
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small-molecule photoswitches for fluorescence bioimaging: engineering and applications
Magdalena
Olesińska-Mönch
and
Claire
Deo
 *
*
Cell Biology and Biophysics Unit, European Molecular Biology Laboratory (EMBL), Heidelberg 69117, Germany. E-mail: claire.deo@embl.de
First published on 9th January 2023
Abstract
Fluorescence microscopy has revolutionised our understanding of biological systems, enabling the visualisation of biomolecular structures and dynamics in complex systems. The possibility to reversibly control the optical or biochemical properties of fluorophores can unlock advanced applications ranging from super-resolution microscopy to the design of multi-stimuli responsive and functional biosensors. In this Highlight, we review recent progress in small-molecule photoswitches applied to biological imaging with an emphasis on molecular engineering strategies and promising applications, while underlining the main challenges in their design and implementation.
Introduction
Fluorescence microscopy is the modality of choice for the investigation of fundamental biological processes, enabling the visualisation of the structure and dynamics of biomolecules in living samples. This ultimately depends on the development of fluorescent probes, tailored to report on biological systems with exquisite spatiotemporal resolution. With the ever-growing complexity of biological questions, more sophisticated controllable and functional reporters are required. Small-molecule photoswitches, which undergo a reversible change in structural and electronic properties upon illumination, provide a unique opportunity to advance fluorescence imaging. Indeed, light is an excellent trigger for biological applications, as it is minimally invasive, offers superior spatiotemporal control and multiplexing capability. The reversible photocontrol of the optical and/or biochemical properties of fluorophores can enable a variety of applications. In particular, photoswitchable fluorophores are highly desirable tools for super-resolution microscopy (SRM), ranging from single molecule localisation-based methods (SMLM), including PALM and STORM to point-scanning techniques such as RESOLFT.1–5 Photoswitchable fluorophores can also facilitate the specific marking of a target of interest in complex samples or the detection of small fluorescence signals, exploiting the signal modulation to separate it from background. Finally, using photoswitches in the context of fluorescent biosensors can lead to sophisticated functional probes acting as controllable sensors and integrators, or able to provide quantitative information. In contrast with photoswitchable proteins,6 synthetic chromophores are small, highly tuneable and usually present superior photophysical properties. Accordingly, considerable effort has been dedicated to adapting small-molecule photoswitches to microscopy.In this Highlight, we provide an overview of recent developments and applications of synthetic photoswitches in fluorescence bioimaging. We focus on engineering strategies for photoswitchable fluorophores and biosensors and discuss the challenges that chemists face in the design of photoswitchable fluorescent systems, due to the numerous requirements of imaging experiments and complex photophysics of such probes.
1. Common classes of synthetic photoswitches for biological applications
Molecular photoswitches are synthetic compounds which undergo a reversible isomerisation upon illumination, forming species with distinct electronic or structural properties. Due to this behaviour, they can be used as building blocks to engineer sophisticated actuators designed to control various properties of molecular systems. Multiple families of photoswitches have been reported, and found applications in material science and pharmacology.7–9 In contrast, there have only been few examples of their use in fluorescence imaging, due to the demanding requirements of this modality. Indeed, to be successfully applied to fluorescence microscopy, photoswitches need to present biocompatibility, efficient photoswitching in aqueous conditions,10 in addition to fluorescence with the brightness and photostability of conventional imaging probes.Several parameters are used to quantify the properties of photoswitches. The wavelength of maximal absorption of the different isomers specifies the illumination required for switching, and the photoisomerisation quantum yield (Φ) quantifies the efficiency of the switching process. Upon illumination, a photostationary state (PSS) is reached, characterised by the proportions of the isomers at equilibrium. Some photoswitches can spontaneously relax to their stable isomer under ambient conditions and are called T-type switches. In contrast, P-type photoswitches present bi-stable isomers, and the reverse reaction can only occur upon illumination at a different wavelength. Generally, all these properties are strongly dependent on the substitution and environment of the photoswitch, which presents both opportunities and challenges for their optimisation. Numerous classes of synthetic photoswitches have been reported. In this section, we only focus on the main classes that have been exploited as fluorescent reporters and their relevant properties in this context, as in-depth reviews on each class of photoswitches are available.
1.1 Photoswitches based on E–Z isomerisation
Several families of switches undergo reversible E–Z photoisomerisation of either C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, NN or CN double bonds. One of their highly attractive features for engineering functional scaffolds is that they undergo large conformation and polarity change upon photoisomerisation.
C, NN or CN double bonds. One of their highly attractive features for engineering functional scaffolds is that they undergo large conformation and polarity change upon photoisomerisation.
Azobenzenes and their heteroaromatic derivatives are composed of two aryl rings connected by a NN double bond (Fig. 1a).11,12 The stable form is the planar E isomer, which converts to the metastable and bent Z isomer upon illumination with UV light. In the unsubstituted azobenzene scaffold, the E isomer displays a characteristic absorption maximum around 320 nm while the Z isomer presents an absorption maximum around 430 nm. Azobenzenes are typically T-type photoswitches, and the reverse isomerisation occurs spontaneously in the dark. However, the photophysical properties of azobenzene derivatives are highly dependent on the nature and substitution of the aryl rings which can result in red-shifting, and in some cases the Z to E transformation can be additionally photochemically induced with visible light. Azobenzenes are among the most widely used photoswitches in biological systems, presenting fast photoswitching, high photoisomerisation quantum yield and fatigue resistance. Their small size, tunability and ease of synthesis make them particularly attractive for this purpose, but azobenzenes can be reduced under physiological conditions which can impair their use in biology.
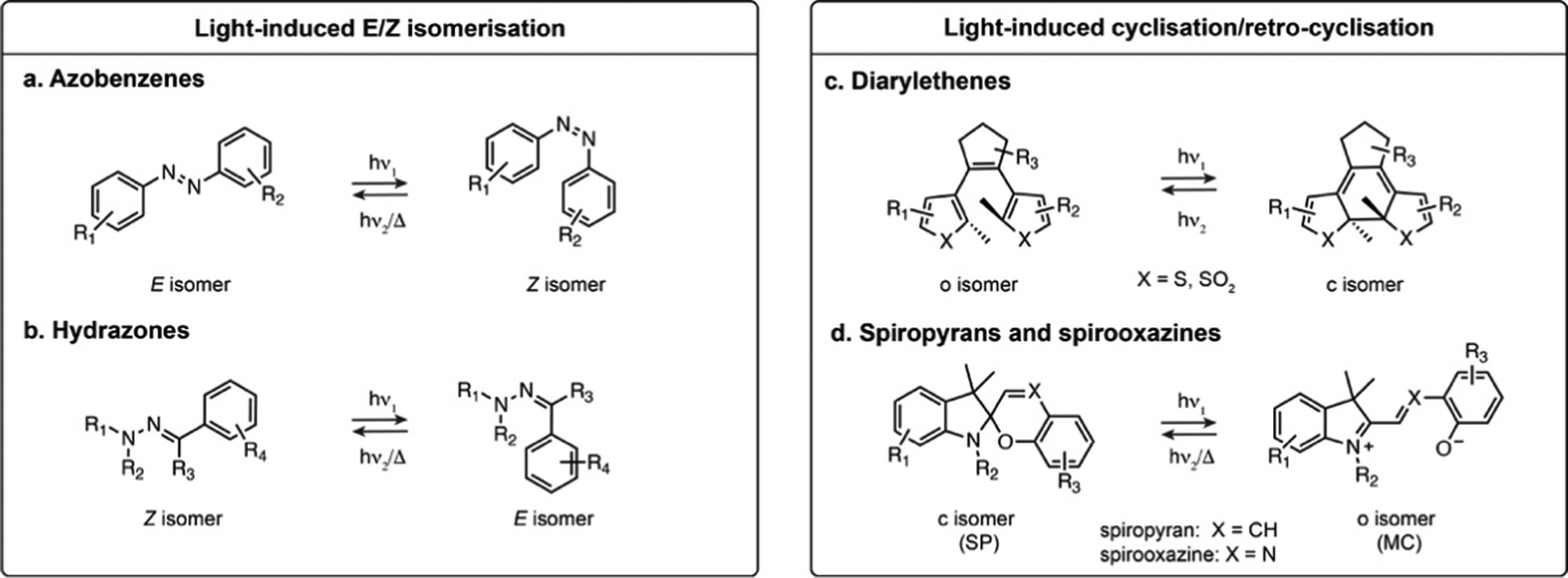 | ||
| Fig. 1 Representative structures and photoisomerisation of most common classes of photoswitches applied in fluorescence microscopy. R1–R4 indicate the most frequent substitution positions. | ||
Hydrazones present a CN–N motif, and upon illumination undergo isomerisation around the CN double bond (Fig. 1b).13,14 Depending on the substituents, hydrazones can be either T- or P-type switches, and exhibit positive or negative photochromism. Their maximal absorption is typically below 400 nm. While it has been shown that they retain photoswitching in aqueous medium, their structure-properties relationship in water has only been scarcely investigated.
1.2 Photoswitches based on cyclisation/retro-cyclisation
The second broadly defined category of photoswitches undergoes a reversible cyclisation upon illumination. Depending on the scaffolds, the change in geometry and polarity upon photoisomerisation can be more or less pronounced, however they have in common a change in electronic conjugation. This results in important spectral differences between the isomers, a particularly attractive feature for achieving near quantitative switching.Diarylethenes (DAEs) are composed of two aromatic rings connected by a CC double bond (Fig. 1c).15–17 The bridged double bond cannot undergo E–Z isomerisation, and upon illumination the compound exhibits an electrocyclisation. The most widely used derivatives are composed of a perfluorocyclopentadiene bridging group and thiophene derived heterocycles, yielding P-type photoswitches with complete thermal bistability. DAEs usually possess high fatigue resistance, fast photoisomerisation kinetics, and can achieve nearly pure PSS compositions. In addition, they can display fluorescence as one of their isomers, albeit with usually low fluorescence quantum yield (ΦF) in the presence of water. These properties make these switches highly attractive for imaging, but their use in biological systems remains scarce due to their high hydrophobicity, making them nearly insoluble in water.
Spiropyrans and their derivatives consist of an indoline and a chromene motifs connected through a spiro carbon atom (Fig. 1d).18,19 Upon illumination, the closed spiropyran (SP) form converts into the open isomer, merocyanine (MC). Although the cyclisation/retro-cyclisation is often presented as the photoisomerisation scheme, the mechanism is complex and additionally features E–Z isomerisation around the CX double bond. The SP form presents no absorption in the visible region, whereas the conjugated MC form absorbs between 500 and 600 nm and typically exhibits weak fluorescence. These T-type photoswitches can display either positive or negative photochromism in aqueous conditions. Their main limitation for biological applications is their moderate stability in water, as the MC form is prone to rapid hydrolysis.20,21
2. Photoswitchable fluorophores based on small-molecule switches
The reversible control of fluorescence emission using light is a very attractive feature for biological imaging. In particular, it can enable the specific marking of cells or subcellular features of interest, to precisely identify and track them in a sample. In addition, light-responsive fluorophores are key components for super-resolution microscopy techniques, to visualise cellular features below the diffraction limit.22,23 Consequently, substantial effort has been dedicated to exploiting established photoswitches for fluorescence control. Several approaches have been explored for this purpose and, in this section, we provide an overview of the different molecular engineering strategies: combining distinct photoswitchable and fluorescent chromophores (Section 2.1); fusing a photoswitch into an established fluorophore scaffold (Section 2.2); or exploiting the intrinsic fluorescence of certain photoswitches (Section 2.3).2.1 Photoswitchable fluorophores based on fluorescent and photoswitchable moieties.
As the vast majority of photoswitches is non-fluorescent, a straightforward approach for fluorescence photoswitching is to combine them with established fluorophores. Most existing systems rely on Förster resonance energy transfer (FRET) for fluorescence modulation, usually exploiting diarylethenes and spiropyrans as acceptors with a variety of fluorophores including small-molecule dyes, fluorescent proteins or luminescent nanomaterials. Only the conjugated isomer of the photoswitch presents an absorption band which overlaps with the fluorophore emission, resulting in efficient energy transfer and concomitant fluorescence quenching. Using this approach, fluorescence photoswitching was achieved in cells and living organisms using diverse nanosystems such as silica nanoparticles, polymeric structures, upconverting nanoparticles or dendrimeric clusters. For example, silica nanoparticles functionalised with cyanines and DAEs could be switched multiple cycles in live HeLa cells (1, Fig. 2a).24 Kim and co-workers developed dendrimers decorated with cyanine fluorophores and cross-linked with DAEs which could be internalised by living cells and zebrafish and showed 50% fluorescence quenching upon isomerisation.25,26 Fluorescence switching using upconversion nanoparticles decorated with DAEs was also shown in C. elegans and mouse.27,28 Recent examples demonstrated the use of photoswitchable FRET nanosystems for the specific marking of cells of interest, enabling their tracking or isolation from a sample pool. Using DAE-decorated fluorescent polymer particles attached to antibodies, Chiu et al. could turn-on fluorescence on the surface of cells of interest upon illumination. The high fluorescence turn-on (150-fold) of the particles allowed separation of target cells from tissue using fluorescence activated cell sorting (2, Fig. 2b).29 In another marking approach, O’Reilly et al. developed a barcoding system based on fluorescence lifetime.30 The authors developed an emissive polymeric structure containing spiropyran switches, and showed that the fluorescence lifetime of the particles could be controlled by photoisomerisation with multiplexing capability, which was then applied to lifetime imaging of mitochondria in living cells. Nanosystems based on FRET were also applied to SRM imaging, including SMLM imaging of lysosomes exploiting a DAE-naphthalimide dyad embedded in a polymeric nanoparticle.31 While most systems display “on-off” fluorescence behaviour, multichromophoric systems can also enable dual-colour fluorescence switching.32,33 Recently, triple colour fluorescence switching of particles was reported by Xu, Tian et al. using spiropyrans attached to a distyrylanthracene and exploiting both one- and two-photon fluorescence.34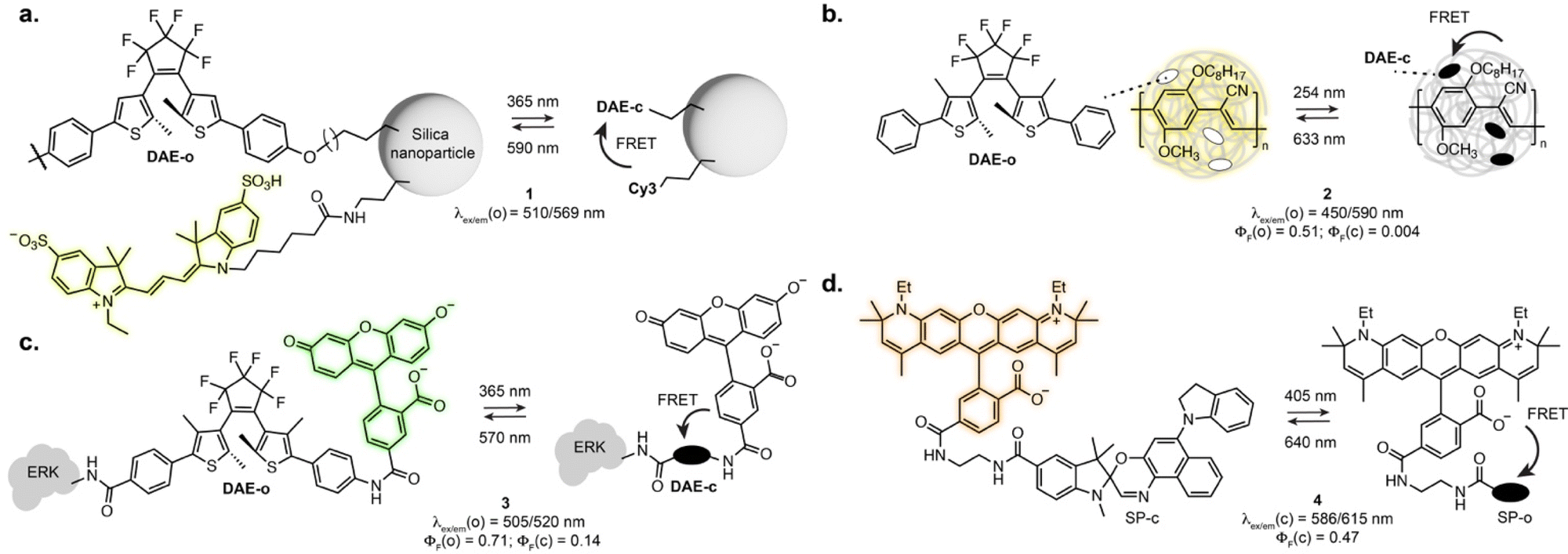 | ||
| Fig. 2 Examples of photoswitchable fluorophores based on inter- or intramolecular FRET between fluorescent and photoswitchable moieties: (a) DAEs and cyanines attached to silica nanoparticles; (b) DAEs embedded in a fluorescent polymeric particle; (c) a DAE-rhodamine dyad attached to an extracellular signal-regulated kinase (ERK); (d) a SP-rhodamine dyad. The properties reported were measured in: aqueous solution for 1 and 2; EtOH for 3; MeOH for 4. The notations (o) and (c) refer to the open and closed isomers, respectively. | ||
While nanostructures present attractive optical properties such as high brightness in the visible range, these systems are limited by poor water solubility, large size, limited cell permeability, and remain scarcely used for specific biomolecule labelling. Single molecule dyads can overcome some of these limitations. In an early example, Imato, Irie et al. demonstrated the use of a DAE-fluorescein dyad bearing a succinimidyl ester moiety to label proteins and studied the photophysical properties of the dyad adduct with a kinase in vitro (3, Fig. 2c).35 Dyads can also be built on small-molecule switches and fluorescent proteins, as demonstrated by the Marriott group that used spiropyran as a FRET acceptor for GFP.36 The SP switch was covalently attached in proximity to the GFP using a SNAP-Tag labelling fusion protein. Using an optical lock in detection approach, fluorescence signal could be detected even at low labelling efficiency. With synthetic fluorophores, several derivatives of rhodamines-spiropyran dyads showed fluorescence switching inside living cells.37–40 Recently, the Anderson group systematically investigated cyanine and rhodamine fluorophores in FRET pair with spiropyrans derivatives. Early examples showed far-red fluorescence photoswitching in live cells, albeit with low fatigue resistance.38 Further optimisation led to dyads suitable for RESOLFT imaging of lipid bilayers in synthetic vesicles with a 2-fold resolution enhancement (4, Fig. 2d).40
In all FRET-based systems, the stability and fatigue resistance of the switch remain major limitations, as the decomposition of the switch progressively increases fluorescence background and lowers achievable contrast. Therefore, it is critical to pursue alternative strategies for robust photoswitchable fluorophores.
2.2 Fusing the photoswitch to a fluorophore scaffold
Directly incorporating established photoswitches into the aromatic scaffold of workhorse fluorophores is an attractive approach to reversibly modulate their emission properties by altering electronic conjugation. A successful example of fluorescence photoswitching was reported by Bossi, Raymo et al. based on an original spirooxazine photoswitch coupled to a coumarin fluorophore (5, Fig. 3a).41 Upon illumination, the ring opening of the oxazine leads to an extension of electronic conjugation, resulting in a bathochromic absorption shift and far-red fluorescence turn-on. The compound was subsequently applied for antibody labelling, and despite low brightness, enabled SMLM of immunostained tubulin filaments with an apparent resolution of 70–80 nm.42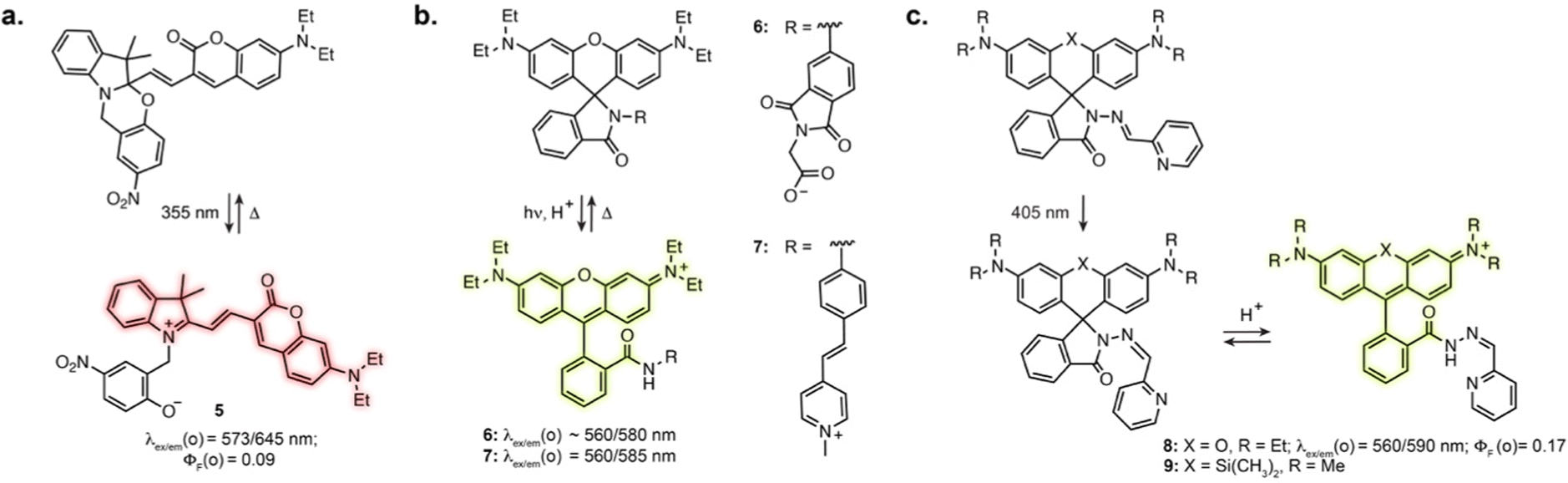 | ||
Fig. 3 (a–c) Representative structures of photoswitchable fluorophores based on a change of electronic conjugation of a fluorophore. The properties reported were measured in: MeCN for 5; H2O for 6; MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 1:1 containing acid for 7; aqueous buffer pH = 5 for 8. The notation (o) refers to the open isomer. :H2O 1:1 containing acid for 7; aqueous buffer pH = 5 for 8. The notation (o) refers to the open isomer. | ||
Other successful examples involve the high contrast open-close equilibrium of rhodamine fluorophores, which can be engineered to be light-responsive. Although not traditionally considered as photoswitches, rhodamine lactams were shown to undergo a light-induced transient ring opening upon illumination. The phthalimide-substituted rhodamine B can be photoactivated using 1- or 2-photon illumination, forming the fluorescent isomer, which spontaneously reverts back to the closed form (6, Fig. 3b).43 This enabled PALM imaging of immunostained tubulin with 50–70 nm resolution. This design was then adapted to multicolour fluorophores by varying rhodamine N-alkylation,44,45 and shown to be applicable to other lactam substitutions.46 In particular, replacement of the phthalimide with a stilbene resulted in activation with visible light (7, Fig. 3b).47 These spirolactam derivatives were used for labelling bacterial cell surface reaching a resolution of 10–40 nm. Recently, Rivera-Fuentes et al. exploited the open-close equilibrium of rhodamines by incorporating a hydrazone photoswitch within the lactam ring (8, Fig. 3c).48 Upon illumination at 405 nm, the hydrazone undergoes E to Z isomerisation. The Z isomer then undergoes spontaneous transient opening which results in blinking. Long-term imaging could be achieved by regularly illuminating the sample, which prompts remaining inactivated fluorophores in their blinking state until bleaching. The initial design enabled SMLM and tracking of acidic compartments such as lysosomes and synaptic vesicles in live neurons. Recently, this scaffold was combined with the HaloTag self-labelling protein, adapting the design to far-red silicon-rhodamine to achieve suitable blinking properties (9, Fig. 3c).49 This was demonstrated with SMLM of the endoplasmic reticulum in living cells. Together, these examples highlight how exploiting and optimising dye-specific mechanisms of fluorescence modulation is an auspicious approach for SRM.
2.3 Intrinsically fluorescent photoswitches
Designing photoswitches that are intrinsically fluorescent as one of their isomers guarantees a structurally small compound. This, however, poses the additional challenge of brightness. Indeed, with a unique chromophore, the photoswitching and fluorescence processes are competitive. Numerous photoswitchable fluorescent DAEs have been reported to date, and the main challenge is their extremely low water solubility. Several examples of fluorescent DAEs in living cells and organisms have been reported,50–54 exploiting strategies to improve solubility such as the introduction of polyethylene glycol chains or charged species. Despite these efforts, only a few of these reports provide characterisation in aqueous conditions, suggesting significant aggregation issues, and the majority function in fluorescence turn-off mode. As such, most of these compounds result in proof-of-principle fluorescence switching experiments but are not well suited for more demanding imaging applications such as SRM. The identification of DAEs fluorescent in the closed state at the single molecule level paved the way for their optimisation towards super-resolution imaging. To date, most examples rely on a DAE bearing two benzothiophene-1,1-dioxide residues attached to a perfluorocyclopentene ring.55,56 Sulfonation of this scaffold resulted in good photoswitching and fluorescence properties in aqueous solution with ΦF = 0.45 in the closed state, retaining photoswitching in living cells (10, Fig. 4a and b).57 Hell, Belov, Irie and co-workers functionalised the DAE scaffold with eight pendant carboxylic acid groups, which could be exploited for antibody labelling using N-hydroxysuccinimide ester chemistry. This compound enabled RESOLFT imaging of immunostained tubulin in fixed Vero cells, reaching 74 nm resolution (11, Fig. 4a and b).58 Similar resolution was achieved with asymmetrical scaffolds, although these displayed significantly lower fluorescence quantum yield (12, Fig. 4a and b) and required long acquisition times.59 Expanding the use of such fluorescent DAEs to SMLM required tuning of the photoisomerisation quantum yield. Accordingly, Hell et al. group appended a methoxy substituent to the pendent phenyl rings, the resulting compound displaying suitable blinking rates for SMLM, demonstrated for vimentin, tubulin and nucleoporin in antibody-labelled cells (13, Fig. 4a and b).60 By systematically evaluating the influence of the carboxylic acid-bearing groups on fatigue resistance, the Hell group identified a novel substitution pattern consisting of four carboxamide branched residues capped with twelve carboxylic acid groups, showing greatly improved photostability compared to the parent compound and able to undergo up to 1000 switching cycles in aqueous buffer.61 Further derivatisation of the structure led to a compound switchable with visible light, and easier to functionalise with protein-labelling moieties (14, Fig. 4a and b).62 Recently, the Hell group reported host-guest DAEs based on ammonium substituents and cucurbit[7]uril CB7 (15, Fig. 4a and b).63 Upon interaction with CB7, the DAE showed an increase in fluorescence quantum yield (1.6-fold) and photostability (>30-fold). Conjugated to antibodies, the probe enabled RESOLFT imaging of microtubules on fixed cells, achieving 70–90 nm spatial resolution. These examples highlight how establishing structure–properties relationship and iterative photoswitch optimisation can lead to advanced probes for SRM applications.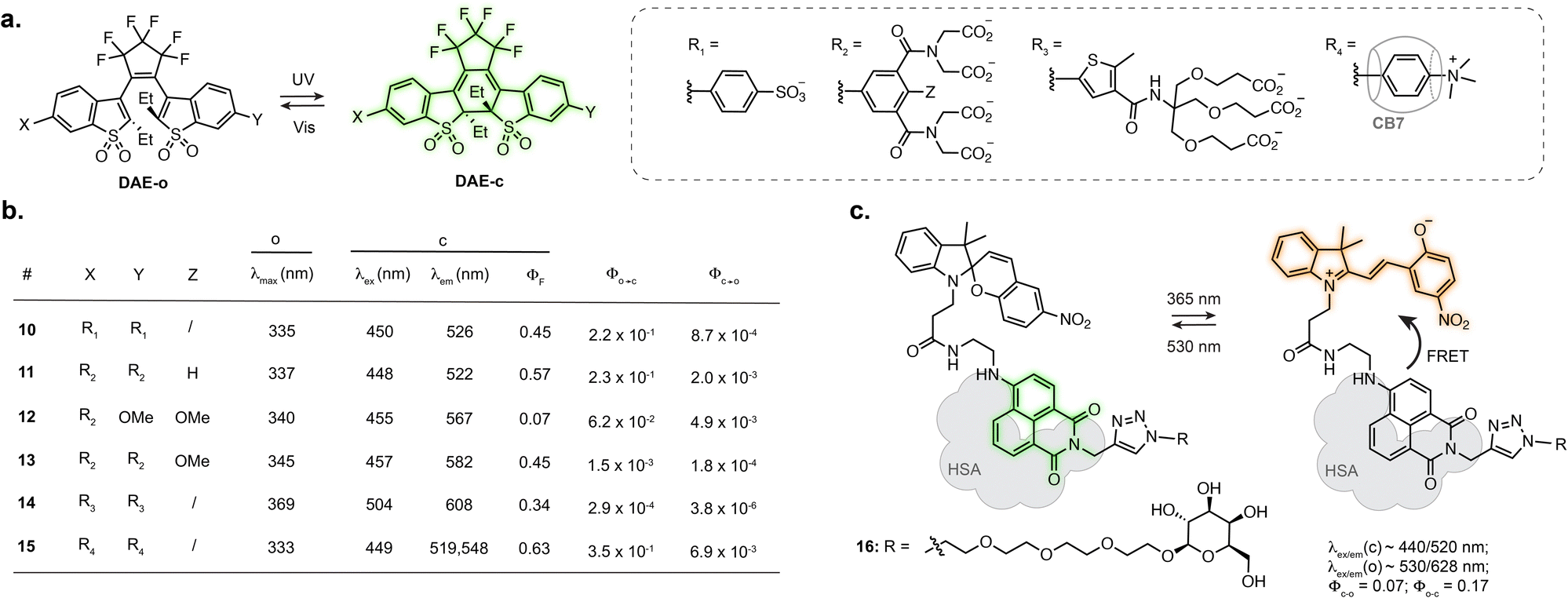 | ||
| Fig. 4 Representative structures of intrinsically fluorescent photoswitches: (a) structure and (b) photophysical properties of fluorescent DAEs, (c) structure of photoswitchable spiropyran-naphtalimide dyad showing fluorogenicity when embedded in human serum albumin (HSA). The properties reported were measured in: aqueous solution for 10 and 15; aqueous solution at pH = 7.0–7.4 for 11, 13, 14, 15; MeOH for 12. The notations (o) and (c) refer to the open and closed isomers, respectively. | ||
While most of the engineering effort has been focused on DAEs, spiropyrans, where the MC isomer presents weak fluorescence in aqueous conditions, have also been investigated. Several examples report fluorescence photoswitching of spiropyrans inside cells, either in solution or embedded into nanostructures.64–69 Interestingly, the photophysical properties of spiropyrans are highly environment dependent, which can lead to a fluorogenic behaviour upon interaction with a biomolecule, therefore augmenting the properties of fluorescent photoswitches for imaging. In particular, an increase in fluorescence quantum yield of the MC isomer can be observed when the switch interacts with a protein, as the hydrophobic protein surface shields the chromophore from the aqueous environment.70 Tian, He, Zhang and co-workers designed a spiropyran-naphthalimide-SP dyad which, when hosted in human serum albumin (HSA), showed high fluorescence increase of the MC isomer, along with an increase in photoisomerisation quantum yield (16, Fig. 4c).71 Combined with a carbohydrate targeting motif, the resulting protein-embedded dyad labelled glycoreceptors in living cells and showed fluorescence photoswitching over several cycles. This dyad:protein adduct was further used for engineering biosensors which will be described in Section 3.2.
3. Photoswitches applied to fluorescence imaging of specific biomolecules, analytes and ions
Numerous examples report the use of small-molecule photoswitches as non-fluorescent binders, in which the reactivity of the probe depends on the isomeric state.72,73 In contrast, there are only a limited number of examples combining photoswitching and biosensing with fluorescence imaging. From an engineering perspective, two main approaches can be identified, either by exploiting the intrinsic properties of the switch or by appending a well-established binding or reactive group to the scaffold. These strategies result in a variety of probes to detect enzyme activity or sensors for biomolecular structures, analytes or ions. In some cases, the resulting probes do not present a photoswitching response and behave as conventional fluorescent biosensors (Section 3.1). In contrast, more sophisticated approaches report photoswitching-gated sensing, or reactivity-gated photoswitching behaviours where the interplay of switching, fluorescence and sensing can lead to multifunctional “smart” reporters (Section 3.2).3.1 Conventional biosensors and reactive probes
Several photoswitches respond to stimuli other than light and display either reversible or irreversible isomerisation in response to a biomolecule, analyte or ion, which in turn modifies the fluorescence properties of the probes. In these systems, the photoswitching capability of the original compound is lost, or photoswitching can be observed but does not impact binding properties. Nevertheless, the reactivity and selectivity of such scaffolds can lead to robust sensors and reactive probes.When introduced into fluorophore scaffolds, azobenzene elicits strong fluorescence quenching. This has been exploited for the design of a variety of biosensors in which the azobenzene is cleaved in response to enzymes, small molecules or ions, through different mechanisms.74 Azobenzenes are highly sensitive to reduction, leading to their widespread application as probes for hypoxia. A rhodamine derivative fused to azobenzene through the aniline of the xanthene core displayed more than 600-fold fluorescence increase under hypoxic conditions (17, Fig. 5a), and was applied to visualise hypoxia during retinal artery occlusion in rat,75 and during osteoarthritis progression in mouse cartilage.76 Azobenzene-quenched probes based on further red-shifted fluorophores were used to monitor hypoxia in living mouse,77–79 and similar scaffolds enabled direct visualisation of azoreductase activity.80,81
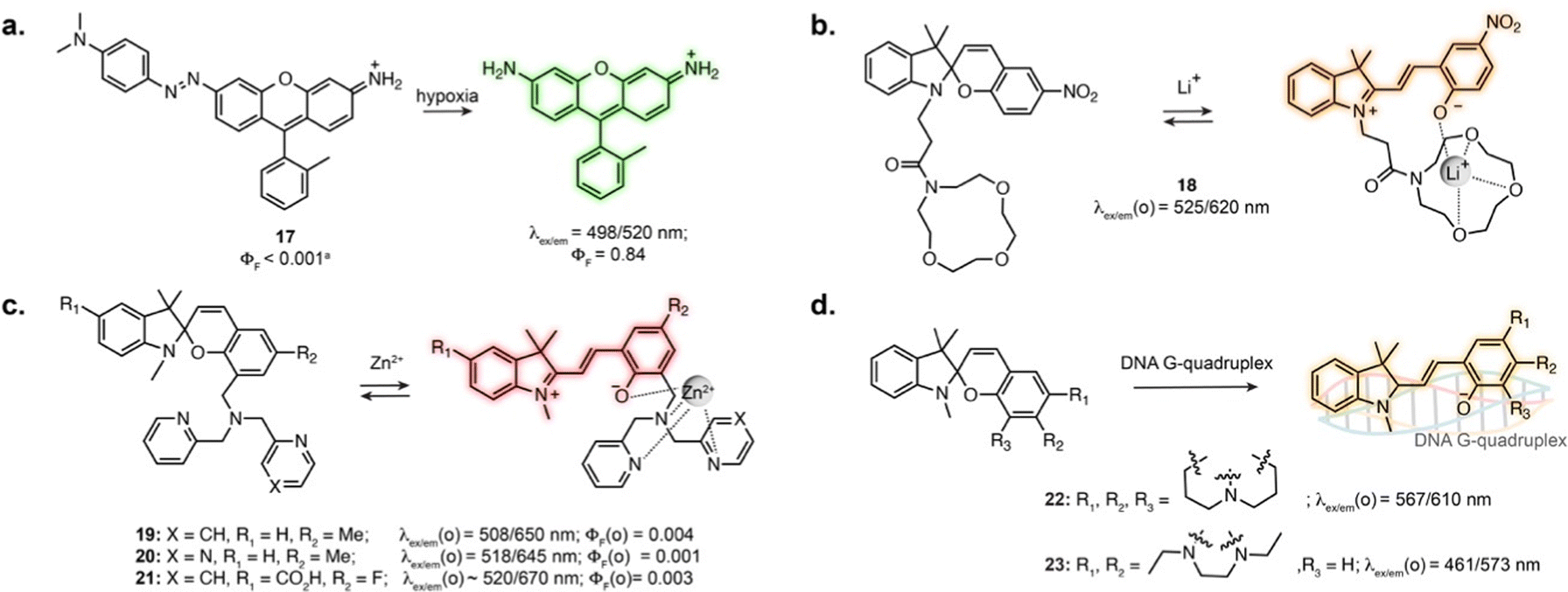 | ||
| Fig. 5 Examples of conventional biosensors and reactive probes based on photoswitches. The properties reported were measured in: aqueous buffer at pH = 7.0–7.4 for 17, 19, 20, 22 and 23; aqueous buffer pH = 7.4 containing 50% MeCN for 18; H2O for 21; The notation (o) refers to the open isomer, after binding or chelation. | ||
The MC isomer of spiropyran derivatives can be used as an ion-binding motif where the phenolate is involved in the chelation, which has been exploited for the design of fluorescent biosensors leveraging its intrinsic fluorescence.82 A general approach for the design of specific biosensors consists in introducing an ion binding motif on the SP structure, leading to probes for various cations applicable in cells.83–91 In particular, several spiropyran derivatives functionalised with crown ethers were shown to undergo isomerisation in the presence of Li+, with a concomitant fluorescence increase.84,85 Compound 18 was used to monitor exogeneous Li+ in zebrafish (Fig. 5b).84 Crown-ether appended spiropyrans were also reported as Ca2+ biosensors, showing fluorescence increase upon ionomycin stimulation in living cells.87 The Lippard group exploited a dipicolylamine chelating group to prepare far-red spiropyran-based zinc sensors (19 and 20, Fig. 5c). Despite low brightness, sensor 19 enabled visualisation of exogeneously applied zinc ion in live cells.88 The affinity of this compound (Kd = 21 pM) was too high to visualise physiological levels of Zn2+. Modifying the chelating unit led to a lower affinity version (Kd = 3.6 nM, 20, Fig. 5c), enabling two-photon imaging of zinc-containing vesicles in brain tissue.89 Sensor 20 could also be applied to compare Zn2+ lysosomal levels upon perturbation in different cell types.90
The SP to MC isomerisation can also be triggered upon binding to specific biomolecular structures. Guo and co-workers reported SP-based probes showing fluorescence turn-on upon binding to DNA G-quadruplexes in vitro (22, 23, Fig. 5d).92,93 These cell-permeable probes showed labelling in living cells, although with low specificity due to significant accumulation in lysosomes and mitochondria.
3.2 Photoswitchable fluorescent biosensors
Certain photoswitch-based biosensors were reported to retain a light-responsive behaviour, where only one isomer presents binding properties, and photoisomerisation triggers binding or unbinding. This offers the possibility of having precise spatial and temporal control of sensor activity. The derivatives of SP-based Li+ and Ca2+ biosensors introduced in Section 3.1 actually report an additional photoswitching behaviour in the bound state.86,87 Similarly, while no photo-response is reported for Zn2+ sensors 19 and 20, the fluorine substituted derivative 21 displayed photoisomerisation (Fig. 5c).91 This results in photoswitchable fluorescence of the ion-bound isomer, although this property has not yet been directly exploited for imaging.The turn-on of the photoswitching or fluorescence properties only following target recognition can however be a highly beneficial property for imaging, as it can greatly reduce background from unbound probes. For example, attaching a tubulin-targeting motif to a spiropyran resulted in fluorescence increase and detectable photoswitching only in the tubulin-bound state, enabling SRM (24, Fig. 6a).94 This behaviour was also achieved by exploiting DAEs appended with recognition motifs. In the unbound state, the DAE adopts an aggregated structure preventing photoswitching. The conformational change elicited by target recognition recovers photoswitching. Yi et al. used this approach to develop a DNA-staining DAE displaying fluorescence turn-on and photoswitching following intercalation with double stranded DNA (25, Fig. 6b).95 Similarly, a DAE-based appended with a cyanoacrylate recognition motif showed specific staining of β-amyloid aggregates in brain sections (26, Fig. 6b).96
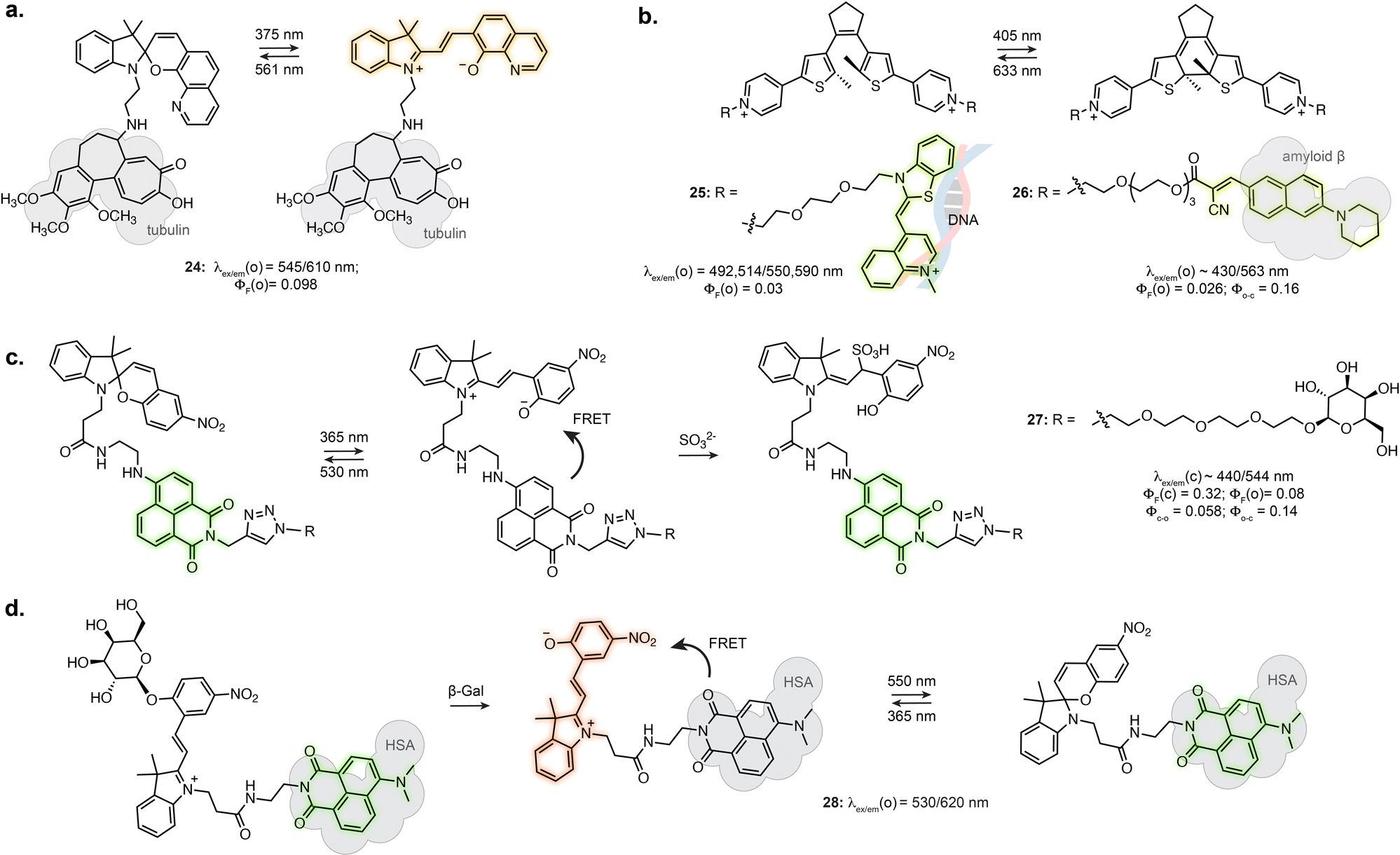 | ||
| Fig. 6 (a–d) Representative examples of fluorescent biosensors and reactive probes based on synthetic photoswitches and presenting light-dependent behaviour. The properties reported were measured in aqueous buffers at pH = 7.2–7.4 for all compounds. The notations (o) and (c) refer to the open and closed isomers of the photoswitchable form, respectively (before reaction with SO32− for (c), after enzymatic cleavage for (d)). | ||
Tian et al. demonstrated the use of a SP-naphthalimide FRET probe as a photoswitchable probe reactive towards sulfite ion (27, Fig. 6c).97 The unreacted probe showed fluorescence switching in cells upon illumination, due to FRET with the formed MC isomer. When attached to D-galactose, the probe specifically labelled glycoreceptors on the cell surface and could be uptaken by endocytosis. The internalised probe retained fluorescence photoswitching upon UV and visible illumination. In addition, the probe could undergo nucleophilic addition of SO32− only as the MC isomer. The non-absorbing MC-SO3 adduct cannot act as a FRET acceptor, resulting in a turn-on of the naphthalimide fluorescence following SO32− addition. In live cell, the probe enabled quantification of endogeneous sulfite release upon stimulation. Recently, the application of this system at other subcellular locations was demonstrated by using a lysosome targeting group.98,99
While in this example the MC in the dyad only acts as FRET acceptor, the far-red fluorescence of the MC isomer can be turned on when hosted in human serum albumin (16, Fig. 4c).71 Zhang et al. engineered an enzyme reactive probe based on this protein-embedded dyad (28, Fig. 6d).100 The spiropyran was locked in the MC form with an acetylated β-galactose group. In the caged form, the probe shows green fluorescence from the naphthalimide. Cleavage of the sugar moiety by β-galactosidase releases the functional merocyanine, showing far-red fluorescence and photoswitching inside the hydrophobic pocket of serum albumin, therefore acting as both fluorophore and FRET acceptor. The probe was used in STORM imaging of β-galactosidase activity in fixed cells.
Overall, these recent examples of photoswitching-gated detection or detection-gated photoswitching behaviours demonstrate how careful engineering of photophysical and biochemical properties can lead to tools enabling precise monitoring of important signalling biomarkers.
Conclusion and outlook
Small-molecule photoswitches offer a unique opportunity for fluorescence microscopy, to precisely control the properties of reporters with exquisite spatial and temporal control. Their implementation and optimisation in this context have led to photoswitchable fluorophores enabling advanced applications such as super-resolution microscopy, and their potential to regulate biosensor function has also been demonstrated. The engineering of such functional reporters remains a challenging task, due to numerous requirements. Indeed, small-molecule photoswitches have primarily been plagued by low water solubility, and strategies to circumvent this limitation by embedding them in nanostructures and biomolecules, or substitution with hydrophilic groups result in cell-impermeability. In addition, optimisation of their photophysical properties (brightness, photostability) in water has proven difficult, which compromises performance and has precluded broad applicability. Furthermore, the design of photoswitchable biosensors and reactive probes comes with additional complexity, as improvements in either photoswitching, fluorescence or sensing properties will impact other critical features of the system. As a result, the development of robust probes is a stepwise and iterative process, and requires the systematic characterisation of structure-properties relationships. While the examples presented in this review show that improvements of the DAE and SP scaffolds have already led to substantial advances, further improvements are needed for applications in living samples. We can also expect that the investigation of emerging water-compatible photoswitchable scaffolds101,102 will open new perspectives for the design of fluorescent reporters. Ultimately, future developments will lead to more functional controllable reporters that can be combined with established biomolecular labelling methods to push forward the investigation of complex biological processes.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Chan Zuckerberg Initiative (Deep Tissue Imaging grant no. 2020-225346).Notes and references
- E. Betzig, G. H. Patterson, R. Sougrat, O. Wolf Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed
.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed
- M. Weber, M. Leutenegger, S. Stoldt, S. Jakobs, T. S. Mihaila, A. N. Butkevich and S. W. Hell, Nat. Photonics, 2021, 15, 361–366 CrossRef CAS PubMed
- M. Lelek, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffie, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev. Methods Primers, 2021, 1, 39 CrossRef CAS PubMed
- K. Nienhaus and G. U. Nienhaus, ACS Nano, 2016, 10, 9104–9108 CrossRef CAS PubMed
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, e1905966 CrossRef PubMed
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymanski, Chem. Sci., 2020, 11, 11672–11691 RSC
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed
- J. Volaric, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 RSC
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2021, 6, 51–69 CrossRef
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS
- D. J. van Dijken, P. Kovaricek, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed
- J. Zhang and H. Tian, Adv. Opt. Mater., 2018, 6, 1701278 CrossRef
- T. Fukaminato, S. Ishida and R. Métivier, NPG Asia Mater., 2018, 10, 859–881 CrossRef CAS
- H. B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, e2108289 CrossRef PubMed
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC
- L. Wang, Y. Hao, J. Huang, Y. He, K. Zeng, J. Li, J. M. Chabu, W. Chen, M. Yang, L. Deng and Y. N. Liu, Anal. Chem., 2016, 88, 9136–9142 CrossRef CAS PubMed
- M. Hammarson, J. R. Nilsson, S. Li, T. Beke-Somfai and J. Andreasson, J. Phys. Chem. B, 2013, 117, 13561–13571 CrossRef CAS PubMed
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed
- F. M. Jradi and L. D. Lavis, ACS Chem. Biol., 2019, 14, 1077–1090 CrossRef CAS PubMed
- H. Y. Jung, S. You, C. Lee, S. You and Y. Kim, Chem. Commun., 2013, 49, 7528–7530 RSC
- Y. Kim, H. Y. Jung, Y. H. Choe, C. Lee, S. K. Ko, S. Koun, Y. Choi, B. H. Chung, B. C. Park, T. L. Huh, I. Shin and E. Kim, Angew. Chem., Int. Ed., 2012, 51, 2878–2882 CrossRef CAS PubMed
- H. Y. Jung, B. Kim, M. H. Jeon and Y. Kim, Small, 2022, 18, e2103523 CrossRef PubMed
- T. Yang, Q. Liu, J. Li, S. Pu, P. Yang and F. Li, RSC Adv., 2014, 4, 15613–15619 RSC
- T. Wu, B. Johnsen, Z. Qin, M. Morimoto, D. Baillie, M. Irie and N. R. Branda, Nanoscale, 2015, 7, 11263–11266 RSC
- C. T. Kuo, A. M. Thompson, M. E. Gallina, F. Ye, E. S. Johnson, W. Sun, M. Zhao, J. Yu, I. C. Wu, B. Fujimoto, C. C. DuFort, M. A. Carlson, S. R. Hingorani, A. L. Paguirigan, J. P. Radich and D. T. Chiu, Nat. Commun., 2016, 7, 11468 CrossRef CAS PubMed
- Y. Xie, M. C. Arno, J. T. Husband, M. Torrent-Sucarrat and R. K. O'Reilly, Nat. Commun., 2020, 11, 2460 CrossRef CAS PubMed
- C. Li, Z. Hu, M. P. Aldred, L.-X. Zhao, H. Yan, G.-F. Zhang, Z.-L. Huang, A. D. Q. Li and M.-Q. Zhu, Macromolecules, 2014, 47, 8594–8601 CrossRef CAS
- D. Kim and T. S. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 34770–34776 CrossRef CAS PubMed
- D. Kim, K. Jeong, J. E. Kwon, H. Park, S. Lee, S. Kim and S. Y. Park, Nat. Commun., 2019, 10, 3089 CrossRef PubMed
- R. Yang, X. Ren, L. Mei, G. Pan, X. Z. Li, Z. Wu, S. Zhang, W. Ma, W. Yu, H. H. Fang, C. Li, M. Q. Zhu, Z. Hu, T. Sun, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2022, 61, e202117158 CAS
- N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206–5208 RSC
- S. Mao, R. K. Benninger, Y. Yan, C. Petchprayoon, D. Jackson, C. J. Easley, D. W. Piston and G. Marriott, Biophys. J., 2008, 94, 4515–4524 CrossRef CAS PubMed
- C. Petchprayoon, Y. Yan, S. Mao and G. Marriott, Bioorg. Med. Chem., 2011, 19, 1030–1040 CrossRef CAS PubMed
- Y. Xiong, P. Rivera-Fuentes, E. Sezgin, A. Vargas Jentzsch, C. Eggeling and H. L. Anderson, Org. Lett., 2016, 18, 3666–3669 CrossRef CAS PubMed
- Y. Xiong, A. Vargas Jentzsch, J. W. M. Osterrieth, E. Sezgin, I. V. Sazanovich, K. Reglinski, S. Galiani, A. W. Parker, C. Eggeling and H. L. Anderson, Chem. Sci., 2018, 9, 3029–3040 RSC
- A. T. Frawley, V. Wycisk, Y. Xiong, S. Galiani, E. Sezgin, I. Urbancic, A. Vargas Jentzsch, K. G. Leslie, C. Eggeling and H. L. Anderson, Chem. Sci., 2020, 11, 8955–8960 RSC
- E. Deniz, M. Tomasulo, J. Cusido, I. Yildiz, M. Petriella, M. L. Bossi, S. Sortino and F. M. Raymo, J. Phys. Chem. C, 2012, 116, 6058–6068 CrossRef CAS
- J. Cusido, S. S. Ragab, E. R. Thapaliya, S. Swaminathan, J. Garcia-Amorós, M. J. Roberti, B. Araoz, M. M. A. Mazza, S. Yamazaki, A. M. Scott, F. M. Raymo and M. L. Bossi, J. Phys. Chem. C, 2016, 120, 12860–12870 CrossRef CAS
- J. Folling, V. Belov, R. Kunetsky, R. Medda, A. Schonle, A. Egner, C. Eggeling, M. Bossi and S. W. Hell, Angew. Chem., Int. Ed., 2007, 46, 6266–6270 CrossRef CAS PubMed
- M. Bossi, J. Foelling, V. N. Belov, V. P. Boyarskiy, R. Medda, A. Egner, C. Eggeling, A. Schoenle and S. W. Hell, Nano Lett., 2008, 8, 2463–2468 CrossRef CAS PubMed
- V. N. Belov, M. L. Bossi, J. Folling, V. P. Boyarskiy and S. W. Hell, Chem. – Eur. J., 2009, 15, 10762–10776 CrossRef CAS PubMed
- Z. Ye, H. Yu, W. Yang, Y. Zheng, N. Li, H. Bian, Z. Wang, Q. Liu, Y. Song, M. Zhang and Y. Xiao, J. Am. Chem. Soc., 2019, 141, 6527–6536 CrossRef CAS PubMed
- M. K. Lee, P. Rai, J. Williams, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2014, 136, 14003–14006 CrossRef CAS PubMed
- E. A. Halabi, D. Pinotsi and P. Rivera-Fuentes, Nat. Commun., 2019, 10, 1232 CrossRef PubMed
- A. Eordogh, A. Martin and P. Rivera-Fuentes, Chem. – Eur. J., 2022, e202202832 CAS
- Y. Zou, T. Yi, S. Xiao, F. Li, C. Li, X. Gao, J. Wu, M. Yu and C. Huang, J. Am. Chem. Soc., 2008, 130, 15750–15751 CrossRef CAS PubMed
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966–15967 CrossRef CAS PubMed
- S. C. Pang, H. Hyun, S. Lee, D. Jang, M. J. Lee, S. H. Kang and K. H. Ahn, Chem. Commun., 2012, 48, 3745–3747 RSC
- F. Hu, L. Jiang, M. Cao, Z. Xu, J. Huang, D. Wu, W. Yang, S. H. Liu and J. Yin, RSC Adv., 2015, 5, 5982–5987 RSC
- G. Naren, W. Larsson, C. Benitez-Martin, S. Li, E. Perez-Inestrosa, B. Albinsson and J. Andreasson, Chem. Sci., 2021, 12, 7073–7078 RSC
- Y. C. Jeong, S. I. Yang, K. H. Ahn and E. Kim, Chem. Commun., 2005, 2503–2505 RSC
- K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 13558–13564 CrossRef CAS PubMed
- S. H. Um, H. J. Kim, D. Kim, J. E. Kwon, J. W. Lee, D. Hwang, S. K. Kim and S. Y. Park, Dyes Pigm., 2018, 158, 36–41 CrossRef CAS
- B. Roubinet, M. L. Bossi, P. Alt, M. Leutenegger, H. Shojaei, S. Schnorrenberg, S. Nizamov, M. Irie, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 15429–15433 CrossRef CAS PubMed
- K. Uno, M. L. Bossi, T. Konen, V. N. Belov, M. Irie and S. W. Hell, Adv. Opt. Mater., 2019, 7, 1801746 CrossRef
- B. Roubinet, M. Weber, H. Shojaei, M. Bates, M. L. Bossi, V. N. Belov, M. Irie and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 6611–6620 CrossRef CAS PubMed
- K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS PubMed
- K. Uno, A. Aktalay, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2100165118 CrossRef PubMed
- D. Kim, A. Aktalay, N. Jensen, K. Uno, M. L. Bossi, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2022, 144, 14235–14247 CrossRef CAS PubMed
- G. Marriott, S. Mao, T. Sakata, J. Ran, D. K. Jackson, C. Petchprayoon, T. J. Gomez, E. Warp, O. Tulyathan, H. L. Aaron, E. Y. Isacoff and Y. Yan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17789–17794 CrossRef CAS PubMed
- L. Wu, Y. Dai, X. Jiang, C. Petchprayoon, J. E. Lee, T. Jiang, Y. Yan and G. Marriott, PLoS One, 2013, 8, e64738 CrossRef CAS PubMed
- D. Hu, Z. Tian, W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 15279–15281 CrossRef CAS PubMed
- Z. Tian, W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2009, 131, 4245–4252 CrossRef CAS PubMed
- N. Xie, K. Feng, B. Chen, M. Zhao, S. Peng, L. P. Zhang, C. H. Tung and L. Z. Wu, J. Mater. Chem. B, 2014, 2, 502–510 RSC
- M.-Q. Zhu, G.-F. Zhang, C. Li, M. P. Aldred, E. Chang, R. A. Drezek and A. D. Q. Li, J. Am. Chem. Soc., 2011, 133, 365–372 CrossRef CAS PubMed
- N. Amdursky, P. K. Kundu, J. Ahrens, D. Huppert and R. Klajn, ChemPlusChem, 2016, 81, 44–48 CrossRef CAS PubMed
- Y. Fu, H. H. Han, J. Zhang, X. P. He, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2018, 140, 8671–8674 CrossRef CAS PubMed
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC
- D. V. Berdnikova, Chem. Commun., 2021, 57, 10819–10826 RSC
- A. Chevalier, P. Y. Renard and A. Romieu, Chem. – Asian J., 2017, 12, 2008–2028 CrossRef CAS PubMed
- W. Piao, S. Tsuda, Y. Tanaka, S. Maeda, F. Liu, S. Takahashi, Y. Kushida, T. Komatsu, T. Ueno, T. Terai, T. Nakazawa, M. Uchiyama, K. Morokuma, T. Nagano and K. Hanaoka, Angew. Chem., Int. Ed., 2013, 52, 13028–13032 CrossRef CAS PubMed
- K. Okada, D. Mori, Y. Makii, H. Nakamoto, Y. Murahashi, F. Yano, S. H. Chang, Y. Taniguchi, H. Kobayashi, H. Semba, N. Takeda, W. Piao, K. Hanaoka, T. Nagano, S. Tanaka and T. Saito, Sci. Rep., 2020, 10, 5425 CrossRef PubMed
- K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2010, 132, 15846–15848 CrossRef CAS PubMed
- X. Tian, Z. Li, Y. Sun, P. Wang and H. Ma, Anal. Chem., 2018, 90, 13759–13766 CrossRef CAS PubMed
- N. Ding, Z. Li, X. Tian, J. Zhang, K. Guo and P. Wang, Chem. Commun., 2019, 55, 13172–13175 RSC
- N. Shin, K. Hanaoka, W. Piao, T. Miyakawa, T. Fujisawa, S. Takeuchi, S. Takahashi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, M. Tanokura, T. Nagano and Y. Urano, ACS Chem. Biol., 2017, 12, 558–563 CrossRef CAS PubMed
- Y. Tian, Y. Li, W. L. Jiang, D. Y. Zhou, J. Fei and C. Y. Li, Anal. Chem., 2019, 91, 10901–10907 CrossRef CAS PubMed
- A. A. Ali, R. Kharbash and Y. Kim, Anal. Chim. Acta, 2020, 1110, 199–223 CrossRef CAS PubMed
- M. Inouye, M. Ueno, K. Tsuchiya, N. Nakayama, T. Konishi and T. Kitao, J. Org. Chem., 1992, 57, 5377–5383 CrossRef CAS
- J. Kang, E. Li, L. Cui, Q. Shao, C. Yin and F. Cheng, Sens. Actuators, B, 2021, 327, 128941 CrossRef CAS
- D. B. Stubing, S. Heng and A. D. Abell, Org. Biomol. Chem., 2016, 14, 3752–3757 RSC
- J. V. Pei, S. Heng, M. L. De Ieso, G. Sylvia, M. Kourghi, S. Nourmohammadi, A. D. Abell and A. J. Yool, Mol. Pharmacol., 2019, 95, 573–583 CrossRef PubMed
- S. Heng, A. M. Mak, R. Kostecki, X. Zhang, J. Pei, D. B. Stubing, H. Ebendorff-Heidepriem and A. D. Abell, Sens. Actuators, B, 2017, 252, 965–972 CrossRef CAS
- P. Rivera-Fuentes and S. J. Lippard, ChemMedChem, 2014, 9, 1238–1243 CrossRef CAS PubMed
- P. Rivera-Fuentes, A. T. Wrobel, M. L. Zastrow, M. Khan, J. Georgiou, T. T. Luyben, J. C. Roder, K. Okamoto and S. J. Lippard, Chem. Sci., 2015, 6, 1944–1948 RSC
- Y. Han, J. M. Goldberg, S. J. Lippard and A. E. Palmer, Sci. Rep., 2018, 8, 15034 CrossRef PubMed
- S. Heng, P. Reineck, A. K. Vidanapathirana, B. J. Pullen, D. W. Drumm, L. J. Ritter, N. Schwarz, C. S. Bonder, P. J. Psaltis, J. G. Thompson, B. C. Gibson, S. J. Nicholls and A. D. Abell, ACS Omega, 2017, 2, 6201–6210 CrossRef CAS PubMed
- X. Tian, J. Li, Y. Zhang, Y. Gao, M. W. Afzal, A. Wang, T. D. James, Y. Bai and Y. Guo, Sens. Actuators, B, 2022, 359, 131618 CrossRef CAS
- J. Li, X. Yin, B. Li, X. Li, Y. Pan, J. Li and Y. Guo, Anal. Chem., 2019, 91, 5354–5361 CrossRef CAS PubMed
- H. Zhang, C. Wang, T. Jiang, H. Guo, G. Wang, X. Cai, L. Yang, Y. Zhang, H. Yu, H. Wang and K. Jiang, Anal. Chem., 2015, 87, 5216–5222 CrossRef CAS PubMed
- K. Liu, Y. Wen, T. Shi, Y. Li, F. Li, Y. L. Zhao, C. Huang and T. Yi, Chem. Commun., 2014, 50, 9141–9144 RSC
- G. Lv, B. Cui, H. Lan, Y. Wen, A. Sun and T. Yi, Chem. Commun., 2015, 51, 125–128 RSC
- J. Zhang, Y. Fu, H. H. Han, Y. Zang, J. Li, X. P. He, B. L. Feringa and H. Tian, Nat. Commun., 2017, 8, 987 CrossRef PubMed
- W. Zhang, F. Huo and C. Yin, Org. Lett., 2019, 21, 5277–5280 CrossRef CAS PubMed
- W. Zhang, F. Huo, Y. Yue, Y. Zhang, J. Chao, F. Cheng and C. Yin, J. Am. Chem. Soc., 2020, 142, 3262–3268 CrossRef CAS PubMed
- X. Chai, H. H. Han, A. C. Sedgwick, N. Li, Y. Zang, T. D. James, J. Zhang, X. L. Hu, Y. Yu, Y. Li, Y. Wang, J. Li, X. P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 18005–18013 CrossRef CAS PubMed
- A. Sailer, J. C. M. Meiring, C. Heise, L. N. Pettersson, A. Akhmanova, J. Thorn-Seshold and O. Thorn-Seshold, Angew. Chem., Int. Ed., 2021, 60, 23695–23704 CrossRef CAS PubMed
- R. Castagna, G. Maleeva, D. Pirovano, C. Matera and P. Gorostiza, J. Am. Chem. Soc., 2022, 144, 15595–15602 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |