
Antiviral supramolecular polymeric hydrogels by self-assembly of tenofovir-bearing peptide amphiphiles†
Maya K.
Monroe
ab,
Han
Wang
ab,
Caleb F.
Anderson
ab,
Meng
Qin
ab,
Chloe L.
Thio
c,
Charles
Flexner
d and
Honggang
Cui
 *abefg
*abefg
aDepartment of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: hcui6@jhu.edu
bInstitute for NanoBioTechnology, The Johns Hopkins University, Baltimore, MD 21218, USA
cDepartment of Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
dDivisions of Clinical Pharmacology and Infectious Diseases, The Johns Hopkins University School of Medicine and Bloomberg School of Public Health, Baltimore, MD 21205, USA
eDepartment of Materials Science and Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA
fDepartment of Oncology and Sidney Kimmel Comprehensive Cancer Center, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
gCenter for Nanomedicine, The Wilmer Eye Institute, The Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA
First published on 24th November 2022
Abstract
The development of long-acting antiviral therapeutic delivery systems is crucial to improve the current treatment and prevention of HIV and chronic HBV. We report here on the conjugation of tenofovir (TFV), an FDA approved nucleotide reverse transcriptase inhibitor (NRTI), to rationally designed peptide amphiphiles (PAs), to construct antiviral prodrug hydrogelators (TFV-PAs). The resultant conjugates can self-assemble into one-dimensional nanostructures in aqueous environments and consequently undergo rapid gelation upon injection into 1× PBS solution to create a drug depot. The TFV-PA designs containing two or three valines could attain instantaneous gelation, with one displaying sustained release for more than 28 days in vitro. Our studies suggest that minor changes in peptide design can result in differences in supramolecular morphology and structural stability, which impacted in vitro gelation and release. We envision the use of this system as an important delivery platform for the sustained, linear release of TFV at rates that can be precisely tuned to attain therapeutically relevant TFV plasma concentrations.
Introduction
A significant challenge in the advancement of chronic antiviral therapeutics is the achievement of long-acting release to prolong dosing intervals.1 There is substantial patient interest in long-acting treatment strategies, including long-acting injectable (LAI) formulations, for the management of chronic infections resulting from hepatitis B virus (HBV) and human immunodeficiency virus (HIV).2–6 Such therapeutic formulations are necessary to meet the diverse needs of the patient population, thereby improving quality of life and treatment adherence.6–11 Tenofovir (TFV) is a crucial frontline treatment for both chronic HBV and HIV, successfully controlling viral replication and consequently slowing progression to severe disease.12,13 Despite this dire medical need, there are no FDA approved long-acting formulations of TFV or any other HBV therapeutic.This is due to the fact that most antiviral LAI delivery strategies employ formulation of therapeutics into nanosuspensions and tenofovir is a highly hydrophilic (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = −2.5) nucleotide reverse transcriptase inhibitor (NRTI) that is consequently unsuitable for nanosuspension formulation.14–17 As a result, much of the research into LAI TFV has focused on masking the hydrophilicity of the parent drug so that it can be successfully converted into nanocrystals or semi-solid nanoparticles for a nanosuspension.18,19 However, the need for potentially irritating excipient materials in such nanosuspensions has led to frequent injection site reactions in both experimental and FDA-approved antiretroviral LAIs.19–21 While these reactions are less severe than the tissue necrosis associated with TFV implants,22 they are still significant enough to dissuade patient interest.20,21 Consequently, a novel formulation strategy is necessary that can form a drug depot in situ without the need for any excipient materials.
P = −2.5) nucleotide reverse transcriptase inhibitor (NRTI) that is consequently unsuitable for nanosuspension formulation.14–17 As a result, much of the research into LAI TFV has focused on masking the hydrophilicity of the parent drug so that it can be successfully converted into nanocrystals or semi-solid nanoparticles for a nanosuspension.18,19 However, the need for potentially irritating excipient materials in such nanosuspensions has led to frequent injection site reactions in both experimental and FDA-approved antiretroviral LAIs.19–21 While these reactions are less severe than the tissue necrosis associated with TFV implants,22 they are still significant enough to dissuade patient interest.20,21 Consequently, a novel formulation strategy is necessary that can form a drug depot in situ without the need for any excipient materials.
Self-assembling peptides represent a potential class of LAI biomaterials, due to their ability to form supramolecular polymeric hydrogels under physiological conditions with great biocompatibility and tunable biodegradability.23–39 Through encapsulation or covalent conjugation of therapeutics, it is possible to use these systems to create a drug depot upon injection for long-acting release.40–49 Peptide amphiphile-based systems are of particular interest for drug delivery due to their robust and consistent self-assembly into filamentous structures, well understood sequence surface display, and robust synthesis protocols.50–58 Furthermore, previous work has demonstrated that the morphology,59–63 stability,63–67 gelation,61,63,68 and release39,69–73 of these self-assembling platforms can be controlled through changes to the peptide sequence, number or type of hydrophobic moiety, linker chemistry, hydrogen bonding propensity, and surface charge. As changes to these supramolecular properties can impact drug delivery outcomes, drug-containing peptide amphiphile systems are especially well suited for adoption as highly tunable, long-acting injectable drug delivery platforms. Beyond being naturally biocompatible, biodegradable, and biomimetic,74–77 such an LAI system has a fixed drug loading content if covalent conjugation is employed, can be applied to a variety of ARVs with differing physicochemical properties, and sequesters the prodrugs within the nanostructures, thereby attaining extremely high concentrations of therapeutic within the hydrogel depot while avoiding the toxicity issues observed in TFV implants. In this context, by conjugating TFV to an amphiphilic peptide sequence, we created a series of self-assembling prodrug hydrogelators that can attain 100% prodrug loading, instantaneous gelation, and sustained release without the need for excipient materials and that exhibit tunable properties for optimization of TFV delivery and release.
Experimental
TFV-etpSS-Pyr and TFV-PA synthesis and purification
TFV-etpSS-Pyr was synthesized using an adaptation of a previously reported procedure (Fig. S2†).78 After evacuating the air and purging the reaction flask with argon, excess oxalyl chloride (0.9 mL, 10.52 mmol) was added dropwise to a stirring solution of DMF (160 μL, 2.09 mmol) and dry tenofovir (0.5 g, 1.74 mmol) dissolved in anhydrous dichloromethane (DCM, 20 mL) and allowed to react at RT under argon for 20 minutes. Excess solvent was then removed by rotary evaporation and the product was redissolved in anhydrous DCM (15 mL) and chilled to 0 °C using an ice bath and purged with argon. 1.1 equivalent of etpSS-Pyr (360 mg, 1.92 mmol) dissolved in anhydrous DCM (5 mL) was added to the chilled stirring solution, followed by the dropwise addition of pyridine (0.84 mL, 10.44 mmol) dissolved in anhydrous DCM (3 mL) under argon. The stirring solution was allowed to react at 0 °C for 15 minutes under inert conditions before being removed from the ice bath and allowed to warm naturally to RT and allowed to react closed to the atmosphere for an additional 3 hours. Water (470 μL, 26.09 mmol) was added to the stirring solution and the mixture was allowed to stir for an additional 30 minutes to quench the reaction. Solvent was removed via rotary evaporation and then the product was air dried for 30 minutes. The product was redissolved in EtOH (25 mL) and allowed to react at 50 °C overnight, vented to the atmosphere. Solvent was then removed by rotary evaporation and the product was redissolved in 0.1% aq. TFA (30 mL) for purification via acidic phase RP-HPLC. The desired product fractions were collected, subjected to rotary evaporation to remove ACN, and lyophilized to give TFV-etpSS-Pyr. 1H-NMR (400 MHz, CD3OD, 25 °C, δ ppm) 8.39 (m, 2H), 8.34 (s, 1H), 7.84 (m, 2H), 7.22 (t, 1H), 4.28–4.48 (m, 2H), 4.12 (m, 2H), 3.98 (dd, 1H), 3.57–3.82 (m, 2H), 3.02 (t, 2H), 1.18 (s, 3H); MS (ESI): 457.2 [M + H]+, 912.9 [2M + H]+, 1368.7 [3M + H]+. The product was stored in a −20 °C freezer for future use.TFV peptide amphiphiles (TFV-PAs) were synthesized by dissolving a 1:2 molar equivalent of PA (17.6–23.6 mg, 0.03 mmol) and TFV-etpSS-Pyr (27.4 mg, 0.06 mmol) in DMSO (1 mL), purging the reaction flask with nitrogen, and allowing the solution to stir for 5 days. The TFV-PAs were then dissolved in 20–30 mL of 70:30 0.1% v/v NH4OH H2O:ACN and purified via basic phase RP-HPLC (samples were run at a gradient of 5% to 55% ACN over 25 min). Collected fractions were analyzed using MALDI-TOF or ESI MS to determine the fraction containing the desired product. Correct fractions were combined, subjected to rotary evaporation, and lyophilized. TFV-PAs were then dissolved in deionized water and the pH of the solutions were tuned to 7.4 prior to aliquoting the solutions into cryo-vials. Purity and concentration of the TFV-PAs was assessed using analytical RP-HPLC. The aliquots were re-lyophilized and subsequently stored in a −20 °C freezer for future use.
