
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanosized metal–organic frameworks as unique platforms for bioapplications
Manuela
Cedrún-Morales
 a,
Manuel
Ceballos
a,
Ester
Polo
b,
Pablo
del Pino
*a and
Beatriz
Pelaz
*c
a,
Manuel
Ceballos
a,
Ester
Polo
b,
Pablo
del Pino
*a and
Beatriz
Pelaz
*c
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Física de Partículas, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: pablo.delpino@usc.es
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Bioquímica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
cCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Inorgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: beatriz.pelaz@usc.es
First published on 9th February 2023
Abstract
Metal–organic frameworks (MOFs) are extremely versatile materials, which serve to create platforms with exceptional porosity and specific reactivities. The production of MOFs at the nanoscale (NMOFs) offers the possibility of creating innovative materials for bioapplications as long as they maintain the properties of their larger counterparts. Due to their inherent chemical versatility, synthetic methods to produce them at the nanoscale can be combined with inorganic nanoparticles (NPs) to create nanocomposites (NCs) with one-of-a-kind features. These systems can be remotely controlled and can catalyze abiotic reactions in living cells, which have the potential to stimulate further research on these nanocomposites as tools for advanced therapies.
Introduction
The huge potential of nanomaterials in biomedicine for diagnosing and treating many human diseases is well-documented.1 During recent decades, the applications of nanotechnology in numerous biology-related areas such as diagnosis, drug delivery, and molecular imaging have gained interest and have been increasingly investigated showing remarkable results.2 In this context, nanoparticle drug delivery systems (DDSs) are extensively used in pharmaceutical research and in clinical settings to improve the efficacy of diagnostic agents and drugs, such as anticancer, antimicrobial, and antiviral drugs.3 The term DDS is employed to describe a carrier that can deliver an active pharmaceutical ingredient (API) to a specific site in the body.4 These systems can overcome some of the main challenges associated with molecular drugs, such as poor stability and solubility, short circulation time in the blood, and low specificity, which can lead to multidrug resistance after long-term exposure.4 Furthermore, the development of new DDSs has led to the possibility of targeting bioactive molecules in specific tissues in the body, avoiding the side effects related to conventional treatments and improving their efficacy. During their synthesis, the selected materials can be tuned to fulfil specific biological needs such as entry routes and loading mechanisms.To overcome DDSs’ limitations such as retention and/or inefficient drug release, DDSs can be further designed to respond to specific stimuli triggering the release of the loaded cargo.5 These stimuli can be divided into two categories, endogenous and external. The endogenous stimuli are referred to as internal conditions of the environment around the nanoparticles (NPs), such as the acidic/hypoxic environment of a tumor and high enzyme levels of matrix metalloproteinases (MMPs),6 among others. On the other hand, external stimuli refer to selected and controlled stimuli outside the body, such as ultrasound, light, magnetic fields, and temperature.5 However, this cargo delivery must occur at the target site to avoid side effects and increase the efficacy of the treatment. NPs can be provided with recognition abilities by modifying them with peptides, nucleic acids, etc., which will optimize the delivery and overcome physiological physical barriers. Despite all these possibilities, there are still some drawbacks associated with these systems that impede their approval for clinical use and that need to be confronted, such as limited targeting abilities and reduced cellular uptake.
Given these challenges, metal–organic frameworks (MOFs) have raised considerable interest as alternative materials in the biomedical field, owing to their unique physicochemical properties. These crystalline and highly porous materials contain ordered pores whose size and chemical features are determined by design.7 As a result, MOFs’ applicability has been exploited for catalysis, gas storage, and sensing, among others.7 Characteristics such as their large pore volumes, high surface areas (up to 8000 m2 g−1), well-defined tunable structures, and permanent porosity make them excellent candidates as DDSs. Moreover, the highly tunable surface functionalities, their ability to host a wide variety of APIs (drugs, enzymes, nucleic acids, etc.) with the corresponding release in a controlled way, and a general trend of a high biocompatible profile allow these materials to overcome some of the main drawbacks mentioned before.8
1. Metal–organic frameworks
MOFs can be defined as extended structures formed by the coordination of metal ions or inorganic polynuclear clusters, also referred to as SBUs (secondary building units), to organic linkers. The SBUs form the structural nodes of MOFs and consist of metal ions (such as Cu(I), Zn(II) or Zr(IV)) and non-metals (such as oxygen or nitrogen). These SBUs are connected through organic linkers, generally in bi-, tri- or tetratopic forms, commonly containing Lewis bases such as carboxylate, phosphonate, pyridyl, imidazolate or other azolate-functional groups (see Fig. 1).9 SBUs determine the large structural diversity of MOFs and serve to classify MOF structures based on their net topology. Remarkably, experimentally described SBUs include most transition metals, several main-group metals, alkali metals, alkaline earth metals, lanthanides, and actinides, and the number of metal atoms in these units can vary from one to eight or even more. For a given SBU, the connectivity and geometry of the ligand will influence the final structure of the MOF and will provide the nanomaterial with features such as permanent porosity, or thermal and chemical stability, relevant for their use as DDSs.9 Furthermore, the bonds between the SBUs and the linkers constitute the most reactive part of the MOF; therefore post-synthesis modifications will be typically happening on these points.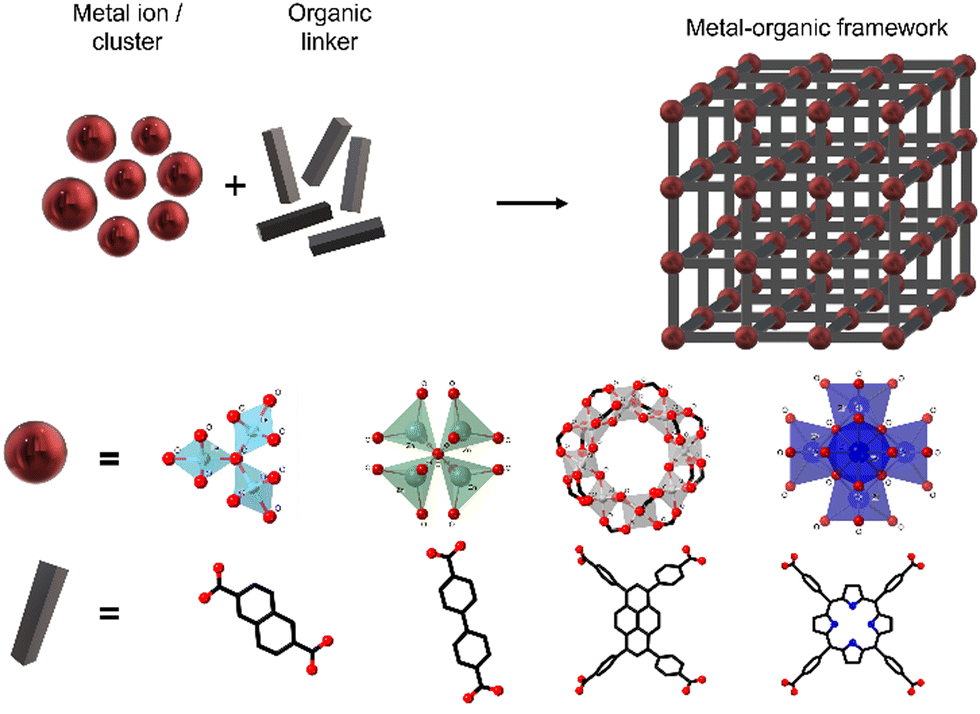 | ||
| Fig. 1 Schematic representation of MOFs, showing some representative SBUs and linkers. | ||
Coordination networks have been known since 1959, using Cu(I) ions as nodes with adiponitrile [NC(CH2)4CN] molecules as linkers.10 Twenty years later, metal ions and neutral donor linkers such as 4,4′-bipyridine11 were employed to develop new coordination networks.
The structures of materials with a single metal are limited by the coordination number and geometries allowed by these nodes. A very common example of MOFs with single-metal nodes is the zeolitic imidazolate framework (ZIF) which has given rise to the synthesis of a huge number of ZIF materials.12 They are a sub-group of MOFs constituted by transition divalent metal ions (M2+) tetracoordinated with imidazolate units or their derivatives. ZIF-8 (Fig. 2a) is one of the most popular materials in the MOF community and is highly employed in different applications due to its versatility and facile and green synthesis,13 in addition to the ability to form composites with a large number of materials.14–16
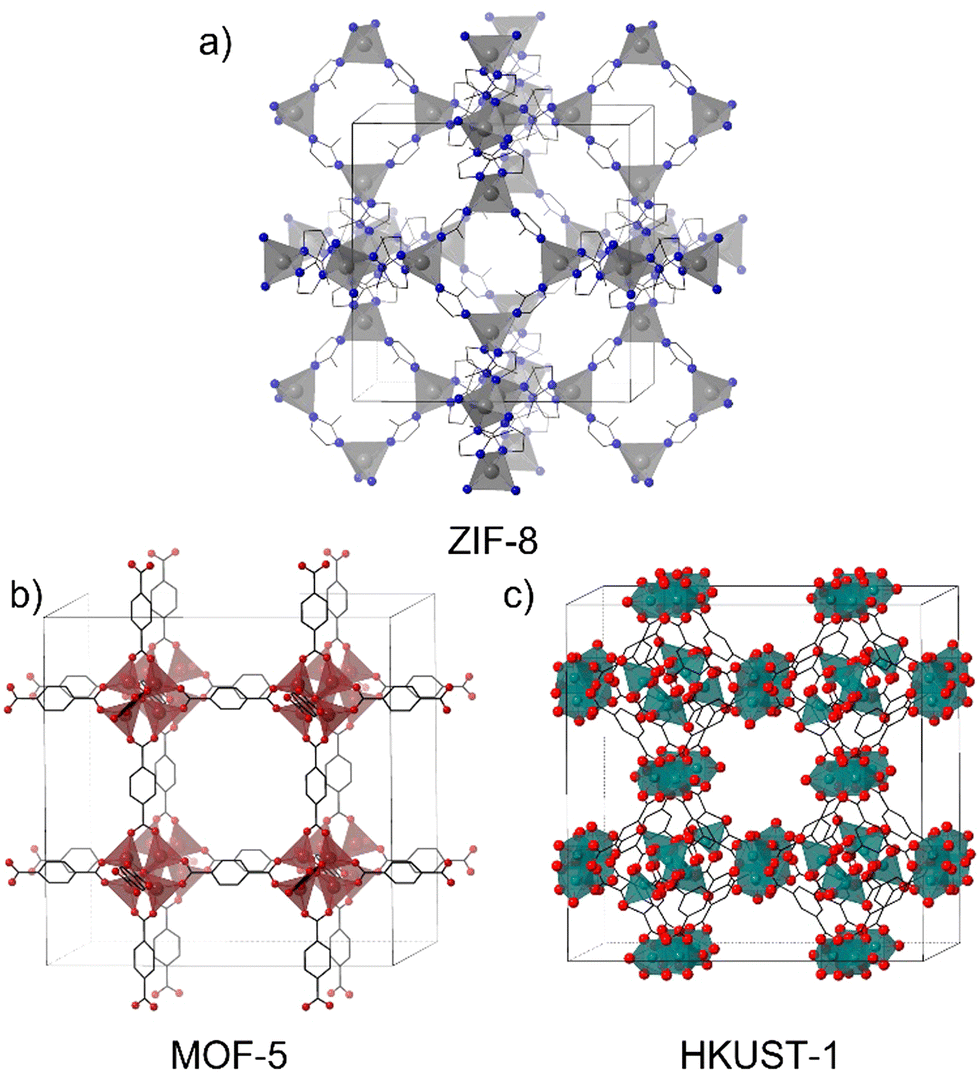 | ||
| Fig. 2 Crystal structures of (a) ZIF-8, (b) MOF-5 and (c) HKUST-1. | ||
On the other hand, MOFs in which the nodes are constituted by clusters of atoms as SBUs have been very attractive since the discovery of MOF-517 and HKUST-118 (Fig. 2b and c) (HKUST stands for Hong Kong University of Science and Technology). These MOFs have greatly contributed to understand the role of the spatial disposition of organic ligands and inorganic moieties in determining the MOF's structure.19
These clusters of atoms can be organized in different geometries such as tetrahedra, trigonal bipyramids, and octahedra, among others, sharing some atoms (generally oxygen) in the vertices of the polyhedra (Fig. 3). The SBU of MIL-88(Fe) (MIL stands for Matériaux de l′Institut Lavoisier) is formed by a trimer of octahedra sharing an oxygen atom per each trimer and an iron atom in the centre of each octahedron.20 In the case of MOF-5, one of the most popular MOFs, its SUB is made by a tetramer of tetrahedra sharing an oxygen atom from a vertex.21 Another example of SBUs is the Zr6 cluster in the UiO MOF family (UiO stands for Universitetet i Oslo), where each Zr is connected to four Zr atoms through a Zr–O–Zr bond, and each Zr atom can be bonded to two bidentate ligands, creating a Zr6O4(OH)4 cluster surrounded by, in theory, twelve linkers.22 One of the most complicated SBUs is that of MOF-520, in which eight aluminum atoms in an octahedral disposition form a ring by sharing an oxygen atom with other aluminum, this spatial organization generates a chiral SBU.23
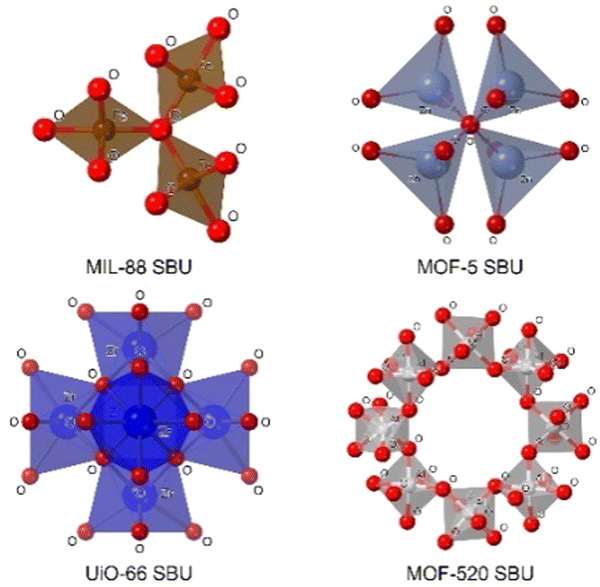 | ||
| Fig. 3 SBUs with different dispositions and geometries. | ||
All these parameters provide us with an idea about the endless possibilities of creating structures that MOFs provide.
As mentioned above, ZIF-8 is one of the most studied MOFs and presents the advantage that its precursors are water-soluble allowing their synthesis under soft synthesis conditions such as room temperature, low reagent concentrations, and short reaction times.24 However, to modify the physical and chemical properties of the material, many other methods such as solvothermal (in dimethylformamide (DMF) or methanol (MeOH)), microwave-assisted, sonochemical, or mechanochemical among others have been employed.25 The synthesis of ZIF-8 is also influenced by the presence of surfactants like hexadecyltrimethylammonium salts (CTA+) and their co-ions (Cl− or Br−).26 However, its synthesis is highly influenced by factors that might have been overlooked such as the gravitational forces.27
In the case of MOFs with SBUs like Zr6, a wide variety of methods can be employed. In general, these methods allow the formation of metal–ligand bonds and also permit the breakage of these bonds and their reorganization to allow for structure propagation. Solvothermal synthesis is one of the most straightforward methods, in which the metal salt(s) and ligand(s) are dissolved in a high boiling point solvent such as DMF, diethylformamide (DEF) or dimethylsulfoxide (DMSO) in a vial or closed vessel, allowing for heating during long periods (i.e., 12 h or longer).28 Usually, organic acids (i.e., acetic acid, formic acid, trifluoroacetic acid, benzoic acid, etc.) are used as modulators to control the particle size.29–31 This is possible as these acids compete with organic linkers for the available Zr sites, preventing the particle growth. Also, inorganic acids such as HCl are employed as modulators protonating the organic linkers and decreasing their reactivity.32
Recently our group has investigated the role of halide ions (Cl−, Br− and I−) as co-modulators in improving not just the shape and size of the UiO-66 NPs, but also the reaction yield. We corroborate the presence of halide ions in the NMOF's structure and the missing-linker defects that the halides generate (Fig. 4).33 Our findings confirmed that by using halides, especially chloride, as co-modulators the production of UiO-66-NH2 and UiO-66 with defined geometries and nanosize is possible.
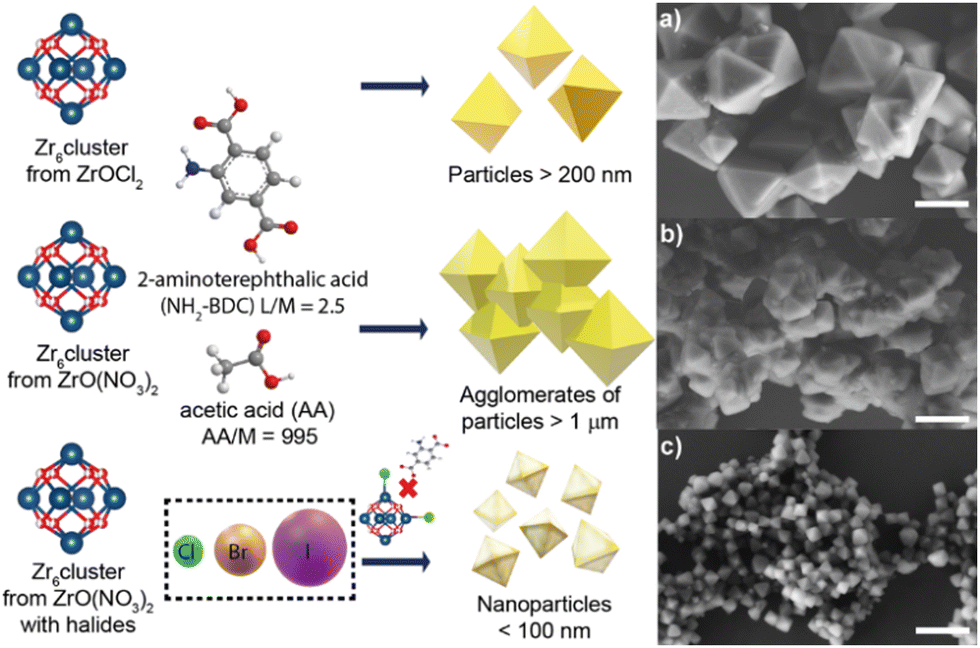 | ||
| Fig. 4 Halide effect in the generation of UiO-66-NH2 and UiO-66 nanocrystals (NMOFs). Representative micrographs of UiO-66-NH2 produced using (a) ZrOCl2 as a precursor without extra halides added, (b) ZO(NO3)2 as a precursor without extra halides added and (c) ZO(NO3)2 as a precursor with extra halides added. Scale bars correspond to 500 μm. Adapted with permission from ref. 33. | ||
There are many different approaches for the synthesis of iNPs@NMOFs, but the most common ones are (i) post-impregnation of the NMOF with metallic salt solutions, followed by the reduction of the metallic ions and (ii) the core–shell approach wherein the mixture of the reaction contains the metal node or cluster, organic linker, and functionalized NPs (Fig. 5).
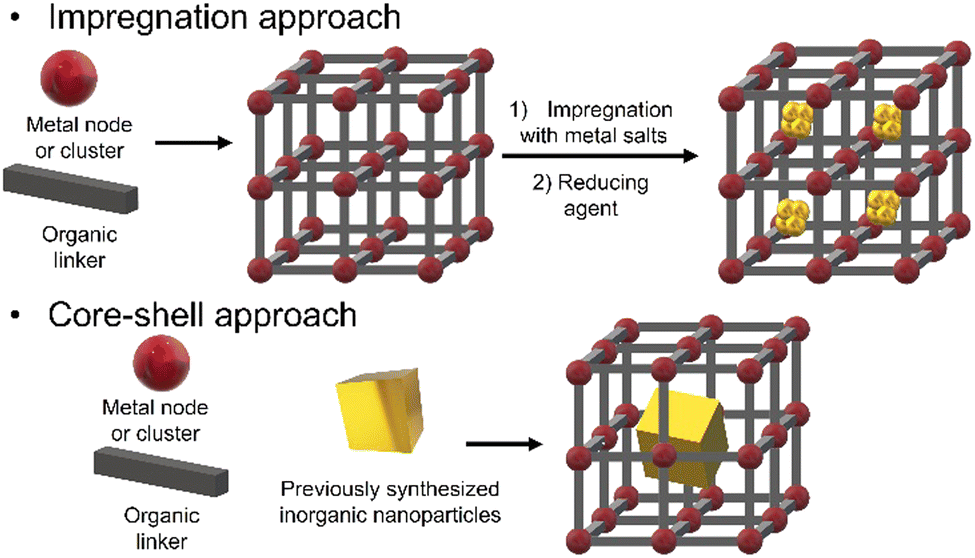 | ||
| Fig. 5 Schematic representation of different approaches to produce iNPs@NMOFs nanocomposites. | ||
The impregnation of NMOFs with metallic salts to reduce them to form small clusters of atoms has been achieved for Cu,36 Ag,38 Ni,39 Au,37 Pt,38 and alloys,39 among others. In most cases, the impregnation step involves the physisorption of metallic species within the MOF matrix, but in some cases, the processes involved the formation of coordinated bonds between an electron donor group from the organic linker and the metal ion. That is the case of UiO-67-bipyridine, where the linker 2,2′-bipyridine-5,5′-dicarboxylic acid has in its structure two nitrogen atoms capable of coordinating redox metals, like Au for the generation of reactive oxygen species (ROS) or Pd40 as a catalyst for Suzuki reactions.41 Also with this methodology, it is possible to use the NMOF's channels as a template to create metallic nanowires. This concept was demonstrated by Duan et. al. by modulating the diffusion rate of the metallic species by the solvent and increasing the redox potential by varying the pH.42 By controlling these parameters and using ultrasonic stirring, they were capable of obtaining well-aligned Au and Pt nanowires inside the NMOF porosity in MOF-545 (Fig. 6a).
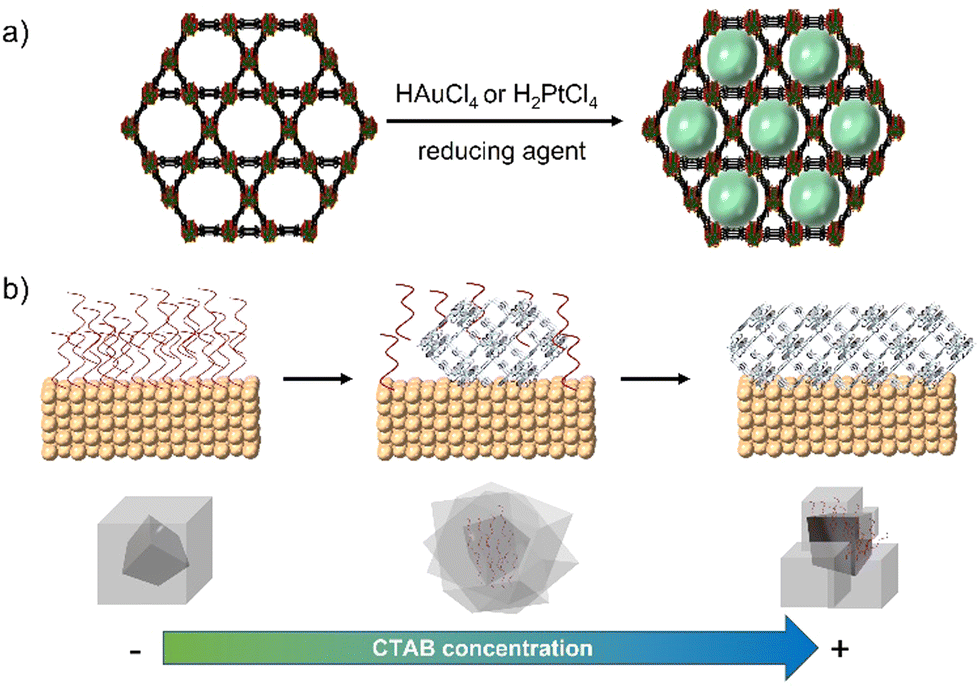 | ||
| Fig. 6 (a) Synthesis of metallic nanowires using the MOF porosity and channels as a template42 and (b) mechanism and effect of CTAB concentration for metal@ZIF-8 nanocomposites.43 | ||
The core–shell approach has not only the limitation that the previously synthesized iNPs must have an affinity not just with the metallic node or cluster and the organic linker, but also must be compatible with the reaction conditions. This last condition is rather critical as many MOF's syntheses are performed at high temperatures and in the presence of organic solvents which are not compatible with the iNPs’ integrity, leading to undesired reshaping or degradation processes. To increase the compatibility with the media, amphipathic polymers like polyethylene glycol (PEG) are used to functionalize iNPs.44 These coatings help to transfer the iNP from aqueous media to organic media.45 PEGylated gold nanorods have been used as seeds to grow core–shell systems for Zr-based NMOFs (NU-901).46,47 In these cases, PEG chains from the nanorods have been proposed to interact with MOF precursors due to their hard oxygen and soft ethylene moieties. Also, PEG chains have been used to functionalize gold nanobipyramids in order to grow ZIF-8 selectively in the middle of the NPs avoiding their tip coverage.48
The use of surfactants such as CTAB has been identified to aid in the production of an adequate surface on colloids, i.e., gold or palladium NPs to grow shells of NMOFs (Fig. 6b).43 This approach has been explored by us to create ZIF-8 shells onto individual NPs to create core@shell composites with controlled size. This approach is suitable to work on different NPs’ sizes and morphologies including anisotropic materials.14–16 Zheng et al. reported the use of different capping agents (i.e., CTAB and tris/hydroxymethyl)aminomethane) to have control over the shape of the ZIF-8's shell to create composites with irregular controlled shapes such as hexapods and burr puzzles.49
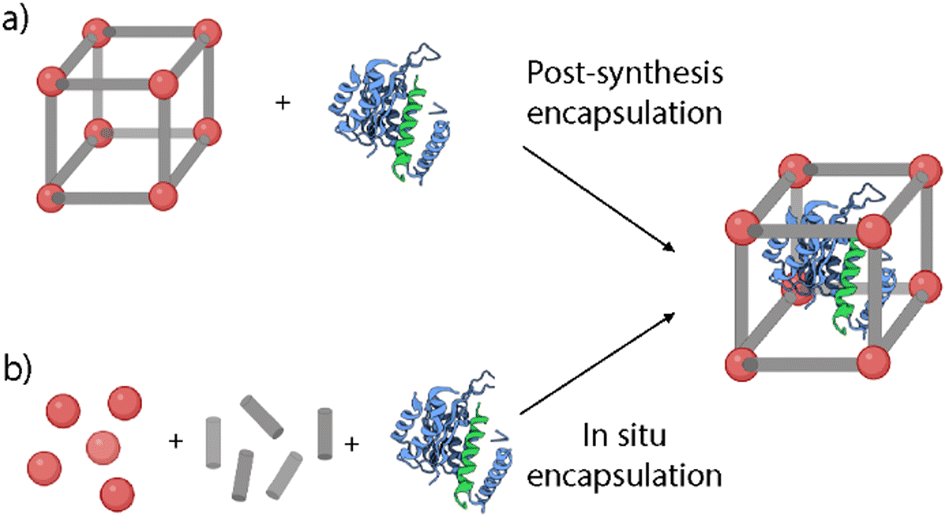 | ||
| Fig. 7 Schematic representation of the cargo encapsulation methods: (a) post-synthesis encapsulation and (b) in situ encapsulation. | ||
Post-synthesis encapsulation. This approach involves the loading of the selected cargoes via noncovalent or covalent interactions, and lately has been successfully performed for the encapsulation of chemotherapeutic drugs, DNA, or enzymes (Fig. 7a).53 The pore and the cargo size play a key role in this approach, as it is necessary to fit these agents inside the MOF's structure. MOFs’ pore sizes can be tailored from micropore (<2 nm) to mesopore (2–50 nm).54 This way, larger pores would allow the encapsulation of larger cargoes and a higher loading capability.53 However, it is critical to ensure the stability of the loading inside the structure to avoid uncontrolled cargo release. To achieve this stable loading, there are some strategies such as the MOF's amorphization via heating or ball-milling treatments, or the further functionalization of the particles with different functionalities to block the pores of the MOFs.14,55
The most used post-synthesis encapsulation approach is based on the infiltration of the selected agents. Here, the loading is achieved by free diffusion of the functional guest molecules through the pore aperture of the pre-synthetized MOFs. This way, the agents will be filling the pores typically with high loading efficiency. One of the main advantages of this method is that the loading can be carried out in mild conditions. Therefore, it enables the incorporation of sensitive cargoes such as enzymes, that may be degraded under synthetic conditions.54 Furthermore, when entrapped inside the MOFs, enzymes can maintain their activity in harsh conditions such as high temperatures or organic solvents.56 For example, Wang et al. recently reported new Zr-based NMOFs (NU-1000 and PCN-222) for the delivery of insulin as a model protein.55 Insulin was loaded via infiltration by mixing the particles with an insulin (0.4 mg mL−1) solution in bis-tris-propane buffer (BTP, pH = 7). The results showed high loading capabilities of the NMOFs with values of 34 and 63 wt% of insulin for NU-1000 and PCN-222 NMOFs, respectively. As another example, Orellana-Tavra et al. reported the loading of a bismuth-based MOF, CAU-740, with two cancer drugs (sodium dichloroacetate (DCA) and α-cyano-4-hydroxycinnamic acid (α-CHC)).57 The results showed maximum loadings of 33.7 wt% (9.8 mol of DCA/mol of CAU-7) and 9.3 wt% (33.2 mol of α-CHC/mol of CAU-7). These differences can be related to the bigger size of α-CHC, which faces more problems in diffusing through the pores of the structure.
This strategy can be applied also for the loading of other relevant active molecules such as photosensitizers (PS). For example, Lou et al. showed the encapsulation of zinc-phthalocyanine (ZnP) PSs inside a Hafnium–Iridium NMOF for highly efficient photodynamic therapy (PDT).58 The loading was achieved by simply mixing the NMOF with ZnP in DMF at 70 °C for 24 h, resulting in loadings of 13.6 wt% of ZnP. The encapsulated cargo presented high light absorption with reduced aggregation-induced quenching and an improvement of 1O2 generation. Moreover, the samples were tested in mouse models and they showed great efficiency in reducing and eradicating colorectal cancer.58
In some other cases, the post-synthesis encapsulation has been employed for the loading of fluorescent probes. As a proof of concept, we loaded a fluorophore inside the pores of ZIF-8-gold nanostar (AuNS) nanocomposites (NCs) to study the controlled cargo release.14 As the cargo we chose Hoechst H 33258 (HOE), a blue fluorescent dye typically used for DNA staining in molecular biology. The loading was performed by immersion of the NCs in a HOE solution in methanol, achieving an average loading of 2.9 × 105 HOE/NC.
However, this method presents some drawbacks as there is a size limitation (cut-off) for the selected loadings that need to diffuse through the pore aperture of the MOFs. This can also lead to some problems such as the leaking of the guest molecules that are smaller than the pore size. As a solution to this problem, Morabito et al. reported a new approach for the encapsulation of large guests inside MOFs by taking advantage of ligand exchange reactions that lead to the “opening” of external framework domains of the pre-synthesized MOF (Fig. 8).59 This ligand exchange is based on the fact that coordinative-saturated metallic centers make the dissociation easier providing the MOFs with temporary vacancies. These vacancies would temporarily expand the pore aperture allowing larger molecules to diffuse inside the NMOFs without interfering with the crystalline structure and morphology. Then, the dissociated linkers can be restored so that the cargo is trapped inside the pores. In 2018, the application of this method was reported for the encapsulation of catalysts inside the structure of robust MOFs such as UiO-66.60 The encapsulation of two molecules was considered, i.e., a fluorescent molecule, Rhodamine 6G (R6G), and a ruthenium complex as a catalyst. The encapsulation process was analyzed under different conditions and proved that the R6G loading was highest at elevated temperatures in polar protic solvents (e.g., methanol), whereas the dye leaking was reduced in aprotic solvents compared to protic solvents. The experiments were repeated in similar conditions for the encapsulation of the ruthenium complex showing coherent results and proving the potential of the method for the encapsulation of transition metal complexes within MOFs based on solvent-dependent, aperture-opening events occurring from dissociative linker exchange reactions in MOFs.
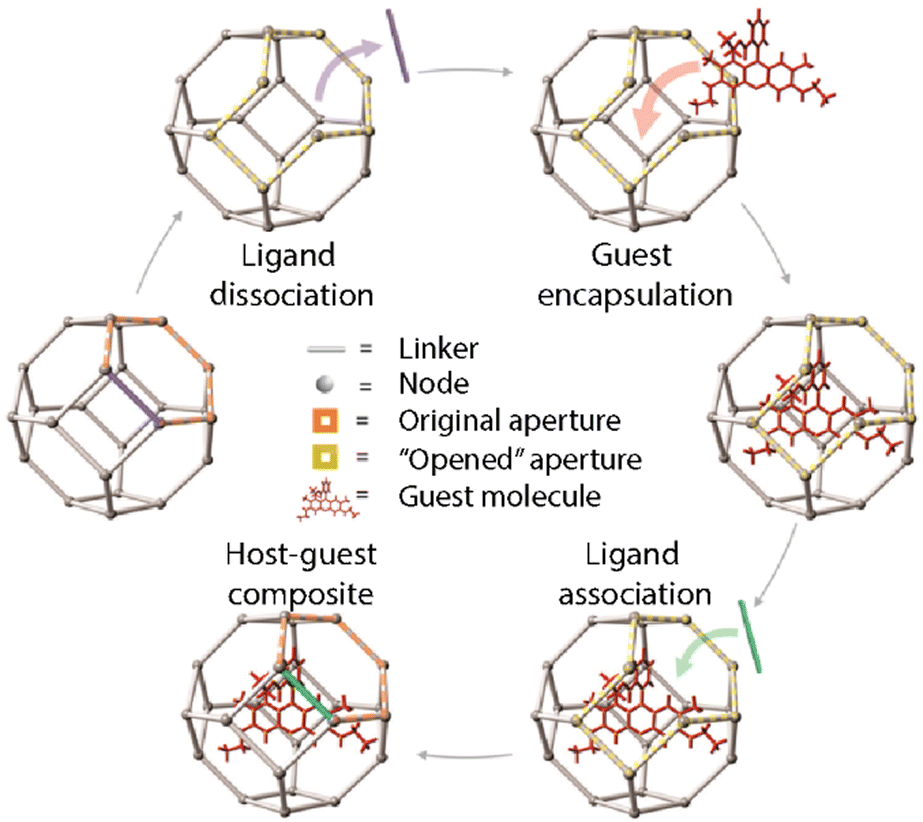 | ||
| Fig. 8 Schematic representation of the post-synthesis encapsulation of a guest molecule taking advantage of the ligand exchange reactions. Adopted with permission from ref. 59. | ||
Adapting this method, we studied the loading capabilities of R6G in a series of UiO-66 MOFs with different synthetic parameters. For the loading, the NMOFs were mixed with a solution of R6G in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Zr:R6G (w:w). The resulting loading ratios were ca. 17–20% w/w for all the synthesized NPs.33 These values show the efficiency of this method to encapsulate the cargo in a post-synthetic method.
:2 Zr:R6G (w:w). The resulting loading ratios were ca. 17–20% w/w for all the synthesized NPs.33 These values show the efficiency of this method to encapsulate the cargo in a post-synthetic method.
In situ encapsulation. This method, also called the de novo approach, involves the encapsulation of guest molecules inside the pores during the MOF growth process (Fig. 7b). The main advantage of this method is the high loading efficiency independent of the pore size.54 Some recent works have studied the encapsulation of enzymes using this approach and proved the reduction of structural changes that usually lead to protein unfolding.61,62 Furthermore, this method can also be applied for the encapsulation of a wide variety of metalorganic compounds and organometallic complexes.63 As an example, Liao et al. employed this method to prove the maintenance of the biological function of enzymes under a wide variety of conditions.61 They reported the encapsulation of catalase inside ZIF-90 and ZIF-8 MOFs. The loading was performed via a water-based mild de novo approach where a zinc nitrate solution was mixed with an aqueous solution containing appropriate amounts of imidazolate-2-carboxaldehyde (ICA), catalase, and a capping agent. The results showed loadings up to 6.0 wt% and the encapsulated catalase showed higher stability and retained its enzymatic activity even under harsh conditions such as urea treatment. Alsaiari et al. reported the first successful encapsulation of CRISPR/Cas9 inside ZIF-8 NMOFs (CC-ZIFs) for its intracellular delivery.64 The co-encapsulation of the Cas9 protein and sgRNA was performed via de novo approach, by mixing Cas9 and sgRNA in a phosphate buffer saline (PBS) solution with the ZIF-8 precursors in water. The NMOFs’ size was characterized by electron microscopy and dynamic light scattering (DLS), presenting sizes of ca. 100 nm. The loading capability was estimated to be 1.2 wt% of Cas9/sgRNA with an efficiency of 17%. Furthermore, the particles showed improved endosomal escape abilities, with a decrease of up to 37% of the gene expression over 4 days. This way, they proved the potential of these ZIF-8 NMOFs as an efficient delivery method of efficient and transient gene editing technology.
However, this encapsulation strategy presents some drawbacks as it requires the stability of the selected guest molecules in the synthetic conditions, which in some cases involve organic solvents or high temperatures. Recently, Cases Díaz et al. have extended the MOF biomineralization in MIL-100 (Fe) using several proteins and enzymes such as bovine serum albumin (BSA), myoglobin, subtilisin Carlsberg, and bovine cytochrome C. This rational synthetic method which also considers the isoelectric point of the protein leads to efficient MOF's protein-loadings (i.e., from 4 to 30 w/w%) while maintaining the enzyme's functionality.62
Overall, guest immobilization techniques still need to be improved to achieve higher loading rates and increased stability of the cargo and loaded MOFs for their application in biomedicine. Some of the main problems to be addressed are related to (i) the microporosity of the structures which avoids the incorporation of macromolecules such as enzymes, (ii) the synthetic conditions that are not biocompatible with the integrity of some cargo (i.e., chemical stability, or maintenance of structure and function of proteins) and (iii) the stability of the cargo once it is encapsulated.54 Therefore, it is necessary to develop new strategies so that these systems can be used in clinics.
There are two main strategies for the surface functionalization of NMOFs. The first one is based on the modification during the NMOF's self-assembly where, as an example, the organic linker can be modified to introduce functional groups to change the final NMOF surface. This method presents an important limitation as the potential of organic molecules that can be introduced as linkers is limited.66,67 The other one is the so-called post-synthetic modification (PSM) that allows the surface functionalization of already synthesized particles. In this case, it is critical to use specific reaction conditions to avoid NMOFs’ degradation and loss of crystallinity and porosity.66 Within this second method, we can find four main approaches (Fig. 9): (i) the attachment of new molecules via coordination to free metal sites (coordinative PSM), (ii) the formation of covalent interactions at reactive sites (covalent PSM), (iii) the “core–shell” strategy based on NMOF coating with polymers or silica and (iv) cell membrane coating.67–69
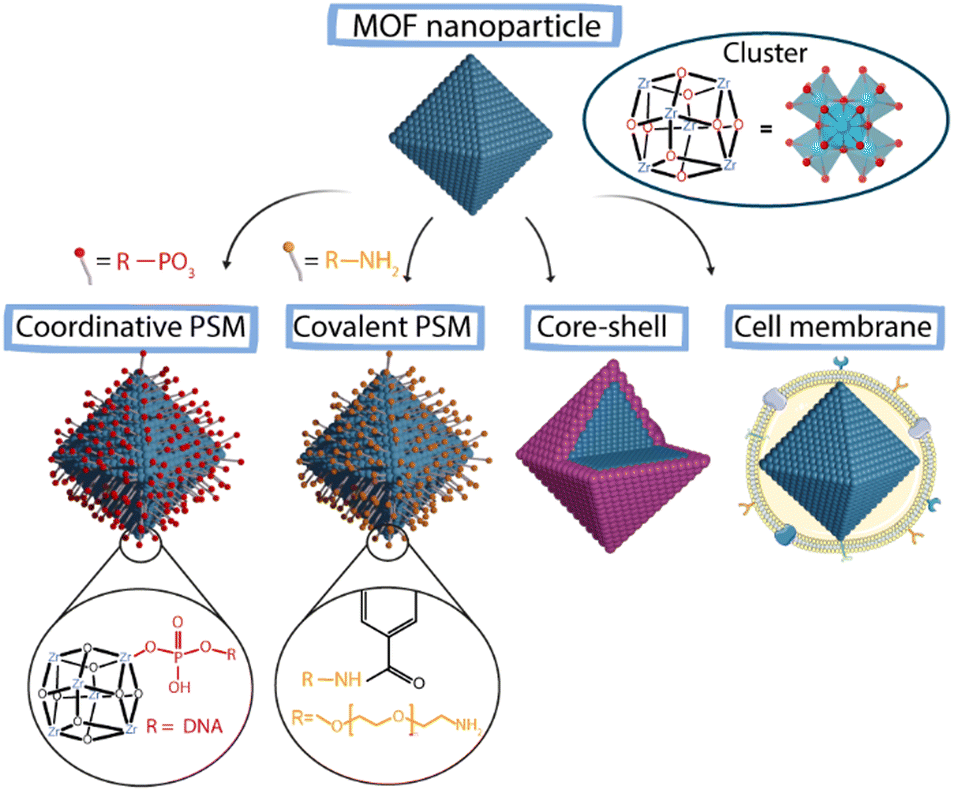 | ||
| Fig. 9 Schematic representation of different PSM approaches to achieve surface modification in MOFs. | ||
Coordinative PSM. This type of PSM takes advantage of the free metal sites present on the superficial SUBs of NMOFs due to structural defects to add new functionalities via coordination chemistry. The most used molecules are chelating molecules such as carboxylates or phosphates. This way, the surface of the NMOFs can be selectively modified while maintaining the porosity and crystallinity of the MOF.70 Using this method, NMOFs can be easily functionalized with biomacromolecules modified with coordinative groups. In the context of biomedical systems, one of the main challenges is the intracellular delivery of biomacromolecules such as proteins or peptides.71 Wang et al. reported the functionalization of the Zr-based NMOF NU-1000 via coordinative PSM with nucleic acids.72 For that, they employed terminal phosphate-modified oligonucleotides achieving a homogeneous and effective coating that increased the NMOF's stability and cellular uptake. Similarly, Chen et al. used the free positions in the Zr present in PCN-222 NMOFs to functionalize their surface with a phosphate-functionalized PEG (5 kDa mPEG-PO3) to increase the stability and delay the release of the encapsulated drug.73
Covalent PSM. The post-synthetic covalent modification of NMOFs’ surface includes two main strategies: (i) the addition of functionalities that do not fit inside the pores of the structure and (ii) the use of reactive moieties in the outer part of the particles to add new functionalities.70 To work, this strategy requires that the selected ligands have specific chemical functionalities that do not interfere with the MOF's structure and allow the modification of the outer and internal surfaces of the MOF.71,74 This MOF functionalization approach can improve the colloidal, chemical and thermal stability of the particles.61,62 Some modifications include N-alkylation, click chemistry or protonation.70 Besides the improvement of the colloidal stability, one of the main purposes of the surface modification of NMOFs in the context of bioapplications is the increment of the blood circulation time of the NPs (stealthiness). Usually, different polymers can be employed for this purpose, such as PEG, which is one of the most typical and common coatings used for nanomaterials.71 As a representative example, Zimpel et al. reported the functionalization of the MOF MIL-100 (Fe) with amino-PEG by the conjugation of the amino group with the NMOF surface groups, achieving increased stability of the NMOFs.75
“Core–shell” strategies. The creation of core–shell structures is an approach widely used for stabilizing colloidal inorganic NPs. Now, this methodology is also being used to stabilize NMOFs, increasing their biocompatibility.70,71 This method is based on coating of the NMOF core with a dense shell. Silica coatings are typical examples of these core–shell strategies, which provide protection to the NMOF against decomposition and can act as a support for further functionalization. The use of organic shells is a very extended strategy to create core–shell systems. Polymers such as poly(vinylpyrrolidone) (PVP) and PEG are widely used to fabricate core–shell nanosystems. To create shells using polymers, amphiphilic polymers such as poly-[isobutylene-alt-maleic anhydride]-graft-dodecyl (PMA) can also be selected. This polymer has been used to stabilize NPs of different materials, shapes and sizes.34,45 Recently, this amphiphilic polymer has been used to coat the surface of ZIF-8 (Fig. 10).14 This way, the MOF was provided with increased water stability while blocking the pores of the structure avoiding the leaking of the cargo. Besides this, the polymer can be modified with functional groups to act as an anchoring point for the further addition of new functionalities. Other examples of this coating strategy include the lipid bi-layer coating and the cell membrane coating, which are usually employed to improve the stability of the particles in biological media and achieve immune evading capacities, leading to an increase in the blood circulation time.76,77
 | ||
| Fig. 10 Scanning electron micrographs obtained by using different detectors to study the stability of AuNS@ZIF-8 NCs in different media: (a) after their synthesis in methanol (first row), (b) after their dispersion in water (middle row) and, (c) in water after their coating with PMA to enhance their water-stability (lower row). Scale bars correspond to 200 nm. Adapted with permission from ref. 14. | ||
Cell membrane coating. The use of cellular membranes to coat colloidal nanomaterials can endow these systems with new properties. Coating of nanomaterials has been carried out using different cell lines including platelets, mesenchymal stem cells, red blood cells, immune cells or cancer cells.69 The final properties of the nanomaterials will be highly influenced by the cellular coating providing them with lymphocyte and dendritic cell activation, tumor targeting, immune escape abilities, endothelial adhesion, or homotypic targeting, respectively.78–81 One of the most commonly used approaches is the use of membranes derived from cancer cells; this coating provides NMOFs with homotypic targeting ability. This term refers to the ability of self-recognition of cancer cells for its cell lineage or the evasion of the immune system.82,83 Therefore, this strategy offers great advantages for its use in areas such as cancer therapy or vaccination.
Whereas NMOFs had been coated before with liposomes or exosomes, this strategy is relatively new, with a few examples available.84,85 Recently, Alyami et al. have developed biomimetic cancer cell-coated zeolitic imidazolate frameworks (ZIFs) for cell targeting and specific delivery of CRISPR/Cas9, following the results previously discussed in Section 1.4.86 They coated the CRISPR/Cas9-loaded ZIF-8 NMOFs (CC-ZIF-8) with human breast adenocarcinoma cell (MCF-7) membranes to improve cell-targeting by the inherent homotypic targeting of tumoral cells. The coating was prepared by extracting the membrane of cancer cells and by mixing them with CC-ZIF-8 under stirring. Finally, the sample was collected by coextrusion with a 200 nm porous polycarbonate membrane, rendering a final uniform population of coated NMOFs of 120 nm. The uptake of CC-ZIF-8 was studied in different cancer cell lines and healthy cells, showing higher uptake for MCF-7 cells and negligible uptake for the healthy ones. This proved the efficiency of this biomimetic approach increasing NMOFs’ selectivity and overcoming one of the main problems in DDSs.
The surface of the nanocrystals constitutes the most reactive domain of the structure and is where the interaction with the components of the surrounding media occurs. As previously mentioned, engineering the surface of NMOFs is critical to determine aspects such as the colloidal stability of the NMOFs in biological media, the formation of the protein corona or the targeting abilities.88 For this reason, it is important to study all the possible interactions on the bio-interface (aggregation, biodistribution, or elimination) to develop stable systems.89
The stability of NMOFs can be divided into colloidal and chemical stability.
1.6.1. Colloidal stability. Colloidal stability refers to the capacity of the particles to remain suspended at equilibrium in a solution. Therefore, the lack of colloidal stability would lead to limitations in the possible administration routes and/or issues related to uncontrolled early drug delivery.90 For this reason, it is critical to consider colloidal stability under long-term storage and further resuspension in solution before their administration.90 Moreover, it is important to analyze the behavior of the particles in physiological environments. This behavior is partially governed by electrostatic interactions. So, when the surface charge of NMOFs is close to neutrality, the repulsion between the particles will be reduced and they will tend to aggregate. Likewise, if the surface charge increases, the colloidal stability will be improved. However, this feature depends on the medium where the particles are placed and the interaction with the components of this medium; for example, proteins tend to adsorb non-specifically forming the so-called protein corona or biocorona which can lead to uncontrolled aggregation processes.91
1.6.2. Chemical stability. The chemical stability of MOFs has received considerable attention during the past few years. Several studies have been performed to understand the decomposition pathways and the stability of the particles in different media.87 Ideally, the chemical stability of NMOFs should synchronize in vivo degradation with the expected functional therapy for a given biomedical application.88 Generally, the chemical stability of MOFs is referred to their ability to retain their structural features after being exposed to chemicals, high temperatures, and mechanical forces.92 This stability depends on several factors such as thermodynamic stability, kinetic stability, or the presence of other elements present in the environment surrounding the particles, all influencing the design of stable framework structures.93,94
Thermodynamic stability. The stability of the MOFs is related to their resistance to chemical treatments (such as water, acid, or salt solutions) to maintain the structure and porosity.95 This stability is mainly related to the strength of the metal–ligand (M–L) coordination bond, which may be predicted by Pearson's (HSAB, hard soft acid-base) theory. This theory considers ligands with relatively high pKa (e.g., azoles like imidazolates or pyrazolates) as soft bases producing robust frameworks with low-valent metal ions (Zn2+, Cu2+, Ni2+ and Mn2+) which can be considered as borderline soft acids. The most typical example of this is ZIF-8 built by the coordination of Zn2+ metal ions and imidazolates. On the other hand, ligands with relatively low pKa (e.g., carboxylic acids) are considered as hard bases and form highly stable MOFs by preferentially binding to high-valent metal ions (Ti4+, Zr4+, Al3+, Fe3+, and Cr3+) which are hard acids.93,95 The Zr4+ MOFs such as UiO-66 ([Zr6O4(OH)4 ((OOCC6H4COO12]) show high stability features. Zr4+ has a high charge density, which creates a strong affinity when the Zr4+ ions interact with carboxylate groups, generating strong Zr–O bonds. The combination of [Zr6O4(OH)4] or [M3OX(CO2)6] (M = Al3+, Cr3+, or Fe3+; X = OH−, F−, or Cl−) clusters with carboxylate linkers with different topologies gives rise to a wide diversity of MOFs with great stability, such as UiO-66, PCN-222, MIL101(Cr), and MOF-808.95
Kinetic stability. The kinetic factors constitute other key points affecting the stability of the MOFs and depend mainly on the rigidity of the linker. The elongation of the ligand has the advantage of the enlargement of the pores, but at the same time, it weakens the framework structure.95 The reason is that the length of the ligand is related to the activation energy of the MOF dissociation process and therefore it affects its degradation.92 As an illustrative example, the UiO isoreticular family of MOFs (UiO-66, UiO-67, and UiO-68) have the same cluster but they differ in the linker length. The coordination bond strength should be equal in the three cases based on the previously explained HSAB theory and the chemical stability should be the same. However, it has been proven that there is a decrease in the stability with increasing linker length directly correlated to its rigidity.87 Dense and rigid frameworks formed by rigid and highly connected building blocks (metal ions/clusters and ligands) generally show higher stability against partial lattice collapse. Another kinetic factor affecting structural stability is the linker hydrophobicity. When the surface of the particle is mainly hydrophobic, the structure tends to be more stable in aqueous media as it avoids the adsorption of water in the pores or its condensation around metal clusters.93 The incorporation of functional groups within the MOF structure will be likely to reduce the pore size and cause the condensation of water vapor at decreased pressures.87 For example, in ZIF-8 the methyl groups on the imidazolates can serve as blocking agents preventing water molecules from approaching the ZnN4 node leading to increased stability.92
Additionally, by selectively designing hydrophobic surfaces or interfaces, water and other guest molecules can be excluded from approaching metal ions.87 The addition of functional groups capable of repelling water from the metal clusters’ surroundings can act as a defense against the weak coordination bonds improving the water stability.93 Furthermore, the structural stability of the particles can be improved by using post-synthetic strategies including post-synthesis exchange, post-synthesis modification or hydrophobic surface treatments.93 Some examples of typical coatings to improve the colloidal and chemical stability of the particles in biological media are polymers, polysaccharides, and phospholipids, where the hydrophobic part can coat homogeneously the outer surface of the NMOFs that would impede the permeation of ions and protect the weak coordination bonds against phosphates present in the medium.90
Despite the efforts, the assessment of the chemical stability in biological media is still not completely addressed. This is a consequence of the high number of variables that should be considered, such as the type of components of the media, their concentration, or the temperature of the solution, among others.92 Generally, different combined techniques such as powder X-ray diffraction (PXRD) and porosity analysis with inert gas are employed to evaluate the stability of the particles. However, although great progress has been made in this matter, some challenges remain unsolved. Therefore, it is important to develop new methods to evaluate and enhance the chemical stability to promote the advancement of NMOFs for bioapplications.
The toxicity of NMOFs, as in the case of other NPs, depends on different parameters that can affect their interaction with living organisms, including chemical composition, structure, size, morphology, and surface properties.88 These materials can be easily internalized into cells and subcellular compartments.96
NPs’ routes of entry inside the cells are determined by their interaction with elements in the extracellular matrix and the outer plasma membrane. Generally, the entry process occurs via endocytosis, where the particles are internalized in the cells via engulfment processes followed by the generation of endocytic vesicles and transported to the particular intracellular compartment.97 Very generally, one can distinguish five types of endocytosis: phagocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, clathrin/caveolae-independent endocytosis, and micropinocytosis.98 Clathrin- and caveolin-mediated endocytosis constitute two main mechanisms of NP uptake.98–100 These interactions are highly affected by the physicochemical properties of NPs, such as the size, shape, surface charge, surface chemistry, and chemical stability (Fig. 11).98,101
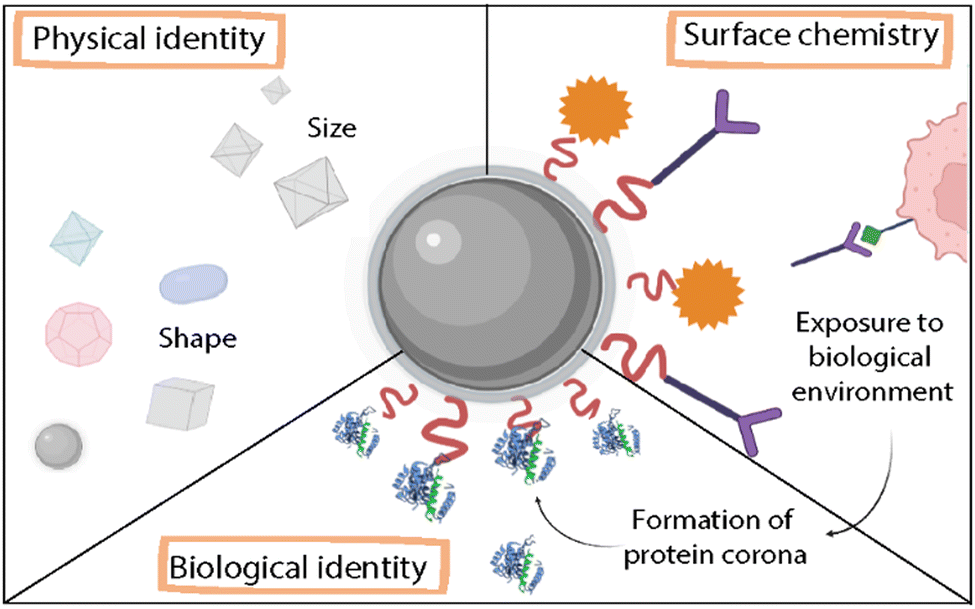 | ||
| Fig. 11 Schematic representation of factors that determine the toxicity of NMOFs. | ||
Size and agglomeration. The size of the NPs plays a key role in the cellular uptake and toxicity of the NPs.98,101 NPs can induce toxicity as their size becomes comparable to the size of the molecular components; their ability to penetrate physiological barriers increases, inducing the interruption of physiological functions and sometimes causing cell death. Furthermore, as mentioned above, when reducing the size of a material to the nanoscale, there is a high increase in the surface area and also in its reactivity. As a result, this increment in the surface area leads to higher exposure to the surrounding media which in case of any possible degradation process can induce the division of the particle into smaller fragments. Like the dimension effects, the agglomeration state of the NPs in biological fluids also affects toxicity. In most cases, the interaction of NMOFs with biological media components leads to an uncontrolled aggregation that produces the formation of bigger agglomerates which changes their biological behavior.
Surface chemistry. As previously mentioned, the surface of NMOFs is their most reactive part. The correct surface engineering can contribute to the stability of the NPs in biological media and to minimize undesired interactions with the surrounding biological environment.88 One important factor is the surface charge of NMOFs. For example, charge affects the electrostatic interaction between the NMOFs and cellular membranes, having a great impact on cellular uptake. Some studies have shown that charged particles, either negative or positive, showed higher uptake than neutral counterparts, due to the attractive/repulsive forces among the cationic/anionic NPs and the negatively charged cell membrane.101
The hydrophobicity will affect the interactions with proteins and the protein corona formation and therefore the way they interact with cells.74 As previously discussed, NMOFs can be functionalized to target specific receptors or molecules by the addition of peptides, antibodies, or proteins.96,98 At the same time, several strategies have been developed to increase the chemical stability and minimize the adsorption of biomolecules such as proteins or lipids, as well as to target the NPs, i.e., PEGylation. Nevertheless, it is worth noticing that despite the surface functionalization of NMOFs, their properties will experience changes when placed in biological media that can be difficult to predict, and these interactions in the bio-interface will define the fate of the NMOFs, and hence, their toxicity.96
Biological chemical stability. The stability under relevant biological conditions is also an important feature to consider before evaluating the final toxicity. MOFs can undergo different degradation processes in biological media. Depending on the nature of MOF components (i.e., metal cations, SBUs or linkers), they will be processed by the organism in a different way and may generate higher toxicity rates. For that, it is important to evaluate both the stability of the structure, choosing MOFs with strong coordination bonds between the metal and the ligand, and the toxicity of the individual components i.e., the toxicity of the free metal, metal clusters, or linkers. Following the concepts discussed in the previous section, for a more acidic environment, it would be convenient to use NMOFs with ‘hard’ constituents to avoid the competition between metal ions and protons from the media. However, in more basic surroundings these MOFs will likely degrade due to the exchange of organic ligands by OH− groups present in the media.96 Depending on the components, the toxicity of the NMOFs would be classified as less toxic for those containing Ca2+, Bi3+ or Eu3+, followed by those with Ti4+, Fe2+, Fe3+, Co3+, Al3+, and Cr3+, and followed by Zr4+, Mg2+, Gd3+, Ni3+, and Zn2+, whereas those with Cu2+ and Mn2+ can be considered the most toxic. Moreover, the hydrophobicity of the linker also plays an important role in the final toxicity of the particles. As an example, hydrophobic ligands interact easily with lipid droplets in the lysosomes, so it takes more time to remove them from the organism, whereas hydrophilic linkers are cleared faster.97 Therefore, the engineering of the particle surface constitutes a key factor for the improvement of the stability, as it would avoid the degradation of the particle and the biodistribution would be mainly influenced by the properties of the MOF (size, surface chemistry, etc.) and not its constituents. The higher the degradation of the MOF, the higher the release of potentially toxic substances.
To assess the biocompatibility of NMOFs, their behavior must be studied carefully. The first step will cover testing their stability and degradation in different biological media, such as phosphate buffer solution (PBS) or cell culture media, at their corresponding pH. These studies will facilitate the prediction of the in vitro behavior of the NPs before performing further toxicity tests. AuNS@ZIF-8 composites were employed for this purpose with and without a PSM based on the use of polymer coating (i.e., PMA).14 The results confirmed that the NMOF-composites were stable in both aqueous and complete cell media upon PSM (Fig. 12a). Recently, we studied the stability and toxicity of NMOFs from the UiO family (UiO-66 and UiO-66-NH2) using different synthetic parameters confirming their high stability, biocompatibility, and reduced toxicity.33 Before the in vitro toxicity assays, the stability of the particles was evaluated in four different media: milli Q water, lysosomal medium (phagolysosomal simulated fluid (PSF), 0.02 M and pH 5), complete cell media (Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS)) and phosphate-buffered saline (PBS, 0.1 M, pH 7.4), showing good stability for all cases except for some aggregation of the particles in PBS, resulting in an increment in the effective diameter after 1 h. Fig. 12b shows the stability of these NMOF in aqueous solution and in lysosomal medium.
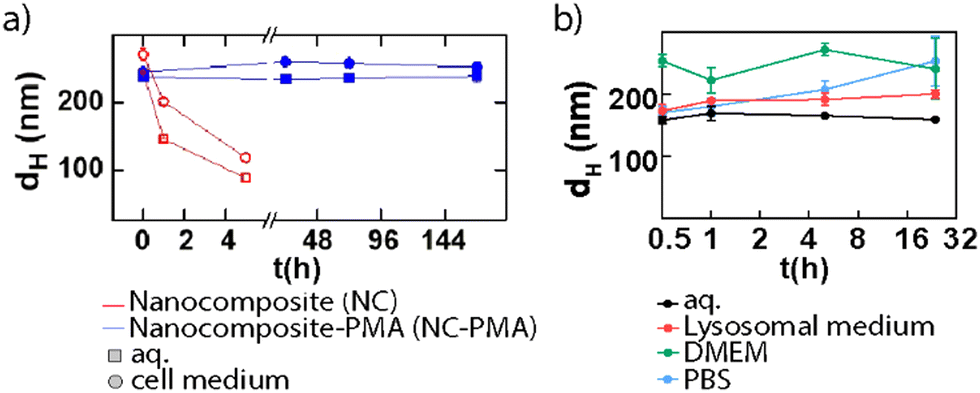 | ||
| Fig. 12 Mean hydrodynamic diameter (dh) for (a) NMOF composites during one week (red) and NMOF composites-PMA (blue) systems in water and cell medium and (b) UiO-66-NH2 NPs during 32 h in different media. Adapted with permission from ref. 14 and 33. | ||
After confirming the stability along time in complex biological media, in vitro studies should be carried out. However, cytotoxicity studies in vitro are generally conducted in cancer lines to prove their potential as cancer treatments, whereas studies in healthy cells are less frequent.74 In these studies, the particles are normally incubated for 24 h to 72 h at different dose rates, and cell viability is measured. The typical parameter evaluated is referred to as IC50 (i.e., half maximal inhibitory concentration), which corresponds to the concentration (in mM or mg mL−1) of the particles that cause 50% inhibition of cell growth.96 For example, we evaluated the in vitro toxicity of the UiO-66-NH2 samples using the resazurin test in A549 adenocarcinoma cells.33 With concentrations of up to 100 μg Zr mL−1, the results showed no significant toxicity for any sample after 24 h. In addition, we studied the cellular uptake by flow cytometry using two different concentration metrics, one based on the particles’ molarity (25 pM) and using equal Zr amount (2.5 μg Zr mL−1). For the samples at 25 pM, the results showed variations in the cell uptake for different sizes of the particles, proving the effect of the size on the fate of the nanocarriers. However, when the samples were evaluated at a concentration of 2.5 μg Zr mL−1 (or 8 μg mL−1 of MOF), the uptake was very similar in all cases independent of the size. These uptake rates were expected based on the physicochemical parameters studied during the characterization of the NMOFs, i.e., size or charge.
2. NanoMOFs for bioapplications
As discussed before, NMOF's unique properties such as the wide variety, their reusability, controllable degradation, the high surface areas, tunable loading capabilities of biological cargoes, or the good biocompatibility of the structures are being appreciated for their application in biomedicine.102 Therefore, they are being used as nanocarriers for the delivery of drugs, imaging agents, biomolecules, etc.; contrast agents for imaging; or for different therapies such as chemotherapy, PTT, or PDT.103 Furthermore, NMOFs constitute a promising material for catalysis in biomedicine, as they can act as heterogeneous catalysts due to the activity of their metallic centres or by synergistically combining with catalytic NPs (Au, Pt, Pd, etc.) forming composites.104The formation of active composites using NMOFs as host carriers allows the incorporation of other functional materials, such as NPs or polymers, creating nanocomposites with improved features with respect to the pristine NMOFs.105 Some typical NPs encapsulated inside NMOF structures are catalytic NPs (Au, Pt, Pd, Cu), magnetic NPs (Fe3O4), or quantum dots. These composites combine both the crystalline and highly porous structures of the NMOFs with the unique properties of the NPs, composing new promising materials for biocatalytic applications, drug delivery, or bio-imaging.105
As DDSs, NMOFs provide intrinsic features such as high loading capabilities or the easily tunable external surface of the particles. They can also be designed as stimuli-responsive systems for the triggered intracellular release of their cargo.107 During the past few years, different cargoes were studied for drug delivery applications of NMOFs. Especially, they have been studied as anticancer treatments based on the delivery of chemotherapeutic drugs (i.e., doxorubicin, cisplatin, or topotecan), siRNA, PS, or light-absorbing dyes.108 On the other hand, NMOFs have been recently explored for the delivery of biomolecules with important roles in biological processes, such as nucleic acids, proteins, lipids, or carbohydrates.103
While there are many possible therapeutic agents that can be efficiently loaded into the pores of NMOFs,109 the high loading capabilities are not enough to create a functional system. For that, it is necessary to control the cargo release to prevent off-target effects and increased cytotoxicity. Stimuli-responsive NMOFs could be a solution to address these problems, especially those responding to given stimuli such as pH, light, magnetic fields, redox, ions, ultrasound, etc. These systems can undergo conformational changes under a determined stimulus that will lead to the release of the cargo.88
pH-responsive NMOFs are the most studied stimuli-responsive systems considering the acidic microenvironment of tumor cells, lysosomes, or endosomes (pH varying from 4.5 to 7.8). Acidic environments can lead to the degradation of some MOFs with an acidic response such as Zn–MOF, Fe–MOF or pH-sensitive bonds as amine or imide, leading to the release of the cargo.5,110 Among them, ZIF-8 and MIL-10× MOFs are widely employed for this purpose due to their pH sensitivity (pH 5.0–6.0).110 Cabrera-García et al. recently reported the design of novel pH-responsive DDSs based on amine-functionalized MIL-100(Fe) and MIL-101(Fe) NMOFs loaded with the cytotoxic molecule 20-(S)-camptothecin (CPT).111 The system showed high stability under physiological pH conditions with no release, whereas when the particles are internalized by the lysosomes, the acidic pH (pH 5) boosted the drug release. This way, they achieved the delivery of the cytotoxic CPT while minimizing the side effects.
Magnetic responsive NMOFs are capable of interacting with an external magnetic field showing a synergistic effect both triggering a controlled drug release and allowing the accumulation of the particles in a target location. This approach is also referred to as magnetophoretic therapy. Furthermore, these systems may be used for MRI, which can lead to enhanced contrast in T2*-weighted images.8,113 Some systems might combine magnetic stimuli-response with other stimuli-responsive features, as in the work reported by Chen et al. where they designed magnetic nanocomposites combining magnetic hyperthermia and chemotherapy treatments (Fig. 13).112 They designed a system composed of a Fe3O4@PDA (polydopamine) core and a ZIF-90 NMOF shell (Fe3O4@PDA@ZIF-90) with an average overall size of 200 nm. The Fe3O4 NPs served as magnetic heating platforms under excitation with an alternating magnetic field, whereas the ZIF-90 shell acted as a protective layer and as a drug container for doxorubicin (DOX) whose release was triggered in a pH-responsive way. After cell uptake, the DOX-loaded Fe3O4@PDA@ZIF-90 NCs showed excellent efficacy in killing tumor cells due to the synergistic effect of magnetic hyperthermia and pH-triggered drug release.
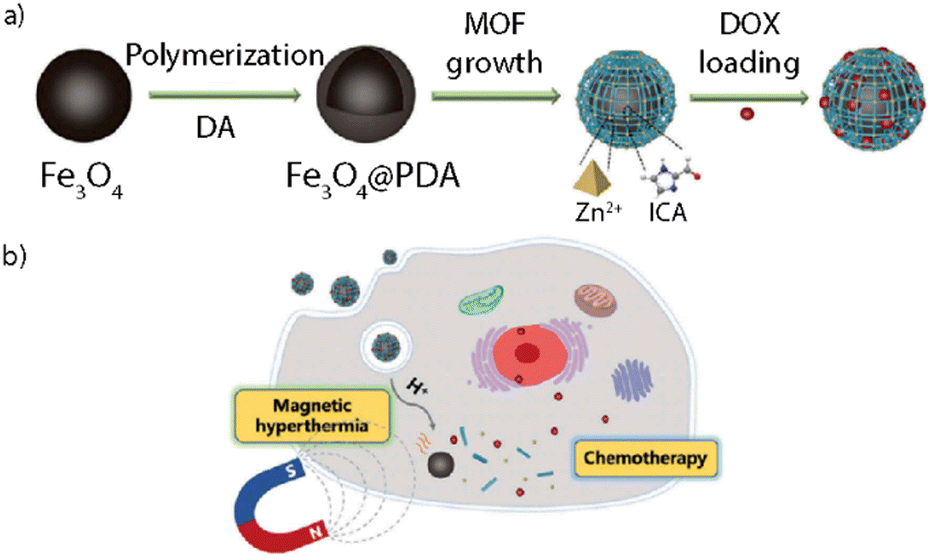 | ||
| Fig. 13 Adapted with permission from ref. 112. | ||
Light-mediated therapy, or phototherapy, also represents a promising strategy for therapeutics and diagnosis as it is non-invasive and involves high spatial precision.109 Light-responsive NMOFs as DDSs aim to control the cargo release via induced conformational changes, chemical bond cleavage, or photothermal conversion of the molecules/materials under illumination.5 Phototherapy aims for cell-killing treatment by employing therapeutic systems capable of reacting under light irradiation. This approach can be divided very generally into two strategies: one based on the in situ generation of reactive oxygen species (ROS), called PDT; and another based on heat generation and temperature increase, called PTT. In both cases, one can differentiate between intrinsic photoresponsive MOFs and MOF composites among the phototherapeutic agents (PTAs) used. PDT constitutes a non-invasive approach for cancer treatment due to the oxidation capabilities of ROS to nucleic acids, enzymes, proteins, and cellular membranes in cancer cells.114 In particular, NMOFs can be used for this type of therapy as a result of the easy diffusion of singlet oxygen and preventable self-quenching.114 Among the different intrinsic photodynamic MOFs used for PDT, porphyrin-based MOFs constitute the most explored structures. In this case, the photo-response of the NMOFs can be tuned by using different porphyrin linkers. Recently, Kan et al. reported a UiO-66 NMOF-based PS (UiO-66-TPP-SH) for PDT.115 NMOFs are based on an S-ethylthiol ester monosubstituted metal-free porphyrin (TPP-SH) incorporated via post-synthetic modifications. The particles, with sizes around 150 nm, were further functionalized with porphyrinic PS that besides maintaining the MOF crystallinity, allowed effective singlet oxygen generation. This way, they produced efficient nanosystems for PDT tumor treatment.
As discussed briefly in Section 1.4, recently, Luo et al. have demonstrated that the encapsulation of PS (i.e., ZnP) in an isolated form inside NMOFs (ZnP@Hf-QC) prevents the PS aggregation and overcomes some solubility limitations while preserving enhanced PDT abilities to treat effectively colorectal cancers in vivo.58
On the other hand, PTT is based on the conversion of near infra-red (NIR) light to heat, known as the photothermal effect, with high precision and efficacy. PTT has the advantage of being non-invasive and easily implemented besides other treatments.114 PTT can further cause the anti-tumor immune response, provoking irreversible cellular damage and subsequent tumor death.116 The main advantage of NIR light-responsive systems versus visible light-responsive ones underlies the biological window range. The biological window(s) corresponds to the light range(s) where light penetration into biological tissues reaches a maximum, i.e., expected penetration depth of 1000–1100 nm. The first biological window comprises wavelengths from 700 nm to 950 nm.117 For this reason, developing systems with strong absorption in the NIR and with high photothermal conversion efficiency is a great strategy to produce PTT platforms. Photothermal MOFs can be fabricated by the encapsulation of PTAs inside the structures.118 Noble metal NPs are excellent materials that fulfill the required features for PTAs and have been widely studied for this application. These materials include Au, Ag, Pt, and Pd and can be excited with light to efficiently produce heat through non-radiative decay processes. However, among these, Au-based NPs (AuNPs) are the most used agents for PTT. Spherical Au NPs have their localized surface plasmon resonance (LSPR) centered around 520 nm, but LSPR can be red-shifted to the NIR by changing the shape and size of the particles, i.e. by producing nanorods, nanostars, nanoplates, nanobipyramids, etc.119 Nanocomposites based on MOFs and AuNPs have a promising future for drug delivery as they combine the advantages of both systems. On the one hand, they have the great features of AuNPs such as excellent tunable photothermal properties and good stability in biological environments. On the other hand, they have the advantages of MOFs such as high porosity, highly tunable surface functionalities, and the ability to host a wide variety of APIs. These composites have been employed for optical sensing, therapeutics, theragnosis, and heterogeneous catalysis with encouraging results.14,102 However, due to the aqueous instability of most of these systems, their application as DDSs should still be optimized.
Recently, Zhou et al. reported the design of a MOF composite that combines the synergistic effect of both PDT and PTT.114 To achieve this, they synthesized a porphyrinic NMOF (PCN-222) shell with a core of plasmonic AuNRs. The singlet oxygen (1O2) generation by the PCN-222 shell was analysed after the exposure of the particles to LED light (640 nm, 20 mW cm−2), by using a fluorescent 1O2 indicator. The results showed an increase in the fluorescence correlated with longer irradiation times. Then, the photothermal activity of the NRs of the composite was evaluated by irradiation with a NIR 808 nm laser at different power values. This led to an increment of the temperature during the first 3 minutes, followed by a plateau after 5 minutes. The plateau temperatures varying from 35 to 60 °C depended on the laser power, confirming the efficient thermal conversion of light (ca. 22.2% conversion efficiency). Finally, the composites were incubated with breast cancer cells and in 4T1 breast tumor-bearing mice proving the potential for enhanced tumor treatment based on the combination of PDT and PTT. Furthermore, these systems can be loaded with functional molecules for application in drug delivery or catalysis.
In 2019, we reported a colloidally stable thermoresponsive carrier composed of a core of AuNSs coated with a nanoMOF ZIF-8 shell, having LSPR centered around 800 nm.14 Transmission electron microscopy (TEM) showed a well-defined core–shell nanocomposite with a single AuNS core and a MOF shell of an average size of 218 nm. X-ray diffraction and thermogravimetric analysis further confirmed the growth of the MOF around the AuNSs. To address the problem of the aqueous instability of the ZIF-8, the authors functionalized the NMOFs with PMA providing the system with stability up to 7 days in aqueous and biological media (Fig. 10 and 12a). To perform in vitro studies and analyse the potential of the systems as drug carriers, the bioactive cargo Hoechst (HOE), a blue-fluorescent dye typically used for DNA staining, was employed. The high loading capabilities were verified by N2-sorption isotherms. Interestingly, the final nanocomposites showed an excellent response to NIR laser illumination inside living cells and high chemical stability with no cargo leakage after the PMA coating. Furthermore, after comparison between the response of the system with and without irradiation under the NIR laser, the authors were able to prove the efficient triggered release of the loading (Fig. 14). These results show the potential of these nanocomposites for their application as intracellular vehicles for drug vectorization.
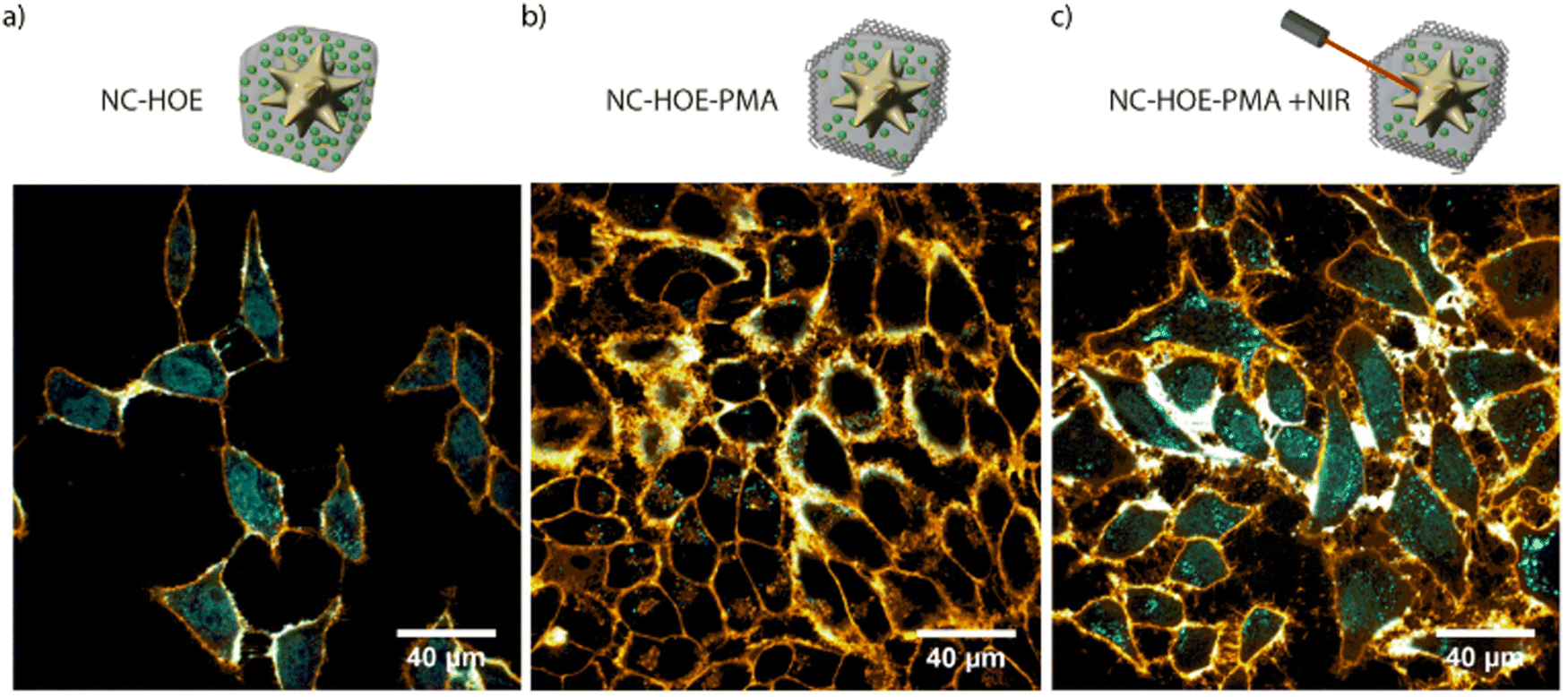 | ||
| Fig. 14 Confocal microscopy image of HeLa cells incubated with NC-HOE (a), NC-HOE-PMA (b), and NC-HOE-PMA after NIR treatment (c). Blue and orange colors represent HOE and cell membrane staining (CellMaskTM Deep Red), respectively. Scale bars correspond to 40 μm. Adapted with permission from ref. 14. | ||
However, some of these transformations can be accomplished by performing abiotic catalysis within living cells using homogeneous transition metal catalysts. These catalysts promote controlled biorthogonal reactions. Biorthogonal reactions can be defined as reactions that take place in aqueous or biologically complex media, usually occurring with high chemo-selectivity and biocompatibility, with minimized off-target reactions.121,122
Transition metal-catalysed reactions have been widely performed inside living cells over the past decade. Before their application, several studies were carried out to solve some of the main problems associated with these materials, such as the poor biocompatibility, the low cellular uptake, or the deactivation of the catalyst in biological media. The main solution to these issues is the use of heterogeneous catalysts such as NPs, i.e., Pd NPs.123 However, some transition-metal NPs’ functionalization and stabilization are difficult and when placed in biological media the NPs tend to aggregate. Also, the high concentration of biomolecules containing thiols or amines can strongly impair the catalytic activity of the particles.15 Beside, if NPs are stabilized with organic coatings the catalytic activity is highly reduced and the particles lose their functionality.123 Another main problem concerning these materials is their synthesis, which is generally performed in organic solvents, and their active surface usually gets deactivated after time. Therefore, there is a necessity to design new systems that can overcome these limitations.
Due to the metal centres, high porosity, and very high surface areas, the NMOFs constitute great candidates for their application as catalyst carriers. They can act as catalysts themselves by using the free metal sites in their surfaces or in synergy with catalytic NPs encapsulated inside the structure as metal and semiconductor oxide NPs.124
However, NMOFs have been mainly studied for biocatalysis due to their ability to load biomolecules, mainly enzymes, where the MOF structure provides protection avoiding the biological degradation of the biomolecule in harsh conditions, such as high temperatures or extreme pH. The combination of activity of loaded catalytic biomolecules with the free metal sites of the NMOFs allows for the improvement of biological activities.125 Furthermore, the easy external modification, stimuli-responsiveness, and some enzyme-mimicking features of MOFs allow their application with enzymes for smart catalysis, as well as their use as versatile nanoplatforms for enzyme delivery and catalytic nanomedicine. Li et al. reported the efficient immobilization of the enzyme lactate dehydrogenase (LDH) in different selected NU-100× MOF structures for their use as a cell-free biosynthetic catalytic system.60 With this work they demonstrated the potential of MOFs as enzyme carriers, as the prepared systems were water stable and showed high enzyme loadings and coenzyme accessibility, making these materials suitable for industrial applications.
Nevertheless, the development of non-natural catalytic reactors that can perform reactions inside living organisms could constitute a new important achievement in the biomedical field. Among different materials, catalytical metal NPs are great candidates, i.e., Au, Pd or Pt NPs. However, as mentioned above, NPs with high surface energy have higher instability and tend to aggregate in biological media losing their catalytic activity.15 In this scenario, NMOFs would act as protective structures to immobilize NPs preventing their aggregation for their use in catalytic processes.124 This is combined with the high external surface areas and porosity of the structure of the NMOF that allows the diffusion of substrates and products minimizing the mass-transfer limit.104 Therefore, the formation of new nanocomposites built from a metal NP encapsulated inside a NMOF structure would produce platforms with improved features for catalysis.124,126
Recently, several NP@MOF composites have been studied for their application in catalysis. One possibility includes the impregnation of NPs through the pores of the structure of the NMOFs. However, due to the small size of the pores, the diffusion of the particles can be difficult. Another effective procedure is called “bottle around the ship” where the MOF is synthesized around the NP, which acts as a nucleation site for the building of the structure, forming the NP@MOF composite. This approach would limit the adsorption of undesired species of the biological milieu to the NP surface, allowing the selective adsorption of substrates.91,124 However, this method has been achieved only with a few MOFs such as ZIF-8 or some Zr MOFs.104
Recently, we reported an innovative water-compatible system based on the core–shell Pd@ZIF-8 nanocomposites that can act as long-lasting bio-orthogonal nanoreactors (NR).15 The nanocomposite is formed by a Pd-nanocube (PdNC) that acts as a catalyst, surrounded by a ZIF-8 shell that protects the PdNC to avoid aggregation or deactivation, and serves as a platform for the diffusion of the reactants. Simultaneously, the ZIF shell acts as a filter for the adsorption of larger biomolecules that would inactivate the Pd. TEM showed a well-defined core–shell nanocomposite with a Pd-core of 24 nm and a shell of 250 nm. The nanocomposite was then coated with the amphiphilic polymer PMA to provide the NMOFs with high stability in aqueous media for up to 7 days. We studied different reacting probes that generate fluorescent products after the removal of the propargylic groups, a reaction that is catalyzed by Pd (Fig. 15). Interestingly, the results showed the efficacy of the NRs to perform catalytic reactions inside living cells, in both 2D monolayers and 3D tumor spheroids, and their reusability by the simple addition of new reactants due to their storage in cytosolic vesicles. These results show the potential of intracellular recurrent NRs and the possible applicability as “catalytic cellular or tissular nanoimplants.”
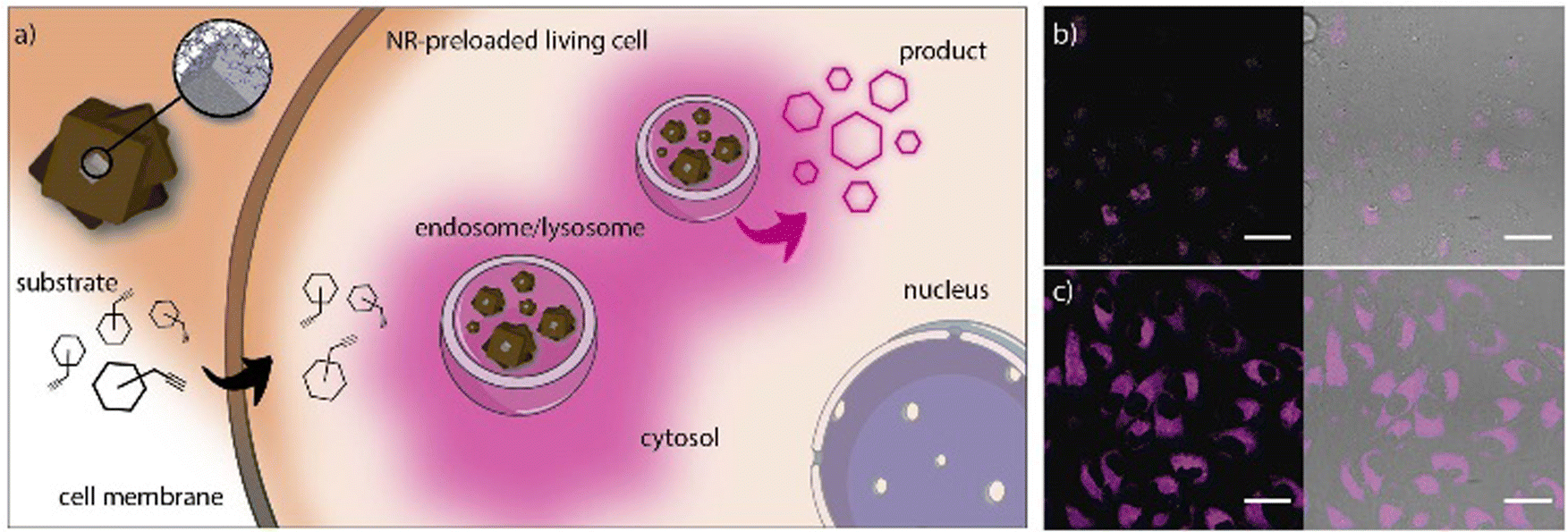 | ||
| Fig. 15 (a) Pd-NCs coated by porous ZIF-8/PMA shell NRs. The NRs tend to accumulate in cytosolic compartments (endosomes/lysosomes) and function as heterogeneous palladium-based NRs capable of processing substrates. (b and c) Confocal microscopy images (60×) after incubation of NR-preloaded cells with different concentrations of substrate 5 (top: 2.5 mM; bottom: 10 mM; the fluorescence corresponds to the intracellular production of cresyl violet). Scale bars correspond to 40 μm. Adapted with permission from ref. 15. | ||
This strategy still has some potential limitations due to the intrinsic reactivity of the reactants or the toxicity problems associated with some metal-based reagents. To avoid this, thermal-driven reactions can serve as alternative processes. Thermal-driven reactions can be triggered by temperature stimuli. However, there are some restrictions when applying this in living organisms, as cells cannot be heated over. We reported a NR based on ZIF-8 NMOFs with a core of AuNSs that could overcome these challenges, AuNS@ZIF-8 (Fig. 16).16 TEM showed a well-defined core–shell nanocomposite with a AuNS of 70 nm and a shell of 215 nm. The plasmonic AuNSs would act as NIR-responsive thermal transducers, achieving localized heating around the AuNSs and addressing the problem of cell overheating. Simultaneously, the ZIF-8 shell would act as a protective barrier avoiding the adsorption of undesired biomolecules such as proteins on the surface of the AuNSs. In this work, they presented a thermal-promoted intramolecular chemical reaction (thermocyclization) performed inside living cells. The ZIF-8 shell of the nanocomposite allows the diffusion of the reactants through the pores of the crystal, close to the AuNS heating source under NIR irradiation. Furthermore, the thermal conductivity of ZIF-8 is rather low, so it avoids the temperature increase in the surrounding media of the NMOFs. The selection of the substrate/nanocomposite pairs was based on the purposes of demonstrating that it is possible to carry out abiotic nucleophilic substitutions in biological media and inside living cells; that the reaction needs heating to be performed; and that a cyclic fluorogenic product can be formed using these NRs. The nanocomposites were tested in mammalian cells with different substrates with two different set-ups a collimated laser or a laser pointer, and in both cases a good level of transformation in living cells was shown. This improvement in the performance of the NSs when the ZIF-8 shell is present is because it facilitates the access of the substrates to the AuNS core while providing protection and control in the flow of reactants and products. This example opens a new avenue of light-promoted intracellular reactions that have many potential applications.
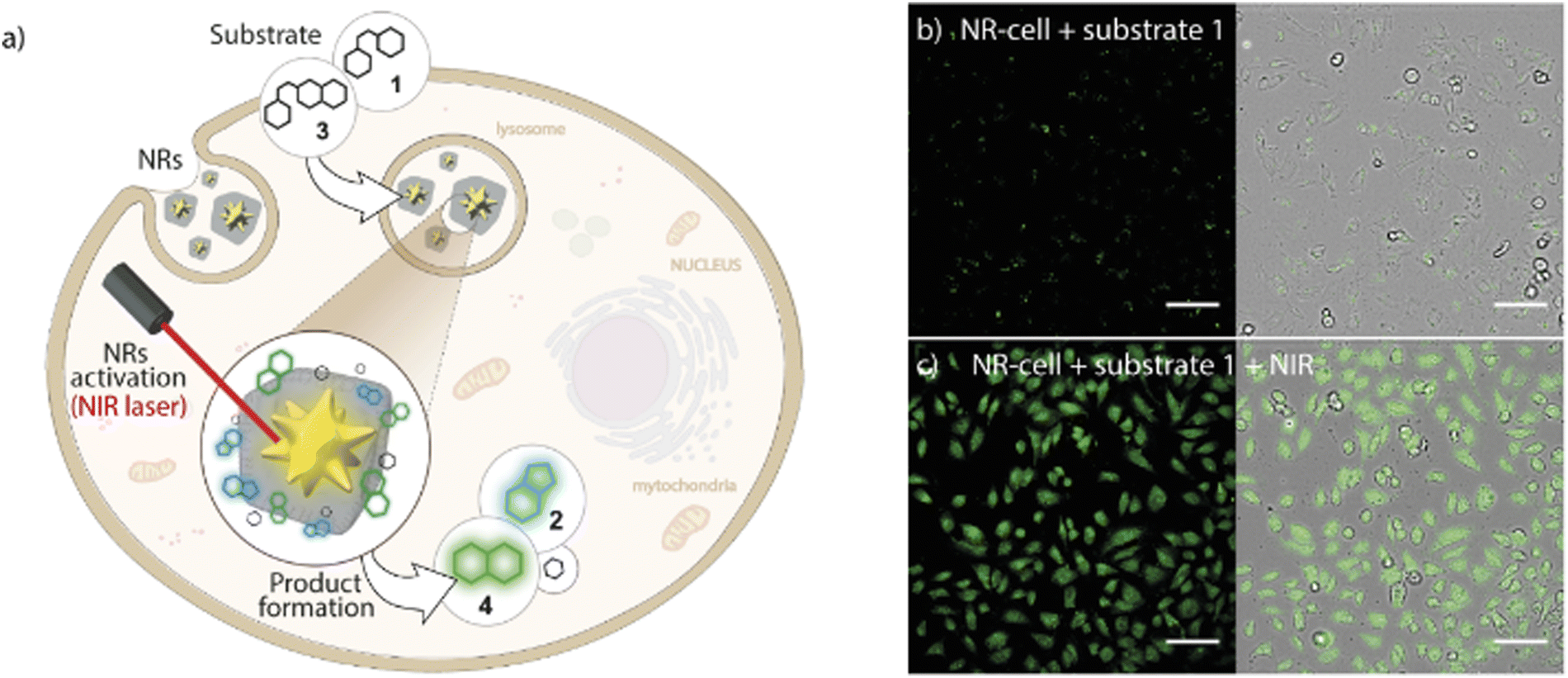 | ||
| Fig. 16 Core–shell MOF-based plasmonic NR for intracellular photothermal reactions. (a) NRs featuring a AuNS coated with a porous polymer-modified ZIF-8 cloak function as heterogeneous photo-gated NRs capable of transforming thermolabile substrates inside living cells (thermocyclization). (b) Confocal microscopy image of NR-preloaded cells incubated with substrate 1 without NIR treatment and (c) after NIR irradiation using the collimated beam system ((10 W cm−2, 1 min). Scale bars correspond to 100 μm. Adapted with permission from ref. 16. | ||
MR imaging is one of the main techniques used for diagnosis, as it can provide morphological and functional information besides being a non-invasive technique.127 However, it requires the use of contrast agents due to the low contrast-to-noise ratio, which implies a considerable reduction in the resolution. Moreover, the conventional contrast agents (i.e., Gd3+-based complexes) used in the clinic suffer from a lack of sensitivity, toxicity, and undesired side effects.130 Some Gd-based NMOFs have been developed to overcome these limitations. In 2006, Lin et al. were the first to report Gd3+-containing NMOFs with improved relaxation times compared to clinical molecules.131 However, the toxicity related to free Gd3+ ions that leach from the structure makes these systems not suitable for clinical use. To solve this, non-Gd3+-based NMOFs have been developed.130
Lin et al. reported a system based on Fe3O4 NPs assembled into responsive ZIF-8 NMOFs, generating T1–T2 changing contrast agents.132 They synthesized 6 nm Fe3O4 NPs that exhibit a T1 contrast enhancement, whereas while embedded in ZIF-8 (forming an assembly), they show a T2 contrast effect (Fig. 17). When the Fe3O4@ZIF-8 nanosystems reach the tumor environment (acidic conditions), the pH and glutathione (GSH) trigger the Fe3O4 NP disassembly recovering the individual NP behavior as T1 contrast agents. In vivo T1 weighted images (mouse liver) showed darkening contrast improvement, while they showed brightening contrast in the tumor sites. This proved the efficiency of these synergic systems to easily distinguish between normal and tumor tissues.
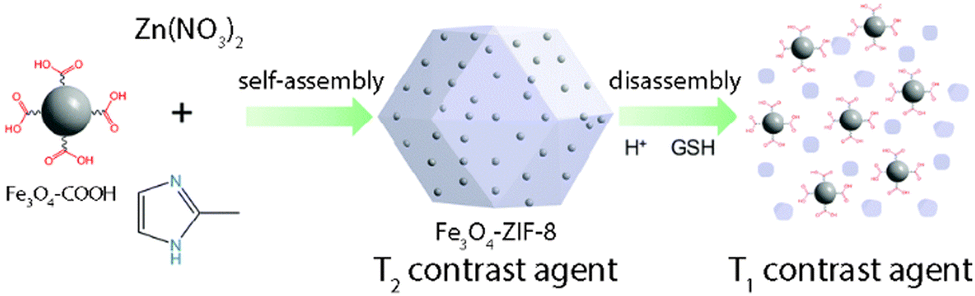 | ||
| Fig. 17 T1–T2 responsive Fe3O4@ZIF-8 nanosystem assembly and disassembly triggered by pH and glutathione (GSH). Adapted from ref. 132. | ||
CT imaging is a non-invasive 3D technique used in diagnosis based on X-ray attenuation by contrast agents. It provides high-resolution images of 3D biological tissues depending on their X-ray absorption abilities. This is applied to visualize mainly bone structures as this technique requires materials with high atomic numbers.127 Lately, Zhang et al. reported the design of UiO-PDT NMOFs as contrast agents for CT imaging.133 They first synthesized UiO-66 NMOFs and then they exchanged the organic ligand with diiodide-substituted monocarboxyl-functionalized BODIPYs (I2-BDP) to obtain UiO-PDT. The resulting NPs, around 70 nm in size, were tested as CT contrast agents using a Micro CT at 80KVp, presenting an excellent X-ray attenuation performance. Moreover, both in vitro and in vivo biocompatibility of these NMOFs was studied, showing no cytotoxicity at high concentrations and a clear accumulation in the tumor site, proving the potential of these systems as CT contrast agents for solid tumors.
PET constitutes another important imaging technique, based on the emission of positrons by radionuclides accumulating in the target organs. When a positron and an electron encounter, annihilation occurs, generating gamma rays that are captured by detector panels, reconstructing the origin source, and forming 3D images.127,130 Chen et al. reported the design of an intrinsic radioactive UiO-66 NMOF with the incorporation of positron-emitting isotope zirconium-89 (89Zr) for in vivo positron emission tomography (PET).128 In this work, they incorporated the PET isotope zirconium-89 (89Zr, t1/2 = 78.4 h) during the synthesis of UiO-66, achieving NMOFs of 90 nm. Then, the particles were functionalized with PEG and a peptide to increase their stability and provide them with tumor-targeting capabilities, respectively. Moreover, the NMOFs were loaded with DOX, and used as a model anticancer drug and a fluorophore to track these nanosystems. In vivo results showed the potential of these systems as image-guidable, tumor-selective cargo delivery nanoplatforms. However, in spite of these promising results, NMOFs for biomedical imaging are still in their infancy and further studies need to be performed to address several aspects of NMOFs in this field.
Recently, a few types of NPs, such as liposomes or microemulsions, have been employed for this purpose due to their versatility and a wide variety of possible functionalizations.134,136 Although few studies have been made, NMOFs represent a promising alternative for detoxifying materials due to some of its features as their high porosity, which make them suitable for selective adsorption and elimination of toxic agents with low in vivo toxicity.134,137
Lately, Rojas et al. reported the pioneering design of a new detoxifying agent based on a single biocompatible MOF for its application for decontamination of drugs, generally related to voluntary poisoning.134 The studied MOF was the MIL-127 structure, based on octahedral iron (III) trimers and 3,3′,5,5′-azobenzenetetracarboxylate anions (TazBz4−). The results of this work showed good biocompatibility, high porosity (1400 m2 g−1) with high adsorption capacities and high chemical stability at different pH values. However, to prevent the intestinal crossing and potential toxicity, the systems were prepared in the micrometre scale (∼20 μm). As a model of overdose pain medication, they used the salicylate derivative aspirin (acetylsalicylic acid, ASA), which is classified among the nine pediatric poisons that can cause death in infants at small doses, and that has no specific antidote. They studied the chemical stability and the adsorption capabilities of the systems both in vitro and in vivo. In vitro results showed high stability and high adsorption capabilities of the MOF, which could reach a maximum ASA loading of 0.14 g g−1 (gram of ASA per gram of MIL-127). Furthermore, after the encapsulation there was no release of the drug, demonstrating the great affinity of the aspirin for the MOF. In vivo results revealed the decrease of the silicates intestinal absorption by over 40-fold. Therefore, this report showed the first experiments concerning the in vivo toxicity and biodistribution of a MOF via oral administration, specifically upon salicylate overdose, with a high reduction of salicylate gastrointestinal absorption and toxicity with outstanding gastrointestinal stability (Fig. 18).103 However, the development of a MOF in the nanoscale was not reported until next year, in 2019, in the work by Rojas et al.,102 where they presented a Ti-based NMOF (MIL-125-NH2) as a drug overload detoxifying agent. The selected NMOF was the tetragonal titanium (IV) aminoterephthalate MIL-125-NH2 (Ti8O8(OH)4(O2C–C6H3NH2–CO2)6). The NMOF presented properties that made them suitable as oral detoxifying agents: the system is based on titanium, which is regarded as one of the most biocompatible metals; it showed very low absorption when orally administered; and it presented high porosity that made the structure an ideal candidate for the encapsulation of drugs. Furthermore, it could be synthesized at the nanoscale, which also opens the window for its further application as a drug delivery agent. The studied drug was again the widely used anti-inflammatory and analgesic ASA. For the experiments, they employed the same quantity (mass) of adsorbent and the same ASA initial concentration under simulated GI conditions. Despite the efficiency of ASA removal of the NMOF, the results are relatively low with respect to the commercial treatment (2–13% using stable MOFs vs 97% using Norit@activated carbon). However, MIL-125-NH2 presented the important advantage of low release after drug adsorption, which is one of the main problems associated with conventional treatments. To confirm the results, MIL-125-NH2 was compared to other common ones (MIL-53(Fe), MIL-53(Fe)-(OH)2, MIL-100-(Fe), UiO-66(Zr), UiO-66(Zr)–NH2, and ZIF-8(Zn)). In vitro results showed the high ASA adsorption capacity of the NMOF (2.59 and 1.64 mol mol−1, ASA/adsorbent ratio = 1:0.5 and 1:2, respectively) after 24 h. After in vitro and in vivo studies, MIL-125-NH2 was demonstrated to be the only MOF capable of resisting intestinal conditions, being an efficient detoxifying agent with the ability to twofold reduce the salicylate concentration peak in blood, while keeping intact its structure and proving good biocompatibility.
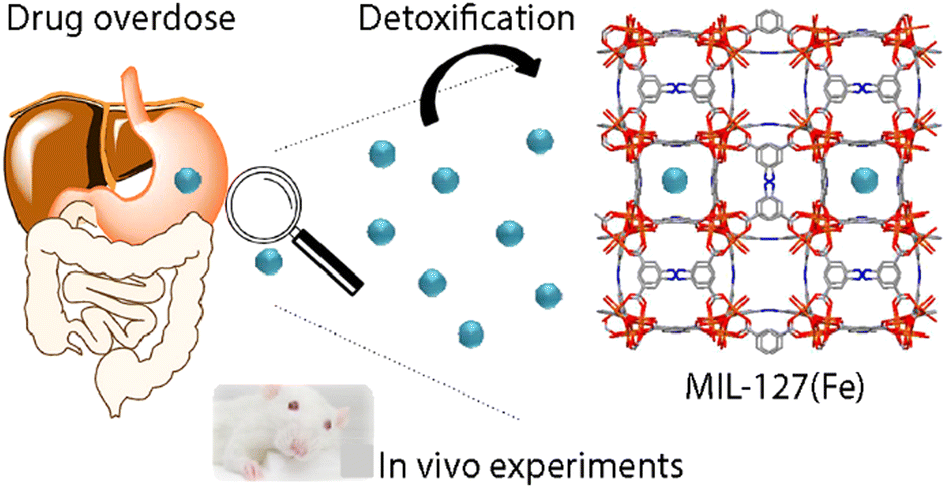 | ||
| Fig. 18 Adapted with permission from ref. 134. | ||
Conclusions
The synthesis of MOFs at the nanoscale has raised considerable interest for their versatility and added value for bioapplications. These systems offer a great chemical variety of MOFs and their unique properties as porous materials. Many of them are of great interest due to their good biocompatibility and their easy functionalization. However, there is still some work to be undertaken to optimize the synthetic methods to yield NMOFs with homogeneous size and morphology. Moreover, the produced NMOF should retain their porosity and surface-chemical reactivity while maintaining their chemical stability in biological media.Another interesting approach is the production of hybrid composites comprising inorganic NPs and MOFs, which may serve as advanced nanomedicines in the near future. These composites combine the properties of their individual components, i.e., NPs and NMOF shells. Here, it is important to highlight the need to create nanocomposites with extremely well-defined structures in order to finely control their physicochemical properties.
These composites can act as remotely activated drug carriers releasing the drug by an external trigger, or act as catalysts to perform non-natural chemical reactions in living cells, producing relevant molecules in situ. We envision that in the following years this field will offer original and robust alternatives to solve the current limitations of therapies based on nanomaterials.
Author contributions
M. C.-M., M. C., E. P., P. d. P. and B. P. contributed to discussion and manuscript writing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support of the MCIN/AEI (PID2019-111218RB-I00, PID2020-119206RB-I00, PID2020-119479RA-I00, RYC-2017-23457, RYC-2019-028238-I), the Xunta de Galicia (ED431F 2017/02, 2021-CP090, EDED431C 2022/018, Centro Singular De Investigación de Galicia Accreditation 2019–2022, and ED431G 2019/03), the European Union (European Regional Development Fund – ERDF; H2020-MSCA-ITN grant agreement no. 860942; and INTERREG V-A Spain–Portugal, project 0624_2IQBIONEURO_6_E), and the European Research Council (starting grant no. 950421). M.C.-M. thanks the AEI (FPU19/03155).References
- L. M. Liz-Marzán, A. E. Nel, C. J. Brinker, W. C. W. Chan, C. Chen, X. Chen, D. Ho, T. Hu, K. Kataoka, N. A. Kotov, W. J. Parak and M. M. Stevens, ACS Nano, 2022, 16, 13257–13259 CrossRef.
- B. Pelaz, C. Alexiou, R. A. A. Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopecek, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. L. Lim, L. M. L. Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X. E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef CAS.
- G. Nabil, K. Bhise, S. Sau, M. Atef, H. A. El-Banna and A. K. Iyer, Drug Discovery Today, 2019, 24, 462–491 CrossRef CAS PubMed.
- P. H. Cortés and S. R. Macías, Metal–organic frameworks in biomedical and environmental field, Springer, 2021 Search PubMed.
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed.
- M. Mitchell, M. Billingsley, R. Haley, M. Wechsler, N. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- E. A. Flügel, A. Ranft, F. Haase and B. V. Lotsch, J. Mater. Chem., 2012, 22, 10119–10133 RSC.
- J. W. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2021, 16, 2000005 CrossRef CAS PubMed.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- Y. Kinoshita, I. Matsubara, T. Higuciu and Y. Saito, Bull. Chem. Soc. Jpn., 1959, 32, 1221–1226 CrossRef CAS.
- S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1995, 34, 2127–2129 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. Okeeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS.
- M. A. Mohamud and A. B. Yurtcan, Int. J. Hydrogen Energy, 2021, 46, 33782–33800 CrossRef CAS.
- C. Carrillo-Carrion, R. Martínez, M. F. N. Poupard, B. Pelaz, E. Polo, A. Arenas-Vivo, A. Olgiati, P. Taboada, M. G. Soliman, U. Catalán, S. Fernández-Castillejo, R. Solà, W. J. Parak, P. Horcajada, R. Alvarez-Puebla and P. D. Pino, Angew. Chem., Int. Ed., 2019, 58, 7078–7082 CrossRef CAS.
- R. Martínez, C. Carrillo-Carrión, P. Destito, A. Alvarez, M. Tomás-Gamasa, B. Pelaz, F. Lopez, J. L. Mascareñas and P. del Pino, Cell Rep. Phys. Sci., 2020, 1, 100076 CrossRef.
- C. Carrillo-Carrión, R. Martínez, E. Polo, M. Tomás-Gamasa, P. Destito, M. Ceballos, B. Pelaz, F. López, J. L. Mascareñas and P. D. Pino, ACS Nano, 2021, 15, 16924–16933 CrossRef.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. Guy Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- V. Guillerm and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 16517–16538 CrossRef CAS PubMed.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Ferey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- S. Y. Zhang, D. Li, D. Guo, H. Zhang, W. Shi, P. Cheng, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2015, 137, 15406–15409 CrossRef CAS PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., 2015, 127, 11314–11319 CrossRef.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- Y. R. Lee, M. S. Jang, H. Y. Cho, H. J. Kwon, S. Kim and W. S. Ahn, Chem. Eng. J., 2015, 271, 276–280 CrossRef CAS.
- S. Wang, L. Furchtgott, K. C. Huang and J. W. Shaevitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 109, E595–604 Search PubMed.
- J. J. Richardson, K. Liang, F. Lisi, M. Björnmalm, M. Faria, J. Guo and P. Falcaro, Eur. J. Inorg. Chem., 2016, 4499–4504 CrossRef CAS.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- J. Xiong, L. Wang, X. Qin and J. Yu, Mater. Lett., 2021, 302, 130427 CrossRef CAS.
- A. Huang, L. Wan and J. Caro, Mater. Res. Bull., 2018, 98, 308–313 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. Eur. J., 2011, 17, 6643–6651 CrossRef CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- M. Ceballos, M. Cedrún-Morales, M. Rodríguez-Pérez, S. Funes-Hernando, J. M. Vila-Fungueiriño, G. Zampini, M. F. Navarro Poupard, E. Polo, P. del Pino and B. Pelaz, Nanoscale, 2022, 14, 6789–6801 RSC.
- J. Hühn, C. Carrillo-Carrion, M. G. Soliman, C. Pfeiffer, D. Valdeperez, A. Masood, I. Chakraborty, L. Zhu, M. Gallego, Y. Zhao, M. Carril, N. Feliu, A. Escudero, A. M. Alkilany, B. Pelaz, P. D. Pino and W. J. Parak, Chem. Mater., 2017, 29, 399–461 CrossRef.
- X. G. Wang, Q. Cheng, Y. Yu and X. Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7836–7840 CrossRef CAS PubMed.
- X. Zhu, H. Huang, H. Zhang, Y. Zhang, P. Shi, K. Qu, S.-B. Cheng, A.-L. Wang and Q. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 32176–32182 CrossRef CAS PubMed.
- S. Goswami, H. Noh, L. R. Redfern, K. I. Otake, C. W. Kung, Y. Cui, K. W. Chapman, O. K. Farha and J. T. Hupp, Chem. Mater., 2019, 31, 1485–1490 CrossRef CAS.
- Y. H. Wang, C. H. Chuang, T. A. Chiu, C. W. Kung and W. Y. Yu, J. Phys. Chem. C, 2020, 124, 12521–12530 CrossRef CAS.
- F. Chen, K. Shen, J. Chen, X. Yang, J. Cui and Y. Li, ACS Cent. Sci., 2019, 5, 176–185 CrossRef CAS PubMed.
- X. Li, B. Zhang, R. Van Zeeland, L. Tang, Y. Pei, Z. Qi, T. W. Goh, L. M. Stanley and W. Huang, Catal. Lett., 2018, 148, 940–945 CrossRef CAS.
- M.-M. Pan, Y. Ouyang, Y.-L. Song, L.-Q. Si, M. Jiang, X. Yu, L. Xu and I. Willner, Small, 2022, 18, 2200548 CrossRef CAS PubMed.
- B. Volosskiy, K. Niwa, Y. Chen, Z. Zhao, N. O. Weiss, X. Zhong, M. Ding, C. Lee, Y. Huang and X. Duan, ACS Nano, 2015, 9, 3044–3049 CrossRef CAS PubMed.
- Y. Li, W. S. Lo, F. Zhang, X. Si, L. Y. Chou, X. Y. Liu, B. P. Williams, Y. H. Li, S. H. Jung, Y. S. Hsu, F. S. Liao, F. K. Shieh, M. N. Ismail, W. Huang and C. K. Tsung, J. Am. Chem. Soc., 2021, 143, 5182–5190 CrossRef CAS PubMed.
- P. Del Pino, F. Yang, B. Pelaz, Q. Zhang, K. Kantner, R. Hartmann, N. M. D. Baroja, M. Gallego, M. Möller, B. B. Manshian, S. J. Soenen, R. Riedel, N. Hampp and W. J. Parak, Angew. Chem., Int. Ed., 2016, 55, 5483–5487 CrossRef CAS PubMed.
- M. G. Soliman, B. Pelaz, W. J. Parak and P. Del Pino, Chem. Mater., 2015, 27, 990–997 CrossRef CAS.
- J. W. M. Osterrieth, D. Wright, H. Noh, C. W. Kung, D. Vulpe, A. Li, J. E. Park, R. P. Van Duyne, P. Z. Moghadam, J. J. Baumberg, O. K. Farha and D. Fairen-Jimenez, J. Am. Chem. Soc., 2019, 141, 3893–3900 CrossRef CAS.
- J. Li, Z. Liu, D. Tian, B. Li, L. Shao and Z. Lou, Nanoscale, 2022, 14, 5561–5568 RSC.
- X. Yang, Y. Liu, S. H. Lam, J. Wang, S. Wen, C. Yam, L. Shao and J. Wang, Nano Lett., 2021, 21, 8205–8212 CrossRef CAS PubMed.
- G. Zheng, Z. Chen, K. Sentosun, I. Pérez-Juste, S. Bals, L. M. Liz-Marzán, I. Pastoriza-Santos, J. Pérez-Juste and M. Hong, Nanoscale, 2017, 9, 16645–16651 RSC.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- M. D. J. Velásquez-Hernández, M. Linares-Moreau, E. Astria, F. Carraro, M. Z. Alyami, N. M. Khashab, C. J. Sumby, C. J. Doonan and P. Falcaro, Coord. Chem. Rev., 2021, 429, 213651 CrossRef.
- B. Maranescu and A. Visa, Int. J. Mol. Sci., 2022, 23, 4458 CrossRef CAS PubMed.
- L. Wang, M. Zheng and Z. Xie, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- N. Ye, X. Kou, J. Shen, S. Huang, G. Chen and G. Ouyang, ChemBioChem, 2020, 21, 2585–2590 CrossRef CAS PubMed.
- X. Chen, Y. Zhuang, N. Rampal, R. Hewitt, G. Divitini, C. A. O'Keefe, X. Liu, D. J. Whitaker, J. W. Wills, R. Jugdaohsingh, J. J. Powell, H. Yu, C. P. Grey, O. A. Scherman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2021, 143, 13557–13572 CrossRef CAS PubMed.
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson and D. A. Gómez-Gualdrón, Chem, 2018, 4, 1022–1034 CAS.
- C. Orellana-Tavra, M. Köppen, A. Li, N. Stock and D. Fairen-Jimenez, ACS Appl. Mater. Interfaces, 2020, 12, 5633–5641 CrossRef CAS.
- T. Luo, G. T. Nash, Z. Xu, X. Jiang, J. Liu and W. Lin, J. Am. Chem. Soc., 2021, 143, 13519–13524 CrossRef CAS PubMed.
- J. V. Morabito, L.-Y. Chou, Z. Li, C. M. Manna, C. A. Petroff, R. J. Kyada, J. M. Palomba, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2014, 136, 12540–12543 CrossRef CAS PubMed.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- F.-S. Liao, W.-S. Lo, Y.-S. Hsu, C.-C. Wu, S.-C. Wang, F.-K. Shieh, J. V. Morabito, L.-Y. Chou, K. C.-W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 2017, 139, 6530–6533 CrossRef CAS PubMed.
- J. Cases Díaz, B. Lozano-Torres and M. Giménez-Marqués, Chem. Mater., 2022, 34, 7817–7827 CrossRef PubMed.
- B. Li, Y. Zhang, D. Ma, T. Ma, Z. Shi and S. Ma, J. Am. Chem. Soc., 2014, 136, 1202–1205 CrossRef CAS PubMed.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS PubMed.
- G. Kaur, S. Øien-Ødegaard, A. Lazzarini, S. M. Chavan, S. Bordiga, K. P. Lillerud and U. Olsbye, Cryst. Growth Des., 2019, 19, 4246–4251 CrossRef CAS.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- R. S. Forgan, Dalton Trans., 2019, 48, 9037–9042 RSC.
- C. V. McGuire and R. S. Forgan, Chem. Commun., 2015, 51, 5199–5217 RSC.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 1706759 CrossRef PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed.
- H. Sohrabi, S. Javanbakht, F. Oroojalian, F. Rouhani, A. Shaabani, M. R. Majidi, M. Hashemzaei, Y. Hanifehpour, A. Mokhtarzadeh and A. Morsali, Chemosphere, 2021, 281, 130717 CrossRef CAS PubMed.
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215–2219 CrossRef CAS PubMed.
- X. Chen, Y. Zhuang, N. Rampal, R. Hewitt, G. Divitini, C. A. O’Keefe, X. Liu, D. J. Whitaker, J. W. Wills and R. Jugdaohsingh, J. Am. Chem. Soc., 2021, 143, 13557–13572 CrossRef CAS PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- A. Zimpel, T. Preiß, R. Röder, H. Engelke, M. Ingrisch, M. Peller, J. O. Rädler, E. Wagner, T. Bein and U. Lächelt, Chem. Mater., 2016, 28, 3318–3326 CrossRef CAS.
- J. Zhuang, H. Gong, J. Zhou, Q. Zhang, W. Gao, R. H. Fang and L. Zhang, Sci. Adv., 2020, 6, eaaz6108 CrossRef CAS PubMed.
- C. Qiao, X. Wang, G. Liu, Z. Yang, Q. Jia, L. Wang, R. Zhang, Y. Xia, Z. Wang and Y. Yang, Adv. Funct. Mater., 2022, 32, 2107791 CrossRef CAS.
- C.-M. J. Hu, R. H. Fang, B. T. Luk, K. N. H. Chen, C. Carpenter, W. Gao, K. Zhang and L. Zhang, Nanoscale, 2013, 5, 2664–2668 RSC.
- L. Rao, L.-L. Bu, Q.-F. Meng, B. Cai, W.-W. Deng, A. Li, K. Li, S.-S. Guo, W.-F. Zhang, W. Liu, Z.-J. Sun and X.-Z. Zhao, Adv. Funct. Mater., 2017, 27, 1604774 CrossRef.
- J. Su, H. Sun, Q. Meng, Q. Yin, P. Zhang, Z. Zhang, H. Yu and Y. Li, Adv. Funct. Mater., 2016, 26, 7495–7506 CrossRef CAS.
- A. Parodi, N. Quattrocchi, A. L. van de Ven, C. Chiappini, M. Evangelopoulos, J. O. Martinez, B. S. Brown, S. Z. Khaled, I. K. Yazdi, M. V. Enzo, L. Isenhart, M. Ferrari and E. Tasciotti, Nat. Nanotechnol., 2013, 8, 61–68 CrossRef CAS PubMed.
- J.-Y. Zhu, D.-W. Zheng, M.-K. Zhang, W.-Y. Yu, W.-X. Qiu, J.-J. Hu, J. Feng and X.-Z. Zhang, Nano Lett., 2016, 16, 5895–5901 CrossRef CAS PubMed.
- E. Soprano, A. Alvarez, B. Pelaz, P. Del Pino and E. Polo, Adv. Biosyst., 2020, 4, 1900260 CrossRef PubMed.
- B. Illes, P. Hirschle, S. Barnert, V. Cauda, S. Wuttke and H. Engelke, Chem. Mater., 2017, 29, 8042–8046 CrossRef CAS.
- S. Wuttke, S. Braig, T. Preiß, A. Zimpel, J. Sicklinger, C. Bellomo, J. O. Rädler, A. M. Vollmar and T. Bein, Chem. Commun., 2015, 51, 15752–15755 RSC.
- M. Z. Alyami, S. K. Alsaiari, Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715–1720 CrossRef CAS PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang and P. Zhang, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS.
- J. Schubert and M. Chanana, Curr. Med. Chem., 2019, 25, 4556 Search PubMed.
- X. Ma, M. Lepoitevin and C. Serre, Mater. Chem. Front., 2021, 5, 5573–5594 RSC.
- P. Del Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
- H. Li, M. Cosio, K. Wang, W. Burtner and H.-C. Zhou, Elaboration and Applications of Metal–Organic Frameworks, 2018, pp. 1–35 DOI:10.1142/9789813226739_0001.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- T. He, X.-J. Kong and J.-R. Li, Acc. Chem. Res., 2021, 54, 3083–3094 CrossRef CAS PubMed.
- R. Ettlinger, U. Lächelt, R. Gref, P. Horcajada, T. Lammers, C. Serre, P. Couvreur, R. E. Morris and S. Wuttke, Chem. Soc. Rev., 2022, 51, 464–484 RSC.
- M. S. de Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- C. Orellana-Tavra, S. Haddad, R. J. Marshall, I. Abánades Lázaro, G. Boix, I. Imaz, D. Maspoch, R. S. Forgan and D. Fairen-Jimenez, ACS Appl. Mater. Interfaces, 2017, 9, 35516–35525 CrossRef CAS PubMed.
- E. Linnane, S. Haddad, F. Melle, Z. Mei and D. Fairen-Jimenez, Chem. Soc. Rev., 2022, 51, 6065–6086 RSC.
- M. Mahmoudi, J. Meng, X. Xue, X. J. Liang, M. Rahmand, C. Pfeiffer, R. Hartmann, P. R. Gil, B. Pelaz, W. J. Parak, P. D. Pino, S. Carregal-Romero, A. G. Kanaras and S. T. Selvan, Biotechnol. Adv., 2014, 32, 679–692 CrossRef CAS PubMed.
- J. Yang and Y. W. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nano-Micro Lett., 2020, 12, 1–29 CrossRef CAS PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef PubMed.
- X. Ge, R. Wong, A. Anisa and S. Ma, Biomaterials, 2022, 281, 121322 CrossRef CAS PubMed.
- B. Nirosha Yalamandala, W. T. Shen, S. H. Min, W. H. Chiang, S. J. Chang and S. H. Hu, Adv. Nanobiomed. Res., 2021, 1, 2100014 CrossRef CAS.
- G. Chedid and A. Yassin, Nanomaterials, 2018, 8, 916 CrossRef PubMed.
- J. Zhou, G. Tian, L. Zeng, X. Song and X. W. Bian, Adv. Healthcare Mater., 2018, 7, 1800022 CrossRef PubMed.
- T. Wen, G. Quan, B. Niu, Y. Zhou, Y. Zhao, C. Lu, X. Pan and C. Wu, Small, 2021, 17, 2005064 CrossRef CAS PubMed.
- Y. Ma, X. Qu, C. Liu, Q. Xu and K. Tu, Front. Mol. Biosci., 2021, 8, 805228 CrossRef CAS PubMed.
- A. Cabrera-García, E. Checa-Chavarria, E. Rivero-Buceta, V. Moreno, E. Fernández and P. Botella, J. Colloid Interface Sci., 2019, 541, 163–174 CrossRef PubMed.
- J. Chen, J. Liu, Y. Hu, Z. Tian and Y. Zhu, Sci. Technol. Adv. Mater., 2019, 20, 1043–1054 CrossRef CAS PubMed.
- W. Cai, J. Wang, C. Chu, W. Chen, C. Wu and G. Liu, Adv. Sci., 2019, 6, 1801526 CrossRef PubMed.
- Z. Zhou, J. Zhao, Z. Di, B. Liu, Z. Li, X. Wu and L. Li, Nanoscale, 2021, 13, 131–137 RSC.
- J.-L. Kan, Y. Jiang, A. Xue, Y.-H. Yu, Q. Wang, Y. Zhou and Y.-B. Dong, Inorg. Chem., 2018, 57, 5420–5428 CrossRef CAS PubMed.
- B. Geng, D. Yang, D. Pan, L. Wang, F. Zheng, W. Shen, C. Zhang and X. Li, Carbon, 2018, 134, 153–162 CrossRef CAS.
- B. Tang, W. L. Li, Y. Chang, B. Yuan, Y. Wu, M. T. Zhang, J. F. Xu, J. Li and X. Zhang, Angew. Chem., 2019, 131, 15672–15677 CrossRef.
- Q. Zheng, X. Liu, Y. Zheng, K. W. Yeung, Z. Cui, Y. Liang, Z. Li, S. Zhu, X. Wang and S. Wu, Chem. Soc. Rev., 2021, 50, 5086–5125 RSC.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- A. Unciti-Broceta, Nat. Chem., 2015, 7, 538–539 CrossRef CAS PubMed.
- P. Destito, C. Vidal, F. López and J. L. Mascareñas, Chem. – Eur. J., 2021, 27, 4789–4816 CrossRef CAS PubMed.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol, 2018, 53, 106–114 CrossRef CAS PubMed.
- S. Yang, L. Peng, S. Bulut and W. L. Queen, Chem. – Eur. J., 2019, 25, 2161–2178 CrossRef CAS PubMed.
- J. Liang and K. Liang, Adv. Funct. Mater., 2020, 30, 2001648 CrossRef CAS.
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef PubMed.
- A. Pandey, N. Dhas, P. Deshmukh, C. Caro, P. Patil, M. Luisa García-Martín, B. Padya, A. Nikam, T. Mehta and S. Mutalik, Coord. Chem. Rev., 2020, 409, 213212 CrossRef CAS.
- D. Chen, D. Yang, C. A. Dougherty, W. Lu, H. Wu, X. He, T. Cai, M. E. Van Dort, B. D. Ross and H. Hong, ACS Nano, 2017, 11, 4315–4327 CrossRef CAS PubMed.
- S. Ambreen, M. Danish, N. D. Pandey and A. Pandey, Beilstein J. Nanotechnol., 2017, 8, 604–613 CrossRef CAS PubMed.
- K. Lu, T. Aung, N. Guo, R. Weichselbaum and W. Lin, Adv. Mater., 2018, 30, 1707634 CrossRef PubMed.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024–9025 CrossRef CAS PubMed.
- J. Lin, P. Xin, L. An, Y. Xu, C. Tao, Q. Tian, Z. Zhou, B. Hu and S. Yang, Chem. Commun., 2019, 55, 478–481 RSC.
- T. Zhang, L. Wang, C. Ma, W. Wang, J. Ding, S. Liu, X. Zhang and Z. Xie, J. Mater. Chem. B, 2017, 5, 2330–2336 RSC.
- S. Rojas, T. Baati, L. Njim, L. Manchego, F. Neffati, N. Abdeljelil, S. Saguem, C. Serre, M. F. Najjar, A. Zakhama and P. Horcajada, J. Am. Chem. Soc., 2018, 140, 9581–9586 CrossRef CAS PubMed.
- S. Rojas, N. Guillou and P. Horcajada, ACS Appl. Mater. Interfaces, 2019, 11, 22188–22193 CrossRef CAS PubMed.
- L. M. Graham, T. M. Nguyen and S. B. Lee, Nanomedicine, 2011, 6, 921–928 CrossRef CAS PubMed.
- J. H. Wang, M. Li and D. Li, Chem. – Eur. J., 2014, 20, 12004–12008 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |