
Personalized demand-responsive biphasic microneedle patch for smart drug administration†
Shuyue
Deng‡
a,
Yao
Shuai‡
b,
Shibo
Zhang
a,
Caixia
Sun
a,
Lei
Chang
c,
Jie
Xu
bd,
Ling
Tong
b,
Qunsheng
Ji
b,
Min
Li
d,
Jianjun
Dai
*ae and
Yanmin
Ju
 *a
*a
aCollege of Pharmacy, China Pharmaceutical University, Nanjing 211198, China. E-mail: jjdai@cpu.edu.cn; juyanmin@cpu.edu.cn
bOncology and Immunology Unit, WuXi AppTec, Nantong 226000, China
cDepartment of Cellular and Molecular Medicine, University of California San Diego School of Medicine, La Jolla, CA, USA
dSchool of Computer Science and Engineering, Central South University, Changsha 410006, China
eMOE Joint International Research Laboratory of Animal Health and Food Safety, Key Laboratory of Animal Bacteriology, Ministry of Agriculture, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, China
First published on 23rd June 2023
Abstract
Many patients, especially those with chronic diseases, would benefit from personalized drugs that could modulate the treatment regimen. Tailored drug delivery via microneedle patches (MNPs) has emerged as a promising technology to address this problem. However, it is still difficult to modulate the treatment regimen in one MNP. Here, multiple treatment regimens were achieved by the same MNP functionalized with modifiable nanocontainers (NCs). The MNPs were biphasic in design, resulting in approximately a twice as high drug loading capacity than that of traditional dissolving MNPs. The drug-loaded NCs could have a zero-order release rate for at least 20 d in vitro. Furthermore, three model MNPs, Type-A (100% drug), Type-B (50% drug and 50% NCs) and Type-C (100% NCs) were generated to simulate various personalized dosing needs. In vivo application of these models could achieve effective therapeutic drug concentrations in the first 12 h and adjusted the duration of effective drug action from 24 h to 96 h and 144 h, respectively, with outstanding biocompatibility. These findings indicate that this device holds significant promise for personalized drug delivery.
Introduction
Conventional drug manufacture for mass production is based on certain specific parameters, including dose intensity, size, shape, and release type.1 However, many drugs, such as anti-tumor drugs, anti-hypertensive drugs, and contraceptives, require multiple medication regimens that depend on the needs of an individual patient, along with medication combinations and the discontinuation of medication.2–4 It is obvious that such mass-produced drugs with a fixed dose intensity would fail to meet the necessary requirements. A personalized drug delivery system (PDDS), also known as an on-demand drug delivery system, provides precise control with regards to the timing, magnitude, and duration of drug release, based on the customized requirements to achieve the desired therapeutic effects.1 This represents a novel drug delivery method and a promising approach with which to address the weaknesses of mass-produced drugs. However, research on personalized drug delivery is still scarce at this stage.5,6Of the currently available smart drug delivery devices, microneedle patches (MNPs) have emerged as a promising candidate for PDDS.7–10 In contrast to other methods currently used for personalized drug delivery, MNPs could be produced in different shapes and tunable dimensions and could be self-administered.11–16 In a previous study, Donnelly et al. developed a dissolving poly(vinyl alcohol) microneedle array for the rapid delivery of Nestorone, which could attain a maximum blood concentration (Cmax of 32 ± 14 ng mL−1) within one hour of skin penetration.17 In another study, Guo et al. used a gelatin/hyaluronic acid particle mixture to load long-acting insulin, which achieved the successful long-term delivery of insulin to address increasing blood glucose levels and effectively reduced fluctuations.18 The results arose from studies relating to person-centric drug delivery and are encouraging; however, these systems still do not allow the adjustable release behavior of the same drug in one MNP, such as how fast or slow the drug is released, or how much, and do not correspond to the true sense of PDDS. It would be far more useful if one MNP could incorporate multiple regimens to achieve modifiable treatments.
Using nanoscale biomaterials to prepare nanomedicine is the other promising approach for regulating drug release behaviors.19,20 In particular, polycaprolactone (PCL), an FDA-approved biodegradable material, has been widely reported for encapsulating drugs to control drug release.21,22 Meanwhile, poloxamer 188 (F68), as an FDA-approved pore-forming agent, could be combined with PCL to prepare PCL/F68 nanoparticles with pores on the surface of the PCL membranes. To achieve a controllable drug release, the number of pores on the PCL/F68 NCs could be regulated by changing the mass ratios of PCL and F68. In a previous study, Ma et al. developed PCL nanoparticles with the antitumor drug paclitaxel for cancer therapy and incorporated different amounts of F68 (from 0% to 40%) in PCL nanoparticles to regulate the drug release rates. The obtained cumulative drug release rates for these nanoparticles ranged from 20% to 40%.23 However, these nanomaterials were always administered intravenously, which not only required professionals, but were also invasive and not conducive to personalized self-administration by patients.19–23 Therefore, a simpler drug delivery method is urgently needed. The introduction of changeable nanomedicines into MNPs is expected to provide improved regulatory effects with regards to differential drug release from the same MNP to facilitate personalized self-administration.
Herein, we developed a personalized biphasic MNP with drug-loaded nanocontainers (NCs) which had the potential to meet changeable medication requirements (Fig. 1). The biphasic MNP, comprising biocompatible hydrophilic PVP24,25 microneedles and a hydrophobic PS base,26,27 demonstrated a superior drug loading capacity and enabled invisible drug delivery through a rapid separation. Next, we used a contraceptive hormone (levonorgestrel (LNG)) as a model drug to verify the feasibility of the system, considering that patients tend to adjust the dosing frequency according to intention and the duration of contraception.28–30 We prepared LNG@PCL/F68 NCs (LPF NCs) with different concentrations of F68 (0%, 40% and 60%) in PCL. The cumulative release rates of LNG from these NCs improved from 68.4% to 85.4%, thus indicating that changing the concentration of F68 could modulate the kinetics of drug release. Since the needs of individuals are too diverse to be fully simulated experimentally, we used three types of MNPs with the same drug dose to simulate typical dosing needs to verify the feasibility of the PDDS: Type-A (100% LNG), Type-B (50% LNG and 50% LPF NCs) and Type-C (100% LPF NCs). In vitro, Type-A reached a plateau of drug release in two days. Interestingly, Type-B and Type-C maintained a controlled rate of drug release with different release kinetics for at least 20 days, thus demonstrating that the release of drugs from the NCs was both modifiable and continuous. The application of the three types of MNPs achieved effective therapeutic drug concentrations in the first 12 h and adjusted the therapeutic effect from 24 h to 96 h and 144 h in model rats, respectively, thus facilitating a variation in medicine demands. In summary, this device provides a potentially convenient strategy for modifiable drug delivery.
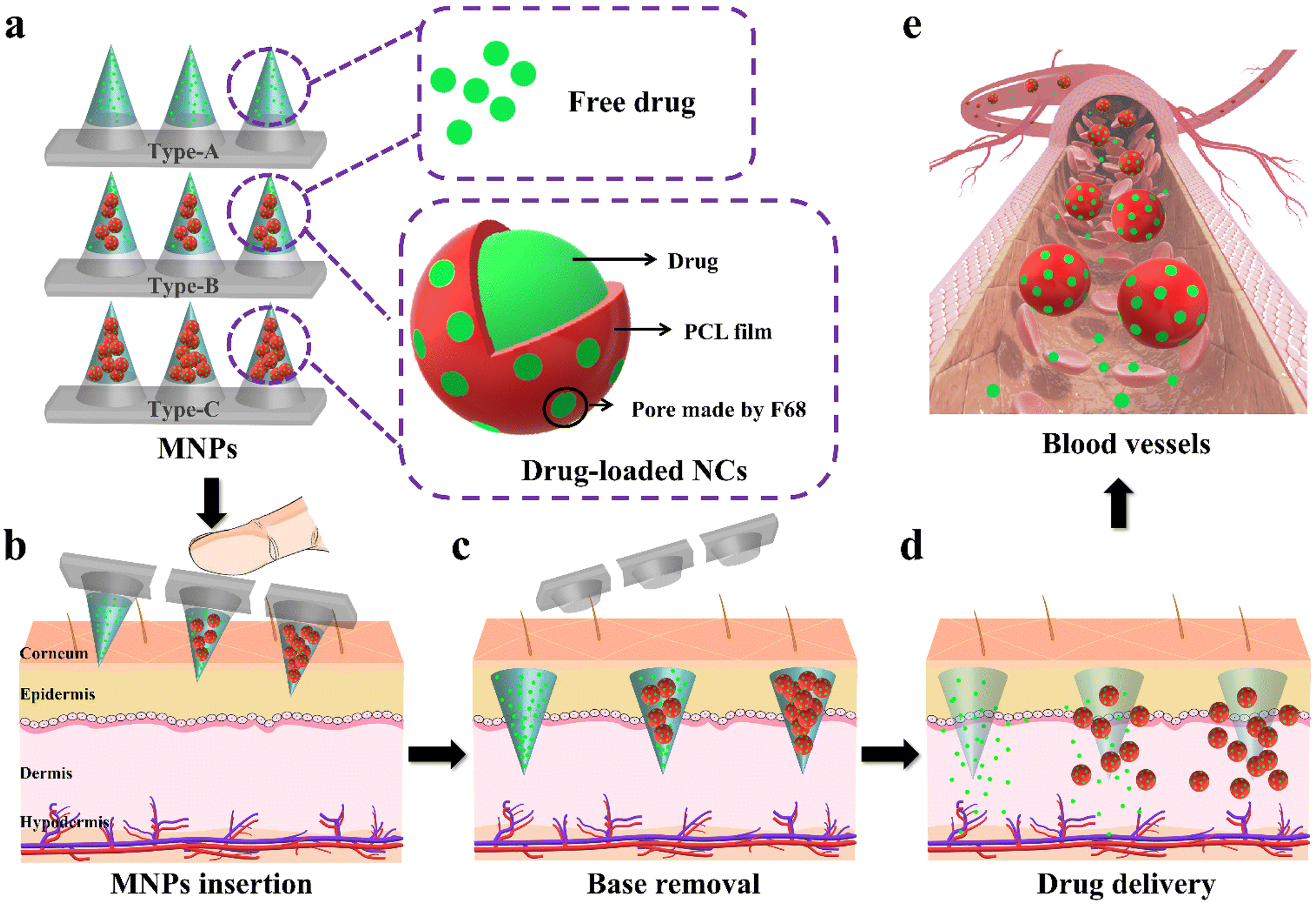 | ||
| Fig. 1 Design and fabrication of multiple biphasic MNPs with tunable drug release behaviors. (a) Schematic diagram of three types of biphasic MNPs (Type-A, Type-B and Type-C) simulating different medication requirements; the magnification views in the box are free drug and drug-loaded NCs. (b) Insertion of MNPs into skin by finger pressure. (c) Immediate separation of the bases and microneedles of MNPs after insertion. (d) Drug and drug-loaded NCs release from the MNPs. (e) Continuous release of drug into the blood circulation through F68 pores of drug-loaded NCs. | ||
Experimental section
Fabrication of drug-loaded MNPs
All MNPs were prepared using a PDMS mold with arrays of conical holes. The dimensions of the PDMS film were 2 mm. Each array contained 10 × 10 microneedles with a 650 μm center-to-center spacing. Each microneedle had an 800 μm needle height and 300 μm width at the base. A two-step solution casting method was used for the fabrication. Microneedle casting solution contained a 40% (w/v) PVP K30 (Macklin, China) solution loaded with 10 mg of LNG powders (Macklin, China). After that, 50 μL of the prepared solution was applied by pipette to the PDMS mold, which was centrifuged for 20 minutes at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to fill the microcavities of the PDMS mold. Superfluous solution left on the surface of the PDMS mold was pipetted away. Then, to cast the hydrophobic base layer, the filled mold was filled with 200 μL of 30% (w/v) PS (J&K Chemical, China) in 1,4-dioxane. After that, the final formulation was dried overnight at room temperature. After desiccation was completed, the microneedle arrays were carefully detached from the PDMS molds with tape and stored in a desiccator until use. In some cases, the MNPs were prepared for comparing water-soluble and water-insoluble bases using the same method as above, except that PS was replaced by PVP in the base layer for the water-soluble base. Also, rhodamine B (R-B) (Aladdin, China) and coumarin 6 (C-6) (Aladdin, China) were used as tracers in the microneedles and base, respectively.
000 rpm to fill the microcavities of the PDMS mold. Superfluous solution left on the surface of the PDMS mold was pipetted away. Then, to cast the hydrophobic base layer, the filled mold was filled with 200 μL of 30% (w/v) PS (J&K Chemical, China) in 1,4-dioxane. After that, the final formulation was dried overnight at room temperature. After desiccation was completed, the microneedle arrays were carefully detached from the PDMS molds with tape and stored in a desiccator until use. In some cases, the MNPs were prepared for comparing water-soluble and water-insoluble bases using the same method as above, except that PS was replaced by PVP in the base layer for the water-soluble base. Also, rhodamine B (R-B) (Aladdin, China) and coumarin 6 (C-6) (Aladdin, China) were used as tracers in the microneedles and base, respectively.
Preparation of LPF NCs
The NCs were formed by using a solvent evaporation method. In summary, 100 mg of PCL, 10 mg of LNG and different contents of F68 (0, 40 and 80 mg) were dissolved in 5 mL of dichloromethane (DCM). The DCM solution was added to 15 mL of a 5% (w/v) PVA (Aladdin, China) aqueous solution and sonicated for 30 minutes in an ice bath to form the O/W emulsion. The DCM in the emulsion was removed by evaporation with stirring for up to 4 hours. After removal of the DCM, the NCs were collected by centrifugation at 11000 rpm for 40 minutes and washed 3 times with distilled water. Then, the NCs were suspended in 2% mannitol solution and freeze-dried to generate LPF NC powders.
Examination of drug distribution in MNPs
The distribution of the drug in the microneedles was examined by confocal laser scanning microscopy (STELLARIS 8 Confocal, Leica, Germany). R-B loaded in the microneedle casting solution was the tracer of the microneedles. C-6 loaded in the second casting solution was the tracer of the base. A single row of microneedles was cut from each MNP and mounted horizontally under a fluorescence microscope for observation.Transdermal penetration of LNG from MNPs in vitro
The Franz diffusion cell (TK-24, Kaikai Science and Technology, China) was used for the transdermal penetration test. The tips of the MNPs were placed facing the rabbit skin cuticle and pressed down with fingers for 2 minutes. Afterward, the applied rabbit skin was transferred to a receiving cell with a stirrer, with the cuticle facing the supply cell and fixed. The receiving cell contained 3 mL of PBS (pH = 7.4) solution with 25% ethanol as the receiving solution. Subsequently, the receiving cell was placed on the transdermal diffuser, and the temperature was set at 37.0 °C. The speed was set at 200 rpm. The receiving solution was removed at predetermined time intervals (0, 2, 4, 8, 12, 24 and 48 h) as the sample solution to be tested and replenished with the same volume of fresh receiving solution. The sample solution was extracted with DCM and the mobile phase was added. The DCM was evaporated and the LNG amount was assayed by HPLC-UV. HPLC-UV experimental conditions: reversed-phase C18 column (250 mm by 4.6 mm; inner diameter, 5 μm particle size). Acetonitrile and distilled water (7:3 ratio, v/v) formed the mobile phase. The injection volume was 20 μL, with a flow temperature of 30 °C and flow rate of 1 mL min−1. The amount of LNG released from MNPs was calculated and the cumulative release curve was plotted.
LNG release from MNPs in vitro
Different types of MNPs (Type-A, Type-B and Type-C) were placed in dialysis bags with a cut-off molecular weight of 10000, clamped, and the bags were placed into centrifuge tubes with 15 mL of PBS (pH = 7.4) solution with 25% ethanol as the release media. Centrifuge tubes containing dialysis bags were placed in a shaker water bath at 37 °C and shaken at 200 rpm. A certain amount of release medium was taken at predetermined time intervals as sample solutions (0, 0.5, 1, 2, 3, 6, 9, 12, 16, 20, and 24 days) to be tested and replenished with the same volume of fresh release medium. The sample solution was extracted with DCM and the mobile phase was added. The DCM was evaporated and the LNG amount was assayed by HPLC-UV. HPLC-UV experimental conditions: reversed-phase C18 column (250 mm by 4.6 mm; inner diameter, 5 μm particle size). Acetonitrile and distilled water (7:3 ratio, v/v) formed the mobile phase. The injection volume was 20 μL, with a flow temperature of 30 °C and a flow rate of 1 mL min−1. The amount of LNG released from MNPs was calculated and the cumulative release curve was plotted. To determine the LNG release kinetics, the release data were plotted on the following four kinetic models, a first-order model, zero-order model, Higuchi model and Korsmeyer–Peppas model. The model that fit best was used to represent the pattern of LNG released from the MNPs.
The mathematical equations for the release kinetic model are:
Zero-order model:
| F = K × t | (1) |
First-order model:
| ln(1 − F) = −K × t | (2) |
Higuchi model:
| F = K × t0.5 | (3) |
Korsmeyer–Peppas model:
| F = Ktn | (4) |
Contraceptive effect of LNG in vivo after being released from MNPs
Under isoflurane anesthesia, the three types of MNPs were applied to the shaved dorsal surface of adult female Sprague-Dawley (SD) rats (200 g, n = 8 rats per group). Blood samples (∼0.5 mL) were collected from the tail vein at 0, 12, 24, 48, 72, 96, 120 and 144 hours after application of the MNP. The plasma was separated by centrifuging at 100 rpm for 10 minutes and analyzed for LNG concentration using an enzyme-linked immunosorbent assay (Thermo Fisher Scientific) according to the manufacturer's instructions. All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University and approved by the Animal Ethics Committee of School of Pharmacy, China Pharmaceutical University.Statistical analysis
All in vitro experiments were performed in experimental triplicate unless otherwise noted. All in vivo experiments were performed with eight experimental replicates unless otherwise noted. Data are presented as means ± SD.Results and discussion
Fabrication and characterization of biphasic MNPs
Considering that traditional dissolving MNPs cannot avoid drug diffusion between the microneedles and base layer during preparation and application,31–34 biphasic MNPs with PVP microneedles and a PS base were fabricated through a simple two-step casting method (Fig. 2a). In brief, the PVP solution was filled into polydimethylsiloxane (PDMS) mold cavities through centrifugation to form microneedles. Afterward, the PS solution was applied to the mold surface to form the base, and the patch was gently peeled off after the MNP was completely dry. The fabricated biphasic MNP was a 10 × 10 array in a base area of 8.9 mm by 8.9 mm (Fig. 2b). Both optical microscopy images (Fig. 2b) and scanning electron microscopy (SEM) images (Fig. 2c) showed uniformly conical microneedles, each 800 μm in height and 300 μm in bottom diameter. The static water contact angle of the PVP film was 72 ± 2°, which was much smaller than that of the PS film (101 ± 2°), indicating that the PVP base was more easily wetted by water (Fig. 2d).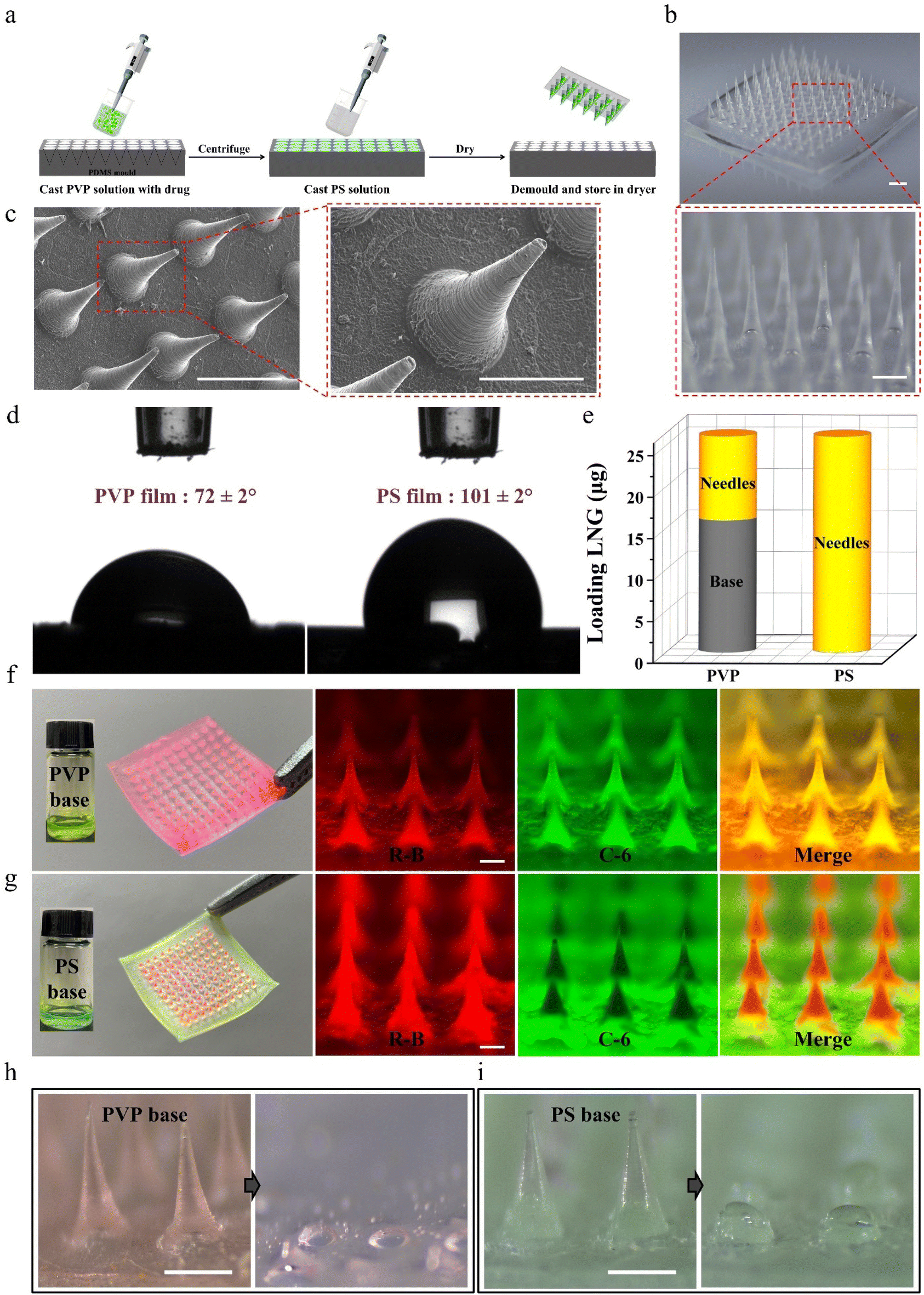 | ||
| Fig. 2 Preparation and characterization of the biphasic MNP. (a) Preparation scheme of drug loaded biphasic MNPs. (b) Optical microscopy images of the MNP. The red dashed box is the corresponding high magnification image. Scale bars are 0.5 mm. (c) SEM images of the MNP. The red dashed box is the corresponding high magnification image. Scale bars are 1 mm. (d) Static water contact angle on the PVP film and PS film. (e) Comparison of drug distribution of the MNPs with the PVP base and PS base (n = 3). (f) Photos and fluorescence microscope images of PVP-based MNPs with dyes loaded. The inset pictures show the original green C-6 casting base solution of PVP. Scale bars are 0.2 mm. (g) Photos and fluorescence microscope images of PS-based MNPs with dyes loaded. The inset pictures show the original green C-6 casting base solution of PS. Scale bars are 0.2 mm. Images comparing the dissolving of PVP-based (h) and PS-based (i) MNPs after being inserted into porcine skin. Scale bars are 0.5 mm. | ||
The effective drug loading between conventional dissolving (PVP-based) MNPs and biphasic (PS-based) MNPs was compared by loading the same concentration of the model drug LNG during the fabrication of PVP microneedles. High performance liquid chromatography (HPLC) was chosen to analyze the content of LNG in MNPs because of its good linearity with a correlation coefficient of 0.9999 (Fig. S1†). The amount of LNG in the microneedles of PVP-based MNPs was 10 ± 1 μg, while that of LNG in the base was 16 ± 2 μg, which would cause a huge drug waste as only two-fifths of the total loaded drug could be injected into skin. Interestingly, the amount of LNG in the microneedles of PS-based MNPs was 27 ± 2 μg, while no LNG was observed in the base, demonstrating that the drug was completely packed in the microneedles (Fig. 2e). Meanwhile, the visible model drugs rhodamine-B (R-B) and coumarin-6 (C-6) were added to the microneedle casting solution and base casting solution to prepare the fluorescent MNPs to observe the drug diffusion between microneedles and base. MNPs with a PVP base and a PS base had a similar morphology under the optical microscope because the same PDMS mold was used in the preparation process (Fig. S2†). However, they showed a different drug distribution in fluorescence microscopy and confocal laser scanning microscopy (CLSM) images. Red R-B and green C-6 were both distributed throughout the microneedles and base of PVP-based MNP, while for PS-based MNP, only red was displayed in the microneedles and only green existed in the base. This clear interface further supported that the biphasic MNP could ensure the complete encapsulation of a drug into microneedles by blocking the drug diffusion effect (Fig. 2f, g and Fig. S3, Videos S1 and S2†). Moreover, PVP-based MNPs showed dissolution of all the microneedles and partial base after being applied to porcine skin, which would create a highly adhesive surface to absorb the partial drug in microneedles and then decrease the drug absorption,26 whereas there was almost no red residue of microneedle dissolution on the green PS base after PS-based MNPs were inserted into the skin because of the hydrophobicity of PS, which facilitated the complete absorption of the drug encapsulated in the microneedles (Fig. 2h, i and Fig. S4†).
These results indicated that the biphasic MNP could not only avoid drug diffusion during the MNP preparation, but also avoid drug adhesion during the MNP application, thus improving the drug delivery efficiency.
Fabrication and characterization of drug-loaded PCL/F68 NCs
To achieve a person-centric customized drug delivery system for PDDS, we selected the hormone LNG, which is commonly used and requires an individualized delivery,28–30 as a model drug and prepared different LNG@PCL/F68 NCs (LPF NCs) for further fabricating MNPs with the ability to satisfy different medication demands. Transmission electron microscope (TEM) images (Fig. 3a, b and Fig. S5†) and scanning electron microscope (SEM) images (Fig. S6†) showed that LNG@PCL NCs displayed a regular spherical shape, whereas LPF NCs displayed a spherical shape with pores on the surface in TEM and drug crystal formation on the surface in SEM, which were caused by the pore-forming effect of F68. The drug encapsulation rates of LNG@PCL NCs and LPF NCs were 74 ± 10% and 70 ± 7% (Fig. 3c). The characteristic stretching band for carbonyl in LNG was found at 1654 cm−1 and the characteristic carbonyl stretching of PCL/F68 NCs was found at 1735 cm−1 in Fourier transform infrared (FTIR) spectroscopy, both of which could be found in LPF NCs (Fig. 3d).35 These results demonstrated the successful LNG loading in NCs. The zeta potential of LPF NCs was −5 ± 1 mV, which was higher than that of LNG@PCL NCs (−8 ± 1 mV) (Fig. S7†), due to non-ionic F68 shifting the shear surface between the PCL/F68 layer and water to a larger distance.21 X-ray diffraction (XRD) of free LNG exhibited characteristic crystalline peaks at 13.95°, 14.60° and 16.13°, which were still observed in the physical mixture sample of LNG and PCL/F68 NCs. However, these peaks disappeared in LPF NCs, indicating that LNG was fully packaged in the PCL/F68 NCs (Fig. 3e).30 Differential scanning calorimetry (DSC) for the melting points of LNG, PCL/F68 and LPF NCs were shown to be at 230 °C, 165 °C and 165 °C, respectively, which similarly indicated that the drug was completely encapsulated in PCL/F68 NCs (Fig. 3f).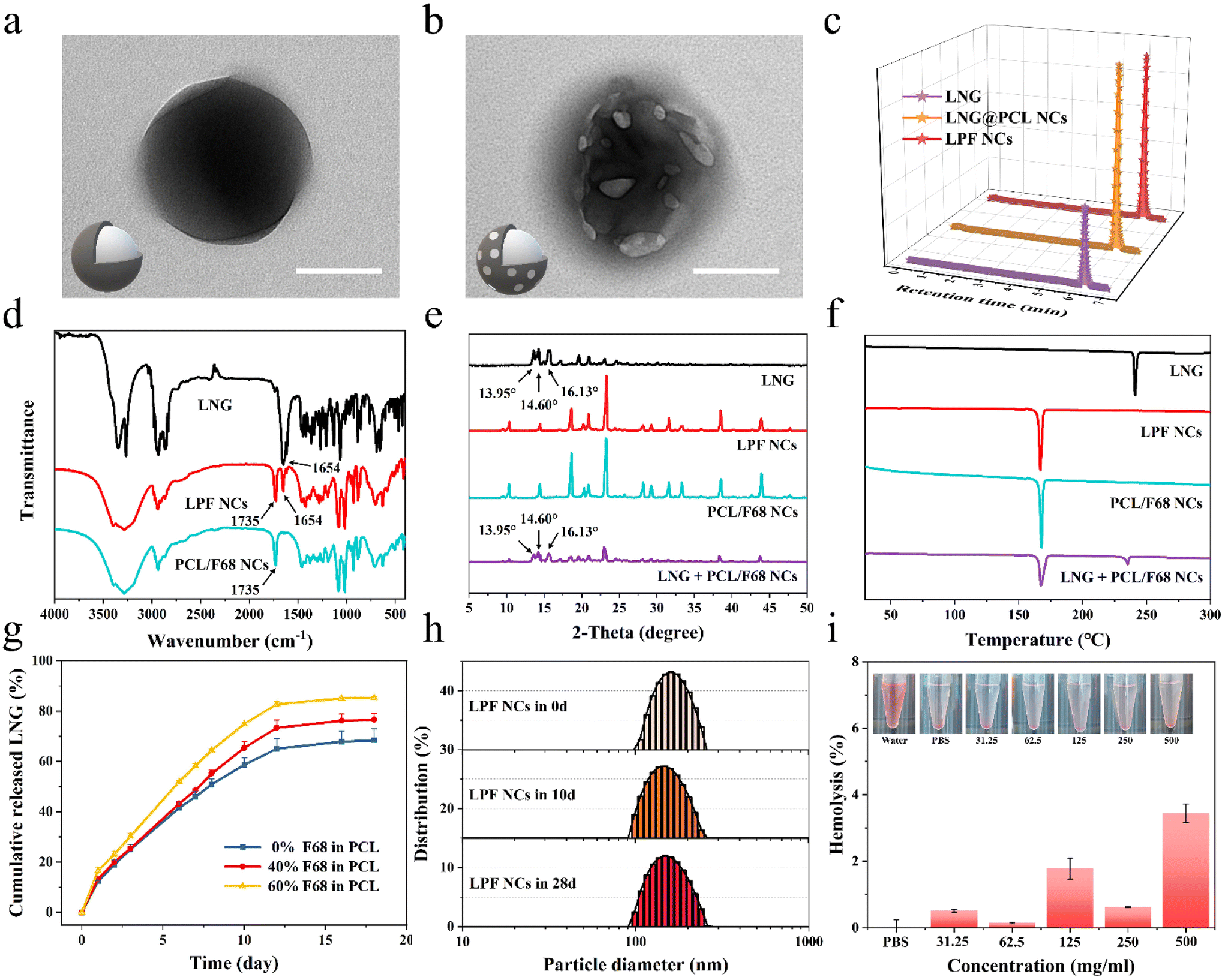 | ||
| Fig. 3 Preparation and characterization of the NCs. TEM images of (a) LNG@PCL NCs and (b) LPF NCs. The inset pictures are schematic diagrams of the corresponding NCs. Scale bars are 100 nm. (c) HPLC of LNG, LNG@PCL NCs and LPF NCs. (d) FTIR spectroscopy of LNG, LPF NCs and PCL/F68 NCs. (e) XRD and (f) DSC of LNG, LPF NCs, PCL/F68 NCs and a mixture of LNG and PCL/F68 NCs. (g) Cumulative LNG release from NCs with different contents of F68 (0%, 40%, and 60%) in PCL in vitro (n = 3). (h) DLS of LPF NCs in 0, 10 and 28 days (n = 3). (i) Hemolysis rates of LPF NCs co-incubated with erythrocytes at different concentrations. The inset pictures show hemolysis of the erythrocytes after co-incubation with different LPF NC concentrations (n = 3). | ||
Drug releasing behaviors from LPF NCs with different F68 amounts (0%, 40%, and 60%) were analyzed by dialysis bags. An initial burst release was observed in all these three LPF NCs during the initial 24 hours before a stable sustained release due to surface-adsorbed, non-encapsulated or loosely bound LNG diffused from the PCL membrane during hydration of the NCs.21 Then, the drug release within these three LPF NCs showed a stable controlled release trend in the next 20 days. The drug release from NCs with different F68 amounts showed a good fit to the Korsmeyer–Peppas release model, indicating that the LNG release was controlled by diffusion and dissolution from the polymeric matrix of PCL/F68 (Fig. S8a–c†). However, their cumulative release rates showed significant differences because of the multiple F68 contents (0%, 40% and 60%) in PCL, at 68.4%, 76.6% and 85.4%, respectively, which confirmed that F68 addition could regulate the drug release rate (Fig. 3g). Meanwhile, the three NCs showed a zero-order release trend in the first 12 days and the rate constant K increased with rising F68 content, which further confirmed that the addition of F68 could increase the pores on the PCL NC surface and regulate the drug release rate (Fig. S8d–f†). LPF NCs with 40% F68 added were chosen as the model NCs for subsequent experiments. The particle size of the LPF NCs was 175.97 nm (PDI = 0.03) on day 0 and 167.48 nm (PDI = 0.07) at day 28 as measured by dynamic light scattering (DLS), and a relatively constant particle size was also observed in LNG@PCL NCs (Fig. 3h and Fig. S9†). This suggested that the produced NCs were uniformly distributed and stable over a long period of time. After testing the hemolysis rates of LPF NCs with different concentrations after co-incubation with erythrocytes for 3 hours, there was no significant hemolysis even when the concentration of LPF NCs was up to 500 mg mL−1, demonstrating the good biocompatibility of the LPF NCs (Fig. 3i). When LPF NCs with different concentrations were incubated with AGS cells for 3 days, more than 90% of the cells could still survive even in the 500 μg mL−1 group, which further demonstrated the safety of the drug-loaded NCs (Fig. S10†). The above results indicated that PCL/F68 NCs could load drugs successfully with a controlled drug release rate, long-term stability and good biocompatibility.
Mechanical properties of drug-loaded MNPs
Next, LPF NCs were integrated into biphasic MNPs for developing user-friendly and on-demand drug-loaded MNPs by the same method mentioned above, after testing the stability of the LPF NCs in the PVP casting solution (Fig. S11 and S12†). To verify the feasibility of the MNPs for transdermal drug delivery, their mechanical properties were tested, including fracture strength and insertion capacity. The fracture strength of the MNPs was first analyzed by loading axial compression from a force test bench (Fig. 4a). The displacement–force curve showed no fracture point even when the force reached the set maximum force of 0.7 N per needle, which was much greater than the force required to penetrate human skin (0.2 N per needle).36 No fracture of the microneedles was observed after this force test, which indicated that the MNPs have sufficient strength to penetrate the skin without breaking (Fig. 4b and c). Parafilms have been widely reported as a standard routine quality control method for MNP mechanical testing.17 As reported, eight-layer parafilms of 127 μm thickness each were used to simulate human skin, and a definite force (10 N) was generated by the force test bench to test the piercing depth of the MNPs (Fig. 4d). Almost 100% of the microneedles penetrated the first two layers and more than 50% of microneedles were able to penetrate the fourth layer (508 μm thickness) with no obvious fractures (Fig. 4e, f and Fig. S13†). Based on the above positive results, the puncture ability of the MNPs was further confirmed. Firstly, a biphasic MNP with dye R-B loaded in the microneedles was applied to a 3% agarose hydrogel for 1 minute, and the base was quickly removed. This showed that the dissolvable microneedles were visually retained in the hydrogel model, with neatly aligned 10 × 10 pinholes after the biphasic microneedles were pierced (Fig. 4g). Next, the R-B MNP was inserted into the skin of Sprague-Dawley (SD) rats. A certain thickness of red fluorescent pinholes was created, which was consistent with the microneedle having a certain height (Fig. 4h). Then, the R-B MNP was inserted into the porcine skin, which is highly similar to human skin.28 The neatly aligned pink pinholes demonstrated the microneedles had the potential to be applied in humans (Fig. 4i). Taken together, the results demonstrated that the biphasic MNPs have the potential to pierce the human stratum corneum and reach the dermis for further effective drug delivery.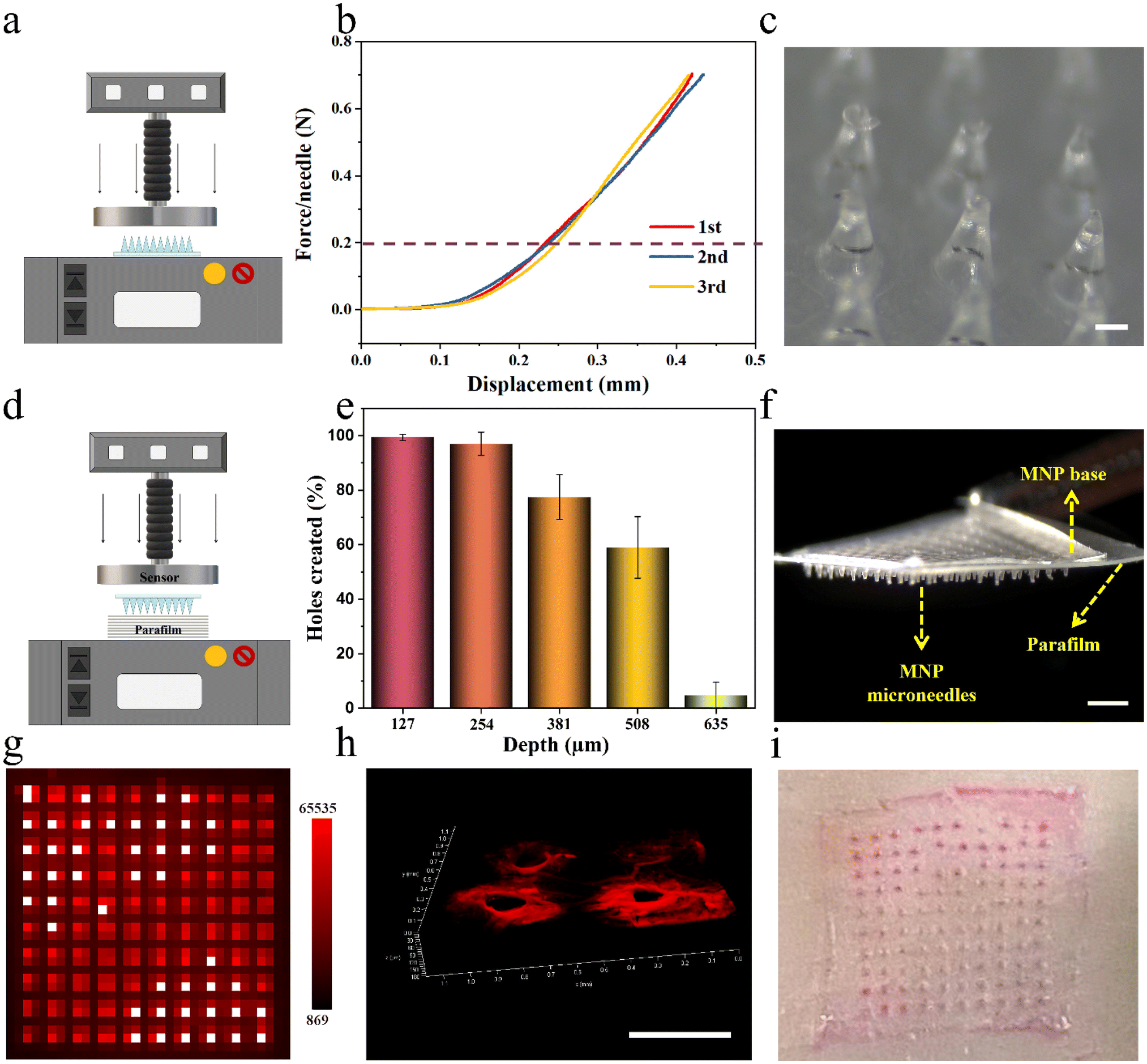 | ||
| Fig. 4 Mechanical properties and drug delivery abilities of drug-loaded MNPs in vitro. (a) Schematic illustration of the mechanical force of the MNPs by a force test bench. (b) Typical force–displacement curves of drug-loaded MNPs. The MNP force required to pierce the human skin is indicated by the purple dashed line. (c) Optical microscope image of the MNP after mechanical force testing. Scale bar is 0.2 mm. (d) Schematic diagram of the MNP piercing into parafilm layers by a force test bench with a compression force of 10 N. (e) Bar graph of the hole numbers created in five parafilm layers after MNP insertion (n = 4). (f) Optical microscope image of MNP puncturing the first layer of parafilm. Scale bar is 0.1 mm. (g) Fluorescence holes created by R-B MNP piercing into agarose hydrogel. (h) CLSM image of fluorescent holes created by R-B MNP on SD rat skin. Scale bar is 0.5 mm. (i) Photo of holes created in porcine skin after R-B MNP insertion. | ||
Drug delivery capabilities of drug-loaded MNPs in vitro
To simulated different needs of individual patients, in the experiments, we chose three MNPs with same dose by altering the ratio of drug and drug-loaded NCs to verify the feasibility of this personalized system, namely Type-A (100% LNG), Type-B (50% LNG and 50% LPF NCs) and Type-C (100% LPF NCs). The three MNPs have similar and sufficient mechanical properties and puncturing abilities for further use because the matrix is the same (Fig. S14†).37,38 The drug transdermal delivery behaviors of the three types MNPs in vitro were examined by Franz diffusion cells (Fig. 5a).39 Free LNG and LPF NCs could be released directly into the diffusion cell, so they would both be detected by HPLC if they were successfully delivered transdermally. The three groups showed a successful delivery of the wrapped cargo at the same rate within 72 hours (Fig. 5b). This was because the transdermal cargo delivery by the MNPs depends on the microneedle matrix17,39 and we prepared the three MNPs with same rapidly dissolvable matrix material-PVP (Fig. S15–S17, and Videos S3 and S4†).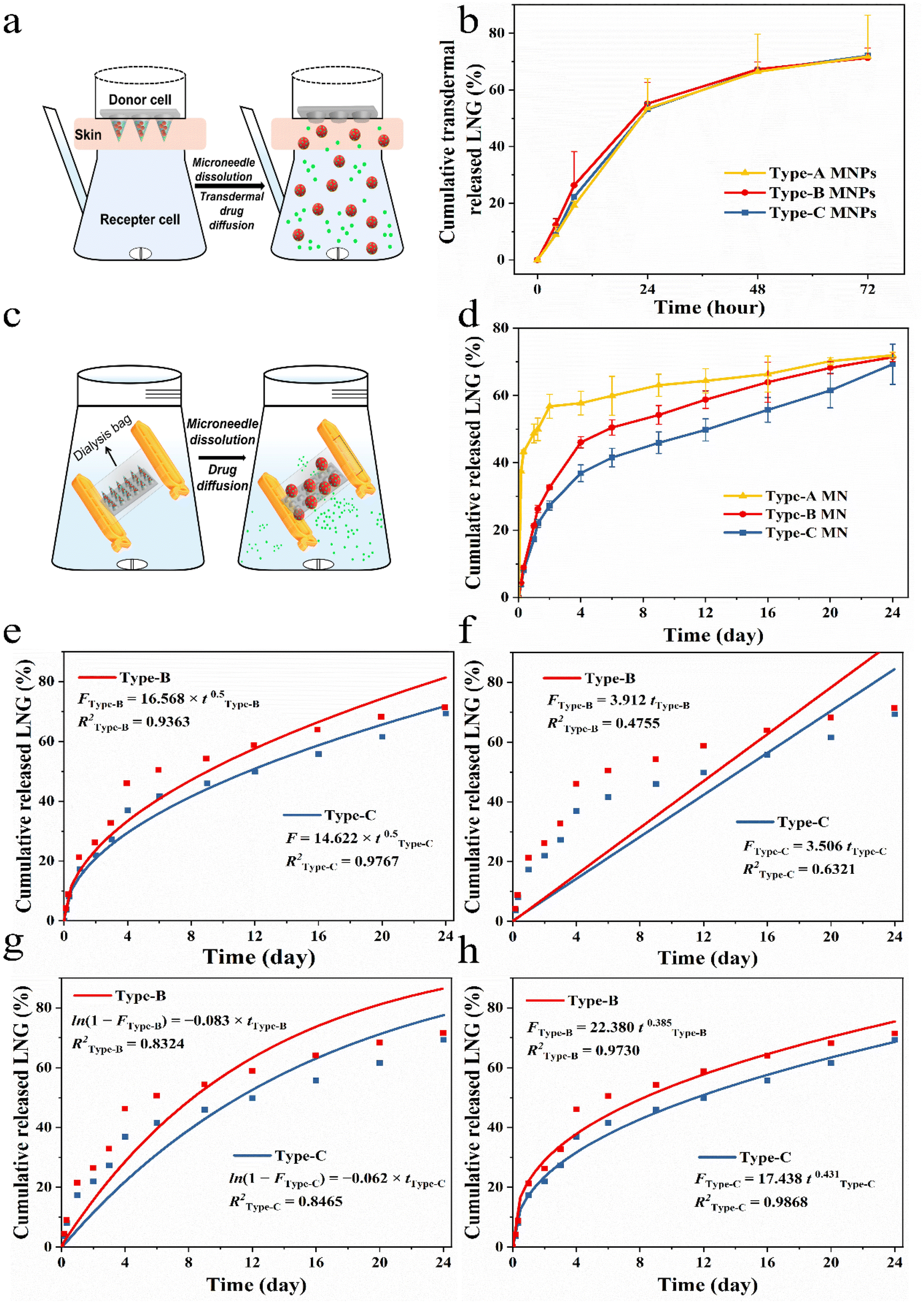 | ||
| Fig. 5 In vitro drug delivery of different MNPs. (a) Schematic diagram of the transdermal drug delivery efficiency of drug-loaded MNPs after puncturing into the skin using a Franz diffusion cell. (b) In vitro transdermal drug delivery profiles of three types of MNPs. (c) Schematic diagram of in vitro drug release from MNPs using dialysis bags (n = 3). (d) Drug release profiles of three types of MNPs (n = 3). (e) Fitting curve of LNG on the first-order model. (f) Fitting curve of LNG on the zero-order model. (g) Fitting curve of LNG on the Higuchi model. (h) Fitting curve of LNG on the Korsmeyer–Peppas model. | ||
Next, the releasing behaviors of the free drug and drug-loaded NCs upon entry into the body were further analyzed by a dialysis bag with a cut-off molecular weight of 10000.18 So, the drug in the NCs could only be detected by HPLC when it was released in the free form and across a dialysis bag (Fig. 5c). The cumulative drug release percentages of the three MNPs were remarkably varied. In the first 4 hours, Type-A showed the fastest drug release rate and released about 40% of the LNG, while Type-C showed the slowest cumulative drug release rate of 3.7% and Type-B demonstrated a release trend between those of Type-A and Type-C. Subsequently, Type-A released 60% of the drug over two days, reaching a plateau, whereas Type-B and Type-C only showed an initial burst LNG release in the first 4 days (Fig. 5d). The initial burst in Type-B and Type-C could be attributed to the rapid PVP dissolution and surface-adsorbed, non-encapsulated or loosely bound LNG diffusion from the membrane surface of PCL during hydration of the NCs.21 In the next 20 days, Type-B and Type-C reached zero-order release, demonstrating that the drug release was controlled and depended on the pore channel action on LPF NCs. The rate constants K of Type-B and Type-C were 1.26 and 1.54, which showed the drug release rate of Type-B was slower than that of Type-C during the controlled release phase. This was due to the number of LPF NCs in Type-B being only half those in Type-C (Fig. S18†).
To further evaluate the drug release kinetics and mechanisms of MNPs with LPF NCs, drug release fitting equations, including the first-order (Fig. 5e), zero-order (Fig. 5f), Higuchi (Fig. 5g), and Korsmeyer–Peppas (Fig. 5h) kinetic models, were used to fit the results. Based on the R2 values of different models. The results suggested that the Korsmeyer–Peppas model fitted the release of LNG from MNPs with LPF NCs best (R2 of Type-B was 0.9730 and R2 of Type-C was 0.9868). These results suggested that release of drug was controlled by diffusion and dissolution from the MNPs with PCL/F68. In summary, the aforementioned MNPs successfully achieved control of the drug release behavior and were beneficial in providing on-demand and stable drug concentrations in vivo.
In vivo transdermal delivery and biocompatibility study of MNPs
Based on the above promising results in vitro, we further used the three types of biphasic MNPs in SD rats to verify their invisible drug delivery ability, contraceptive efficiency and biocompatibility. Firstly, the separation capability of the base from the microneedles was visualized in rats by MNPs with R-B microneedles and a C-6 base. A SD rat was administered the MNP placed on the skin for 1 minute, and then the green base was removed. A clear array of red holes could be seen at the application site in the rat (Video S5†). This again demonstrated the ideal in vivo separation between microneedles and base, showing that biphasic design could achieve an invisible drug delivery for contraception. The invisibility could remain only in the drug-loading microneedles in the body to exert the drug effects and eliminate the discomfort and embarrassment of users wearing MNPs for long periods of time.To assess the LNG pharmacokinetics of different biphasic MNPs in vivo, adult female SD rats were divided into three groups, Type-A MNPs, Type-B MNPs, and Type-C MNPs. Each SD rat was administered with the corresponding MNP and this MNP was kept on the skin for 1 minute before removing the PS base. Blood samples were collected from the rats at predetermined intervals and the LNG concentration in the rat plasma was measured by enzyme-linked immunosorbent assay (ELISA) kits (Fig. 6a). As the results demonstrated, Type-A MNPs showed an effective contraceptive concentration in the first 12 hours, higher than the human therapeutic threshold of 200 pg mL−1, and reached a peak LNG concentration (Cmax) of 470 ± 30 pg mL−1 at 24 hours.40 This then rapidly declined below the human treatment threshold, only maintaining an effective concentration for the first 24 hours. Type-B MNPs reached the human therapeutic threshold upon application to rats immediately within the first 12 hours just like Type-A. Thereafter, the plasma LNG concentration declined gradually, remaining at the human therapeutic threshold for 96 hours. In contrast, Type-C MNPs did not reach the human therapeutic threshold in the initial 12 hours because of the absence of free LNG, reaching Cmax of 350 ± 50 pg mL−1 (mean ± SD) at 48 hours after sustained drug release through LPF NCs, and maintaining the effective drug concentration at the human therapeutic threshold level for at least 144 hours (Fig. 6b). These results suggested that the duration of drug action could be adjusted according to the demand and actual situation (like physiological status and age) of the customers by varying the F68 content in the LPF NCs or the ratio of the drug and tunable NCs in the MNPs. This facilitates the individualization of the desired dosing intervals, the days of drug action and the drug release effective concentration to best satisfy the requirements of PDDS. Also, people of various ages were asked about their preference for different contraceptive methods through a questionnaire. 72.5% of the participants indicated that they preferred individualized on-demand MNPs compared to other contraceptive methods (Fig. 6c). This indicated that an on-demand drug delivery system by MNPs was more acceptable than long-acting contraceptives or short-acting pills in the small population of the reproductive age population studied.
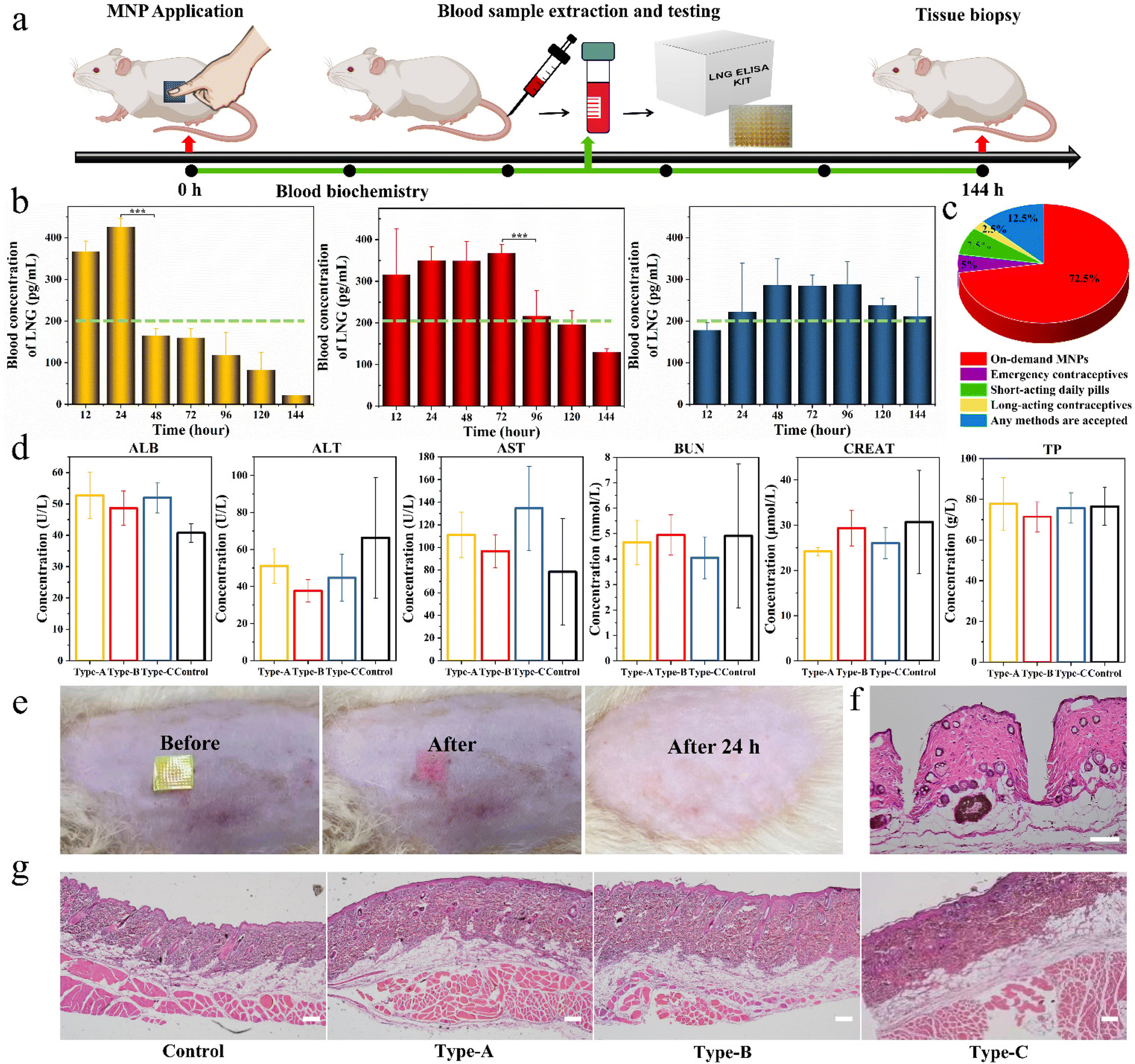 | ||
| Fig. 6 Transdermal delivery profiles and safety of different biphasic MNPs in vivo. (a) Schematic illustration of the experimental design for drug-loaded MNP application. (b) Rat plasma concentrations of the model drug after administration of biphasic Type-A (yellow), Type-B (red) and Type-C (blue) MNPs. The therapeutic LNG level in humans is indicated by the green dashed line. All the data are presented as mean ± SD (n = 8). (c) Preference of human participants for different contraceptive methods (n = 40). (d) Serum biochemical indexes of ALB, ALP, ALT, AST, BUN, CREAT and TP concentrations on day 2 after the application of MNPs. All the data are presented as mean ± SD (n = 8). (e) Representative images of rat skin before and after separation of the base of biphasic MNP. (f) H&E staining image of rat skin penetrated by MNP. Scale bar is 0.1 mm. (g) H&E staining images of skin and subcutaneous adipose tissue after application with different types of MNPs for day 6. Scale bars are 0.2 mm. | ||
The toxicity of the above MNPs to the liver and kidney was evaluated via serum biochemical analysis on day 2 after the application of MNPs, including serum albumin (ALB), alanine aminotransferase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN), creatinine (CREAT) and total protein (TP) levels. No significant differences were observed between different groups and all were within the normal range (Fig. 6d).41 After removal of the green base, a clear patch array of red holes could be seen on the back of the SD rat, and the site could be observed as needle-like holes by a hematoxylin–eosin (H&E) staining analysis, which indicated that MNP successfully pierced into the skin. Also, the red holes at the administration site completely dissolved and disappeared within 24 h, and neither edema nor erythema were observed at the administration site (Fig. 6e and f). After the experiments, the skin at the application site was again analyzed with H&E and the results showed that all the groups were intact without needle-like holes, indicating that the holes initially produced had healed spontaneously. Meanwhile, the administration site showed no changes in inflammatory cells, skin architecture, or other signs of tissue damage, which indicated the fantastic bio-safety of the prepared MNPs (Fig. 6g).
Overall, these results demonstrated that the biphasic MNP could rapidly separate base from the skin, achieving an effective contraceptive concentration multiple times and having an excellent biocompatibility in vivo.
Conclusion
In this work, we have fabricated biphasic MNPs with an adjustable drug release ability for diverse personalized medication needs by regulating the ratio of drug and NCs. The biphasic design could encapsulate the drug 100% in microneedles and allow an immediate separation between the microneedles and the base for invisible drug delivery, which eliminates the discomfort and social embarrassment of users. Then, three model MNPs, Type-A (100% LNG), Type-B (50% LNG and 50% LPF NCs) and Type-C (100% LPF NCs) were used to verify the feasibility of this personalized system. In vitro, Type-A reached a plateau rapidly within the first two days. However, Type-B and Type-C could keep zero-order release at different rates for at least 20 days. In vivo, Type-A could rapidly reach the effective human therapeutic threshold after administration and stay active for only 24 hours. Type-B and Type-C could adjust the effective drug duration from 24 hours to 96 and 144 hours, respectively, to meet the various needs of users. The novel PDDS is user-friendly and valuable for effectively delivering drugs for patients with various needs and preferences.Author contributions
S. D. conceived and fabricated the MNPs, contributed to the characterization of the MNPs, participated in animal experiments and analyzed the results. J. D. and Y. J. led and supervised the project. Y. S., J. X., L. T., Q. J., M. L., and J. D. provide administrative, technical, or material support. S. D., Y. S., J. X., L. T., Q. J., and M. L. conducted rat animal experiments and data analysis. S. Z. and C. S. contributed to the data analysis. S. D., Y. S., and Y. J. discussed the results. S. D., and Y. J. contributed to the manuscript writing. All the authors have read and approved the final manuscript.Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52002402 and 32172855) and Natural Science Foundation of Jiangsu Province (Grant No. BK20200574). The authors acknowledge all the colleagues in the Oncology and Immunology Unit, WuXi AppTec, for their kind help.References
- D. Raijada, K. Wac, E. Greisen, J. Rantanen and N. Genina, Adv. Drug Delivery Rev., 2021, 176, 113857 CrossRef CAS PubMed.
- M. W. Wilson, L. Martini and A. Clarke, Novel manufacturing technologies for the production of patient-centric drug products, Springer International Publishing, 2016, vol. 26, pp. 485–515 Search PubMed.
- K. Goutsouliak, J. Veeraraghavan, V. Sethunath, C. De Angelis, C. K. Osborne, M. F. Rimawi and R. Schiff, Nat. Rev. Clin. Oncol., 2020, 17, 233–250 CrossRef PubMed.
- P. Suryavanshi, V. S. Chaudhari and S. Banerjee, Drug Delivery Transl. Res., 2022, 12, 758–791 CrossRef PubMed.
- J. Zhou, A. V. Kroll, M. Holay, R. H. Fang and L. Zhang, Adv. Mater., 2020, 32, e1901255 CrossRef PubMed.
- A. Radhakrishnan, G. Kuppusamy, S. Ponnusankar and N. K. Shanmukhan, Pharmacogenomics J., 2020, 20, 1–18 CrossRef CAS PubMed.
- J. Yang, J. Yang, X. Gong, Y. Zheng, S. Yi, Y. Cheng, Y. Li, B. Liu, X. Xie, C. Yi and L. Jiang, Adv. Healthcare Mater., 2022, 11, e2102547 CrossRef PubMed.
- B. Cai, Y. Gong, Z. Wang, L. Wang and W. Chen, Theranostics, 2021, 11, 10012–10029 CrossRef CAS PubMed.
- J. H. Gong, L. J. Chen, X. Zhao and X. P. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 17142–17152 CrossRef CAS PubMed.
- M. Lu, L. Fan, H. Chen, Z. Zhang and Y. Zhao, Small, 2022, 18, e2201889 CrossRef PubMed.
- D. M. Selene, B. M. Napolitano, F. P. Antonio, S. Luigia, A. Eugenio, M. Alfonso, D. G. Paolo, S. Antonino, D. Principia, D. S. Luca and D. I. Stefania, Biomater. Adv., 2022, 142, 213169 CrossRef PubMed.
- A. H. Sabri, Y. Kim, M. Marlow, D. J. Scurr, J. Segal, A. K. Banga, L. Kagan and J. B. Lee, Adv. Drug Delivery Rev., 2020, 153, 195–215 CrossRef CAS PubMed.
- M. Guo, Y. Wang, B. Gao and B. He, ACS Nano, 2021, 15, 15316–15327 CrossRef CAS PubMed.
- W. Li, J. Y. Chen, R. N. Terry, J. Tang, A. Romanyuk, S. P. Schwendeman and M. R. Prausnitz, J. Controlled Release, 2022, 347, 489–499 CrossRef CAS PubMed.
- Y. J. Chen, Y. P. Hsu, Y. L. Tain, N. S. Li, H. H. Pang, S. W. Kuo and H. W. Yang, Biosens. Bioelectron., 2022, 208, 114234 CrossRef CAS PubMed.
- P. P. Pan, Q. Liu, L. Wang, C. X. Wang, L. Hu, Y. J. Jiang, Y. H. Deng, G. S. Li and J. D. Chen, Adv. NanoBiomed Res., 2023, 3, 2200126 CrossRef CAS.
- E. Altuntaş, I. A. Tekko, L. K. Vora, N. Kumar, R. Brodsky, O. Chevallier, E. McAlister, Q. Kurnia Anjani, H. O. McCarthy and R. F. Donnelly, Int. J. Pharm., 2022, 614, 121422 CrossRef PubMed.
- B. Z. Chen, L. Q. Zhang, Y. Y. Xia, X. P. Zhang and X. D. Guo, Sci. Adv., 2020, 6, eaba7260 CrossRef CAS PubMed.
- S. Koutsopoulos, Adv. Drug Delivery Rev., 2012, 64, 1459–1476 CrossRef CAS PubMed.
- C. Moya-Lopez, A. Juan, M. Donizeti, J. Valcarcel, J. A. Vazquez, E. Solano, D. Chapron, P. Bourson, I. Bravo, C. Alonso-Moreno, P. Clemente-Casares, C. Gracia-Fernández, A. Longo, G. Salloum-Abou-Jaoude, A. Ocaña, M. M. Piñeiro, C. Hermida-Merino and D. Hermida-Merino, Pharmaceutics, 2022, 14, 1138 CrossRef CAS PubMed.
- B. Rabha, K. K. Bharadwaj, D. Baishya, T. Sarkar, H. A. Edinur and S. Pati, Polymers, 2021, 13, 1322 CrossRef CAS PubMed.
- P. Davoodi, L. Y. Lee, Q. Xu, V. Sunil, Y. Sun, S. Soh and C. H. Wang, Adv. Drug Delivery Rev., 2018, 132, 104–138 CrossRef CAS PubMed.
- G. Ma and C. Song, J. Appl. Polym. Sci., 2007, 104, 1895–1899 CrossRef CAS.
- Q. K. Anjani, A. H. B. Sabri, E. Utomo, J. Domínguez-Robles and R. F. Donnelly, Mol. Pharm., 2022, 19, 1191–1208 CrossRef CAS PubMed.
- T. Ning, F. Yang, D. Chen, Z. Jia, R. Yuan, Z. Du, S. Liu, Y. Yu, X. Dai, X. Niu and Y. Fan, Adv. Healthcare Mater., 2022, 11, e2102180 CrossRef PubMed.
- S. Li, D. Xia and M. R. Prausnitz, Adv. Funct. Mater., 2021, 31, 2103359 CrossRef CAS PubMed.
- J. Jeevanandam, S. Pan, J. Rodrigues, M. A. Elkodous and M. K. Danquah, Biomater. Sci., 2022, 10, 4107–4118 RSC.
- J. J. Yland, K. A. Bresnick, E. E. Hatch, A. K. Wesselink, E. M. Mikkelsen, K. J. Rothman, H. T. Sørensen, K. F. Huybrechts and L. A. Wise, BMJ [Br. Med. J.], 2020, 371, m3966 Search PubMed.
- W. Li, R. N. Terry, J. Tang, M. R. Feng, S. P. Schwendeman and M. R. Prausnitz, Nat. Biomed. Eng., 2019, 3, 220–229 CrossRef CAS PubMed.
- G. Yao, G. Quan, S. Lin, T. Peng, Q. Wang, H. Ran, H. Chen, Q. Zhang, L. Wang, X. Pan and C. Wu, Int. J. Pharm., 2017, 534, 378–386 CrossRef CAS PubMed.
- M. B. Perez Cuevas, M. Kodani, Y. Choi, J. Joyce, S. M. O'Connor, S. Kamili and M. R. Prausnitz, Bioeng. Transl. Med., 2018, 3, 186–196 CrossRef CAS PubMed.
- E. Caffarel-Salvador, S. Kim, V. Soares, R. Y. Tian, S. R. Stern, D. Minahan, R. Yona, X. Lu, F. R. Zakaria, J. Collins, J. Wainer, J. Wong, R. McManus, S. Tamang, S. McDonnell, K. Ishida, A. Hayward, X. Liu, F. Hubálek, J. Fels, A. Vegge, M. R. Frederiksen, U. Rahbek, T. Yoshitake, J. Fujimoto, N. Roxhed, R. Langer and G. Traverso, Sci. Adv., 2021, 7, eabe2620 CrossRef CAS PubMed.
- X. Zhao, X. Li, P. Zhang, J. Du and Y. Wang, J. Controlled Release, 2018, 286, 201–209 CrossRef CAS PubMed.
- J. Yu, J. Wang, Y. Zhang, G. Chen, W. Mao, Y. Ye, A. R. Kahkoska, J. B. Buse, R. Langer and Z. Gu, Nat. Biomed. Eng., 2020, 4, 499–506 CrossRef CAS PubMed.
- O. S. Manoukian, M. R. Arul, N. Sardashti, T. Stedman, R. James, S. Rudraiah and S. G. Kumbar, J. Appl. Polym. Sci., 2018, 135, 46068 CrossRef PubMed.
- M. Jang, B. M. Kang, H. Yang, J. Ohn, O. Kwon and H. Jung, Adv. Healthcare Mater., 2021, 10, e2001691 CrossRef PubMed.
- H. Shi, J. Zhou, Y. Wang, Y. Zhu, D. Lin, L. Lei, S. Vakal, J. Wang and X. Li, Small, 2022, 18, e2104657 CrossRef PubMed.
- Y. Yuan, C. Hua, Z. Yan, X. Ling, L. Rui, Z. Hui, Y. Tai and L. Zhou, Nano Res., 2022, 15, 8336–8344 CrossRef.
- Y. Lee, W. Li, J. Tang, S. P. Schwendeman and M. R. Prausnitz, J. Controlled Release, 2021, 337, 676–685 CrossRef CAS PubMed.
- W. Li, J. Tang, R. N. Terry, S. Li, A. Brunie, R. L. Callahan, R. K. Noel, C. A. Rodríguez, S. P. Schwendeman and M. R. Prausnitz, Sci. Adv., 2019, 5, eaaw8145 CrossRef CAS PubMed.
- J. M. Lee, M. A. Lee, H. N. Do, Y. I. Song, R. J. Bae, H. Y. Lee, S. H. Park, J. S. Kang and J. K. Kang, Lab. Anim. Res., 2012, 28, 115–121 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00780d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |