
Facile synthesis of poly(disulfide)s through one-step oxidation polymerization for redox-responsive drug delivery†
Ruhe
Zhang‡
a,
Tianqi
Nie‡
b,
Liying
Wang
c,
Danni
He
d,
Yang
Kang
*e,
Chao
Zhang
 *a and
Jun
Wu
*fg
*a and
Jun
Wu
*fg
aSchool of Biomedical Engineering, Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, China. E-mail: zhchao9@mail.sysu.edu.cn
bGuangzhou Twelfth People's Hospital, Guangzhou 510620, China
cDepartment of Hematology, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen, 518107, China
dDepartment of Medical Ultrasonics, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen 518107, China
eScientific Research Center, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen 518107, China. E-mail: kangy26@mail.sysu.edu.cn
fBioscience and Biomedical Engineering Thrust, The Hong Kong University of Science and Technology (Guangzhou), Nansha, Guangzhou 511400, China. E-mail: junwuhkust@ust.hk
gDivision of Life Science, The Hong Kong University of Science and Technology, Hong Kong SAR, China
First published on 18th April 2023
Abstract
Poly(disulfide)s-based systems with repetitive disulfide bonds in their backbones are emerging as promising tumor microenvironment responsive platforms for drug delivery. However, complicated synthesis and purification processes have restricted their further application. Herein, we developed redox-responsive poly(disulfide)s (PBDBM) by one-step oxidation polymerization of a commercially available monomer, 1,4-butanediol bis(thioglycolate) (BDBM). PBDBM can self-assemble with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol)3400 (DSPE-PEG3.4k) by the nanoprecipitation method and be formulated into PBDBM NPs (sub 100 nm). It can also be loaded with docetaxel (DTX), a first-line chemotherapy agent for breast cancer, to form DTX@PBDBM NPs with a loading capacity of 6.13%. DTX@PBDBM NPs with favorable size stability and redox-responsive capability exhibit superior antitumor activity in vitro. In addition, owing to the different glutathione (GSH) levels in normal and tumor cells, PBDBM NPs with disulfide bonds could synergistically increase intracellular ROS levels, further inducing apoptosis and cell cycle arrest in the G2/M phase. Moreover, in vivo studies revealed that PBDBM NPs could accumulate in tumors, suppress 4T1 tumor growth, and significantly attenuate the systemic toxicity of DTX. Thus, a novel redox-responsive poly(disulfide)s nanocarrier was successfully and facilely developed for cancer drug delivery and effective breast cancer therapy.
Introduction
The emergence of nanotechnology formulations, including liposomes, polymeric micelles, biomolecular nanoparticles, and inorganic nanoparticles, has brought different kinds of delivery systems for poorly soluble chemotherapeutic agents.1–8 The unique size of nanoparticles provides them with passive targeting capability through enhanced permeability and retention effects, thereby increasing tumor site retention and mitigating the toxic side effects of free drugs due to nonspecific distribution.9,10 Enhanced drug solubilization and further modification also have prolonged the blood circulation time and improved the pharmacokinetic performance.11–13 However, low targeting efficiency, uncontrolled drug release, and early leakage of drugs during blood circulation are the disadvantages of conventional nanocarriers.14 Notably, nanocarriers are supposed to release drugs at the tumor site with high specificity and speed to maximize the antineoplastic effect and minimize side effects.15 Therefore, further research and development of innovative drug carriers are needed.Compared to the normal physiological environment, the heterogeneous tumor microenvironment (TME) contains, but is not limited to, an acidic environment, overexpressed enzymes, and elevated redox levels.16 The pathological environment can induce tumor resistance and accelerate tumor progression and metastasis.17 Hence, taking advantage of the metabolic abnormalities in the TME, TME-responsive drug delivery systems, including pH, enzyme, and redox stimuli-responsive systems, have been exploited recently.18,19 In particular, redox-responsive systems have received extensive attention owing to the different redox states of cancer cells and normal cells.20–22 Specifically, cancer cells with hyperproliferation and metabolism in comparison with normal cells may produce reactive oxygen species (ROS) that are detrimental to cellular components. The glutathione (GSH) levels in cancer cells are thus enhanced correspondingly and compensated to detoxify ROS.23 In general, the GSH level in the blood is micromolar (2–10 μM), while the GSH level in the cytoplasm of tumor cells is millimolar (2–10 mM), which is at least four-fold the level of normal cells.24–26 Thus, redox-responsive drug delivery systems could remain intact in blood circulation while degrading in the reductive cancer cell cytoplasm to fulfill on-demand controlled drug release.27 In addition, together with the GSH-depleting ability, redox-responsive systems can also synergistically amplify oxidative stress during their dissociation to induce cell apoptosis.28,29
Among the redox-responsive delivery systems, poly(disulfide)s-based systems incorporating repetitive disulfide bonds in their backbone are emerging as promising systems for drug delivery. These systems are biodegradable and can be easily modified, which further enhance their versatility.30,31 To date, they have been successfully applied to deliver small-molecule drugs, nucleic acids, quantum dots, and proteins.22,32–34 Oxidation polymerization of dithiols has been reported using various oxidizing agents, including iodine, hydrogen peroxide, and dimethyl sulfoxide (DMSO). However, these methods require high temperatures (up to 160 °C) or triethylamine as a catalyst.35,36 An alternative approach, such as anionic ring-opening polymerization of lipoic acid derivatives as propagators, was developed by Matile et al. Although this method has been widely utilized for drug delivery, propagators and monomers require multistep synthesis.37 Poly(disulfide)s with more functional groups can be synthesized by step-growth polymerization of disulfide bond-containing bifunctional monomers. For example, Zhong et al. developed poly(disulfide urethane)s by the polycondensation of bis(ethyl L-serinate) with an α-amino acid-based L-lysine ethyl ester diisocyanate. However, the cumbersome bis(ethyl L-serinate) preparation process increases the cost and is not environmentally friendly.38 To this end, a novel, simple, and environmentally friendly approach is required to develop poly(disulfide)s-based drug delivery systems.
It has always been our hope to obtain bioactive drug delivery carriers quickly and easily by one-step polymerization.1,39 Hence, in this study, hydrophobic and biodegradable poly(disulfide)s (PBDBM) were facilely synthesized by one-step oxidative polymerization of the commercially available monomer 1,4-butanediol bis(thioglycolate) (BDBM) and exploited as a redox-responsive nano-drug delivery platform. Amphiphilic DSPE-PEG3.4k was added to improve the stability of the nano drug delivery system.40 DTX was chosen as the model drug, and different feed ratios were screened and optimized by the classical nanoprecipitation self-assembly method (Scheme 1). The screened systems, hereafter referred to as PBDBM NPs and DTX@PBDBM NPs (DTX-loaded PBDBM NPs), were further investigated for their redox responsiveness, cytotoxicity, and effect on modulating the cellular ROS level, cell cycle, apoptosis, in vivo biodistribution, and tumor suppression potency.
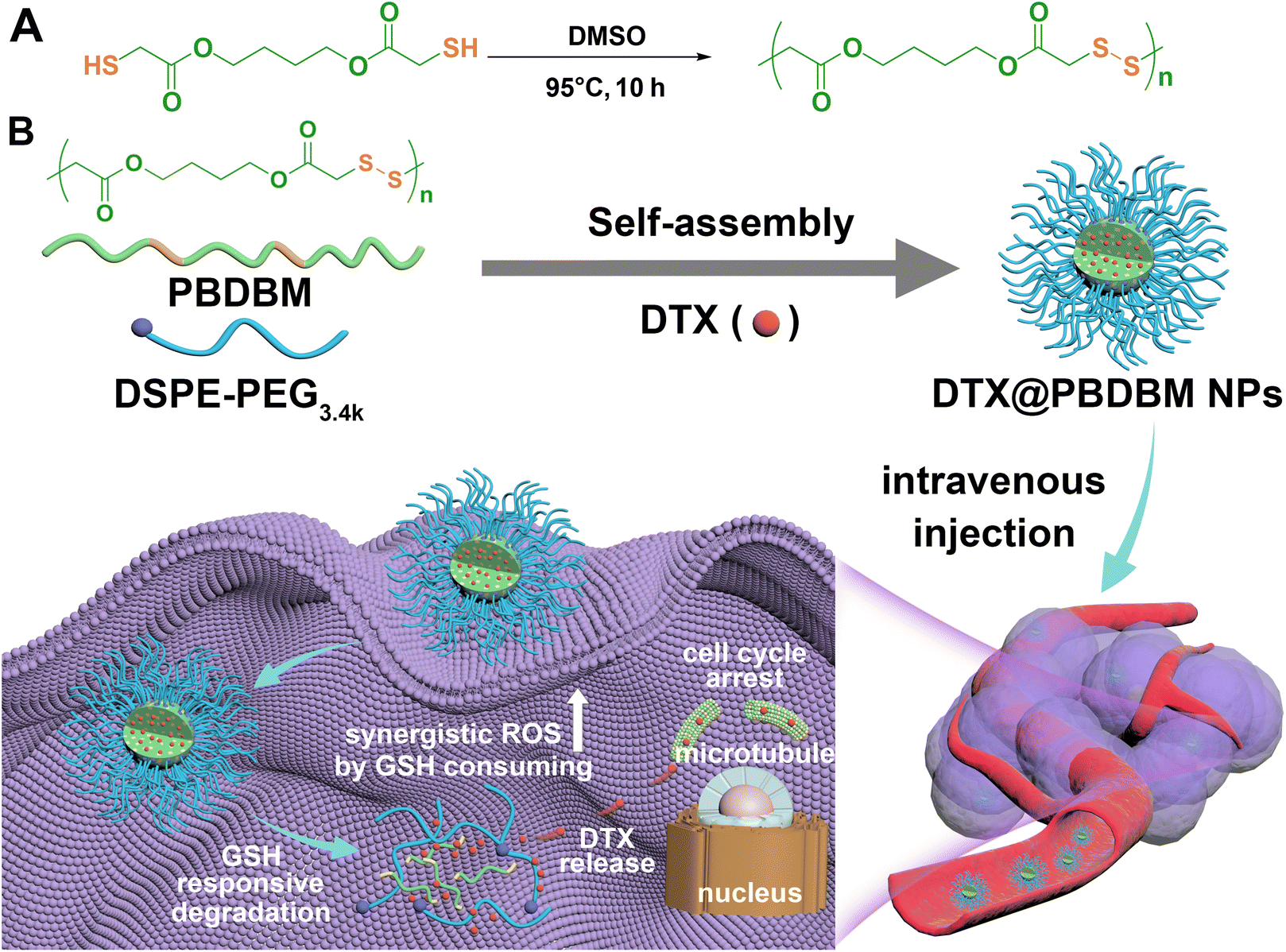 | ||
| Scheme 1 (A) Synthetic procedure of redox-responsive poly(disulfide)s (PBDBM). (B) Scheme illustration of the composition and preparation of DTX@PBDBM NPs and their redox-responsive degradation after intravenous injection and cell uptake. | ||
Results and discussion
Preparation and characterization of poly (disulfide)s based on 1,4-butanediol bis(thioglycolate)
Redox-responsive poly(disulfide)s (PBDBM) were facilely synthesized by a one-step oxidation polymerization technique. 1,4-Butanediol bis(thioglycolate) was oxidized by DMSO at 95 °C for 10 h, which is depicted in Scheme 1A. The obtained polymer PBDBM was first confirmed by 1H NMR spectroscopy. As shown in Fig. 1A, the proton signal of the No. 3 carbon atom in the polymer shifted downfield, whereas the proton signals of the No. 1 and 2 carbon atoms did not change. In addition, it is worth noting that the peak observed at 2.0 ppm, which was attributed to the sulfhydryl proton of the monomer, was not present in the polymer. Moreover, the stretching vibration peak of –SH– at approximately 2567 cm−1 disappeared, and the stretching vibration peaks of –S–S– at approximately 445 cm−1 appeared (FT-IR spectrum shown in Fig. 1B). These results suggested that the monomer's thiol group reacted during the polymerization process, leading to the successful formation of disulfide bonds and the corresponding polymer PBDBM. The molecular weight of PBDBM was determined to be Mn ∼ 8200, Mw ∼ 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 with a PDI (Mw/Mn) of ∼ 1.8 via gel permeation chromatography (GPC) (Fig. 1C). The redox-responsive degradation of PBDBM was also characterized by GPC after incubation with dithiothreitol as a reducing agent. GSH was not selected because it contains amino and carboxyl groups and is not suitable for GPC measurement. As revealed in Fig. 1C, PBDBM was gradually degraded with the extension of incubation time with dithiothreitol, indicating its potential as a redox-responsive drug delivery carrier.
900 with a PDI (Mw/Mn) of ∼ 1.8 via gel permeation chromatography (GPC) (Fig. 1C). The redox-responsive degradation of PBDBM was also characterized by GPC after incubation with dithiothreitol as a reducing agent. GSH was not selected because it contains amino and carboxyl groups and is not suitable for GPC measurement. As revealed in Fig. 1C, PBDBM was gradually degraded with the extension of incubation time with dithiothreitol, indicating its potential as a redox-responsive drug delivery carrier.
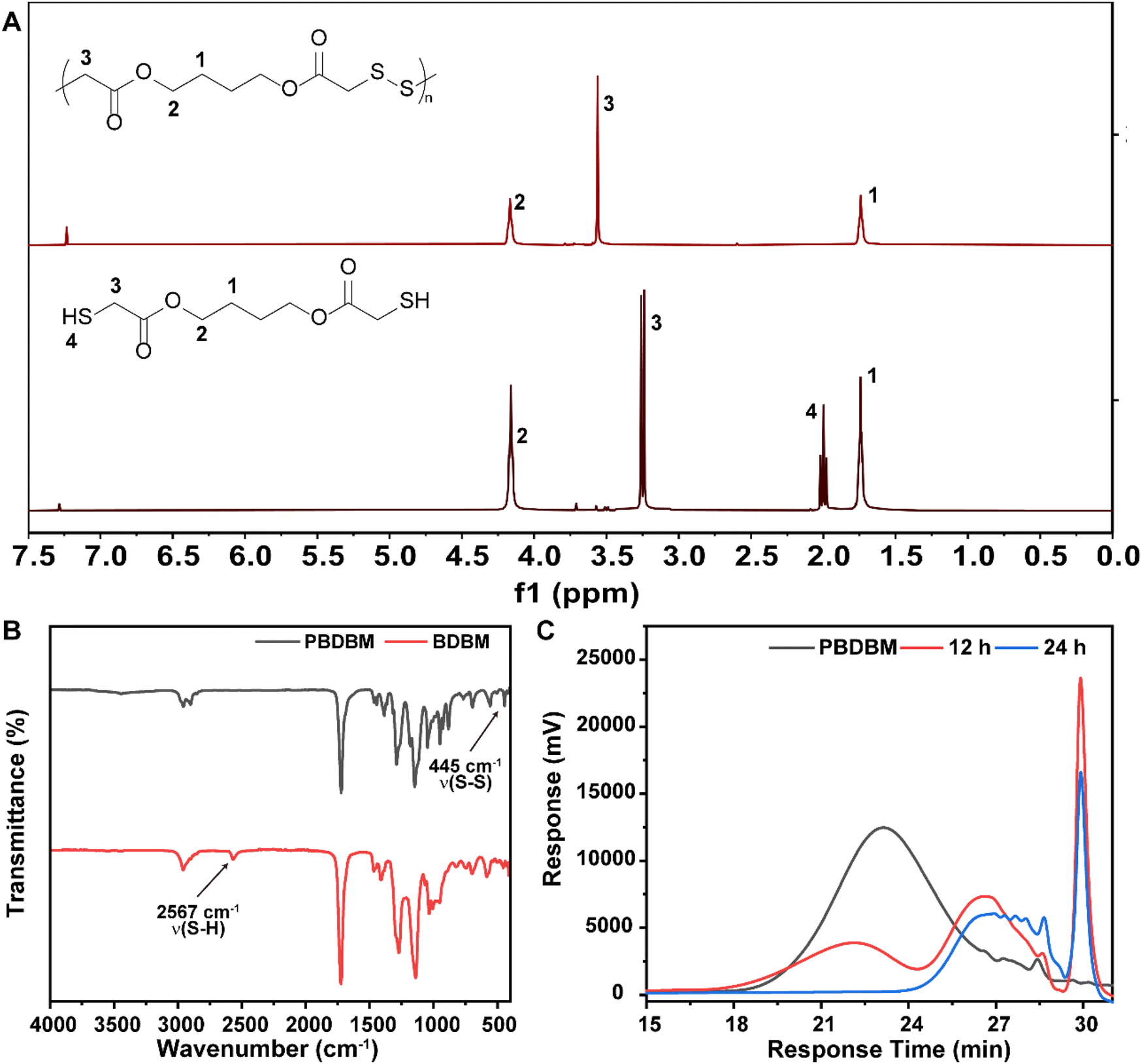 | ||
| Fig. 1 Characterization of poly(disulfide)s (PBDBM). (A) 1H NMR spectra of PBDBM and BDBM. (B) FT-IR spectra of PBDBM and BDBM. (C) GPC profiles of PBDBM and PBDBM after incubation with dithiothreitol for 12 and 24 h. | ||
Preparation and characterization of PBDBM-based NPs
PBDBM NPs were prepared by PBDBM and DSPE-PEG3.4k self-assembly through the classic nanoprecipitation method (Scheme 1). By adjusting the oil:water ratio and the DTX feed ratio, the preparation of DTX@PBDBM NPs was optimized in terms of the Z-average size, polydispersity index (PDI), zeta potential, loading capacity (LC), and loading efficiency (LE) (Table S1†). The final oil:water ratio chosen was 1:10, and the DTX feed ratio was 15 wt% of PBDBM. DTX@PBDBM NPs prepared by the optimized parameters exhibited the smallest size (Fig. 2A) and higher LC (6.13%) and LE (46.99%). The Z-average size of DTX@PBDBM NPs was 143 ± 2.3 nm with a PDI of 0.153 ± 0.041, which was larger than that of PBDBM NPs, which was 78 ± 1.3 nm with a PDI of 0.242 ± 0.01 (Fig. S1A†). TEM images shown in Fig. S1B† and Fig. 2B revealed that the morphology of both the PBDBM NPs and DTX@PBDBM NPs was spherical in shape. The sizes of both NPs were slightly smaller than those acquired by DLS measurements, which might be the result of the drying process during TEM analysis sample preparation.41 PBDBM NPs and DTX@PBDBM NPs possessed negative zeta potential values of −24.9 mV (Fig. S1C†) and −28.4 mV (Fig. 2C), respectively. To evaluate the size stability of both NPs, PBDBM NPs and DTX@PBDBM NPs were stored for one week at 25 °C and showed no significant change in the size. This stability could be associated with the moderate zeta potential of DSPE-PEG, which likely reduced their aggregation (Fig. S1D† and Fig. 2D).42
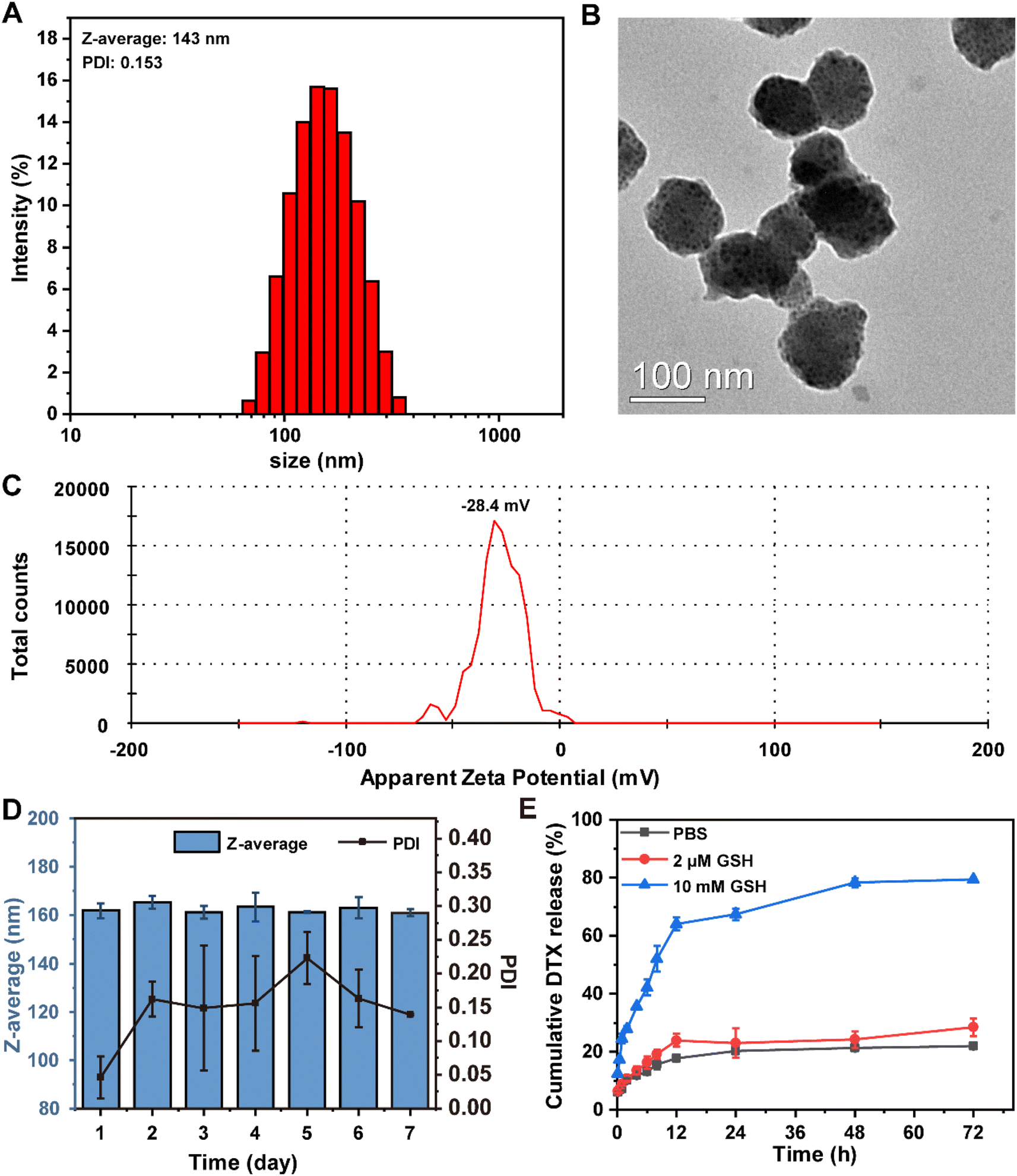 | ||
| Fig. 2 Synthesis and characterization of DTX@PBDBM NPs. (A) Z-average size of DTX@PBDBM NPs. (B) TEM image of DTX@PBDBM NPs. (C) Zeta potential of DTX@PBDBM NPs. (D) Stability of DTX@PBDBM NPs. (E) Release of DTX from DTX@PBDBM NPs at different reducing concentrations. All data are expressed as mean ± S.D. (n = 3). | ||
To investigate the ability of DTX@PBDBM NPs to release DTX within the reductive microenvironment of tumor cells, we first evaluated the size variation of DTX@PBDBM NPs under 10 mM GSH. As shown in Fig. S2,† the size of DTX@PBDBM NPs significantly increased after the addition of 10 mM GSH, indicating that the degradation and aggregation of DTX@PBDBM NPs were triggered by a high level of GSH. Subsequently, we analyzed the in vitro release profiles of DTX@PBDBM NPs under different reducing concentrations. PBS with 10 mM GSH was applied to simulate the high redox tumor microenvironment, and PBS with 2 μM GSH was applied to simulate the redox conditions of the blood. As shown in Fig. 2E, the release curves of DTX indicated that the cumulative release of DTX reached 67.3% after 24 h of incubation in PBS with 10 mM GSH, but only 22.9% and 20.2% of DTX was released in PBS with 2 μM GSH and in the absence of GSH at the same time, respectively. This different release pattern was attributed to high GSH-induced cleavage of disulfide bonds. With the extension of time, the release of DTX in PBS with 10 mM GSH gradually increased and reached 79.2% at 72 h, which may be ascribed to the gradual degradation of PBDBM. Therefore, the above-mentioned results confirmed that DTX@PBDBM NPs with redox-responsive ability have great potential for anticancer drug delivery in a controlled manner.
Cellular uptake of DTX@PBDBM NPs
To track the cell uptake process of PBDBM NPs, coumarin6 (C6) labeled PBDBM NPs (C6@PBDBM NPs, Fig. S3†) were prepared with a size comparable to that of DTX@PBDBM NPs and subjected to confocal laser scanning microscopy (CLSM) and flow cytometry with 4T1 cells. As illustrated in Fig. 3A, treatment with C6@PBDBM NPs resulted in a time-dependent cellular uptake process, as evidenced by a gradual increase in the green fluorescence of C6 over time. Conversely, treatment with free C6 showed faster uptake within the first hour, but no significant increase in the fluorescence intensity could be observed at subsequent time points. After 6 h of exposure, comparable intensities were observed between the two groups. Further flow cytometry analysis also quantitatively confirmed the time-dependent process of C6@PBDBM NPs and the different fluorescence changes over time (Fig. 3B–D). The difference may be related to the different cellular uptake mechanisms. In specific, C6 is a small, lipophilic molecule that is taken up by cells through free diffusion, which is concentration-dependent. Therefore, the uptake of C6 gradually slowed down after 1 h. However, after being encapsulated in PBDBM NPs, its internalization is mainly through endocytosis, which is time-dependent and energy-dependent regardless of the concentration gradient. Thus, it can be observed that its uptake gradually increased with time. The above results demonstrated that PBDBM NPs could be efficaciously captured by 4T1 cells.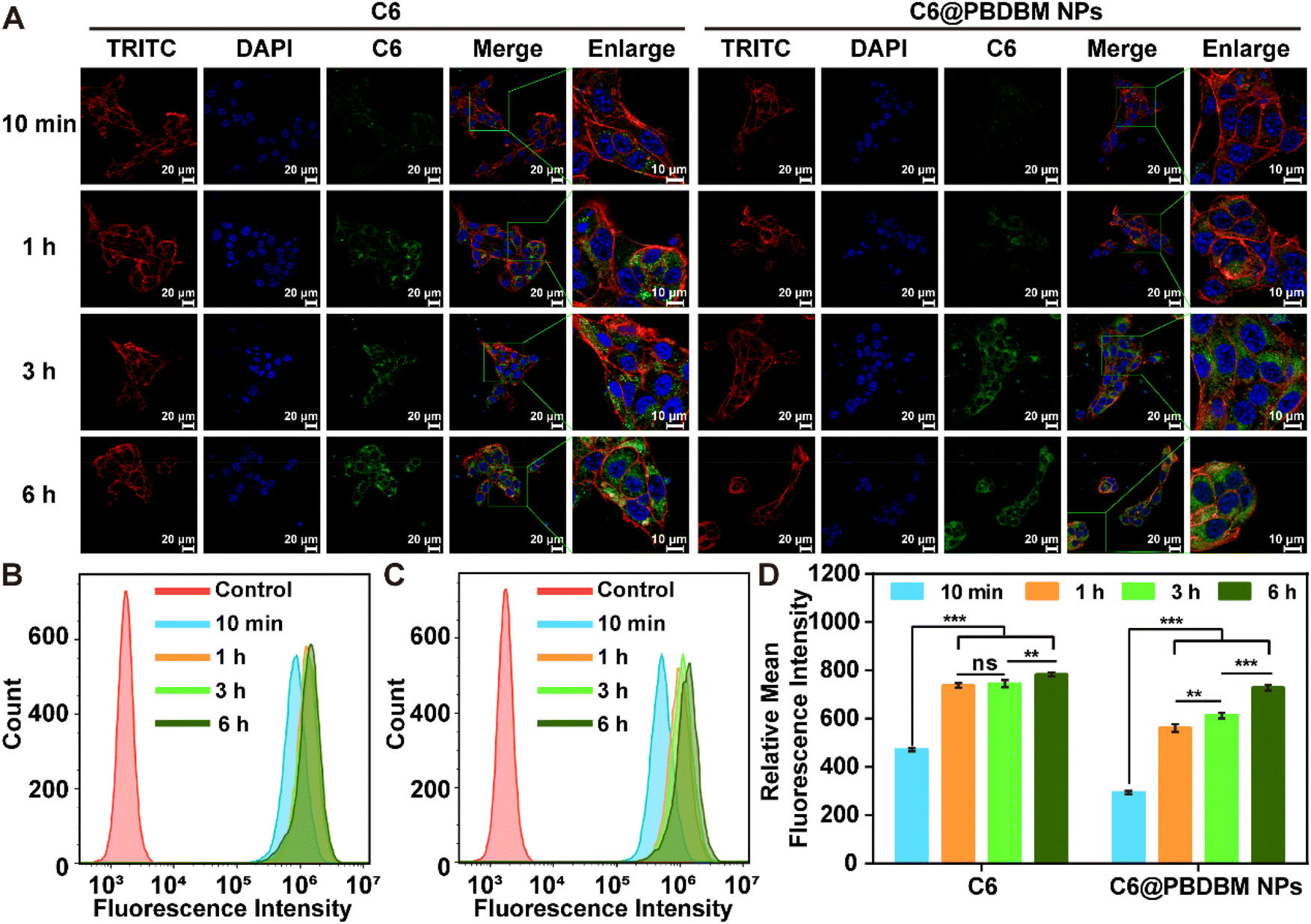 | ||
| Fig. 3 Qualitative and quantitative study on the cellular uptake of free C6 and C6@PBDBM NPs. (A) CLSM images (qualitative study) of 4T1 cells after exposure with C6 (left) and C6@PBDBM NPs (right) for 10 min, 1 h, 3 h, or 6 h. Flow cytometric analysis of 4T1 cells after treatment with (B) C6 and (C) C6@PBDBM NPs for 10 min, 1 h, 3 h, or 6 h. (D) Relative mean fluorescence intensity (quantitative study) of 4T1 cells treated as (B) and (C). Untreated 4T1 cells was used as a control (n = 3, **p < 0.01; ***p < 0.001). | ||
Antitumor effect of DTX@PBDBM NPs in vitro
Given the favorable cellular uptake performance, we next investigated the in vitro antitumor effects of PBDBM NPs, DTX, and DTX@PBDBM NPs in 4T1 cells using an MTT assay. The equivalent DTX concentration range was from 0.001 μg mL−1 to 2 μg mL−1. As shown in Fig. 4A, dose-dependency of cell viability in PBDBM NPs, free DTX, and DTX@PBDBM NPs treatments could be observed. Notably, DTX@PBDBM NPs suppressed the proliferation of 4T1 cells more significantly with drug concentrations over 0.01 μg mL−1. To further explore the superior antitumor effects of DTX@PBDBM NPs, their antitumor mechanism was preliminarily investigated.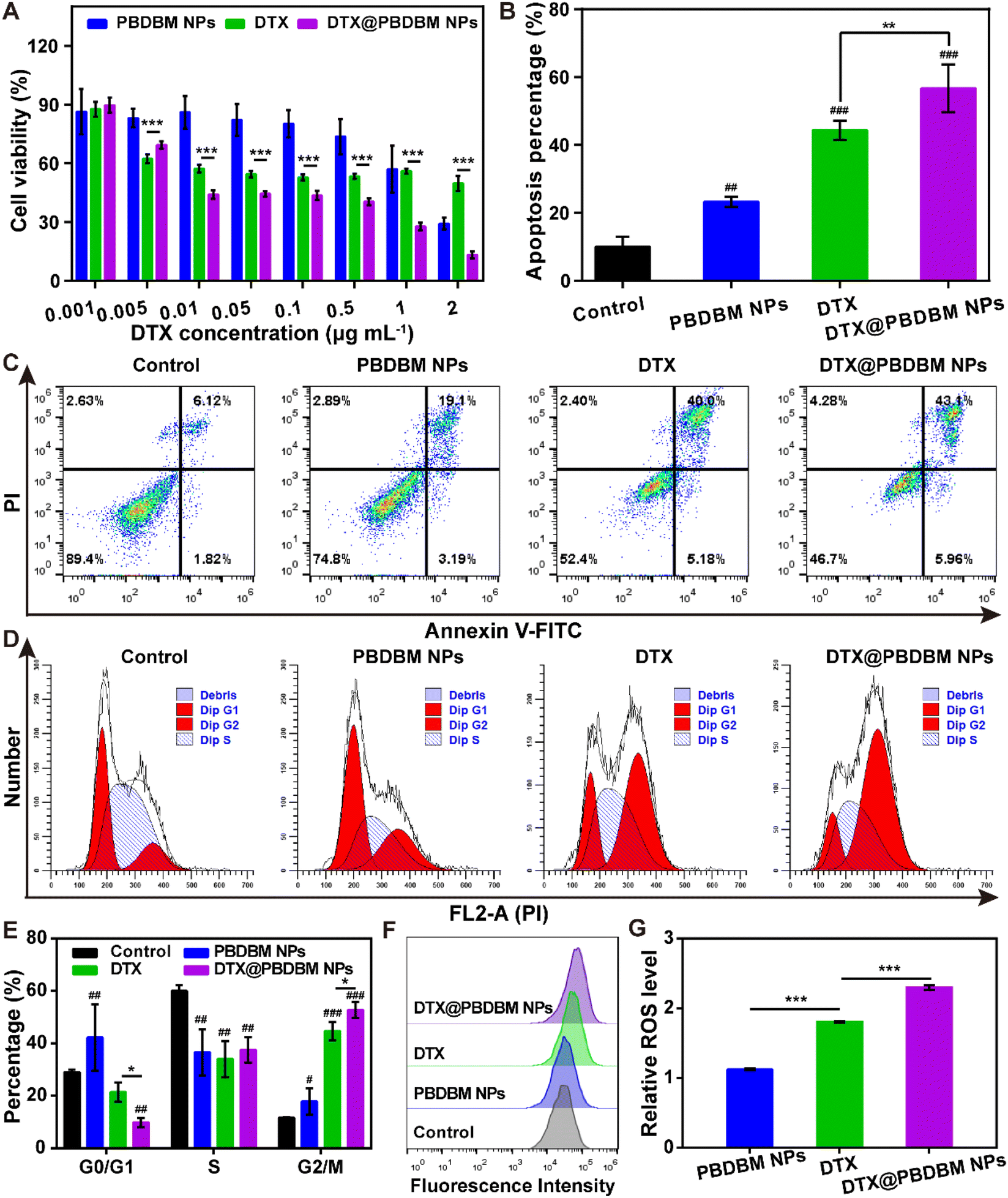 | ||
| Fig. 4 In vitro cellular anticancer evaluation of DTX@PBDBM NPs. (A) Cell viability of 4T1 cells exposed to PBDBM NPs, DTX, and DTX@PBDBM NPs at different concentrations for 48 h (n = 6). (B) Apoptosis percentage of 4T1 cells following treatment with 1640 (control), PBDBM NPs, DTX, and DTX@PBDBM NPs for 48 h (n = 3). (C) Representative flow scatter plots of 4T1 cell apoptosis. (D) Representative flow cytometry histograms of the cell cycle of 4T1 cells treated with 1640 (control), PBDBM NPs, DTX, and DTX@PBDBM NPs for 24 h. (E) Quantitative analysis of 4T1 cell cycle distribution (n = 3). (F) Representative fluorescence intensity histograms of 4T1 cells after treatment with 1640 (control), PBDBM NPs, DTX, and DTX@PBDBM NPs for 12 h and incubated with DCFH-DA for 30 min. (G) Relative ROS level compared with the control group calculated by flow cytometry analysis (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 vs. DTX group; ##p < 0.01; ###p < 0.001 vs. control group. | ||
First, the apoptosis of PBDBM NPs, DTX, and DTX@PBDBM NPs was evaluated with an Annexin V-FITC/PI kit by flow cytometry (Fig. 4B and C). After treatment with DTX, PBDBM NPs, and DTX@PBDBM NPs for 48 h with equivalent DTX concentrations (0.25 μg mL−1), the apoptosis rate of each group was detected. As shown in the flow scatter plots (Fig. 4C), 4T1 cells treated with PBDBM NPs showed a slightly elevated apoptosis rate (22.29%) in comparison with the control group (7.94%), whereas the DTX group and DTX@PBDBM NP group showed significantly increased apoptosis rates of 45.18% and 49.06%, respectively. The quantitative results shown in Fig. 4B also clearly demonstrated that DTX@PBDBM NPs could more significantly induce 4T1 cell apoptosis, verifying the superior antitumor effects of DTX@PBDBM NPs.
DTX is known to inhibit microtubule depolymerization, inducing G2/M cell cycle arrest. Therefore, the effects of different formulations on the cell cycle distribution of 4T1 cells were evaluated after 24 h of treatment. According to the flow cytometric results shown in Fig. 4D and E, PBDBM NPs slightly induced cycle arrest in G2/M. DTX and DTX@PBDBM NPs significantly induced 4T1 cell cycle arrest in G2/M, with the proportion increasing from 11.1% (control group) to 44.6% and 52.7%, respectively. The significant cell cycle arrest was due to DTX treatment. The enhanced cell cycle arrest effect of DTX@PBDBM NPs compared to DTX may be due to the active endocytic transport of DTX@PBDBM NPs, which was independent of the concentration and corresponded to previous cellular uptake results. In addition, this effect may be partially related to the redox-responsive PBDBM NPs, which have GSH-depleting and ROS-amplifying abilities.29
To further clarify its mechanism for inducing cell cycle arrest, we next detected the intracellular ROS level after 12 h of different treatments, as a redox-responsive system could synergistically amplify oxidative stress.28 DCFH-DA, a ROS fluorescent probe, was utilized in this study. As shown in Fig. 4F and G, PBDBM NPs with redox-sensitive poly(disulfide)s induced a slightly higher ROS level, DTX induced an almost twofold increase in the ROS level, and DTX@PBDBM NPs induced an over twofold increase in the ROS level compared to the control group. It has been reported that disulfide groups in a polymer can react with GSH and consume GSH, thereby amplifying oxidative stress.29 Thus, it is reasonable to speculate that PBDBM containing disulfide bonds can deplete GSH in high reducing intracellular environments, thereby synergizing with DTX to increase ROS production. Notably, the overload of ROS can damage intracellular biomolecules such as DNA, further inducing cell apoptosis and cell cycle arrest.43 Overall, except for the effect of DTX on cell cycle arrest, the better in vitro antitumor effect of DTX@PBDBM NPs than DTX also benefited from the GSH-depletive property of PBDBM, which may synergistically amplify ROS levels and further induce cell apoptosis and cell cycle arrest.
Blood compatibility of DTX@PBDBM NPs
Blood compatibility is imperative for nanocarriers, considering that they may finally be administered intravenously. Herein, a hemolysis assay was employed using mouse red blood cells (RBC) to evaluate the blood compatibility of PBDBM NPs and DTX@PBDBM NPs. Fig. 5A shows the RBC suspensions after exposure to PBDBM NPs and DTX@PBDBM NPs at gradient concentrations for 3 h followed by centrifugation. Although hemolysis was slightly concentration-dependent with both NPs, the maximal hemolysis of both NPs was less than the permissible level of 10% across all the tested concentrations, which was quantified by measuring hemoglobin absorption in the supernatant (Fig. 5B). These results suggest that both PBDBM NPs and DTX@PBDBM NPs have good blood compatibility and are suitable for systemic administration.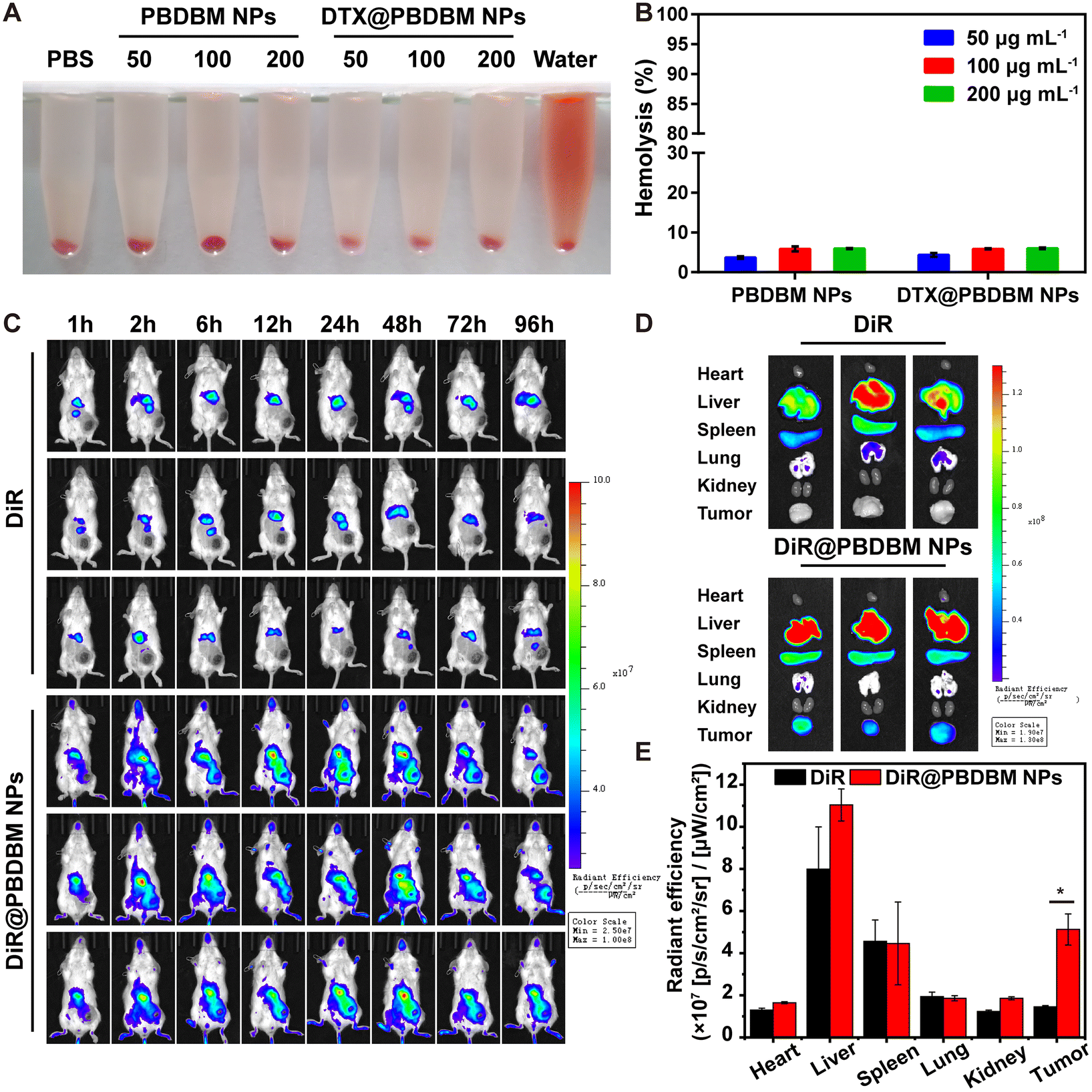 | ||
| Fig. 5 Blood compatibility and in vivo biodistribution of DTX@PBDBM NPs. (A) Photograph of RBC suspensions after exposure to PBS, water, PBDBM NPs, and DTX@PBDBM NPs at different concentrations for 3 h followed by centrifugation. (B) Hemolysis percentage of PBDBM NPs and DTX@PBDBM NPs at various concentrations (n = 3) (C) Biodistribution of DiR and DiR@PBDBM NPs in 4T1 orthotopic tumor-bearing mice post-intravenous injection at different time points. (D) Fluorescence images of the harvested tumors and vital organs at 96 h post-intravenous injection. (E) Quantitative analysis of the average radiant efficiency of the DiR in tumors and primary organs from (D) (n = 3, *p < 0.05). | ||
Biodistribution of DTX@PBDBM NPs
The biodistribution of PBDBM NPs in vivo was evaluated with DiR-loaded PBDBM NPs (DiR@PBDBM NPs, Fig. S4†), which were injected into 4T1 orthotopic tumor-bearing mice via the tail vein. A fluorescent DiR probe, which mimicked DTX, was injected as the control group. Noninvasive imaging of the fluorescence of DiR in mice was checked using an IVIS Spectrum in vivo imaging system. As demonstrated in Fig. 5C, the fluorescence of free DiR accumulated rapidly in the liver tissue and then decayed quickly. In contrast, although the fluorescence signal of DiR@PBDBM NPs can also be detected in the liver area, the fluorescence intensity in the tumor area increased significantly with time. Moreover, fluorescence signals could still be observed even after 96 h of tail vein injection, which indicated that the DiR@PBDBM NPs could achieve longer blood circulation, passive tumor targeting, and accumulation through the enhanced permeability and retention effect. The fluorescence of organs and tumors ex vivo at 96 h post-injection was also measured. As illustrated in Fig. 5D and E, the fluorescence intensity of tumors in the DiR@PBDBM NP group was notably greater than that in the free DiR group, which matched the results of in vivo imaging and thus further indicated the potential of DTX@PBDBM NPs to improve the antitumor efficacy in vivo.In vivo antitumor efficacy of DTX@PBDBM NPs
To test the in vivo antineoplastic efficacy of PBDBM NPs, 4T1 orthotopic tumor-bearing mice were administered with different formulations (PBS, PBDBM NPs, DTX, and DTX@PBDBM NPs) via the tail vein with an equivalent DTX dose of 5 mg kg−1. As shown in Fig. 6A, the tumors in mice treated with PBS grew sharply, whereas the tumors in mice treated with PBDBM NPs, free DTX, and DTX@PBDBM NPs displayed different tumor inhibition effects. Specifically, the tumor volume of the PBDBM NP group was slightly decreased and contained compared with that of the control group, which might be due to GSH depletion and oxidative stress amplification. In addition, free DTX and DTX@PBDBM NPs displayed significant tumor suppression, with tumor suppression rates of 46.3% and 54.4%, respectively. The tumors harvested at the termination of the study were imaged (Fig. 6C) and weighed (Fig. 6D), which further confirmed the tumor-inhibiting effect of different administrations. To further understand the anticancer effect of DTX@PBDBM NPs, H&E staining and TUNEL detection were performed with tumors harvested at the termination of the study. As shown in Fig. 6E, the tumor cells in the PBS group and the PBDBM NP group were tightly organized, and only a small amount of tumor cells in the PBDBM NP group underwent apoptosis. However, DTX and DTX@PBDBM NP treatments caused apparent cell apoptosis. Notably, although there was no significant difference in the tumor volume after treatments with DTX and DTX@PBDBM NPs, DTX@PBDBM NPs showed a stronger apoptosis-inducing effect (Fig. S5†).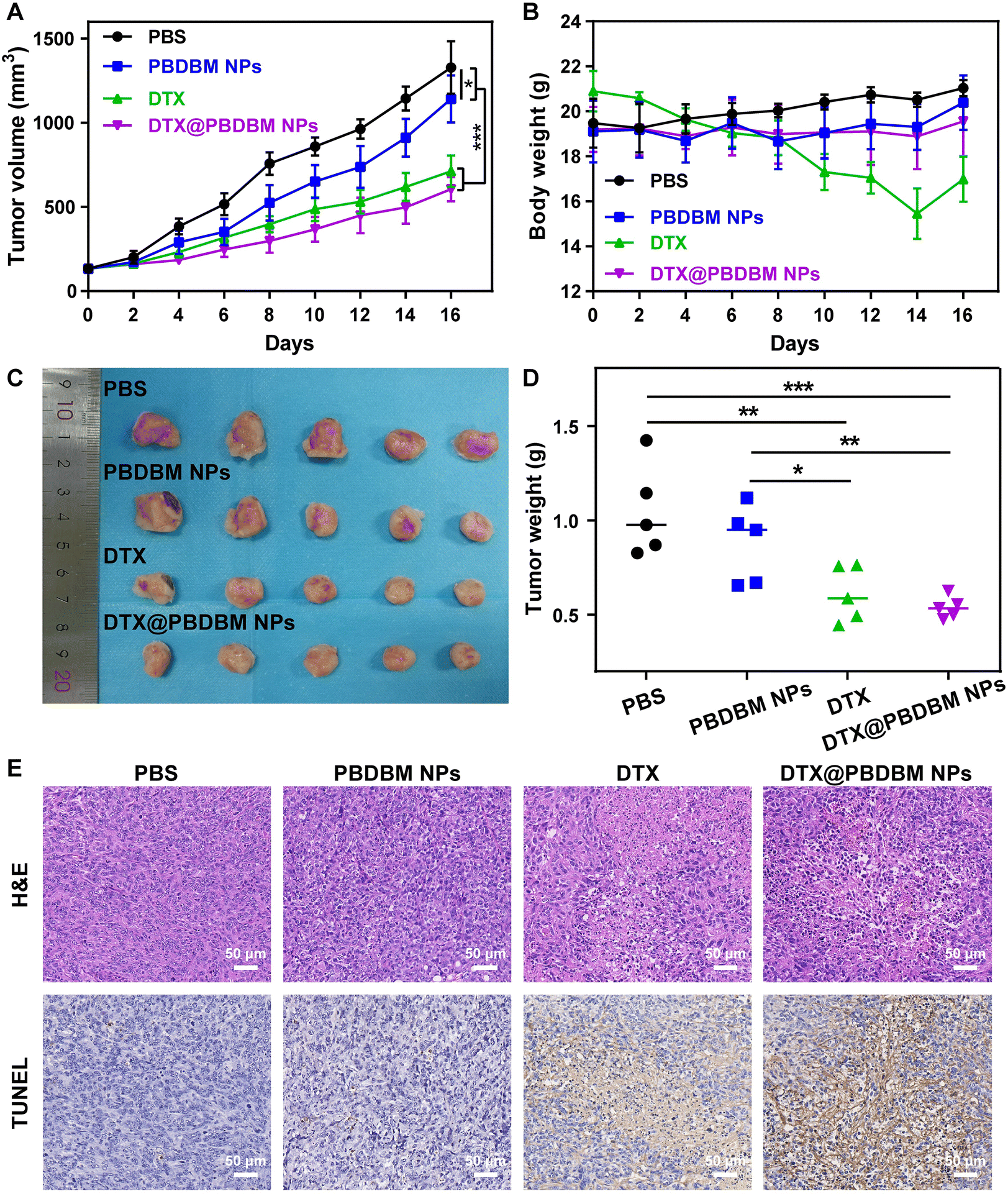 | ||
| Fig. 6 In vivo anticancer efficiency. (A) Tumor volume curves and (B) body weight of 4T1 orthotopic tumor-bearing mice treated with different formulations with an equivalent DTX dose of 5 mg kg−1, including PBS, PBDBM NPs, DTX, and DTX@PBDBM NPs (n = 5). (C) Representative photograph and (D) weights of the harvested tumors from the mice following different administrations at the termination of the study. *p < 0.05, **p < 0.01, ***p < 0.001. (E) Representative H&E and TUNEL images of tumor sections after systemic administration of PBS, PBDBM NPs, DTX, and DTX@PBDBM NPs. Magnification factor: 20×. | ||
In addition, the body weight of mice administered with free DTX showed a sustained and dramatic decrease until the end of treatment, whereas mice administered with PBDBM NPs and DTX@PBDBM NPs maintained the body weight in a small range (Fig. 6B). To further investigate the biosafety of PBDBM NPs, blood biochemical analysis of liver and kidney functions (Fig. S6†) and major tissue section analysis (Fig. S7†) were performed. Compared with the control group, the ALP level of the DTX group exhibited a significant decrease, and the BUN and CREA levels of the DTX group exhibited a significant increase, revealing that DTX treatment had a certain degree of acute toxicity to the liver and kidneys. In addition, the H&E images in Fig. S7† show an incomplete liver structure with vacuolar degeneration in the PBS and DTX groups, which might be related to tumor metastasis. However, no significant difference in blood biochemical levels could be observed in the PBDBM NP and DTX@PBDBM NP groups. Although some tumor metastasis could also be discovered in the livers of the PBDBM NP and DTX@PBDBM NP groups, vacuolar degeneration was alleviated. In addition, in the slices of the other major organs, pathological or inflammation sites could hardly be observed. An evident improvement in liver and kidney function impairment may be attributed, on the one hand, to the good biocompatibility and histocompatibility of DTX@PBDBM NPs and, on the other hand, to the controllable redox-responsive release of DTX@PBDBM NPs, which results in a relatively reduced drug release in nontarget tissues such as the liver and kidneys. Overall, the above results demonstrated that DTX@PBDBM NPs had better antitumor effects and negligible systemic toxicity.
Conclusions
In summary, redox-responsive and biodegradable poly(disulfide)s nano-carriers were facilely and successfully prepared by one-step oxidative polymerization. The prepared DTX@PBDBM NPs exhibited a suitable particle size (143 nm), a favorable surface potential (−28.4 mV), a high drug loading capacity (6.13%), and great size stability. In addition, DTX@PBDBM NPs could be captured by 4T1 cells and reacted with GSH, which synergistically increased ROS levels and further induced cell apoptosis and cell cycle arrest, exhibiting a superior anticancer effect compared to free DTX. Moreover, in vivo studies revealed that the nanoparticle system can prolong the blood circulation time and accumulate in tumors, resulting in significant suppression of 4T1 tumor growth and evident alleviation of systemic toxicity by DTX. Therefore, this study provides a convenient one-step strategy for the synthesis of a novel redox-responsive biocompatible poly(disulfide)s nanocarrier. Compared with traditional drug delivery systems, this nanocarrier has higher efficiency and control in drug delivery and can also reduce the problem of systemic toxicity caused by non-specific distribution, which is a very important advancement.Ethical statement
All animal experiments were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sun Yat-sen University and approved by the Experimental Laboratory Animal Committee of Sun Yat-sen University (approval number: SYSU-IACUC-2021-000901).Author contributions
Ruhe Zhang: validation, investigation, data curation, formal analysis, visualization, writing – original draft, and review and editing. Tianqi Nie: investigation, data curation, formal analysis, writing – original draft, and review and editing. Liying Wang: investigation and data curation. Danni He: data curation. Yang Kang: conceptualization, supervision, and writing – review and editing. Chao Zhang: conceptualization, supervision, writing – review and editing, and project administration. Jun Wu: conceptualization, supervision, writing – review and editing, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate the financial support of the National Natural Science Foundation of China (No. 52173150 and 51973243), the Shenzhen Basic Research Project (JCYJ20190807155801657), and the key international (regional) cooperative research projects of the National Natural Science Foundation of China (5181001045).References
- X. R. You, L. Y. Wang, L. Wang and J. Wu, Adv. Funct. Mater., 2021, 31, 2100805 CrossRef CAS.
- M. Lu, X. Huang, X. H. Cai, J. J. Sun, X. M. Liu, L. Y. Weng, L. Zhu, Q. Q. Luo and Z. P. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 20551–20565 CrossRef CAS PubMed.
- V. Kushwah, D. K. Jain, A. K. Agrawal and S. Jain, Colloids Surf., B, 2018, 172, 213–223 CrossRef CAS PubMed.
- Q. Yang, H. L. Chen, Y. Bai, Y. Cao, W. J. Hu and L. K. Zhang, Adv. Healthcare Mater., 2018, 7, 1700816 CrossRef PubMed.
- Y. Zhu, Y. Q. Wu, S. S. Li, X. L. Yuan, J. Shen, S. Y. Luo, Z. S. Wang, R. Gao, J. Wu and L. Ge, Chem. Eng. J., 2022, 446, 137321 CrossRef CAS.
- V. Kushwah, S. S. Katiyar, C. P. Dora, A. K. Agrawal, D. A. Lamprou, R. C. Gupta and S. Jain, Acta Biomater., 2018, 73, 424–436 CrossRef CAS PubMed.
- H. Choudhury, B. Gorain, M. Pandey, S. A. Kumbhar, R. K. Tekade, A. K. Iyer and P. Kesharwani, Int. J. Pharm., 2017, 529, 506–522 CrossRef CAS PubMed.
- X. R. You, L. Y. Wang, J. F. Zhang, T. Tong, C. L. Dai, C. Chen and J. Wu, Chin. Chem. Lett., 2023, 34, 107720 CrossRef CAS.
- S. H. Wang, K. M. Cheng, K. Chen, C. Xu, P. W. Ma, G. H. Dang, Y. Q. Yang, Q. Lei, H. Y. Huang, Y. Yu, Y. Fang, Q. Y. Tang, N. Jiang, H. L. Miao, F. A. Liu, X. Zhao and N. Li, Nano Today, 2022, 45, 101512 CrossRef CAS.
- T. B. Chen, S. T. Xu, W. Huang and D. Y. Yan, Biomater. Sci., 2022, 10, 3901–3910 RSC.
- N. Bertrand, J. Wu, X. Y. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- J. Tao, W. H. Shi, K. Chen, W. Lu, A. J. Elbourne, L. Bao, L. X. Weng, X. D. Zheng, X. D. Su, Z. G. Teng and L. H. Wang, Biomater. Sci., 2023, 11, 822–827 RSC.
- R. X. Chen, P. Ouyang, L. C. Su, X. Xu, P. H. Lian, Y. Q. Li, Q. Gao, Y. F. Zhang, S. Nie, F. Luo, R. Q. Xu, X. D. Zhang, X. X. Li, Y. Cao, P. Y. Gao, J. J. Kang, J. Wu and L. Li, Chin. Chem. Lett., 2022, 33, 4610–4616 CrossRef CAS.
- Q. Y. He, J. Chen, J. H. Yan, S. D. Cai, H. J. Xiong, Y. F. Liu, D. M. Peng, M. Mo and Z. B. Liu, Asian J. Pharm. Sci., 2020, 15, 416–448 CrossRef PubMed.
- R. Cheng, F. H. Meng, C. Deng and Z. Y. Zhong, Nano Today, 2015, 10, 656–670 CrossRef CAS.
- Z. Y. Shen, N. Ma, F. Wang, J. M. Ren, C. X. Hou, S. Chao, Y. X. Pei and Z. C. Pei, Chin. Chem. Lett., 2022, 33, 4563–4566 CrossRef CAS.
- T. T. Sun, J. Xu, T. B. Chen, C. L. Tu, L. J. Zhu and D. Y. Yan, Biomater. Sci., 2022, 10, 997–1007 RSC.
- L. Y. Wang, C. L. Dai, Y. F. Fang, X. R. You and J. Wu, Nano Res., 2022, 15, 4544–4551 CrossRef CAS.
- X. D. Jing, H. Hu, Y. Z. Sun, B. Yu, H. L. Cong and Y. Q. Shen, Small Methods, 2022, 6, 2101437 CrossRef CAS PubMed.
- D. D. Li, R. H. Zhang, G. T. Liu, Y. Kang and J. Wu, Adv. Healthcare Mater., 2020, 9, 2000605 CrossRef CAS PubMed.
- S. H. Tang, G. H. Li, H. Zhang, Y. J. Bao, X. D. Wu, R. Yan, Z. Q. Wang and Y. X. Jin, Biomater. Sci., 2023, 11, 3128–3143 RSC.
- H. L. Chen, Z. M. Liu, B. Wei, J. Huang, X. R. You, J. Y. Zhang, Z. L. Yuan, Z. L. Tang, Z. Y. Guo and J. Wu, Bioact. Mater., 2021, 6, 655–665 CrossRef CAS PubMed.
- M. A. Badgley, D. M. Kremer, H. C. Maurer, K. E. DelGiorno, H. J. Lee, V. Purohit, I. R. Sagalovskiy, A. Ma, J. Kapilian, C. E. M. Firl, A. R. Decker, S. A. Sastra, C. F. Palermo, L. R. Andrade, P. Sajjakulnukit, L. Zhang, Z. P. Tolstyka, T. Hirschhorn, C. Lamb, T. Liu, W. Gu, E. S. Seeley, E. Stone, G. Georgiou, U. Manor, A. Iuga, G. M. Wahl, B. R. Stockwell, C. A. Lyssiotis and K. P. Olive, Science, 2020, 368, 85–89 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- C. V. Smith, D. P. Jones, T. M. Guenthner, L. H. Lash and B. H. Lauterburg, Toxicol. Appl. Pharmacol., 1996, 140, 1–12 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- H. L. Sun, Y. F. Zhang and Z. Y. Zhong, Adv. Drug Delivery Rev., 2018, 132, 16–32 CrossRef CAS PubMed.
- D. C. Qi, L. Xing, L. J. Shen, W. S. Sun, C. Cai, C. H. Xue, X. W. Song, H. Yu, H. L. Jiang, C. J. Li, Q. R. Jin and Z. Q. Zhang, Chin. Chem. Lett., 2022, 33, 4595–4599 CrossRef CAS.
- Y. X. Xiong, C. Xiao, Z. F. Li and X. L. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC.
- E. K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS PubMed.
- X. Y. Zhang, K. Hong, Q. M. Sun, Y. Q. Zhu and J. Z. Du, Biomater. Sci., 2021, 9, 5275–5292 RSC.
- Y. H. Wang, Y. H. Jiang, D. S. Wei, P. Singh, Y. J. Yu, T. Lee, L. P. Zhang, H. K. Mandl, A. S. Piotrowski-Daspit, X. Y. Chen, F. Li, X. Li, Y. Y. Cheng, A. Josowitz, F. Yang, Y. Zhao, F. Y. Wang, Z. W. Zhao, A. Huttner, R. S. Bindra, H. H. Xiao and W. M. Saltzman, Nat. Biomed. Eng., 2021, 5, 1048–1058 CrossRef CAS PubMed.
- X. J. Xu, H. L. Tang, J. J. Guo, H. H. Xin and Y. Ping, Signal Transduction Targeted Ther., 2022, 7, 269 CrossRef PubMed.
- E. J. Goethals and C. Sillis, Macromol. Chem., 1968, 119, 249–251 CrossRef CAS.
- R. L. Whistler and D. J. Hoffman, J. Polym. Sci., Part A: Polym. Chem., 1967, 5, 2111–2117 CrossRef CAS.
- G. Gasparini, E. K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- W. T. Lu, X. X. Wang, R. Cheng, C. Deng, F. H. Meng and Z. Y. Zhong, Polym. Chem., 2015, 6, 6001–6010 RSC.
- Y. B. Meng and J. Wu, Chin. J. Polym. Sci., 2022, 40, 1016–1027 CrossRef CAS.
- B. J. Sun, C. Luo, H. Yu, X. B. Zhang, Q. Chen, W. Q. Yang, M. L. Wang, Q. M. Kan, H. T. Zhang, Y. J. Wang, Z. G. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- D. J. Wu, A. Ensinas, B. Verrier, C. Primard, A. Cuvillier, G. Champier, S. Paul and T. Delair, J. Mater. Chem. B, 2016, 4, 5455–5463 RSC.
- S. D. Li and L. Huang, Biochim. Biophys. Acta, 2009, 1788, 2259–2266 CrossRef CAS PubMed.
- J. Y. Tang, O. Y. Fu, M. F. Hou, H. W. Huang, H. R. Wang, K. T. Li, S. Fayyaz, C. W. Shu and H. W. Chang, Semin. Cancer Biol., 2019, 58, 109–117 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00461a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |