
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic hyaluronic acid coating preserves the phenotypes of lymphatic endothelial cells†
Sanjoy
Saha
 a,
Fei
Fan
a,
Laura
Alderfer
a,
Francine
Graham
b,
Eva
Hall
a and
Donny
Hanjaya-Putra
*abc
a,
Fei
Fan
a,
Laura
Alderfer
a,
Francine
Graham
b,
Eva
Hall
a and
Donny
Hanjaya-Putra
*abc
aDepartment of Aerospace and Mechanical Engineering, Bioengineering Graduate Program, University of Notre Dame, IN 46556, USA. E-mail: dputra1@nd.edu
bDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, IN 46556, USA
cHarper Cancer Research Institute, University of Notre Dame, IN 46556, USA
First published on 19th September 2023
Abstract
Lymphatic endothelial cells (LECs) play a critical role in the formation and maintenance of the lymphatic vasculature, which is essential for the immune system, fluid balance, and tissue repair. However, LECs are often difficult to study in vivo and in vitro models that accurately mimic their behaviors and phenotypes are limited. In particular, LECs have been shown to lose their lymphatic markers over time while being cultured in vitro, which reflect their plasticity and heterogeneity in vivo. Since LECs uniquely express lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), we hypothesized that surface coating with hyaluronic acid (HA) can preserve LEC phenotypes and functionalities. Dopamine conjugated hyaluronic acid (HA–DP) was synthesized with 42% degree of substitution to enable surface modification and conjugation onto standard tissue culture plates. Compared to fibronectin coating and tissue culture plate controls, surface coating with HA–DP was able to preserve lymphatic markers, such as prospero homeobox protein 1 (Prox1), podoplanin (PDPN), and LYVE-1 over several passages in vitro. LECs cultured on HA–DP expressed lower levels of focal adhesion kinase (FAK) and YAP/TAZ, which may be responsible for the maintenance of the lymphatic characteristics. Collectively, the HA–DP coating may provide a novel method for culturing human LECs in vitro toward more representative studies in basic lymphatic biology and lymphatic regeneration.
Introduction
Lymphatic endothelial cells (LECs) play a vital role in the immune system, serving as the gatekeepers for lymphocyte trafficking and the maintenance of immune homeostasis.1,2 They are also important for the removal of interstitial fluid and waste products from tissues.3,4 In a variety of diseases, such as cancer and chronic inflammation, the integrity of LECs is disrupted, leading to lymphatic dysfunction and immune suppression.5,6 To understand many of these mechanisms and pathophysiology, it is imperative to have a reliable in vitro culture system that can preserve the phenotype and characteristics of LECs. While prospero homeobox protein (Prox1) is considered as the master regulator to maintain lymphatic identity,7,8 other lymphatic markers are differentially expressed in vivo. Lymphatic capillaries express high levels of lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1). Pre-collecting and collecting vessels express low level of LYVE-1, but high level of podoplanin.9,10 Hence, it is important to have a culture system that recapitulate the lymphatic phenotypes and heterogeneity observed in vivo.Another important aspect of having a robust in vitro culture system for LECs is tissue engineering. Nowadays there have been tremendous efforts to generate lymphatic vessels using natural and synthetic materials.10–13 Often it is not realized that just having a more complex and physiologically relevant 3D system will not result in better tissue engineering unless rudimentary 2D culture is optimized as well. Therefore, it is important to be able to culture LECs on a coating that supports their proliferation, survival, and functional activity over extended periods of time. For decades, the isolation and growth of cells in vitro under controlled conditions has been one of the most utilized experimental approaches in the field of cell biology.14 As a result, various extra cellular matrix (ECM) coatings both natural and synthetic have been established.15
Given the plasticity and heterogeneity of LECs, various ECM coatings have been used to culture LECs.10 Currently, LECs are cultured on tissue culture plastics or on conventional coatings like fibronectin or collagen. Fibronectin is one of the most used ECMs for in vitro culture;16–22 it was shown that LECs adhered and proliferated differently on a fibronectin coated plate compared to tissue culture plastic.19 Previous study reveals that the ligand for integrin α5β1, selectively promoted the growth of LECs through vascular endothelial growth factor receptor-3 (VEGFR-3), which is one of the transmembrane receptors responsible for lymphatic endothelial migration, survival and proliferation.23–25 Another recent study also shows that the adhesion and migration of LECs stimulated by vascular endothelial growth factor-C (VEGF-C) or VEGF-D are α9β1-dependent, which is present in some mutant version of fibronectin.21,26–28 Similarly, Collagen-I and Collagen-IV have been used in some studies for culturing LECs as a monolayer or as an embryoid body (EB) culture.22,29 Though it was observed that Collagen-IV induces migration, cell alignment, proliferation, and differentiation into mature lymphatic capillaries in vivo,30 another study found that Collagen-IV did not favor LEC differentiation in vitro.31 Collagen-I has been used for monolayer culture17,22,30,32,33 of primary LECs and supporting lymphatic vessel like structure.34 Laminin, among the natural ECMs, is the least utilized coating for LECs. It is reported that α4-laminins, such as 411 (formerly laminin-8), 421 (formerly laminin-9), and 423 (formerly laminin-14), are expressed by vascular and lymphatic endothelial cells.29,35,36 Overall, the outlook on using collagen or laminin as coating for culturing LEC is still in contention, while fibronectin is considered as a benchmark in this regard.
Compared to ECM proteins, the use of hyaluronic acid or hyaluronan (HA) has been shown to hold promise in culture of blood endothelial cells (ECs).37–39 HA is an abundant component of the ECM that binds to various receptors and influences activities of ECs. Low molecular weight (LMW) HA was proved to have the ability to interact with its receptors, such as CD44 or receptor for hyaluronan-mediated motility (RHAMM), triggering series of intracellular signal transduction and promoting angiogenesis.40–43 Although CD44 and RHAMM are reported as the main receptors on vascular ECs, they are mostly absent from lymphatic vessels; wherein the only known receptor for HA is LYVE-1, a homolog of CD44.44,45 LYVE-1 is thus likely to play a major role in the regulation of HA on biological behaviors of LECs. Indeed, it was shown in several studies that LMW HA induces lymphangiogenesis through LYVE-1 mediated signaling pathways.46–48 One recent study also shows that HA-binding peptide modulates EC spreading and migration through focal adhesion kinase (FAK).49 Despite the unique expression of HA receptors by LECs, the effect of HA on the regulation of lymphatic characteristic markers has not been explored yet.
In this work, we describe the development of a novel synthetic coating based on HA to preserve the phenotypes and characteristics of LECs. We synthesized dopamine-conjugated HA (HA–DP), which can be conjugated onto the surface of tissue culture plates. Compared to other conventional ECM based coating with fibronectin, we demonstrated that HA–DP can preserve lymphatic phenotypes over several passages of LECs culture in vitro. Moreover, LECs cultured on HA–DP exhibited reduced FAK, which may be responsible for the maintenance of the lymphatic characteristics.
Experimental
Synthesis of dopamine-conjugated hyaluronic acid (HA–DP)
HA (1.026 g, 2.56 mmol disaccharide unit) was dissolved in 85.5 mL MES buffer (0.1 M, pH 5.5) and 28.5 mL ethanol was added. The reaction mixture was brought to equilibrium at room temperature, DMTMM (2.9 g, 4 equivalents) was added to activate HA at room temperature. After 30 minutes of incubation, dopamine (484.7 mg, 1 equivalent) was added to the reaction mixture, followed by stirring at room temperature overnight. HA–DP was purified by dialysis against deionized water at 4 °C for 4 days, lyophilized, and stored at −20 °C until use. HA–DP was characterized by 1H NMR in D2O showing peaks at 1.99 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH3 in HA and 7.3–7.4 (aromatic protons in DP). The degree of substitution was calculated as 42%.
O)CH3 in HA and 7.3–7.4 (aromatic protons in DP). The degree of substitution was calculated as 42%.
Coating of hyaluronic acid–dopamine (HA–DP)
HA–DP was coated on tissue culture plates through polymerization of dopamine under basic conditions. HA–DP was dissolved in deionized water at 5 mg ml−1. Prior adding HA–DP to wells for coating, 20 μL of 10 M NaOH was added per 1 mL of HA–DP. After incubating at 37 °C overnight, the wells were vigorously washed with deionized water and cell culture medium for cell culture.Characterization of HA coating
HA–DP coating was characterized and quantified using toluidine blue assay, as previously described.50 Briefly, serial dilution of free HA in DI water was prepared to create a standard curve. Then, both free HA solution and HA–DP coated plate were incubated with 1 ml Toluidine blue O (TBO, Sigma 198161-5G). In the case of free HA solution, supernatant was washed off carefully after centrifugation. For coated six-wells plate, the supernatant was aspirated directly. Both samples were washed properly with 10 mM NaOH. At this stage, a picture was taken to show TBO staining of coated wells compared to non-coated ones. Then, 50% acetic was added to the samples and incubated at room temperature for 20 minutes. Finally, the solutions were collected and ran for absorbance in microplate reader at 634 nm. Absorbance of free HA solutions of different concentration were used to create standard curve, then absorbance of HA–DP coated sample was fit to the standard curve to quantify amount of HA in the coating.Cell culture
Human juvenile lymphatic endothelial cells (C-12216) of four donors (PromoCell, Heidelberg, Germany) were expanded and used for experiments between passages 4 and 8, as previously described.15,51 Briefly, LECs were maintained at 37 °C with 5% CO2 in Endothelial Cell Growth Medium (EGM-MV2, C-22022, PromoCell). To keep the cell passaging constant throughout experiments, cells were passaged every 5 days at a 1 to 3 ratio. Human LECs were characterized for the positive expression of CD31, LYVE-1, Prox1, and podoplanin throughout the experiments. All cell lines were routinely tested for mycoplasma contamination and were negative throughout this study.FACS analysis
Human LECs were analyzed for lymphatic markers using flow cytometry (FACS) following standard procedure.52 Briefly, cells were trypsinized and centrifuged following resuspension in FACS buffer. Suspended cells (1 × 106 cells) were stained with the antibodies (1 μg mL−1) for 30 minutes at room temperature: anti-LYVE-1 antibody (R&D systems, FAB20892A), anti-PDPN antibody APC (Biolegend, 337004), as well as their corresponding IgG isotype controls (ESI Table 1†). For intracellular staining, the cells were fixed and permeabilized with Foxp3/Transcription Factor Staining Buffer Set (Thermo, 00-5523-00) and then incubated with anti-Prox1 antibody FITC (Novus Biologicals, NBP1-30045AF488) for 30 minutes. The cells were washed twice and resuspended in FACS buffer for analysis. Then, the cells were analyzed using flow cytometry (BD LSR FortessaX-20) and the metadata were analyzed using FlowJo.Gene expression
To analyze the effect of different ECM on lymphatic phenotypes, LECs were cultured on tissue culture plastic, fibronectin, and HA–DP coatings for 5 days in EGM-MV2 media. Three biological replicates (n = 3) were collected per condition and analyzed with real-time qRT-PCR with triplicate readings as previously described.15 RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher) according to the manufacturer's protocol. cDNA was then used with the TaqMan Universal PCR Master Mix and Gene Expression Assays for LYVE-1, Prox1, PDPN, VEGFR3, YAP, TAZ, MYC, CTGF, and GAPDH (ESI Table 2†). Each sample was prepared in triplicate and the relative expression was normalized to GAPDH and analyzed using the ΔΔCt method.Immunofluorescence
To visualize the lymphatic protein expression, LECs were seeded on tissue culture plastic, fibronectin, and HA–DP coatings for 5 days. Samples were fixed with 4% paraformaldehyde, blocked with 1% BSA, permeabilized with 0.1% Triton-X, and stained for LYVE-1, Prox1, CD144, ERG and podoplanin (ESI Table 3†). To visualize focal adhesion kinase and F-actin distribution FAK antibody (Sigma, 2 μg ml−1) and phalloidin (Abcam, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) were used. Samples were rinsed twice in PBS and counterstained with DAPI (Thermo Fischer, 300 nM). All samples were imaged in Nikon AX-R confocal at 40× magnification.
:1000) were used. Samples were rinsed twice in PBS and counterstained with DAPI (Thermo Fischer, 300 nM). All samples were imaged in Nikon AX-R confocal at 40× magnification.
FAK and F-actin quantification
The thresholding tool in ImageJ was used to identify the FAKs. Then analyze particles was used with size restricted to 4–20 μm to count the number of focal adhesions. Ten fluorescent images were taken per coating at 40× using the Nixon AXR confocal microscope and analyzed with the FIJI Directionality Plug-in.53 A statistical analysis of the dispersion was performed using GraphPad Prism 9 (GraphPad Software Inc., La Jolla, CA).Statistical analysis
Statistical analysis was performed with GraphPad Prism. For each coating condition, at least three independent experiments were performed with three biological replicates. Statistical comparisons were made using Student's t test for paired data, analysis of variance (ANOVA) for multiple comparisons, and with Tukey post hos analysis for parametric data. Specifically, Student's t test was used to analyze differences between protein expression and gene expression on different coatings. Significance levels were set at the following: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.Results and discussion
LECs exhibit a decrease in lymphatic markers in vitro
While previous studies have reported that blood endothelial cells lose their characteristics over time,54,55 little is known about the phenotypes of LECs during in vitro culture. Therefore, we aimed to characterize the phenotypes of LECs during in vitro culture. Human dermal LECs from single donor (n = 3) were cultured on tissue culture plates from passage 5 to 7 and their lymphatic phenotypes were characterized using FACS, real-time qRT-PCR, and immunofluorescent image analysis (Fig. 1). We confirmed using immunofluorescent (Fig. 2A–C) and FACS analysis (Fig. 2D–F and ESI Fig. 1–3†) that LECs are heterogenous in nature and consistently express Prox-1 from passage 5 to 7. Since Prox-1 is known to be the master lymphatic regulator,56,57 these observations confirmed that LECs maintained their lymphatic identity (Fig. 2G). However, we observed a gradual decrease in the expression of key lymphatic markers podoplanin (Fig. 2H) and LYVE-1 (Fig. 2I). Overall, these results suggest that while LECs maintained their identity in culture by expressing transcription factor Prox1, they started to lose their lymphatic phenotypes by gradual decrease in key lymphatic markers podoplanin and LYVE-1. This observation is consistent with the lymphatic heterogeneity and plasticity that can be found in vivo.58–60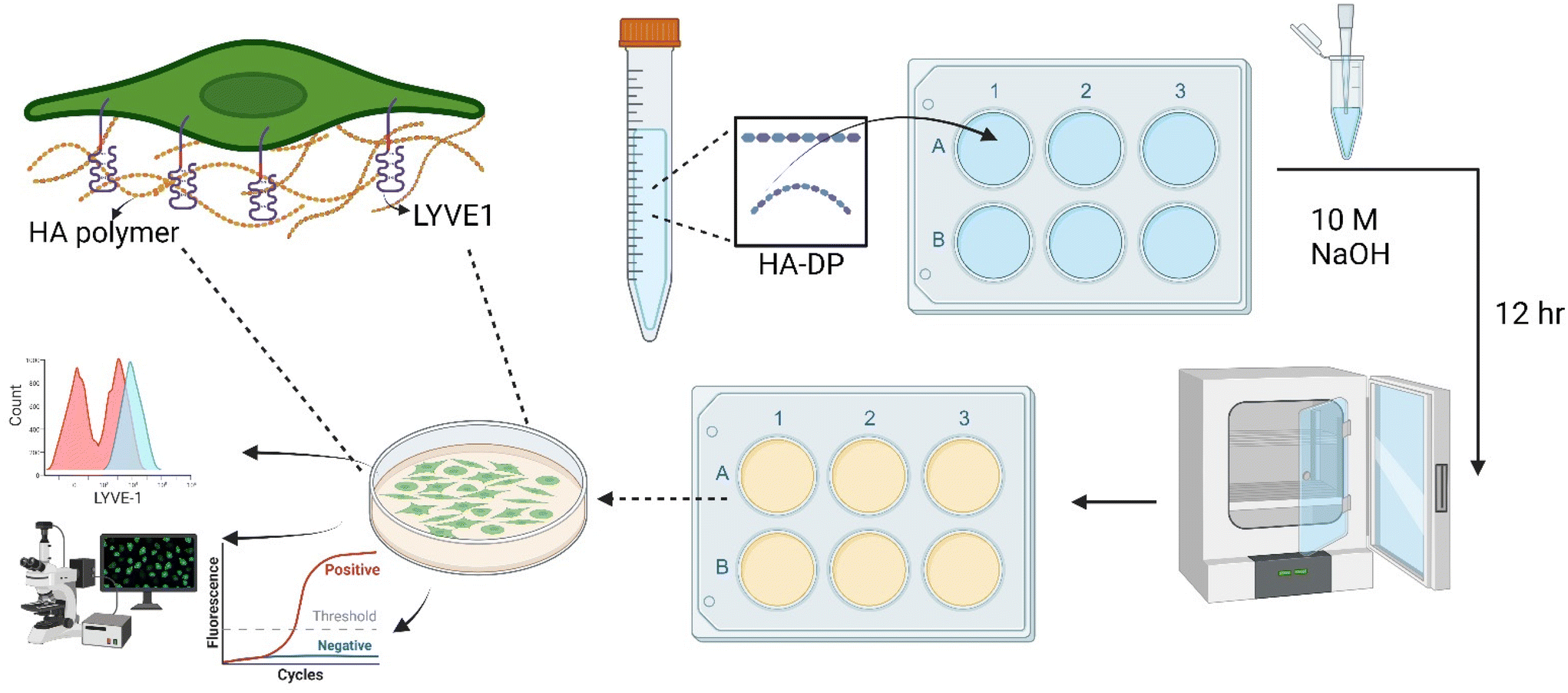 | ||
| Fig. 1 Schematic of experimental procedure to investigate cell–ECM interaction on HA–DP coating. The HA–DP polymer was dissolved in DI water, then added to culture plate in the presence of 10 M NaOH. The plates were incubated overnight and washed with DI water before seeding the cells. An enlarged image on top left shows interaction between HA polymer and LYVE-1 receptor, one of the key glycoprotein receptors of LECs. Lymphatic phenotypes were analysed using flow cytometry, immunofluorescent imaging, and real-time RT-PCR. | ||
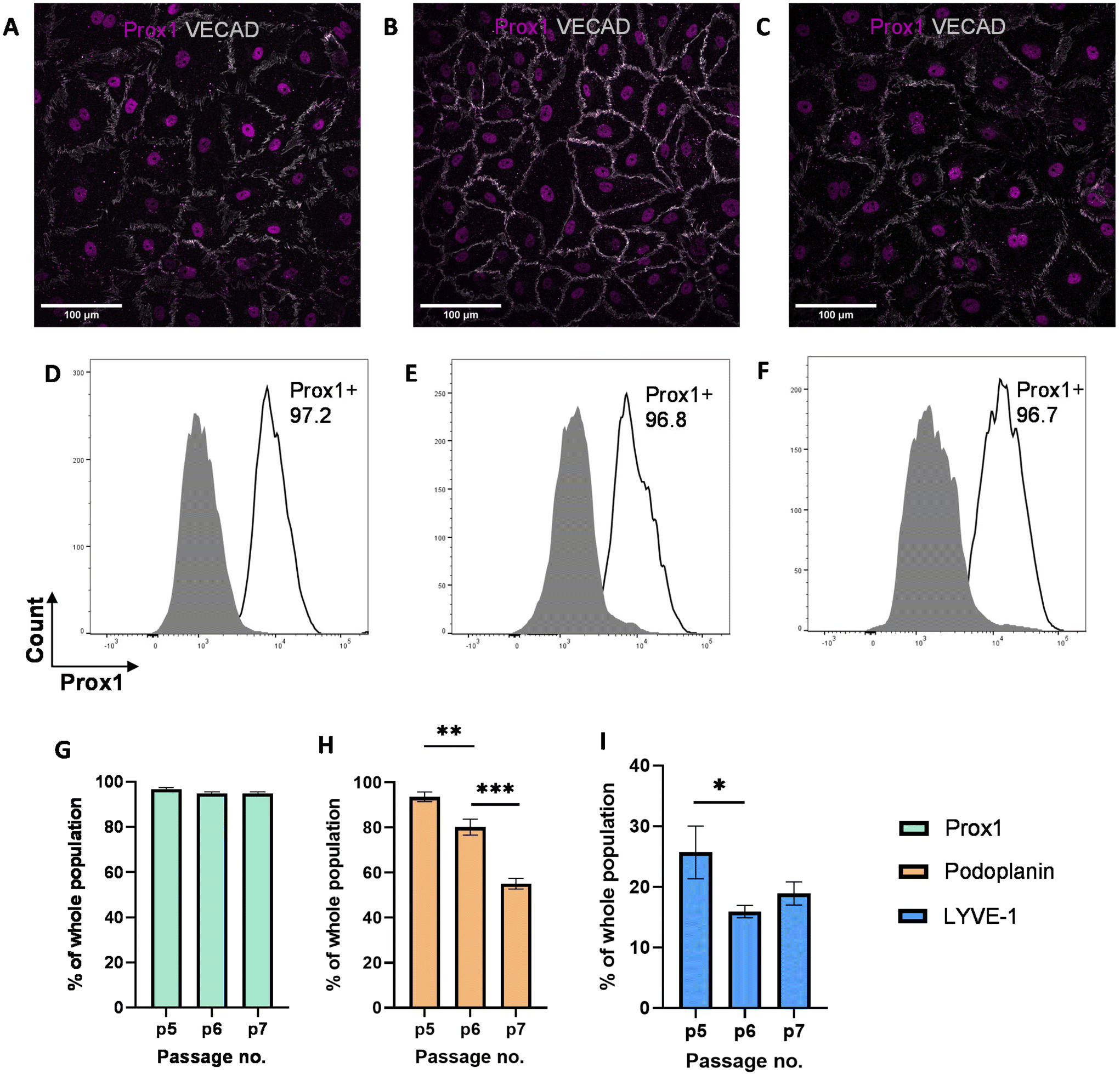 | ||
| Fig. 2 Expression of lymphatic markers by LECs cultured on tissue culture plates. Representative immunofluorescent images of LECs cultured on tissue culture plates at (A) P.5, (B) P.6, and (C) P.7 stained for VE-CAD (white) and Prox-1 (magenta). Scale bars are 100 μm. LECs cultured on tissue culture plates were analyzed for Prox-1 using flow cytometry. Representative flow cytometry analysis indicating Prox-1 expression by LECs (white histogram) at (D) P.5, (E) P.6, and (F) P.7 compared to isotype control (grey histogram). Flow cytometry data for LECs at P.5, P.6, and P.7 were analysed for percentage of (G) Prox1+, (H) podoplanin+, and (I) LYVE-1+ cells population. Data represents mean ± stdev., n = 4 per condition. Significance levels were set at: *P < 0.5, **P < 0.01, and ***P < 0.001. | ||
Synthesis and characterization of HA–DP
After validating the heterogeneity and plasticity of LECs, we hypothesized that the conventional coating system might not be optimized for culturing LECs. Since LECs uniquely express LYVE-1 to bind to HA and activate intracellular signaling to promote lymphangiogenesis,61 we postulated that tunable synthetic HA coating can serve as a supportive matrix for LEC culture in vitro. Since HA is a negatively charged polysaccharide, we have to modify HA with dopamine group to enable its conjugation to the surface of the tissue culture plate.62 The dopamine group was conjugated with the carboxyl group of HA to generate HA–DP polymer (Fig. 3A). 1H-NMR indicated dopamine peaks at 7.47–7.21 ppm and N-acetyl groups of HA peaks at 1.99 ppm, which confirmed the conjugation of dopamine group to HA (Fig. 3B). We synthesized HA–DP polymer with various degree of substitution (DS) and discovered that HA–DP with 42% of DS to be optimum for surface coating (data not shown). Therefore, HA–DP with 42% DS was used to coat tissue culture plate for our subsequent studies. Toluidine blue assay was used to quantify the amount HA–DP conjugated to the tissue culture plate. The presence of HA–DP was evident through toluidine blue assay in HA–DP coated plate compared to the control (Fig. 3C). Quantification using a standard curve revealed 9.5 ± 0.2 μg cm−2 was present in the HA–DP coated plate (Fig. 3D and ESI Fig. 4†).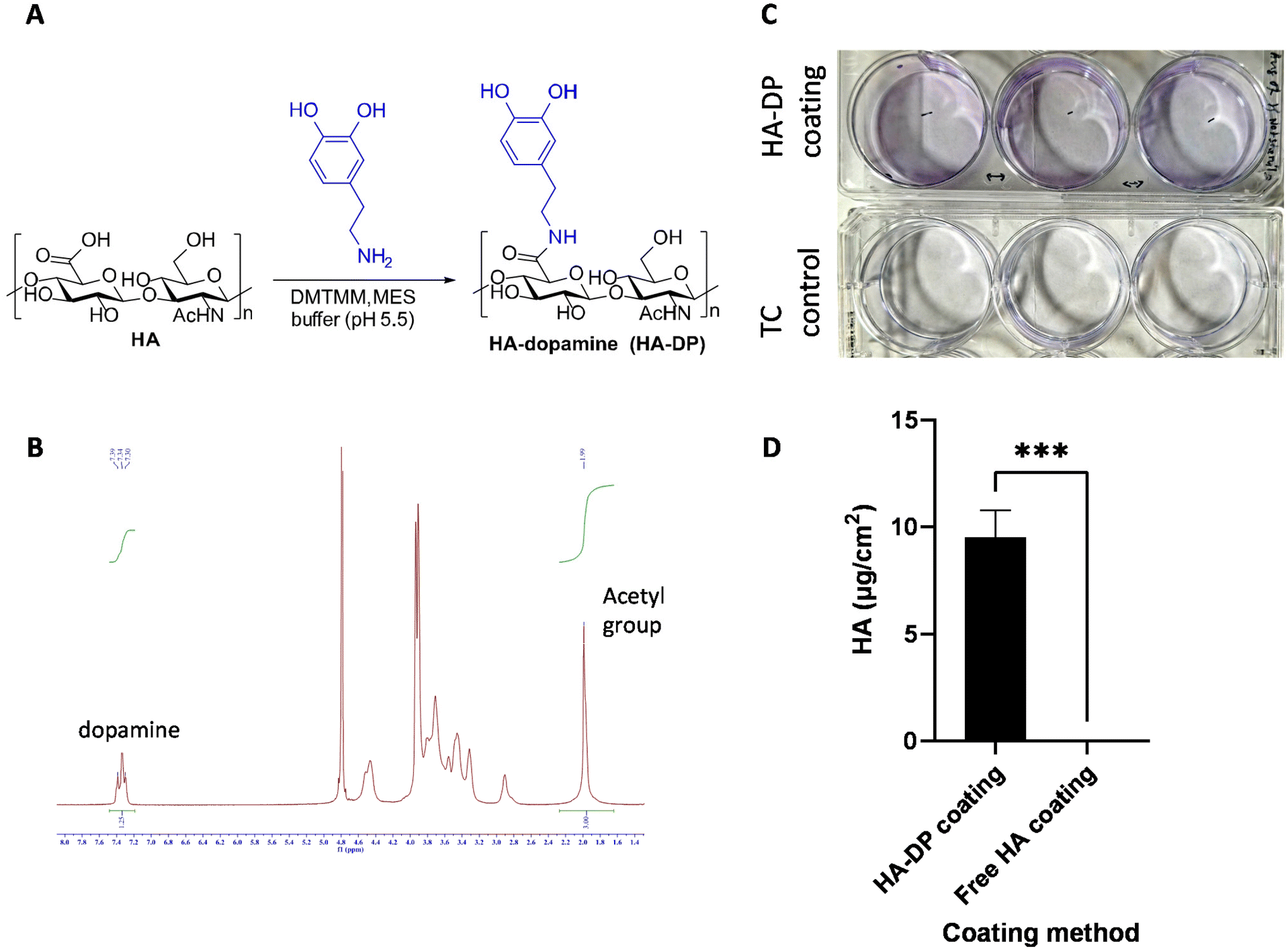 | ||
| Fig. 3 Synthesis and characterization of HA–DP. (A) Schematic of chemical reaction to generate HA–DP. The dopamine was conjugated with the carboxyl group of HA using DMTMM, MES buffer (pH 5.5). (B) The conjugation of dopamine group to HA was confirmed using 1H-NMR. The protons of the aromatic ring of DP showed peaks at 7.4–7.2 ppm and those of N-acetyl groups of HA showed 2.0 ppm of the spectrum of 1H-NMR confirming an appropriate conjugation of DP to HA. 1H-NMR spectra analysis confirms 42% of degree of substitution. (C) Representative photo taken after TBO staining and washing as described in characterization method, a control tissue culture plastic was used to show difference in purple blue stain due to staining. (D) The difference between amount of bound HA on the surface between two coating method. The left column of the graph represents the coating with HA–DP and right one represents coating with free HA solution. We found no presence of HA from free solution-based coating compared to HA–DP based coating. | ||
Preservation of LEC phenotype though HA–DP coating
To test the hypothesis that HA–DP can preserve lymphatic phenotypes, we cultured LECs on HA–DP, as well as tissue culture plate and fibronectin-coated plate as controls. LECs were analyzed for their lymphatic phenotypes using FACS and real-time qRT-PCR analysis. To evaluate lymphatic phenotypes at gene expression levels among different coating conditions, we decided to evaluate them at early passage (P.5) and late passage (P.7), where their gene expression levels were known to be different. At early passage (P.5), we noticed that LECs cultured on HA–DP expressed 1.70 ± 0.18-fold more expression of LYVE-1 and 2.24 ± 0.19-fold more expression of PDPN compared to LECs cultured on tissue culture plate (Fig. 4A and B). LECs cultured on fibronectin coated plate expressed 1.39 ± 0.20-fold more expression of PDPN compared to LECs cultured on tissue culture plate (Fig. 4B). While the expression levels of others lymphatic markers Prox1 and VEGFR3 were not significantly different among different coating conditions (Fig. 4C and D). At higher passage (P.7), compared to LECs cultured on tissue culture plate, LECs cultured on HA–DP express higher levels of lymphatic markers LYVE-1 (4.67 ± 0.56-fold), PDPN (4.33 ± 0.33-fold), Prox1 (3.64 ± 0.30-fold), and VEGFR3 (2.0 ± 0.64-fold) (Fig. 4E–H). LECs cultured on fibronectin coated plate also expressed higher lymphatic markers LYVE-1 (3.85 ± 0.57-fold), PDPN (1.94 ± 0.38-fold), Prox1 (2.63 ± 0.49-fold), and VEGFR3 (1.28 ± 0.63-fold) compared to LECs cultured on tissue culture plates (Fig. 4E–H). Overall, LECs cultured on HA–DP express higher lymphatic markers compared to LECs cultured on tissue culture plates and fibronectin coated plates. To further validate this trend at the protein level, we performed FACS analysis on LECs cultured on different coating conditions from passage 5 to 7. For LECs cultured on tissue culture plate, we noticed a decrease in the percentage of LYVE-1+ cells (Fig. 4I) and podoplanin+ cells (Fig. 4J) from passage 5 to 7. LECs cultured on fibronectin coated plates also demonstrated a decrease in the percentage of LYVE-1+ cells (Fig. 4I) and podoplanin+ cells (Fig. 4J) at a lesser extent. Interestingly, LECs cultured on HA–DP coated plates maintained the percentage of LYVE-1+ cells (Fig. 4I) and demonstrated less decrease in the percentage of podoplanin+ cells (Fig. 4J). Overall, these results suggested that HA–DP coating preserves lymphatic phenotypes during in vitro culture of LECs for at least until passage 7.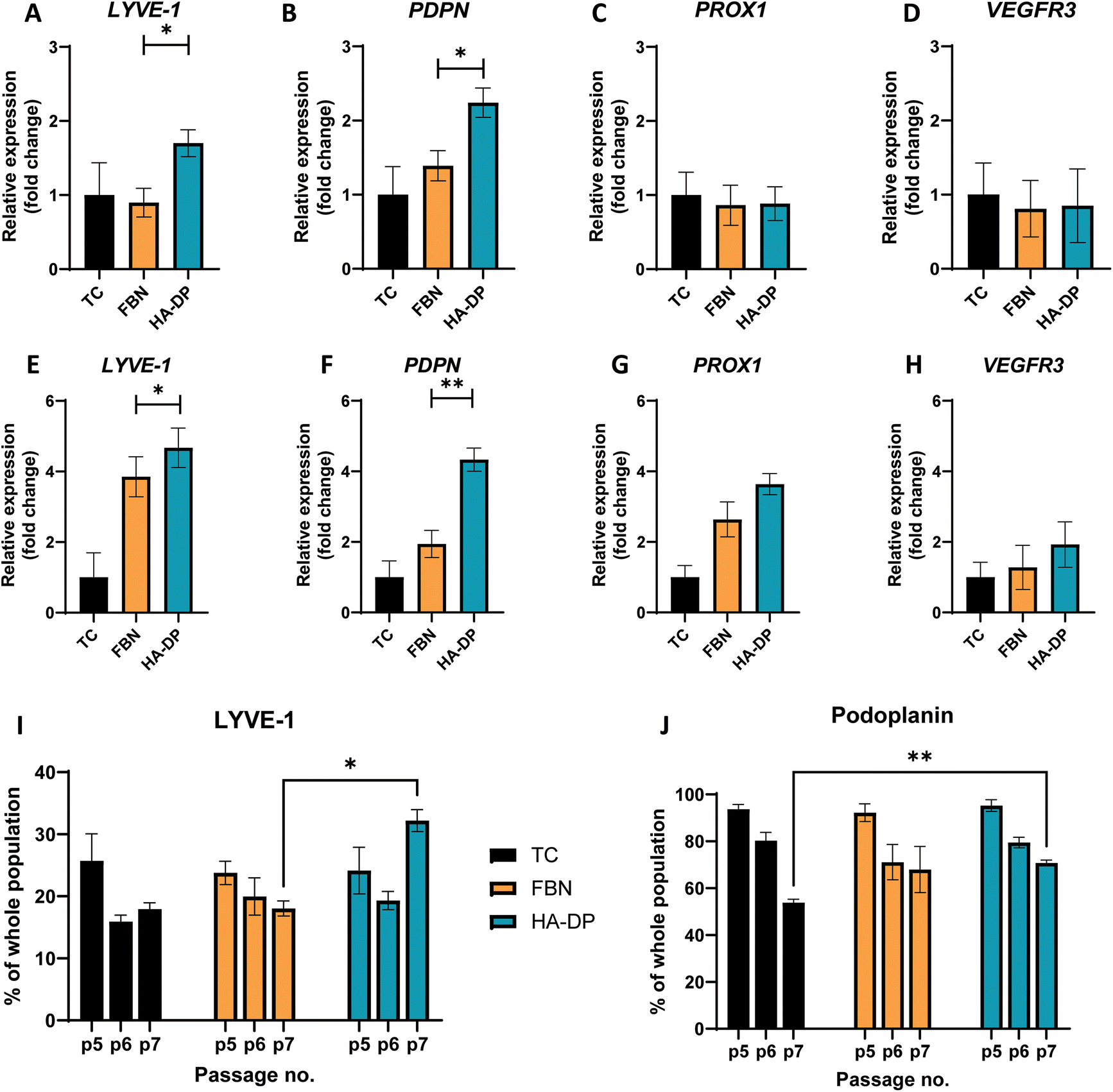 | ||
| Fig. 4 Protein and gene expression of LEC cultured on different coatings. Real-time qRT-PCR for early passage (P.5) LECs cultured on tissue culture plate (black), fibronectin-coated plate (yellow), and HA–DP (blue) coated plate for (A) LYVE-1, (B) PDPN, (C) Prox1, and (D) VEGFR3. Real-time qRT-PCR for late passage (P.7) LECs cultured on tissue culture plate (black), fibronectin-coated plate (yellow), and HA–DP (blue) coated plate for (E) LYVE-1, (F) PDPN, (G) Prox1, and (H) VEGFR3. Three biological replicates (n = 3) were collected per condition and analysed with real-time qRT-PCR with triplicate readings. Flow cytometry analysis for LECs (P.5–7) cultured on tissue culture plate (black), fibronectin-coated plate (yellow), and HA–DP (blue) coated plate indicating percentage of cells that are (I) LYVE-1+ and (J) podoplanin+. Data represents mean ± stdev., n = 4 per condition. Significance levels were set at: *P < 0.5 and **P < 0.01. | ||
Differential expression of YAP/TAZ and FAK on fibronectin and HA–DP coated plates
Since HA–DP can preserve lymphatic phenotypes better than the standard of culture on fibronectin-coated plates, we decided to further investigate the differences in mechanotransduction among culture conditions. Difference in ECM component often induces mechanotransduction on the cells independent of matrix stiffness.55 Though there might not be much difference in stiffness between fibronectin and HA–DP, we were interested in to investigate focal adhesion kinase (FAK) and F-actin. At low passage (P.5), we did not observe significant expression of FAK on LECs cultured on fibronectin and HA–DP (ESI Fig. 5†). However, at late passage (P.7) LECs cultured on fibronectin-coated plate express higher FAK (0.03 ± 0.004 count per μm2) compared to LECs cultured on HA–DP (0.013 ± 0.0004 count per μm2) (Fig. 5A and B). Similarly, F-actin density and alignment was more pronounced in LECs cultured on fibronectin compared to HA–DP (Fig. 5C and D). Since FAK can also activate the “Hippo Pathway”, we also investigated YAP/TAZ and its downstream genes CTGF and MYC. At low passage (P.5), though not significant, LECs cultured on HA–DP express lower CTGF, YAP, and TAZ (ESI Fig. 6†). But, at high passage (P.7), LECs cultured on HA–DP express lower TAZ (0.71 ± 0.1-fold), CTGF (0.34 ± 0.12-fold), and MYC (0.76 ± 0.07-fold) compared to LECs cultured on fibronectin coated plates (Fig. 6A–D). These observations were also confirmed using immunofluorescent imaging of YAP/TAZ (Fig. 6E and F). Furthermore, quantification of the fluorescent signal indicates that culturing LECs on HA–DP led to a more cytoplasmic localization of YAP/TAZ compared to LECs cultured on fibronectin coated plates (Fig. 6G and H). Therefore, culturing LECs on HA–DP enabled cytoplasmic degradation of YAP/TAZ, which subsequently enhance transcription of lymphatic master regulator Prox1, including its targets LYVE-1, podoplanin, and VEGFR3. The differences in local and global expression of YAP/TAZ in both FBN and HA–DP explains our results and suggests that HA–DP coated plate preserve lymphatic phenotypes by downregulating FAK and YAP/TAZ pathways (Fig. 7). Overall, these observations are consistent with previous studies that show YAP/TAZ negatively regulate Prox1 during development and in vitro culture.15,63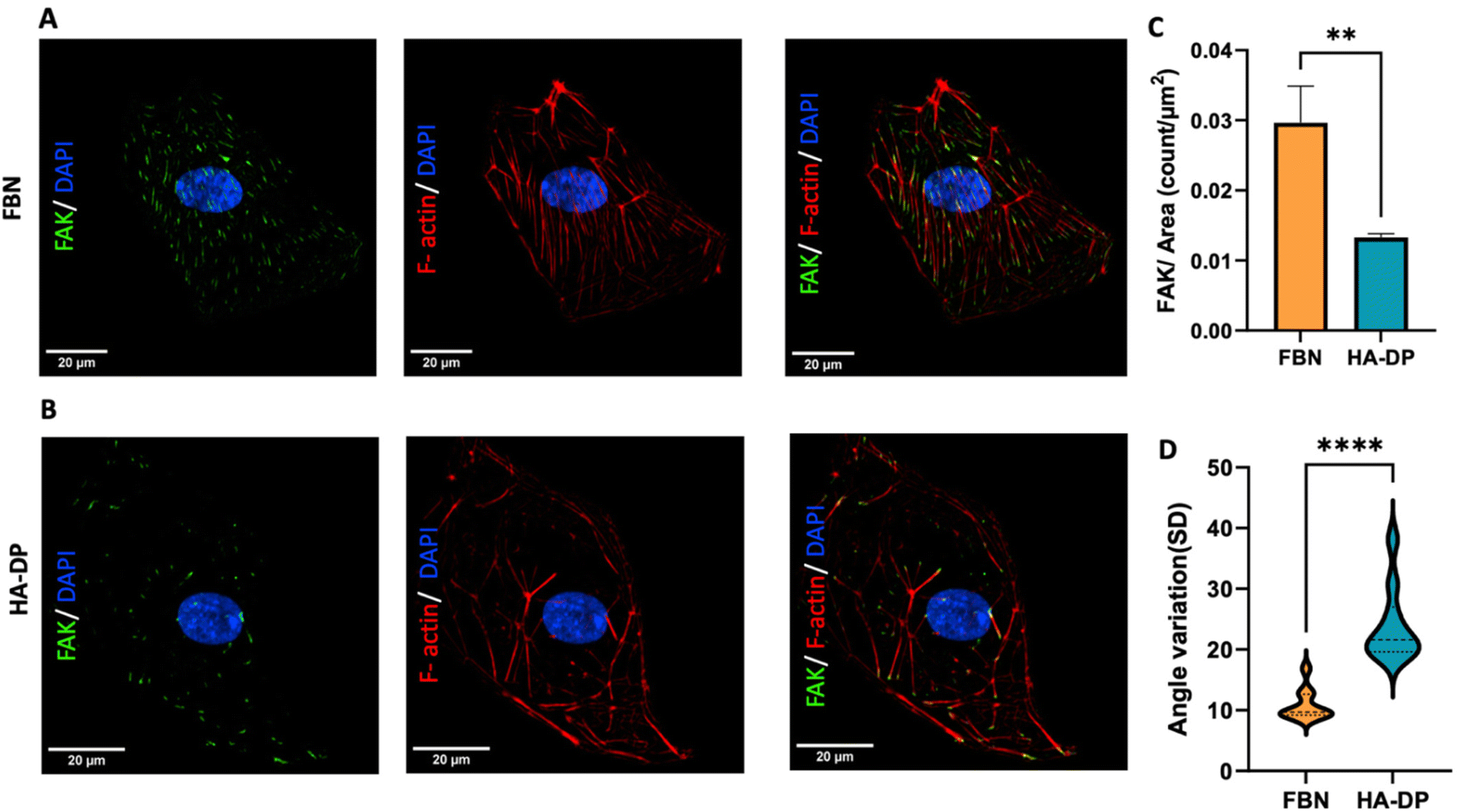 | ||
| Fig. 5 Focal adhesion kinase (FAK) expression of LECs cultured on fibronectin and HA–DP coated plates. Representative immunofluorescent images of LECs (P.7) cultured on (A) fibronectin and (B) HA–DP coated plates stained for FAK (green), F-actin (red), and nuclei (blue). Scale bars are 20 μm. (C) The number of FAK per surface area was analysed for LECs cultured on fibronectin and HA–DP coated plates. (D) The orientation of F-actin was analysed using angle variation (SD) for LECs cultured on fibronectin and HA–DP coated plates. Data represents mean ± stdev., n = 4 per condition. Significance levels were set at: **P < 0.05 and ****P < 0.001. | ||
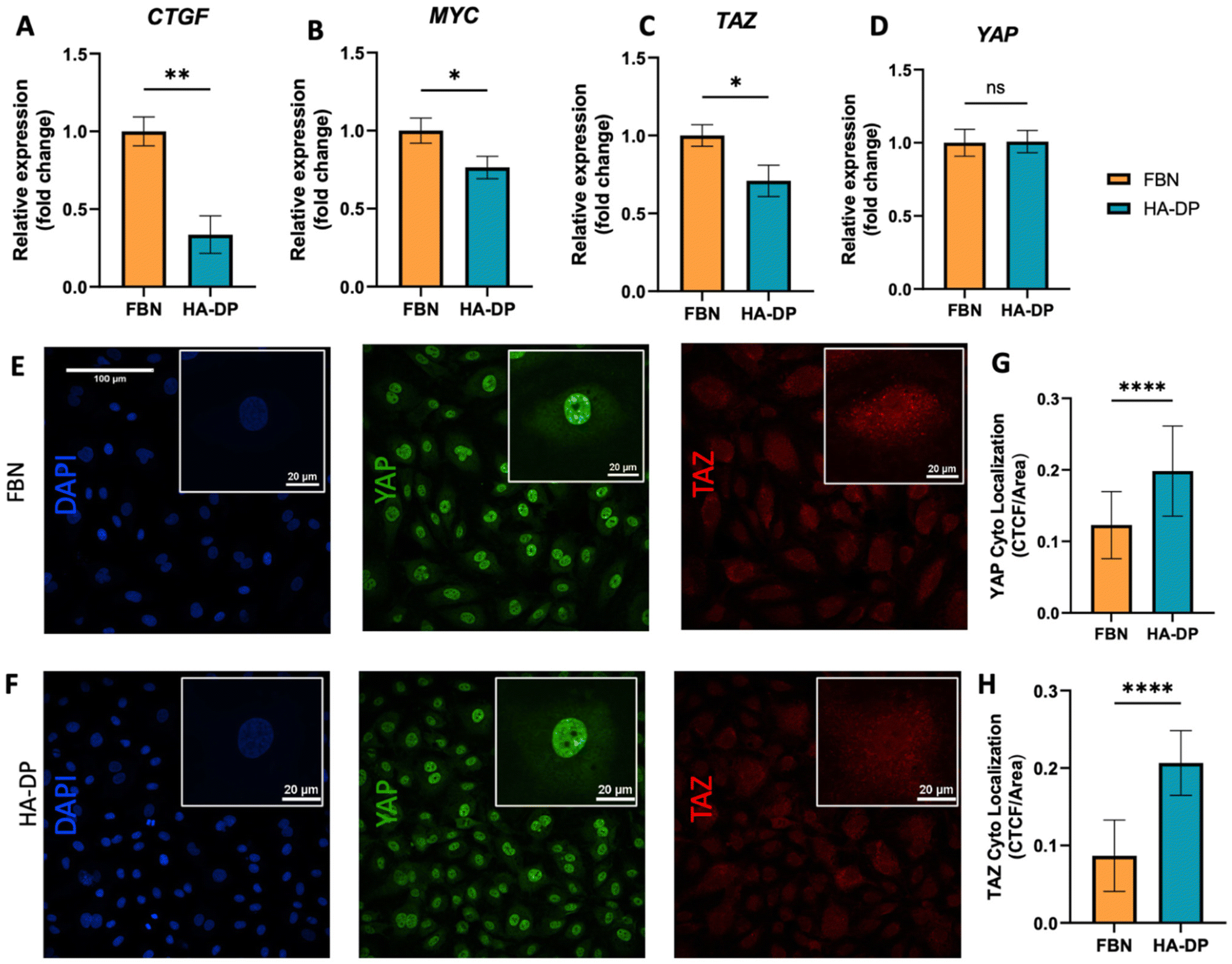 | ||
| Fig. 6 YAP/TAZ expression for LECs cultured on HA–DP and fibronectin coated plates. Real-time qRT-PCR data for (A) CTGF, (B) MYC, (C) YAP, and (D) TAZ expressed by LECs (P.7) cultured on HA–DP and fibronectin coated plates. Three biological replicates (n = 3) were collected per condition and analysed with real-time qRT-PCR with triplicate readings. Representative confocal z-stacks of LECs (P.7) cultured on (E) fibronectin and (F) HA–DP coated plates stained with YAP (green), TAZ (red) and nuclei (blue). Scale bars are 100 μm. Insets indicate the localization of YAP (green) and TAZ (red). Scale bars are 20 μm. Fluorescent intensity quantification demonstrates (G) an increase in cytoplasmic localization for YAP and (H) an increase in cytoplasmic localization of TAZ for LECs cultured on HA–DP compared to fibronectin. CTCF, corrected total cell fluorescence. Data represents mean ± stdev., n = 4 per condition. Significance levels were set at: **P < 0.5, **P < 0.05 and ****P < 0.001. | ||
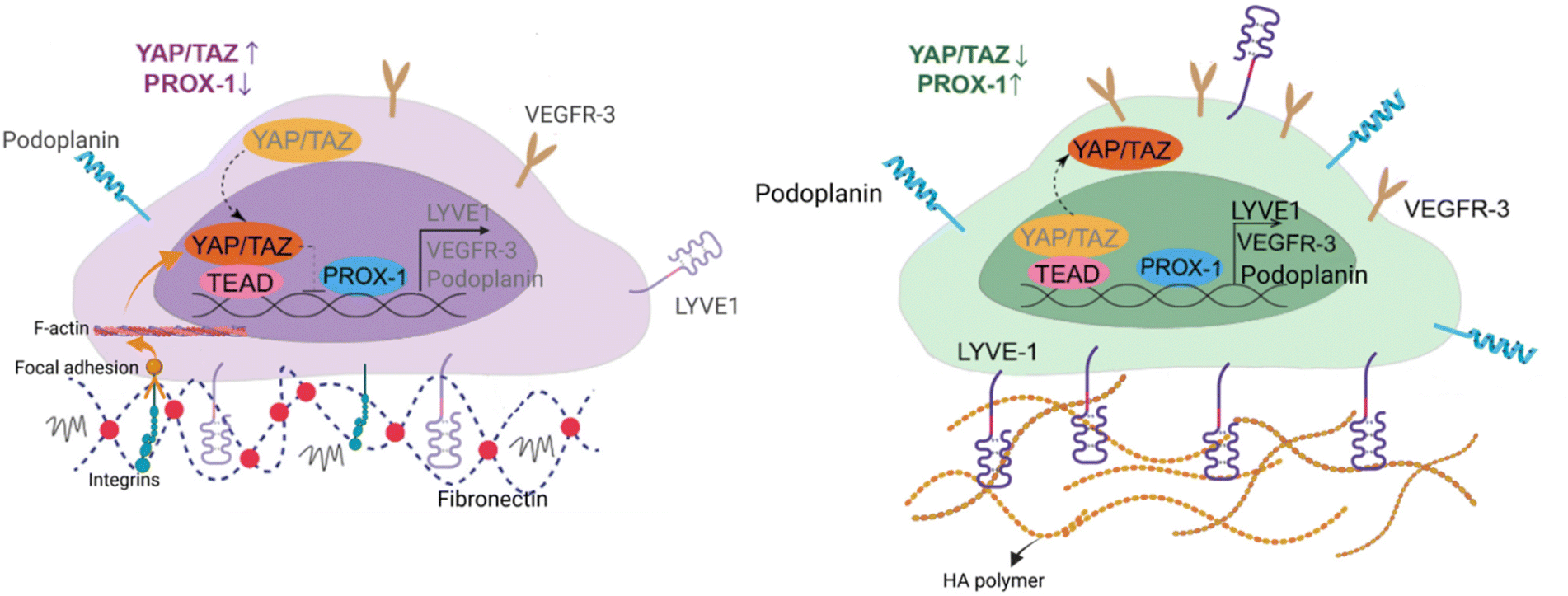 | ||
| Fig. 7 Schematic diagram depicting the role of HA–DP in preserving lymphatic phenotypes. HA–DP was able to preserve key lymphatic markers, including Prox1, LYVE-1, podoplanin, and VEGFR3. When LECs are cultured on fibronectin coated plates, YAP/TAZ enter the nucleus and bind to the PROX-1 promoter, inhibiting its transcription, including its targets, such as LYVE-1, podoplanin, and VEGFR3. In contrast, culturing LECs on HA–DP coated plates enables YAP/TAZ to undergo cytoplasmic degradation, which subsequently enhance transcription of Prox1, including its targets, such as LYVE-1, podoplanin, and VEGFR3. | ||
Discussion
The discovery of unique lymphatic markers, such as Prox1, LYVE-1, and PDPN have allowed the isolation and cultured of LECs, which paved the way for many studies for better understanding of lymphatic biology. While it is challenging to isolate and maintain murine LECs in culture, human primary LECs can be isolated from adult and juvenile tissues. Once isolated, these LECs can be cultured on regular tissue culture plate, as well as on plates coated with fibronectin or collagen. While there is no agreement on the best culture condition to culture LECs in vitro, it has been recognized that LECs may start to lose their lymphatic expression overtime during culture.9,64–67 One of these investigations even mentions reduction in Prox1 as well. However, in this study, we confirmed that LECs can be cultured from P.5–7, while maintaining their Prox1 expression. But they started to lose other key lymphatic markers, such as LYVE-1 and podoplanin. Both LYVE-1 and podoplanin are surface receptor uniquely expressed by LECs. LYVE-1 is important for leukocytes trafficking and highly abundant in lymphatic capillaries.68,69 While podoplanin is responsible for blood-lymphatic separation and highly abundant in lymphatic pre-collecting and collecting vessels.51,70–72 Lymphatic capillaries are LYVE1high and podoplaninlow, because of the abundant hyaluronic acid used by leukocytes to enter the lymphatic capillaries.73,74 However, it is still unclear if other matrix proteins are responsible for the pre-collecting and collecting vessels to express LYVE1low and podoplaninhigh. Therefore, understanding how different matrix proteins can be used to enrich certain population of LECs in vitro is important and can be used for future studies in vascular and lymphatic biology.We demonstrated that a simple coating with HA–DP can effectively preserve key lymphatic markers over several passages in vitro. In fact, key lymphatic makers were maintained on high passage (P.7) of LECs cultured on HA–DP, compared to LECs cultured on fibronectin coated plates. LECs uniquely express LYVE-1, a specific receptor for HA, and provide a unique advantage for engineered matrices containing HA. Other than LYVE-1, VEGFR3 is the only receptor that can preserve lymphatic phenotypes in the presence of VEGF-C. But a growth factor independent, ECM-based interaction can be modulated through HA–DP coating, which opens many opportunities to modify culture media based on different applications. Therefore, future studies can use HA–DP to mimic human physiology and pathology depending on the intended applications (i.e., lymphatic capillaries vs. collecting vessels).
Moreover, we showed that LECs on HA–DP coating expressed reduced FAK and F-actin stress fibres. FAK is considered one of the key mechanotransductory components and one of the first molecules recruited to focal adhesions in response to external mechanical stimuli. FAK is also a regulator of F-actin dynamics.75,76 Which explains our result from Fig. 5. As cells experience more mechanical stress, more focal adhesion assembly occurs and in turn causes increased amount of F-actin stress fibre and their alignment. The FAK is a pivotal mediator of cell mechanosignaling and relays these stimuli to other mechanotransducers like YAP/TAZ.77 Simultaneously, Hippo effectors YAP/TAZ act as mechanosensing switches whose expression is analogous to the encountered mechanotransduction.78 All these concludes into the fact that LECs experience reduced mechanotransduction on HA–DP coating compared to fibronectin.
The “Hippo Pathway” and YAP/TAZ are critically involved in initial LEC specification, differentiation, and sprouting during early lymphatic development and in maintaining lymphatic integrity during adulthood.63 Recent studies described that YAP/TAZ work as stress-mediated mechanotransducers in LECs like that of BECs but different in that YAP/TAZ regulate Prox1 transcriptional activity in LECs.79,80 Strikingly, lymphatic YAP/TAZ negatively regulate Prox1 transcription, and they modulate Prox1 activity and lymphatic plasticity.63 Consistent to previous findings, our data also suggest that culturing LECs on HA–DP coated plates causes cytoplasmic degradation of YAP/TAZ, which subsequently enhances transcription of lymphatic master regulator Prox1, including its targets LYVE-1, PDPN, and VEGFR3.
Collectively, we demonstrated that a simple HA–DP can be used to preserve lymphatic phenotypes during in vitro culture of primary LECs. Mechanistically, HA–DP caused downregulation of YAP/TAZ, which upregulate Prox1 and therefore maintain lymphatic phenotypes, consistent with previous findings. We hope that these results would further improve subsequent in vitro studies and inspire other researchers to adapt this technology for other applications (i.e., culturing mouse LECs, which has been known to be very difficult to culture in vitro). Overall, this simple yet effective HA–DP coating may be useful for culturing human LECs in vitro for applications in basic lymphatic biology and lymphatic regeneration.
Author contributions
S. S., F. F., L. A., and D. H.-P. conceived the ideas, designed the experiments, interpreted the data, and wrote the manuscript. S. S., F. F., F. G., and E. H. conducted the experiments and analysed data. All authors have approved the manuscript.Conflicts of interest
The authors have declared that no conflict of interest exists.Acknowledgements
We acknowledge support from the University of Notre Dame through “Advancing Our Vision” Initiative in stem cell research, Harper Cancer Research Institute – American Cancer Society Institutional Research Grant (IRG-17-182-04), American Heart Association through Career Development Award (19-CDA-34630012 to D. H.-P.), National Science Foundation (2047903 to D. H.-P.), and National Institute of Health (R35-GM-143055 to D. H.-P.). We would like to thank Center for Environmental Science and Technology for help with material characterization.References
- T. Tammela and K. Alitalo, Cell, 2010, 140, 460–476 CrossRef CAS PubMed.
- K. Alitalo and P. Carmeliet, Cancer Cell, 2002, 1, 219–227 CrossRef CAS PubMed.
- H. Wiig and M. A. Swartz, Physiol. Rev., 2012, 92, 1005–1060 CrossRef CAS PubMed.
- J. B. Dixon, S. Raghunathan and M. A. Swartz, Biotechnol. Bioeng., 2009, 103, 1224–1235 CrossRef CAS PubMed.
- Y. Wang and G. Oliver, Genes Dev., 2010, 24, 2115–2126 CrossRef CAS PubMed.
- S. Liao and T. P. Padera, Lymphatic Res. Biol., 2013, 11, 136–143 CrossRef CAS PubMed.
- N. C. Johnson, M. E. Dillard, P. Baluk, D. M. McDonald, N. L. Harvey, S. L. Frase and G. Oliver, Genes Dev., 2008, 22, 3282–3291 CrossRef CAS PubMed.
- M. G. Bixel and R. H. Adams, Genes Dev., 2008, 22, 3232–3235 CrossRef CAS PubMed.
- M. H. Ulvmar and T. Mäkinen, Cardiovasc. Res., 2016, 111, 310–321 CrossRef CAS PubMed.
- L. Alderfer, E. Hall and D. Hanjaya-Putra, Acta Biomater., 2021, 133, 34–45 CrossRef CAS PubMed.
- L. Alderfer, A. Wei and D. Hanjaya-Putra, J. Biol. Eng., 2018, 12, 32 CrossRef CAS PubMed.
- S. Landau, A. Newman, S. Edri, I. Michael, S. Ben-Shaul, Y. Shandalov, T. Ben-Arye, P. Kaur, M. H. Zheng and S. Levenberg, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101931118 CrossRef CAS PubMed.
- J. S. T. Hooks, F. C. Bernard, R. Cruz-Acuna, Z. Nepiyushchikh, Y. Gonzalez-Vargas, A. J. Garcia and J. B. Dixon, Biomaterials, 2022, 284, 121483 CrossRef CAS PubMed.
- M. A. Gimbrone Jr., R. S. Cotran and J. Folkman, J. Cell Biol., 1974, 60, 673–684 CrossRef CAS PubMed.
- L. Alderfer, E. Russo, A. Archilla, B. Coe and D. Hanjaya-Putra, FASEB J., 2021, 35, e21498 CrossRef CAS PubMed.
- A. A. A. Alghamdi, C. J. Benwell, S. J. Atkinson, J. Lambert, R. T. Johnson and S. D. Robinson, Front. Cell Dev. Biol., 2020, 8, 395 CrossRef PubMed.
- M. Irigoyen, E. Anso, E. Salvo, J. Dotor de las Herrerias, J. J. Martinez-Irujo and A. Rouzaut, Cell. Mol. Life Sci., 2008, 65, 2244–2255 CrossRef CAS PubMed.
- S. Lutter, S. Xie, F. Tatin and T. Makinen, J. Cell Biol., 2012, 197, 837–849 CrossRef CAS PubMed.
- T. Makinen, T. Veikkola, S. Mustjoki, T. Karpanen, B. Catimel, E. C. Nice, L. Wise, A. Mercer, H. Kowalski, D. Kerjaschki, S. A. Stacker, M. G. Achen and K. Alitalo, EMBO J., 2001, 20, 4762–4773 CrossRef CAS PubMed.
- M. Mitsi, M. M. Schulz, E. Gousopoulos, A. M. Ochsenbein, M. Detmar and V. Vogel, PLoS One, 2015, 10, e0145210 CrossRef PubMed.
- A. Sabine, Y. Agalarov, H. Maby-El Hajjami, M. Jaquet, R. Hagerling, C. Pollmann, D. Bebber, A. Pfenniger, N. Miura, O. Dormond, J. M. Calmes, R. H. Adams, T. Makinen, F. Kiefer, B. R. Kwak and T. V. Petrova, Dev. Cell, 2012, 22, 430–445 CrossRef CAS PubMed.
- E. Garrafa, L. Trainini, A. Benetti, E. Saba, L. Fezzardi, B. Lorusso, P. Borghetti, T. Bottio, E. Ceri, N. Portolani, S. Bonardlli, S. M. Giulini, G. Annibale, A. Corradi, L. Imberti and A. Caruso, Lymphology, 2005, 38, 159–166 CAS.
- X. Zhang, J. E. Groopman and J. F. Wang, J. Cell Physiol., 2005, 202, 205–214 CrossRef CAS PubMed.
- J. F. Wang, X. F. Zhang and J. E. Groopman, J. Biol. Chem., 2001, 276, 41950–41957 CrossRef CAS PubMed.
- T. Dietrich, J. Onderka, F. Bock, F. E. Kruse, D. Vossmeyer, R. Stragies, G. Zahn and C. Cursiefen, Am. J. Pathol., 2007, 171, 361–372 CrossRef CAS PubMed.
- N. E. Vlahakis, B. A. Young, A. Atakilit and D. Sheppard, J. Biol. Chem., 2005, 280, 4544–4552 CrossRef CAS PubMed.
- E. Bazigou, S. Xie, C. Chen, A. Weston, N. Miura, L. Sorokin, R. Adams, A. F. Muro, D. Sheppard and T. Makinen, Dev. Cell, 2009, 17, 175–186 CrossRef CAS PubMed.
- F. Tatin, A. Taddei, A. Weston, E. Fuchs, D. Devenport, F. Tissir and T. Makinen, Dev. Cell, 2013, 26, 31–44 CrossRef CAS PubMed.
- A. M. Foskett, U. R. Ezekiel, J. P. Trzeciakowski, D. C. Zawieja and M. Muthuchamy, Front. Physiol., 2011, 2, 103 CAS.
- B. Sauter, D. Foedinger, B. Sterniczky, K. Wolff and K. Rappersberger, J. Histochem. Cytochem., 1998, 46, 165–176 CrossRef CAS PubMed.
- T. Kono, H. Kubo, C. Shimazu, Y. Ueda, M. Takahashi, K. Yanagi, N. Fujita, T. Tsuruo, H. Wada and J. K. Yamashita, Arterioscler., Thromb., Vasc. Biol., 2006, 26, 2070–2076 CrossRef CAS PubMed.
- E. Garrafa, G. Alessandri, A. Benetti, D. Turetta, A. Corradi, A. M. Cantoni, E. Cervi, S. Bonardelli, E. Parati, S. M. Giulini, B. Ensoli and A. Caruso, J. Cell Physiol., 2006, 207, 107–113 CrossRef CAS PubMed.
- P. S. Yoo, A. L. Mulkeen, A. Dardik and C. H. Cha, J. Surg. Res., 2007, 143, 94–98 CrossRef PubMed.
- F. Laco, M. H. Grant and R. A. Black, J. Biomed. Mater. Res., Part A, 2013, 101, 1787–1799 CrossRef PubMed.
- T. Ishikawa, Z. Wondimu, Y. Oikawa, G. Gentilcore, R. Kiessling, S. Egyhazi Brage, J. Hansson and M. Patarroyo, Matrix Biol., 2014, 38, 69–83 CrossRef CAS PubMed.
- C. Schuster, M. Mildner, A. Botta, L. Nemec, R. Rogojanu, L. Beer, C. Fiala, W. Eppel, W. Bauer, P. Petzelbauer and A. Elbe-Burger, Am. J. Pathol., 2015, 185, 2563–2574 CrossRef PubMed.
- D. Hanjaya-Putra, V. Bose, Y. I. Shen, J. Yee, S. Khetan, K. Fox-Talbot, C. Steenbergen, J. A. Burdick and S. Gerecht, Blood, 2011, 118, 804–815 CrossRef CAS PubMed.
- D. Hanjaya-Putra, K. T. Wong, K. Hirotsu, S. Khetan, J. A. Burdick and S. Gerecht, Biomaterials, 2012, 33, 6123–6131 CrossRef CAS PubMed.
- M. T. Ngo and B. A. Harley, Adv. Healthc. Mater., 2017, 6, 1700687 CrossRef PubMed.
- L. Sherman, J. Sleeman, P. Herrlich and H. Ponta, Curr. Opin. Cell Biol., 1994, 6, 726–733 CrossRef CAS PubMed.
- S. Ibrahim and A. Ramamurthi, J. Tissue Eng. Regener. Med., 2008, 2, 22–32 CrossRef CAS PubMed.
- D. C. West, I. N. Hampson, F. Arnold and S. Kumar, Science, 1985, 228, 1324–1326 CrossRef CAS PubMed.
- P. W. Noble, Matrix Biol., 2002, 21, 25–29 CrossRef CAS PubMed.
- E. Turley, M. Hossain, T. Sorokan, L. Jordan and J. Nagy, Glia, 1994, 12, 68–80 CrossRef CAS PubMed.
- A. Aruffo, I. Stamenkovic, M. Melnick, C. B. Underhill and B. Seed, Cell, 1990, 61, 1303–1313 CrossRef CAS PubMed.
- M. Yu, H. Zhang, Y. Liu, Y. He, C. Yang, Y. Du, M. Wu, G. Zhang and F. Gao, Exp. Cell Res., 2015, 336, 150–157 CrossRef CAS PubMed.
- M. Wu, Y. Du, Y. Liu, Y. He, C. Yang, W. Wang and F. Gao, PLoS One, 2014, 9, e92857 CrossRef PubMed.
- C. H. Antoni, Y. McDuffie, J. Bauer, J. P. Sleeman and H. Boehm, Front. Bioeng. Biotechnol., 2018, 6, 25 CrossRef PubMed.
- X. Pang, W. Li, L. Chang, J. E. Gautrot, W. Wang and H. S. Azevedo, ACS Appl. Mater. Interfaces, 2021, 13, 25792–25804 CrossRef CAS PubMed.
- S. Lee, S. Kim, J. Park and J. Y. Lee, Int. J. Biol. Macromol., 2020, 151, 1314–1321 CrossRef CAS PubMed.
- D. P. Jeong, E. Hall, E. Neu and D. Hanjaya-Putra, Cell. Mol. Bioeng., 2022, 15, 467–478 CrossRef CAS PubMed.
- L. Bui, S. Edwards, E. Hall, L. Alderfer, K. Round, M. Owen, P. Sainaghi, S. Zhang, P. D. Nallathamby, L. S. Haneline and D. Hanjaya-Putra, Commun. Biol., 2022, 5, 635 CrossRef CAS PubMed.
- K. M. Varberg, R. O. Garretson, E. K. Blue, C. Chu, C. R. Gohn, W. Tu and L. S. Haneline, Am. J. Physiol.: Cell Physiol., 2018, 315, C502–C515 CrossRef CAS PubMed.
- M. Wagner, B. Hampel, D. Bernhard, M. Hala, W. Zwerschke and P. Jansen-Dürr, Exp. Gerontol., 2001, 36, 1327–1347 CrossRef CAS PubMed.
- J. L. Zhang, J. M. Patel and E. R. Block, Mech. Ageing Dev., 2002, 123, 613–625 CrossRef CAS PubMed.
- Y. K. Hong, N. Harvey, Y. H. Noh, V. Schacht, S. Hirakawa, M. Detmar and G. Oliver, Dev. Dyn., 2002, 225, 351–357 CrossRef CAS PubMed.
- J. T. Wigle and G. Oliver, Cell, 1999, 98, 769–778 CrossRef CAS PubMed.
- J. Kang, J. Yoo, S. Lee, W. Tang, B. Aguilar, S. Ramu, I. Choi, H. H. Otu, J. W. Shin, G. P. Dotto, C. J. Koh, M. Detmar and Y. K. Hong, Blood, 2010, 116, 140–150 CrossRef CAS PubMed.
- S. Lee, I. Choi and Y. K. Hong, Semin. Thromb. Hemostasis, 2010, 36, 352–361 CrossRef PubMed.
- W. Ma and G. Oliver, Physiology, 2017, 32, 444–452 CrossRef CAS PubMed.
- D. G. Jackson, R. Prevo, S. Clasper and S. Banerji, Trends Immunol., 2001, 22, 317–321 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Cho, J. Kim, J. H. Ahn, Y.-K. Hong, T. Mäkinen, D.-S. Lim and G. Y. Koh, Circ. Res., 2019, 124, 225–242 CrossRef CAS PubMed.
- J. W. Robering, A. Weigand, R. Pfuhlmann, R. E. Horch, J. P. Beier and A. M. Boos, J. Cell. Mol. Med., 2018, 22, 3740–3750 CrossRef CAS PubMed.
- S. Amatschek, E. Kriehuber, W. Bauer, B. Reininger, P. Meraner, A. Wolpl, N. Schweifer, C. Haslinger, G. Stingl and D. Maurer, Blood, 2007, 109, 4777–4785 CrossRef CAS PubMed.
- K. L. Jablon, V. L. Akerstrom, M. Li, S. E. Braun, C. E. Norton and J. A. Castorena-Gonzalez, Microcirculation, 2023, 30, e12778 CrossRef CAS PubMed.
- S. F. Schoppmann, A. Soleiman, R. Kalt, Y. Okubo, C. Benisch, U. Nagavarapu, G. S. Herron and S. Geleff, Microcirculation, 2004, 11, 261–269 CrossRef CAS PubMed.
- L. A. Johnson, S. Banerji, W. Lawrance, U. Gileadi, G. Prota, K. A. Holder, Y. M. Roshorm, T. Hanke, V. Cerundolo, N. W. Gale and D. G. Jackson, Nat. Immunol., 2017, 18, 762–770 CrossRef CAS PubMed.
- D. G. Jackson, Matrix Biol., 2019, 78–79, 219–235 CrossRef CAS PubMed.
- P. R. Hess, D. R. Rawnsley, Z. Jakus, Y. Yang, D. T. Sweet, J. Fu, B. Herzog, M. Lu, B. Nieswandt, G. Oliver, T. Makinen, L. Xia and M. L. Kahn, J. Clin. Invest., 2014, 124, 273–284 CrossRef CAS PubMed.
- R. Bianchi, E. Russo, S. B. Bachmann, S. T. Proulx, M. Sesartic, N. Smaadahl, S. P. Watson, C. D. Buckley, C. Halin and M. Detmar, Arterioscler., Thromb., Vasc. Biol., 2017, 37, 108–117 CrossRef CAS PubMed.
- D. P. Jeong, D. Montes, H. C. Chang and D. Hanjaya-Putra, Phys. Biol., 2023, 20, 045004 CrossRef PubMed.
- L. A. Johnson, S. Banerji, W. Lawrance, U. Gileadi, G. Prota, K. A. Holder, Y. M. Roshorm, T. Hanke, V. Cerundolo, N. W. Gale and D. G. Jackson, Nat. Immunol., 2017, 18, 762–770 CrossRef CAS PubMed.
- D. G. Jackson, Matrix Biol., 2019, 78–79, 219–235 CrossRef CAS PubMed.
- S. Y. Li, D. D. Mruk and C. Y. Cheng, Spermatogenesis, 2013, 3, e25385 CrossRef PubMed.
- M. Schober, S. Raghavan, M. Nikolova, L. Polak, H. A. Pasolli, H. E. Beggs, L. F. Reichardt and E. Fuchs, J. Cell Biol., 2007, 176, 667–680 CrossRef CAS PubMed.
- D. Lachowski, E. Cortes, B. Robinson, A. Rice, K. Rombouts and A. E. Del Rio Hernandez, FASEB J., 2018, 32, 1099–1107 CrossRef CAS PubMed.
- G. Nardone, J. Oliver-De La Cruz, J. Vrbsky, C. Martini, J. Pribyl, P. Skladal, M. Pesl, G. Caluori, S. Pagliari, F. Martino, Z. Maceckova, M. Hajduch, A. Sanz-Garcia, N. M. Pugno, G. B. Stokin and G. Forte, Nat. Commun., 2017, 8, 15321 CrossRef CAS PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 CrossRef CAS PubMed.
- B. Cha, S. Moon and W. Kim, BMB Rep., 2021, 54, 285–294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00873h |
| This journal is © The Royal Society of Chemistry 2023 |