions gave teriflunomide 4. The main adjustments in the reaction conditions consisted in selecting the liquid additive, the base, the temperature of the barrel and the reaction time (Table 3). The use of CDI (entry 1) gave a very good result (77% isolated overall yield), provided that CH3CN was used as a liquid additive at a temperature of 50 °C. These results compare favorably in terms of reaction time to the ones previously obtained by ball-milling (5 h).7 Reaction with EDC.HCl/DIPEA was much slower and increasing the reaction temperature resulted in a mixture of products (entry 2). Adding oxyma (entry 3) reduced the reaction time and provided a good yield of 4. Reaction with COMU was slow (entry 4). Noteworthily, the recovery of teriflunomide from the reaction with CDI is facilitated (simple precipitation in water) because of the absence of a base or additives. In the case of a reaction where oxyma is present, more careful recrystallization was needed to remove the by-products. In contrast to these results, CDI was not suitable for the preparation of moclobemide. Either with EtOAc or CH3CN as a liquid additive, recirculation was difficult and resulted in lower conversion and yield than in the case of teriflunomide (entry 5). Again, increasing the temperature to accelerate the reaction with EDC.HCl/DIPEA resulted in a mixture of products (entry 6). The presence of oxyma dramatically improved the results with a complete conversion obtained within 5 min (entry 7). By switching to K2CO3 (entry 8), the transformation could be carried out in 1 min in a continuous mode to furnish 87% yield of moclobemide. The same yield was obtained when scaling-up was performed and 11 g of moclobemide was produced in 2 min. Conversion and yields with COMU were excellent but the process needed recirculation (entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry (product) | Coupling agent | Base (equiv.) | Liquid additive (η)/additive (equiv.) | T (°C), t (min) | Conv.b % (yieldc, %) |
| a In the case of CDI, activation + reaction time. b Determined by HPLC (see ESI). c In the case of teriflunomide, isolated yield was calculated on the final product after acidic treatment. d A mixture of products was obtained. | |||||
| 1 (4) | CDI | — | CH3CN (0.6) | 50 (10 + 20)a | >99 (77) |
| 2 (4) | EDC.HCl | DIPEA (0.33) | CH3CN (0.6) | 80 (120) | 67d |
| 3 (4) | EDC.HCl | DIPEA (0.3) | CH3CN (0.6)/oxyma (1) | 50 (60) | 99 (75) |
| 4 (4) | COMU | DIPEA (1.1) | CH3CN (0.6) | 30 (120) | 85 (80) |
| 5 (5) | CDI | — | EtOAc (0.6) | 30 (1 + 2)a | 88 (68) |
| 6 (5) | EDC.HCl | DIPEA (0.33) | CH3CN (0.6) | 80 (10) | 85d |
| 7 (5) | EDC.HCl | DIPEA (0.33) | CH3CN (0.6)/oxyma (1) | 30 (5) | >99 |
| 8 (5) | EDC.HCl | K2CO3 (0.3) | CH3CN (0.6)/oxyma (1) | 30 (1) | 95 (87) |
| 9 (5) | COMU | DIPEA (1.1) | CH3CN (0.6) | 30 (10) | 99 (95) |
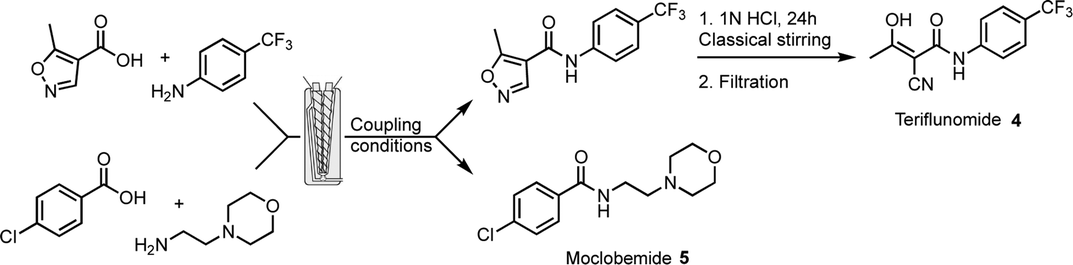
As expected, the optimal conditions for the synthesis of the two APIs, teriflunomide and moclobemide, by TSE, depend on the chemical reactivity of the starting materials (the aniline involved in the reaction of teriflunomide is less reactive than the primary amine in the case of moclobemide) but also on the physico-chemical properties of the reaction medium with the necessity to obtain a homogeneous mixture, which can be extruded at the end of the process. Considering the coupling systems reported herein, and the possibility to adjust various parameters, optimal conditions should be obtained for any coupling reaction. In the case of moclobemide, continuous mode could be performed (entry 8) because the reaction time was short. In the other cases, longer reaction time required recirculation. Developing a continuous process, for scaling up the synthesis, would require in further studies a switch to a twin-screw extruder that could accommodate longer lengths of screw.12b
In conclusion, we developed a general amidation by TSE, starting from a carboxylic acid and an amine. This method easily accommodates solids and was applied successfully to the efficient synthesis of teriflunomide and moclobemide, complementing the toolbox developed for the preparation of APIs by twin-screw extrusion. As recently demonstrated,13c drastically decreasing the amount of solvent used, combined with an improved efficiency of the amidation reaction by engaging a quasi stoichiometric amount of starting material results in a more sustainable and cost saving approach. This paves the way for the dissemination of the technology of reactive extrusion for organic synthesis and in industry.
The Centre National de la Recherche Scientifique (CNRS), University of Montpellier, and Chimie Balard Cirimat Carnot Institute (through the ANR programme N°16 CARN 0008-01) are acknowledged for financial support. We thank Marcelin Lamaty for his contribution to the design of the graphical abstract.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- R. M. deFigueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- J. Magano, Org. Process Res. Dev., 2022, 26, 1562–1689 CrossRef CAS.
- (a) D. Tan, L. Loots and T. Friscic, Chem. Commun., 2016, 52, 7760–7781 RSC; (b) E. Colacino, A. Porcheddu, C. Charnay and F. Delogu, React. Chem. Eng., 2019, 4, 1179–1188 RSC; (c) M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef; (d) T. Friscic, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef CAS PubMed; (e) W. I. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868–21874 CrossRef CAS PubMed; (f) O. Bento, F. Luttringer, T. Mohy El Dine, N. Petry, X. Bantreil and F. Lamaty, Eur. J. Org. Chem., 2022, e202101516 CAS.
- T.-X. Métro, J. Martinez and F. Lamaty, ACS Sustainable Chem. Eng., 2017, 5, 9599–9602 CrossRef.
- T.-X. Métro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781–11783 RSC.
- T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. – Eur. J., 2015, 21, 12787–12796 CrossRef PubMed.
- V. Štrukil, B. Bartolec, T. Portada, I. Đilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100–12102 RSC.
- (a) A. Wróblewska, P. Paluch, E. Wielgus, G. Bujacz, M. K. Dudek and M. J. Potrzebowski, Org. Lett., 2017, 19, 5360–5363 CrossRef PubMed; (b) C. Duangkamol, S. Jaita, S. Wangngae, W. Phakhodee and M. Pattarawarapan, RSC Adv., 2015, 5, 52624–52628 RSC; (c) T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703–15715 CrossRef CAS.
- (a) V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318–9321 CrossRef CAS PubMed; (b) J. G. Hernandez and E. Juaristi, J. Org. Chem., 2010, 75, 7107–7111 CrossRef CAS PubMed; (c) T.-X. Métro, E. Colacino, J. Martinez and F. Lamaty, RSC Green Chem. Ser., 2015, 31, 114–150 Search PubMed; (d) C. Bolm and J. G. Hernandez, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed; (e) V. Porte, M. Thioloy, T. Pigoux, T.-X. Métro, J. Martinez and F. Lamaty, Eur. J. Org. Chem., 2016, 3505–3508 CrossRef CAS; (f) M. Anselmi, P. Stavole, E. Boanini, A. Bigi, E. Juaristi and L. Gentilucci, Future Med. Chem., 2020, 12, 479–491 CrossRef CAS PubMed; (g) Y. Yeboue, M. Jean, G. Subra, J. Martinez, F. Lamaty and T.-X. Métro, Org. Lett., 2021, 23, 631–635 CrossRef CAS PubMed.
- (a) R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC; (b) D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 19, 1507–1518 RSC.
- (a) D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS; (b) N. B. Patil, R. S. Atapalkar, S. P. Chavan and A. A. Kulkarni, Chem. – Eur. J., 2021, 27, 17695–17699 CrossRef CAS PubMed; (c) O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- (a) Y. Yeboue, B. Gallard, N. Le Moigne, M. Jean, F. Lamaty, J. Martinez and T.-X. Métro, ACS Sustainable Chem. Eng., 2018, 6, 16001–16004 CrossRef CAS; (b) K. J. Ardila-Fierro, D. E. Crawford, A. Körner, S. L. James, C. Bolm and J. G. Hernández, Green Chem., 2018, 20, 1262–1269 RSC.
- S. R. Manne, B. G. de la Torre, A. El-Faham and F. Albericio, Synthesis, 2020, 3189–3210 CAS.
- A. El-Faham and F. Albericio, J. Pept. Sci., 2010, 16, 6–9 CrossRef CAS PubMed.
- P. O'Connor, J. S. Wolinsky, C. Confavreux, G. Comi, L. Kappos, T. P. Olsson, H. Benzerdjeb, P. Truffinet, L. Wang, A. Miller and M. S. Freedman, N. Engl. J. Med., 2011, 365, 1293–1303 CrossRef PubMed.
- C. N. Stefanis, B. H. Alevizos and G. N. Papadimitriou, Int. Pharmacopsychiatry, 1982, 17, 43–48 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization of compounds and copies of 1H and 13C NMR. See DOI: https://doi.org/10.1039/d2cc06934b |
| ‡ CDI: 1,1′-Carbonyldiimidazole, EDC.HCl: N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride), COMU: 1-[(1-(Cyano-2-ethoxy-2 oxoethylideneaminooxy) dimethylaminomorpholino)] uronium hexafluorophosphate, HOBt: 1-Hydroxybenzotriazole, Oxyma: Ethyl hydroxyiminocyanoacetate. |
| § η is defined as the ratio between the volume of liquid (in mL) divided by the total mass of solids (in grams). |
| This journal is © The Royal Society of Chemistry 2023 |