
Transdermal delivery of allopurinol to acute hyperuricemic mice via polymer microneedles for the regulation of serum uric acid levels†
Rui
Wang
a,
Han
Wang
b,
Guohua
Jiang
 *ac,
Yanfang
Sun
*d,
Tianqi
Liu
a,
Lei
Nie
ef,
Amin
Shavandi
f,
Khaydar E.
Yunusov
g,
Uladzislau E.
Aharodnikau
h and
Sergey O.
Solomevich
h
*ac,
Yanfang
Sun
*d,
Tianqi
Liu
a,
Lei
Nie
ef,
Amin
Shavandi
f,
Khaydar E.
Yunusov
g,
Uladzislau E.
Aharodnikau
h and
Sergey O.
Solomevich
h
aSchool of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou, 310018, China. E-mail: ghjiang_cn@zstu.edu.cn
bWenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, China
cInternational Scientific and Technological Cooperation Base of Intelligent Biomaterials and Functional Fibers, Hangzhou, 310018, China
dCollege of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, Zhejiang 310018, China. E-mail: katherineyfs@zzstu.edu.cn
eCollege of Life Science, Xinyang Normal University, Xinyang, China
f3BIO-BioMatter, École polytechnique de Bruxelles, Université Libre de Bruxelles (ULB), Brussels, Belgium
gInstitute of Polymer Chemistry and Physics, Uzbekistan Academy of Sciences, Tashkent, Uzbekistan
hResearch Institute for Physical Chemical Problems, Belarusian State University, Minsk, Belarus
First published on 21st December 2022
Abstract
Allopurinol (AP) is widely used to treat hyperuricemia which may cause severe side effects upon oral administration. Alternative means for the treatment of hyperuricemia are demanded to simultaneously facilitate drug absorption, patient compliance, and fewer side effects. In this study, a new polymer microneedle (MN) system was developed for the transdermal delivery of AP to acute hyperuricemic mice. This study aims to achieve the controllable regulation of serum uric acid (SUA) levels with fewer side effects compared with oral administration. The matrix of polymer MNs consisted of polyvinylpyrrolidone (PVP) and polycaprolactone (PCL), in which the rapid dissolution of PVP offers a rapid dissolution of AP into the blood and the biodegradability of PCL resulting in a sustainable drug release behavior. An in vivo study demonstrated that the AP-loaded MN system can effectively reduce the SUA levels as oral administration with lower side effects, which will be conducive to reducing the adverse reactions and improving the bioavailability of AP. This MN-mediated strategy can facilitate transcutaneous hyperuricemia treatment and provide a new alternative for the exploration of clinical treatment of hyperuricemia and improvement of patient compliance.
Introduction
Gout, a common chronic disease caused by the deposition of monosodium urate (MSU) crystals in the articular and non-articular structures, has a substantial disease burden and is increasing among different ethnic groups and regions, which may become a modern gout epidemic analogous to the obesity epidemic.1–6 The progression of gout can be divided into multiple stages.7,8 The first is the development of hyperuricemia, which is an essential but not sufficient step for the development of gout.2,9 The majority of people with hyperuricemia will not suffer from gout, even after prolonged periods of observation.7 In addition, it is estimated that hyperuricaemia prevalence in China is 13.3%, while that of gout is 1.1%. Hence, hyperuricemia may not necessarily develop into gout, but the probability of gout will be greatly increased with sustained hyperuricemia, which is a physiological prerequisite for gout.1,2,10 Inspired by this, we tried to prevent people from suffering from gout by treating hyperuricemia in advance.Non-drug therapy, such as diet, reduced intake of purine-rich foods (beer, meat, and seafood) and increased water consumption, provides physical barriers for hyperuricemia patients but these barrier approaches generally have high failure rates due to poor patient compliance and acceptance.11–13 Controlling uric acid (UA) production is the best way to prevent and treat hyperuricemia through drug therapy.2 In the past decades, AP has been used as the most anti-hyperuricemia drug14 to control hyperuricemia and gout and reduce the complications of cardiovascular disease and renal failure.15 However, using drugs for the treatment of hyperuricemia may give rise to serious side effects such as gastrointestinal intolerance, liver/kidney damage, allergy, and central nervous system disorders.16–18 Therefore, it is urgent to find a way to accelerate drug absorption, avoid the first-pass effect and increase drug efficacy, which will improve the bioavailability of anti-hyperuricemia drugs.19–23
Microneedles (MNs) are micrometre-scale structures that penetrate the stratum corneum of the skin to deliver drugs in a minimally invasive manner and can be utilized as a new drug delivery system.24–32 Given the potential of MNs for their minimal invasiveness and self-administration without professional training, the application of MNs has grown rapidly. Hitherto, MNs have been used for the delivery of drugs, which include insulin,33,34 vaccines,35–37 protein, and various high- and low-molecular-weight compounds.30,38 Several diagnostic applications of MNs have been reported which are largely based on using MNs for drawing blood or other fluids for biomarkers detection.39,40 Given that MNs can effectively penetrate the epidermis and deliver drugs, MNs have also been reported for skin wound healing.41,42
Among the several different types of MNs, a matrix of dissolvable MNs comprised of biocompatible and biodegradable polymers, such as polyvinylpyrrolidone (PVP),43–45 a water-soluble polymer widely used in the pharmaceutical industry, and polycaprolactone (PCL), a biodegradable and biocompatible polymer with a low melting temperature of 59–64 °C,46,47 are approved by US FDA for use in a variety of medical devices.48–50
In this study, we present a new MN system for the transdermal delivery of AP to acute hyperuricemic mice. This study aims to achieve a controllable regulation of serum uric acid (SUA) levels with fewer side effects when administered by MNs. The matrix of MNs consists of PVP and PCL; the rapid dissolution of PVP enables the rapid dissolution of AP into the blood and the biodegradability of PCL results in a sustainable drug release behavior. In addition, the mechanical strength of the MNs can be further improved by mixing PCL with the matrix of needle tips. The water solubility of PVP increases the probability of PCL contact with tissue fluid, thus promoting the hydrolysis process and achieving rapid degradation of PCL in vivo.51 The chemical, morphological and mechanical properties of the MNs were investigated, and then in vivo studies were performed to compare the efficacy and side effects of oral and MN treatment on SUA levels. We envisage that this AP-loaded MN system can provide a new avenue to increase acute-hyperuricemic patients’ compliance and reduce side effects.
Experimental
Materials
Polyvinylpyrrolidone (PVP Mw = 58 kDa), polycaprolactone (PCL Mw = 45 kDa), polyvinyl alcohol (PVA, 1788 type, alcoholysis = 87.0–89.0%), potassium oxonate (98%, Mw = 195.18) and hypoxanthine (99%) were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). Allopurinol (AP, 98%) and rhodamine 6G (R6G) were obtained from Aladdin Chemistry Co. Ltd (Shanghai, China). Serum uric acid (UA) (Art. No. E-BC-K016-M), serum creatinine (SCr) (Art. No. E-BC-K188-M), nitric oxide (NO) colorimetric assay kit (Art. No. E-BC-K035-M) and adenosine deaminase (ADA) activity assay kit (Art. No. E-BC-K197-M) were obtained from Elabscience Biotechnology Co., Ltd (Wuhan, China). Strat-M membranes with a diameter of 25 mm and thickness of 300 μm were purchased from Shanghai Xuanyi Environmental Protection Technology Co., Ltd (Shanghai, China).Design and fabrication of MNs
The fabrication process of AP-loaded MNs is shown in Fig. 1a. PCL and PVP with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were dissolved in a solvent (dichloromethane/ethanol, 50/50 in v/v). Under magnetic stirring at 400 rpm for 60 min, a homogeneous solution with a concentration of ∼40 wt% was formed. Then, AP was dissolved in the homogeneous solution of PVP and PCL to achieve a concentration of ∼40.2 wt%. The as-obtained solution was magnetically stirred at 400 rpm for 2 h to ensure that the drug is uniformly distributed in the matrix solution. In addition, to ensure the distribution of drugs in each MN, the drug content, stirring time, stirring speed and vacuum pumping times were kept the same for the preparation of all MN patches. The as-obtained solution was filled into the PDMS mold (20 × 20 arrays) with a maximum drug loading capacity of ∼100 mg by vacuum pumping, and the excess solution on the surface was scraped off. Thereafter, an aqueous solution of PVA/PVP (20 wt%) was pipetted onto the mold to form the patch backing layer. After completely drying for 12 h at 45 °C, the dried MNs were carefully peeled off and finally sealed and placed in a vacuum incubator for further analysis. The structure, morphology, and geometric dimensions of the fabricated MNs were visualized by scanning electron microscopy (SEM, Vltra55, Zeiss, Germany) and three-dimensional laser scanning confocal microscopy (LSCM, C2, Nikon Corporation, Japan).
:3 were dissolved in a solvent (dichloromethane/ethanol, 50/50 in v/v). Under magnetic stirring at 400 rpm for 60 min, a homogeneous solution with a concentration of ∼40 wt% was formed. Then, AP was dissolved in the homogeneous solution of PVP and PCL to achieve a concentration of ∼40.2 wt%. The as-obtained solution was magnetically stirred at 400 rpm for 2 h to ensure that the drug is uniformly distributed in the matrix solution. In addition, to ensure the distribution of drugs in each MN, the drug content, stirring time, stirring speed and vacuum pumping times were kept the same for the preparation of all MN patches. The as-obtained solution was filled into the PDMS mold (20 × 20 arrays) with a maximum drug loading capacity of ∼100 mg by vacuum pumping, and the excess solution on the surface was scraped off. Thereafter, an aqueous solution of PVA/PVP (20 wt%) was pipetted onto the mold to form the patch backing layer. After completely drying for 12 h at 45 °C, the dried MNs were carefully peeled off and finally sealed and placed in a vacuum incubator for further analysis. The structure, morphology, and geometric dimensions of the fabricated MNs were visualized by scanning electron microscopy (SEM, Vltra55, Zeiss, Germany) and three-dimensional laser scanning confocal microscopy (LSCM, C2, Nikon Corporation, Japan).
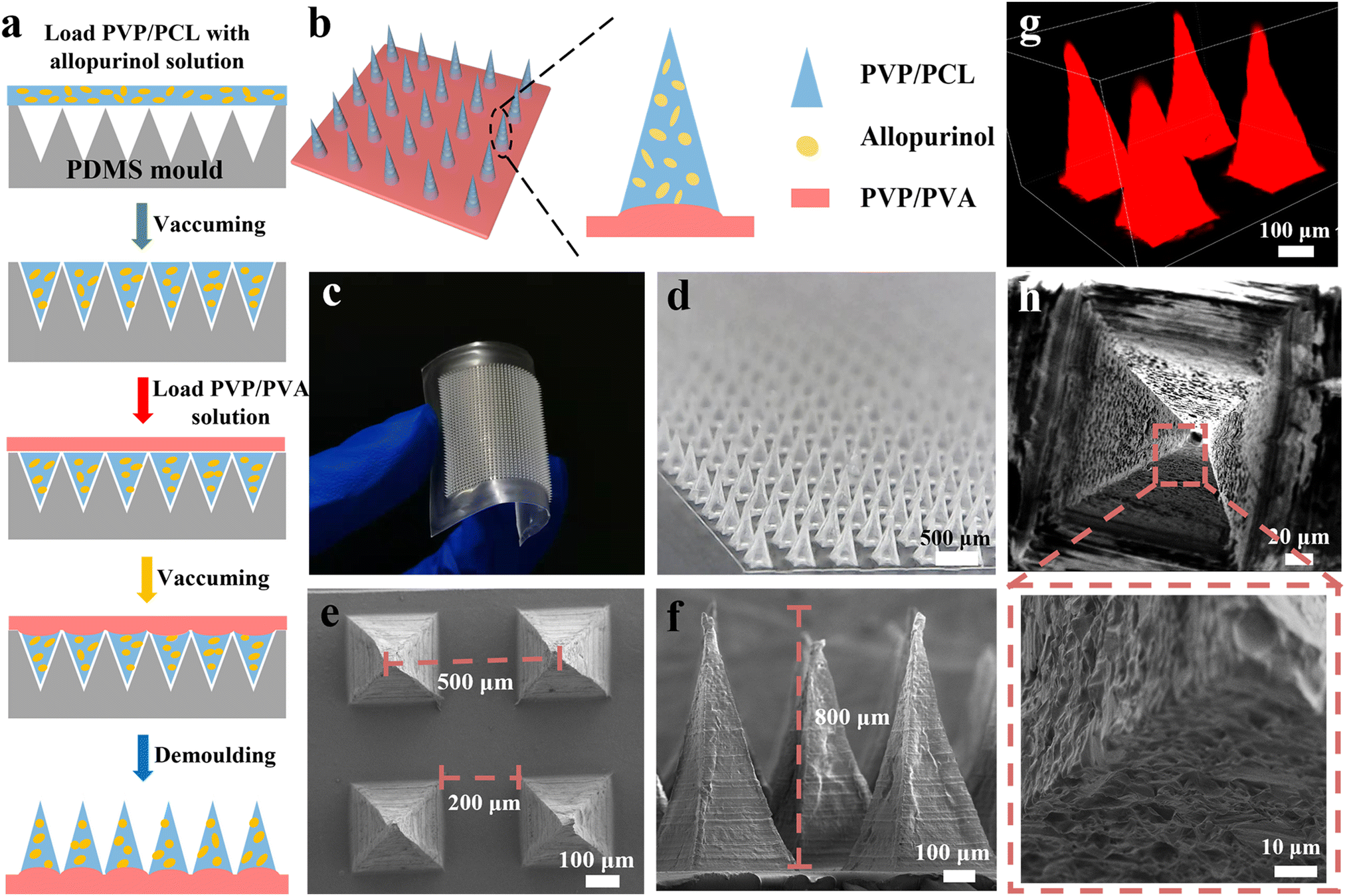 | ||
| Fig. 1 Schematic of the fabrication process of MNs (a), schematic structure of MNs (b), digital images of the as-fabricated MNs (c and d), SEM images of PVP/PCL MNs (e and f), the confocal image of MN arrays (g), and the porous structure of MNs formed by the rapid dissolution of PVP (h). | ||
Mechanical strength measurement
To explore whether the MNs have sufficient strength to pierce the skin, the mechanical properties of the MNs were evaluated by a compression test.17 Before and after the compression test, the morphological changes of the MNs were observed by an optical microscope (OM, Imager A1m). In addition, the skin embedding and insertion potentials were examined by an ex vivo skin insertion test.In vitro drug release study
The prepared MNs were put into vials containing 20 mL of PBS and the vials were incubated in a water bath at 37 °C. Thereafter, at distinct time intervals, a 3 mL aliquot was taken out from each sample and supplemented with an equal volume of PBS. Eventually, the amounts of AP released from the MNs were quantitatively analyzed by a ultraviolet-visible spectrophotometer (UV, TU-1901, Beijing Purkinje General Instrument Co., Ltd, China).To further investigate drug diffusion and absorption through the skin, a Franz diffusion cell system was used for in vitro transdermal absorption detection. Briefly, 25 mL of PBS was added to the receiving chamber (the level of PBS buffer was exactly 1 mm higher than the surface of the skin in vitro to ensure stable contact between the PBS and the mouse skin during the sampling process). Then, the Strat-M membrane inserted with the MN patch was used as a carrier and fixed between the receiving chamber and supply chamber of the Franz diffusion cell. The as-assembled Franz diffusion cell was incubated in a water bath at 37 °C under magnetic stirring at 350 rpm. At distinct intervals, a 3 mL aliquot was taken out from the sampling port and replenished with an equal amount of PBS. Ultimately, the amount of the drug was determined by UV spectrophotometry.
In vivo pharmacodynamic study
ICR mice (male, 20–22 g, Wenzhou Research Institute, Chinese Academy of Sciences) were used in this study. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Wenzhou Research Institute, Chinese Academy of Sciences and approved by the Animal Ethics Committee of WIUCAS22121604. The trends of the SUA levels of the two induction protocols at predetermined time intervals were monitored to select an appropriate hyperuricemia mice model: (1) intraperitoneal injection of hypoxanthine (500 mg kg−1) combined with subcutaneous injection of potassium oxonate (100 mg kg−1) to obtain a hyperuricemia mice model and (2) an intraperitoneal injection of hypoxanthine (500 mg kg−1) combined with a subcutaneous injection of potassium oxonate (400 mg kg−1) to get another model of hyperuricemic mice. Three mice per group were used to do the statistical study.At distinct intervals, the mice were bled from the orbit and placed in a refrigerator at 4 °C overnight. The serum was obtained by centrifuging the collected blood at 3000 rpm for 10 min and placing it in a refrigerator at 4 °C to determine the amount of serum uric acid (SUA), serum creatinine (SCr), and nitric oxide (NO) by colorimetry. A specific operation process is used for the microplate reader to detect the absorbance of the standard hole and the measuring hole under specified conditions at the optimal detection wavelength and then use the formula on the biochemical assay kit to obtain the results.
Histological and immunohistochemical analysis
The mice were sacrificed and their organs (heart, liver, spleen, lung, and kidneys) were retrieved. Subsequently, a part of the liver tissue was taken, instantly snap-frozen in liquid nitrogen, and then stored at −80 °C to measure the activity of ADA in the liver. Briefly, after weighing the liver tissue block, 9 times the volume of pre-cooled 0.9% sodium chloride solution was added to prepare a 10% tissue homogenate. Afterward, the supernatant was obtained by centrifuging the tissue homogenate at 2500 rpm for 10 min, which was used to measure the ADA activity of the tissue homogenate using colorimetry.The retrieved kidney tissues were fixed with paraformaldehyde (4%) overnight for histological studies. First, the formol-fixed kidney tissue was dehydrated in graded alcohol (75%, 85%, 95%, and 100%), then cleared in xylene, and finally embedded in paraffin. Then, small sections (∼5 μm in thickness) were stained with standard hematoxylin–eosin (H&E) solution and examined closely with an optical microscope to identify the histopathological lesions.
Statistical analysis
Statistical analysis of the experimental data was conducted using GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA) and a statistical comparison of the analytical values was obtained by one-way ANOVA analysis. The values of p < 0.05 (*), 0.01 (**), 0.001 (***) and 0.0001 (****) were considered as statistically significant differences. The values were presented as means with standard deviation (S.D.).Results and discussion
Fabrication of MN patches
To achieve a sustained anti-hyperuricemia effect, a new MNs system is developed for the transdermal delivery of AP for the regulation of SUA levels in acute hyperuricemic mice. The matrix of MNs consists of PVP and PCL, in which the rapid dissolution of PVP offers rapid delivery of AP into blood,45,52,53 the biodegradability of PCL results in the sustainable drug release, and offers the necessary mechanical strength for MNs.32 Firstly, the mixed homogeneous solution of PCL, PVP, and AP is coated on the PDMS MN mold to form the needle tips. Then the solution of PVA/PVP is poured onto the mold to form the dissolving back patching (Fig. 1b). The resultant PVP/PCL MNs contain ∼0.2 mg of AP, which is determined by converting the body surface area of the humans and ICR mice. The drug loading in MNs can be precisely controlled by adjusting the concentration of AP (Fig. S1 and S2†).Fig. 1c and d display the digital microscopic images of the prepared microneedle patch, indicating a flexible back patch and a regular microneedle array. The SEM images (Fig. 1e and f) of the as-fabricated MNs exhibit a pyramidal shape, which is also confirmed by the stereomicroscope images (Fig. S3†). The base width of pyramidal MNs and the spacing between the adjacent needle tips of MNs are ∼200 and ∼500 μm, respectively. The height of the MNs is ∼800 μm. To further investigate the geometrical structure of the MNs, R6G was mixed with the homogeneous solution of the needle tips when filled into the cavities of the mold, so that the MNs arrays with red needle tips can be observed using LSCM (Fig. 1g). In addition, the porous structure of MNs can also be observed after the immersion of MN patches into PBS solution for 5 min due to the high solubility of PVP (Fig. 1h and S4a†), which is beneficial for rapidly releasing AP and accelerating the biodegradation of PCL in the body fluid.
Mechanical properties and drug release of MNs
The mechanical properties of the as-fabricated MNs are tested by a compression test. Fig. 2a shows the force-displacement graph of the PVP/PCL and PVP MNs. The pure PVP MNs could tolerate compressive forces of 0.12 N per needle with a displacement of 600 μm. In addition, the PVP/PCL MNs could withstand a maximum compressive force of ∼0.23 N per needle. Although the mechanical strength of pure PVP MNs is sufficient to penetrate the skin,54 it can be further improved by mixing PCL into the matrix of the needle tips. Moreover, the insets in Fig. 2a and S4b† show the morphology of the PVP/PCL MNs after the compression test, and there are no signs that correspond to the puncture sites after the insertion and removal of MNs. The H&E-stained histological section image clearly exhibits that the MNs can penetrate the skin with a depth of ∼500 μm (Fig. 2c). The difference between the size of the pinhole in the histological image and MNs is due to the skin surface deformation during the insertion of MNs.55Fig. 2d shows the fluorescence image of the histological sections after insertion by MNs containing R6G. Strong red fluorescence can be observed around the puncture area. In addition, many microchannels with a depth of ∼300–500 μm can be observed in Fig. 2e, which verifies that the prepared MNs can efficiently penetrate the skin tissue. Moreover, red fluorescence signals are also found around the puncture sites (Fig. 2f), which further verified that the drugs can be released into the skin tissue due to the dissolution of the MN polymer matrix.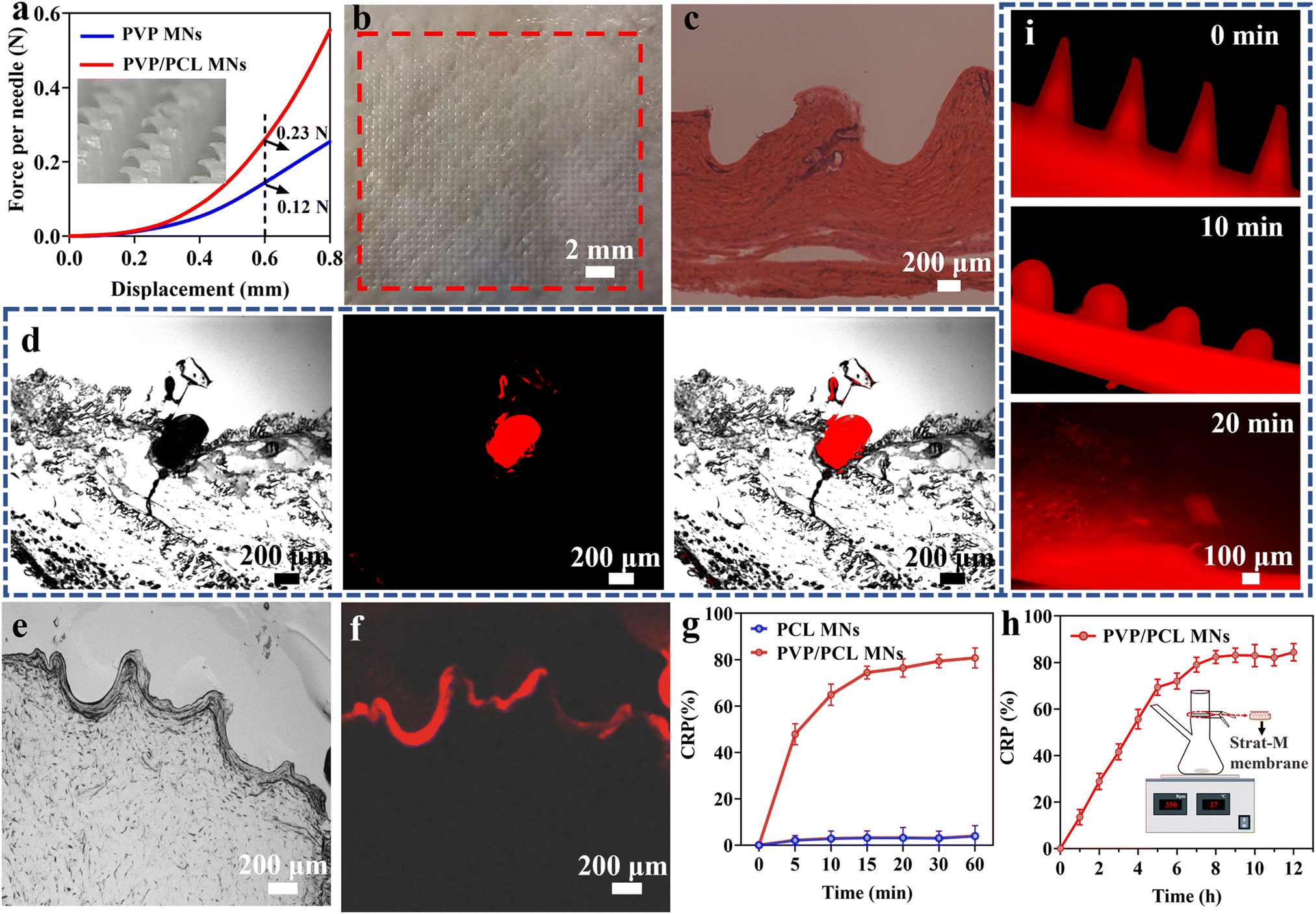 | ||
| Fig. 2 The force–displacement graph of PVP/PCL and PVP MNs (a), digital images of porcine skin after the insertion and removal of MNs (b), H&E-stained histological section images obtained by an optical microscope (c), fluorescent images of histological sections after insertion by MNs containing R6G (d), histological images of porcine skin after the removal of MNs ex vivo (e and f), in vitro AP release curves of MNs in PBS solution (g), and the in vitro skin drug release profile of AP-loaded PVP/PCL MNs (h), and the digital images of MN dissolution in mouse cadaver skin ex vivo for 1, 10, and 20 min (i). | ||
To evaluate the drug release behaviours, the cumulated release percentage (CRP) of AP from AP-loaded MNs is tested in vitro using PBS solution (pH = 7.4) as the release media. AP can be rapidly released from MNs within 15 min as shown in Fig. 2g. However, almost no drug release can be observed from the PCL MNs due to the slow degradation of PCL. However, when PVP/PCL MNs penetrate the skin, the water solubility of PVP increases the probability of PCL contact with tissue fluid, thus promoting the hydrolysis process and achieving rapid degradation of PCL in vivo.51 Moreover, the degradation rate of PCL will be further accelerated due to the synergistic effect of hydrolysis and enzyme.56 In addition, it can be seen from Fig. 2g that AP is rapidly released from PVP/PCL MNs which is close to 80% within 15 min. After that, the release of AP stabilizes due to the slow degradation of PCL. Therefore, we consider that 80% of the drugs are loaded in PVP and 20% in the PCL portions (Fig. S5†). To more accurately reflect the penetration and diffusion of the drugs in the skin, a Franz diffusion cell was used to conduct an in vitro skin drug release test (Fig. S6†). The schematic of the Franz diffusion cell is shown in the inset of Fig. 2h. The AP-loaded PVP/PCL MNs are applied to the Strat-M membrane, which is fixed in the Franz diffusion cell by pressing a pinch clamp. Samples were taken from the sampling port and the amount of AP in each sample is quantitated at different intervals. The AP release profiles of the MNs show a steady increase within the first 7 h, and ∼70% of AP can be released after 7 h after application. It indicates the drug in the MNs can be effectively delivered through the micropores formed by the microneedles penetrating the skin. In addition, PVP/PCL MN arrays containing R6G were inserted into mouse cadaver skin (Fig. S4c†), and its dissolution over time was monitored. Fig. 2i shows the fluorescence morphological changes of MNs after the insertion of PVP/PCL MNs into the skin for 0, 10, and 20 min. The clear sharp MN edges observed before insertion become round after insertion for 10 min and disappear after insertion for 20 min.
Regulation of SUA in vivo
To investigate the regulation of SUA in vivo, an ICR mice hyperuricemia model was established by intraperitoneal injection of hypoxanthine and subcutaneous injection of potassium oxonate. The SUA level can reach the maximum level of ∼520 μmol L−1, 3 times higher than the normal state, 2 h after the intraperitoneal injection of hypoxanthine (∼500 mg kg−1) and the subcutaneous injection of potassium oxonate (100 mg kg−1). Then, the SUA level decreased slowly to ∼250 μmol L−1 6 h after the injection due to the presence of the urate oxidase (UOX) gene in mice, which can oxidize SUA into allantoin. Allantoin is more soluble in water than SUA and easier to be excreted with urine57 (Fig. S7†). However, the SUA level can be maintained at higher levels by intraperitoneal injection of hypoxanthine (∼500 mg kg−1) and subcutaneous injection of potassium oxonate (400 mg kg−1). The maximum level (∼650 μmol L−1) can be obtained 1.5 h after the injection, which can still be maintained at ∼500 μmol L−1 after 6 h (Fig. S8†). Therefore, the latter method was adopted for establishing the hyperuricemia model.The therapeutic efficiency of the delivery of AP via MNs was evaluated in vivo using the hyperuricemia model. The hyperuricemia mice were randomly divided into 4 groups after modeling: (I) control group (health), (II) model group (without any treatment), (III) oral group (treatment by oral AP), (IV) MN group (treatment by AP-loaded PCL or PVP/PCL MNs), as shown in Fig. 3a. Note that, when AP-loaded MNs were manually placed on in vivo mice skin and gently pressed, the MNs penetrated the skin and embedded completely under the skin surface (Fig. S9a†). Fig. 3b shows the profiles of SUA levels against time of the different groups after treatment. The SUA level can be kept stable at ∼150 μmol L−1 in the health group. In the case of the model group, the SUA level can be decreased slowly from ∼650 μmol L−1 to ∼500 μmol L−1 after 5 h. After oral treatment by AP (∼10 mg kg−1), the SUA level can be decreased slowly from ∼600 μmol L−1 after 1 h to ∼300 μmol L−1 after 5 h. However, the SUA level shows a slight decrease (from ∼650 to 480 μmol L−1) for the AP-loaded PCL MNs, although with the same dose amount as the oral group. It can be attributed to the slow degradation of PCL in the body. The hyperuricemia model treated by AP-loaded PVP/PCL MNs exhibits the best treatment among all groups. After treatment for 1 h, the SUA level becomes ∼550 μmol L−1. It further decreased to the lowest concentration of ∼157.49 ± 23.46 μmol L−1 after 3 h which is close to the level of the health group. The SUA levels still can be maintained at the therapeutic level (∼150 μmol L−1) even after 5 h, thus exhibiting a longer inhibition time against increasing SUA levels and the best treatment effect among all groups. It is mainly attributed to the composition of the matrix of MNs. Some drugs are released rapidly following the dissolution of PVP, resulting in the rapid diffusion of AP into blood similar to the oral delivery route. The biodegradability of PCL can also be accelerated due to the dissolution of PVP. The AP loaded in PCL can serve as a drug reservoir for sustained drug release.
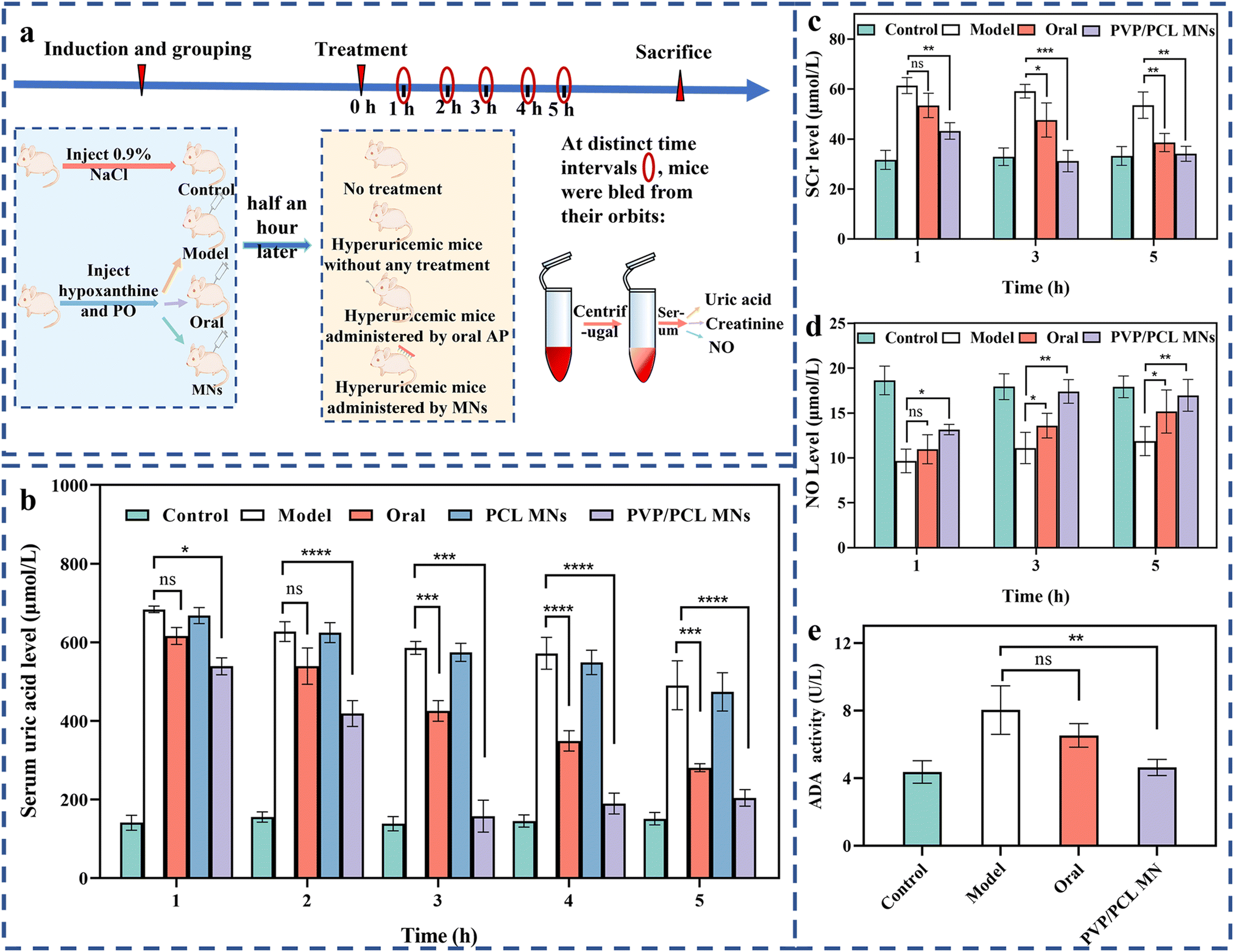 | ||
| Fig. 3 The animal experiment process via polymer MNs for the regulation of SUA levels (a), the SUA (b), SCr (c) and NO (d) levels of the mice treated by AP-loaded PVP/PCL MNs compared with the healthy, model, oral and PCL MN groups, and the ADA activity of the liver tissue of the mice treated with AP-loaded PVP/PCL MNs compared with the healthy, model and oral groups (e). | ||
Blood and immunohistochemical analysis
Serum creatinine (SCr) is the most commonly used screening test for kidney and renal functions, and its high concentration is associated with renal function injury.58,59 Therefore, the SCr levels can be used to determine renal function. Fig. 3c shows the SCr levels of all groups. The SCr of the model group shows the highest levels among all groups at different intervals, indicating the most severe kidney and renal damage due to no drug treatment. In the case of the oral group, the SCr levels show a slow decreasing trend after 1 to 5 h of treatment. Although the SCr levels exhibit a similar trend in the PVP/PCL MN group, they are lower than those of the oral groups at all intervals. Importantly, the SCr levels (∼32 μmol L−1) are close to the health group after 5 h of treatment. It indicates that negligible kidney and renal function damage is induced by MN treatment because of the lower AP concentration in the blood compared with that in the oral group.In addition, NO (also known as an endothelial-derived relaxing factor, EDRF), as an important mediator regulating vascular tone, is also affected by the decreasing renal blood flow that caused the impairment of the vasodilation function.59,60 Therefore, the level of NO is another biomarker for hyperuricemia.61Fig. 3d shows the NO levels among all groups at different intervals. At 1 h, no significant difference was found among all experimental groups. However, the higher NO levels of the PVP/PCL MNs groups enrolled in this study, compared to the model and oral groups, reach the normal level after 5 h of treatment.
Adenosine deaminase (ADA) is an important enzyme of nucleoside metabolism.62 The increase in ADA activity will promote the catabolism of nucleic acid and lead to an increase in SUA production.62 Therefore, the inhibition of ADA activity can efficiently reduce the concentration of UA.63 To determine the hepatic activity of ADA, ADA activities from the liver tissue are tested using a commercial ADA activity assay kit. As shown in Fig. 3e, after 5 h, compared with the control group, the ADA activity of the model group increased significantly. However, significant inhibition of the hepatic ADA activity occurred in the oral and PVP/PCL MNs groups compared with the model group. In particular, the hepatic ADA activity of the PVP/PCL MN group remains at a relatively low level that is close to the healthy group and significantly lower than that of the model group. Taken together, AP-loaded MN treatment can reverse liver and kidney damage by effectively reducing the production of SUA, thereby promoting the secretion of SCR and NO and inhibiting the activity of ADA in mice. The relatively higher ADA activity in the oral group may be attributed to the liver damage caused by oral administration.
Histopathology analysis
At the end of 6 hours of treatment, the treated mice were sacrificed and the heart, liver, spleen, lung, and kidney tissues were retrieved (Fig. 4a). No tissue lesions were observed in the heart, spleen, and lung (Fig. S9b†). No significant drastic liver and kidney changes are found in the PVP/PCL MNs groups compared with the control group, suggesting that the MN administration did not cause remarkable liver damage and renal impairment. In contrast, acute liver and kidney damage was found in the model group, which was confirmed by the increase in the SCr and ADA levels. In addition, traditional oral drug administration also showed severe liver toxicity due to the first-pass effect and slow clearance. Hence, the MN-mediated strategy explored in this study can be used as an attractive anti-hyperuricemia treatment with lower damage and toxicity and a longer therapeutic effect.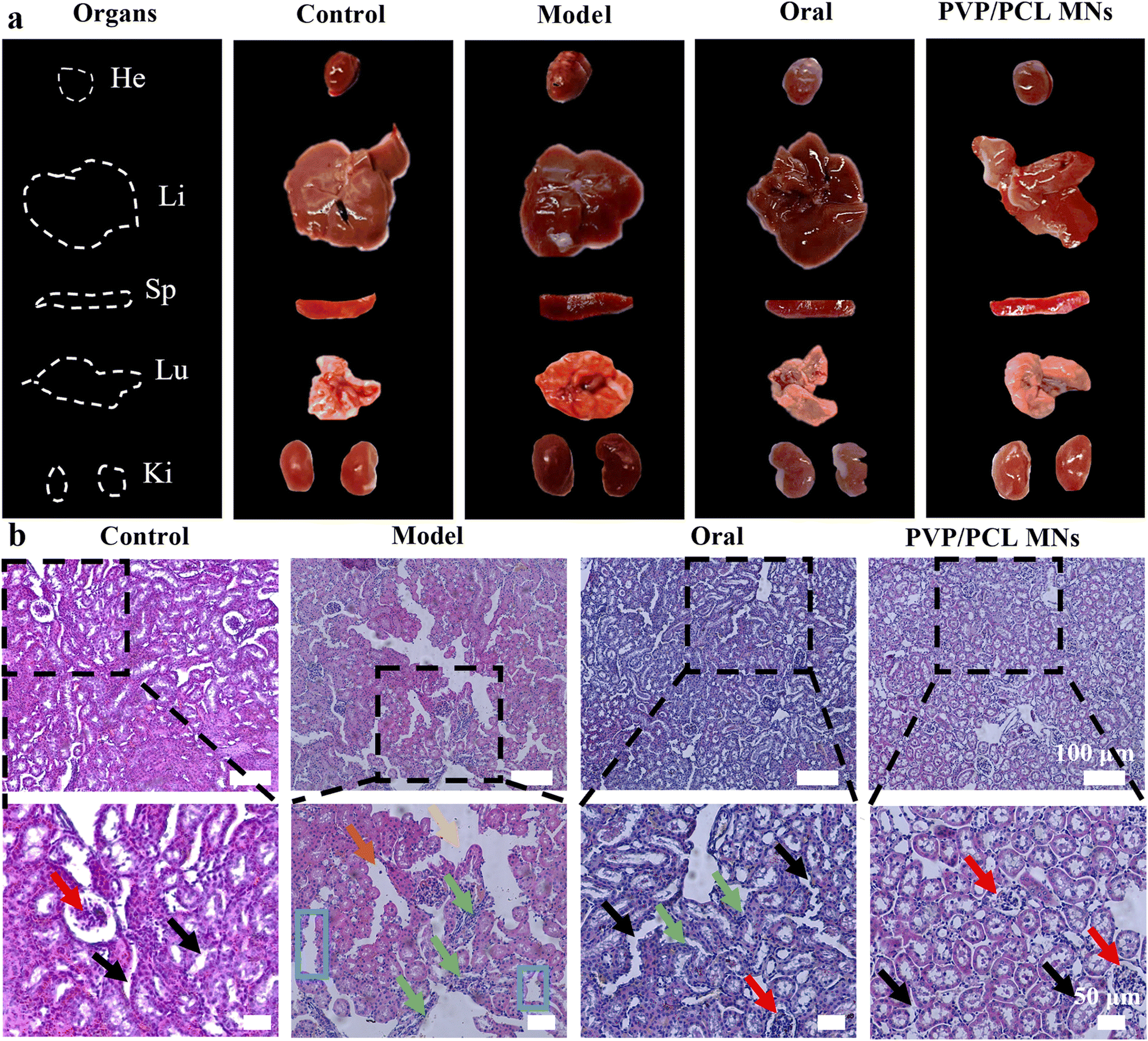 | ||
| Fig. 4 The images of major organs (heart, liver, spleen, lung, and kidneys) retrieved from the mice (a) and H&E staining images of kidney tissue slices from treated mice (b) (red arrow: normal glomerulus morphology, black arrow: complete brush border structure, yellow arrow: dilatation of renal tubule, orange arrow: renal tubular epithelial cell necrosis, green arrow: renal interstitial inflammatory cell infiltration, blue square frame: the disappearance of the brush border). | ||
The kidneys are an important functional organ for urine production. However, high SUA levels can cause damage to the glomerulus, renal tubules, and epithelial cells of the renal tubular.64 For this reason, histological studies of renal parenchyma were performed. As shown in Fig. 4b, the kidney section of the blank control shows regular cell arrangement, normal hepatic architecture, and glomerulus morphology without inflammation or necrosis. However, the hyperuricemic model group depicts mild tubular edema. In addition, the dilatation of renal tubules, the degeneration of tubular epithelial cells, and the disappearance of the brush border of the epithelial cells were observed in the model group. In the oral group, some whites were observed on the surface of the renal tissue, revealing severe nephrotoxicity. Meanwhile, the phenomenon of inflammatory cell infiltration in the renal interstitium was also observed, which indicates that oral administration worsens kidney damage. However, the kidney section of the PVP/PCL MN group shows normal glomerulus morphology and a complete brush border structure, which was similar to the control group, suggesting that it could validly alleviate the renal histopathological damages in hyperuricemic mice. In short, MN-mediated strategies not only have a significant hypouricemic effect but also have protective effects on liver and kidney tissues.
Conclusions
In view of the limited clinical efficacy and severe side effects of conventional oral AP in the treatment of hyperuricemia and gout, herein, we proposed AP encapsulated in PVP/PCL composite MNs by the mold casting method to regulate SUA levels, realize sustained drug release and reduce or even reverse the damage to the kidneys. The mechanical strength of the as-fabricated MNs provides sufficient potential to pierce the skin and release AP in a sustained pattern due to the dissolution and degradation differences of the raw materials in the MN matrix. An in vivo study demonstrated that the AP-loaded MNs system can effectively reduce SUA levels similar to oral administration but with lower side effects, which will contribute to reducing liver and kidney damage and improve the bioavailability of AP. Thus, this MN-mediated strategy can facilitate transcutaneous hyperuricemia treatment and provide a new alternative for the exploration of clinical treatment of hyperuricemia.Author contributions
R. W., H. W. and T. L. produced MNs and performed cell and animal experiments; R. W., T. L. and G. J. analyzed the results and wrote the draft of the manuscript; G. J. and Y. S. conceived the idea and designed the experiments; L. N. and A. S. provided suggestions and commented on the manuscript; K. E. Y., U. E. A. and S. O. S. reviewed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Huadong Medicine Joint Funds of the Zhejiang Provincial Natural Science Foundation of China under Grant No. LHDMZ23H300003 and the National Natural Science Foundation of China under Grant No. 51873194.References
- N. Dalbeth, A. L. Gosling, A. Gaffo and A. Abhishek, Lancet, 2021, 397, 1843 CrossRef CAS PubMed.
- N. Dalbeth, H. K. Choi, L. A. B. Joosten, P. P. Khanna, H. Matsuo, F. Perez-Ruiz and L. K. Stamp, Nat. Rev. Dis. Primers, 2019, 5, 69 CrossRef PubMed.
- J. A. Singh, I. Herbey, A. Bharat, J. E. Dinnella, S. Pullman-Mooar, S. Eisen and N. Ivankova, Arthritis Care Res., 2017, 69, 1724 CrossRef PubMed.
- M. Toprover, S. Krasnokutsky and M. H. Pillinger, Curr. Rheumatol. Rep., 2015, 17, 70 CrossRef PubMed.
- M. Chen-Xu, C. Yokose, S. K. Rai, M. H. Pillinger and H. K. Choi, Arthritis Rheumatol., 2019, 71, 991 CrossRef PubMed.
- C.-F. Kuo, M. J. Grainge, W. Y. Zhang and M. Doherty, Nat. Rev. Rheumatol., 2015, 11, 649 CrossRef PubMed.
- N. Dalbeth, P.-G. Amanda, C. Frampton, T. Neogi, W. J. Taylor and T. R. Merriman, Ann. Rheum. Dis., 2018, 77, 1048 CrossRef CAS PubMed.
- N. Dalbeth and L. Stamp, Ann. Rheum. Dis., 2014, 73, 1598 CrossRef PubMed.
- N. Dalbeth, M. E. House, O. Aati, P. Tan, C. Franklin, A. Horne, G. D. Gamble, L. K. Stamp, A. J. Doyle and F. M. McQueen, Ann. Rheum. Dis., 2015, 74, 908 CrossRef PubMed.
- A. Shiozawa, S. M. Szabo, A. Bolzani, A. Cheung and H. K. Choi, J. Rheumatol., 2017, 44, 388 CrossRef CAS PubMed.
- H. K. Choi, S. Liu and G. Curhan, Arthritis Rheum., 2005, 52, 283 CrossRef PubMed.
- H. K. Choi, K. Atkinson, E. W. Karlson, W. Willett and G. Curhan, Lancet, 2004, 363, 1277 CrossRef PubMed.
- H. Nan, Q. Qiao, Y. H. Dong, W. G. Gao, B. Tang, R. L. Qian and J. Tuomilehto, J. Rheumatol., 2006, 33, 1346 Search PubMed.
- N. Dalbeth, K. Billington, A. Doyle, C. Frampton, P. Tan, O. Aati, J. Allan, J. Drake, A. Horne and L. K. Stamp, Arthritis Rheumatol., 2019, 71, 1739–1746 CrossRef CAS PubMed.
- F. Mozaffari, S. M. H. Razavian and M. A. Ghasemzadeh, J. Pharm. Innovation, 2022 DOI:10.1007/s12247-022-09624-2.
- F. K. Aldawood, A. Andar and S. Desai, Polymers, 2021, 13, 2815 CrossRef CAS PubMed.
- M. A. Sawon and M. F. Samad, J. Drug Delivery Sci. Technol., 2021, 63, 102477 CrossRef CAS.
- S. Mdanda, P. Ubanako, P. P. D. Kondiah, P. Kumar and Y. E. Choonara, Polymers, 2021, 13, 2405 CrossRef CAS PubMed.
- B. Xu, Q. Y. Cao, Y. Zhang, W. J. Yu, J. Y. Zhu, D. P. Liu and G. H. Jiang, ACS Biomater. Sci. Eng., 2018, 4, 2473 CrossRef CAS PubMed.
- G. Song, G. H. Jiang, T. Q. Liu, X. Y. Zhang, Z. Y. Zeng, R. F. Wang, P. F. Li and Y. H. Yang, ACS Biomater. Sci. Eng., 2020, 6, 4116 CrossRef CAS PubMed.
- B. Homayun, X. T. Lin and H.-J. Choi, Pharmaceutics, 2019, 11, 129 CrossRef CAS PubMed.
- L. M. Ruan, G. Song, X. Y. Zhang, T. Q. Liu, Y. F. Sun, J. L. Zhu, Z. Y. Zeng and G. H. Jiang, Biomater. Sci., 2021, 9, 6830 RSC.
- H. Juster, B. van der Aar and H. de Brouwer, Polym. Eng. Sci., 2021, 59, 877 CrossRef.
- X. X. Zhang, G. P. Chen, L. Y. Sun, F. F. Ye, X. Shen and Y. J. Zhao, Chem. Eng. J., 2021, 406, 126741 CrossRef CAS.
- Y. Zhang, G. H. Jiang, W. J. Yu, D. P. Liu and B. Xu, Mater. Sci. Eng., C, 2018, 85, 18 CrossRef CAS PubMed.
- Z. J. Fan, Y. Wei, Z. R. Yin, H. F. Huang, X. Z. Liao, L. Y. Sun, B. Liu and F. Z. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 40278 CrossRef CAS PubMed.
- R. K. Mishra, K. Y. Goud, Z. H. Li, C. Moonla, M. A. Mohamed, F. Tehrani, H. Teymourian and J. Wang, J. Am. Chem. Soc., 2020, 142, 5991 CrossRef CAS PubMed.
- R. Jamaledin, C. Di Natale, V. Onesto, Z. B. Taraghdari, E. N. Zare, P. Makvandi, R. Vecchione and P. A. Netti, J. Clin. Med., 2020, 9, 542 CrossRef CAS PubMed.
- R. Jamaledin, C. K. Y. Yiu, E. N. Zare, L.-N. Niu, R. Vecchione, G. J. Chen, Z. Gu, F. R. Tay and P. Makvandi, Adv. Mater., 2020, 32, 2002129 CrossRef CAS PubMed.
- R. Wang, G. H. Jiang, U. E. Aharodnikau, K. Yunusov, Y. F. Sun, T. Q. Liu and S. O. Solomevich, Macromol. Rapid Commun., 2022, 43, 2200037 CrossRef CAS PubMed.
- Z. Y. Zeng, G. H. Jiang, T. Q. Liu, G. Song, Y. F. Sun and X. Y. Zhang, Bio-Des. Manuf., 2021, 4, 902 CrossRef CAS.
- G. Song, Y. F. Sun, T. Q. Liu, X. Y. Zhang, Z. Y. Zeng, R. F. Wang, P. F. Li, C. H. Li and G. H. Jiang, Chem. Eng. J., 2021, 426, 130790 CrossRef CAS.
- Y. Q. Zhang, J. C. Yu, A. R. Kahkoska, J. Q. Wang, J. B. Buse and Z. Gu, Adv. Drug Delivery Rev., 2018, 139, 51 CrossRef PubMed.
- X. Jin, D. D. Zhu, B. Z. Chen, M. Ashfaq and X. D. Guo, Adv. Drug Delivery Rev., 2018, 127, 119 CrossRef CAS PubMed.
- Y. Yin, W. Su, J. Zhang, W. P. Huang, X. Y. Li, H. X. Ma, M. X. Tan, H. H. Song, G. L. Cao, S. J. Yu, D. Yu, J. H. Jeong, X. Zhao, H. Li, G. J. Nie and H. Wang, ACS Nano, 2021, 15, 14347 CrossRef CAS PubMed.
- K. van der Maaden, W. Jiskoot and J. Bouwstra, J. Controlled Release, 2012, 161, 645 CrossRef CAS PubMed.
- P. C. DeMuth, J. J. Moon, H. Suh, P. T. Hammond and D. J. Irvine, ACS Nano, 2012, 6, 8041 CrossRef CAS PubMed.
- S. Kusama, K. Sato, Y. Matsui, N. Kimura, H. Abe, S. Yoshida and M. Nishizawa, Nat. Commun., 2021, 12, 658 CrossRef CAS PubMed.
- Z. Y. Wang, J. Y. Luan, A. Seth, L. Liu, M. L. You, P. Gupta, P. Rathi, Y. X. Wang, S. S. Cao, Q. S. Jiang, X. Zhang, R. Gupta, Q. J. Zhou, J. J. Morrissey, E. L. Scheller, J. S. Rudra and S. Singamaneni, Nat. Biomed. Eng., 2021, 5, 64 CrossRef CAS PubMed.
- X. X. Zhang, G. P. Chen, F. K. Bian, L. J. Cai and Y. J. Zhao, Adv. Mater., 2019, 31, 1902825 CrossRef PubMed.
- J. J. Chi, X. X. Zhang, C. W. Chen, C. M. Shao, Y. J. Zhao and Y. A. Wang, Bioact. Mater., 2020, 5, 253 CrossRef PubMed.
- X. X. Zhang, G. P. Chen, Y. X. Liu, L. Y. Sun, L. Y. Sun and Y. J. Zhao, ACS Nano, 2020, 14, 5901 CrossRef CAS PubMed.
- S.-J. Yang, J.-O. Jeong, Y.-M. Lim and J.-S. Park, Mater. Des., 2021, 201, 109485 CrossRef CAS.
- S. P. Sullivan, D. G. Koutsonanos, M. D. P. Martin, J. W. Lee, V. Zarnitsyn, S.-O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Nat. Med., 2010, 16, 915 CrossRef CAS PubMed.
- M. Z. Xing, X. Wang, L. C. Zhao, Z. Q. Zhou, H. Liu, B. R. Wang, A. Cheng, S. H. Zhang and Y. H. Gao, Int. Pharm., 2021, 600, 120406 CrossRef CAS PubMed.
- H. X. Wang, W. S. Wang, C. P. Li, A. Xu, B. S. Qiu, F. F. Li and W. P. Ding, Chem. Eng. J., 2022, 428, 131913 CrossRef CAS.
- Y. Hao, Y. W. Chen, X. L. He, F. Yang, R. X. Han, C. L. Yang, W. Li and Z. Y. Qian, Bioact. Mater., 2020, 5, 542 CrossRef PubMed.
- P. C. DeMuth, Y. Min, B. Huang, J. A. Kramer, A. D. Miller, D. H. Barouch, P. T. Hammond and D. J. Irvine, Nat. Mater., 2013, 12, 367 CrossRef CAS PubMed.
- X. F. Li, Q. A. Xu, J. Wang, P. Zhang, Y. X. Wang and J. Ji, J. Mater. Chem. B, 2021, 9, 5528 RSC.
- N. H. Khan, M. Mir, L. Qian, M. Baloch, M. F. A. Khan, A. Rehman, E. E. Ngowi, D.-D. Wu and X.-Y. Ji, J. Adv. Res., 2022, 36, 223 CrossRef CAS PubMed.
- D. Huang, Z. D. Hu, Y. Ding, G. X. Wang, B. Lu, J. H. Ji and Z. C. Zhen, Polym. Degrad. Stab., 2019, 263, 195 CrossRef.
- N. N. Aung, T. Ngawhirunpat, T. Rojanarata, P. Patrojanasophon, P. Opanasopit and B. Pamornpathomkul, AAPS PharmSciTech, 2020, 21, 25 CrossRef CAS PubMed.
- R. Obaidat, F. BaniAmer, S. M. Assaf and A. Yassin, AAPS PharmSciTech, 2021, 22, 253 CrossRef CAS PubMed.
- W. C. Sun, Z. Araci, M. Inayathullah, S. Manickam, X. X. Zhang, M. A. Bruce, M. P. Marinkovich, A. T. Lane, C. Milla, J. Rajadas and M. J. Butte, Acta Biomater., 2013, 9, 7767 CrossRef CAS PubMed.
- J. W. Lee, J. H. Park and M. R. Prausnitz, Biomaterials, 2008, 29, 2113e24 Search PubMed.
- B. Lu, G. X. Wang, D. Huang, Z. L. Ren, X. W. Wang, P. L. Wang, Z. C. Zhen, W. Zhang and J. H. Ji, Polym. Degrad. Stab., 2018, 150, 133–139 CrossRef CAS.
- J. Lu, N. Dalbeth, H. Y. Yin, C. G. Li, T. R. Merriman and W.-H. Wei, Nat. Rev. Rheumatol., 2019, 15, 413 CrossRef PubMed.
- D.-C. Tarng, H.-Y. Lin, M.-L. Shyong, J.-S. Wang, W.-C. Yang and T.-P. Huang, Am. J. Nephrol., 1995, 15, 31 CrossRef CAS PubMed.
- F. Perez-Ruiz, J. S. Sundy, J. N. Miner, M. Cravets and C. Storgard, Ann. Rheum. Dis., 2016, 75, 1074 CrossRef CAS PubMed.
- D. Pitocco, E. Di Stasio, F. Romitelli, F. Zaccardi, B. Tavazzi, A. Manto, S. Caputo, T. Musella, C. Zuppi, S. A. Santini and G. Ghirlanda, Diabetes-Metab. Res., 2008, 24, 318 CrossRef CAS PubMed.
- R. Dhouibi, H. Affes, M. Ben Salem, D. Moalla, R. Marekchi, S. Charfi, S. Hammami, Z. Sahnoun, K. Jamoussi, K. M. Zeghal and K. Ksouda, Life Sci., 2021, 268, 118998 CrossRef CAS PubMed.
- M. X. Pang, Y. Y. Fang, S. H. Chen, X. X. Zhu, C. W. Shan, J. Su, J. J. Yu, B. Li, Y. Yang, B. Chen, K. L. Liang, H. M. Hu and G. Y. Lv, Med. Sci. Monit., 2017, 23, 1129 CrossRef CAS PubMed.
- Y. Zhang, L. L. Deng, C. M. Wu, L. J. Zheng and G. Zhong, J. Funct. Foods, 2018, 48, 566 CrossRef CAS.
- Y. Lee, P. Werlinger, J.-W. Suh and J. H. Cheng, Microorganisms, 2022, 10, 851 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01836e |
| This journal is © The Royal Society of Chemistry 2023 |