
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkaline hydrogel electrolyte from biosourced chitosan to enhance the rate capability and energy density of carbon-based supercapacitors†
Sirine
Zallouz
ab,
Jean-Marc
Le Meins
ab and
Camélia
Matei Ghimbeu
 *abc
*abc
aInstitut de Science des Matériaux de Mulhouse (IS2M), Université de Haute-Alsace, CNRS UMR 7361, F-68100 Mulhouse, France. E-mail: camelia.ghimbeu@uha.fr; Tel: +33 (0) 3 89 60 87 43
bUniversité de Strasbourg, F-67081 Strasbourg, France
cRéseau sur le Stockage Electrochimique de l’Energie (RS2E), FR CNRS 3459, 80039 Amiens Cedex, France
First published on 14th November 2022
Abstract
This paper reports the development of a safe carbon-based supercapacitor, which is based on a green biodegradable hydrogel electrolyte that is prepared from chitosan biopolymer and KOH as the electrolyte source. The impact of electrolyte solution ageing time on electrolyte gel formation is investigated. A critical time of 2 days is necessary to obtain gel electrolytes mechanically exploitable. This is associated with the gel structural modification, as observed by FTIR and 1H/13C NMR. Between 2 and 4 days, the capacitance increases from 76 to 95 F g−1 and remains stable up to 21 days. Good rate handling is achieved (62%) with a capacitance of 59 F g−1 at 10 A g−1. Remarkably, the developed gel exhibits good stability when the cell voltage is increased from 0.8 V to 1.3 V. The voltage window extension allows to obtain for the C–C device, a high energy density (5.1 W h kg−1) at a power density of 32.5 W kg−1, which is almost 3 times higher than that delivered by liquid 2 M KOH at 0.8 V. The gel electrolyte could be used with pseudocapacitive materials, C/Co3O4 and voltage window extension is achieved along with significant increase in energy density from 1.66 to 6.31 W h kg−1. Better capacitance retention is obtained by the chitosan–KOH gel electrolyte than by liquid KOH. Advantageously, the gel electrolyte prevents the electrode degradation and positive current collector from undergoing corrosion.
1. Introduction
Supercapacitors are electrochemical energy storage (EES) devices that are characterized by a high-power density and long cycle life. However, their energy density still needs to be improved, which is why many studies have been devoted to this topic.1 A supercapacitor is typically composed of two electrodes with a high surface area and an electrolyte composed of positive and negative ions. The most common systems are electrochemical double layer capacitors (EDLCs), which rely on carbonaceous materials as electrodes. When a voltage is applied, opposite charges of electrolyte ions accumulate on each side of the electrodes and the supercapacitor is charged. The nature and duration of the interaction between the electrolyte and electrode defines the time response, capacitance, energy and power density as well as the cycle life of the device.2 That is why the choice of the electrode material and the electrolyte is crucial to achieve a good energy storage. Carbon is the most commonly used electrode material owing to its high specific surface area, good stability, good conductivity and low cost.3 The most commonly used electrolytes in traditional supercapacitors are aqueous liquid electrolytes due to their accessibility, high ionic conductivity and satisfactory performance. However, these electrolytes exhibit some risks, including leakage of harmful liquids, corrosion and even the emission of hazardous gas.4 These drawbacks have driven researchers to investigate gel or solid-like electrolytes, which might limit these problems. For two decades, many works have reported the formation and use of hydrogel electrolytes,5 gel polymer electrolytes6 and solid-state electrolytes7 in different energy storage devices, i.e., batteries,8 supercapacitors9 and fuel cells.10 In particular, their application in dye-sensitized solar cells is emerging.11,12 More recently, a photonic hydrogel for self-monitoring of solid-state electrolytes in zinc–air batteries that allows real time check of the state of the battery has been reported.13Hydrogels and gel electrolytes are particularly promising, as they exhibit the diffusive properties of liquids and cohesive properties of solids, which provides them structural, thermal and mechanical stability as well as good ion transport properties.14 Gel polymer electrolytes can be classified based on the type of host source (synthetic or natural polymer), polymeric composition, type of cross-linking (chemical or physical crosslinking) and physical appearance.15 For electrochemical capacitors, the commonly used polymers have a synthetic origin. For example, poly(vinyl)alcohol (PVA)16 is the most employed polymer in combination with different types of aqueous acid electrolytes, such as H2SO417 and H3PO4.18 It exhibits a good chemical stability and high water absorbance due to its hydroxyl (–OH) groups. Other synthetic polymers, such as poly(ethylene oxide) (PEO)19 and poly(acrylic) acid (PAA),20 have also been adopted as polymer hosts. However, the handling of synthetic base matrices again introduces environmental problems and biodegradability issues. Moreover, the fabrication process requires multiple steps and involves safety problems and high fabrication costs. All of these aspects hamper the reliable and scalable use of these polymers.
To address this concern, attempts to use biodegradable gel electrolytes that are fabricated from natural polymer hosts have been reported. For instance, a microsupercapacitor was fabricated using a biodegradable hydrogel electrolyte that was agarose based with NaCl salt as the ion source.21 In another work, a cross-linked sodium alginate hydrogel mixed with KCl was tested and exhibited self-healing abilities.22 Moreover, hydroxyethyl cellulose was used with H3PO4 as a solid polymer electrolyte for the supercapacitor.23 Other natural hosts, such as gelatine and dough, have also been reported.
Another proposed biopolymer was chitosan, which is obtained by the deacetylation of chitin, a natural polymer composed of polysaccharides. Chitosan is among the most abundant natural polymers in nature and can be found in the exoskeletons of arthropods, such as shrimps, crabs and lobsters.24 Chitosan has been investigated in many fields, such as medicine,25 tissue engineering,26 water treatment27 and food packaging.28 Furthermore, chitosan was also used as a green carbon precursor and has been explored in tuning carbon properties.29 According to some sources, chitosan is the first biopolymer that was used for polymer electrolyte applications.30
Chitosan is formed of 1,4-linked 2-deoxy-2-aminoglucose and therefore contains several polar groups, such as hydroxyl and amino groups (NH2). It is weakly alkaline and is soluble in a dilute aqueous solution of acetic acid (CH3COOH). Chitosan functional groups play a role as electron donors to interact with inorganic salts.31 The nitrogen atom acts as a complexation site for cation coordination and generally forms a gel by chemical crosslinking. Chitosan can form a polymer hydrogel either by chemical or ionic crosslinking using a cross-linking reagent or suitable ions, respectively. For example, Choudhury et al. prepared a chitosan hydrogel membrane electrolyte using a neutral Na2SO4 electrolyte, which acted as ionic crosslinking and was applied for fuel cell applications.32 In another work, Fazdallah et al. reported a low-cost chitosan–oxalic acid-based proton conducting membrane, which was obtained by a solution casting method for fuel cells.33 The combination of chitosan with ionic liquids has also proven to be promising. We can cite Yamagata et al., who investigated a nonaqueous gel electrolyte based on chitosan with 1-ethyl-3-methylimidazole tetrafluoroborate (EMImBF4) in an EDLC device up to 2.5 V. They obtained a specific capacitance of 131 F g−1 at a 2.5 mA cm−2 current rate, which is slightly higher than that of its ionic liquid in its liquid state counterparts (128 F g−1).34 Yang et al. proposed a supramolecular carboxylated chitosan with Li2SO4 as hydrogel electrolyte in carbon supercapacitors that delivered 31.89 F g−1 at 0.5 A g−1 at 1.4 V potential window and had a stability of 32% of capacitance retention after 400 cycles.35 In another work, an alkali hydrogel electrolyte is prepared based on chitosan and FeCl3 as a crosslinking agent. The quasi-solid state supercapacitors exhibited a capacitance of 39.11 F g−1 at 0.5 A g−1 and a rate capability of 38.5% at 5 A g−1.36
Efforts have been devoted to increasing the ionic conductivity of gel electrolytes by using plasticizers. For instance, Kumer et al. produced a chitosan–lithium perchlorate complex gel electrolyte using ethylene carbonate and propylene carbonate with a conductivity of 5.5 10−3 S cm−1 and the device exhibited a capacitance of 130 F g−1 at 10 mV s−1.37 All mentioned reports indicate that there is still room for performance improvement when using chitosan as a host agent for supercapacitors, because capacitance is moderate and stability is challenging. Moreover, long-cycle performance is not systematically provided, which raises questions about the cycle life of such devices.
