
Plasmonic quenching and enhancement: metal–quantum dot nanohybrids for fluorescence biosensing
Niko
Hildebrandt
 *,
Mihye
Lim
,
Namjun
Kim
,
Da Yeon
Choi
and
Jwa-Min
Nam
*
*,
Mihye
Lim
,
Namjun
Kim
,
Da Yeon
Choi
and
Jwa-Min
Nam
*
Department of Chemistry, Seoul National University, Seoul 08826, South Korea. E-mail: niko.hildebrandt@snu.ac.kr; jmnam@snu.ac.kr
First published on 19th January 2023
Abstract
Plasmonic metal nanoparticles and semiconductor quantum dots (QDs) are two of the most widely applied nanomaterials for optical biosensing and bioimaging. While their combination for fluorescence quenching via nanosurface energy transfer (NSET) or Förster resonance energy transfer (FRET) offers powerful ways of tuning and amplifying optical signals and is relatively common, metal–QD nanohybrids for plasmon-enhanced fluorescence (PEF) have been much less prevalent. A major reason is the competition between fluorescence quenching and enhancement, which poses important challenges for optimizing distances, orientations, and spectral overlap toward maximum PEF. In this feature article, we discuss the interplay of the different quenching and enhancement mechanisms (a mixed distance dependence of quenching and enhancement – “quenchancement”) to better understand the obstacles that must be overcome for the development of metal–QD nanohybrid-based PEF biosensors. The different nanomaterials, their combination within various surface and solution based design concepts, and their structural and photophysical characterization are reviewed and applications toward advanced optical biosensing and bioimaging are presented along with guidelines and future perspectives for sensitive, selective, and versatile bioanalytical research and biomolecular diagnostics with metal–QD nanohybrids.
 Niko Hildebrandt | Niko Hildebrandt received his PhD in Chemistry from the University of Potsdam and worked as a group leader at the Fraunhofer Institute for Applied Polymer Research. He has been a Full Professor at the University of Paris Saclay (2010–2022) and the University of Rouen (since 2022) and is currently a Visiting Professor in the Department of Chemistry at Seoul National University. His main research interests are nanotechnology-based FRET and luminescence for biosensing and bioimaging. |
 Mihye Lim | Mihye Lim received her BS degree in Chemistry from Kyungpook National University in 2015 and MS degree in Chemistry from POSTECH in 2017. During her MS degree, she researched the synthesis and applications of quantum dots. She joined the laboratory of Professor Jwa-Min Nam at Seoul National University as a PhD student in 2022. Her current research focus is the hybrid structure of metal nanoparticles and quantum dots. |
 Namjun Kim | Namjun Kim received his BS degree in Chemistry from Kwangwoon University in 2018. He joined the research group of Prof. Jwa-Min Nam at Seoul National University as a PhD student in 2018. His research interests include supported lipid bilayer-based sensing and plasmonic applications using plasmonic nanoprobes. |
 Da Yeon Choi | Da Yeon Choi received her BS degree in Chemistry from Yonsei University in 2016. She has been a PhD student in Prof. Jwa-Min Nam's research group since 2016. Her major interests include design, synthesis, and biomedical applications of plasmonic nanostructures. |
 Jwa-Min Nam | Jwa-Min Nam received his PhD in Chemistry from Northwestern University and worked as a postdoctoral fellow at the University of California, Berkeley. He is currently a Full Professor of Chemistry and a vice chair of the Department of Chemistry at Seoul National University. His major research interests are plasmonic nanostructures and nanoprobes for biosensing, bioimaging, therapeutics, nanoparticle-tethered lipid bilayer systems, and cell–nanostructure interfaces. |
1. Introduction
Owing to their unique physical, chemical, and optical properties, advanced synthesis and fabrication, and versatile biofunctionalization, plasmonic and luminescent nanoparticles (NPs) have had an enormous influence on biosensing and bioimaging over the last two decades.1–10 In particular, gold NPs (AuNPs) with their exceptional plasmonic properties and biocompatibility and semiconductor quantum dots (QDs) with their extremely bright, stable, and size-tunable fluorescence have played an outstanding role in the improvement of existing applications and the development of new bioanalytical applications.11–14 If a biomolecule or a biomolecular interaction changes the distance of an optically active partner (e.g., a dye or a NP), such that it strongly or weakly interacts with the plasmonic or luminescent NPs, the resulting energy transfer, caused by optical enhancement or deactivation (quenching) of the optically active partner, can be used for a quantitative analysis of the biomolecule or biomolecular interaction with high sensitivity and high spatial resolution. Plasmonic NPs can enhance Raman signals via surface-enhanced Raman scattering (SERS),15,16 quench or enhance photoluminescence (PL) via nanosurface energy transfer (NSET),17,18 or enhance PL via radiative rate enhancement (Purcell effect)19 of the interacting partner. QDs can engage in Förster resonance energy transfer (FRET), NSET, electron transfer, or charge transfer with the interacting partner.20,21Although both plasmonic metal NPs and QDs have been widely applied by using all those different interactions with various optically active molecules, their combination in metal–QD nanohybrids has mainly focused on PL quenching via NSET (also often referred to as FRET) from QDs to AuNPs.21 Exploitation of the PL enhancement of QDs via plasmonic NPs has been much more limited, in particular, when it comes to biosensing or bioimaging. In this Feature article, we first describe and explain the different mechanisms responsible for PL quenching and enhancement induced by plasmonic NPs. Instead of revisiting the theory of plasmonic enhancement and quenching, which has been treated in many other reviews and textbooks, we focus on the phenomena and concomitant modifications of optical properties in an equation-free approach with the aim of making plasmonic quenching and enhancement (i.e., “quenchancement”) accessible to both newcomers and experts from different fields of chemistry, biology, physics, and engineering. After providing a brief overview of fluorescence biosensing applications using plasmonic metal NPs or QDs alone, we review various applications of plasmon-quenched and plasmon-enhanced PL in metal–QD nanohybrids with a focus on plasmon-enhanced PL because this field has not been reviewed before. For all examples, we discuss advantages and disadvantages for biosensing. Finally, we conclude by summarizing the status-quo of metal–QD nanohybrids for biosensing and suggesting future requirements, directions, and trends for advanced bioanalysis.
2. Purcell vs. Förster. Plasmonic enhancement and quenching by metal nanoparticles
2.1 Localized surface plasmons
Localized surface plasmons (LSPs) are electron oscillations at the surface of metal NPs that interface with the surrounding medium (a dielectric).22–25 The frequency of these oscillations is strongly dependent on the dielectric constants (i.e., the refractive index) of the metal and the medium.9 LSPs can be induced by light (i.e., an external oscillating electromagnetic field), whose frequency (or wavelength) is in resonance with the electron oscillation frequency. This LSP resonance (LSPR) is usually relatively broad (many tens to a few hundreds of nm) and leads to an extinction of the incident light in the resonant wavelength range due to absorption and scattering. The extinction of small metal NPs (diameters below circa 30 nm) is mainly dominated by absorption, whereas larger metal NPs (diameters above circa 80 nm) mainly scatter and show slightly red-shifted extinction spectra. The LSPs, in turn, strongly enhance the electric field in very close proximity (a few tens of nm) of the NP surface, which can result in enhanced photophysical properties of optically active species placed in that enhanced electric field. Both resonance wavelength and local electric field enhancement are also dependent on the size and shape of the NP as well as their interactions.26 Thus, the resonance wavelength ranges of simple nanospheres can be shifted and extended by more complicated structures and extreme enhancements can be created in so-called plasmonic hotspots at the apex of pointed structures (e.g., triangles) or at the interface between two or more nanomaterials.27–29Fig. 1 provides an overview of LSPR wavelength ranges of typical metal nanostructures, which are mainly based on gold (Au), silver (Ag), copper (Cu), and aluminum (Al), and more sophisticated nanostructures that can be used to shift the LSPR to higher wavelengths (e.g., NIR-II) or the design of plasmonic hotspots.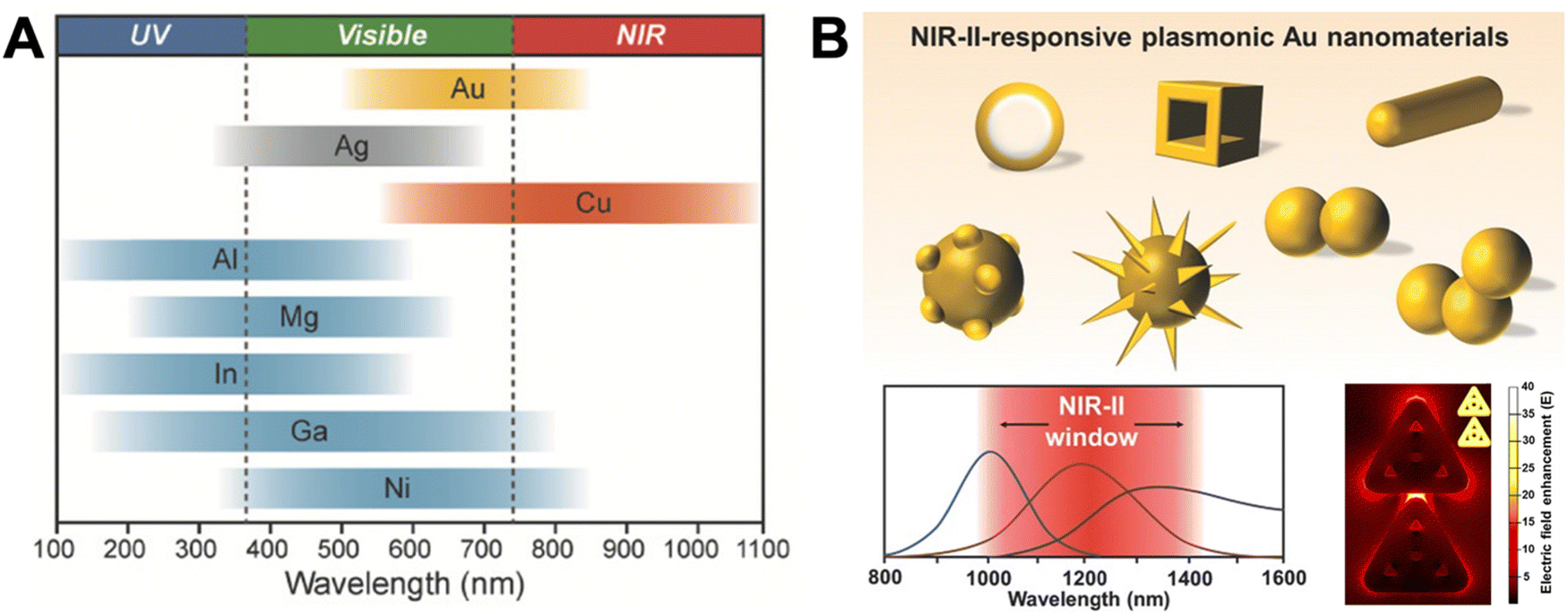 | ||
| Fig. 1 (A) The spectral ranges of different plasmonic metal NPs. Reproduced with permission from ref. 28. Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA. (B) Different sophisticated plasmonic Au nanostructures (top), their possible extension of plasmon resonance wavelengths to NIR-II (bottom left), and their possible electric field enhancement in inter and intra NP hotspots (shown in the example of ring-in-a-triangle nanoframes). Reproduced with permissions from ref. [27] (copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA) and ref. 29 (copyright 2022 American Chemical Society). | ||
Whether a fluorophore near a plasmonic NP will be quenched or enhanced mainly depends on the spectral overlap of the LSPR spectrum with the absorption and/or emission spectra of the fluorophore and the distance (d) between the fluorophore and plasmonic NP. The quenching efficiency is the highest for direct contact with the NP surface and decays with ∼d−4 until circa 20 nm. It requires overlap of the fluorophore emission with the LSPR extinction spectrum. The enhancement efficiency is the highest for short distances of approximately 5 to 10 nm (energy losses inside the metal are very strong at shorter distances) and decays with ∼d−3 until several tens of nanometers. Both the absorption and emission of the fluorophore can be enhanced, which means that the LSPR extinction spectrum, and in particular, the scattering part should overlap with one or with both.
There is a distance range (between circa 5 and 15 nm), in which PL is both quenched and enhanced and this competition must be taken into account when designing a sensor based on quenching or enhancement. The competition between quenching and enhancement and the simultaneous decay of their efficiencies with distance are the main reasons, why the design of plasmon enhanced fluorescent biosensors is much more complicated than the design of quenched fluorescent biosensors. For very short distances both quenching and energy losses in the metal are present, which creates a win–win situation. For longer distances quenching and enhancement compete and the enhancement strongly attenuates with distance (at very long distances enhancement is guaranteed but it is very weak), which creates a loss–loss situation. The mechanisms for both quenching and enhancement and the related distances are summarized in Scheme 1. The following sections will explain the details of quenching (via NSET) and enhancement (via LSP-induced electric field enhancement).
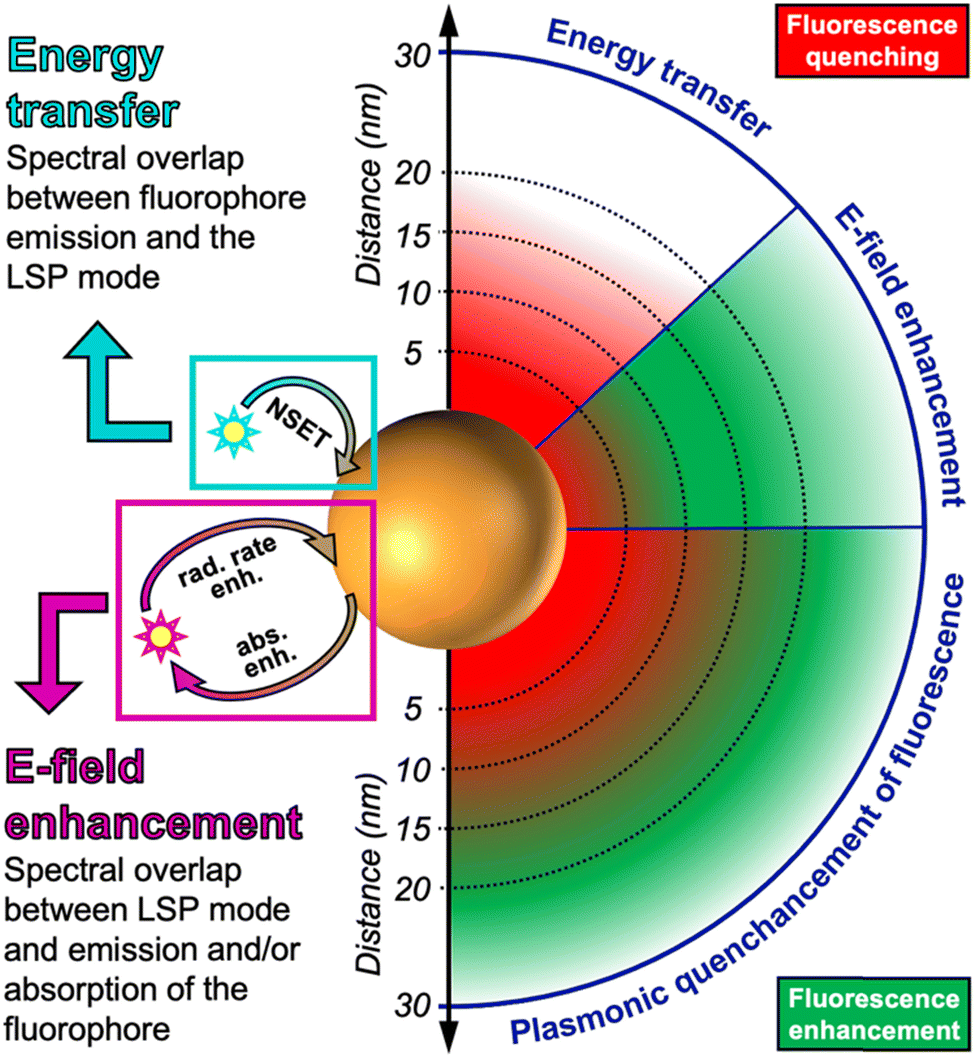 | ||
| Scheme 1 Nanosurface energy transfer (NSET) between the transition dipole moment of the fluorophore emission and the LSPs on the plasmonic NP results in distance-dependent (∼d−4) quenching in the range of circa 1 to 20 nm. Electric field (E-field) enhancement via the LSPs on the plasmonic NP results in distance-dependent (∼d−3) absorption enhancement (abs. enh.) and/or radiative rate enhancement (rad. rate enh.) of the fluorophore up to several tens of nanometers (up to ∼30 nm shown here). Due to strong energy losses by scattering of electrons in the metal, fluorescence is quenched very close to the plasmonic NP. Usually both NSET and E-field enhancement occur simultaneously, leading to a mixed distance dependence of quenching and enhancement (i.e., “quenchancement”). | ||
2.2 Luminescence quenching via nanosurface energy transfer (NSET)
Similar to FRET, NSET is a resonance energy transfer mechanism.18,20,30–34 In FRET, both the energy donor and energy acceptor are considered as point dipoles and the FRET rate decays with an inverse sixth power dependence (d−6) because the dipole–dipole coupling can be approximated as Coulomb coupling (coupling of two charges, which is proportional to d−3) and after Fermi's golden rule the FRET rate is proportional to the square of the coupling between the donor and acceptor. In NSET, the acceptor consists of LSPs on the surface of a relatively large metal NP, which can be approximated as a collection of many point dipoles oscillating with the same frequency. Based on Fermi's golden rule, Persson and Lang approximated the point-dipole-surface-dipole distance dependence as the product of dipole coupling (d−3) and a surface damping function proportional to d−1 (they also found that the volume damping function was distance independent, i.e. ∼1).35 Thus, the NSET distance dependence (surface damping) was found to be proportional to d−4 (and volume damping ∼d−3). The difference in FRET (∼d−6) and NSET (∼d−4) distance dependence can be mainly translated into a longer distance range of NSET (circa 1 to 40 nm) compared to FRET (circa 1 to 20 nm).One must keep in mind that all these energy transfer rate calculations are based on approximations and there exist also various uncertainties, including the NP size (e.g., small NPs may also be approximated as point dipoles or even volume dipoles because of the stronger curvature of the surface), the dielectric constants (i.e., refractive indices) of the metal and the surrounding medium, the LSPR spectrum (absorption and scattering may be considered equally or separate for calculating the spectral overlap with donor emission), and the orientations of the dipoles. Therefore, experimental results may deviate from the ideal approximation. Although the majority of studies that investigated the distance dependence of metal NP-quenched PL found the NSET mechanism (∼d−4) to be the best fit with the experimental results, others found FRET (∼d−6) to be a better fit.18 The competition between plasmonic quenching and enhancement at intermediate distances from the metal NP surface (between circa 5 and 40 nm) is another important uncertainty and the signal change of a plasmonic-enhanced PL biosensor is certainly different when using the FRET or the NSET distance range. Because energy transfer-based biosensors usually rely on very small (sub-nanometers to a few nanometers) distance changes, such uncertainties make predictions, and thus, the design of the biosensor, extremely delicate. In addition, the luminescent donor may also significantly deviate from the point-dipole approximation. While it will most probably be an excellent approximation for small fluorophores, QDs with diameters of up to tens of nanometers may not be ideal point dipoles. Although calculations showed that the dipole approximation works very well for small (3.9 nm) spherical QDs,36 most QDs are not spherical and can be significantly larger. The same shape considerations hold true for the metal NPs, which may also strongly deviate from hard spheres.
Despite all material-related and experimental uncertainties, one can keep in mind some guidelines when designing plasmonic-quenched PL biosensors (using QDs and other fluorophores): (i) the closer the distance between the fluorophore and metal NP, the more efficient the PL quenching; (ii) the energy transfer efficiency decays most probably with a d−4 distance dependence (NSET); (iii) the distance to be considered is the one between the surface of the metal NP and the center of the fluorophore; (iv) distances between roughly 5 and 15 nm should be avoided because of the competition between quenching and enhancement; and (v) all those guidelines are based on spherical NPs and if they are not, deviations, such as varying plasmonic hotspots, should be kept in mind. Finally, these guidelines only provide a good starting point and experimental validation and optimization will always be necessary to design an efficient plasmonic-quenched PL biosensor.
2.3 Luminescence enhancement via absorption and radiation rate enhancement
Compared to PL quenching, for which the relatively simple rule “the closer the better” can be applied, PL enhancement is significantly more complicated. Intuitively, one may think that the extremely large enhancement factors of more than 10 orders of magnitude for Raman scattering should also be applicable for PL enhancement. However, normal (or nonresonant) Raman scattering is purely enhanced (and not quenched) at close distances to the metal NP surface and the very inefficient Raman process can therefore be extremely efficiently improved.37 In contrast, PL (as well as resonant Raman scattering) is very efficiently quenched at close distances to the metal NP. In fact, one can consider normal Raman enhancement as PL enhancement with zero absorption and zero quantum yield. In other words, fluorophores with very low absorption cross sections and quantum yields can also be very efficiently enhanced, whereas the PL of good absorbers with high quantum yields can only be weakly or moderately enhanced (Fig. 2).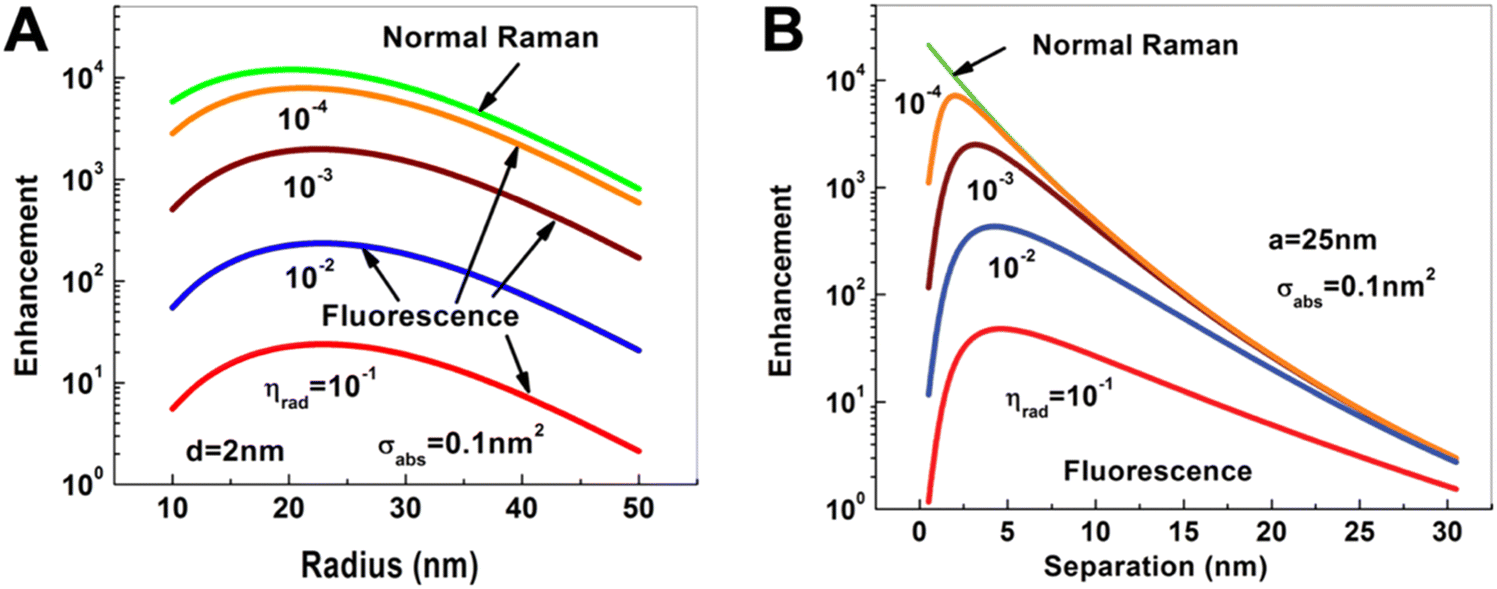 | ||
| Fig. 2 Dependence of Raman (green) and fluorescence (red, blue, brown, and orange) enhancement: (A) on the radius of a spherical Au NP when the Raman or fluorescent molecule is placed at a distance of d = 2 nm from the Au NP surface; (B) on the distance of the Raman or fluorescent molecule from the surface of a Au NP with a radius of a = 25 nm. The fluorescent molecule has an absorption cross section of σabs = 0.1 nm2 and quantum yields (ηrad) ranging from 10−4 to 10−1. Excitation for both processes at dipole mode resonance (ωex = ω1 = 2.562 eV) and emission at the same Stokes frequency (ωS = 2.462 eV). Reproduced with permission from ref. 37. Copyright 2012 American Physical Society. | ||
Similar to SERS, plasmonic enhancement of PL has been used and investigated for more than four decades and several reviews have described and discussed the underlying mechanisms as well as applications for biosensing.38–40 Khurgin and Sun developed an analytical model that can be used to modify the different properties of a plasmonic metal NP and its interaction with a nearby fluorophore. They used this model to study the enhancement of absorption,41,42 electroluminescence,41 and PL,37,41,43–45 losses in metal NPs,46,47 the influence of more complex metal nanostructures,48,49 and the comparison of Raman vs. PL enhancement.37,44 These very interesting articles (including 231 equations for those who want to dig into the details) nicely illustrate the possibilities and limitations of PL enhancement. Here, we review the main results concerning material and distance properties of metal NPs and fluorophores with the aim to provide some guidelines (similar to quenching – vide supra) of important aspects that need to be considered for the development of plasmon-enhanced fluorescent biosensors. Similar to the NSET discussion, the described phenomena are valid for spherical NPs and small fluorophores and different shapes and sizes will lead to deviations from the ideal model.
Concerning PL enhancement via LSPR, one needs to distinguish different LSP eigenmodes (modes of collective oscillation in the NP). Only the 1st mode possesses a dipole moment that can couple to an external radiation field (e.g., light excitation or a nearby fluorophore). The higher order modes are nonradiative. The metal NP plays a dual role, namely as an “antenna” for in and out coupling of energy and as a “cavity” for energy concentration by an increased local density of states (LDS). Both roles are somewhat contradictory because an efficient antenna requires a large dipole for strong coupling with radiation over a long distance (several tens of nanometers), whereas an efficient cavity requires a highly concentrated and small space (a few nanometers). The 1st mode is a great antenna because it couples well with the “outside world” of the metal NP (for interacting with fluorophores over several tens of nanometers). However, it is a relatively weak cavity because its effective volume is large (low LDS). The higher order modes are very weak antennas because they concentrate the energy (high LDS) very close to the NP surface. However, this strong energy concentration makes them great cavities. Unfortunately, the energy dissipates extremely quickly (on the femtosecond time scale) by the scattering of electrons in the metal, which makes losses (i.e., quenching) very efficient at distances close to the NP surface. Therefore, only fluorophore–surface distances above ∼5 nm can result in appreciable enhancement. Because large NPs scatter more and the dipole scales with the NP volume, large NPs couple stronger with radiation and are better antennas than small NPs.
PL requires the absorption and emission of light and both processes can be enhanced by proximal metal NPs. For absorption enhancement, the LSPR scattering spectrum must overlap with the absorption spectrum of the fluorophore. Absorption enhancement contains two processes. The first one is coupling of the excitation light into the LSP dipole mode and the second is the absorption of the energy in the LSP dipole mode by the fluorophore. For each combination of NP size (with radius a), fluorophore distance from the NP surface (d), and absorption cross section (σa, with σa in nm2 corresponding to ∼3.82 × 10−7 times the molar absorptivity in M−1 cm−1) there is a specific enhancement. The lower the initial absorption cross section of the fluorophore, the higher the relative enhancement. The distance should be close but not too close because of the strong energy losses below circa 5 nm. The optimal NP size depends on the material and the absorption cross section. For example, for an absorbing molecule with a total absorption cross section (which is σa multiplied by the number of molecules) of σa = 1 nm2, placed at a distance of 5 nm from a AgNP with a radius of ∼15 nm (and embedded in GaN), a maximum theoretical absorption enhancement of ∼35-fold can be reached (Fig. 3A). For σa = 100 nm2 (which would be more relevant for the development of detectors than for biosensing), the maximum absorption enhancement factor would be ∼10 for a = 20 nm and d = 5 nm.
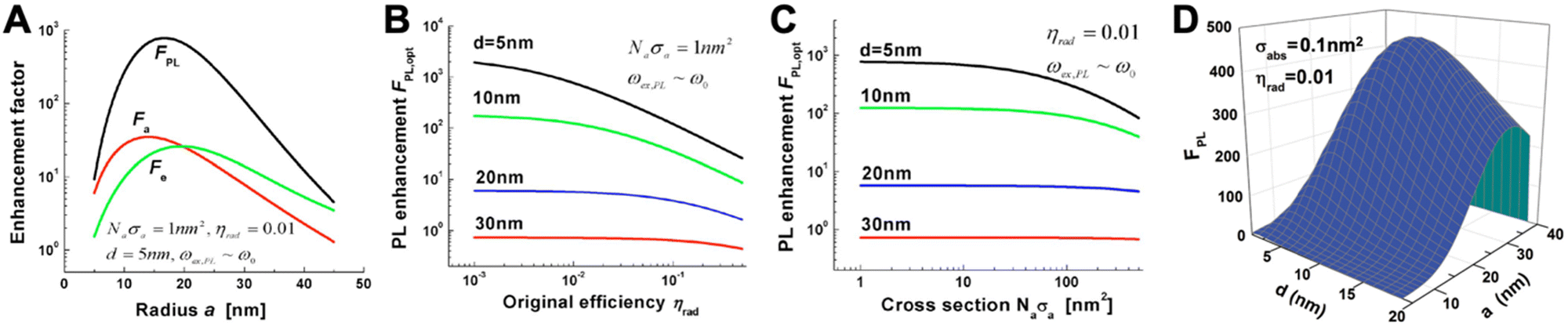 | ||
| Fig. 3 (A) Absorption (Fa-red), emission (Fe-green), and PL (FPL-black) enhancement factors as a function of metal sphere radius for a AgNP placed in GaN. The overall absorption cross section (Naσa) is 1 nm2, the initial PL quantum yield of the fluorophore (ηrad) is 1%, and the fluorophore–surface distance is 5 nm. Optical excitation and emission wavelengths are very close to the LSPR wavelength (ωex,PL = ω0). (B) Maximum PL enhancement (for optimized NP radius a) as a function of ηrad for different fluorophore–surface distances (d). (C) Maximum PL enhancement (for optimized NP radius a) as a function of Naσa for different d. (D) PL enhancement dependence on the NP radius (a) and on the fluorophore–surface distance (d) for a fluorophore with σabs = 0.1 nm2 and ηrad = 0.01 and a AuNP placed in air. The optical excitation is in resonance with the LSPR frequency ωex = ω0 = 2.562 eV and the emission is at ωPL = 2.462 eV. A to C reproduced with permission from ref. 41. Copyright 2009 The Optical Society D adapted with permission from ref. 37. Copyright 2012 American Physical Society. | ||
The emission of a fluorophore can also be enhanced by the strong local electric field (high LDS) caused by the LSPs. In this case, the LSPR scattering spectrum must overlap with the emission spectrum of the fluorophore. The radiative decay rate of a fluorophore is dependent on its environment. If a fluorophore is placed in a plasmon-enhanced electric field, the radiative decay rate can be enhanced, which means that the quantum yield (Φ or ηrad) increases and the radiative decay time (which is the inverse of the decay rate) decreases. This phenomenon is the so-called Purcell effect. Similar to absorption, emission enhancement is the best for inefficient emitters, i.e., fluorophores with initially small quantum yields. Taking the same example of a fluorophore placed at d = 5 nm from a AgNP embedded in GaN, a quantum yield of ηrad = 1% would lead to a maximum enhancement of ∼30 fold for a NP radius of ∼20 nm. The maximum overall PL enhancement would then be ∼800-fold for a NP with a radius of ∼17 nm (Fig. 3A). For all distances, the dependence of the enhancement factor on the quantum yield (Fig. 3B) is stronger than on the absorption cross section (Fig. 3C). Owing to the compromise between the antenna and cavity, the PL enhancement strongly depends on both NP size and fluorophore-NP distance. The NP should be large enough for strong radiative in and out coupling of the dipole mode (antenna) but also small enough to provide a small effective mode volume for sufficient Purcell enhancement (cavity). The distance must consider the nonradiative higher-order LSP modes that are confined at the NP surface (d < 5 nm). Fluorophores placed very close to the NP surface will couple to these modes and their energy will dissipate as metal losses. The overall result is shown in Fig. 3D for a AuNP placed in air and a fluorophore with an absorption cross section of 0.01 nm2 (corresponding to x fluorophores with an absorptivity of ∼260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/x M−1 cm−1 per fluorophore) and a PL quantum yield of 1%.
000/x M−1 cm−1 per fluorophore) and a PL quantum yield of 1%.
Again, these models are based on spherical NPs and small fluorophores and the experimental results (in particular for different metal nanostructures and QDs) may deviate from the ideal approximation. Still, the general guidelines for designing plasmonic-enhanced PL biosensors (using QDs and other fluorophores) are the following: (i) fluorophores with small absorption cross sections and low quantum yields (with higher impact for the quantum yield) exhibit the highest relative PL enhancement; (ii) close distances (below ∼5 nm) lead to strong PL quenching and should be avoided; (iii) the enhancement efficiency decays with an approximate d−3 distance dependence; (iv) the distance to be considered is the one between the surface of the metal NP and the center of the fluorophore; and (v) the distance for the maximum enhancement depends on the NP size. Similar to NSET quenching, these guidelines only provide a good starting point and experimental validation and optimization are necessary to design an efficient plasmon-enhanced PL biosensor.
3. Plasmonic metal nanostructures and quantum dots for fluorescence biosensing
Both plasmonic metal nanostructures and QDs have been widely used in combination with small molecular fluorophores to develop fluorescent biosensors. Before delving into their combination as metal–QD nanohybrids, here, we briefly review these more common small fluorophore-based plasmon-enhanced fluorescence (PEF) and energy transfer (NSET or FRET) biosensors and show some recent applications.3.1 Plasmonic metal nanostructures for fluorescence biosensing
PL quenching by plasmonic AuNPs is arguably the most applied approach for combining plasmonics and fluorescence within biosensing and Au NPs have been used as quenchers for many different fluorophores and for the detection of various biomolecules and biomarkers.4,9,13,14,24,39 However, also more sophisticated and advanced nanostructures and materials other than Au, such as Ag or nonnoble metals, have been used for biosensor development.9,28,50 Although FRET is most often cited when referring to energy transfer from fluorophores to AuNPs, NSET is the more probable mechanism.18 However, for the final objective of biosensor development based on efficient fluorescence quenching, the actual energy transfer mechanism is of minor importance. Significant advantages of using metal NPs for fluorescence quenching include the very broad LSPR bands (which can cover the PL spectra of various acceptors), the very high extinction coefficients (for high energy transfer efficiency), the relatively long distance range of up to circa 20 to 40 nm (NSET with a d−4 distance dependence), and the lack of PL (no background PL from acceptor emission). Because the LSPR spectrum of very large NPs is dominated by scattering rather than absorption, the application of smaller NPs (below circa 80 nm in diameter) with a stronger absorption component is recommended. The biological targets and biosensing methods are almost boundless, including many different types of immunoassays, protein-binding assays, nucleic acid hybridization assays, aptasensors, and environmental sensors, and we refer interested readers to the many review articles cited above for more information about those biosensing approaches.Owing to the more sophisticated design, including the competition of quenching and enhancement and the relatively small zone (distance to the NP) of efficient PL enhancement (cf. Section 2.3), PEF, also called metal enhanced fluorescence (MEF) or surface enhanced fluorescence (SEF), with metal NPs is much less developed than plasmon-quenched fluorescence.51–53 Discussions about PEF in review articles related to plasmon-enhanced biosensing are relatively short compared to SERS and FRET/NSET.4,9,24,39,54,55 In many cases, enhanced PL quenching by NSET to plasmonic NPs is also termed as plasmon-enhanced PL (even if the enhancement concerns enhanced quenching of PL), which can be a little confusing when searching for the right literature. Here, we use PEF only for enhanced (and not for quenched) PL.
Because efficient electric field enhancement is paramount for PEF, the use of sophisticated nanostructures, such as plasmonic gap nanostructures,56 or the combination of different metals with nonmetal high-refractive-index materials to mitigate the metal losses at distances close to the NP surface,28 can be highly beneficial for efficient local PL enhancement.50,57 While single-metal spherical NPs usually provide relatively moderate experimental PL enhancement factors (between approximately 1 and 30),38,39,51,58,59 the enhancement can be significantly improved by exploiting both quenching and enhancement (the overall enhancement is then calculated as the ratio of enhanced and quenched PL intensities),58,59 by modifying the shapes of metal NPs or adding metal or dielectric shells,38,60 or by designing plasmonic hotspots via plasmonic gap nanostructures.38,50,56 For example, DNA was used to specifically place fluorophores in the quenching (very close to the NP surface) or in the enhancement (beyond ∼10 nm) zones, such that the overall enhancement was between 40 and 100 fold.58,59 Concerning sophisticated design and material combination, we recently developed Au–Ag nanocuboids consisting of DNA-coated Au nanorods coated with a thin Ag shell. By labeling an Alexa Fluor 647 dye to the opposite end of the DNA and coating the DNA-nanocuboids with a silica shell, the dyes could be fixed at a distinct distance of ∼10 nm from the Au surface, which resulted in an ∼186-fold PL enhancement.60 The silica shell also served for bioconjugation with antibodies, such that the fluorescence-amplified nanocuboids (FANCs) could be applied for microarray-based quantification of the microRNA miR-134 in the concentration range of 100 aM to 1 pM with an estimated limit of detection (LOD) of 1 fM or 40 mol (Fig. 4). On the high end of PL enhancement, inter-particle gaps, NPs on mirror constructs, or other structures and combinations of metal NPs to design very high electric field enhancement in plasmonic gaps, could reach experimental enhancement factors of up to several thousands.38,56 Various fluorescence biosensors have been developed with these different PEF approaches.38–40,50,55–57
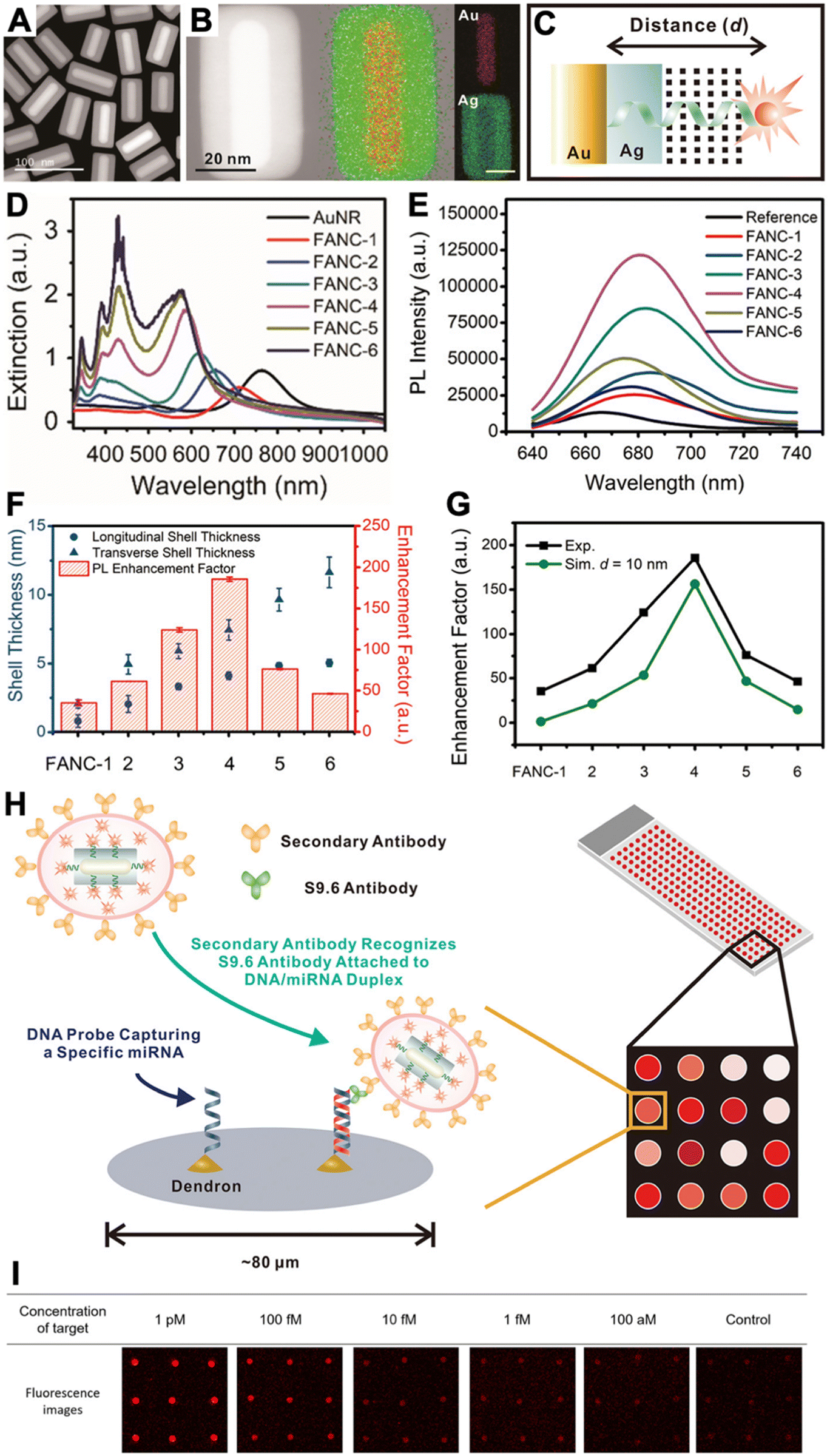 | ||
| Fig. 4 (A) Dark-field transmission electron microscopy image of FANCs. (B) Energy dispersive spectrometry elemental mapping of FANCs. (C) Schematic of dye-FANC distance tuning via the Ag shell and DNA. (D) Absorption spectra of AuNRs and FANCs with different shell thicknesses. (E) Fluorescence spectra (λex = 620 nm) of dye (reference) and different FANCs, showing maximum dye fluorescence amplification for FANC-4. (F) Shell thickness-dependent EHFs for the different FANCs. (G) Comparison of experimental and simulated EHFs at d = 10 nm. (H) Schematic principle of a microarray-based microRNA (miRNA) assay, in which secondary antibody-conjugated FANCs bind to miRNA-induced DNA/RNA duplex-binding S9.6 antibodies on a microarray spot. (I) Analysis of the microarray fluorescence images resulted in miRNA quantification in the 100 aM to 1 pM concentration range. Reproduced with permission from ref. 60. Copyright 2021 American Chemical Society. | ||
3.2 Semiconductor quantum dots for fluorescence biosensing
Although many luminescent NPs are available,7 QDs have arguably been most applied for biosensing and bioimaging and the same counts for energy transfer based applications. Several recent reviews have discussed the theory and applications of such QD and QD-FRET based biosensors.12,21,61–64 Advantages of QDs compared to other fluorophores include their narrow and symmetric PL bands, broad and strong absorption, color tunability by both size and material, high quantum yields and brightness, high chemical, physical, and photo stability, versatility of surface functionalization strategies, and commercial availability.QDs can be used as both FRET donors and acceptors.21 The QD donor approach is relatively straightforward because QDs can be excited at any wavelength shorter than their emission spectrum and their emission can be tuned in order to well overlap with the absorption of most fluorescent dyes or proteins. Thus, excitation can be performed at a wavelength that does not directly excite the acceptor and the spectral overlap can be optimized for efficient FRET and for the minimum overlap of the QD and acceptor PL. The QD acceptor approach is a little more complex because of the spectrally broad and strong absorption of QDs. This condition makes it impossible to find a conventional fluorescent donor, whose PL spectrum overlaps with the QD absorption and that can be excited at a wavelength, which does not simultaneously excite the QD. Thus, for a FRET experiment both the fluorescent donor and the QD acceptor get excited by the excitation light source and FRET, which requires an excited-state donor and a ground-state acceptor, cannot occur. Nevertheless, several ways to overcome this QD-FRET-acceptor dilemma have been proposed, including the use of luminescent lanthanide donors with extremely long excited-state lifetimes (while the lanthanide donor remains in its excited state, directly excited QDs can decay back to the ground state, after which lanthanide-to-QD FRET can occur),65 the use of bioluminescent donors (no direct QD excitation by light),66 the use of a very high excess of fluorophore donors (increased probability that the QD acceptor directly decays back to the ground state before all donors),67 and the use of upconversion NPs (excitation in the NIR does not directly excite the QD).68
Many different biosensors have been developed with QDs as FRET donors and/or acceptors and their special photochemical, photophysical, and nanomaterial properties provided unique FRET concepts, including spectral (different QD colors) and temporal (different distances between QDs and the interacting FRET partner) multiplexing,61,69 concentric FRET (for which several donors or acceptors are positioned around a central QD),70 FRET photonic wires,71,72 and long-lifetime FRET in vivo probes.73 One important feature of QDs is their relatively large surface area, which allows for interaction with many alike or different biomolecules and FRET partners. In a recent example, we exploited the nanometric surfaces of QDs for the development of a new type of background-free time-gated immunoassay that required only a single FRET-donor labeled antibody and a QD with a thin organic ligand coating (Fig. 5).74 A small genetically engineered antibody with a size of approximately 15 kDa (nanobody) and specific for the epidermal growth factor receptor EGFR was labeled with a terbium complex (Tb). A hexahistidine (His6) tag on the opposite end of the EGFR binding site of the nanobody allowed for efficient polyhistidine-metal affinity mediated self-assembly to a 625 nm emitting core–shell QD, which served as a FRET acceptor for the Tb FRET donor. This Tb-to-QD FRET was disrupted by the presence of EGFR, which bound to the nanobody and displaced it from the QD due to steric hindrance. The resulting FRET signal-decrease with increasing EGFR concentration was used for the quantification of EGFR in the sub to low nanomolar concentration range and with a LOD of ∼80 pM. The wash-free displacement assay required only a single Tb-labeled nanobody and a non-bioconjugated QD, which made this assay approach significantly simpler compared to conventional immunoassays.
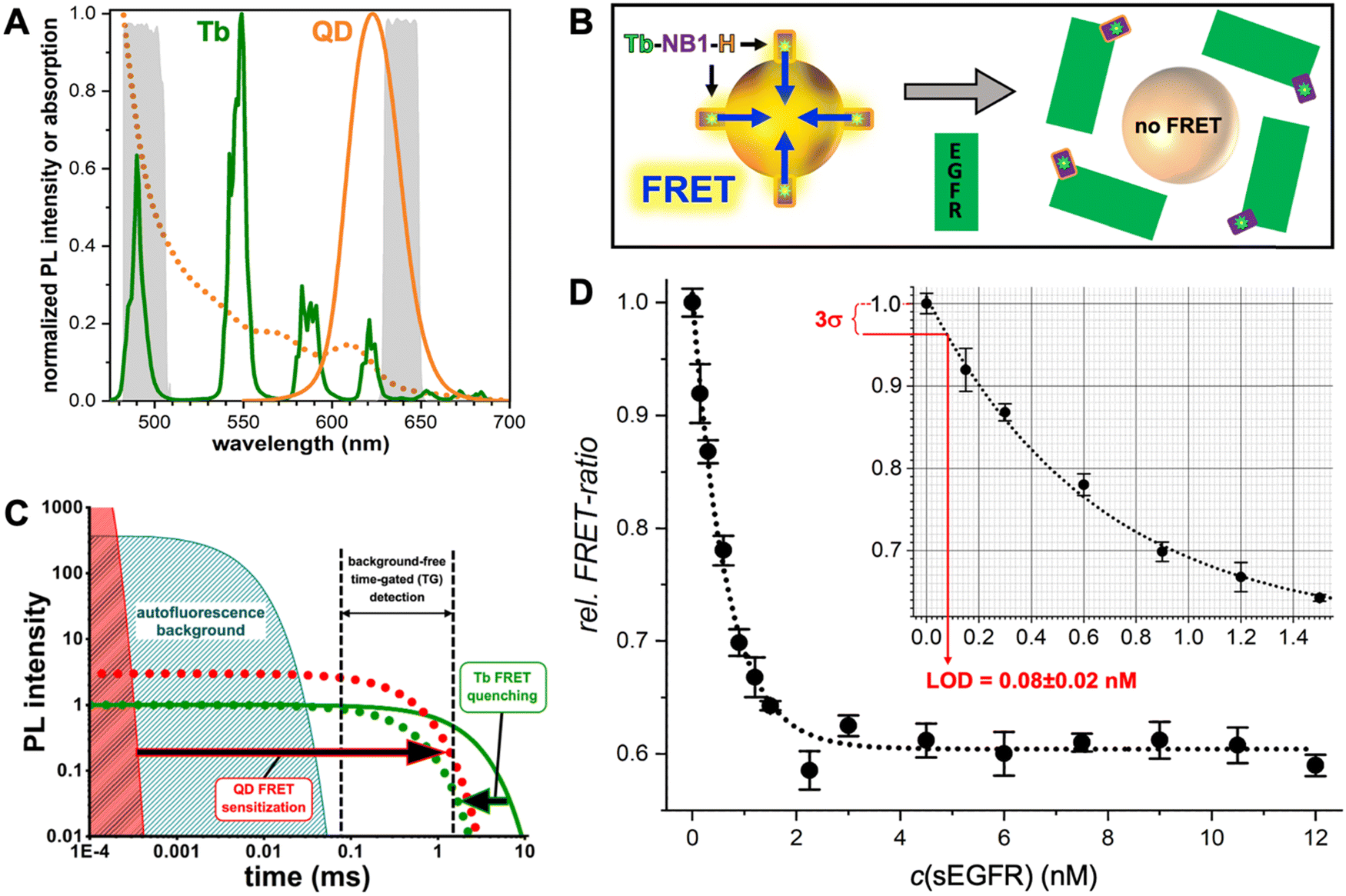 | ||
| Fig. 5 (A) PL spectra of the Tb3+ complex (Tb-maximum excitation wavelength λex = 339 nm) donor and the QD acceptor used for the single-nanobody displacement FRET immunoassay (principle shown in B). The dotted line shows the QD absorption spectrum that overlaps with the Tb PL spectrum for efficient FRET. The gray spectra in the background present the spectral ranges of the Tb and QD detection channels. (B) A Tb-labeled nanobody (Tb-NB1), attached to the surface of the core–shell QD via polyhistidine (H) mediated self-assembly to the ZnS shell, is displaced by EGFR binding to the nanobody. Despite the opposite ends of the EGFR binding site and hexahistidine tag on the nanobody, the larger sizes of EGFR and QDs compared to the nanobody resulted in successful nanobody displacement and a concomitant disruption of FRET. (C) In Tb-to-QD TG-FRET immunoassays, FRET from Tb with a very long (ms) PL lifetime (green line) is transferred to the QD with a short (ns) PL lifetime (red area), such that both have the same long (ms) lifetime after FRET (dotted lines). Pulsed excitation and TG intensity detection after the decay of directly excited QDs and autofluorescence background results in background-free biosensing. (D) The displacement assay calibration curve showed decreasing FRET signals with increasing EGFR concentrations. EGFR could be quantified in the sub to low nanomolar concentration range with an LOD of 80 ± 20 pM. (A–D) Reproduced with permission from ref. 74. Copyright 2022 The Authors (CC BY-NC 4.0). Angewandte Chemie International Edition published by Wiley-VCH GmbH. | ||
4. Plasmon-quenched fluorescence (PQF) in metal–QD nanohybrids
Similar to PQF (i.e., NSET) of conventional fluorophores (cf. Section 3.1), PQF of QDs via metal nanomaterials has been largely dominated by AuNPs. Many review articles that discuss and summarize biosensing via NSET from different fluorophores (including QDs) to AuNPs13,14,75–80 and, in particular, from QDs to AuNPs18,21,61,81–84 have been published over the last 10 years. Recently, we also reviewed the actual energy transfer mechanism and the use of both terms (NSET or FRET) within different bioanalytical studies using QDs and other fluorophores as donors.18 Energy transfer to fluorescent Au nanoclusters was also characterized in detail and in addition to NSET, nanovolume energy transfer (NVET) was proposed as a possible mechanism.85,86 Considering that numerous biosensing applications using QD-to-AuNP NSET have already been reviewed in detail (cf. review papers cited above), we limit this section to the discussion of a few representative and interesting recent examples, in which metal–QD nanohybrids were used for NSET biosensing.In one recent study, Yoo et al. developed a rather unconventional NSET strategy, which employed target-specific in vitro and in vivo click chemistry for switching QD PL.87 The NSET system consisted of AuNPs (diameter ∼27 nm) as acceptors and CdSSe/ZnS QDs (λem ∼ 520 nm) as donors (Fig. 6A). AuNPs were surface-functionalized with two different thiol-PEG chains, one containing a terminal azide (N3) and the other containing a terminal carboxylate (AuNP@PEG). An additional methoxy PEG chain terminated with a matrix metalloproteinase-2 (MMP-2)-cleavable peptide (mPEG-MMP CP) was attached to the PEG-carboxylate, resulting in shielding of the N3via the steric hindrance of the long mPEG-MMP CP chains (AuNP@[PEG]2). QDs were surface-functionalized with dibenzocyclooctyne (DBCO) for copper-free click chemistry with the N3 on the AuNPs. In the absence of the MPP-2 target (Fig. 6B(i)), a mixture of QD-DBCO and AuNP@[PEG]2 showed strong QD PL (PL on) because the azide was not accessible for the DBCO and thus, QDs and AuNPs were separated. In the presence of MPP-2 (Fig. 6B(ii)), the mixture resulted in QD PL quenching (PL off) because MMP-2 cleaved mPEG-MMP CP, such that the N3 became available for binding to QD-DBCO, which brought QDs and AuNPs in a close proximity for NSET. High concentrations of MMP-2 resulted in the formation of AuNP-QD clusters (Fig. 6C) that further reduced the QD PL intensity. First, the MMP-2 detection principle was shown to be functional in vitro, for which the QD PL intensity quenching was proportional to the number of cells. Afterwards, also in vivo compatibility (on mice) was demonstrated via tumor size-dependent QD PL quenching (Fig. 6D). The authors claimed that larger tumors have higher MMP-2 concentrations and that their metal–QD nanohybrids may become a useful tool for tumor progression prediction.
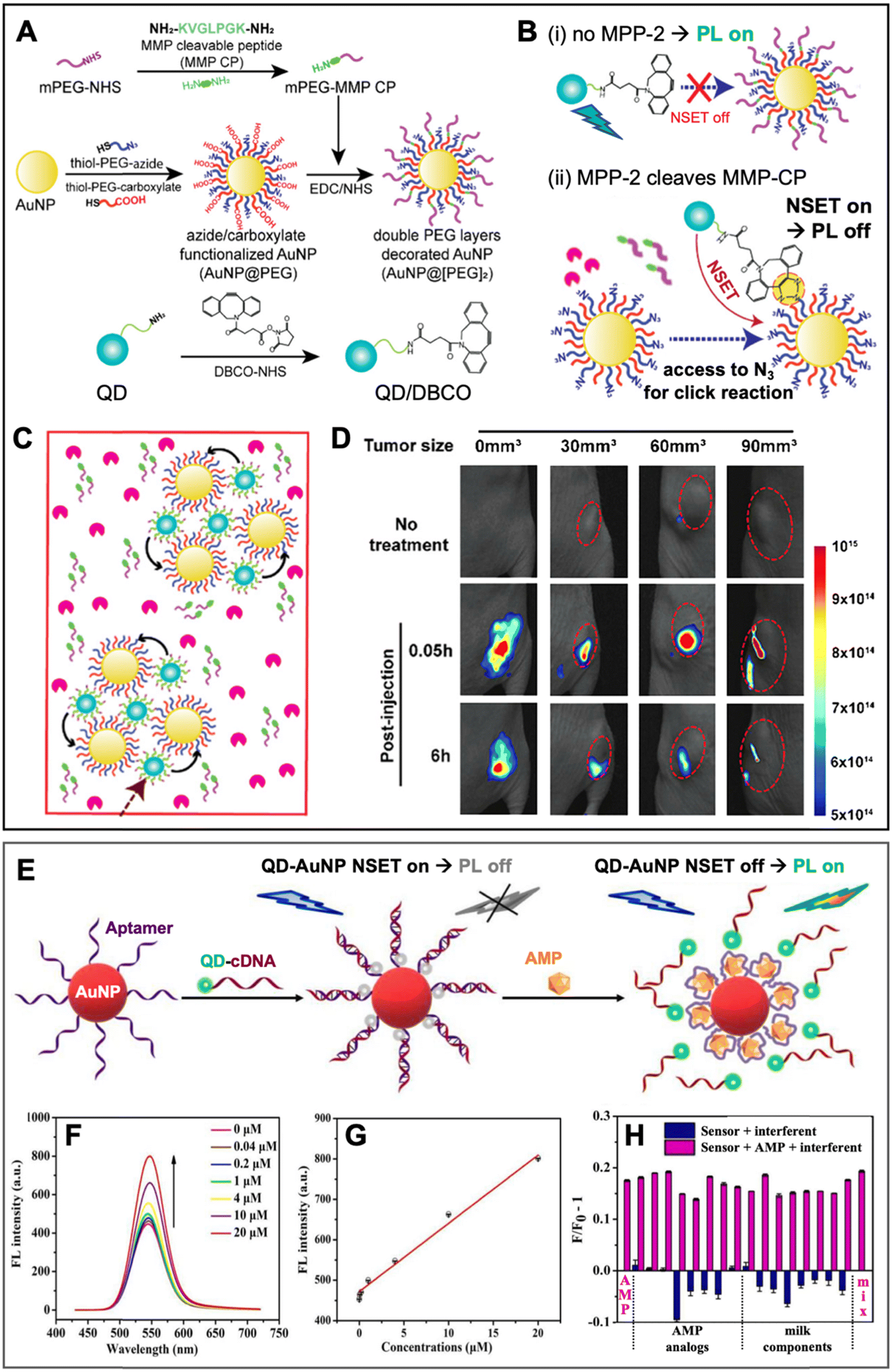 | ||
| Fig. 6 Examples of metal–QD nanohybrids for NSET biosensing. (A) Schematic of the preparation of MMP-2-responsive AuNPs (AuNP@[PEG]2) functionalized with a shorter PEG-N3 chain and a longer PEG-peptide (MMP-CP) chain and of DBCO-functionalized QDs (bottom). (B) Schematic of the MM2-2 detection principle. (i) Without MMP-2, the N3 group on the AuNPs is shielded, AuNPs and QDs are spatially separated, and QDs can emit PL. (ii) Cleavage of the MMP-CP and copper-free click reaction between DBCO-QDs and AuNPs by MMP-2 becomes possible, leading to NSET and QD PL quenching. (C) High concentrations of MMP-2 result in the formation of QD–AuNP clusters. (D) In vivo PL imaging (excitation and emission wavelengths were not mentioned for these experiments) of mice bearing tumors of different sizes (0 mm3 corresponds to no tumor) without injection (no treatment) and with injection of AuNP@[PEG]2 and QD/DBCO in the tumors. Whereas the normal tissue did not result in PL reduction over time, the tumors show size-dependent PL reduction over time due to MMP-2-mediated QD–AuNP cluster formation (cf. C). A to D adapted with permission from ref. [87] Copyright 2022 American Chemical Society. (E) Schematic principle of a QD–AuNP NSET aptasensor for AMP quantification. AMP-aptamer binding displaced QD-cDNA from the AuNP, which resulted in PL recovery (NSET off, PL on). (F) Increasing AMP concentrations resulted in increasing QD PL intensity (λex = 400 nm). (G) AMP assay calibration curve (maximum QD PL intensities over AMP concentration). (H) Selectivity of AMP assay tested against different AMP analogs and milk components. E to H adapted with permission from ref. 88. Copyright 2022 Elsevier B.V. | ||
Yang and Dai et al. applied QD–AuNP NSET for ampicillin (AMP) sensing in liquid samples.88 Their metal–QD nanohybrids comprised AuNPs (diameter ∼15 nm) surface-functionalized with DNA aptamers against AMP and CdTe QDs (diameter ∼4 nm, λem ∼ 530 nm) surface-functionalized with complementary DNA (cDNA) (Fig. 6E). In the absence of AMP, QD-cDNA hybridized to the AuNP-aptamer and the close QD–AuNP distance resulted in NSET and concomitant quenching of QD PL. AMP-aptamer binding (in the presence of AMP) displaced QD-cDNA, which disrupted NSET and led to AMP-dependent QD PL recovery in the concentration range from circa 0.04 to 20 μM (Fig. 6F and G). The AMP sensor showed relatively good selectivity for AMP, though most AMP analogs (oxytetracycline, chlortetracycline, vancomycin hydrochloride, and neomycin sulfate) or components found in milk (L-tryptophan, L-cysteine, glutamic acid, glucose, Ca2+, Cl−, and Mg2+) resulted in additional QD quenching, whereas a mixture of all interferents resulted in a slightly increased PL signal (Fig. 6H). Despite these interferences, AMP could even be quantified in 3-fold diluted, filtered, dried, and reliquified AMP-spiked milk samples with concentration recovery rates between 88 and 99%. Such relatively quick and simple PL sensors have the potential to replace more complicated or time-consuming analytical techniques, such as high-performance liquid chromatography (HPLC) or enzyme-linked immunosorbent assays (ELISA).
A similar aptamer-based PL turn-on NSET sensor was recently proposed by Yüce et al. for the quantification of bacteria (E. coli and S. typhimurium) in liquid samples.89 They used aptamer-functionalized Au nanorods (AuNRs, ∼25 nm × 46 nm) and cDNA-functionalized QDs (λem ∼ 514 nm), which were disassembled by E. coli binding to the aptamer (Fig. 7A). The E. coli concentration-dependent PL intensity recovery of the QDs (Fig. 7B) was functional in the concentration range from ∼10 to 106 CFU mL−1 with an LOD of ∼5 CFU mL−1. By combining the AuNR-QD E. coli NSET probe with a nanohybrid NSET probe for S. typhimurium that consisted of nanourchins (AuNU) and upconversion nanoparticles (UCNP), duplexed quantification (Fig. 7C) became possible via 350 nm excitation combined with 620 nm emission detection for AuNR-QD and 980 nm excitation combined with 545 nm detection for AuNU-UCNP. The simultaneous quantification of pathogens is of potential interest for food analysis and biomedical diagnostics.
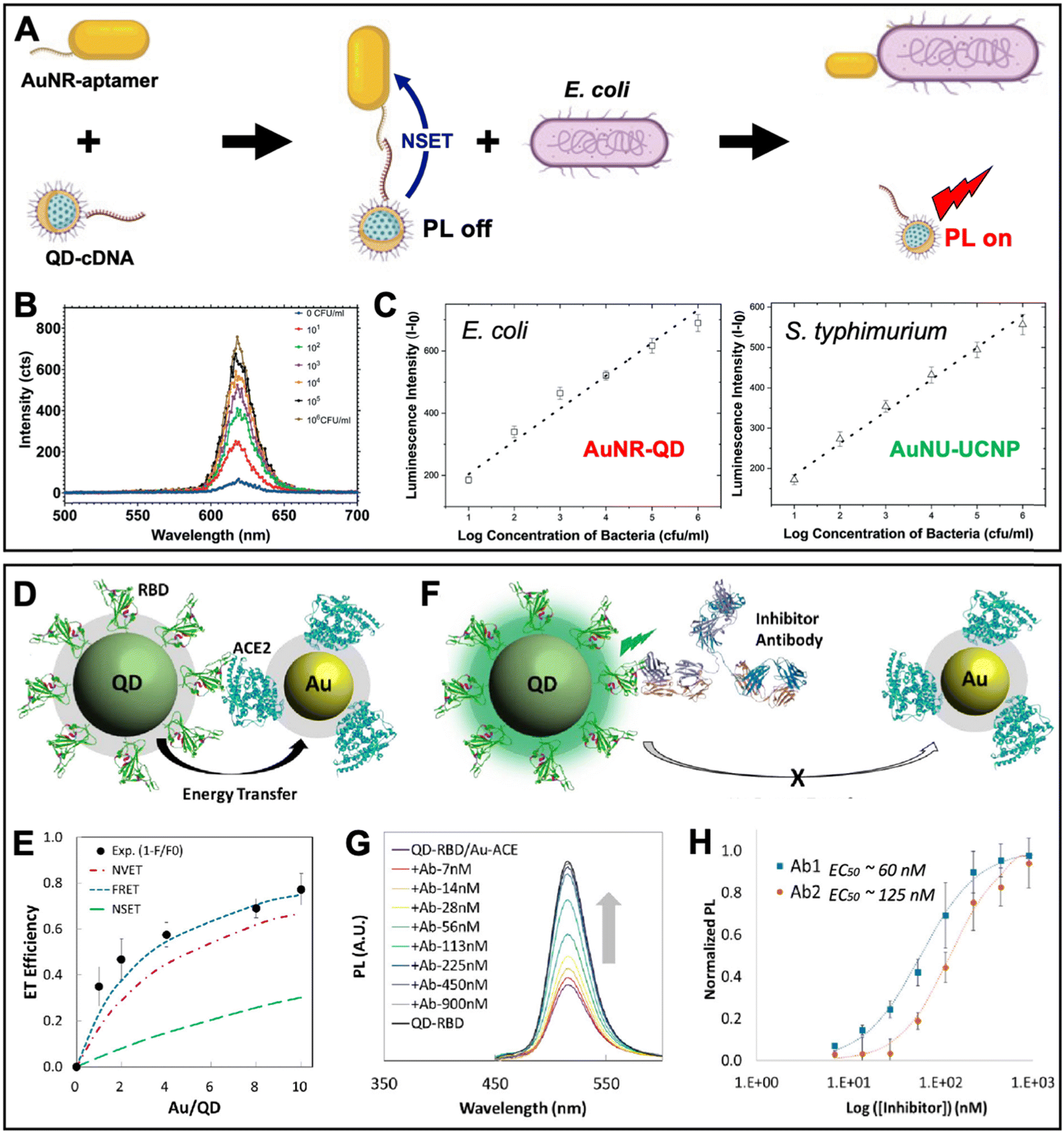 | ||
| Fig. 7 Examples of metal–QD nanohybrids for NSET biosensing. (A) Schematic principle of a QD–AuNR NSET aptasensor for pathogen (E. coli) quantification. E. coli-aptamer binding displaced QD-cDNA from the AuNR, which resulted in PL recovery (NSET off, PL on). (B) Increasing E. coli concentrations resulted in increasing QD PL intensity (λex = 350 nm). (C) Combination of AuNR-QDs (λex = 350 nm; λem: peak intensities at 620 nm) with AuNU-UCNPs (Au nanourchins and upconversion nanoparticles – λex = 980 nm and λem: peak intensities at 545 nm) can be used for duplexed (dual color) quantification of E. coli and S. typhimurium. A to C adapted with permission from ref. 89. Copyright 2022 The Authors. Published by the Royal Society of Chemistry (Creative Commons license CC-BY-NC 3.0). (D) Schematic diagram of the QD-RBD to AuNP-ACE2 NSET assay for analyzing the SARS-CoV-2 Spike protein to host cell transmembrane enzyme binding. (E) Assembly of several AuNPs around a single QD increased the ET efficiency. Experimental data and photophysical assumptions gave a best fit to the FRET model. (F) Schematic principle of an inhibition assay in which a neutralizing antibody displaced QDs and AuNPs (NSET off, PL on). (G) Increasing antibody concentrations resulted in increasing QD PL recovery (λex = 395 nm). (H) Inhibition assay calibration curves for two different neutralizing antibodies (Ab1 and Ab2). D to H adapted with permission from ref. 90. Copyright 2020 American Chemical Society (Creative Commons license CC-BY-NC-ND 4.0). | ||
Considering the maturity of QD-to-AuNP NSET biosensing, it is not surprising that the approach was also used to study the SARS-CoV-2 coronavirus infection. Gorshkov and Oh et al. investigated the binding of the SARS-CoV-2 Spike protein S1 subunit receptor binding domain (RBD) to the angiotensin converting enzyme 2 (ACE2 – a transmembrane enzyme expressed on the plasma membrane surface of SARS-CoV-2 host cells) (Fig. 7D).90 This study is one of the rare examples, in which relatively small AuNPs (diameter ∼6 nm) were used, such that several of them could be assembled around a larger ZnSe/CdZnS/ZnS QD (diameter ∼8.2 nm, λem ∼ 514 nm). Increasing the number of acceptors around a single donor in a centrosymmetric donor–acceptor system increases the ET efficiency, which was experimentally confirmed and resulted in a maximum efficiency of ∼80% (Fig. 7E). Surprisingly, the authors found that under their experimental conditions and photophysical estimations the FRET model provided the best fit to the experimental data, followed by NVET and NSET. While a detailed analysis of the ET was not the major aim of this study, the result demonstrated that using the number of acceptors as the only variable within an otherwise constant system (the same donor–acceptor distance) is not necessarily well suited to determine the ET mechanism. More importantly, the ET experiments confirmed the binding of RBD on the QDs to ACE2 on the AuNPs, which was utilized for analyzing the inhibitory activity of neutralizing antibodies (Fig. 7F) via the antibody concentration-dependent recovery of QD PL (Fig. 7G). Half-maximal effective concentrations (EC50) of 60 nM and 125 nM were found for SARS-CoV-2 (Ab1) and RBD (Ab2) specific antibodies, respectively (Fig. 7H). Such NSET sensors provide the potential for simple and rapid biochemical screening of drugs, antibodies, or other biologics to prevent SARS-CoV-2 infection. Notably, the QD-RBD conjugates (but without Au NPs) were also used for live-cell imaging of RBD-ACE2 binding, endocytosis, and their inhibition by neutralizing antibodies.
5. Plasmon-enhanced fluorescence (PEF) in metal–QD nanohybrids
PEF of QDs was experimentally demonstrated more than 20 years ago91,92 and since these initial proofs of concept, many different metal–QD hybrid nanostructures, including various materials and architectures, have been developed for PEF of QDs (Tables 1–3). As the PEF of QDs is not a specific research field, various different acronyms (including PEF, MEF, and SEF), names for QDs (including quantum dots, nanocrystals, and semiconductor NPs), and the term “enhancement” for both PL intensity and lifetime quenching and increase have been used, and some studies did not investigate enhancement but rather reduced blinking or increased QD stability, it is not simple to find all literature related to PEF of QDs. While we believe that our review provides a thorough overview of the field, we apologize to those authors, whose work is not presented here. Although many different approaches have been developed, the large majority of metal–QD nanohybrids focused on Au and Ag nanostructures, CdSe/ZnS QDs, and the distance control between plasmonic NPs and QDs via surface-based layers separated via SiO2 spacers. Metal nanorods were used to exploit both the transversal and longitudinal LSPR, metal NP multimers or sophisticated nanostructures were employed to create plasmonic hotspots, both absorption and emission of QDs were enhanced, the distance dependence of PEF was investigated, single QDs and ensemble measurements were performed, PL intensity and PL lifetime enhancement were scrutinized, and enhancement factors (EHFs) between a few percent up to several orders of magnitude were found. In general, as expected from the theoretical assumptions (vide supra), plasmonic hotspot architectures and QDs with relatively low initial PL quantum yields resulted in the strongest enhancement. Nevertheless, the almost unlimited possibilities of materials, sizes, shapes, distances, photophysical properties, and experimental conditions have resulted in a large diversity of results and despite the many experimental and theoretical studies, a clear prediction of the ideal metal–QD hybrid nanostructure for PEF is not possible. This lack of prediction is most probably one of the major reasons why the PEF of QDs has only been rarely used for biosensing.| Year | Plasmonic nanomaterials | QDs | Max. PL (intensity) enhancement factor | Max. Purcell (lifetime) enhancement factor | Comments | Ref. |
|---|---|---|---|---|---|---|
| 2002 | Au NPs (∅ 12–15 nm) a monolayer with polyelectrolyte spacers to QDs | CdSe/ZnS (∅ ∼ 8 nm); λem ∼ 645 nm | ∼3 | — | Distance dependence was investigated | 91 |
| 2002 | Etched Au surface (with 10–50 nm valleys and peaks) | CdSe/ZnS; λem ∼ 600 nm | ∼5 | ∼1400 | Single QD measurements; reduced QD blinking | 92 |
| 2005 | Ag nanodisks (∅ ∼ 160 nm, height ∼50 nm) | CdSe/ZnS (doped in PMMA in between the Ag islands), λem ∼ 655 nm | ∼52 | ∼10 | Quantum yield enhancement also estimated | 98 |
| 2006 | Ag nanoislands with a PVA spacer (∼10 nm) to QDs | CdTe; λem ∼ 660 nm | ∼5 | ∼3 | Single QD measurements; reduced QD blinking | 101 |
| 2006 | Au nanoprisms (∼200 nm edge length and ∼40 nm thickness) | CdSe/ZnS doped in the PMMA matrix as a spacer; λem ∼ 580 nm | ∼33 | — | — | 102 |
| 2006 | 3 Monolayers of Au NPs (∅ ∼ 7 nm) coated with a PE layer-by-layer spacer (∼1.4 to 11.7 nm thickness) and capped by QD monolayers | CdTe (λem ∼ 556, 612, 667, and 757 nm) | ∼10 | ∼1.9 | Distance dependence and QY enhancement (23 fold) investigated; QD emission redshifted from the LSPR band are best enhanced; red shift and broadening of QD emission | 103 |
| 2007 | Au nanoprisms (∼100 nm and 200 nm edge length) and Au nanocylinders (∅ ∼ 100 nm and 200 nm) with ∼35 nm thickness | CdSe/ZnS QDs (∅ ∼ 5 nm) and CdSe nanorods (∼10 nm × 3 nm) doped in the PMMA matrix as a spacer; λem ∼ 580 nm (QDs) and ∼ 567 nm (nanorods) | ∼30 (QDs) | — | Distance dependence was investigated | 104 |
| ∼19 (nanorods) | ||||||
| 2007 | Ag nanoislands (∅ ∼ 100 nm) coated with the SiO2 spacer (∼10 nm) coated with drop cast QD films. | CdSe/ZnS (core ∅ ∼ 1.9 nm); λem ∼ 492 nm | ∼15.1 | — | Red shifted QD emission | 105 |
| 2008 | Ag nanoprisms (∼100 nm edge length), Au NPs (∅ ∼ 80 and 100 nm), or Ag cubes (∼50 nm) overcoated with a thin QD-doped PMMA layer | CdSe/CdS/CdZnS/ZnS (λem ∼ 625 nm) | ∼10 | — | Enhancement depends on the plasmonic structure (Ag nanoprisms enhance best); excitation enhancement contributes more than emission enhancement | 106 |
| 2009 | Patterns of Au NPs (∅ ∼ 18 nm) or Ag nanoprisms (∼100 nm edge length and ∼12 nm thickness) on GaAs with polymer layer-by-layer spacers to QDs | CdSe (∅ ∼ 4 nm and ∼5.5 nm); λem ∼ 585 nm and ∼640 nm | ∼2 | — | Distance dependence was investigated | 107 |
| 2009 | Au nanodisk (∅ ∼ 50, 100, or 200 nm, height ∼50 nm) array (interdisk distance ∼50, 100, 150 or 200 nm) coated on a Si wafer and streptavidin/biotin spacer layers (∼9, 16 or 20 nm) | CdSe/ZnS (λem ∼ 605 nm) coated with streptavidin | ∼15 | — | Biomolecules (streptavidin) used for nanohybrid assembly; distance dependence (lateral and vertical) was investigated; EHF calculated relative to QDs on a planar Au substrate | 93 |
| 2010 | QDs sandwiched between Au nanodisks (see ref. 93) and Au NPs (∅ ∼ 10 nm) using streptavidin/biotin spacer layers (∼10.5 or 17.5 nm) | CdSe/ZnS (λem ∼ 605 nm) coated with biotin | ∼15 | — | Similar study (the same group) to ref. 93; surface roughness was investigated | 94 |
| 2010 | Au film (∼200 nm thickness and ∼2.2 nm roughness) coated with the SiO2 spacer layer (∼18 to 300 nm) coated with QDs. | CdSe/ZnS (λem ∼ 565 nm) | ∼2.4 | ∼6.1 | Distance dependence was investigated; single QD experiments | 108 |
| 2010 | Au NPs (∅ ∼ 25, 45, 65, and 85 nm) annealed on a glass slide and coated with a QD-doped PMMA layer | CdSe (λem between ∼532 and 612 nm) | ∼10 | — | Enhancement depends on excitation (Au NP) and emission (QD) wavelengths and Au NP size (small one quenches due to stronger absorption and large one enhances due to stronger scattering) | 109 |
| 2010 | Ag nanoprisms (∼50 to 100 nm edge length and ∼10 to 15 nm thickness) deposited (drop casting) on QD monolayers | CdSe/CdS/ZnS (λem ∼ 598 nm and ∼625 nm) and CdSe/ZnS (λem ∼ 550 nm) | ∼1.6 | ∼10 | Enhancement depends on emission (QD) and scattering (Ag) wavelengths (overlap), Ag scattering intensity, and QD quantum yields | 110 |
| 2011 | Ag nanoprisms (∼50 to 100 nm edge length and ∼10 to 15 nm thickness) deposited (drop casting) on QD monolayers | CdSe/CdS/ZnS (λem ∼ 625 nm) | ∼1.9 | ∼6.1 | Enhancement depends on excitation wavelength; QD quantum yields are considered for rate enhancement calculation | 111 |
| 2011 | Ag nanoislands (∅ ∼ 50 nm, thickness ∼37 nm) coated with the SiO2 spacer (∼10 nm); QDs spin coated on top | CdSe/ZnS (λem ∼ 625 nm) | ∼160 | — | Up to 240-fold enhancement in hotspots | 112 |
| 2012 | Ag nanorods (∅ ∼ 50 nm, length ∼197 nm) on the Si wafer coated with the PMMA layer; QDs spin coated on top | CdSe/ZnS (λem ∼ 515 nm) | ∼6 | ∼6.4 | PL intensity enhancement was determined against PL intensity on a smooth Ag surface; PL lifetime enhancement against QDs in solution | 113 |
| 2012 | Wormlike Au nanoislands (∼100 to 400 nm length) coated with QD-doped PMMA | CdTeSe (∅ ∼ 6.7 nm, λem ∼ 580 to ∼765 nm) | ∼4 | — | — | 114 |
| 2012 | Au disk (∅ ∼ 90 nm; thickness ∼40 nm) monomers or dimers (∼14 or 30 nm gap size); QDs attached via BSA (∅ ∼ 8 nm) | CdSe/CdS/ZnS (∅ ∼ 10 nm, λem ∼ 610 nm) | ∼2.7 | ∼11.1 | Single QD measurements; excitation enhancement (∼16 fold) competed with emission quenching | 97 |
| 2012 | Ag films (∼60 nm thick) composed of densely packed Ag NPs (∅ ∼ 50 nm). QD layer (∼50 to 100 nm) coated above. | CdSe (∅ between ∼2.1 and 3.1 nm); λem between ∼479 and 560 nm | ∼16.5 | ∼0.56 | Enhancement depends on the QD emission band (surface state or band-edge emission), QD size, and sample annealing; PL lifetime increases for plasmon-enhanced emission | 115 |
| 2012 | Au NP clusters (∼50 nm to 1 μm) annealed on a glass slide from ∼50 nm Au NPs. Polymer layer (∼1 μm thick with embedded QDs coated on top. | CdSe/ZnS; λem ∼ 655 nm | ∼3 | — | Competition between excitation and emission enhancements experimentally shown | 116 |
| 2013 | AuNPs (∅ ∼ 20 nm) inside polymer NPs (∅ ∼ 150 nm) co-doped with Si QDs. | Si QDs (∅ ∼ 5 nm) | ∼15 | — | Different AuNP doping concentrations investigated; composite polymer NPs used for inkjet printing | 117 |
| 2013 | Ag NPs (∅ ∼ 40 nm) on a glass slide and coated with the Al2O3 spacer layer (∼2 to 20 nm) | CdSe (∅ ∼ 5.5 nm); λem ∼ 590 nm | ∼24 | Lifetime not significantly shortened | Distance and excitation wavelength dependence were investigated | 118 |
| 2014 | Nanoporous Au films (pore size ∼7 to 71 nm) with streptavidin (on QDs) as the spacer | CdSe/ZnS (∼10 nm × 5 nm and ∼4 nm × 3 nm); λem ∼ 605 nm and 525 nm | ∼100 (QD525), ∼70(QD605) | — | Single QD measurements | 119 |
| 2014 | Au NP (∅ ∼ 15 nm) layers coated with the TiO2/SiO2 spacer (∼7 to 65 nm) for changing the dielectric constant | CdSe/ZnS (∅ ∼ 4.5 nm); λem ∼ 608 nm | ∼3.4 | — | Distance and dielectric constant dependence were investigated | 120 |
| 2014 | Vertically aligned Au nanorods (∅ ∼ 40 nm, length ∼101 nm) with ∼7.7 nm edge-to-edge distance; SiO2 spacer (∼5 to 50 nm) and QD monolayer on top | CdSe/ZnS (core ∅ ∼ 8 nm); λem ∼ 614 nm | ∼10.4 | ∼4.6 | Distance dependence was investigated; experimental and theoretical comparison | 100 |
| 2015 | Au NPs (∅ ∼ 5 nm) coated on the sides of polymer nanocylinders (thickness ∼10 nm) | CdSe/ZnS in polymer nanocylinder arrays; λem ∼ 605 nm | ∼7 | — | Nanocylinders of different diameters were investigated | 121 |
| 2015 | Au films with an ∼1 nm PAH layer at the bottom and Ag nanocubes (∼75 nm) with ∼3 nm PVP coating on top; QDs in between | CdSe/ZnS (∅ ∼ 6 nm); λem ∼ 625 nm | ∼2300 | ∼880 | Single QD measurements | 122 |
| 2015 | Au nanocones (∼125 nm height and ∼160 nm base ∅) | CdSe/ZnS with an ∼3 nm organic ligand and thiol layer; λem ∼ 650 nm | ∼5 | ∼7.6 | Single QD measurements; blinking of QDs on cone tips was observed | 123 |
| 2015 | Au NPs (∅ ∼ 9 nm) in a ZnO film (∼30 nm) coated on a QD layer | CdSe/ZnS QD layer (∼20 nm); λem ∼ 550 nm | ∼4.5 | — | — | 124 |
| 2015 | Thermally treated crystalline Ag films (∼30 to 200 nm thick); QDs directly spincoated on the films | CuInZnS (∅ ∼ 10 nm); λem ∼ 630 nm | ∼45 | ∼1.7 | Distance dependence was investigated | 125 |
| 2015 | Ag nanoboxes (∼100 nm × 100 nm × 35 nm); between quantum wells and QDs in the PMMA layer | InGaN quantum well FRET donors (∼2 nm thick, λem ∼ 516 nm) and CdSe/ZnS QD (∅ ∼ 6.3 nm, λem ∼ 650 nm) acceptors in an ∼80 nm PMMA layer | ∼1.6 (QD enhancement) | — | FRET enhancement and quenching were investigated by PL intensity and lifetime | 126 |
| 2016 | Ag NP (∅ ∼ 51 or 72 nm) monolayers coated with the PMMA spacer (∼44 nm); QDs spin-coated on top | PbS and PbS/CdS (∅ ∼ 5 nm); λem between ∼1100 and 1500 nm | ∼2.8 | ∼1.4 | Excitation wavelength dependent enhancement; excitation enhancement dominates emission enhancement | 127 |
| 2016 | Ag nanotips (∅ ∼ 50 nm) at different distances from single QDs | CdSe/ZnS (∅ ∼ 5.2 nm); λem ∼ 605 nm | ∼2.5 | — | Single QD measurements; on and off resonance investigated | 128 |
| 2016 | Triangular Au NP (tip-to-tip length ∼133 to 218 nm, thickness ∼50 nm) arrays coated with biotinylated BSA; streptavidin coated QDs bind to biotin on BSA (total spacer thickness ∼12 nm) | Ag2S (∅ ∼ 2.7 nm and ∼4.1 nm); λem ∼ 1100 nm and ∼1200 nm | ∼103 | — | Au NP and QD size dependence of enhancement was investigated. | 96 |
| 2016 | Random Au nanoislands (∼11 nm thick and ∼10–200 nm long) with intragaps of 7 nm and 10 nm; QDs bound to nanoislands | CdSe (λem ∼ 610 nm) and PbS (λem ∼ 850 nm) | ∼240 | — | Theoretical modeling combined with absorption spectroscopy; wavelength-dependent absorption enhancement and spectral broadening; PL not investigated | 129 |
| 2016 | Ag nanostructures (∼35 nm thick boxes ∼100 nm, rings ∼150 nm/250 nm inner/outer ∅, or discs ∼70 nm ∅) between quantum wells and QDs in the PMMA layer | InGaN quantum well FRET donors (∼2 nm thick, λem ∼ 516 nm) and CdSe/ZnS QD (∅ ∼ 6.3 nm, λem ∼ 650 nm) acceptors in an ∼80 nm PMMA layer | ∼1.7 (QD enhancement) | — | FRET enhancement and quenching were investigated by PL intensity and lifetime | 130 |
| 2016 | Au NP (∅ ∼ 5 nm) monomers or dimers attached via DNA (∼12 and ∼18 nm) to QDs | CdSe (∅ ∼ 5 nm); λem ∼ 520 nm | ∼4.3 (monomer), ∼6.8 (dimer) | ∼1.8 (monomer), ∼2.6 (dimer) | Distance dependence was investigated | 95 |
| 2017 | Au layers with the Al2O3 spacer at the bottom and Ag NPs (∅ ∼ 150 nm) with SiO2 coating (∼2 nm to 20 nm) on top. QDs in between | CdSe (∅ ∼ 3.4 nm); λem ∼ 600 nm | ∼1130 | Investigated but not quantified | Distance dependence investigated; halide perovskite QDs also investigated | 99 |
| 2018 | Open Ag nanoring (depth ∼100 nm, width ∼100 nm) array | CdSe/ZnS QDs (∅ ∼ nm) embedded in an ∼30 nm thick polymer layer above the nanoring array; λem ∼ 540 nm and 610 nm | ∼40 | — | Broad SPR spectrum results in multicolor enhancement (of both QDs); biosensing was performed with molecular beacon probes and dyes (no QDs), which exploited both quenching and enhancement | 131 |
| 2019 | Ag NPs (∅ ∼ 13 nm) in PVP nanofibres | Perovskite QDs (∅ ∼ 3.4 nm) in PVDF nanofibres; λem ∼ 407 nm, ∼456 nm, and ∼527 nm | ∼4.8 | — | Membranes via layer-by-layer electrospinning of fibres | 132 |
| 2020 | Au micropyramids (∼3.7 μm base length and ∼200 nm shell thickness) in liquid crystals. QDs positioned on the apex of Au micropyramids. | CdSe/ZnS (core ∅ ∼ 10 nm); λem ∼ 620 nm | — | ∼3 to 8 | Single QD measurements; reduced QD blinking | 133 |
| Year | Plasmonic nanomaterials | QDs | Max. PL (intensity) enhancement factor | Max. Purcell (lifetime) enhancement factor | Comments | Ref. |
|---|---|---|---|---|---|---|
| 2006 | SiO2-coated Au NPs (∅ ∼ 45 nm) | CdSe (∅ ∼ 6 nm); λem ∼ 630 nm | ∼1.8 | — | In THF, distance dependence was investigated | 138 |
| 2008 | Biotinylated Ag NPs (∅ ∼ 45 nm) coupled to streptavidin-coated QDs. | CdSe/ZnS (∼10 nm × 20 nm long ellipsoids); λem ∼ 655 nm | ∼2.6 | ∼2.6 | In PBS buffer, single QD measurements; reduced blinking | 134 |
| 2010 | Au NPs (∅ ∼ 10 nm) | CdTe (λem ∼ 525 nm) | ∼3 | ∼1.6 | In water, QDs and Au NPs mixed in solution (distance tuned via the concentration ratio) | 139 |
| 2013 | Au nanorods (∼50 nm × 20 nm) with thin organic coating (∼1.8 nm or ∼2.5 nm) to bind QDs; complete system encapsulated in a thick SiO2 shell | CdSe/ZnS (∅ ∼ 5 nm); λem ∼ 650 nm | ∼5 | — | In water, QD–Au nanorod ratios investigated | 140 |
| 2013 | Au NPs (∅ ∼ 54 nm) coated with SiO2 (∼7 to 38 nm) coated with QDs | CdSe/ZnS (λem ∼ 610 nm) | ∼1.2 | — | In ethanol, distance dependence was investigated | 141 |
| 2014 | Ag NPs (∅ ∼ 15 nm) mixed with QDs at different concentration ratios | CdTe (∅ ∼ 2 nm); λem ∼ 500 nm | ∼3.3 | ∼1.4 | In water, red shift and spectral broadening of QDs observed | 142 |
| 2014 | Au NPs (∅ ∼ 30 nm) coated with SiO2 (∼5 to 12 nm) coated with QDs coated with SiO2 (∼5 to 10 nm) coated with dyes | CdSe QDs (λem ∼ 580 nm) as FRET donors and S101 dyes (λem ∼ 600 nm) as acceptors | ∼1.7 | — | In water, PL decays measured but lifetimes not analyzed; distance dependence and QD-to-dye FRET were investigated; quantitative FRET analysis not convincing | 143 |
| 2015 | Au nanourchins (∅ ∼ 50 to 170 nm) with SiO2 shells (thickness ∼5 to 20 nm) | PbS (∅ ∼ 4.7 nm); λem ∼ 1225 nm | ∼1.6 | ∼15 | In water, remote NIR imaging (distance ∼30 to 70 cm) was performed; intensity EHF in reference to quenched nanohybrids was ∼5.6; simulated PL EHF was ∼4 to 16 | 144 |
| 2015 | SiO2 (∼35 nm) and Au (∼20 nm) shells on QDs | CdSe/CdS (core ∅ ∼ 6 nm, total ∅ ∼ 30 nm); λem ∼ 675 nm | — | ∼6 | In water, single QD measurements; completely suppressed QD blinking; high photostability | 145 |
| 2016 | Se-doped CuS nanodisks (∅ ∼ 31 nm, aspect ratio ∼4.5) | Same CuS nanodisks (both plasmonic and luminescent), λem ∼ 500 nm | ∼3 (2 PE PL enhancement) | — | In chloroform, 2 photon excitation PL of nanodisks is enhanced by their own in-plane LSPR mode | 137 |
| 2016 | AgNPs (∅ ∼ 20 nm) coated with SiO2 (∼9.5 to 58.5 nm) and functionalized with QDs | Bi2S3 QDs (∅ ∼ 5 nm; λem ∼ 425 nm) | ∼5.7 | — | In ethanol–toluene mixture, distance dependence was investigated; enhancement leads to emission redshift | 146 |
| 2018 | Au nanostars (∅ ∼ 50 nm) coated with the SiO2 shell (∼5 to 36 nm) | CdSe/ZnS; λem ∼ 550 nm | ∼4.8 | ∼16 | In ethanol, distance dependence was investigated; enhancement is nanostar-shape dependent | 136 |
| 2019 | Au NPs (∅ ∼ 15 nm) coated with CdS or ZnS shells (∼2 to 16 nm) | CdSe/CdS (∅ ∼ 8 nm); λem ∼ 620 nm | ∼2.5 to 3.5 | — | In ethanol, distance dependence was investigated | 147 |
| 2020 | Au nanorods (∼45 nm × 19 nm) coated with the CTAB bilayer (∼3.2 nm) and with polyelectrolyte (layer-by-layer deposition) spacers (∼2 to 6 nm) | CdSe/CdZnS; λem ∼ 620 nm | ∼11 | — | In water, distance dependence, QD per Au nanorod ratio, and QD position (on the Au nanorod tip or side) were investigated | 148 |
| 2021 | Au nanorods (∼40 nm × 16 nm) with SiO2 shells (∼3 to 50 nm) | CdSe (∅ ∼ 4.6 nm); λem ∼ 620 nm | ∼3.3 | ∼2.7 | In water, distance dependence was investigated | 135 |
| 2021 | Au nanorods (∼40 nm × 21 nm) with SiO2 shells (∼6 to 20 nm) | Si QDs as FRET donors (∅ ∼ 3.1 nm; λem ∼ 612 nm) and acceptors (∅ ∼ 4.9 nm; λem ∼ 690 nm) | ∼18 (for donors) and ∼12 (for acceptors) | — | In chloroform, distance dependence and FRET enhancement were investigated | 149 |
| Year | Plasmonic nanomaterials | QDs | Max. PL (intensity) enhancement factor | Max. Purcell (lifetime) enhancement factor | Bioapplication, comments | Ref. |
|---|---|---|---|---|---|---|
| 2010 | Ag nanoprisms (∼70 nm edge length) and polymer (layer-by-layer deposition) spacers (∼8 nm) | CdSe (∅ ∼ 5.5 nm); λem ∼ 640 nm | ∼2.5 | — | Surface-based; extension of ref. 107 to Cu2+ sensing (LOD ∼5 nM; dynamic range up to ∼100 μM; reaction time ∼5 min); photobrightening with UV-ozone irradiation resulted in 11-fold intensity enhancement; Cu2+ quenched the QDs due to Cu2+–Cd2+ cation exchange | 152 |
| 2011 | DNA-functionalized Au NPs (∅ ∼ 16 nm) | DNA-functionalized CdSe/ZnS QDs; λem ∼ 531 nm | ∼2.3 | — | Solution-based; ssDNA sensing (LOD ∼15 nM) via competition of target ssDNA and ssDNA-QD for hybridization to ssDNA-AuNP as determined by electrophoresis; distance dependence was investigated | 156 |
| 2014 | Amine-functionalized Au nanodendrites (∅ ∼ 62 nm) with sharp tips as plasmonic hotspots | Carboxyl-functionalized CdTe/CdS QDs (λem ∼ 545, 600, and 625 nm) | ∼3 | ∼2.5 | Solution-based; TNT sensing (LOD ∼50 pM); TNT sensing in spiked tap and lake water; displacement of QDs by TNT | 157 |
| 2014 | Au nanoflowers (∅ ∼ 40, 80, 120, 130, and 150 nm) with amino-functionalized SiO2 shells (thickness ∼16 nm); | CdTe/CdS/ZnS (λem ∼ 615 nm) | ∼1.4 | — | Cell-based; cell membrane targeting by specific antibodies; imaging of breast cancer cells; Au nanoflowers were used for photothermal conversion | 158 |
| 2015 | Au nanopillar or nanohole (∼50 nm thick/deep squares of 140 nm × 140 nm) arrays; reduced antibody, antigen, antibody, and streptavidin as spacers (∼10 to 20 nm thick) | streptavidin-coated CdSe/ZnS (∼13 nm × ∼6 nm nanorods); λem ∼ 655 nm | — | — | Surface-based; PSA sandwich immunoassay (LOD < 100 pg mL−1) using a conventional fluorescence microscope for QD PL detection | 154 |
| 2015 | Au nanohole (∼50 nm deep squares of 140 nm × 140 nm) arrays; DNA-aptamer, antigen, DNA-aptamer, and streptavidin as spacers (∼17 nm thick) | streptavidin-coated CdSe/ZnS (∼13 nm × ∼6 nm nanorods); λem ∼ 655 nm | — | — | Surface-based; similar work to ref. [154] but using DNA-aptamers instead of antibodies; thrombin sandwich aptamer assay (LOD ∼1 ng mL−1 or ∼27 pM) | 155 |
| 2016 | Au NPs (∅ ∼ 11 to 104 nm) coated with the SiO2 shell (∼12 nm thick) | CdTe (∅ ∼ 2.5 nm); λem ∼ 543 nm | ∼15.5 | — | Solution-based; FRET between QDs and NO2−-sensitive dye “neutral red” (electrostatically attached to the nanohybrids) resulted in NO2−-concentration dependent PL ratio change (LOD ∼60 nM); NO2− detection in tap water, lake water, bacon lixivium, and cell supernatant | 159 |
| 2016 | Ag NPs (∅ ∼ 50 nm) with the SiO2 shell (∼7 nm thick) | CdTe/CdS (∅ ∼ 6 nm); λem ∼ 690 nm | ∼4 | — | Solution-based; hyaluronidase quantification (LOD ∼2 pg mL−1) by enzymatic digestion, followed by magnetic separation, followed by plasmonic enhancement; detection in serum and urine | 160 |
| 2020 | Au NPs (∅ ∼ 20 nm) mixed (1:1:1 vol. ratio) with QDs and fumed SiO2 and then deposited on the SiO2 microsphere (∅ ∼ 5 μm) array |
Amino-modified CdSe/ZnS QDs (∅ ∼ 4.3 nm; λem ∼ 620 nm) adsorbed to fumed SiO2 | ∼2 | — | Surface-based; gaseous formaldehyde quantification (0.5 to 2.0 ppm) via nonradiative electron transfer from QDs to carbonyl of formaldehyde, which led to QD PL quenching | 153 |
| 2020 | Ag NPs (∅ ∼ 43 nm) with the SiO2 shell (∼9 nm thick) | Carboxyl-functionalized CdTe QDs (∅ ∼ 3 nm; λem ∼ 539 nm) | ∼3.9 | — | Solution-based; quantification of tetracycline (∼0.2 to 400 μM) via electron transfer from QDs, which led to QD PL quenching; detection in spiked milk samples; distance dependence was investigated | 161 |
In this section, we first review typical surface and solution based metal–QD nanohybrid structures and their characterization concerning PEF. Then, we present metal–QD nanohybrids that were used for actual biosensing applications. Overview tables including the properties of metal nanostructures and QDs and both PL intensity and PL lifetime enhancement factors (Tables 1–3) provide an efficient tool for finding, comparing, and interpreting different PEF approaches and applications.
5.1 Typical surface-based metal–QD nanohybrid architectures for PEF
Owing to the high controllability of depositing multiple very thin layers with different materials on solid supports, such as glass slides or Si wafers, surface-based metal–QD nanohybrid architectures, in which a nanostructured metal layer and a QD layer are separated via an optically inactive spacer layer (e.g., SiO2) of specific thickness, were the first and have so far been the most investigated metal–QD nanohybrids for PEF. Surface-based nanostructures do not only allow for a specific and steady positioning of the NPs, such that single NP analysis becomes easier to perform. They are also highly advantageous for accomplishing specific orientations, such that plasmonic hotspots (e.g., between NP multimers or between NPs and surfaces) or specific LSPR bands (e.g., longitudinal and transversal LSPR bands in nanorods) can be designed at the position of the QDs for optimized PL enhancement. When looking at the overview Table 1, it becomes clear that the surface-based PEF of QDs has been largely dominated by Au and Ag nanostructures, such as nanopatterned surfaces, nanoislands, NPs, nanorods, nanoprisms, nanodisks, nanocones, nanocubes, nanoboxes, nanotips, or nanorings, and Cd-based (i.e., CdSe and CdTe) QDs. Spacer layers have been mainly composed of SiO2 or poly(methyl methacrylate) (PMMA) between the metal and QD layers or with embedded metals or QDs. However, with the aim of going toward biosensing, some studies also used biomolecules as spacers.93–97 Both PL intensity (absorption and/or emission) enhancement and PL lifetime (emission only) enhancement have been investigated on the ensemble and single NP levels with EHFs ranging from a few percent up to several thousands. Notably, there have been major differences between intensity and lifetime enhancement in many studies, most probably caused by the competition of quenching and enhancement, which both contribute to PL lifetime shortening. However, even a reliable determination of intensity EHFs can be difficult because small changes in reproducibility or parameters of the metal–QD nanohybrids can modify the enhancement, as exemplified by two studies of Jen et al., in which the use of the same Au nanodisk array, the same QDs, and the same streptavidin–biotin spacers led to intensity EHFs of ∼15 (streptavidin-coated QDs and Au–QD distance of 16 nm) and ∼4 (biotin-coated QDs and Au–QD distance of 17.7 nm), respectively.93,94 Very large enhancements (EHF >100) were usually attained by specific plasmonic hotspots and for single QDs, whereas simpler nanostructures and ensemble QD EHFs provided EHFs in the range of 2 to 20. Many studies investigated the dependence of PEF on metal–QD distances. Distances between ca. 5 and 15 nm were usually found to provide the best enhancement, whereas shorter distances most often resulted in efficient quenching and longer distances resulted in only minor or no enhancement. Another interesting aspect of the PEF of QDs is related to spectral investigations. Most QDs showed red-shifted PEF and overlap of the LSPR spectrum with the excitation spectrum (absorption or excitation enhancement) of QDs contributed stronger to PEF than overlap of the LSPR spectrum with the emission spectrum (emission enhancement) of QDs. Because both excitation and emission enhancements depend on electric field enhancement by the metal nanostructures, overlap with the scattering part (and not the absorption part) of the LSPR was most important. Thus, nanostructures with higher scattering fractions (e.g., larger NPs) were found to be better suited for the PEF of QDs. As also expected from the theoretical estimations (cf. Section 2), dim QDs with low PL quantum yields were much better suited for PEF compared to bright QDs with high quantum yields. Concerning single QD studies, PEF most often resulted in photostabilization and reduced blinking.In the following paragraphs, we discuss some representative examples of surface-based PEF metal–QD nanohybrids that utilized different nanostructures, investigated the distance dependence of PEF, and used both steady-state and time-resolved PL measurements for scrutinizing the different enhancement and quenching mechanisms. Many other examples including the most important information concerning the metal–QD hybrid systems are summarized in Table 1.
In one of the early experimental works, Nurmikko et al. realized the PEF of QDs via 50 nm thick QD-doped PMMA films spin coated on indium tin oxide (ITO) coated glass.98 After the generation of holes with approximately 100 to 160 nm diameter (Fig. 8A), a 200 to 400 nm thick Ag film was deposited on the patterned substrate, such that ∼50 nm high Ag nanodisks were surrounded by PMMA doped with 655 nm emitting CdSe/ZnS core/shell QDs (Fig. 8B). Excitation and emission from the bottom through the glass resulted in Ag-nanodisk size-dependent PL enhancement with PL intensity EHFs of up to 52 (Fig. 8C) and PL lifetime EHFs of up to 10 (PL lifetime decreased from 10 ns to ∼1 ns – Fig. 8D) for the 50 nm high and 160 nm diameter disk array with a lattice constant of 300 nm (which overlapped best with the QD PL spectrum). Using the quantum yield of the QDs in free space (0.3) and the relation between PL quantum yield, PL lifetime, and emission rate, the authors found a quantum yield enhancement of ∼3.1 and an emission rate enhancement of ∼30. Because this value did not correspond to the 52-fold PL intensity enhancement, they further investigated arrays with and without the continuous Ag layer on top of the Ag nanodisks and found that only with the nanodisk-connecting layer, the interplay of coupled localized and propagating surface plasmon polaritons could lead to strong PEF with EHFs of up to 52. This study showed that careful design of plasmonic nanostructures is essential to strongly enhance the efficiency of QD-based light-emitting devices.
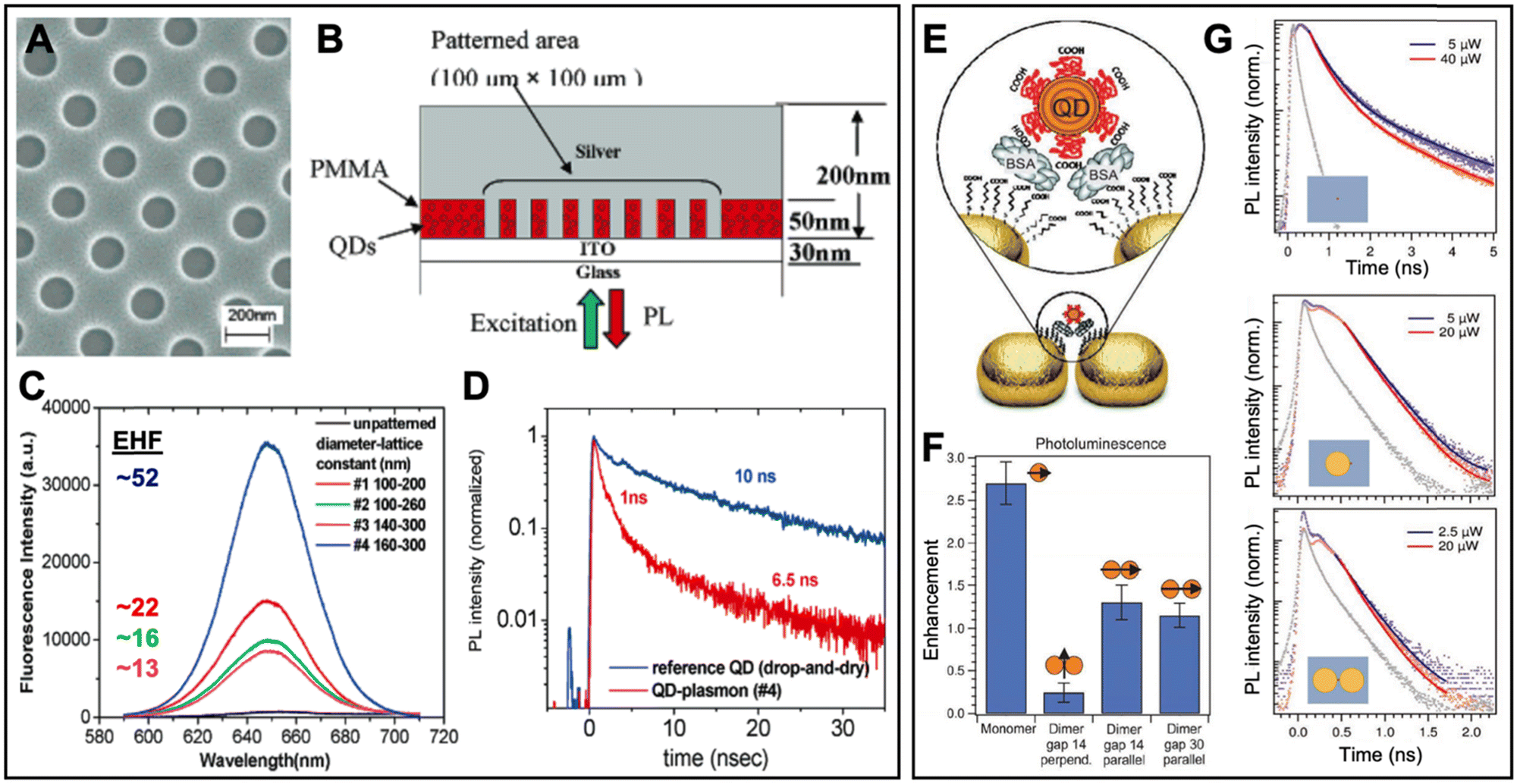 | ||
| Fig. 8 Examples of surface-based metal–QD nanohybrids for PEF. (A) SEM image (top view) of a nanoarray of holes with diameters of ∼160 nm and a lattice constant of ∼300 nm in PMMA. (B) Schematic (side view) of the finished Ag–QD nanostructure. (C) QD PL spectra (λex = 532 nm) and EHFs from arrays with different nanodisk diameters and lattice constants. (D) QD PL lifetime measurement of QDs (λex = 545 nm; λem = 655 nm) on a glass substrate (blue) and QDs in the 160 nm diameter and 300 nm lattice constant nanoarray (red). A to D adapted with permission from ref. 98. Copyright 2005 American Chemical Society. (E) PEG-coated QDs were precisely attached in the gap region of Au nanodisk dimers via binding to BSA. (F) Integrated PL EHFs of different Au nanodisk monomer and dimer structures with different gap sizes and perpendicular or parallel excitation polarization. (G) PL decay curves (λex = 636 nm; λem: 650–690 nm) for QDs on the ITO substrate (top), a QD coupled to a Au nanodisk monomer (center), and a QD coupled to a dimer antenna with a 14 nm gap (bottom). The gray curves show the instrument response function. Note the different time scales for QDs alone and QDs coupled to Au nanodisks. (E–G) Adapted with permission from ref. 97. Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Wenger et al. used beam lithography to position Au nanodisks of 90 nm diameter and 40 nm thickness on ITO, such that they were separated by a gap of 14 or 30 nm.97 CdSe/CdS/ZnS core/shell/shell QDs coated with polyethylene glycol (PEG) and emitting at around 610 nm were then attached in the center above the dimer gap antennas (Fig. 8E) via bovine serum albumin (BSA). Au nanodisk monomers resulted in the highest PL intensity EHFs (∼2.7), whereas the nanogap antennas with parallel excitation polarization resulted in an EHF of only 1.3 (Fig. 8F). This finding was rather surprising because excitation enhancement was 15.9 fold for the nanogap dimers and 5.1 fold for the monomers. In addition, the PL lifetime EHFs of QDs were 11.1 for both dimers and monomers (Fig. 8G). The differences were attributed to competing enhancement and quenching effects, which also resulted in decreased quantum yields (quantum yield EHFs ∼0.5 for the monomers and <0.1 for the dimers). Parallel excitation polarization was found to have significantly better enhancement compared to perpendicular polarization, whereas the difference in gap size (14 nm vs. 30 nm) had only minor influence on the PL enhancement. Similar to the study of Nurmikko (vide supra), this work showed that careful investigation of metal–QD nanohybrids by steady-state and time-resolved spectroscopy is necessary to understand and distinguish the different competing enhancement and quenching effects for optimizing the PEF of QDs.
One of the highest PL intensity EHFs of approximately 1130 was found by Zhang and Li et al., who were also using gap mode plasmonic nanostructures for the PEF of QDs.99 The authors coated an ultrathin Al2O3 layer on a Cr/Au (5 nm/200 nm) film and attached CdSe QDs via amino groups on the Au/Al2O3 substrate. Ag NPs with a diameter of circa 150 nm and coated with SiO2 of different thicknesses (∼2 nm to 20 nm) formed a uniform film on top of the QD layer (Fig. 9A). Simulations predicted a strong electric field enhancement in the gap (formed by the SiO2 layer) between the Ag NPs and the Au film (Fig. 9B). The different SiO2 layer thicknesses resulted in very different PL intensity EHFs of 10 (2 nm SiO2), 40 (16 nm SiO2), 270 (10 nm SiO2), and 1130 (6 nm SiO2), which showed that only a few nanometers can make a strong difference for the PEF of QDs (Fig. 9C). Notably, QDs on a glass substrate (which were used as a reference for calculating the EHFs) displayed extremely low PL intensities, which was most probably one of the reasons for the very strong relative enhancement. PL lifetime measurements (Fig. 9D) revealed both quenching and enhancement because the system with the lowest PL intensity (EHF ∼10 for 2 nm SiO2) resulted in the strongest decrease of PL lifetime, whereas thicknesses from 6 to 20 nm did not result in strong reduction of the PL lifetime. Unfortunately, the authors did not quantitatively analyze the PL lifetime data. Nevertheless, the competition between PL enhancement and quenching was clearly observed by measuring both PL intensities and lifetimes.
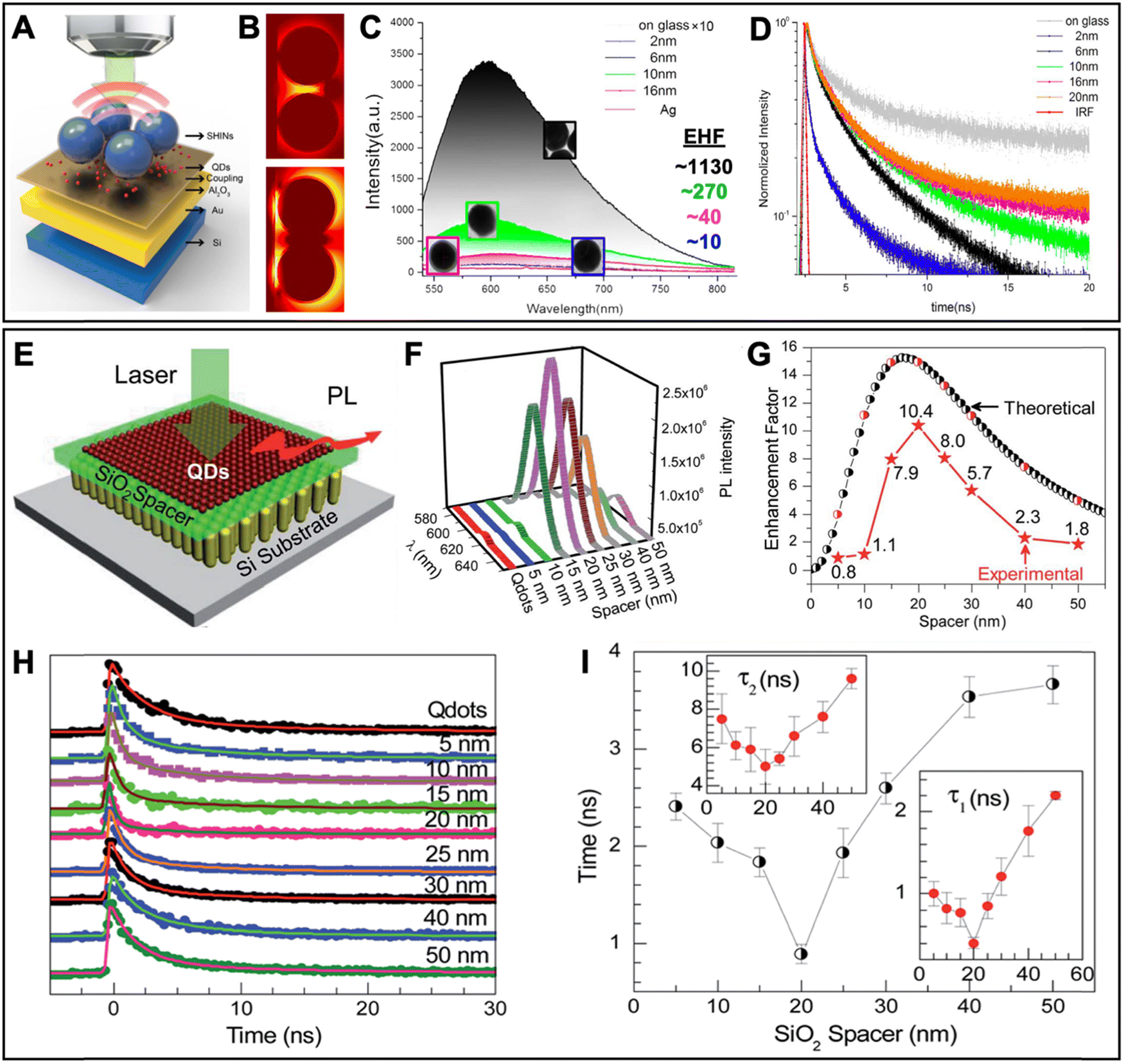 | ||
| Fig. 9 Examples of surface-based metal–QD nanohybrids for PEF. (A) Schematic of a metal–QD nanohybrid system with QDs in a gap mode between SiO2-coated Ag NPs and a Au film. (B) Simulation of the electromagnetic field amplitude distribution for a transverse electromagnetic wave at the scattering peak wavelength excited at 532 nm. (C) PEF spectra (λex = 532 nm) of CdSe films with Ag NPs of different SiO2 shell thicknesses. EHFs were calculated in reference to the CdSe film on glass without Ag NPs. Uncoated AgNPs resulted in strong quenching of QD emission. (D) PL decay curves (λex = 532 nm; λem = 600 ± 35 nm) of CdSe films with Ag NPs of different SiO2 shell thicknesses. IRF: Instrument response function. A to D adapted with permission from ref. 99. Copyright 2017 Elsevier Ltd. (E) Schematic of a metal–QD nanohybrid consisting of a vertically aligned Au nanorod monolayer and CdSe/ZnS QDs monolayer films. A SiO2 spacer layer controls the distance between Au nanorods and QDs. (F) PL spectra (λex = 532 nm) of QD films on Au nanorods with different SiO2 spacer thicknesses. (G) Theoretical (circles) and experimental (stars) EHFs as a function of SiO2 spacer thickness. (H) PL decays (excitation and emission wavelengths for these experiments were not provided) of QD films on Au nanorods with different SiO2 spacer thicknesses. Solid lines are fits to the data using double-exponential decay functions. (I) Average lifetime (insets show the single lifetimes) as a function of the SiO2 spacer thickness. (E–I) Reproduced with permission from ref. 100. Copyright 2014 The Royal Society of Chemistry. | ||
In another interesting example of the PEF of QDs, Demir and Xiong et al. exploited the strong longitudinal LSPR of vertically aligned Au nanorods (∅ ∼ 40 nm, length ∼101 nm, and ∼7.7 nm edge-to-edge distance) on Si substrates (Fig. 9E).100 Due to the interaction between the Au nanorods within their monolayer, the strong longitudinal plasmon band at 717 nm (in aqueous solution) was shifted to 610 nm, which corresponded well to the QD emission maximum at 614 nm. The distance dependence of PEF was investigated with SiO2 spacer thicknesses ranging from 5 to 50 nm using both steady-state (Fig. 9F) and time-resolved (Fig. 9H) PL measurements. For both PL intensity (Fig. 9G) and lifetime (Fig. 9I) a thickness of 20 nm was found to provide the largest EHFs, in agreement with theoretical calculations (Fig. 9G). The difference between an EHF of 10.4 for PL intensity and 4.6 for PL lifetime was most probably caused by a combination of radiative rate enhancement (the Purcell effect contributes to both PL intensity enhancement and PL lifetime quenching) and absorption enhancement (contributes to PL intensity enhancement only). Again, only the combination of steady-state and time-resolved PL experiments could reveal the different enhancement mechanisms. In addition, the use of theoretical calculations to estimate an optimal distance between the metal and QD layers was demonstrated.
5.2 Typical solution-based metal–QD nanohybrid architectures for PEF
Solution-based metal–QD nanohybrid architectures have been developed much less frequently, most probably due to the lower controllability of placing plasmonic metal nanostructures and QDs at a specific distance and orientation to each other. In addition, it is significantly more difficult to design and fabricate plasmonic hotspots by well-defined gap structures in solution and to investigate the metal–QD nanohybrids on the single molecule level. Thus, only very few examples with hotspots on pointed metal nanostructures exist and EHFs above 10 have been very rare because they are usually averaged on the ensemble level and single hotspots with extremely large EHF cannot be separated. Considering that the high impact of today's literature is often related to high quantitative improvements, the investigation of solution-based PEF of QDs is also less rewarding in academic research. When taking into account the many design parameters and synthesis as well as characterization methods that need to be tested and optimized for accomplishing metal–QD hybrid nanostructures with PEF, it is not really surprising that only a few studies focused on the challenging endeavor of fabricating and characterizing reproducible solution-based nanostructures for the PEF of QDs. Nevertheless, for biosensing, EHFs of 2 or 3 may already present a considerable improvement and it is therefore important to investigate, understand, and optimize solution-based metal–QD hybrid nanostructures. Table 2 summarizes solution-based PEF of QDs. Most EHFs were below 6 and the large majority of nanostructures was based on SiO2-coated Au NPs and CdSe-based QDs. In the following paragraphs, we discuss some representative examples of solution-based PEF metal–QD nanohybrids that utilized different nanostructures and assembly approaches. Several other examples including the most important information concerning the metal–QD hybrid systems are summarized in Table 2.Although most solution-based metal–QD nanohybrids have been developed within the last 10 years, Lakowicz et al. have fabricated and characterized a system based on AgNPs and QDs already in 2008.134 The nanohybrids were prepared by simply mixing biotinylated AgNPs (∅ ∼ 45 nm) and streptavidin-coated QDs and the strong biotin–streptavidin interaction resulted in nanocomposites that mainly consisted of one AgNP and one QD (Fig. 10A). Whereas the assembly was performed in aqueous buffer, the solution was spun-cast on microscope slides for single QD PL measurements. Compared to bare QDs, the QD/Ag nanocomposites showed significantly reduced blinking and an average PL intensity that was approximately 2.6-fold higher (Fig. 10B). PL lifetime measurements of single QDs and single QD/Ag nanohybrids (Fig. 10C) confirmed the enhancement with circa 2.6-fold lifetime quenching (Fig. 10D). Although ensemble PL measurements were not performed, the self-assembly via biotin–streptavidin in PBS showed the functionality of the system in solution and represented already a first proof-of-concept for measuring biological recognition via the PEF of QDs.
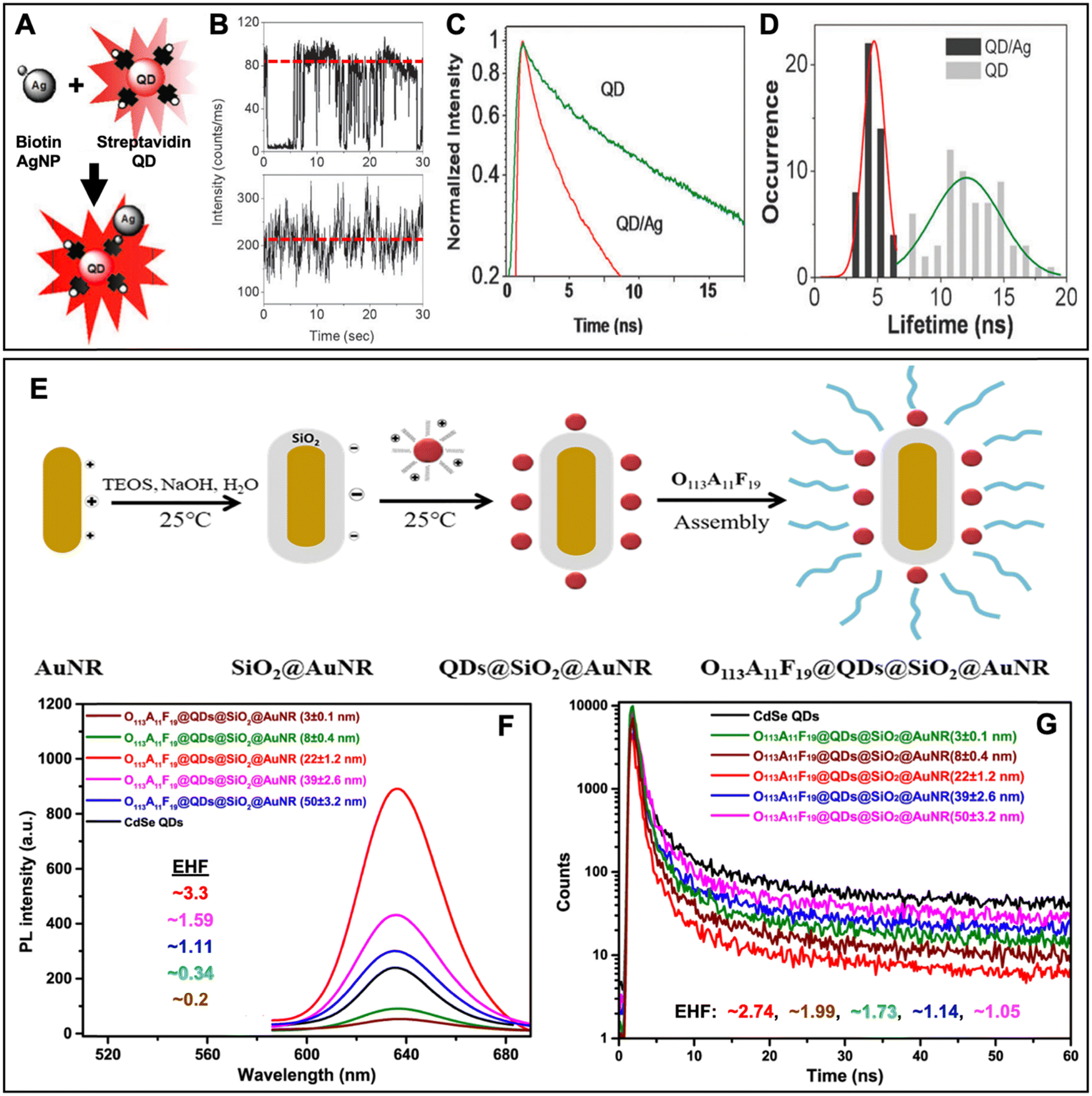 | ||
| Fig. 10 Examples of solution-based metal–QD nanohybrids for PEF. (A) Schematic of a solution-based nanohybrid consisting of streptavidin-coated QDs and biotinylated AgNPs. (B) Typical intensity trajectories from a single QD (top) or single QD/Ag nanocomposite (bottom) on glass substrates. The red dotted lines present the maximum occurrence of single QD intensities, which is circa 2.6-fold higher for the QD/Ag nanohybrids. (C) PL decays of QD and QD/Ag nanohybrids. (D) On average, single QD/Ag nanohybrids showed a circa 2.6-fold faster PL lifetime compared to bare QDs. All experiments were performed with λex = 470 nm and λem = 655 ± 10 nm. A to D adapted with permission from ref. 134. Copyright 2009 The Royal Society of Chemistry. (E) Fabrication procedure of aqueous metal–QD nanohybrids consisting of SiO2-coated AuNRs, CdSe QDs, and a co-block polymer coating (O113A11F19 corresponds to the polymerization degree of blocks O, A, and F). (F) PL spectra (λex = 520 nm) of bare QDs and QD/AuNR nanohybrids with different SiO2 shell thicknesses. EHFs correspond to the ratios of QD/AuNR and bare QD PL intensities. (G) PL decay curves (λex = 520 nm) of bare QDs and QD/AuNR nanohybrids with different SiO2 shell thicknesses. EHFs correspond to the ratios of bare QD and QD/AuNR PL lifetimes. E to G adapted with permission from ref. 135. Copyright 2021 Elsevier B.V. | ||
Wen et al. coated Au nanorods (AuNR, ∼40 nm × 16 nm) with SiO2 shells of different thicknesses (∼3 to 50 nm) and investigated the AuNR to QD distance dependence of PEF in water with steady-state and time-resolved PL spectroscopy.135 Positively charged QDs could bind via electrostatic interaction to the negatively charged SiO2 shells and the QD/AuNR nanohybrids were then encapsulated in an amphiphilic co-block polymer coating for water solubility (Fig. 10E). The maxima of the longitudinal LSPR band of the AuNRs and the emission band of the QDs overlapped very well around 640 nm. 3 and 8 nm thick SiO2 shells resulted in PL intensity quenching, whereas 22, 39, and 50 nm thick shells enhanced the PL intensity with a maximum EHF of 3.3 for the 22 nm thick SiO2 shell (Fig. 10F). Because PL quenching also leads to reduced PL lifetimes (similar to many of the surface-based metal–QD nanohybrids – see Section 5.1), the lifetime EHFs were all positive and different from the PL intensity EHFs (Fig. 10G). The PL lifetime EHFs of the 22, 39, and 50 nm thick SiO2 shell systems were slightly lower than the intensity EHFs, which indicated a combination of absorption enhancement and radiative rate enhancement. The stability and biocompatibility of these nanohybrids were other interesting aspects for a potential implementation into biosensing applications.
In one of the few examples that exploited plasmonic hotspots for the PEF of QDs in solution, Zhang et al. fabricated Au nanostars (∅ ∼ 50 nm) with different apex lengths that were coated with amino-functionalized SiO2 shells (∼5 to 36 nm), to which carboxyl-functionalized QDs were attached in ethanol (Fig. 11A).136 Longer apexes resulted in longer LSPR peak wavelengths and stronger electric fields at the plasmonic hotspots of the apexes (Fig. 11B). The PL intensity EHFs increased with the apex length (Fig. 11C), which showed that the electric field strength was more important than the resonance peak wavelength because the longer the apex lengths the worse the spectral overlap with the QD emission. Interestingly, the PL lifetime EHFs of the nanohybrids with longer apexes were significantly larger than the PL intensity EHFs (e.g., 16 vs. 4.8 for AuNS5), whereas the PL lifetime of the nanohybrid with the shortest apexes was only slightly quenched (Fig. 11D). Although this finding was unfortunately not discussed in detail, it would most probably mean that for the short apexes the absorption enhancement dominated radiative rate enhancement and for the longer apexes absorption and emission quenching was competing with radiative rate enhancement. Similar to the surface-based metal–QD nanohybrids discussed above, this study demonstrated that both steady-state and time-resolved PL measurements are necessary to better understand the PEF mechanisms and that competing electric field enhancement and spectral overlap require a careful design of such LSPR hotspot systems. From the biosensing point of view, a combination of PL intensity increase with strong PL lifetime quenching could be very useful for designing high-sensitivity sensing approaches.
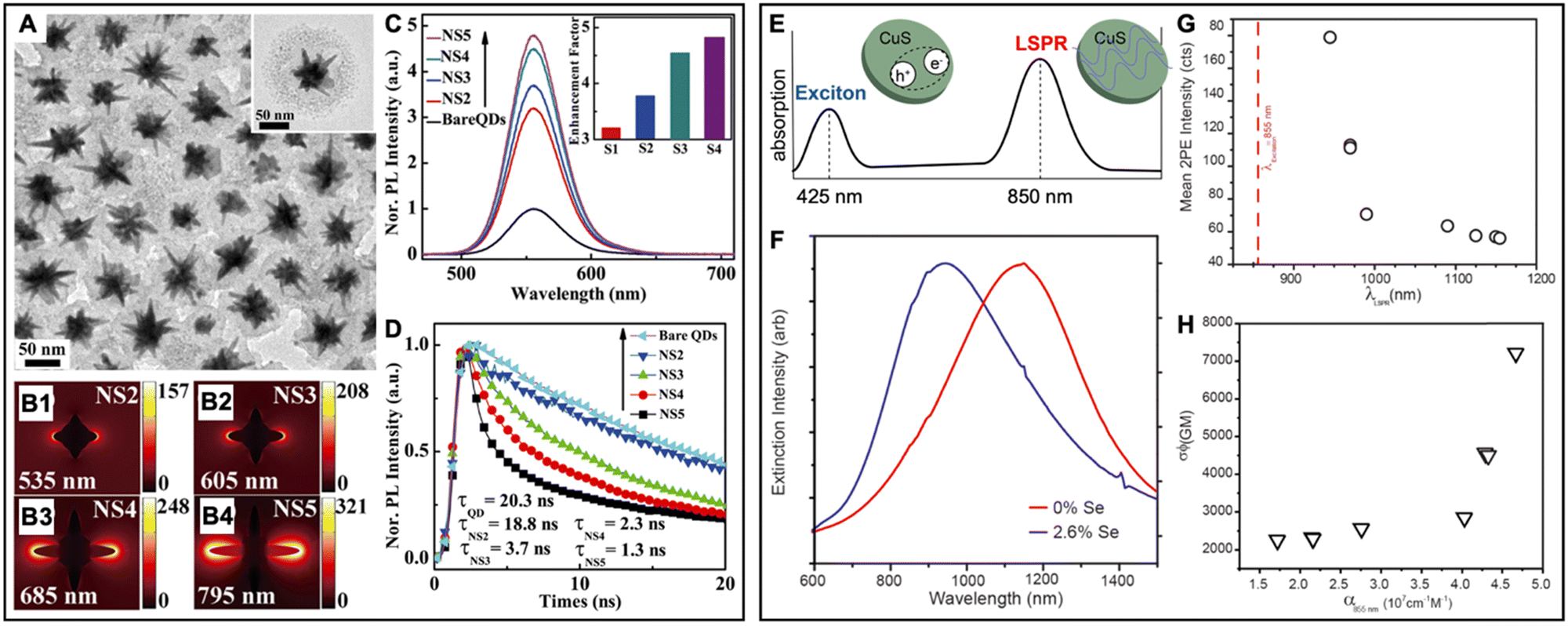 | ||
| Fig. 11 Examples of solution-based metal–QD nanohybrids for PEF. (A) TEM images of Au nanostars (NS) coated with SiO2 and QDs (inset provides better visibility of QDs coated to the SiO2 shell). (B) Simulated electric field distributions and resonance peak wavelengths of AuNSs with different apex lengths. (C) PL spectra (λex = 375 nm) of QDs and AuNS/QD hybrids. The inset shows EHFs (ratio of nanohybrids to QD PL intensities) of the different AuNS/QD nanohybrids. (D) PL decay curves (λex = 375 nm; λem: 550–560 nm) of QDs and AuNS/QD nanohybrids. A to D reproduced with permission from ref. 136. Copyright 2018 Authors. Published by AIP Publishing (Creative Commons license CC-BY 4.0). (E) Schematic principle of the absorbance profile for plasmon-assisted 2PA of CuS nanodisks. (F) Extinction spectra of 2.6% Se-doped and undoped CuS nanodisks. (G) Dependence of 2PA PL intensity on LSPR peak wavelength. (H) 2PA cross section as a function of CuS nanodisk molar absorptivity at 855 nm. (E–H) adapted with permission from ref. 137. Copyright 2016 American Chemical Society. | ||
A very unique and unusual plasmon-enhanced QD fluorescence system was developed by Tao et al.137 The authors used Se-doped CuS nanodisks (∅ ∼ 31 nm, aspect ratio ∼4.5) in chloroform and the same nanodisks provided both LSPR and luminescence (thus, strictly speaking not actually a hybrid system and not a QD). Moreover, considering that the LSPR was in the NIR at ∼1150 nm, it enhanced the nanodisks’ two-photon absorption (2PA) instead of the conventional one-photon absorption below ∼500 nm (Fig. 11E) or the nanodisk PL at ∼505 nm. Because the 2PA maximum wavelength of the CuS nanodisks was found around 855 nm, Se doping was used to shift the LSPR from 1150 nm to 945 nm, which led to higher molar absorptivities of the nanodisks at 855 nm (Fig. 11F). The blue-shifted LSPR spectra also resulted in significantly higher two-photon excitation PL intensities (Fig. 11G) and 2PA cross sections (Fig. 11H) with EHFs reaching ∼3. Because there was negligible spectral overlap of the NIR LSPR band with the emission around 500 nm, 2PA excitation enhancement was considered as the only possible mechanism of PEF. This study demonstrated that colloidal PEF nanosystems can be designed as “all-in-one” platforms and that PEF can also be used for enhancing 2PA PL, which may become very beneficial for bioimaging applications.
5.3 Metal-QD nanohybrids for PEF biosensing
Despite the many examples of surface or solution based PEF of QDs in metal–QD nanohybrids, including the use of biomolecules as metal–QD spacers, actual biosensing applications have remained rare (Table 3). This lack of advancing the metal–QD nanohybrids to the next level may be related to two major problems. First, it is already very challenging to design, prepare, combine, position, and orient two different well-defined nanosystems (plasmonic metal structures and QDs) and merging those metal–QD nanohybrids with a third biological nanosystem that includes specific biological recognition with a target molecule adds significant complication to the overall design and accomplishment of target-specific PEF. Second, the attainable PL EHFs remain relatively low (usually well below one order of magnitude), especially for QDs with high PL quantum yields. One needs to keep in mind that QDs are most often more stable and brighter than organic fluorophores. Thus, using a very bright QD without PEF may result in almost the same biosensing performance and the added value of PEF becomes insignificant when related to the experimental effort. Nevertheless, an EHF of 2 may already be highly significant for a bioanalytical application and optimizing an already very good system is always more difficult than optimizing a bad one. As an example from real life, one may imagine the effort of improving the speed of a car from 50 to 100 km h−1 or from 200 to 400 km h−1. Both have an EHF of 2 but the latter is obviously more challenging. To harness the photophysical advantages of metal–QD nanohybrids for improved biosensing, both a better control and understanding of the nanohybrids and more efforts in their combination with biosensing approaches are necessary.The PEF of QDs was already exploited for biosensing in 2004, when Knoll et al. used a combination of surface plasmon field enhancement and fluorescence spectroscopy to detect DNA hybridization and to analyze QD blinking.150,151 However, their approach did not make use of LSPR and metal–QD nanohybrids but used a conventional SPR instrument to excite surface plasmon waves in the normal Kretschmann configuration (a sample placed on a thin gold film, which is attached to a high refractive index prism) and detected QD fluorescence photons emitted normal from the prism base plane. Arguably the first actual biosensing application based on PEF by metal–QD nanohybrids was realized in 2010, when Batteas et al. extended their surface-based QD-coated Ag nanoprism array approach107 to Cu2+ sensing via Cu2+-induced quenching of QD PL.152
The quantification of ions or small molecules that quenched the PL of plasmon-enhanced QDs has remained the main strategy for biosensing. For example, Xing et al. developed a surface-based metal–QD nanohybrid biosensor for gaseous formaldehyde.153 The metal–QD nanohybrids were embedded in a sensing film that consisted of a mixture of AuNPs (∅ ∼ 20 nm), CdSe/ZnS QDs (∅ ∼ 4.3 nm), and fumed silica (FS), which was spin coated on a SiO2 microsphere array (Fig. 12A). The distance between QDs and AuNPs was adjusted by the concentration of the different components and the best film resulted in a PL intensity EHF of ∼2. In a gas chamber, the small formaldehyde sensor was exposed to different gas mixtures and the PEF QD spectra were measured using a spectrograph (Fig. 12B). Within the sensing film, formaldehyde molecules bound to the amino groups on the QDs and the carbonyl groups in the attached formaldehyde acted as acceptors for electrons in the conduction band of excited QDs (Fig. 12C). This photoinduced electron transfer resulted in formaldehyde-induced QD PL quenching (Fig. 12D) with a linear concentration-dependence from 0 to 2 ppm (Fig. 12E). The novel sensor could quantify low concentrations of gaseous formaldehyde by an anhydrous process at room temperature with relatively short response (∼100 s) and recovery (∼80 s) times.
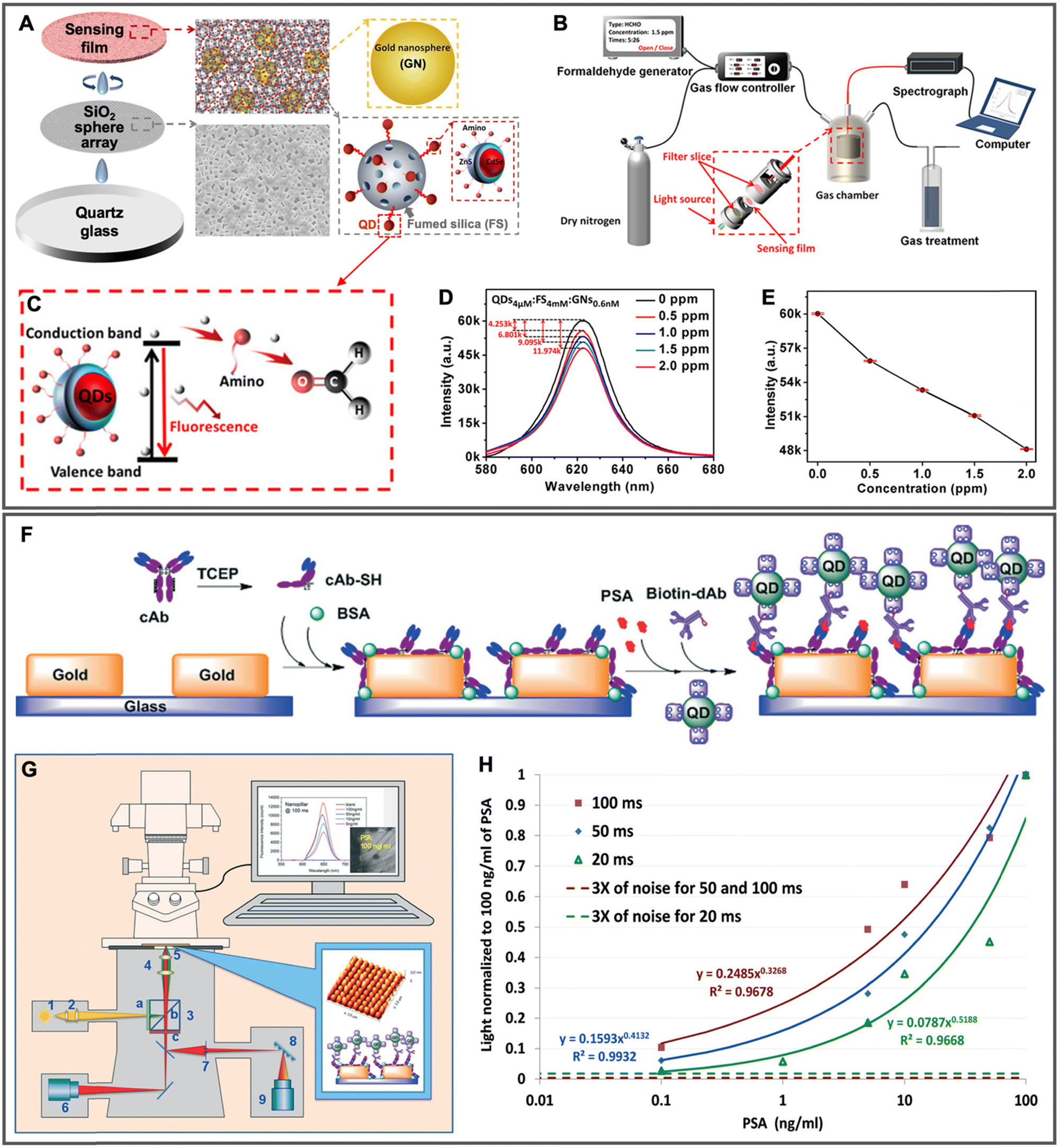 | ||
| Fig. 12 Examples of metal–QD nanohybrids for biosensing. (A) Schematic of the fabrication process of the QD/FS/GN sensing film deposited on a silica sphere array. (B) Schematic of the formaldehyde sensor. (C) Schematic of the QD PL quenching mechanism. (D) PL spectra (λex = 520 nm) of sensing films after injecting different concentrations of gaseous formaldehyde for 500 s. (E) PL intensity (peak intensities from D) as a function of formaldehyde concentration. A to E adapted with permission from ref. 153. Copyright 2020 American Chemical Society. (F) Schematic of PSA sandwich immunoassay on the Au nanoarray for the PEF of QDs. (G) Schematic of the detection set-up using a standard fluorescence microscope (1, 2: excitation; 3: filter cube with different filters (a, b) and beam splitter (c); 4: objective; 5: sample; 6: EMCCD camera; 7 and 8: polychromator; 9: CCD detector). λex = 540 ± 13 nm; λex = 605 ± 28 nm. (H) Immunoassay calibration curves for different integration times (20, 50, and 100 ms). (F–H) adapted with permission from ref. 154. Copyright 2015 The Royal Society of Chemistry. | ||
The more interesting but also more challenging concept of altering the actual PEF via biological binding (modification of metal–QD distance) has been much less employed for biosensing. Zhou et al. used antibody-prostate specific antigen (PSA)-antibody or aptamer–thrombin–aptamer sandwich complexes to target-specifically adjust the distance between Au nanopillar arrays and QDs, such that the PEF of the QDs could be used for target quantification.154,155 Au nanopillar (∼50 nm × 140 nm × 140 nm) arrays (320 nm pitch) were fabricated by e-beam writing on glass, followed by resist development, Au film coating, and resist lift-off. For the PSA immunoassay, reduced (using tris(2-carboxyethyl)phosphine, TECP) PSA capture antibodies (cAb) were self-assembled on the Au nanopillars via the free sulfhydryl groups and BSA was used for blocking the free spaces on the array. Biotinylated PSA detection antibodies (dAb) were self-assembled to streptavidin-functionalized QDs. In the presence of PSA (and only then), cAb-PSA-dAb sandwich complexes brought Au nanopillars and QDs within the distance range of circa 10 to 20 nm (Fig. 12F), such that the PL of QDs could be enhanced. Unbound QDs could be flushed away by a simple washing step. Using a conventional fluorescence microscope coupled to a CCD spectrometer (Fig. 12G), the authors accomplished a LOD below 100 pg mL−1 PSA with only 100 ms integration time (Fig. 12H). Unfortunately, EHFs of ∼4 to 8 at the top rims and ∼11 to 20 at the bottom rims of the nanopillars were only simulated and not experimentally verified. Although the relatively high sensitivity and low LOD confirmed an excellent assay performance, it would have been interesting to compare the Au nanopillar enhanced QD PL sensor to a QD sensor on a glass slide without Au nanopillars. Nevertheless, the incorporation of such plasmon-enhanced metal–QD nanoarray chips into microfluidic setups may become a useful tool for point-of-care diagnostics.
6. Conclusions and future perspectives
Metal–QD nanohybrids confine two different worlds of optics (plasmonics and luminescence) within the three-dimensional nanospace of a few tens of nanometers in every direction. Controlling the photophysical properties of metal–QD nanohybrids requires to place, orient, and possibly move the distinct metal and QD components with nanometric or even sub-nanometric precision. While both components influence each other's optical properties, they already have a complicated optical life on their own, where wavelength (or energy), intensity (or cross section), and lifetime (or rate) of absorption, scattering, and luminescence are defined by their size, shape, structure, composition, and environment. These sophisticated properties of metal–QD nanohybrids are two sides of the same coin. On the one side, they offer a myriad of possibilities to design new hybrid materials with properties that are unreachable within a single material and to use them for advancing methods, devices, and technologies in physics, chemistry, and biology. On the other side, understanding, control, reliability, reproducibility, and improvement of the new properties are extremely difficult to accomplish and require an enormous amount of experimental and theoretical work.Clearly, controlled enhancement and/or quenching of QD fluorescence via plasmonic metal nanostructures is a highly versatile and interesting field of research. Many experimental studies have confirmed the basic theoretical expectations, i.e., close distances (below circa 5 nm) lead to efficient quenching, maximum enhancement is found within a distance range of approximately 5 to 30 nm, plasmonic hotspots result in significantly stronger enhancement, bad absorbers and emitters are easier to enhance than good ones, and spectral overlap of LSPR and absorption and/or fluorescence spectra influences both enhancement and quenching. The current state of knowledge concerning fluorescence quenching via energy transfer from QDs to metal is significantly more advanced than the one related to plasmon-enhanced QD fluorescence. The former is a relatively simple “The closer the better and maximum spectral overlap, please!” issue, whereas the latter could probably be described by a more complicated “Close is great but not too great because there is strong quenching. So please move further away but not too far because enhancement strongly decreases with distance! And can you please overlap the LSPR band with both absorption and emission spectrum. And only the scattering part of the LSPR band please, because the absorption part will also lead to quenching!”. In other words, the sweet-spot of fluorescence enhancement is difficult to predict concerning distance, wavelength, and intensity (enhancement factor). Moreover, the distance range for efficient enhancement can be very narrow and must be experimentally determined for every new metal–QD nanohybrid.
With those considerations in mind, it is not very surprising that fluorescence quenching of QDs is mainly performed with one single material (Au) and one single nanostructure (spherical NP), whereas enhancement employs a large variety of both materials and nanostructures as well as their combinations. Surface-based metal–QD nanohybrids offer the best control of position and orientation and are therefore the system of choice for designing plasmon-enhanced QD fluorescence devices. Because hotspots are localized on only a small fraction of entire metal nanostructures, large enhancements are most likely to be detected for single to a few QDs that are localized at exactly those spots. Selectively measuring those spots is significantly easier on surface-based nanostructures compared to metal–QD nanohybrids in solution, which usually provide measurements on the ensemble level. Fixing positions and orientations of both metals and QD nanomaterials in solution is also more challenging. Consequently, solution-based metal–QD nanohybrids for plasmon-enhanced fluorescence have been developed much less frequently than surface-based ones.
Combining the nanoworld of plasmonic fluorescence enhancement with biomolecular analysis is intriguing because many chemical and biological molecules and functions have the same nanometric dimensions. However, when the inorganic and organic nanocosmoses are combined with the aim of exploiting the benefits of both, things usually get even more complicated and elusive.162 Although biomolecules have been used as spacers to separate metal and QD nanostructures at defined distances for optimal fluorescence enhancement, biosensing requires the measurement of a binding event or a structural modification of a biomolecule. Such biosensing concepts are relatively easy to realize for fluorescence quenching (NSET and FRET biosensors) but somewhat more challenging for fluorescence enhancement. Using the biomolecular interaction to move the plasmon enhanced fluorescence in and out (high signal to noise ratio) of the enhancement sweet-spot, which is already difficult to determine for a fixed metal–QD nanohybrid, is an ambitious task that has so far been realized by only a few studies and with only moderate enhancement factors. Employing a fixed metal–QD nanohybrid and measuring the interaction of a chemical or biological target with the QDs via changes in the enhanced fluorescence has been a more common approach.
Despite all the challenges and problems discussed above, plasmon-enhanced fluorescence of QDs has come a long way and the development of biosensing concepts is only in its infancy particularly for practical applications. Bringing enhancement to the same or better application level than quenching will require a more profound analysis of metal–QD distances and orientations for different nanohybrid systems and with different biomolecules. In particular DNA will be well suited for such studies because distance (e.g., number of based) and rigidity (e.g., single and double stranded) can be relatively easily controlled. But also defined peptide systems or small protein binders may be amenable to investigate the influence of distance and orientation under biological conditions. Another intriguing path will be the combination of quenching and enhancement. With full biomolecular control of the distance and orientation between metals and QDs, the binding or conformational event could in principle move the system from total quenching into maximum enhancement, which would result in extremely high overall enhancement factors without the necessity of using hotspots. Thus, strong enhancement could also be implemented into ensemble measurements. Single-molecule experiments are extremely useful for the analysis of biological structures and dynamics and enhanced fluorescence signals in plasmonic hotspots would be well suited to make such measurements faster and more sensitive. In addition to adapting established metal–QD nanohybrids to biosensing and bioimaging for a better understanding and improvement of technologies, the development of new materials and material combinations is clearly very promising. The advancement of nanotechnology over the last few decades and the possibility of fabricating more and more complicated inorganic nanostructures (e.g., our recent development of interlocked Au nanocatenanes)163 holds enormous potential to improve both enhancement and quenching of metal–QD nanohybrids as well as to move into optimized wavelength ranges (e.g., near infrared) for bioimaging. The nanospace is small but still offers unlimited possibilities of organic and inorganic material design, which will continue to make the development and application of plasmon-enhanced fluorescence of QDs and other luminescent nanomaterials a highly interesting field in the future.
Abbreviations
| 2PA | Two-photon absorption |
| 2PE | Two-photon excitation |
| Ab | Antibody |
| ACE2 | Angiotensin converting enzyme 2 |
| AgNP | Silver nanoparticle |
| AMP | Ampicillin |
| AuNP | Gold nanoparticle |
| AuNR | Gold nanorod |
| AuNU | Gold nanourchin |
| BSA | Bovine serum albumin |
| cAb | Capture antibody |
| cDNA | Complementary DNA |
| CP | Cleavable peptide |
| CTAB | Cetyltrimethylammonium bromide |
| dAb | Detection antibody |
| DBCO | Dibenzocyclooctyne |
| DNA | Deoxyribonucleic acid |
| EGFR | Epidermal growth factor receptor |
| EHF | Enhancement factor |
| ELISA | Enzyme-linked immunosorbent assay |
| FANC | Fluorescence-amplified nanocuboid |
| FRET | Förster resonance energy transfer |
| His6 | Hexahistidine |
| HPLC | High-performance liquid chromatography |
| IRF | Instrument response function |
| ITO | Indium tin oxide |
| LDS | Local density of states |
| LOD | Limit of detection |
| LSP | Localized surface plasmons |
| LSPR | Local surface plasmon resonance |
| MEF | Metal-enhanced fluorescence |
| miR | microRNA |
| MMP2 | Matrix metalloproteinase-2 |
| NB | Nanobody |
| NIR | Near infrared |
| NP | Nanoparticle |
| NSET | Nanosurface energy transfer |
| NVET | Nanovolume energy transfer |
| PBS | Phosphate-buffered saline |
| PE | Polyelectrolyte |
| PEF | Plasmon-enhanced fluorescence |
| PEG | Polyethylene glycol |
| PL | Photoluminescence |
| PMMA | Poly(methyl methacrylate) |
| PQF | Plasmon-quenched fluorescence |
| PSA | Prostate-specific antigen |
| PVA | Polyvinyl alcohol |
| PVP | Polyvinylpyrrolidone |
| QD | Quantum dot |
| RBD | Receptor binding domain |
| RNA | Ribonucleic acid |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SEF | Surface enhanced fluorescence |
| SERS | Surface-enhanced Raman scattering |
| TECP | Tris(2-carboxyethyl)phosphine |
| UCNP | Upconversion nanoparticle |
| UV | Ultraviolet |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Brain Pool program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (2021H1D3A2A0204958912) and the National Research Foundation of Korea (NRF) grants funded by the Korean government (MIST) (NRF-2017R1A5A1015365 and NRF-2021R1A2C3010083) for financial support.References
- J. Probst, S. Dembski, M. Milde and S. Rupp, Expert Rev. Mol. Diagn., 2012, 12, 49 CrossRef CAS PubMed.
- S. Zeng, D. Baillargeat, H.-P. Ho and K.-T. Yong, Chem. Soc. Rev., 2014, 43, 3426 RSC.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743 RSC.
- A. Csáki, O. Stranik and W. Fritzsche, Expert Rev. Mol. Diagn., 2018, 18, 279 CrossRef PubMed.
- J.-F. Masson, Analyst, 2020, 145, 3776 RSC.
- H. Chang, W.-Y. Rho, B. S. Son, J. Kim, S. H. Lee, D. H. Jeong and B.-H. Jun, Adv. Exp. Med. Biol., 2021, 1309, 133 CrossRef CAS PubMed.
- W. R. Algar, M. Massey, K. Rees, R. Higgins, K. D. Krause, G. H. Darwish, W. J. Peveler, Z. Xiao, H.-Y. Tsai, R. Gupta, K. Lix, M. V. Tran and H. Kim, Chem. Rev., 2021, 121, 9243 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS PubMed.
- L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- G. Sun, Y. Xie, L. Sun and H. Zhang, Nanoscale Horiz., 2021, 6, 766 RSC.
- S. Silvi and A. Credi, Chem. Soc. Rev., 2015, 44, 4275 RSC.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792 RSC.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed.
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946 CrossRef CAS PubMed.
- H. Kwee Lee, Y. Hong Lee, C. S. Lin Koh, G. Chuong Phan-Quang, X. Han, C. Leng Lay, H. Y. Fan Sim, Y.-C. Kao, Q. An and X. Yi Ling, Chem. Soc. Rev., 2019, 48, 731 RSC.
- T. L. Jennings, M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2006, 128, 5462 CrossRef CAS PubMed.
- C. Chen and N. Hildebrandt, TrAC Trends Anal. Chem., 2020, 123, 115748 CrossRef CAS.
- M. Pelton, Nat. Photonics, 2015, 9, 427 CrossRef CAS.
- FRET-Förster Resonance Energy Transfer: From Theory to Applications, ed. I. L. Medintz, N. Hildebrandt, John Wiley & Sons, 2013 Search PubMed.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Diaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267 CrossRef CAS PubMed.
- T. Teranishi, M. Eguchi, M. Kanehara and S. Gwo, J. Mater. Chem., 2011, 21, 10238 RSC.
- M. Couture, S. S. Zhao and J.-F. Masson, Phys. Chem. Chem. Phys., 2013, 15, 11190 RSC.
- Handbook of Surface Plasmon Resonance, ed. Richard B. M. Schasfoort, The Royal Society of Chemistry, 2017 Search PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef CAS.
- J.-E. Park, M. Kim, J.-H. Hwang and J.-M. Nam, Small Methods, 2017, 1, 1600032 CrossRef.
- S. Kim, J.-M. Kim, J.-E. Park and J.-M. Nam, Adv. Mater., 2018, 30, 1704528 CrossRef PubMed.
- S. Go, S. Yoo, J. Son, S. Lee, J. Lee, S. Lee, J. Kim, M. Park, W. Park, J.-M. Kim, J.-M. Nam and S. Park, Nano Lett., 2022, 22, 1734 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, 2006 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013 Search PubMed.
- N. Hildebrandt, in FRET – Förster Reson. Energy Transf. Theory Appl. ed. I. Medintz, N. Hildebrandt, Wiley VCH, Weinheim, 2013, pp. 105–164 Search PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815 CrossRef CAS PubMed.
- B. N. J. Persson and N. D. Lang, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 5409 CrossRef CAS.
- C. Curutchet, A. Franceschetti, A. Zunger and G. D. Scholes, J. Phys. Chem. C, 2008, 112, 13336 CrossRef CAS.
- G. Sun and J. B. Khurgin, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 85, 063410 CrossRef.
- M. Bauch, K. Toma, M. Toma, Q. Zhang and J. Dostalek, Plasmonics, 2014, 9, 781 CrossRef CAS PubMed.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2014, 140, 386 RSC.
- J.-F. Li, C.-Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962 RSC.
- J. B. Khurgin and G. Sun, J. Opt. Soc. Am. B, 2009, 26, B83 CrossRef CAS.
- J. B. Khurgin, G. Sun and R. A. Soref, Appl. Phys. Lett., 2009, 94, 071103 CrossRef.
- G. Sun and J. B. Khurgin, IEEE J. Sel. Top. Quantum Electron., 2011, 17, 110 CAS.
- G. Sun, J. B. Khurgin and D. P. Tsai, Opt. Lett., 2012, 37, 1583 CrossRef PubMed.
- G. Sun, J. B. Khurgin and R. A. Soref, Appl. Phys. Lett., 2009, 94, 101103 CrossRef.
- J. B. Khurgin, Nat. Nanotechnol., 2015, 10, 2 CrossRef CAS PubMed.
- J. B. Khurgin and G. Sun, Opt. Express, 2015, 23, 30739 CrossRef CAS PubMed.
- G. Sun, J. B. Khurgin and A. Bratkovsky, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045415 CrossRef.
- J. B. Khurgin and G. Sun, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 88, 053838 CrossRef.
- M. Ha, J.-H. Kim, M. You, Q. Li, C. Fan and J.-M. Nam, Chem. Rev., 2019, 119, 12208 CrossRef CAS PubMed.
- Y. Jeong, Y.-M. Kook, K. Lee and W.-G. Koh, Biosens. Bioelectron., 2018, 111, 102 CrossRef CAS PubMed.
- R. Knoblauch and C. D. Geddes, Review of Advances in Metal-Enhanced Fluorescence, Springer, New York, 2019 Search PubMed.
- S. M. Fothergill, C. Joyce and F. Xie, Nanoscale, 2018, 10, 20914 RSC.
- I. A. Larmour and D. Graham, Analyst, 2011, 136, 3831 RSC.
- M. T. Yaraki and Y. N. Tan, Chem. – Asian J., 2020, 15, 3180 CrossRef CAS PubMed.
- J.-M. Kim, C. Lee, Y. Lee, J. Lee, S.-J. Park, S. Park and J.-M. Nam, Adv. Mater., 2021, 33, 2006966 CrossRef CAS PubMed.
- J.-E. Park, J. Kim and J.-M. Nam, Chem. Sci., 2017, 8, 4696 RSC.
- Y. Cheng, T. Stakenborg, P. Van Dorpe, L. Lagae, M. Wang, H. Chen and G. Borghs, Anal. Chem., 2011, 83, 1307 CrossRef CAS PubMed.
- Z. Zhu, P. Yuan, S. Li, M. Garai, M. Hong and Q.-H. Xu, ACS Appl. Bio Mater., 2018, 1, 118 CrossRef CAS.
- J.-H. Hwang, S. Park, J. Son, J. W. Park and J.-M. Nam, Nano Lett., 2021, 21, 2132 CrossRef CAS PubMed.
- M. Cardoso Dos Santos, W. R. Algar, I. L. Medintz and N. Hildebrandt, TrAC Trends Anal. Chem., 2020, 125, 115819 CrossRef CAS.
- P. T. Snee, TrAC Trends Anal. Chem., 2020, 123, 115750 CrossRef CAS.
- A. Soldado, L. C. Barrio, M. Díaz-Gonzalez, A. de la Escosura-Muñiz and J. M. Costa-Fernandez, in Adv. Clin. Chem., ed. G. S. Makowski, Elsevier, 2022, pp. 1–40 Search PubMed.
- L.-L. Chen, L. Zhao, Z.-G. Wang, S.-L. Liu and D.-W. Pang, Small, 2022, 18, 2104567 CrossRef CAS PubMed.
- N. Hildebrandt, L. J. Charbonnière, M. Beck, R. F. Ziessel and H.-G. Löhmannsröben, Angew. Chem., Int. Ed., 2005, 44, 7612 CrossRef CAS PubMed.
- M.-K. So, C. Xu, A. M. Loening, S. S. Gambhir and J. Rao, Nat. Biotechnol., 2006, 24, 339 CrossRef CAS PubMed.
- M. J. Roberti, L. Giordano, T. M. Jovin and E. A. Jares-Erijman, Chem. Phys. Chem., 2011, 12, 563 CrossRef CAS PubMed.
- L. Mattsson, K. D. Wegner, N. Hildebrandt and T. Soukka, RSC Adv., 2015, 5, 13270 RSC.
- W. J. Peveler and W. R. Algar, ACS Chem. Biol., 2018, 13, 1752 CrossRef CAS PubMed.
- H.-Y. Tsai, H. Kim, M. Massey, K. D. Krause and W. R. Algar, Methods Appl. Fluoresc., 2019, 7, 042001 CrossRef CAS PubMed.
- C. M. Spillmann, M. G. Ancona, S. Buckhout-White, W. R. Algar, M. H. Stewart, K. Susumu, A. L. Huston, E. R. Goldman and I. L. Medintz, ACS Nano, 2013, 7, 7101 CrossRef CAS PubMed.
- S. A. Díaz, G. Lasarte Aragonés, S. Buckhout-White, X. Qiu, E. Oh, K. Susumu, J. S. Melinger, A. L. Huston, N. Hildebrandt and I. L. Medintz, J. Phys. Chem. Lett., 2017, 8, 2182 CrossRef PubMed.
- M. Cardoso Dos Santos, I. Colin, G. Ribeiro Dos Santos, K. Susumu, M. Demarque, I. L. Medintz and N. Hildebrandt, Adv. Mater., 2020, 32, 2003912 CrossRef CAS PubMed.
- R. Su, Y.-T. Wu, S. Doulkeridou, X. Qiu, T. J. Sorensen, K. Susumu, I. L. Medintz, P. M. P. van Bergen en Henegouwen and N. Hildebrandt, Angew. Chem., Int. Ed., 2022, 61, e202207797 CAS.
- T. Sen and A. Patra, J. Phys. Chem. C, 2012, 116, 17307 CrossRef CAS.
- P. Wang, Z. Lin, X. Su and Z. Tang, Nano Today, 2017, 12, 64 CrossRef CAS.
- Y. Zhang, G. Wang, L. Yang, F. Wang and A. Liu, Coord. Chem. Rev., 2018, 370, 1 CrossRef CAS.
- P. F. Gao, Y. F. Li and C. Z. Huang, TrAC Trends Anal. Chem., 2020, 124, 115805 CrossRef CAS.
- L. Tu, Gold Bull., 2022, 55, 169 CrossRef CAS.
- N. H. Anh, M. Q. Doan, N. X. Dinh, T. Q. Huy, D. Q. Tri, L. T. N. Loan, B. V. Hao and A.-T. Le, RSC Adv., 2022, 12, 10950 RSC.
- J. E. Kim, J. H. Choi, M. Colas, D. H. Kim and H. Lee, Biosens. Bioelectron., 2016, 80, 543 CrossRef CAS PubMed.
- L. Cui, C. Li, B. Tang and C. Zhang, Analyst, 2018, 143, 2469 RSC.
- M. Chern, J. C. Kays, S. Bhuckory and A. M. Dennis, Methods Appl. Fluoresc., 2019, 7, 012005 CrossRef CAS PubMed.
- Z. S. Pehlivan, M. Torabfam, H. Kurt, C. Ow-Yang, N. Hildebrandt and M. Yuce, Microchim. Acta, 2019, 186, 563 CrossRef CAS PubMed.
- E. Oh, A. L. Huston, A. Shabaev, A. Efros, M. Currie, K. Susumu, K. Bussmann, R. Goswami, F. K. Fatemi and I. L. Medintz, Sci. Rep., 2016, 6, 35538 CrossRef CAS PubMed.
- S. Yamazaki, E. Oh, K. Susumu, I. L. Medintz and A. M. Scott, J. Phys. Chem. C, 2021, 125, 12073 CrossRef CAS.
- O.-V. Pham-Nguyen, J. Shin, Y. Park, S. Jin, S. R. Kim, Y. M. Jung and H. S. Yoo, Biomacromolecules, 2022, 23, 3130 CrossRef CAS PubMed.
- W. Yu, A. Hao, Y. Mei, Y. Yang and C. Dai, Microchem. J., 2022, 179, 107454 CrossRef CAS.
- M. Torabfam, H. Kurt, M. K. Bayazıt and M. Yüce, Sens. Diagn., 2022, 1, 765 RSC.
- K. Gorshkov, K. Susumu, J. Chen, M. Xu, M. Pradhan, W. Zhu, X. Hu, J. C. Breger, M. Wolak and E. Oh, ACS Nano, 2020, 14, 12234 CrossRef CAS PubMed.
- O. Kulakovich, N. Strekal, A. Yaroshevich, S. Maskevich, S. Gaponenko, I. Nabiev, U. Woggon and M. Artemyev, Nano Lett., 2002, 2, 1449 CrossRef CAS.
- K. T. Shimizu, W. K. Woo, B. R. Fisher, H. J. Eisler and M. G. Bawendi, Phys. Rev. Lett., 2002, 89, 117401 CrossRef CAS PubMed.
- M. T. Zin, K. Leong, N.-Y. Wong, H. Ma, M. Sarikaya and A. K.-Y. Jen, Nanotechnology, 2009, 20, 015305 CrossRef PubMed.
- K. Leong, Y. Chen, D. J. Masiello, M. T. Zin, M. Hnilova, H. Ma, C. Tamerler, M. Sarikaya, D. S. Ginger and A. K.-Y. Jen, Adv. Funct. Mater., 2010, 20, 2675 CrossRef CAS.
- M.-X. Li, W. Zhao, G.-S. Qian, Q.-M. Feng, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2016, 52, 14230 RSC.
- I. G. Theodorou, Z. a R. Jawad, H. Qin, E. O. Aboagye, A. E. Porter, M. P. Ryan and F. Xie, Nanoscale, 2016, 8, 12869 RSC.
- E. Bermúdez Ureña, M. P. Kreuzer, S. Itzhakov, H. Rigneault, R. Quidant, D. Oron and J. Wenger, Adv. Mater., 2012, 24, OP314 Search PubMed.
- J.-H. Song, T. Atay, S. Shi, H. Urabe and A. V. Nurmikko, Nano Lett., 2005, 5, 1557 CrossRef CAS PubMed.
- H. Yin, J. Yi, Z.-W. Yang, Z.-Y. Xu, S.-J. Xie, L. Li, C.-Y. Li, J. Xu, H. Zhang, S.-J. Zhang, J.-F. Li and Z.-Q. Tian, Nano Energy, 2017, 42, 232 CrossRef CAS.
- B. Peng, Z. Li, E. Mutlugun, P. L. Hernandez Martinez, D. Li, Q. Zhang, Y. Gao, H. V. Demir and Q. Xiong, Nanoscale, 2014, 6, 5592 RSC.
- K. Ray, R. Badugu and J. R. Lakowicz, J. Am. Chem. Soc., 2006, 128, 8998 CrossRef CAS PubMed.
- P. P. Pompa, L. Martiradonna, A. D. Torre, F. D. Sala, L. Manna, M. De Vittorio, F. Calabi, R. Cingolani and R. Rinaldi, Nat. Nanotechnol., 2006, 1, 126 CrossRef CAS PubMed.
- V. K. Komarala, Y. P. Rakovich, A. L. Bradley, S. J. Byrne, Y. K. Gun’ko, N. Gaponik and A. Eychmueller, Appl. Phys. Lett., 2006, 89, 253118 CrossRef.
- P. P. Pompa, L. Martiradonna, A. D. Torre, L. Carbone, L. L. del Mercato, L. Manna, M. De Vittorio, F. Calabi, R. Cingolani and R. Rinaldi, Sens. Actuators B Chem., 2007, 126, 187 CrossRef CAS.
- I. M. Soganci, S. Nizamoglu, E. Mutlugun, O. Akin and H. V. Demir, Opt. Express, 2007, 15, 14289 CrossRef CAS PubMed.
- Y. Chen, K. Munechika, I. Jen-La Plante, A. M. Munro, S. E. Skrabalak, Y. Xia and D. S. Ginger, Appl. Phys. Lett., 2008, 93, DOI:10.1063/1.2956391.
- Y.-H. Chan, J. Chen, S. E. Wark, S. L. Skiles, D. H. Son and J. D. Batteas, ACS Nano, 2009, 3, 1735 CrossRef CAS PubMed.
- C. Vion, P. Spinicelli, L. Coolen, C. Schwob, J.-M. Frigerio, J.-P. Hermier and A. Maître, Opt. Express, 2010, 18, 7440 CrossRef CAS PubMed.
- L. Lu, D. Chen, F. Sun, X. Ren, Z. Han and G. Guo, Chem. Phys. Lett., 2010, 492, 71 CrossRef CAS.
- K. Munechika, Y. Chen, A. F. Tillack, A. P. Kulkarni, I. Jen-La Plante, A. M. Munro and D. S. Ginger, Nano Lett., 2010, 10, 2598 CrossRef CAS PubMed.
- K. Munechika, Y. Chen, A. F. Tillack, A. P. Kulkarni, I. Jen-La Plante, A. M. Munro and D. S. Ginger, Nano Lett., 2011, 11, 2725 CrossRef CAS PubMed.
- H. Langhuth, S. Frédérick, M. Kaniber, J. J. Finley and U. Rührmair, J. Fluoresc., 2011, 21, 539 CrossRef CAS PubMed.
- S. R. Ahmed, H. R. Cha, J. Y. Park, E. Y. Park, D. Lee and J. Lee, Nanoscale Res. Lett., 2012, 7, 438 CrossRef PubMed.
- L. Liu, X. Xu, T. Luo, Y. Liu, Z. Yang and J. Lei, Solid State Commun., 2012, 152, 1103 CrossRef CAS.
- L. Hu, H. Wu, C. Cai, T. Xu, B. Zhang, S. Jin, Z. Wan and X. Wei, J. Phys. Chem. C, 2012, 116, 11283 CrossRef CAS.
- S. H. Choi, B. Kwak, B. Han and Y. L. Kim, Opt. Express, 2012, 20, 16785 CrossRef CAS.
- N. A. Harun, M. J. Benning, B. R. Horrocks and D. A. Fulton, Nanoscale, 2013, 5, 3817 RSC.
- S. Jin, E. DeMarco, M. J. Pellin, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Phys. Chem. Lett., 2013, 4, 3527 CrossRef CAS.
- L. Zhang, Y. Song, T. Fujita, Y. Zhang, M. Chen and T.-H. Wang, Adv. Mater., 2014, 26, 1289 CrossRef CAS PubMed.
- B.-T. Liu, T.-H. Liao, S. Tseng and M.-H. Lee, Appl. Surf. Sci., 2014, 292, 615 CrossRef CAS.
- S.-Y. Cho, H.-J. Jeon, H.-W. Yoo, K. M. Cho, W.-B. Jung, J.-S. Kim and H.-T. Jung, Nano Lett., 2015, 15, 7273 CrossRef CAS PubMed.
- T. B. Hoang, G. M. Akselrod, C. Argyropoulos, J. Huang, D. R. Smith and M. H. Mikkelsen, Nat. Commun., 2015, 6, 7788 CrossRef CAS PubMed.
- A. J. Meixner, R. Jaeger, S. Jaeger, A. Braeuer, K. Scherzinger, J. Fulmes, S. zur, O. Krockhaus, D. A. Gollmer, D. P. Kern and M. Fleischer, Faraday Discuss., 2015, 184, 321 RSC.
- N.-Y. Kim, S.-H. Hong, J.-W. Kang, N. Myoung, S.-Y. Yim, S. Jung, K. Lee, C. W. Tu and S.-J. Park, RSC Adv., 2015, 5, 19624 RSC.
- W. Kong, B. Zhang, R. Li, F. Wu, T. Xu and H. Wu, Appl. Surf. Sci., 2015, 327, 394 CrossRef CAS.
- L. J. Higgins, V. D. Karanikolas, C. A. Marocico, A. P. Bell, T. C. Sadler, P. J. Parbrook and A. L. Bradley, Opt. Express, 2015, 23, 1377 CrossRef CAS PubMed.
- H. Y. Liang, H. G. Zhao, Z. P. Li, C. Harnagea and D. L. Ma, Nanoscale, 2016, 8, 4882 RSC.
- H. Takata, H. Naiki, L. Wang, H. Fujiwara, K. Sasaki, N. Tamai and S. Masuo, Nano Lett., 2016, 16, 5770 CrossRef CAS PubMed.
- T. A. R. Purcell, M. Galanty, S. Yochelis, Y. Paltiel and T. Seideman, J. Phys. Chem. C, 2016, 120, 21837 CrossRef CAS.
- L. J. Higgins, C. A. Marocico, V. D. Karanikolas, A. P. Bell, J. J. Gough, G. P. Murphy, P. J. Parbrook and A. L. Bradley, Nanoscale, 2016, 8, 18170 RSC.
- A. Kannegulla, Y. Liu, B. Wu and L.-J. Cheng, J. Phys. Chem. C, 2018, 122, 770 CrossRef CAS.
- K. Gu, H. Peng, S. Hua, Y. Qu and D. Yang, Nanomaterials, 2019, 9, 770 CrossRef CAS PubMed.
- H. Mundoor, E. M. Cruz-Colon, S. Park, Q. Liu, I. I. Smalyukh and J. Van De Lagemaat, Opt. Express, 2020, 28, 5459 CrossRef PubMed.
- Y. Fu, J. Zhang and J. R. Lakowicz, Chem. Commun., 2009, 313 RSC.
- K. Yang, X. Yin, Y. Yan, K. Yang, P. Pi, S. Xu, J. Fang, Q. Li and X. Wen, Chem. Eng. J., 2021, 426, 131571 CrossRef CAS.
- F. Shan, D. Su, W. Li, W. Hu and T. Zhang, AIP Adv., 2018, 8, 025219 CrossRef.
- B. C. Marin, S.-W. Hsu, L. Chen, A. Lo, D. W. Zwissler, Z. Liu and A. R. Tao, ACS Photonics, 2016, 3, 526 CrossRef CAS.
- N. Liu, B. S. Prall and V. I. Klimov, J. Am. Chem. Soc., 2006, 128, 15362 CrossRef CAS PubMed.
- R. Li, S. Xu, C. Wang, H. Shao, Q. Xu and Y. Cui, Chem. Phys. Chem., 2010, 11, 2582 CrossRef CAS PubMed.
- D. Nepal, L. F. Drummy, S. Biswas, K. Park and R. A. Vaia, ACS Nano, 2013, 7, 9064 CrossRef CAS PubMed.
- H. Naiki, A. Masuhara, S. Masuo, T. Onodera, H. Kasai and H. Oikawa, J. Phys. Chem. C, 2013, 117, 2455 CrossRef CAS.
- A. E. Ragab, A.-S. Gadallah, T. Da Ros, M. B. Mohamed and I. M. Azzouz, Opt. Commun., 2014, 314, 86 CrossRef CAS.
- S. T. Kochuveedu, T. Son, Y. Lee, M. Lee, D. Kim and D. H. Kim, Sci. Rep., 2014, 4, 4735 CrossRef PubMed.
- K. Wu, J. Zhang, S. Fan, J. Li, C. Zhang, K. Qiao, L. Qian, J. Han, J. Tang and S. Wang, Chem. Commun., 2015, 51, 141 RSC.
- B. Ji, E. Giovanelli, B. Habert, P. Spinicelli, M. Nasilowski, X. Xu, N. Lequeux, J.-P. Hugonin, F. Marquier, J.-J. Greffet and B. Dubertret, Nat. Nanotechnol., 2015, 10, 170 CrossRef CAS PubMed.
- M. Peng, G. Bi, C. Cai, G. Guo, H. Wu and Z. Xu, Opt. Lett., 2016, 41, 1466 CrossRef CAS PubMed.
- A. K. Tobias and M. Jones, J. Phys. Chem. C, 2019, 123, 1389 CrossRef CAS.
- L. Trotsiuk, A. Muravitskaya, O. Kulakovich, D. Guzatov, A. Ramanenka, Y. Kelestemur, H. V. Demir and S. Gaponenko, Nanotechnology, 2020, 31, 105201 CrossRef CAS PubMed.
- J. Cao, H. Zhang, X. Pi, D. Li and D. Yang, Nanoscale Adv., 2021, 3, 4810 RSC.
- R. Robelek, L. Niu, E. Schmid and W. Knoll, Anal. Chem., 2004, 76, 6160 CrossRef CAS PubMed.
- R. Robelek, F. D. Stefani and W. Knoll, Phys. Status Solidi – Appl. Mater. Sci., 2006, 203, 3468 CrossRef CAS.
- Y.-H. Chan, J. Chen, Q. Liu, S. E. Wark, D. H. Son and J. D. Batteas, Anal. Chem., 2010, 82, 3671 CrossRef CAS PubMed.
- S. Xue, X.-F. Jiang, G. Zhang, H. Wang, Z. Li, X. Hu, M. Chen, T. Wang, A. Luo, H.-P. Ho, S. He and X. Xing, ACS Sens., 2020, 5, 1002 CrossRef CAS PubMed.
- H. Y. Song, T. I. Wong, A. Sadovoy, L. Wu, P. Bai, J. Deng, S. Guo, Y. Wang, W. Knoll and X. Zhou, Lab Chip, 2015, 15, 253 RSC.
- H. Y. Song, T. I. Wong, S. Guo, J. Deng, C. Tan, S. Gorelik and X. Zhou, Sens. Actuators, B, 2015, 221, 207 CrossRef CAS.
- Y.-Q. Li, L.-Y. Guan, H.-L. Zhang, J. Chen, S. Lin, Z.-Y. Ma and Y.-D. Zhao, Anal. Chem., 2011, 83, 4103 CrossRef CAS PubMed.
- H. Chen and Y. Xia, Anal. Chem., 2014, 86, 11062 CrossRef CAS PubMed.
- T. Jiang, N. Yin, L. Liu, J. Song, Q. Huang, L. Zhu and X. Xu, RSC Adv., 2014, 4, 23630 RSC.
- Y. Liu, N. Kang, X. Ke, D. Wang, L. Ren and H. Wang, RSC Adv., 2016, 6, 27395 RSC.
- S. Gong and Y. Xia, Chem. Commun., 2016, 52, 9660 RSC.
- X. Wang, L. Zhang, A. Hao, Z. Shi, C. Dai, Y. Yang and H. Huang, ACS Appl. Nano Mater., 2020, 3, 9796 CrossRef CAS.
- J. Xu, X. Qiu and N. Hildebrandt, Nano Lett., 2021, 21, 4802 CrossRef CAS PubMed.
- Y. Kim and J.-M. Nam, Nat. Synth., 2022, 1, 649 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |