
Visible-light-induced dehydrogenative amidation of aldehydes enabled by iron salts†
Han
Gao
 a,
Lin
Guo
a,
Yining
Zhu
a,
Chao
Yang
*a and
Wujiong
Xia
*ab
a,
Lin
Guo
a,
Yining
Zhu
a,
Chao
Yang
*a and
Wujiong
Xia
*ab
aState Key Lab of Urban Water Resource and Environment, School of Science, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China. E-mail: xyyang@hit.edu.cn; xiawj@hit.edu.cn
bSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
First published on 7th February 2023
Abstract
A direct dehydrogenative amidation reaction of aldehydes and amines under a visible light mediated ligand-to-metal charge transfer (LMCT) process was described. In this protocol, aldehyde substrates were activated by photoinduced hydrogen atom abstraction (HAA), generating acyl chloride intermediates followed by nucleophilic addition of amines. The synthetic method furnishes good functional group tolerance and broad substrate scope toward both aliphatic and aromatic components.
Amide compounds widely exist in various organic molecules and materials.1 Developing effective synthetic routes of amides has been considered as one of the most important subjects in organic and medicinal chemistry.2 Traditional transformations employing carboxylic acids and amines have been well established toward the synthesis of amides,3 yet high temperature is required in most of the cases. Transition metal-catalysed cross-coupling reactions between carboxylic acid derivatives and amines have recently become viable pathways.4 As an important alternative to the preparation of amides, aldehyde amidation reaction keeps attracting chemists’ attention due to its atom economy and selectivity.5 The usage of transition metals (Scheme 1a),6N-heterocyclic carbenes (NHC) (Scheme 1b),7 or peroxides (Scheme 1c)8 was developed through different pathways in the dehydrogenative process, while high temperature was still necessary, which might restrict further development of the transformations. A visible-light-induced mild construction pathway of amide bonds from aldehydes was accomplished by the in situ generation of hydrogen peroxide, combined with different photosensitizers (PS) in most cases (Scheme 1d).9 The hemiaminal intermediate was oxidized by hydrogen peroxide and then formed amide bonds. However, the elegant pathway would somehow gain limitations in the scope of aldehydes and amines, most of which only worked with secondary alkyl amines and aryl aldehydes. Recently, Pandey and co-workers developed a cross-dehydrogenating coupling (CDC) pathway to form amide bonds.10 Amines were oxidized, followed by N radical addition to aldehydes to form the hemiaminal radical intermediate (Scheme 1e). For another approach, the hydrogen atom abstraction (HAA) process of aldehyde C–H was developed employing multiple photocatalysts to build new C–C bonds, achieving cross-coupled products.11 Different pathways were also reported applying the HAA method to amide synthesis.12 However, it seemed underdeveloped directly using amine substrates to approach one-pot amide synthesis by activating alkyl/aryl aldehydes through the HAA process under light irradiation.13
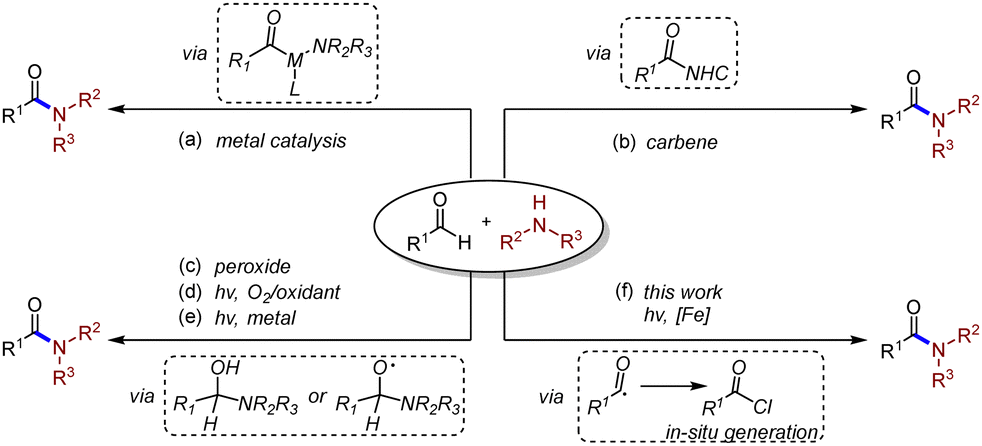 | ||
| Scheme 1 Aldehyde amidation reaction. | ||
Ligand to metal charge transfer (LMCT), as a common photoexcitation pathway, has been rekindled in organophotocatalysis during the past few years.14 The oxidized ligand could be unleashed from the metal complex as a free radical species and participated in subsequent organic transformations. Our group recently reported a ring-opening process combining LMCT decarboxylation with singlet oxygen.15 Combining LMCT and HAA processes has brought unique opportunities for the design of new transformations, among which the release of chlorine radicals plays the critical role to activate C–H bonds. In 2019, Rovis and co-workers successfully achieved the direct activation of C–H bonds, generating carbon radicals followed by the addition of Michael acceptors under visible-light mediated copper (II) catalysed LMCT conditions.16 Soon after, the same group realized a similar HAT process using Ferric chloride to proceed with LMCT transformation.17 Very recently, Zeng and co-workers reported a Fe-LMCT amidation transformation using aldehydes and nitroarenes.18 In their work, trace product was detected if aniline was applied as a substrate instead of nitrobenzene. Yoon and co-workers discovered an elegant decarboxylative amination process to form C–N bonds using excess copper (II) salt as an LMCT initiator and oxidant.19 As part of our ongoing interest in the development of photoinduced transformations, we herein reported a photoinduced dehydrogenative amidation reaction, building C–N bonds enabled by iron salts (Scheme 1f).
Following this thought, we initially set up the optimization study using benzaldehyde (1) and N-methyl-1-phenylmethanamine (2) as model substrates with ferric chloride as a catalyst (Table 1). With the addition of ferrous chloride (1.0 equiv.) and potassium fluoride (2.0 equiv.) into the system, 73% yield of product 3 was isolated (Table 1, entry 1). Employment of 450 nm LEDs decreased the yield (entry 2). If the reaction was performed in dry DCM, the yield dramatically dropped (entry 3). If using dry MeCN instead, the yield increased to 57% (entry 4). Reduced iron(III) loading did not give a positive effect on the yield (entry 5). When no base or other bases such as NaHCO3 and CsF were employed, the reaction efficiency dropped (entries 6–8). Moreover, reducing the amount of aldehyde substrate negatively affected the reaction yield (entry 9), and switching the additive from ferrous chloride to NH4Cl provided less efficiency (entry 10). Furthermore, if the reaction was performed open air, only a small amount of product was isolated (entry 11). The control reaction showed that light was crucial (entry 12).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield%b |
a Reaction conditions: 1 (0.3 mmol), 2 (0.1 mmol), FeCl3 (15 mol%), KF (2.0 equiv.), FeCl2 (1.0 equiv.) in DCM:MeCN = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 W 390 nm LEDs at room temperature for 12 hours.
b Yields of isolated product. N.R.: no reaction.
c Oxygen less than 2 ppm. :1, 10 W 390 nm LEDs at room temperature for 12 hours.
b Yields of isolated product. N.R.: no reaction.
c Oxygen less than 2 ppm.
|
||
| 1 | None | 73 |
| 2 | 450 nm 10 W LEDs | Trace |
| 3 | Dry DCM as solvent | 33 |
| 4 | Dry MeCN as solvent | 57 |
| 5 | 5 mol% of FeCl3 | 56 |
| 6 | Without KF | 33 |
| 7 | NaHCO3 instead of KF | 62 |
| 8 | CsF instead of KF | 18 |
| 9 | 1.0 equiv. of aldehyde 1 | 35 |
| 10 | NH4Cl instead of FeCl2 | 43 |
| 11 | Open to air | 16 |
| 12 | In the dark | N.R. |
| 13 | Without FeCl3 | 35 |
| 14 | Without FeCl2 | 60 |
| 15 | Glove box setupc | 8 |
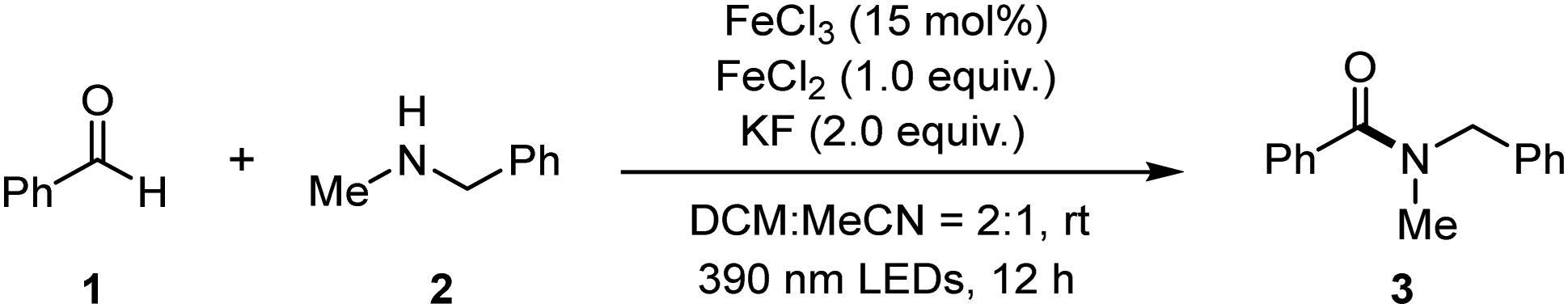
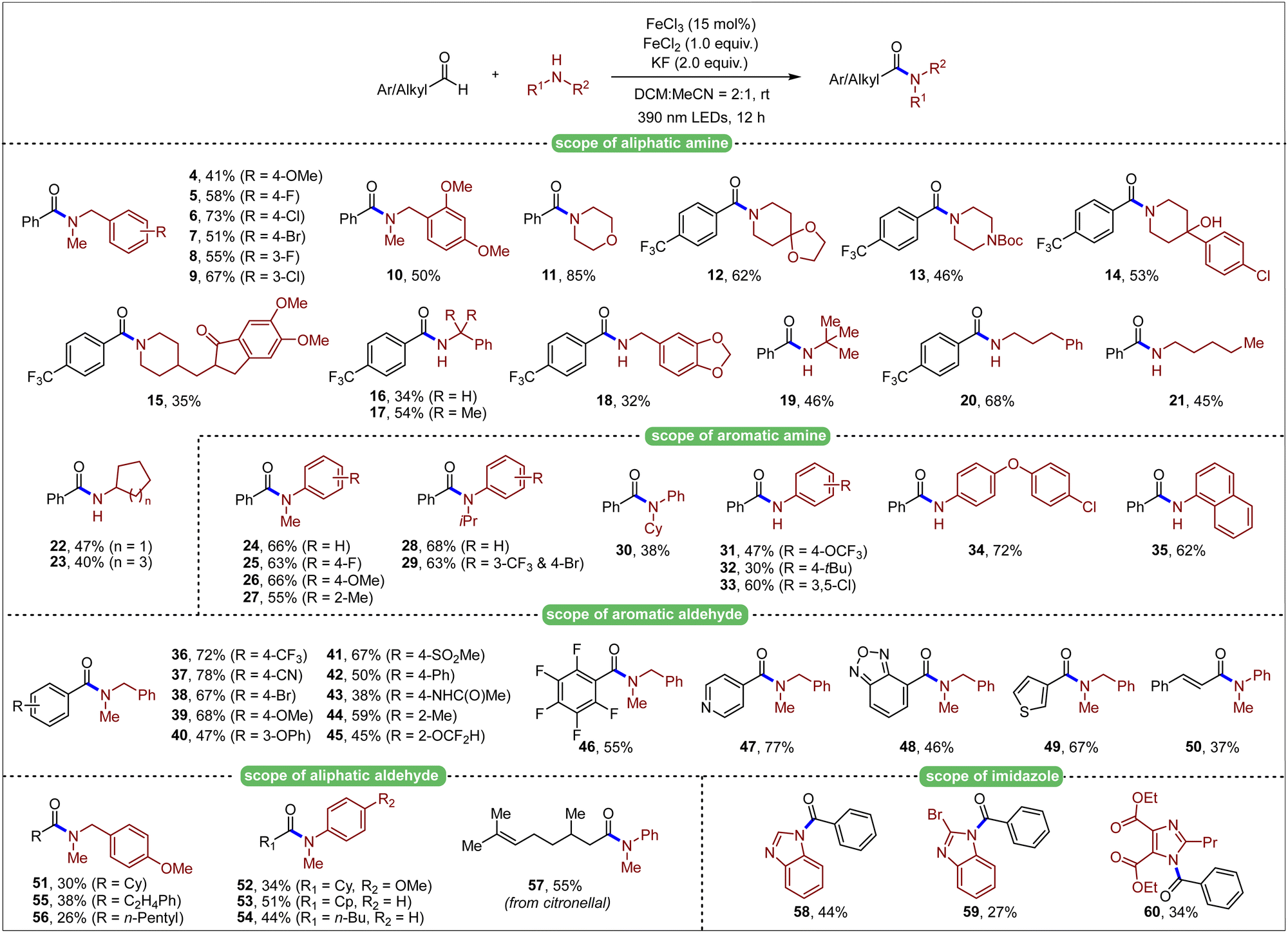
After obtaining the optimized conditions, we next investigated the scope with respect to amine components (Table 2). Secondary N-methyl benzylamine derivatives bearing different aromatic substituents were firstly investigated and proved to be suitable for the reaction (4–10). Other secondary alkyl amines performed well, giving the desired products (11–13) in moderate to good yields. Commercially available pharmaceutical molecules, such as haloperidol and donepezil, were also amenable to amidation for their derivatives (14, 15), where the tertiary hydroxyl group was compatible under this transformation. Primary alkyl amines including benzylamines (16–18) and more general alkyl amines (19–23), showed moderate reactivity with aldehydes under photoinduced conditions. Encouraged by above results, we continued investigating aromatic amines, which were relatively obscure in most of the amidation reactions under light irradiation. Various secondary N-alkyl substituted anilines proved to be suitable under the optimized conditions (24–30). Additionally, unprotected aniline derivatives with different aromatic substitutions were explored, which showed descend to good reactivity to give the desired amide products in 30–72% yields (31–35).
| Reaction conditions: aldehyde (0.6 mmol), amine (0.2 mmol), FeCl3 (15 mol%), KF (2.0 equiv.), FeCl2 (1.0 equiv) in DCM:MeCN = 2:1, 10 W 390 nm LEDs at room temperature for 12 hours. Yields of isolated product. |
|---|
|
After investigation in amine substrates, we then explored the aldehyde scope (Table 1). Various substituted benzaldehydes were readily reacted with N-methyl-1-phenylmethanamine (36–46). Heteroaryl aldehydes were also well tolerated (47–49). Moreover, vinyl aldehyde was also examined, and 37% yield of the product was obtained (50). Aliphatic aldehydes, in which generated acyl radicals were not as stable as aromatic ones, were then investigated. Corresponding amide products were both successfully generated no matter whether alkyl or aryl amines were employed (51–56). Citronellal was also introduced as the substrate, generating the corresponding amide product (57) in moderate yield. Furthermore, imidazole substrates were also compatible and successfully formed subsequent amide bonds (58–60), which indicated potential application in further development of this method. To illustrate the capability of the transformation, a gram-scale setup (10 mmol of amine 2) was delivered. Gratifyingly, 61% of product 3 was isolated after 54 h, indicating the potential application of this transformation (Scheme 2).
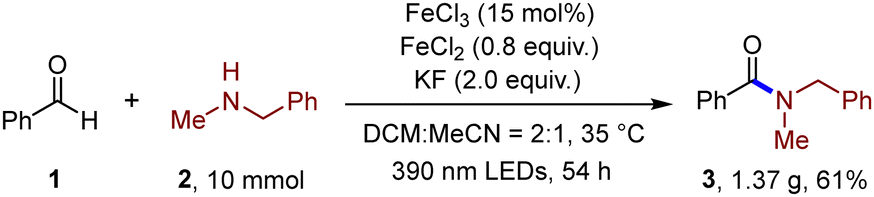 | ||
| Scheme 2 Gram-scale reaction. | ||
To further understand the reaction mechanism, control experiments were carried out, as shown in Scheme 3. The addition of TEMPO as a typical radical scavenger significantly hindered the generation of amide, which proved that the reaction might go through a radical process (Scheme 3A). To further prove the existence of an acyl radical intermediate, Michael acceptor (61) was introduced in the absence of amines. Radical addition-hydrogenation product (62) was subsequently isolated in 12% yield, revealing the generation of an acyl radical (Scheme 3B). Acyl chlorides (63) and a small amount of acyl fluoride (64) were detected by high resolution mass spectrometry (HRMS) under standard conditions in the absence of amines after 6 h, which indicated the existence of acyl chloride intermediates (Scheme 3C).20
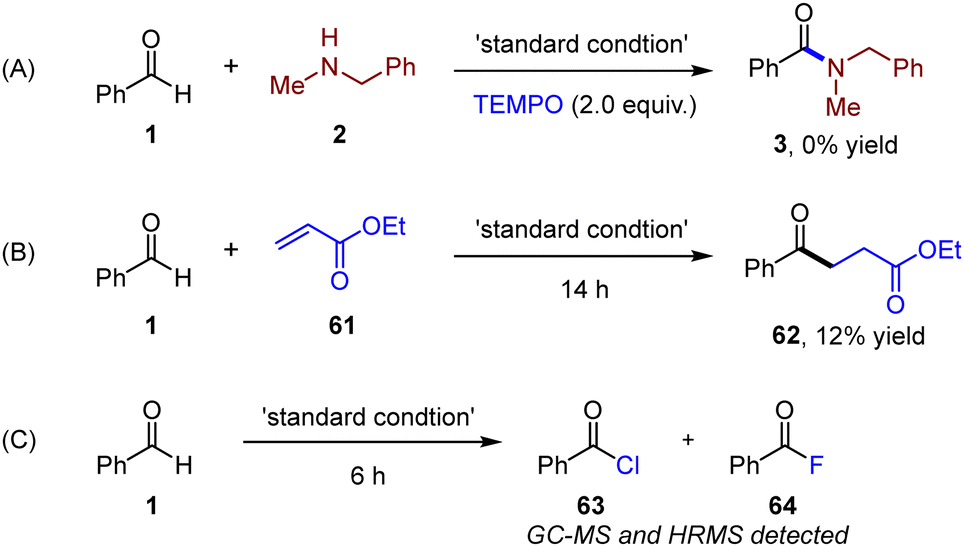 | ||
| Scheme 3 Control reaction and radical trapping experiment. | ||
Based on the above discovery, a plausible mechanism was proposed for this reaction, as shown in Scheme 4. Tervalent iron chloride proceeded through the LMCT process under irradiation of purple light (390 nm), generating a Cl radical and Fe(II) species. Fe(III) salts would be refilled by the generated Fe(II), which was oxidized by a trace amount of O2 which might be dissolved in the solvents.21 The additive FeCl2 could also be oxidized and participated in the reaction minorly. The Cl radical abstracted a hydrogen atom from the aldehyde substrate, generating a carbonyl radical and hydrogen chloride. The carbonyl radical then captured a Cl radical, which was extruded from the chloride intermediate, and would minorly react with KF, generating an acyl fluoride intermediate. Finally, the acyl halide intermediate was nucleophilically attacked by the amine to form the final amide product, hydrogen chloride and hydrogen fluoride.
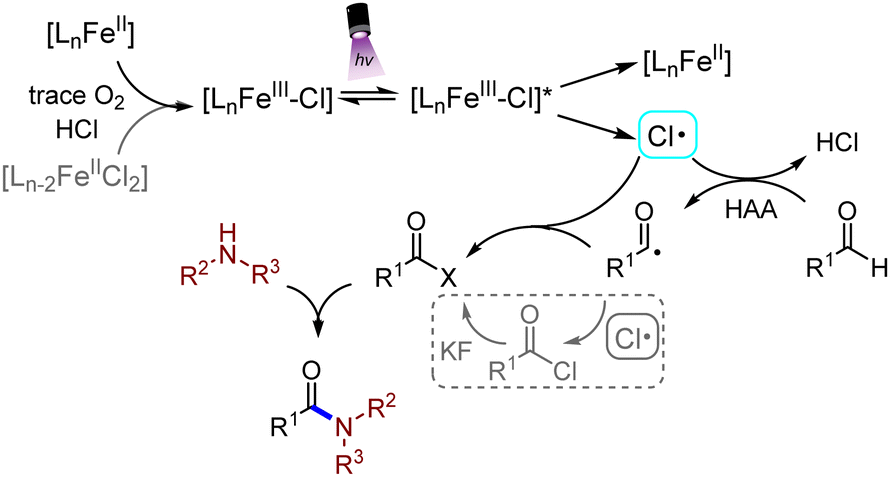 | ||
| Scheme 4 Proposed reaction mechanism. | ||
In conclusion, we have developed an amidation reaction directly using aldehydes and amines/imidazoles as starting materials under mild photoinduced LMCT conditions. Broad scope and great functional group tolerance of both aldehydes and amines showed the potential application prospects of this reaction. Moreover, control experiments and radical trapping experiments revealed that the reaction underwent acyl radical mediated nucleophilic addition upon visible light irradiation.
We are grateful for the financial support from the Science and Technology Plan of Shenzhen (No. JCYJ20210324133001004 and JCYJ20210324132803009), Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515220069), State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2022TS24), and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765 CAS; (c) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS; (d) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) A. S. Santos, A. M. S. Silva and M. M. B. Marques, Eur. J. Org. Chem., 2020, 2501 CrossRef CAS; (b) B. Lu, W.-J. Xiao and J.-R. Chen, Molecules, 2022, 27, 517 CrossRef CAS PubMed; (c) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (d) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- M. Todorovic and D. M. Perrin, Pept. Sci., 2020, 112, e24210 CAS.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed.
- (a) A. M. Whittaker and V. M. Dong, Angew. Chem., Int. Ed., 2015, 54, 1312 CrossRef CAS PubMed; (b) S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002 CrossRef CAS PubMed; (c) O. P. S. Patel, D. Anand, R. K. Maurya and P. P. Yadav, Green Chem., 2015, 17, 3728 RSC.
- (a) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798 CrossRef CAS PubMed; (b) R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, Org. Lett., 2016, 18, 1198 CrossRef CAS PubMed; (c) C. Zheng, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 6940 CrossRef CAS PubMed.
- (a) X. Liu and K. F. Jensen, Green Chem., 2012, 14, 1471 RSC; (b) R. Fu, Y. Yang, J. Zhang, J. Shao, X. Xia, Y. Ma and R. Yuan, Org. Biomol. Chem., 2016, 14, 1784 RSC.
- (a) D. Leow, Org. Lett., 2014, 16, 5812 CrossRef CAS PubMed; (b) A. Monga, A. P. Pandey and A. Sharma, Adv. Syn. Catal., 2019, 361, 3554 CrossRef CAS; (c) A. Hassan Tolba, M. Krupička, J. Chudoba and R. Cibulka, Org. Lett., 2021, 23, 6825 CrossRef CAS PubMed.
- G. Pandey, S. Koley, R. Talukdar and P. K. Sahani, Org. Lett., 2018, 20, 5861 CrossRef CAS PubMed.
- (a) K. Zhao, X.-C. Zhang, J.-Y. Tao, X.-D. Wu, J.-X. Wu, W.-M. Li, T.-H. Zhu and T.-P. Loh, Green Chem., 2020, 22, 5497 RSC; (b) L. Wang, T. Wang, G.-J. Cheng, X. Li, J.-J. Wei, B. Guo, C. Zheng, G. Chen, C. Ran and C. Zheng, ACS Catal., 2020, 10, 7543 CrossRef CAS; (c) P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180 CrossRef CAS PubMed; (d) T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366 CrossRef CAS PubMed; (e) S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723 CrossRef CAS PubMed; (f) V. Murugesan, A. Ganguly, A. Karthika and R. Rasappan, Org. Lett., 2021, 23, 5389 CrossRef CAS PubMed; (g) A. Chinchole, M. A. Henriquez, D. Cortes-Arriagada, A. R. Cabrera and O. Reiser, ACS Catal., 2022, 12, 13549 CrossRef CAS.
- (a) T. Wang, Z. Wen, G. Laudadio, L. Capaldo, R. Lammers, J. A. Rincón, P. García-Losada, M. O. Frederick, R. Broersma and T. Noël, ACS Cent. Sci., 2022, 8, 51 CrossRef PubMed; (b) Q. Li, P. Dai, H. Tang, M. Zhang and J. Wu, Chem. Sci., 2022, 13, 9361 RSC.
- (a) S. Gaspa, I. Raposo, L. Pereira, G. Mulas, P. C. Ricci, A. Porcheddu and L. De Luca, New J. Chem., 2019, 43, 10711 RSC; (b) N. F. Nikitas, M. K. Apostolopoulou, E. Skolia, A. Tsoukaki and C. G. Kokotos, Chem. – Eur. J., 2021, 27, 7915 CrossRef CAS PubMed.
- (a) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100 CrossRef CAS PubMed; (b) T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296 CrossRef CAS PubMed; (c) N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6163 CrossRef CAS PubMed; (d) P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349 CrossRef CAS PubMed; (e) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543 CrossRef PubMed.
- J.-L. Tu, H. Gao, M. Luo, L. Zhao, C. Yang, L. Guo and W. Xia, Green Chem., 2022, 24, 5553 RSC.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729 CrossRef CAS PubMed.
- Y. C. Kang, S. M. Treacy and T. Rovis, ACS Catal., 2021, 11, 7442 CrossRef CAS PubMed.
- N. Xiong, Y. Dong, B. Xu, Y. Li and R. Zeng, Org. Lett., 2022, 24, 4766 CrossRef CAS PubMed.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94 CrossRef CAS PubMed.
- (a) M. Tryniszewski and M. Barbasiewicz, Synthesis, 2022, 1446 CAS; (b) E. D. Kalkman, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 19458 CrossRef CAS PubMed; (c) See ESI†.
- (a) L. Zhang, X. Si, Y. Yang, S. Witzel, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, ACS Catal., 2019, 9, 6118 CrossRef CAS; (b) S. Pagoti, S. Surana, A. Chauhan, B. Parasar and J. Dash, Catal. Sci. Tech., 2013, 3, 584 RSC; (c) L. Ilies, Y. Itabashi, R. Shang and E. Nakamura, ACS Catal., 2017, 7, 89 CrossRef CAS; (d) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS PubMed; (e) Z. Zhang, G. Zhang, N. Xiong, T. Xue, J. Zhang, L. Bai, Q. Guo and R. Zeng, Org. Lett., 2021, 23, 2915 CrossRef CAS PubMed; (f) S. Rana, J. P. Biswas, S. Paul, A. Paik and D. Maiti, Chem. Soc. Rev., 2021, 50, 243 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06507j |
| This journal is © The Royal Society of Chemistry 2023 |