Anti-HBV efficacy studies
Anti-HBV efficacy was evaluated on HepAD38 cells, using an adaptation of a previously reported procedure.79 HepAD38 cells were plated on collagen coated 96-well flat-bottomed plates at a density of 6 × 104 cells per well and grown for 3 days in the presence of 200 μL of Ham's F-12K (Kaighn's) medium supplemented with 10% fetal bovine serum (FBS), 1% of an antibiotic solution (penicillin and streptomycin), and 0.3 mg mL−1 tetracycline at 37 °C in 5% carbon dioxide. On day zero the cells were washed with PBS and treated with tetracycline-free F-12K medium containing TFV, TFV-PA2, or TFV-PA3 (180 μL of medium supplemented with 20 μL of 10× concentration therapeutic solution to give 200 μL of medium at the final therapeutic concentration). Each therapeutic was screened at a minimum of five concentrations in quadruplicate. Water treated cells (180 μL of medium supplemented with 20 μL of H2O) were used as the negative control. On day three the medium was removed and replaced with 200 μL of fresh tetracycline-free medium containing the test compound at the appropriate concentration. On day four, 150 μL of cell supernatant was collected and supplemented with 50 μL of nuclease free H2O, and DNA was extracted and eluted in 50 μL of nuclease-free water using QIAamp DNA blood mini kits (Qiagen, Germantown, MD) following the manufacturer provided spin protocol for DNA purification from blood or body fluids. HBV DNA was quantified by qPCR, using Integrated DNA Technologies PrimeTime Gene Expression Master Mix (Integrated DNA Technologies, Coralville, IA) and HBV TaqMan primer/probe (20×, Assay ID Vi03453405_s1, ThermoFisher Scientific, Pittsburgh, PA). A serial dilution of gBlocks Gene Fragments (Integrated DNA Technologies, Coralville, IA) of known HBV copy number was used as the standard for the absolute quantification of DNA copy number from cycle threshold values. All qPCR runs used the following cycling parameters for amplification of 2 μL of aqueous DNA: a preamplification cycle at 95 °C for 10 minutes, 50 cycles of 95 °C and 60 °C, followed by a melt curve, in accordance with the manufacturer's protocol. Percent HBV production for each therapeutic concentration was determined based on average number of copies of HBV DNA produced by water treated cells and averaged between the three biological repeats of the assay before being plotted against concentration. GraphPad Prism software was used to calculate the IC50 values (“[inhibitor] vs. normalized response” non-linear regression equation).Results and discussion
Molecular design
Given the highly hydrophilic nature of TFV, a peptide amphiphile design with distinct hydrophilic and hydrophobic segments, a robust intermolecular hydrogen bonding sub-domain, and a relatively long alkyl tail was deemed to be necessary.22,23,50,62–64 In light of these considerations, we developed three molecular designs that incorporated one to three valines at the N-terminus, in an attempt to balance increased TFV content with enhanced intermolecular associative interaction forming propensity (Fig. 1a).63,80–83 The tenofovir moiety was covalently conjugated to a cysteine residue near the C-terminus of the peptide using a reducible disulfonyl-ethyl phosphonate linker (etpSS) to create the TFV-bearing peptide amphiphiles (TFV-PAs, named TFV-PA1, TFV-PA2, and TFV-PA3, respective to the number of valines incorporated in the sequence). For all designs, palmitic acid was incorporated at the N-terminus as the hydrocarbon tail of the peptide amphiphile, and glutamic acid was included at the C-terminus to provide distinct amphiphilicity to the molecules. The TFV-PAs self-assemble into filamentous structures in aqueous conditions due to hydrophobic collapse of the alkyl tails, with intermolecular hydrogen bonding of the peptide backbones driving axial growth (Fig. 1b & c).50 In the presence of physiologically-relevant salt concentrations, the supramolecular structures can physically entangle to form a hydrogel drug depot in situ (Fig. 1b & d). These TFV-bearing peptide amphiphiles differ from previous drug amphiphiles produced by our lab in that the therapeutic is not the hydrophobic component of the self-assembling system and is consequently not the major driving force behind supramolecular polymerization. While TFV still contributes to self-assembly behavior, by linking it to a peptide-amphiphile with strong self-assembling propensity, we can, to an extent, decouple self-assembly from therapeutic content. This enables us to modify design considerations such as number and type of therapeutic moieties, without significantly impacting self-assembly, gelation, and release of the LAI drug delivery system. Such a coupling strategy results in a more general drug delivery platform that allows for future alterations so that the therapeutic-bearing peptide amphiphile design can be applied to other hydrophilic therapeutics.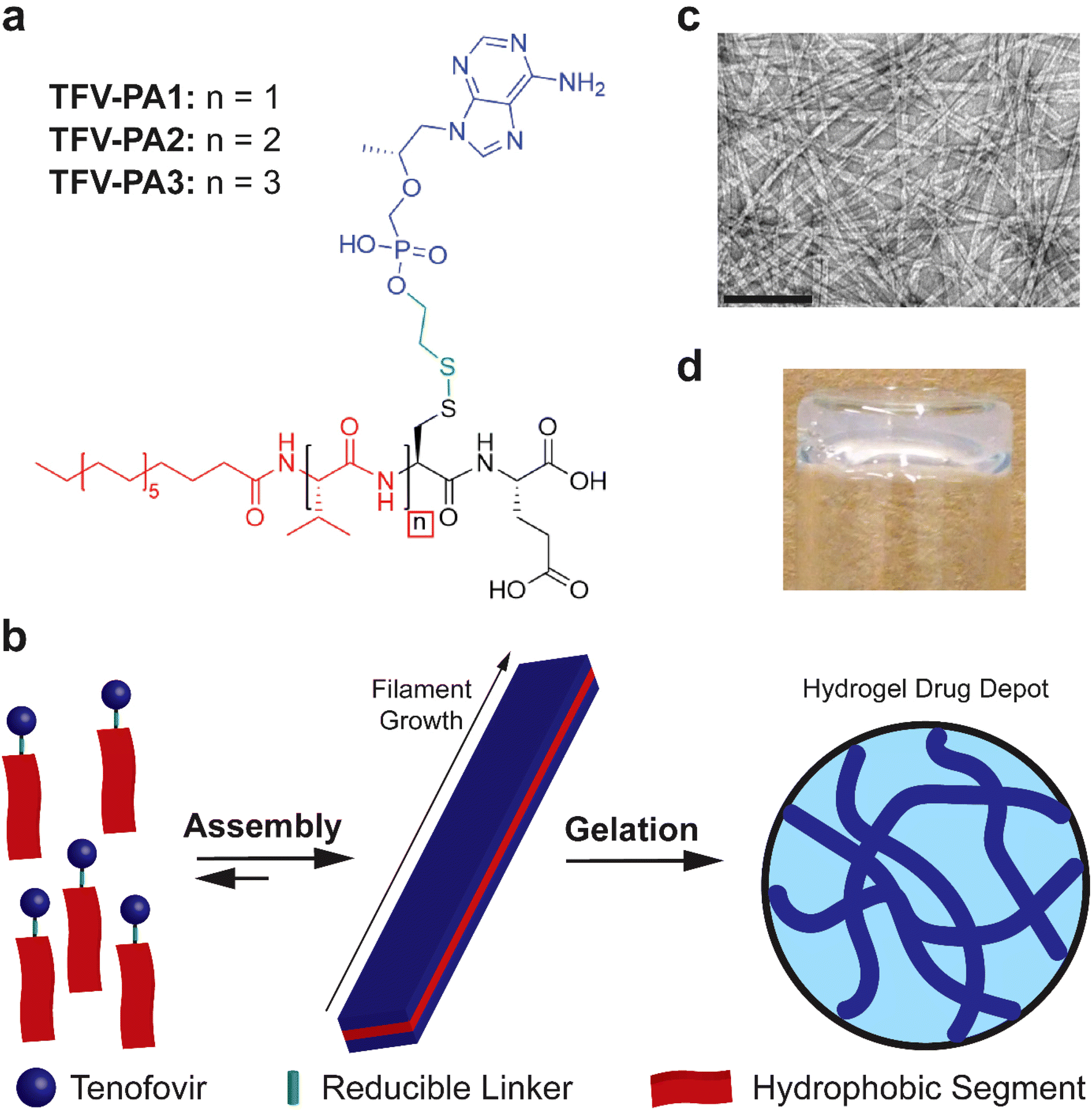 | ||
| Fig. 1 Tunable platform to direct the self-assembly and release of phosphonate-containing antiviral prodrugs. (a) Molecular platform for the design of TFV-PAs: blue text and bonds represent the tenofovir (TFV) portion of the PA, green text and bonds represent the reducible etpSS linker, red text and bonds indicate the hydrophobic portion of the molecule (including the alkyl chain and valine residues), red boxes indicate points in the design where we made modifications for this design series. (b) Schematic showing the proposed mechanism of self-assembly and systemic self-delivery for our platform. Intermolecular hydrogen bonding between the peptide backbones of the TFV-PAs drives axial filament growth, hydrophobic collapse of the valine side chains drives lateral growth of the filaments in water. Under physiological conditions, the filaments entangle to form a self-supporting gel due to salt-screening, resulting in the formation of a hydrogel drug depot following injection. (c) Transmission electron microscopy image of TFV-PA3 aqueous solution (pH 7.4, conc. 1 mM), scale bar is 200 nm. (d) Picture of hydrogel formed from 10 mM TFV-PA3 solution following the addition of 10× PBS at a 10:1 ratio (to achieve 1× PBS). | ||
Self-assembly and gelation
As supramolecular morphology, stability, packing, and gelation are all relevant characteristics for clinical adaptation of the TFV prodrug hydrogelators, it was necessary to fully characterize these features of the different TFV-PA designs. All three designs were shown to form nanobelts more than 1 micron in length at 1 mM concentration, with structures increasing in width from 10.0 ± 2.5, 11.8 ± 1.3, to 18.9 ± 3.0 nm for TFV-PA1, TFV-PA2, and TFV-PA3, respectively (Fig. 2 and Fig. S6†). This self-assembling propensity of all three TFV-PA designs is notable, as covalent conjugation of organophosphorus-based groups to peptide sequences has traditionally been used to impede self-assembly or gelation, with cleavage of these groups in response to a trigger offering a way to engineer controlled delivery systems.32,43,84–89 Consequently, the creation of PAs that incorporate a phosphonate group while still undergoing self-assembly and gelation in physiologically-relevant conditions is a significant accomplishment. | ||
| Fig. 2 Supramolecular morphology and gelation of the prodrug designs. (a–c) Negatively stained transmission electron microscopy images of (a) TFV-PA1, (b) TFV-PA2, and (c) TFV-PA3, scale bars represent 200 nm. (d–f) Negatively stained transmission electron microscopy images of (d) TFV-PA1, (e) TFV-PA2, and (f) TFV-PA3, scale bars represent 100 nm. Nanobelt widths of the different designs were measured to be 10.0 ± 2.5 nm, 11.8 ± 1.3 nm, and 18.9 ± 3.0 nm for TFV-PA1, TFV-PA2, and TFV-PA3, respectively (data are given as mean ± SD, n = 35). (Insets) Images of gels formed by the different designs by adding 10 μl of 10× PBS to 100 μl of 5 mM prodrug solution. The critical gelation concentrations (assessed by a simple inversion test) were between 5 and 10 mM for TFV-PA1, between 2 and 5 mM for TFV-PA2, and between 1 and 2 mM for TFV-PA3. | ||
We hypothesize that the difference in width between the supramolecular structures is a result of hydrophobic interactions between valines contributing to lateral growth of the filaments, with increased number of valines resulting in a corresponding increase in lateral growth and wider structures.60,90 This increase in nanobelt width also corresponded to an observable increase in persistence length of the filamentous structures. Surprisingly, despite an established correlation between flexibility of supramolecular structures and gelation propensity,61,91,92 the designs displayed improved gelation with increasing number of valines (Fig. 2d–f insets and Fig. S8†). The addition of phosphate buffered solution (PBS) (final concentration 1× PBS to mimic physiological salt concentrations in the extracellular environment)93,94 to 5 mM concentration solutions of the three molecular designs resulted in only partial gel formation for TFV-PA1, weak gel formation for TFV-PA2, and robust gel formation for TFV-PA3. The critical gelation concentrations (CGCs) of the different designs were assessed by adding PBS (final concentration 1× PBS) to different concentrations of PA solutions and then performing a simple inversion test to demonstrate the formation of a gel (Fig. S8†). The CGCs of the different designs were determined to be between 5 and 10 mM for TFV-PA1, between 2 and 5 mM for TFV-PA2, and between 1 and 2 mM for TFV-PA3. Considering the desirability of instantaneous gelation for LAI formulation, 10 mM solutions of TFV-PA2 and TFV-PA3 were allowed to assemble in the presence of 10 mM Nile red and injected into 10× PBS (ESI Videos S1 and S2†). Both designs formed gels instantly upon injection, TFV-PA1 was not tested due to its inferior gelation properties.
To understand the unexpected trend of gelation propensity, we investigated the stability of the supramolecular structures. The critical micellization concentrations (CMCs) of the different designs were determined by a Nile red encapsulation assay, which uses a fluorometer to identify the differential fluorescence of encapsulated Nile red (635 nm) versus free Nile red; an increase in the intensity at 635 nm suggests the presence of nanostructures. A lower CMC would correspond to enhanced stability of the structures and increased numbers of filaments, as more of the TFV-PAs are partitioned into supramolecular structures rather than existing as free monomers in solution, since the concentration threshold for supramolecular assembly is lower. Consequently, designs with a lower CMC would be expected to have improved gelation propensity. In correspondence with our hypothesis that the lower CGC of the three-valine design was a result of its improved supramolecular stability, TFV-PA3 had the lowest CMC of the three designs, with the CMCs for TFV-PA1, TFV-PA2, and TFV-PA3 calculated as 54, 51, and 8 μM, respectively (Fig. 3a–c). CMC differences between designs can be partially attributed to differences in the supramolecular packing/hydrogen bonding within the structures. We employed circular dichroism, which uses the differential absorption of left- and right-handed circularly polarized light, to determine the secondary structure of the peptide portion of the TFV-PAs. The CD spectra for the different designs showed increasing hydrogen bonding with increased number of valines, as indicated by the increased depth of the negative peak in the 216 nm range (Fig. 3d and Fig. S7†). This is logical, as valines are known to be amino acids with a high propensity to form associative interactions through hydrogen bonding.80,81 Furthermore, we speculate that the hydrogen bonding differences between TFV-PA1 and TFV-PA2 could explain the different CGC values between these designs, despite their similar CMCs, as the intermolecular associations might influence gelation.
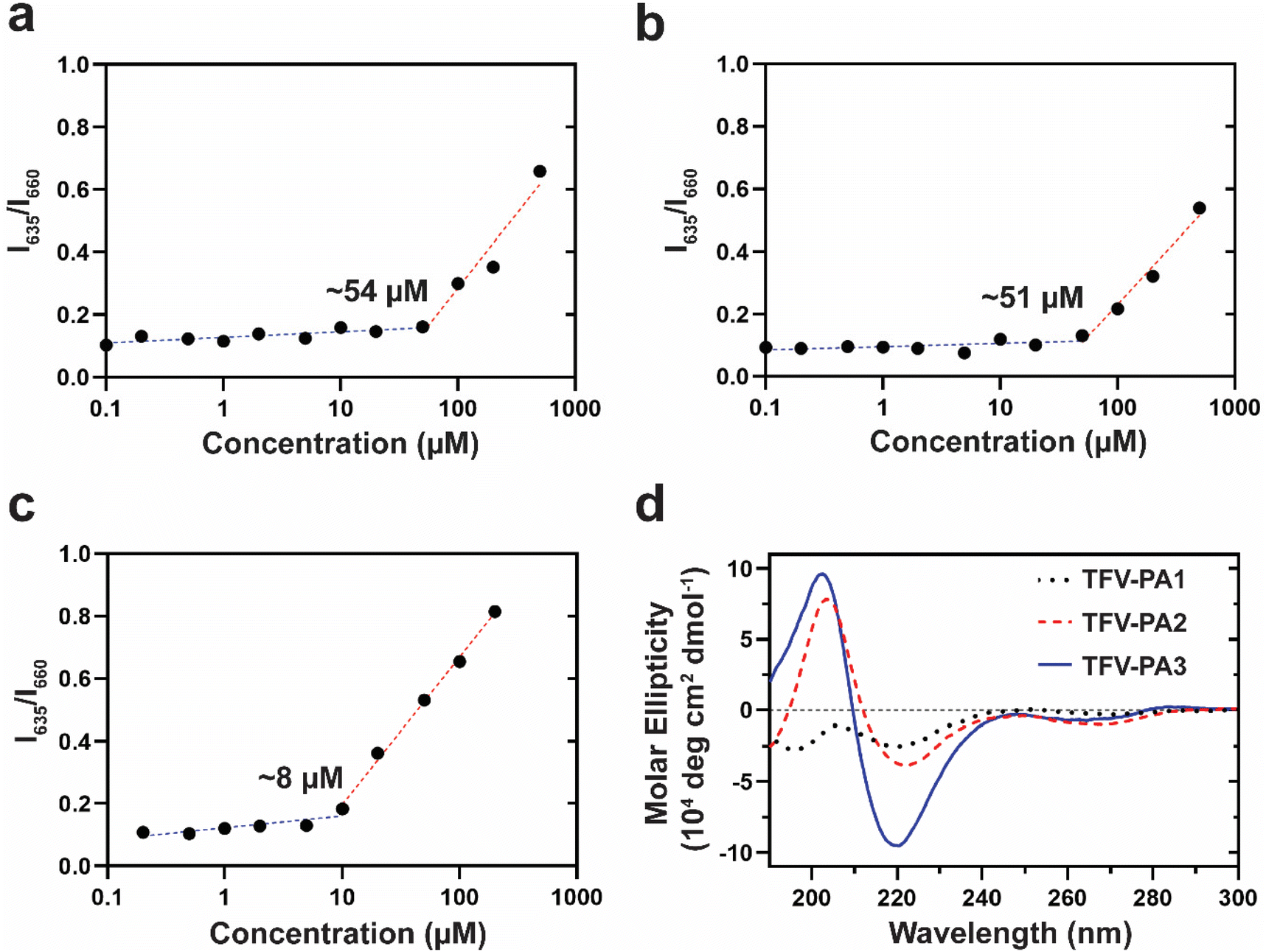 | ||
| Fig. 3 Supramolecular stability and molecular ellipticity of the three designed TFV prodrugs in aqueous solutions. The critical micellization concentration of the different designs, as determined by a shift in the ratio of fluorescent intensity at 635 nm (indicating encapsulated Nile red) versus 660 nm (indicating free Nile red) for (a) TFV-PA1 (54 µM), (b) TFV-PA2 (51 µM), and (c) TFV-PA3 (8 µM) (data are given as mean ± SD, n = 3). (d) Circular dichroism spectra of assembled solutions for TFV-PA1–3 designs (100 μM in H2O), indicating the strength of associative hydrogen bonding between molecules. | ||
Consequently, by adding hydrophobic amino acids we can increase the intermolecular interactions between our TFV-PAs, lowering the concentration of building units necessary for self-assembly and resulting in more stable supramolecular structures with slower release behavior. As anticipated, the design with the largest intermolecular hydrogen bonding peak, TFV-PA3, displayed the lowest CMC, indicating that the design could form nanostructures at lower concentrations and that are correspondingly more robust. These results suggest that we can manipulate the associative interactions between supramolecular building units by varying the number of hydrophobic amino acids, in this case valines, in our peptide sequences. The different assembly characteristics of the TFV-PA designs are significant for clinical development as previous work suggests that supramolecular stability has a significant impact on release, efficacy, and toxicity of self-assembling prodrugs.66,73 As monomer disassociation from supramolecular structures and diffusion of those monomers from the PA hydrogel into the surrounding medium partially governs the TFV-PA release rate, it is possible to prolong the release profile by modifying the CMC of the PA design.73 In this way, we believe it is possible to attain and subsequently tune long-acting release by simple design modifications of the TFV-PAs, such as increasing the number of valines.
Therapeutic release
We conducted in vitro gel release experiments to quantify release rates of free TFV-PAs from hydrogel depots. Hydrogels were formed at two different concentrations and then aged at 37 °C with a fixed volume of PBS solution to act as the release medium. The release supernatant was collected and replaced at predetermined time points and sample concentrations were analyzed using RP-HPLC. Only the two and three valine designs were explored, as the gels formed by TFV-PA1 solutions were not robust enough to last beyond a few days. As expected from the CMC of the designs, the TFV-PA2 hydrogels exhibited significantly faster release, with about 50% of the initial prodrugs released by day 14, than the TFV-PA3 hydrogels, which still retained more than 80% of the initial prodrugs on day 30 (Fig. 4a). It is worth noting that the time scale of release for both TFV-PAs considerably outperformed previously explored, physically loaded TFV hydrogels and polymer fibers, which reported approximately 95% TFV release within 24 hours.95,96 The TFV-PA2 percent release rate was independent of hydrogel concentration, with both the 5 and 10 mM gels showing similar percent release (48% and 56% on day 14 for the 10 and 5 mM gels respectively, Fig. 4a), and the 10 mM gels releasing twice the total number of nanomoles of prodrug released by the 5 mM gels (217 ± 12 nanomoles and 126 ± 7 nanomoles released on day 14, respectively). This, in combination with the observation that the average concentration in the release supernatant is significantly higher than the CMC calculated for the design (361 ± 91 μM for 5 mM gels and 619 ± 268 μM for 10 mM gels, versus 51 μM for the CMC), suggests that release from TFV-PA2 hydrogels is controlled by physical breakdown of the gel at the interface (surface erosion) rather than the CMC through disassociation of supramolecular structures and subsequent diffusion into the release media of the monomers (bulk erosion). The dominance of surface erosion necessitates that entire fibers are released from the gel rather than monomers of the conjugate. In accordance with this, TEM imaging of the release supernatant revealed dense networks of filaments > 1 micron in length (Fig. S10a & b†). In contrast, the percent release rate of the TFV-PA3 5 mM hydrogels was approximately twice that of the TFV-PA3 10 mM hydrogels, with 16.3 ± 1.3% (36.7 ± 3.0 nanomoles) of the 5 mM hydrogels and 6.1 ± 0.5% (27.3 ± 2.2 nanomoles) of the 10 mM hydrogels released on day 30 (Fig. 4a). TEM imaging of the release supernatant revealed only short plaques of filaments for both the 5 and 10 mM TFV-PA3 hydrogels (Fig. S10c & d†), indicating that bulk erosion was more dominant than surface erosion for both concentrations. Additionally, it is worth noting that the average concentration of release supernatant from day 8 onwards for the TFV-PA3 10 mM gels was 13.3 ± 4.2 μM, which is only slightly higher than the calculated CMC of 8 μM. These results suggest that different mechanisms of gel dissolution are involved in release for the different designs, with the TFV-PA2 gels predominantly displaying surface erosion and the TFV-PA3 gels predominantly displaying bulk erosion. This indicates that significant modulation of monomer release behavior can be attained by altering the number of hydrophobic amino acids in the TFV-PA designs.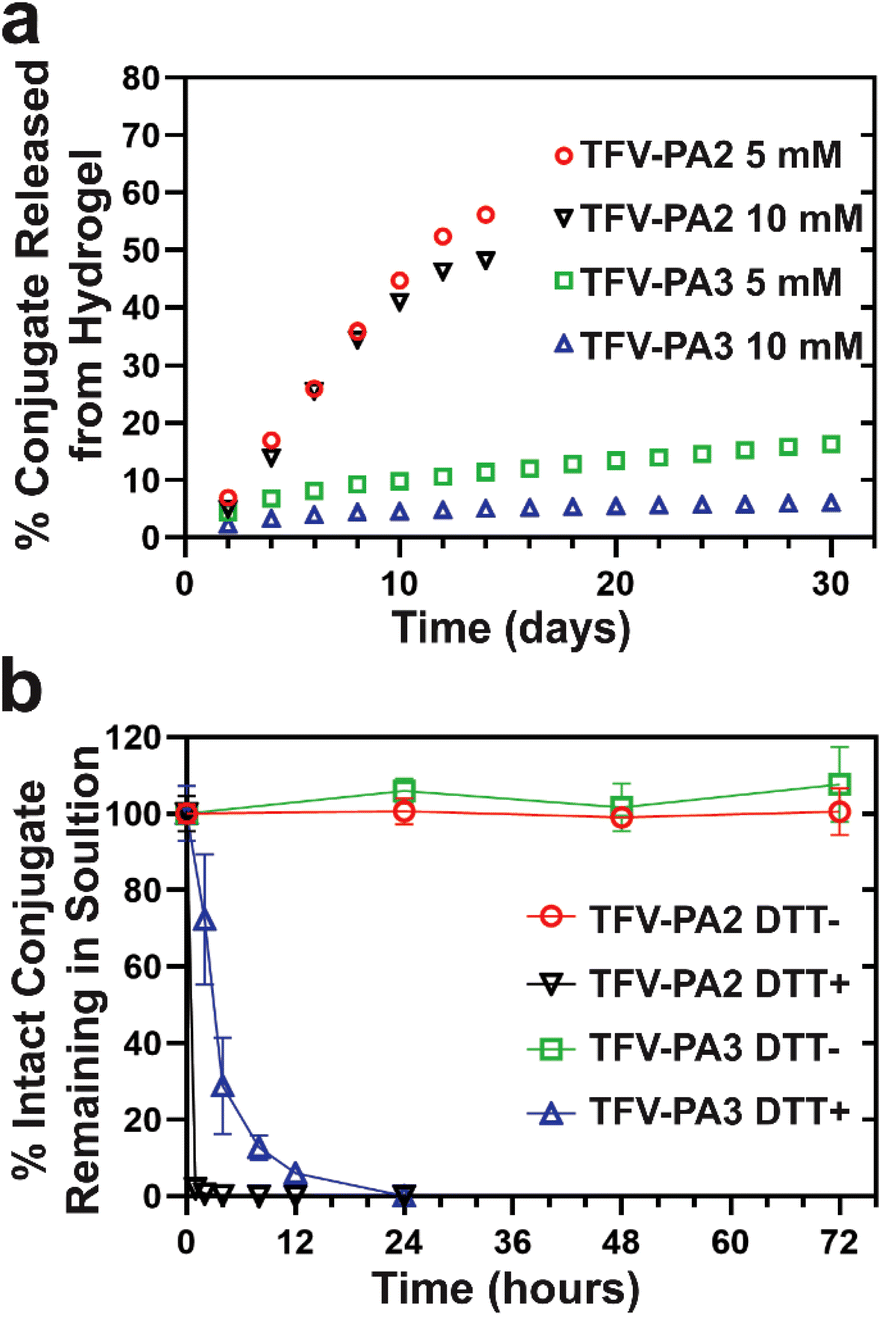 | ||
| Fig. 4 Release behavior of the designed prodrugs and hydrogels. (a) Graph of in vitro prodrug release rate (as measured by analytical HPLC) from hydrogels made from 5 and 10 mM solutions of TFV-PA2 and TFV-PA3, represented as percentage of conjugate contained in the original gels (data are given as mean ± SD, n = 3). (b) Graph of release of free TFV from 100 μM solutions of TFV-PA2 and TFV-PA3 in the presence and absence of 10 mM dithiothreitol (DTT) in 1× PBS, represented as percentage of intact TFV-PA conjugate contained in the solutions at t = 0 (data are given as mean ± SD, n = 3). | ||
We preformed free drug release experiments to demonstrate that TFV is cleaved from the peptide amphiphile under reductive conditions. Such studies are crucial to demonstrate the ability of the TFV-PAs to function as an effective LAI delivery system, as the parent drug must be released upon cellular uptake and then converted into its active diphosphate form by cellular enzymes, which is then incorporated into viral DNA via reverse transcription by viral enzymes.1 By incubating 100 μM 1× PBS solutions of TFV-PA2 and TFV-PA3 in the presence or absence of 10 mM of dithiothreitol (DTT), a known reducing agent, and then quantifying remaining prodrug using reverse-phase analytical HPLC, we showed that the prodrugs are stable at 37 °C and 1× PBS for 3 days but that TFV is quickly released in the presence of DTT, with more than 90% of TFV released after 1 hour for TFV-PA2, more than 70% of TFV released after 4 hours for TFV-PA3, and all TFV released by 24 hours for both designs (Fig. 4b). We used ESI mass spectrometry to further confirm the release of the parent drug, with no prodrug remaining visible in the spectra of the 24 hour release samples (Fig. S9†). Instead, cleaved peptide was noticeable in the spectra of all DTT positive samples, confirming the reduction of the disulfide bond. Intact prodrug was visible in the spectra of the 2–8 hour TFV-PA3 release samples, further supporting the incomplete conversion observed by RP-HPLC for samples incubated for less than 12 hours. The mechanism of free TFV release is believed to correspond to that previously reported by Pradere et al.97
Antiviral efficacy
Next, we investigated the impact of peptide conjugation on antiviral efficacy of the prodrug hydrogelators. To assess the anti-HBV efficacy of our prodrugs, HepAD38 cells (from an immortal cell line stably transfected with the HBV genome) were treated with varying concentrations of TFV, TFV-PA2, or TFV-PA3, with water treated cells as the negative control, following the established protocol for the cell-line.79 The cell supernatant was collected and subjected to qPCR analysis to quantify the copies of HBV DNA released from the cells. A dose response – inhibition curve was fit to the data to calculate the IC50 values for each therapeutic from the averaged biological repeats (Fig. 5). The extrapolated IC50 values for TFV, TFV-PA2, and TFV-PA3 were 1.1 (95% CI: 0.79–1.4), 1.5 (95% CI: 0.70–3.4), and 0.25 (95% CI: 0.16–0.41) μM. The determined IC50 value for TFV falls within the range of values for the HepAD38 cell line reported in literature (0.14 to 5.46 μM).98–101 Critically, the antiviral studies demonstrate that peptide conjugation does not negatively impact antiviral efficacy. This is significant as previous work from our lab has demonstrated that filamentous self-assembly can reduce cellular uptake of conjugates.73,102 Furthermore, there was a consistent trend in all biological repeats of lower IC50 values for TFV-PA3, suggesting that conjugation of TFV to this peptide amphiphile possibly improves efficacy. As antiviral efficacy is a consequence of many complicated factors, we cannot be sure of the exact cause for the lower IC50 values for TFV-PA3, but we speculate that the trend might result from the increased hydrophobicity of the three valine prodrug, which should improve membrane permeability. However, further experiments would be necessary to confirm the mechanism of cellular entry and the membrane permeability of the TFV-PAs, which are beyond the scope of this paper.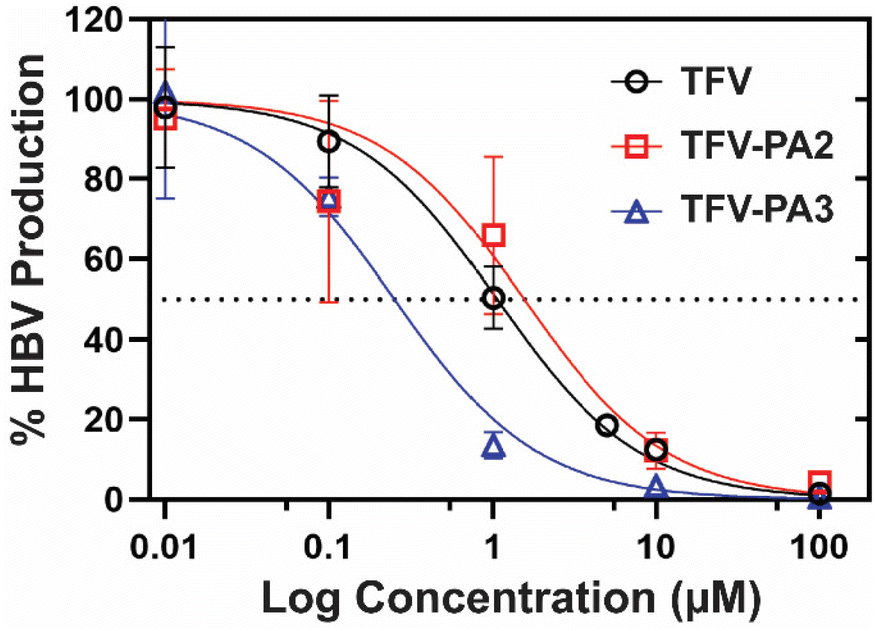 | ||
| Fig. 5 In vitro antiviral efficacy assay. Percent reduction of HBV production, as quantified by qPCR of cellular supernatant, for HepAD38 cells treated with varying concentrations of TFV and TFV prodrugs. Water treated cells were used as a negative control. Data are given as mean ± SD (p > 0.05 repeated measures one-way ANOVA, n = 3). IC50 values were calculated as 1.1, 1.5, and 0.25 µM for TFV, TFV-PA2, and TFV-PA-3, respectively. | ||
Conclusions
In this work, we created a first-of-its-kind NRTI hydrogel delivery platform through the conjugation of TFV to amphiphilic peptide sequences to create antiviral prodrug hydrogelators. To the best of our knowledge, this work represents the first excipient-free, injectable delivery system for tenofovir and is an important extension of the current peptide amphiphile system that is capable of supramolecular assembly with an intact phosphonate group. We demonstrated that modifying the number of valines in the peptide sequence impacts supramolecular assembly and gelation of the designs, which in turn affects release. Hydrogels formed from TFV-PA3 display near-linear release in vitro for more than a month, with more than 80% of prodrugs remaining at day 30. Furthermore, prodrug conversion does not negatively impact the antiviral activity of the TFV-PAs and free TFV is released under reducing conditions. Consequently, this work represents an important first step in the development of an LAI TFV formulation. The TFV-PA3 design is particularly promising for further optimization for in vivo exploration and eventual clinical development, as it can form a hydrogel drug depot in situ capable of sustained release.Author contributions
Maya Monroe contributed to the conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, validation, visualization, and writing (original draft) necessary for the manuscript. Han Wang participated in the conceptualization, investigation, methodology, validation, and writing (review and editing). Caleb Anderson contributed to the investigation and writing (review and editing) of the publication. Meng Qin assisted with investigation for the manuscript. Chloe Thio provided resources, aided in the development of methodology, and contributed to the writing (review and editing). Charles Flexner contributed to conceptualization, funding acquisition, supervision, and writing (review and editing). Honggang Cui was responsible for the conceptualization, funding acquisition, project administration, resources, supervision, and writing (review and editing) of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was made possible by financial support from the Johns Hopkins Center for AIDS Research (JHU CFAR) under NIH/NIAID P30AI094189 and the NSF Graduate Research Fellowship Program, DGE-1746891. Chloe Thio is supported by R01AI138810 from NIAID. Charles Flexner is supported by the Long Acting/Extended Release Antiretroviral Research Resource Program, R24AI118397 from NIH/NIAID. Additionally, we would like to thank Hyon Hwang of the Thio Lab for his significant contribution of expertise towards the antiviral studies. We would also like to thank the Kokkoli Lab (Chemical and Biomolecular Engineering, JHU) for the use of their Infinity 1260 RP-HPLC.References
- M. Monroe, C. Flexner and H. Cui, Bioeng. Transl. Med., 2018, 3(2), 102–123 CrossRef PubMed.
- K. Meyers, Y. Wu, H. Qian, T. Sandfort, X. Huang, J. Xu, J. Zhang, W. Xia, D. Glidden, H. Wu and H. Shang, AIDS Behav., 2018, 22, 1217–1227 CrossRef PubMed.
- A. Van Der Straten, M. K. Shapley-Quinn, K. Reddy, H. Cheng, J. Etima, K. Woeber, P. Musara, T. Palanee-Phillips, J. M. Baeten and E. T. Montgomery, AIDS Patient Care STDS, 2017, 31(7), 305–314 CrossRef.
- K. A. Christopoulos, J. Colasanti, M. O. Johnson, M. Diaz Tsuzuki, X. A. Erguera, R. Flores, J. Kerman, K. Dance, J. A. Sauceda, T. B. Neilands, S. E. Dilworth, K. A. Koester, J. Gutierrez, J. A. Schneider, E. Montgomery and M. C. McNulty, Open Forum Infect. Dis., 2022, 9(7), ofac293 CrossRef PubMed.
- R. C. Bollinger, C. L. Thio, M. S. Sulkowski, J. McKenzie-White, D. L. Thomas and C. Flexner, Lancet HIV, 2020, 7, e443–e448 CrossRef PubMed.
- J. Williams, H. R. Sayles, J. L. Meza, P. Sayre, U. Sandkovsky, H. E. Gendelman, C. Flexner and S. Swindells, Nanomedicine, 2013, 8, 1807–1813 CrossRef CAS PubMed.
- E. Dolgin, Nat. Med., 2014, 20, 323–324 CrossRef CAS PubMed.
- R. M. Gulick and C. Flexner, Annu. Rev. Med., 2019, 70, 137–150 CrossRef CAS PubMed.
- Joint United Nations Programme on HIV/AIDS, Miles To Go: Closing Gaps Breaking Barriers Righting Injustices, 2018.
- J. M. Jacobson and C. W. Flexner, Curr. Opin. HIV AIDS, 2017, 12, 343–350 CrossRef CAS PubMed.
- R. J. Landovitz, R. Kofron and M. McCauley, Curr. Opin. HIV AIDS, 2016, 11, 122–128 CrossRef CAS PubMed.
- N. A. Terrault, A. S. F. Lok, B. J. McMahon, K. M. Chang, J. P. Hwang, M. M. Jonas, R. S. Brown, N. H. Bzowej and J. B. Wong, Hepatology, 2018, 67, 1560–1599 CrossRef PubMed.
- Department of Health and Human Services: Panel on Antiretroviral Guidelines for Adults and Adolescents, Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV, 2019.
- M. N. Arimilli, C. U. Kim, J. Dougherty, A. Mulato, R. Oliyai, J. P. Shaw, K. C. Cundy and N. Bischofberger, Antiviral Chem. Chemother., 1997, 8, 557–564 CrossRef CAS.
- C. Flexner, in Goodman & Gilman's: The Pharmacological Basis of Therapeutics, ed. L. Brunton, B. Chabner and B. Knollmann, McGraw-Hill, 12th edn, 2009, pp. 1623–1663 Search PubMed.
- B. Edagwa, J. E. McMillan, B. Sillman and H. E. Gendelman, Expert Opin. Drug Delivery, 2017, 14, 1281–1291 CrossRef CAS PubMed.
- M. Malamatri, K. Taylor, S. Malamatrix, D. Douroumis and K. Kachrimanis, Drug Discovery Today, 2018, 23(3), 534–547 CrossRef PubMed.
- J. J. Hobson, A. Al-khouja, P. Curley, D. Meyers, C. Flexner, M. Siccardi, A. Owen, C. F. Meyers and S. P. Rannard, Nat. Commun., 2019, 10, 1413 CrossRef PubMed.
- D. A. Cobb, N. Smith, S. Deodhar, A. N. Bade, N. Gautam, B. L. D. Shetty, J. E. McMillan, Y. Alnouti, S. M. Cohen, H. E. Gendelman and B. Edagwa, Nat. Commun., 2021, 12, 5458 CrossRef CAS PubMed.
- C. Orkin, K. Arasteh, M. Górgolas Hernández-Mora, V. Pokrovsky, E. T. Overton, P. M. Girard, S. Oka, S. Walmsley, C. Bettacchi, C. Brinson, P. Philibert, J. Lombaard, M. St. Clair, H. Crauwels, S. L. Ford, P. Patel, V. Chounta, R. D'Amico, S. Vanveggel, D. Dorey, A. Cutrell, S. Griffith, D. A. Margolis, P. E. Williams, W. Parys, K. Y. Smith and W. R. Spreen, N. Engl. J. Med., 2020, 382, 1124–1135 CrossRef CAS PubMed.
- S. Swindells, J. F. Andrade-Villanueva, G. J. Richmond, G. Rizzardini, A. Baumgarten, M. Masiá, G. Latiff, V. Pokrovsky, F. Bredeek, G. Smith, P. Cahn, Y. S. Kim, S. L. Ford, C. L. Talarico, P. Patel, V. Chounta, H. Crauwels, W. Parys, S. Vanveggel, J. Mrus, J. Huang, C. M. Harrington, K. J. Hudson, D. A. Margolis, K. Y. Smith, P. E. Williams and W. R. Spreen, N. Engl. J. Med., 2020, 382, 1112–1123 CrossRef CAS PubMed.
- J. T. Su, S. M. Simpson, S. Sung, E. B. Tfaily, R. Veazey, M. Marzinke, J. Qiu, D. Watrous, L. Widanapathirana, E. Pearson, M. M. Peet, D. Karunakaran, B. Grasperge, G. Dobek, C. M. Cain, T. Hope and P. F. Kiser, Antimicrob. Agents Chemother., 2020, 64, 1–17 CrossRef PubMed.
- P. Berndt, G. B. Fields and M. Tirrell, J. Am. Chem. Soc., 1995, 117, 9515–9522 CrossRef CAS.
- Y. C. Yu, P. Berndt, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1996, 118, 12515–12520 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- D. J. Adams, K. Holtzmann, C. Schneider and M. F. Butler, Langmuir, 2007, 23, 12729–12736 CrossRef CAS PubMed.
- Y. Zhu, L. Wang, Y. Li, Z. Huang, S. Luo, Y. He, H. Han, F. Raza, J. Wu and L. Ge, Biomater. Sci., 2020, 8, 5415–5426 RSC.
- L. Tang, C. Xu, A. Xuan, Z. Zhu and D. Ruan, Biomater. Sci., 2022, 10, 5134–5145 RSC.
- Y. Wu, S. H. Kelly, L. Sanchez-Perez, J. H. Sampson and J. H. Collier, Biomater. Sci., 2020, 8, 3522–3535 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- T. Shimada, S. Lee, F. S. Bates, A. Hotta and M. Tirrell, J. Phys. Chem. B, 2009, 113, 13711–13714 CrossRef CAS PubMed.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC.
- M. O. Guler, R. C. Claussen and S. I. Stupp, J. Mater. Chem., 2005, 15, 4507–4512 RSC.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS PubMed.
- J. E. P. Sun, B. Stewart, A. Litan, S. J. Lee, J. P. Schneider, S. A. Langhans and D. J. Pochan, Biomater. Sci., 2016, 4, 839–848 RSC.
- J. Li, X. Li, Y. Kuang, Y. Gao, X. Du, J. Shi and B. Xu, Adv. Healthcare Mater., 2013, 2, 1586–1590 CrossRef CAS PubMed.
- R. Lin, A. G. Cheetham, P. Zhang, Y. A. Lin and H. Cui, Chem. Commun., 2013, 49, 4968–4970 RSC.
- F. Wang, D. Xu, H. Su, W. Zhang, X. Sun, M. Monroe, R. Chakroun, Z. Wang, W. Dai, R. Oh, H. Wang, F. Wan and H. Cui, Sci. Adv., 2020, 6(18), eaaz8985 CrossRef PubMed.
- P. Schiapparelli, P. Zhang, M. Lara-Velazquez, H. Guerrero-Cazares, R. Lin, H. Su, R. W. Chakroun, M. Tusa, A. Quiñones-Hinojosa and H. Cui, J. Controlled Release, 2020, 319, 311–321 CrossRef CAS PubMed.
- G. Cinar, A. Ozdemir, S. Hamsici, G. Gunay, A. Dana, A. B. Tekinay and M. O. Guler, Biomater. Sci., 2017, 5, 67–76 RSC.
- M. K. Monroe, H. Wang, C. F. Anderson, H. Jia, C. Flexner and H. Cui, J. Control Release, 2022, 348, 1028–1049 CrossRef CAS PubMed.
- Y. S. Velichko, S. I. Stupp and M. O. De La Cruz, J. Phys. Chem. B, 2008, 112, 2326–2334 CrossRef CAS PubMed.
- O. S. Lee, S. I. Stupp and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3677–3683 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- S. I. Stupp, R. H. Zha, L. C. Palmer, H. Cui and R. Bitton, Faraday Discuss., 2013, 166, 9–30 RSC.
- M. J. Webber, J. Tongers, M. A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3–11 CrossRef CAS PubMed.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- T. Shimada, N. Sakamoto, R. Motokawa, S. Koizumi and M. Tirrell, J. Phys. Chem. B, 2012, 116(1), 240–243 CrossRef CAS PubMed.
- Y. C. Yu, V. Roontga, V. A. Daragan, K. H. Mayo, M. Tirrell and G. B. Fields, Biochemistry, 1999, 38, 1659–1668 CrossRef CAS PubMed.
- M. Black, A. Trent, Y. Kostenko, J. S. Lee, C. Olive and M. Tirrell, Adv. Mater., 2012, 24, 3845–3849 CrossRef CAS PubMed.
- E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 8819–8821 CrossRef CAS PubMed.
- T. J. Moyer, H. Cui and S. I. Stupp, J. Phys. Chem. B, 2013, 117(16), 4604–4610 CrossRef CAS PubMed.
- H. Cui, A. G. Cheetham, E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2014, 136(35), 12461–12468 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- J. H. Ortony, C. J. Newcomb, J. B. Matson, L. C. Palmer, P. E. Doan, B. M. Hoffman and S. I. Stupp, Nat. Mater., 2014, 13, 812–816 CrossRef CAS PubMed.
- C. F. Anderson, R. W. Chakroun, H. Su, R. E. Mitrut and H. Cui, ACS Nano, 2019, 13(11), 12957–12968 CrossRef CAS PubMed.
- H. Su, F. Wang, W. Ran, W. Zhang, W. Dai, H. Wang, C. F. Anderson, Z. Wang, C. Zheng, P. Zhang, Y. Li and H. Cui, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4518–4526 CrossRef CAS PubMed.
- Y. Shi, D. S. Ferreira, J. Banerjee, A. R. Pickford and H. S. Azevedo, Biomater. Sci., 2019, 7, 5132–5142 RSC.
- M. Isik, C. C. Eylem, T. Haciefendioglu, E. Yildirim, B. Sari, E. Nemutlu, E. Emregul, B. O. Okesola and B. Derkus, Biomater. Sci., 2021, 9, 8270–8284 RSC.
- M. J. Webber, J. B. Matson, V. K. Tamboli and S. I. Stupp, Biomaterials, 2012, 33, 6823–6832 CrossRef CAS PubMed.
- A. G. Cheetham, P. Zhang, Y. A. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- A. G. Cheetham, Y. C. Ou, P. Zhang and H. Cui, Chem. Commun., 2014, 50, 6039–6042 RSC.
- Z. Chen, P. Zhang, A. G. Cheetham, J. H. Moon, J. W. Moxley, Y. A. Lin and H. Cui, J. Controlled Release, 2014, 191, 123–130 CrossRef CAS PubMed.
- R. W. Chakroun, F. Wang, R. Lin, Y. Wang, H. Su, D. Pompa and H. Cui, ACS Nano, 2019, 13(7), 7780–7790 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 6638–6663 RSC.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- K. E. Giesler and D. C. Liotta, J. Med. Chem., 2016, 59, 10244–10252 CrossRef CAS PubMed.
- S. K. Ladner, M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. U. T. Guo, C. Seeger and R. W. King, Antimicrob. Agents Chemother., 1997, 41(8), 1715–1720 CrossRef CAS PubMed.
- C. A. Kim and J. M. Berg, Nature, 1993, 362, 267–270 CrossRef CAS PubMed.
- D. L. Minor and P. S. Kim, Nature, 1994, 367, 660–663 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- Z. Song, X. Chen, X. You, K. Huang, A. Dhinakar, Z. Gu and J. Wu, Biomater. Sci., 2017, 5, 2369–2380 RSC.
- Z. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC.
- Z. Yang and B. Xu, Adv. Mater., 2006, 18, 3043–3046 CrossRef CAS.
- S. Winkler, D. Wilson and D. L. Kaplan, Biochemistry, 2000, 39, 12739–12746 CrossRef CAS PubMed.
- H. T. West, C. M. Csizmar and C. R. Wagner, Biomacromolecules, 2018, 19, 2650–2656 CrossRef CAS PubMed.
- Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS PubMed.
- K. Thornton, A. M. Smith, C. L. R. Merry and R. V. Ulijn, Biochem. Soc. Trans., 2009, 37, 660–664 CrossRef CAS PubMed.
- H. Cui, T. Muraoka, A. G. Cheetham and S. I. Stupp, Nano Lett., 2009, 9(3), 945–951 CrossRef CAS PubMed.
- J. M. Godbe, R. Freeman, J. A. Lewis, I. R. Sasselli, M. H. Sangji and S. I. Stupp, Acta Biomater., 2021, 135, 87–99 CrossRef CAS PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- G. L. Ackerman, in Clinical Methods: The History, Physical, and Laboratory Examinations, ed. H. K. Walker, W. D. Hall and J. W. Hurst, Butterworths, Boston, 3rd edn, 1990 Search PubMed.
- B. F. Palmer and D. J. Clegg, Adv. Physiol. Educ., 2016, 40, 480–490 CrossRef PubMed.
- S. S. Timur, A. Şahin, E. Aytekin, N. Öztürk, K. H. Polat, N. Tezel, R. N. Gürsoy and S. Çalış, Pharm. Dev. Technol., 2018, 23, 301–310 CrossRef CAS PubMed.
- E. A. Krogstad and K. A. Woodrow, Int. J. Pharm., 2014, 475, 282–291 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- Y. Zhu, M. Curtis, X. Qi, M. D. Miller and K. Borroto-Esoda, Antiviral Chem. Chemother., 2009, 19, 165–176 CrossRef PubMed.
- Gilead, Section 2.6 Nonclinical Summary: Emtricitabine/Rilpivirine/Tenofovir Disoproxil Fumarate Fixed-Dose Combination, 2010.
- C. Niu, H. Bao, T. Tolstykh, H. M. Micolochick Steuer, E. Murakami, B. Korba and P. A. Furman, Antiviral Ther., 2010, 15, 401–412 CrossRef CAS PubMed.
- J. L. Dienstag, Hepatology, 2009, 49, S112–S121 CrossRef CAS PubMed.
- L. L. Lock, C. D. Reyes, P. Zhang and H. Cui, J. Am. Chem. Soc., 2016, 138, 3533–3540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Gelation videos, additional experimental protocols, mass spectrometry data, purity data, NMR data, hydrogel images, and gel release and cryogenic TEM images. See DOI: https://doi.org/10.1039/d2bm01649d |
| This journal is © The Royal Society of Chemistry 2023 |