
Circularly polarized near-infrared phosphorescence of chiral chromium(III) complexes†
Yang
Cheng
,
Jiang
He
,
Wenjie
Zou
,
Xiaoyong
Chang
,
Qingqing
Yang
 * and
Wei
Lu
*
* and
Wei
Lu
*
Department of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China. E-mail: yangqq3@sustech.edu.cn; luw@sustech.edu.cn
First published on 18th January 2023
Abstract
Homoleptic Cr(III) complexes containing anionic tridentate 1,8-(bisoxazolyl)carbazolide ligands are phosphorescent in deaerated solutions with peak maxima in the range of 813–845 nm. The ligand carbon-centred chirality has been transferred to the helical chirality of the complexes and hence induced circularly polarized NIR-emissions with dissymmetry factor in the scale of 2.0 × 10−3.
Circularly polarized luminescence (CPL) active materials have recently attracted considerable attention due to their potential applications, such as in stereoscopic displays, smart sensors/probes, organic light-emitting diodes (OLEDs) and advanced security inks.1 In particular, extending and consolidating circularly polarized emission in the near infrared (NIR) region is highly desirable for versatile applicability of biological imaging or photonics.2 However, the development of NIR-active chiroptical luminogens remains a considerable challenge when compared with the classical visible CPL emitters.1h,3 The emission in the NIR region is expected to yield lower quantum yield (Φ) due to efficient non-radiative deactivation pathways, recognized as the ‘energy gap law’.4 So far, only a few studies with NIR-CPLs over 700 nm have been reported,3,5 thus portending the great challenges and tremendous prospects of the NIR-CPL emitters.
CPL emitters are generally evaluated by the luminescence dissymmetry factor (glum)1a and emission quantum yield (Φ). Due to the trade-off between these two parameters, materials with both large |glum| and Φ values are still limited. Accordingly, the electronically dipole-forbidden and magnetically allowed transitions such as intrashell f–f and d–d are promising solutions to provide high glum. Recently, Cr(III) complexes that exhibit strong emission in the red to NIR region have been achieved by an introduction of strong-field ligands.6 Due to the d3 electronic configuration and the strong ligand field environments, pseudo-octahedral Cr(III) complexes display magnetically-allowed but electric-dipole forbidden spin-flip Cr(2E/2T1 → 4A2) emission, which makes them ideal candidates for NIR-CPL materials,7 alternative to precious metals (Ru, Ir, Pt, or Au)8 and lanthanides.1b,9 In this way, several attempts have been made recently. A recent example was the chiral resolution of [Cr(dqp)2]3+ (dqp = 2,6-di(quinolin-8-yl)pyridine), which achieved high glum of 0.2 at 749 nm.5f Moreover, the functionalization of the dqp ligand with a methoxy group has been proven to improve the chiral resolution, provide high Φ (17%) and maintain the high glum.5g The molecular ruby [Cr(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′′-dipyridin-2-ylpyridine-2,6-diamine) can also be resolved into enantiopure materials by chiral HPLC with glum ≈ 0.093 at 775 nm.5e However, the neutral dqp and ddpd ligands lead to the emission of Cr(2E/2T1→4A2), which are only slightly tuneable. In addition, all these CPL-active Cr(III) complexes rely on chiral resolution processes, which become problematic in large scale preparation.
Our group has an interest in the employment of carbon-centred chiral ligands to induce helical chirality complexes.10 More recently, anionic tridentate N-donor ligands have been proven effective to lead to tuneable NIR emission in Cr(III) complexes, and the combination of π-donating amido with pyridine coordination units can achieve NIR-II luminescence at 1067 nm in frozen solutions at 77 K by increasing metal-ligand bond covalence.4c,11 We herein report a series of Cr(III) complexes bearing carbazolyl-based tridentate ligands with carbon centred chirality in the hope of circumventing the reported homo-chiral helicities from the non-chiral ligand twist. In this way, we can achieve helical chiral Cr(III) complexes without the tedious chiral resolutions. We choose 1,8-(bisoxazolyl)carbazolide as the tridentate ligand,12 in which the chiral bisoxazolyls are readily implanted from commercially available chiral aminoalcohols. For comparison, complexes with ligands exhibiting different positions of the chiral centres and non-chiral ligands were also prepared (Scheme 1 and ESI†).
 | ||
| Scheme 1 Syntheses and chemical structures of 1S/1R, 2S/2R and 3. | ||
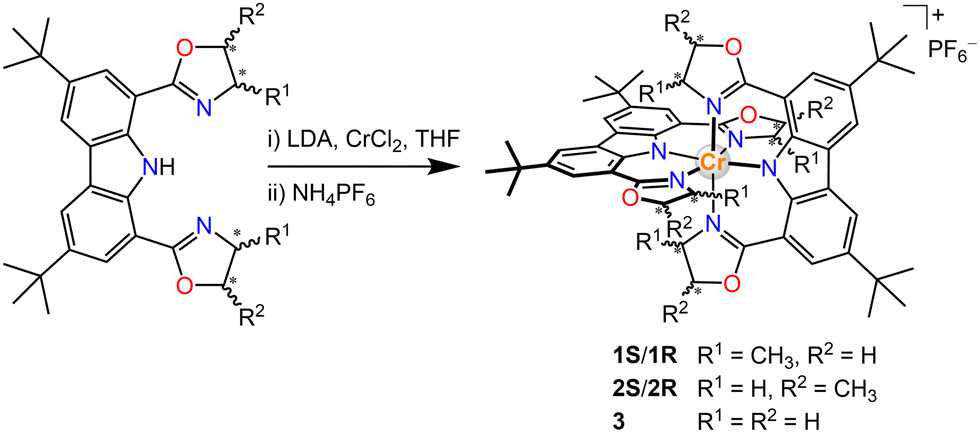
Single crystals of 1S/1R and 3 were obtained by slow diffusion of n-hexane into the respective dichloromethane solution at ambient temperature and their structures were determined by X-ray crystallography (Fig. 1). The carbazole-based N(imido)–Cr–N(imido) bond angles (Table S4, ESI†) are 179.33(12)°–178.58(9)°, and the N(oxazolyl)–Cr–N(oxazolyl) bond angles are 176.55(11)°–174.87(9)°, revealing a more octahedral geometry than the reported [Cr(dpc)2]+.11a The Cr–N(imido) bond lengths (1.988(2)–1.999(2) Å) are shorter than the Cr–N(oxazolyl) bond lengths (2.040(2)–2.086(2) Å), which corresponds to the covalent nature of the Cr–N(imido) bond in the present structure. The two tridentate ligands coordinate in a meridional fashion to the central Cr(III) ion. The unit cell of 3 contained D2d symmetrical enantiomeric PP and MM. In sharp contrast, due to the orientation of the chiral-methyl groups, the symmetry of 1S/1R results in lower D2 symmetry. For 1R, only the MM configuration was observed while in 1S, only the PP configuration was observed.
 | ||
| Fig. 1 (a) Illustration of the chiral structures. Crystal structures of complexes (c) 1S/1R and (b) 3, drawn in 50% probability ellipsoids with H-atoms, counter-ions and solvate molecules omitted for clarity. | ||
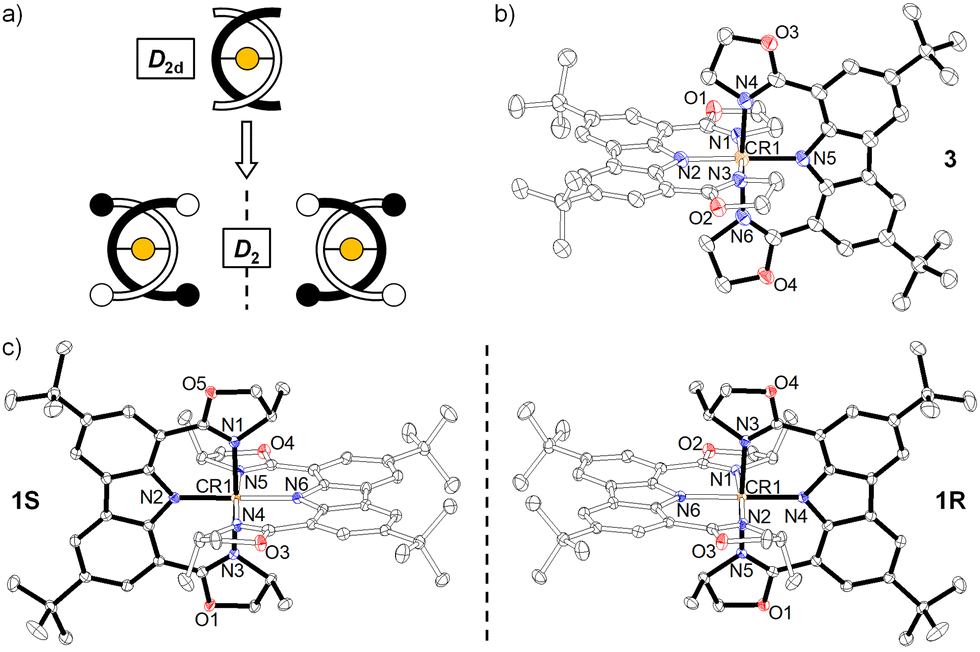
Complexes 1–3 show distinct X-band electron paramagnetic resonance (EPR) spectra which were measured in frozen acetonitrile at 100 K. Both 2 and 3 showed a prominent derivative signal at g ≈ 2 with some faint features at higher g values (Fig. 2), which is characteristic of Cr(III) species with small zero field splitting (ZFS, D < hν, D and E/D are the axial and orthorhombic zero-field parameters).13 The weak signal at g ≈ 5.9 is assigned to the almost forbidden “Δm = 3” transition within the ms = ±3/2 Kramers doublet.13 Interestingly, a broad derivative band at higher g values with extremely weak signal at g ≈ 2 was observed for 1 from the frozen solution. These features are in qualitative accordance with some Cr(III) species, which feature an electronic spin S = 3/2 with moderately large zero field splitting D and small rhombicity E (D > hν, E/D ≈ 0).13,14 The spin-lattice interaction increases with the ZFS and therefore the signal diminishes at relatively low temperatures.15 The g value of these Cr(III) species depends on spin–orbit interaction with excited spin quartet terms due to t2g → eg excitations. It has been pointed out that the major contribution to the ZFS parameter D in the distorted octahedral symmetry comes from the 2Eg excited doublet states.14 As a result, D is sensitive to the strength of the equatorial ligands. The small rhombicity observed of 1 surprisingly exhibits a different EPR signal compared with 2 and 3.16 In such a case, the EPR signals of 1–3 are primarily related to the stereoelectronic effect from the changes of the chiral steric groups.17
 | ||
| Fig. 2 EPR spectra of 1S, 2R, and 3 recorded in frozen acetonitrile at 100 K (concentration ∼3.0 × 10−3 M). | ||
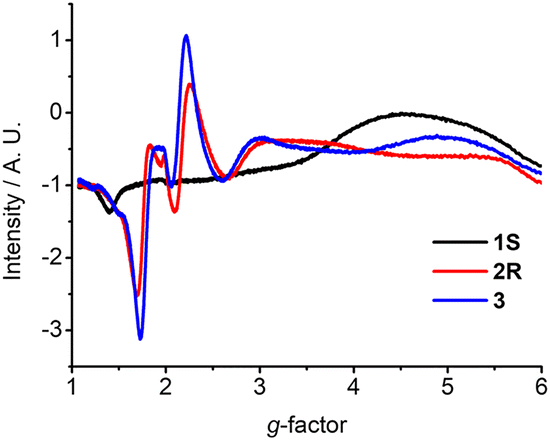
The photophysical properties of Cr(III) complexes 1–3 were explored using the ultraviolet-visible (UV-vis) absorption spectra and the fluorescence spectra. As expected, 1–3 display a similar pattern with two dominant bands between 250 and 350 nm (ε ∼ 4–8 × 104 M−1 cm−1) which were assigned to ligand-centred π–π* transitions (Fig. 3a). The bands between 350 and 600 nm (ε ∼ 1–3 × 104 M−1 cm−1), corresponding to the lowest energy transitions, were tentatively assigned to metal-centred (MC) transition mixing with some ligand-to-metal charge transfer (LMCT), according to previous studies.4c,11a Upon excitation at 450 nm in degassed acetonitrile at 298 K, the emission peaks are at 845, 820 and 813 nm, respectively (Fig. 3d). The emission maximum of these Cr(III) complexes is insensitive to the polarity of the solvents (Fig. S19, ESI†). Unlike the sharp emission bands of other Cr(III) complexes which correspond to the spin-flip Cr(2E/2T1 → 4A2) transitions, the broad emission bandwidth further implies spin-flip transition mixing with charge-transfer characters.11a The decay curves (Fig. S20, ESI†) of emission intensity were fitted mono-exponentially to give the emission lifetime (τ) of 56.6, 259.4 and 266.7 μs for 1–3, respectively (Table S5, ESI†). The quantum yield (Φ) was determined to be 2.4%, 7.5% and 8.2% for 1–3, respectively, in degassed acetonitrile at 298 K.
 | ||
| Fig. 3 (a) Absorption spectra of 1S, 2S, and 3 in deaerated acetonitrile at 298 K (1.0 × 10−5 M). (b) Electronic circular dichroism of 1S/1R and 2S/2R (1.0 × 10−5 M) in deaerated acetonitrile at 298 K. (c) Plots of absorption dissymmetry factor gabs of 1S/1R and 2S/2R. (d) Emission spectra of 1S, 2S, and 3 in deaerated acetonitrile at 298 K (1.0 × 10−5 M). (e) CPL spectra of 1S/1R and 2S/2R (1.0 × 10−4 M) in deaerated acetonitrile at 298 K. (f) CPL spectra of 1S/1R in the solid state at 298 K. | ||
Although both 1S/1R and 2S/2R possess carbon-centred chirality, their electronic circular dichroism (ECD) signals show dramatic differences in the wavelengths of the peaks and corresponding intensity. The ECD spectra of 1S/1R exhibit an evident dichroic band at the lowest energy of 580 nm (Δε = +7.2 M−1 cm−1 for 1S and −11.1 M−1 cm−1 for 1R) with an absorption dissymmetry factor |gabs| of 0.007 and 0.012 (Fig. 3b and c). The strong Cotton effects of 1S/1R at 510 and 580 nm are attributed to LMCT and metal-centred transitions, while the peaks below 350 nm correspond to ligand-centred transitions (π–π*). In the present case, the Cotton effect for 1 is more significant below 400 nm but with a lower |gabs|. Meanwhile, 2S and 2R only show some weak ECD signals below 450 nm with infinitesimal |gabs|.
Consistent with the ECD spectra, such an aspect is also clearly observed in the CPL spectra of these Cr(III) complexes. Upon nonpolarized excitation at 450 nm, the 1S and 1R display a pair of mirror NIR-CPL spectra between 700 and 1000 nm both in diluted deaerated acetonitrile and the solid state (Fig. 3e and f). The circularly polarized emission bands at 845 nm exhibit a glum value of +0.002 for 1S and −0.002 for 1R in degassed solution (Fig. S23, ESI†). In the solid state, the glum values of +0.003 for 1S and −0.003 for 1R were also obtained (Fig. S23, ESI†). By contrast, the glum for complexes 2S/2R was almost zero in degassed solution. The sharp difference could be rationalised by the less crowded metal centre and less twisted helical chirality of 2S/2R when compared with those of 1S/1R. These experimental results revealed that the carbon-centred chirality-induced steric hindrance determines the twist degree of helical chirality as well as the glum, which also indicates that the glum can be further improved by using a more bulky carbon-centred chiral group. In addition, compared to the reported glum of Cr(2E/2T1 → 4A2),5e–g the lower glum of complexes 1S/1R further confirmed the mixing of electronically dipole-allowed charge-transfer transitions in the excited states.
Accordingly, the emissions of 1–3 are essentially metal-based transitions with some LMCT character, which is mainly dependent on the electronic effect of the ligand substituents. As shown in the X-ray photoelectron spectra (XPS), the Cr 2p1/2/2p3/2 binding energies (eV) of 1–3 are 587.14/577.33, 586.84/576.96, and 586.78/577.02, respectively (Fig. 4a and Fig. S24, ESI†). The methyl groups at the oxazolyl units in complex 1 are closer to the metal centre, thus slightly increasing the electron binding energy of the Cr(III) ion and hence rendering it harder to be oxidized. Cyclic voltammetry (CV) also revealed a similar tendency (Fig. 4b). The first reduction potentials of 1–3 are reversible at around −2.0 V vs. Fc+/0 which can be assigned to [Cr(L)2]3+/2+, and are much more negative than in [Cr(dpc)2]+ (E1/2 = −1.51 V vs. Fc+/0).11a,18 The cathodically shifted potentials reflect the higher electron density at the metal centre, which is attributed to the stronger σ-donating nature of the oxazolyl units than pyridine. The oxidation potentials appear at +0.70 V vs. Fc+/0 corresponding to the formal oxidations of [Cr(L)2]4+/3+, while the peak at ca. 0.90 V is assigned to the ligand oxidation, according to the CV measurement of the ligand precursor (Fig. S26–S28, ESI†). Combined with the modified structures, 1S shows a 0.1 V more positive potential, evidently reflecting the utilisation of the secondary coordination sphere on the oxazolyl subunit. And this is in conformity with the results of XPS measurements. By using ultraviolet photoelectron spectroscopy (UPS) (Fig. S25, ESI†), the HOMO energies were calculated to be −5.24, −5.44 and −5.46 eV for 1–3, respectively, which is also consistent with the electrochemical studies.19
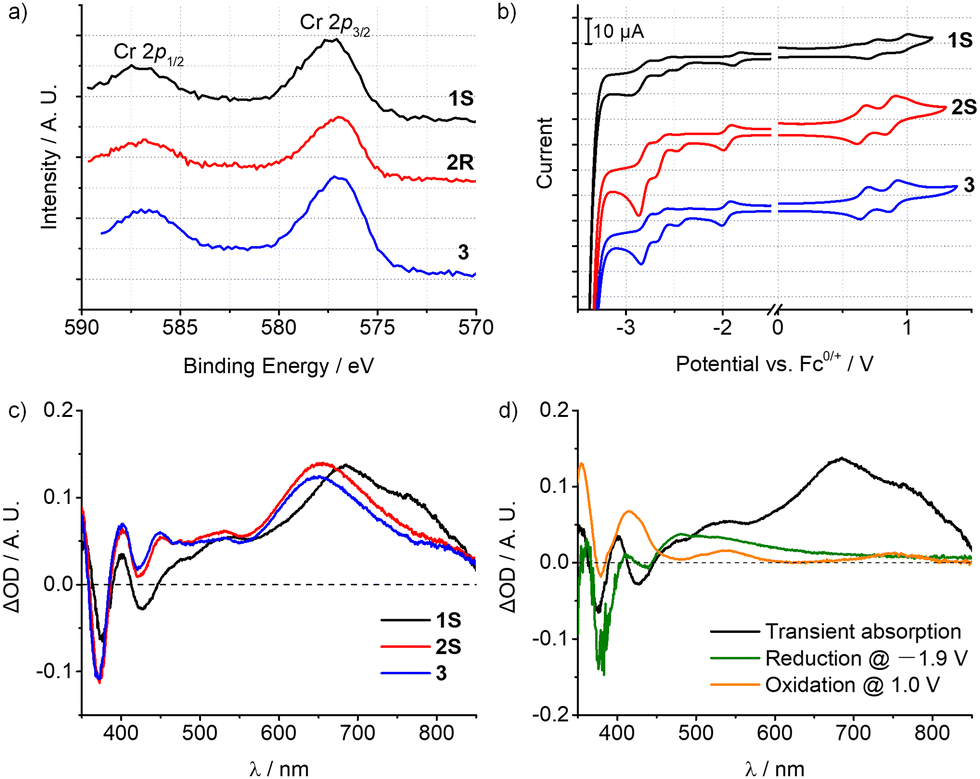 | ||
| Fig. 4 (a) X-ray photoelectron spectra of 1S, 2R, and 3 showing the scan of the Cr 2p states. (b) Cyclic voltammograms of 1S, 2S, and 3 recorded in deaerated acetonitrile at 298 K (0.1 M nBu4NPF4 as supporting electrolyte, scanning rate 100 mV s−1). (c) Nanosecond transient absorption spectrum of 1S, 2S, and 3 (1 × 10−4 M in deaerated acetonitrile) at time zero after laser excitation. (d) Differential UV-vis absorption spectra of 1S in spectro-eletrochemical experiments upon reduction at −1.9 V vs. Fc+/0 and upon oxidization at 1.0 V vs. Fc+/0, respectively, and their comparison with the nanosecond transient absorption spectrum of 1S. | ||
The nanosecond transient absorption (TA) spectroscopy with spectro-electrochemical investigations further corroborates the admixture of LMCT character of the lowest energy excited state. Upon excitation at 355 nm with a nanosecond laser, the patterns of the TA spectra of 1–3 are very similar (Fig. 4c), featuring an excited-state absorption (ESA) band peaking at 685 nm for 1 (655 nm for 2 and 650 nm for 3) and a weaker ESA band near 400 and 550 nm. Furthermore, a strong ground state bleach (GSB) at around 375 nm for 1–3 and a weak GSB at 425 nm for 1 were observed. For 1S, spectro-electrochemistry (Fig. 4d) at a potential of −1.9 V vs. Fc+/0 (inducing reduction of Cr3+/2+) yielded bleaches of the original absorption at around 375 nm and gave an additional weak absorption at 445–850 nm. Oxidation spectro-electrochemistry was performed by applying a potential of 1.0 V vs. Fc+/0, corresponding to the oxidation of the ligand and the metal centre. The band at 380 nm is slightly bleached, and at the same time, strong absorption bands at 355 and 415 nm, and weak absorptions at 540 and 750 nm were also observed. The characteristic peaks of the TA spectra largely overlapped with the spectro-electrochemical difference spectra superposition of Cr3+/2+ reduction and ligand oxidation in the 350–600 nm range,20 suggesting that the lowest-energetic excited state of 1 involves some LMCT character. However, the maximum ESA band peaking at 685 nm was missing in the spectro-electrochemical spectra.
In summary, Cr(III) complexes 1S/1R, 2S/2R and 3 have been prepared through the introduction of chiral oxazolyl in the backbones of the carbazole tridentate ligand. By simply changing the position of the carbon-centred chirality at the oxazolyl units, 1S/1R and 2S/2R show a dramatic difference of the degree of helical chirality, which is also reflected in gabs and glum. The 1S/1R enantiomers show mirror-image CPL signals in the NIR region with the maximum glum value of 2.0 × 10−3, while 2S/2R have almost no chiral optical activity. These results underline the interest of carbon-centred chiral ligands as a means to access chiral Cr(III) complexes. Our study provides an approach towards chiral Cr(III) complexes, which could achieve relatively high glum but avoid chiral resolution processes for NIR-CPL emitters.
This work was supported by the National Natural Science Foundation of China (21873044) and the Guangdong Major Project of Basic and Applied Basic Research (2019B030302009).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS; (b) G. Muller, Dalton Trans., 2009, 9692 RSC; (c) X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918 CrossRef CAS PubMed; (d) L. Cerdán, F. Moreno, M. Johnson, G. Muller, S. de la Moya and I. García-Moreno, Phys. Chem. Chem. Phys., 2017, 19, 22088 RSC; (e) K. Staszak, K. Wieszczycka, V. Marturano and B. Tylkowski, Coord. Chem. Rev., 2019, 397, 76 CrossRef CAS; (f) D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331 RSC; (g) L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109 CrossRef CAS; (h) X. Li, Y. Xie and Z. Li, Adv. Photonics Res., 2021, 2, 2000136 CrossRef.
- (a) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC; (b) A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- P. Vázquez-Domínguez, O. Journaud, N. Vanthuyne, D. Jacquemin, L. Favereau, J. Crassous and A. Ros, Dalton Trans., 2021, 50, 13220 RSC.
- (a) R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CrossRef CAS; (b) S. F. Wang, B. K. Su, X. Q. Wang, Y. C. Wei, K. H. Kuo, C. H. Wang, S. H. Liu, L. S. Liao, W. Y. Hung, L. W. Fu, W. T. Chuang, M. Qin, X. Lu, C. You, Y. Chi and P. T. Chou, Nat. Photonics, 2022, 16, 843 CrossRef CAS; (c) Y. Cheng, Q. Yang, J. He, W. Zou, K. Liao, X. Chang, C. Zou and W. Lu, Dalton Trans., 2022 10.1039/d2dt02872g.
- (a) M. Penconi, M. Cazzaniga, S. Kesarkar, P. R. Mussini, D. Ceresoli and A. Bossi, Photochem. Photobiol. Sci., 2017, 16, 1220 CrossRef CAS PubMed; (b) T. Biet, T. Cauchy, Q. Sun, J. Ding, A. Hauser, P. Oulevey, T. Bürgi, D. Jacquemin, N. Vanthuyne, J. Crassous and N. Avarvari, Chem. Commun., 2017, 53, 9210 RSC; (c) J. Song, M. Wang, X. Zhou and H. Xiang, Chem. – Eur. J., 2018, 24, 7128 CrossRef CAS PubMed; (d) F. Zinna, L. Arrico and L. Di Bari, Chem. Commun., 2019, 55, 6607 RSC; (e) C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 1307 RSC; (f) J. R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244 CrossRef PubMed; (g) J. R. Jiménez, M. Poncet, S. Míguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095 CrossRef PubMed.
- (a) O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522 CrossRef CAS PubMed; (b) C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860 CrossRef CAS PubMed.
- M. Poncet, A. Benchohra, J. R. Jiménez and C. Piguet, ChemPhotoChem, 2021, 5, 880 CrossRef CAS.
- C. Li and P. Duan, Chem. Lett., 2021, 50, 546 CrossRef CAS.
- (a) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673 RSC; (b) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1 CrossRef CAS PubMed.
- J. Lin, M. Xie, X. Zhang, Q. Gao, X. Chang, C. Zou and W. Lu, Chem. Commun., 2021, 57, 1627 RSC.
- (a) N. Sinha, J. R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722 CrossRef CAS PubMed; (b) N. Sawicka, C. J. Craze, P. N. Horton, S. J. Coles, E. Richards and S. J. A. Pope, Chem. Commun., 2022, 58, 5733 RSC.
- T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538 CrossRef CAS PubMed.
- N. Shaham, H. Cohen, D. Meyerstein and E. Bill, J. Chem. Soc., Dalton Trans., 2000, 18, 3082 RSC.
- T. Weyhermüller, T. K. Paine, E. Bothe, E. Bill and P. Chaudhuri, Inorg. Chim. Acta, 2002, 337, 344 CrossRef.
- N. Clément, C. Toussaint, G. Rogez, C. Loose, J. Kortus, L. Brelot, S. Choua, S. Dagorne, P. Turek and R. Welter, Dalton Trans., 2010, 39, 4579 RSC.
- Y. Abe and H. Ogino, Bull. Chem. Soc. Jpn., 1989, 62, 56 CrossRef CAS.
- M. Jäger and H. B. Stegmann, J. Chem. Soc., Perkin Trans. 2, 1998, 697 RSC.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572 CrossRef CAS PubMed.
- L. Zheng, T. Zhu, W. Xu, L. Liu, J. Zheng, X. Gong and F. Wudl, J. Mater. Chem. C, 2018, 6, 3634 RSC.
- N. Sinha, B. Pfund, C. Wegeberg, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 9859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: characterization data and additional spectra. CCDC 2223181, 2223182 and 2223185. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06548g |
| This journal is © The Royal Society of Chemistry 2023 |