In aqueous electrolytes, the potential window is generally limited by water electrolysis to 1.23 V. However, it was shown that liquid neutral electrolytes can reach higher maximum cell voltages up to 1.638 and even 1.9 V.39 This is due to the better stability of porous carbon, which also avoids the corrosive feature encountered when using alkali or acidic electrolytes. It was demonstrated that the performance deterioration of carbon–carbon capacitors during floating with 1 M Li2SO4 at 1.6 V is caused by the oxidation of the positive electrode and the corrosion of the positive current collector.40 It can be concluded that most of the reported studies on expanded voltage windows use aqueous neutral electrolytes, such as K2SO441 and Na2SO4,38 and to our knowledge no study on alkaline or hydroxide electrolytes has been reported. Therefore, this route is auspicious, as it offers the possibility to be investigated on pseudocapacitive materials that contain metal-based electrodes, in which the cycling stability is limited to alkaline media.
In this study, we report on the preparation of a chitosan–KOH hydrogel electrolyte and its electrochemical performance in carbon–carbon and carbon–metal oxide supercapacitors. The optimization shows the importance of the synthesis conditions; drying, stirring and ageing were shown to be crucial to the gelling process and were attentively controlled. The modification of the chitosan solution with ageing time was followed by NMR analysis, while the resulting chitosan–KOH gel electrolyte was analysed by FTIR. The electrolyte gel cross-linking was found to influence the electrochemical performance of the electrodes. An improved capacitance compared to that of liquid KOH is observed. Moreover, with the hydrogel, the voltage window can extend up to 1.3 V using an alkaline electrolyte, which is reported for the first time to our knowledge. This led to a significant increase in energy density for both C–C and C/Co3O4 devices and higher capacity retention during long-term cycling than with liquid electrolyte. Post-mortem analyses performed on positive and negative electrodes showed limited degradation at 1.3 V with the gel-electrolyte. Moreover, the chitosan–KOH gel electrolyte inhibited corrosion after cycling.
2. Experimental section
2.1. Chemicals
Low molecular weight chitosan, glyoxylic acid monohydrate (98%) and acetic acid 100% were purchased from Sigma-Aldrich. Activated carbon R3 extra (Norit, Amersfoort, The Netherlands) was used as the active material for the electrochemical study. Qualitative filter paper no. 413 was purchased from VWR. All chemicals were used without further purification. Chitosan was stored in the absence of humidity to prevent ageing and dissolution problems.2.2. Electrolyte gel preparation and characterization
The chitosan–KOH gel electrolytes were synthesized as schematically presented in the flow chart in Fig. 1. First, a solution of 1 wt% chitosan was prepared by dissolving chitosan (0.997 g) in 97.99 mL of an aqueous solution of acetic acid (1.030 g) (1% wt) as the solvent. To ensure good solubility, the solution was rigorously stirred for ∼1 h 30 min in a beaker closed with para-film, followed by the addition of glyoxylic acid cross-linker (1.5 g). The solution was then stirred at 26 °C for a time period between 0 and 21 days to investigate its impact on gel formation (Fig. 1, step a). Afterwards, 15 mL of this solution were mixed with 15 mL of 2 M KOH for another 24 h under stirring to homogenize the mixture (Fig. 1, step b). After this step, the pH increased from ∼2.3 to ∼13.7. Subsequently, 10 mL of the as-obtained solution were cast in a 10 cm diameter Petri dish and maintained under a fume hood for ∼2 h to allow the solvent to evaporate slowly and form the gel at a temperature of ∼23 °C and a relative humidity of 26.8% (Fig. 1, step c). Notably, the drying process strongly depends on the conditions (temperature and humidity) and, if they are modified, the drying time should be adjusted. A disk with a diameter of 12 mm was cut with a cutter and used as an electrolyte when constructing the 2-electrode cell. Real images of the whole synthesis process can be viewed in Fig. S1, ESI.†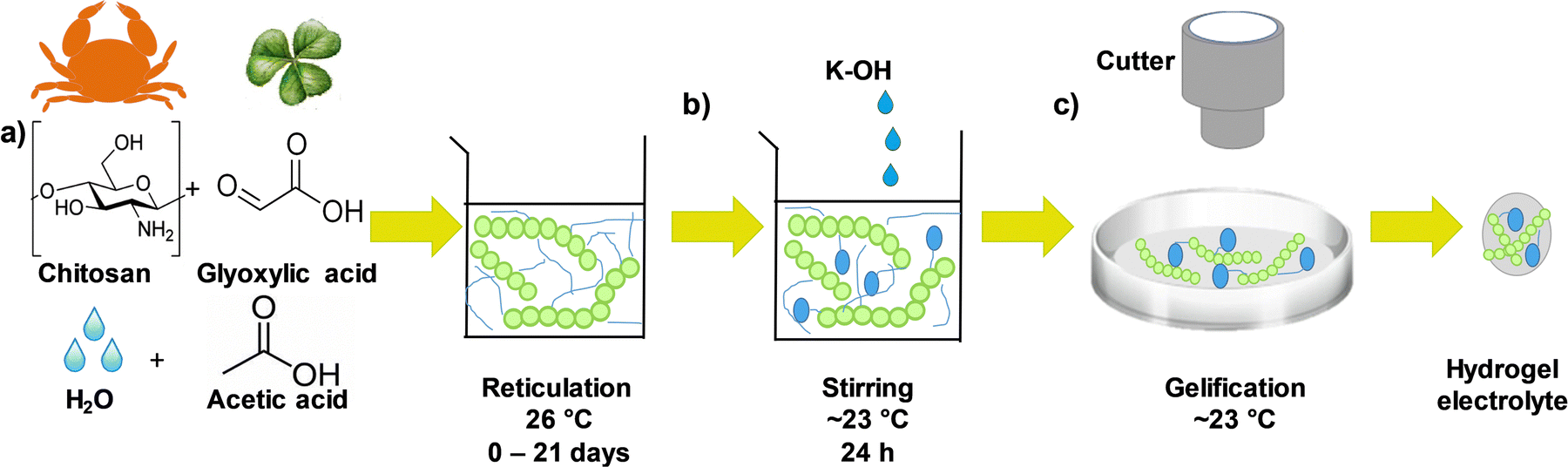 | ||
| Fig. 1 Schematic representation of the preparation of the chitosan–KOH gel electrolyte including the following steps: (a) polymer host + solvent solution, (b) addition of KOH aqueous electrolyte, (c) solution casting in a petri dish. | ||
The chemical nature of the gel electrolytes prepared under different conditions was characterized by 1H NMR and 13C NMR, which were recorded on a Varian Mercury 300 MHz spectrometer with deuterated chloroform (CDCl3) as the solvent at room temperature for the chitosan solution at different ageing days (step a of Fig. 1). Fourier transform infrared spectroscopy (FTIR) spectrometer, Thermo Fisher Scientific iS50, with the ATR (attenuated total reflectance) mode was performed on the circular ready-to-use gel electrolyte (step c of Fig. 1).
Physicochemical analyses of the carbon material were performed prior to the electrochemical measurements. The textural properties were evaluated using a porosimeter system, i.e., ASAP 2020 from Micromeritics, by N2 adsorption at 77 K. Details of the procedure can be found in the ESI.† Cycled electrodes were washed in 50 mL distilled water and then dried at 80 °C for 8 h. Afterwards, Raman analysis was performed on pristine and cycled electrodes with a Raman microspectrometer LabRAM BX40 (Horiba 300) using a wavelength of 532 nm. Mapping on 9 different spots was conducted and the average values were used to obtain representative information on the materials.
2.3. Electrochemical characterization
Electrodes were prepared by mixing a commercial porous carbon (Norit R3 Extra, 95%) and PTFE (5%) as binder in ethanol until the solvent evaporated. Then, the paste was rolled on a plastic film to reach a thickness of 150–250 μm. Circular self-standing electrodes were then cut (∼9 mg weight, 0.785 cm2 surface) and dried at 120 °C for 24 h under vacuum. Electrochemical tests were performed with a multichannel VSP300 potentiostat (Biologic, France). A two-electrode Teflon Swagelok cell was built using qualitative filter paper as a separator, the hydrogel electrolyte on each side of the separator and in contact with both the positive and negative electrodes, and stainless-steel current collectors. Cyclic voltammetry (CV), galvanostatic charge with potential limitation (GCPL) and electrochemical impedance spectroscopy (EIS) were carried out for these devices. A Hg/HgO reference electrode was added to the two-electrode device to perform standard three-electrode cell investigations for monitoring electrode potentials. The potential values are presented vs. Hg/HgO electrode. A three-electrode cell using 2 M KOH liquid electrolyte was constructed for comparison purposes. The specific capacitance was calculated from GCPL according to eqn (1) as follows: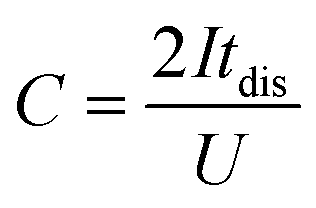 | (1) |
The energy density and power density were determined based on the following equations:
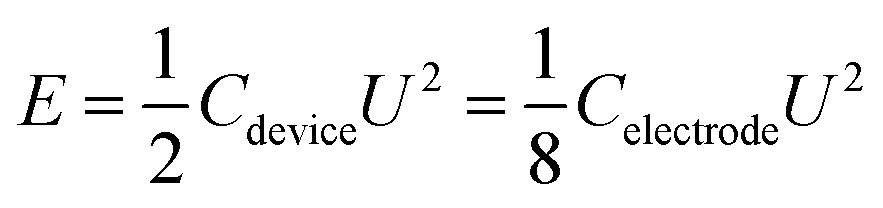 | (2) |
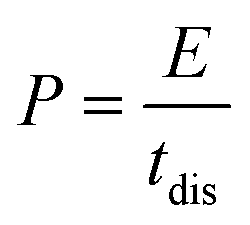 | (3) |
3. Results and discussion
3.1. Carbon characterization
The textural and structural properties of activated carbon are presented in Fig. 2. The N2 adsorption/desorption isotherm (Fig. 2a) is mainly type I with a very small hysteresis. Thus, the carbon is highly microporous with a small portion of mesopores. The calculated specific surface area of Norit R3 Extra is 1224 m2 g−1, while the micropore and mesopore volumes are 0.47 cm3 g−1 and 0.09 cm3 g−1, respectively (Table S1, ESI†). The mesopore volume is negligible compared to the microporous volume. The pore size distribution (PSD) (inset Fig. 2a) determined by 2D-NLDFT shows a main peak that corresponds to a pore width of 0.7 nm and another small contribution from the pores centred at 1.5 nm. Mesopores are not observed in the pore size distribution due to their small volume.43 Both types of pores are important in the charge storage mechanism. Micropores assure that charge accumulation and ion adsorption occurrence, while mesopores contribute to ion transport between the bulk of the electrode and the electrode/electrolyte interface.44 Raman spectroscopy shows the structure of the activated carbon, as seen in Fig. 2b. The spectrum deconvoluted according to the procedure proposed by Sadezky et al.45 shows several contributions and typically, the D1 and G bands corresponding to graphitic lattice vibration mode with A1g symmetry (defects) and to ideal graphitic lattice vibration mode with E2g symmetry, are observed at ca. 1337 cm−1 and 1588 cm−1. The ID1/IG value calculated from the area under the curve is 1.64, which indicates that the material has a disordered structure.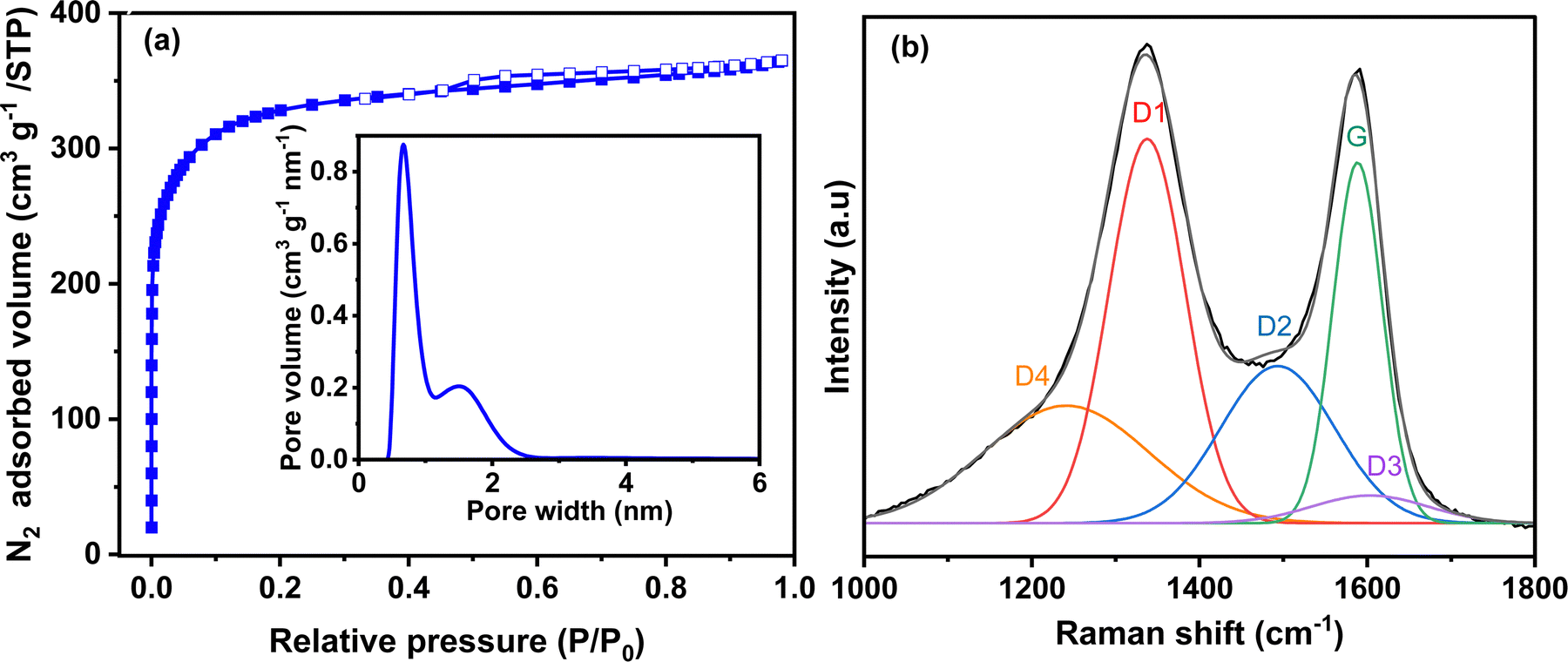 | ||
| Fig. 2 (a) N2 adsorption/desorption isotherm (inset: pore size distribution calculated from the 2D-NLDFT heterogeneous surface carbon model for N2 adsorption) and (b) deconvoluted Raman spectrum of activated carbon Norit R3 Extra. The description of each peak contribution is provided elsewhere.45 | ||
3.2. Gel electrolyte solution characterization
The chitosan solution obtained in step (a) by a mixture of chitosan and glyoxylic acid in acetic acid solution was analysed at different time intervals (0, 4 and after 7 days) by 1H NMR and 13C liquid NMR (Fig. 3). The chemical structure of chitosan is presented in Fig. S2, ESI,† to facilitate the interpretation. For comparison purposes, the 1H and 13C liquid NMR spectra of single precursors are provided in Fig. S3, ESI.† The precursor NMR spectra are comparable to those reported in the literature.46 Concerning 1H NMR, the peak at 4.75 ppm originates from the deuterium oxide solvent (Fig. 3a and Fig. S3, ESI†). Characteristic proton peaks of chitosan appear between 3 and 4 ppm relative to the hydrogen bonded to C3, C4 and C5 in the chitosan solution (Fig. 3a).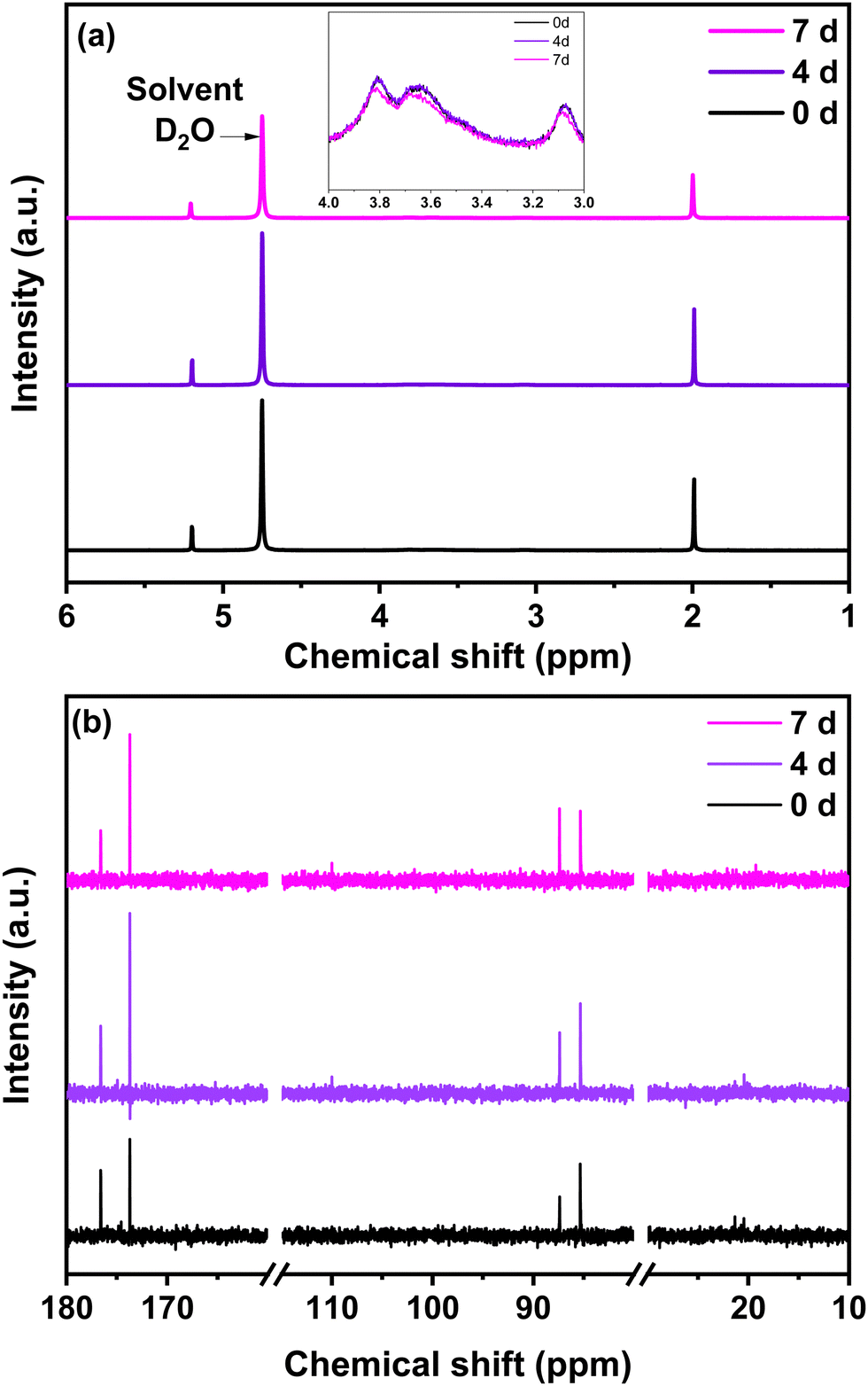 | ||
| Fig. 3 (a) 1H NMR (inset: 1H NMR from 3–4 ppm) (b) 13C liquid NMR of the chitosan solution at different ageing days, i.e., 0, 4 and 7 days. | ||
The acetic acid signature seen in Fig. S3a (ESI†) remains present in the NMR spectrum of the chitosan solution. This is also the case for glyoxylic acid. Together, both acids contribute to catalyse the chitosan reticulation. Indeed, the measured pH was approximately 2, therefore acidic favouring the crosslinking and polymerization, according to the literature.47 The 1H NMR provides valuable information, but is not sufficient for understanding the crosslinking of chitosan in the presence of aldehydes. For 13C NMR, the characteristic peaks of the individual precursors are shown in Fig. S3b, ESI.† For the chitosan solution (Fig. 3b), the small signals that occur at 20 ppm and the intense signal at 176 ppm belong to acetic acid. The signals located at 80–85 ppm and 174 ppm originated from glyoxylic acid. An increase in the 174 ppm peak of glyoxylic acid to the detriment of the 176 ppm peak of acetic acid is observed with the increase in ageing time from 0 to 7 days. Similar behaviour is observed when comparing the evolution of the 88 ppm and 85 ppm peaks of glyoxylic acid over time. Regarding the chitosan contribution, its peaks are less visible because it is difficult to obtain a high concentration of chitosan in solution, observation which corresponds with other works.48 A small peak in 13C NMR starts to be visible at ∼110 ppm beginning from day 4 (Fig. 3b). As proposed in the literature, this peak is associated to C1 of the chitosan structure after crosslinking.48 This result indicates that crosslinking also occurs at C1 of the chitosan level.
The degree of conversion of the aldehyde into imine linkages increased in the first week of solution preparation, proving that the reaction equilibrium of the imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds) formed in water was reached by this time.49 Numerous factors determine the path of crosslinking and hydrogel formation,50i.e., the pH and the polymer concentration. For instance, a low concentration of polymer leads to a parallel crosslinking hydrogel without perpendicular cross-linking.47 As an example, oxalic acid was used to form a polymer chitosan membrane based on the same mechanism.33
N bonds) formed in water was reached by this time.49 Numerous factors determine the path of crosslinking and hydrogel formation,50i.e., the pH and the polymer concentration. For instance, a low concentration of polymer leads to a parallel crosslinking hydrogel without perpendicular cross-linking.47 As an example, oxalic acid was used to form a polymer chitosan membrane based on the same mechanism.33
To obtain more information on the chemical evolution of the self-standing circular chitosan–KOH gel electrolytes (step c) of (Fig. 1) with the solution ageing time, FTIR was performed (Fig. 4).
First, it can be observed that the Schiff base and O–H stretching groups located at ∼3400 cm−1 overlap and increase with the ageing time.51 This can have the following origins: (i) the presence of glyoxylic acid in water, in which the carbonyl converts to a geminal diol (two hydroxyl O–H functional groups bound to the same carbon)52 and (ii) the reaction of chitosan and glyoxylic acid leading to a Schiff base53 (imines with a characteristic CN bonds) formation, which is accompanied by the formation of H2O molecules. According to the literature, the Schiff base is present between 3200 and 3400 cm−1.54,55 The increase in the Schiff base signature demonstrates the progressive reticulation of chitosan, which is in accordance with the NMR results presented above.
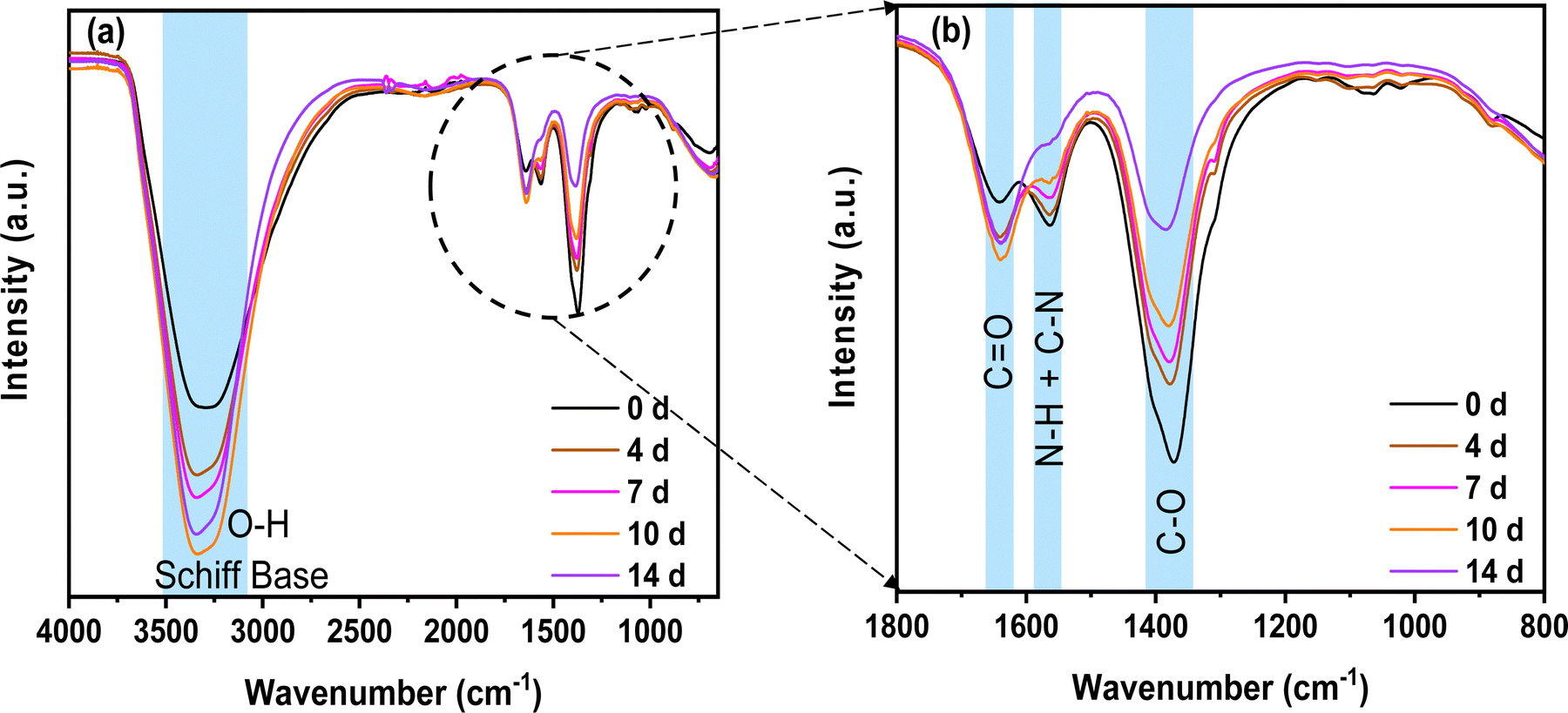 | ||
| Fig. 4 FTIR spectra of the different chitosan–KOH gel electrolytes prepared with solutions at different ageing days: (a) spectra from 4000 to 700 cm−1 and (b) magnified views of the spectra from 1800 to 800 cm−1. | ||
As we discussed above, the imine formation and polymer reticulation are slow and take almost 7 days, which explains the progressive increase in OH groups with ageing.53 Concomitantly, the C–O stretching groups at 1375 cm−1 decreased significantly over time, probably due to the H-bonding of the C–O groups of chitosan and the carboxyl group of glyoxylic acid in water. The bands matching N–H and C–N stretching (1560 cm−1) decrease as well. This behaviour might be associated with the deprotonation of the primary amine group (NH2) of chitosan in the solution.56 Water evaporation during gelling comes from condensation reactions. This results in a slight increase in CO groups at 1640 cm−1. All of these modifications suggest a possible crosslinking of the different precursors in the solution. Nonetheless, it is necessary to remember that the analysis was done on the gel and not on the solution. Hence, most of the solvent was already removed at the time of the analysis and the mixture was concentrated by evaporation to form a film.
Another key issue is the manipulation and mechanical stability of the gel electrolytes obtained. It has been observed that they exhibit disparate mechanical behaviour. At 0 d, the formation of a polymeric reticulated film was not possible on a large surface. After the evaporation step, only small fragments were formed. At 2 and 3 d, the circular gel is extremely fragile, breaks easily, and can barely be called self-standing. Starting from 4 d, the circular gel is mechanically stable and easy to handle for further use. The reason for these peculiar mechanical characteristics may be related to the different degree of reticulation of chitosan as shown by FTIR and NMR analyses. Therefore, this result suggests that a certain period of time is necessary to cross-link the chitosan, and that the quality of the gel might influence the electrochemical performance, as will be investigated in the next section.
An important characteristic when formulating a gel electrolyte is the ionic conductivity. In our case, the ionic conductivity was calculated according to a procedure described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 The value was estimated to be 0.45 × 10−2 S cm−1 (Table 1), which is comparable to the values reported in the literature.34,37 A high ionic conductivity is imperative to achieve good electrochemical performance. This will be further discussed in the next section.
57 The value was estimated to be 0.45 × 10−2 S cm−1 (Table 1), which is comparable to the values reported in the literature.34,37 A high ionic conductivity is imperative to achieve good electrochemical performance. This will be further discussed in the next section.
| Thickness (cm) | Conductivity (S cm−1) | |
|---|---|---|
| chitosan–KOH gel electrolyte | 0.03 | 0.45 × 10−2 |
3.3. Electrochemical characterization
Several capacitors were built with activated carbon and electrolyte gels and tested using different techniques. To investigate the effect of the ageing time, the voltage window used in the first tests was 0.8 V to stay in the safe zone with the KOH electrolyte. Fig. 5 shows the performance of different chitosan–KOH gel electrolytes prepared with chitosan solutions with different ageing times. The performance using 2 M KOH liquid electrolyte is also provided as a reference. The cyclic voltammograms at 10 mV s−1 are presented in Fig. 5a. As mentioned above, when stirring the chitosan–KOH solution, with ageing times less than 2 days, it is not possible to form a gel upon evaporation. This was associated to the low degree of cross-linking of the chitosan, as illustrated by the FTIR and NMR analyses. As a result such gels are not suitable to be used in the device. Starting from two days of ageing (2 d), the gels were formed and it can be noted that the CV shape is rectangular for all the electrolytes; thus, the behaviour follows a double layer capacitance.2 A net increase in performance was observed from 2 to 4 days, after which the increase was rather steady. In particular, the voltammograms for the chitosan–KOH gels after 6 d and 21 d presented a slightly larger area, suggesting a better capacitance. Additionally, the liquid electrolyte has a smaller area under the curve and slower EDL formation.58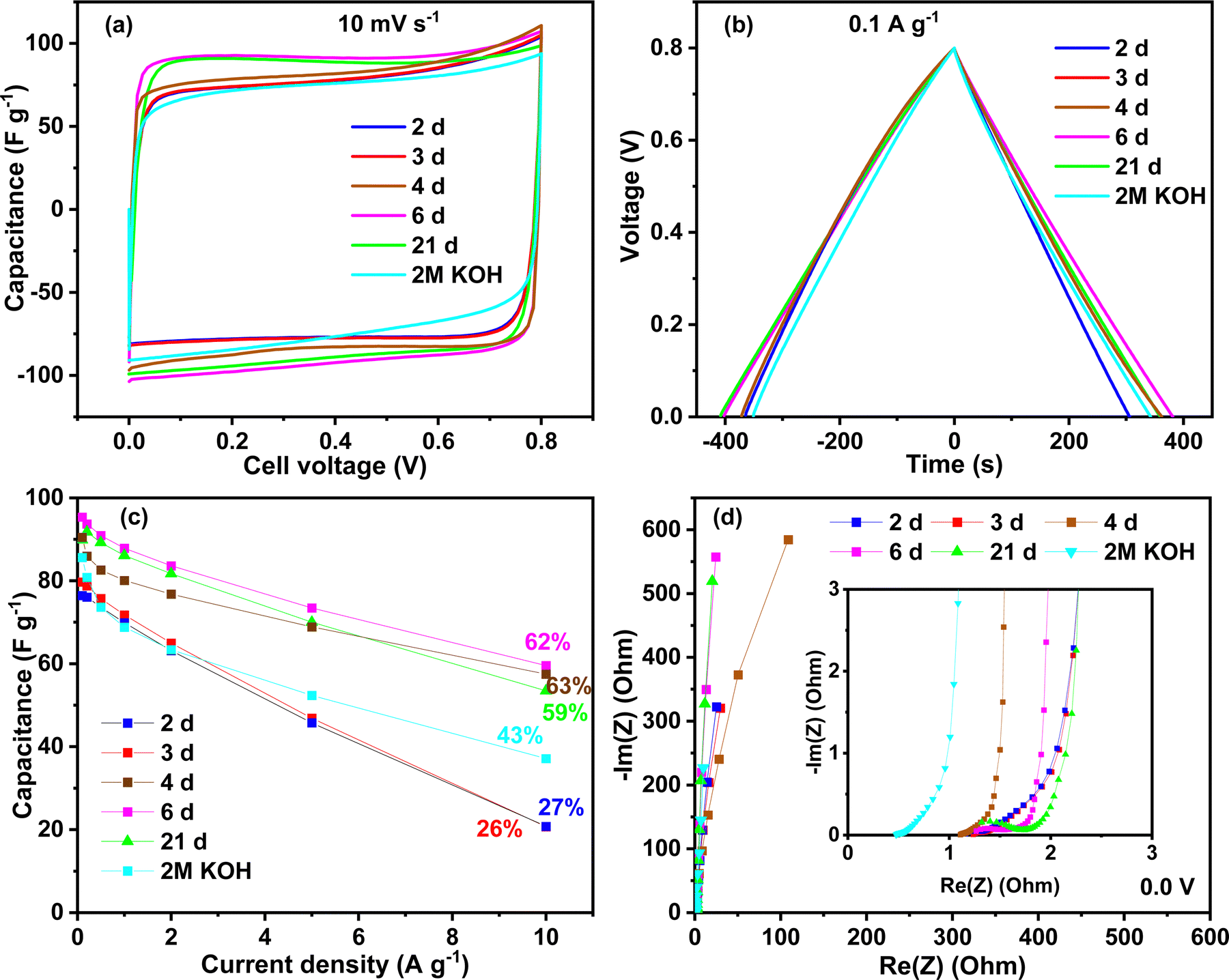 | ||
| Fig. 5 Electrochemical performance of activated carbon symmetric capacitors using chitosan–KOH gel electrolyte obtained by different ageing times of the solution. (a) Cyclic voltammetry at 10 mV s−1 (b) galvanostatic charge discharge at 0.1 A g−1. (c) Rate capability up to 10 A g−1. (d) Electrochemical impedance spectroscopy at 0.0 V (inset: EIS at high frequency). | ||
A similarity in the charge discharge time for all the electrolytes at 0.1 A g−1 was observed for GCPL (Fig. 5b). Solely, the gel used after 2 d shows a smaller discharging time compared to that of the others.
Charge discharge tests were conducted up to 10 A g−1 to evaluate the capacitance behaviour at high current regimes (Fig. 5c). Similar to the CV, two families in the gel electrolytes are distinguished, i.e., for 2 d and 3 d gel electrolytes, the performance is moderate and the rate capability is approximately 27% at 10 A g−1. Starting from the fourth day (4 d), the capacitance retention increased, reaching approximately 60% until day 21. These values are higher than those obtained in liquid 2 M KOH, which maintains 43% of its initial capacitance at 10 A g−1. Moreover, the initial capacitance is 95 F g−1 at 0.1 A g−1 for the 6 d gel electrolyte, which is higher than 2 M KOH (85 F g−1 at 0.1 A g−1). This suggests a crucial influence of the chemical and/or mechanical properties of the chitosan–KOH gel electrolytes on the performance. Low cross-linking and mechanical stability of gels (up to day 3) results in low-moderate performance while higher cross-linking allow self-standing gel electrolytes with good performance to be achieved (starting from day 4). These results lead to the conclusion that the synthesized gel herein can be used from the fourth day up to at least 3 weeks, without any deficiency in the performance at both low and high current densities.
Fig. 5d presents the Nyquist plot from the EIS measurement at 0.0 V. All the materials tested with different aged chitosan-derived electrolytes present a vertical line almost parallel to the Y axis, demonstrating capacitive behaviour.59 The inset presents the data at high frequencies. Since all the devices differ only by the electrolyte nature, the electrical series resistance (ESR) represented by the intersection of the curve with the real axis in the high frequency region can mainly be related to the bulk electrolyte conductivity.60 The gel electrolytes exhibit values of ∼1.1 Ω, whereas the liquid 2 M KOH electrolyte exhibits, as expected, a smaller ESR value of ∼0.5 Ω. It is known that aqueous electrolytes have a high ionic conductivity;61 however, other factors contribute to the improvement in performance, such as diffusion and the presence of functional groups.62 To thoroughly exploit the impedance analysis, the capacitance vs. frequency at 0.0 V is presented in Fig. S4, ESI,† and compares the time response for the different devices. The capacitance retention is calculated based on the specific capacitance ratio at 1 Hz and the initial capacitance at 1 mHz.63 At a frequency equal to 1 Hz, the capacitance retention is 54% for the device using the 2 d- and 3 d-aged gels, while for the gel electrolytes at 4 d and 6 d the retention is higher than 84%. For the gel at 21 d, the retention has an intermediate value of 77%. In addition, a nice plateau is seen for the gel electrolytes at 4 d, 6 d and 21 d between 1 mHz and 200 mHz, suggesting that the charge stored in the system is almost constant. These results are in accordance with the other electrochemical techniques and show a better response starting from 4 days of ageing of the chitosan solution.
To confirm the applicability of the chitosan–KOH gel electrolyte in supercapacitor devices, tests under different conditions must be performed. The analyses were conducted twice to have reliable data. The error between the different electrochemical tests was estimated to max. 15%. Fig. S5, ESI,† presents the performance of a chitosan–KOH gel electrolyte (21 d) using CV at different scanning rates (Fig. S5a, ESI†). The rectangular shape is conserved even at high sweep rates, demonstrating the good response and cyclability of the gel. The GCPL study under different current densities from 0.1 to 10 A g−1 is presented in Fig. S5b, ESI.† The triangular shape of the charge discharge is maintained at high current regimes in which no visible IR drop is observed, which validates the efficiency of the chitosan–KOH gel electrolyte and its capability to operate under rough circumstances.2 Last, EIS (Fig. S5c and d, ESI†) confirms the low ESR value and good time response of the gel electrolyte.
With the aim of increasing the energy density, the maximum operating voltage can be extended for liquid electrolytes and, in earlier works, this was proven for neutral electrolytes.38 Herein, we tried this approach on the basic electrolyte and, to our knowledge, there is no report on voltage window extension for alkaline electrolytes. To illustrate this possible window widening, Fig. 6 presents the positive E+ and negative E− electrode potentials as well as the electrode potential when the voltage is set to 0 V. The E0V values correspond to the electrode potential when the voltage applied to the cell was 0 V. The figure presents the test performed for the chitosan–KOH gel electrolyte as well as for the liquid 2 M KOH. It illustrates the values at the maximum cell voltage ranging from 0.6 to 1.4 V. Several attempts to register the value at 1.5 V were conducted, but the formation of bubbles prohibits any valuable values and inform about the maximal voltage limit. Three different analyses were performed and confirmed similar results. In general, the two figures show similar behaviour. The potential of the positive electrode does not vary much with increasing cell voltage and, more importantly, it does not exceed the theoretical oxygen evolution potential value for both liquid and gel electrolytes. Concerning the negative electrode potential E−, it decreases gradually until reaching the hydrogen storage potential, which is not harmful to capacitive storage, as it has been stated elsewhere.64 In fact, reversible electrosorption of nascent hydrogen inside the pores of activated carbon contributes to the capacitance and can increase the efficiency of the device.65 It can be noted that the E0V profile decreases with increasing cell voltage. According to the literature, this behaviour may be due to a local pH change within the cell.66 By monitoring the potential at 0 V, the positive electrode potential is prevented from reaching the O2 decomposition limit, thus the full potential range that is available is exploited. From this analysis, it can be concluded that a voltage of 1.4 V already achieves the limit of the device; thus 1.3 V seems to be a better suitable voltage for safer use.
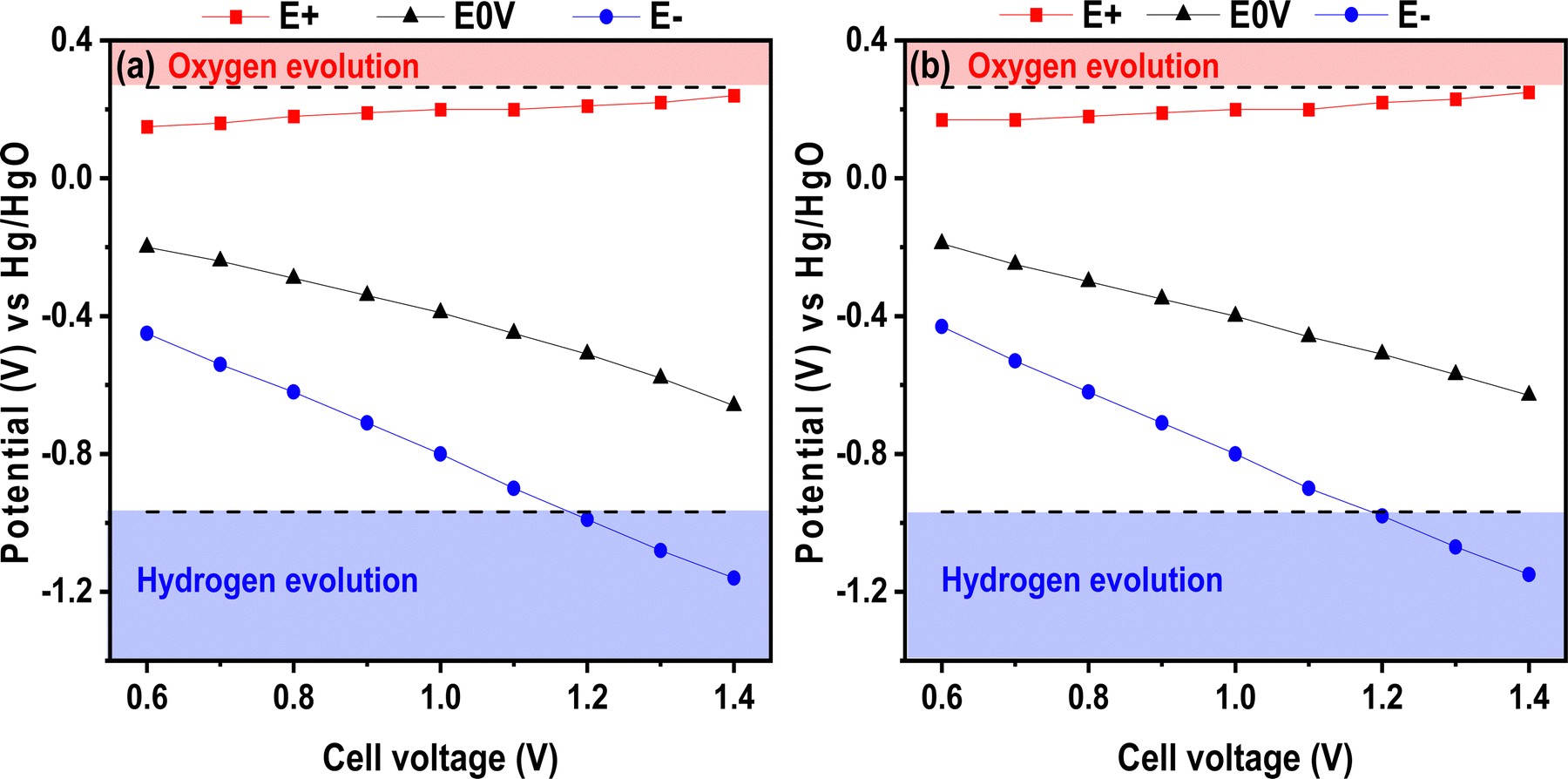 | ||
| Fig. 6 Potential over cell voltage from 0.6 to 1.4 V in a 3-electrode cell using (a) liquid 2 M KOH and (b) chitosan–KOH gel electrolyte as electrolytes. | ||
Fig. 7 presents the carbon cyclic voltammograms and the charge discharge from 0.6 to 1.5 V for liquid 2 M KOH and chitosan–KOH gel electrolyte, respectively. The cyclic voltammograms (Fig. 7a and c) reveal different shapes. In the case of liquid 2 M KOH, the rectangular shape is maintained until a voltage of 0.8 V is reached, while some distortions are observed for higher potentials. Starting from 0.9 V, there is a small increase in current at the end of the CV and a progressive significant decrease in the current values (counterpart) during the negative sweep. These phenomena probably come from redox reactions in the system,67i.e., oxidation of carbon and nascent hydrogen produced at the negative electrode.68 At negative sweep, the hydrogen storage contributes to the capacitance. Moreover, this is not detrimental for the ageing and long life of supercapacitors since capacitance is conserved during ageing.69 Notably, these distortions are not observed for the chitosan-gel electrolyte, in which a quasi-rectangular shape is conserved up to a cell voltage of 1.3 V is obtained.
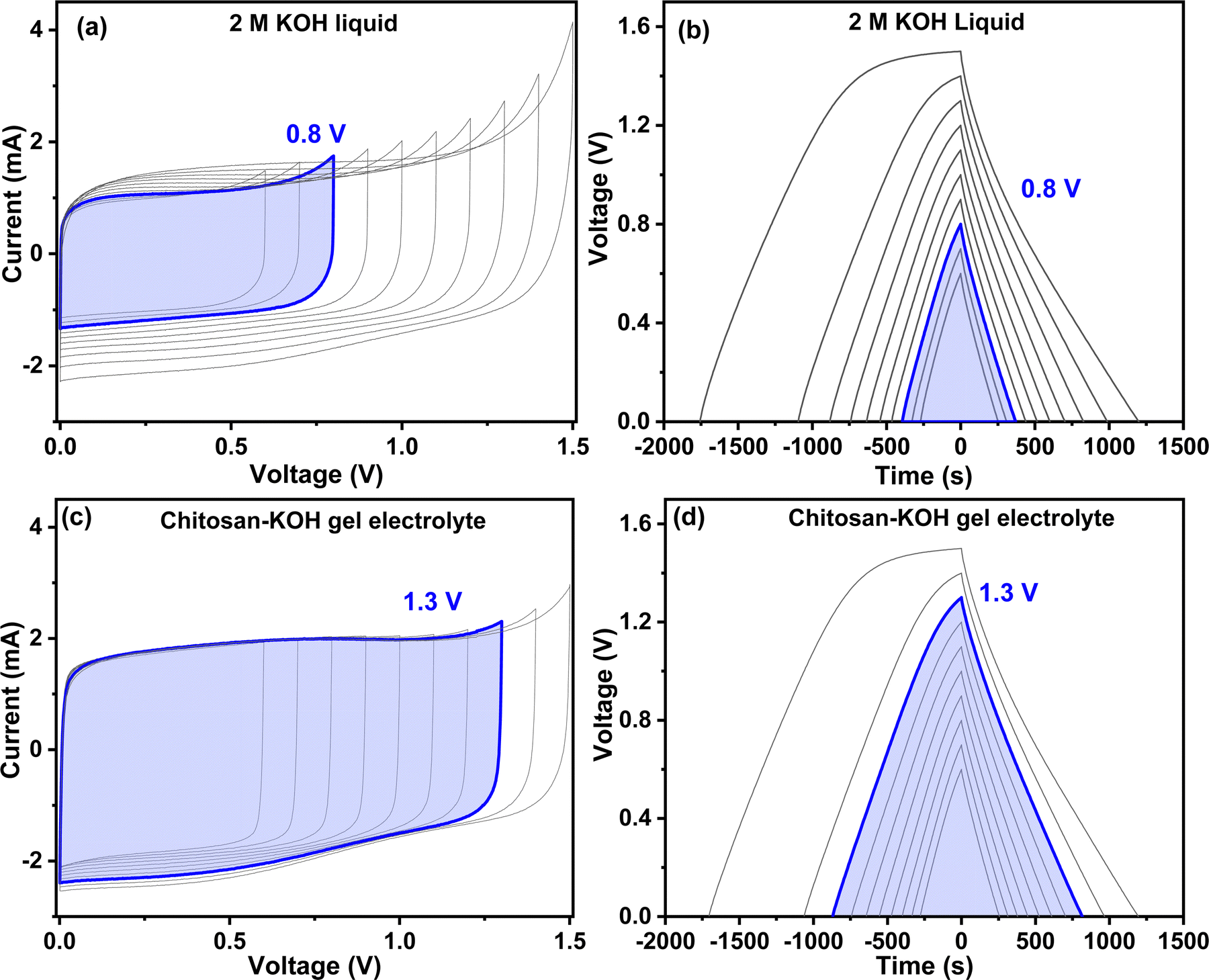 | ||
| Fig. 7 (a) Cyclic voltammetry at 0.1 mV s−1, (b) galvanostatic charge discharge at 0.1 A g−1 of activated carbon using liquid 2 M KOH from 0.6 to 1.5 V, (c) cyclic voltammetry at 0.1 mV s−1 and (d) galvanostatic charge discharge at 0.1 A g−1 using chitosan–KOH gel electrolyte. | ||
The charge discharge profiles (Fig. 7b and d) confirm the CV results, as the rectangular shape typical for EDL is seen at 1.3 V for chitosan–KOH gel. To obtain an overall view of the behaviour of the gel electrolyte, a full analysis of a symmetric device at 1.3 V was performed (Fig. S6, ESI†). It can be noted that the CV rectangular shape is also maintained at high scanning rates (Fig. S6a, ESI†). The charge discharge profiles are triangular and quite reversible with a rate capability of ∼70% at 10 A g−1 (Fig. S6b, ESI†), which is an encouraging result. Moreover, the Nyquist plot (Fig. S6c, ESI†) shows a low ESR and a vertical line at high frequencies specific to EDL capacitors.67 The capacitance versus frequency profiles (Fig. S6d, ESI†) exhibit a good capacitance retention as well, reaching 93% at 0.0 V. These different tests at 1.3 V show that the symmetric device using the chitosan–KOH gel electrolyte exhibits a maintained response under the different conditions, which is very promising. It can be suggested that 1.3 V is the optimal voltage for chitosan–KOH gel electrolyte.
To obtain a more thorough understanding of the carbon electrode state after the voltage window extension measurement in both liquid 2 M KOH and chitosan–KOH gel electrolyte, post mortem analyses were performed (Fig. 8). Two different cells were built for each system to provide sufficient materials for analyses. The N2 adsorption and Raman spectroscopy results are shown in Fig. 8 and Table 2. Fig. 8a and b shows that the cycled electrodes (positive and negative) undergo a serious decrease in surface area when used with both liquid and gel electrolytes compared with that of the pristine electrode. This indicates that there is an important change in the textural properties after charging and discharging up to 1.4 V. Nevertheless, the positive electrodes have a smaller SBET than that of the negative electrodes. Previous works associated this decrease in porosity with oxidation phenomena, leading to porosity collapse or pore blocking with functional groups.40 This will be further supported by EDX analysis, which will be discussed later. In addition, the isotherm shape is similar for positive electrodes at a higher P/P0 range, whereas for negative electrodes, it looks different. If we compare the positive electrodes between them (Fig. 8a), they have a similar profile with a slightly larger SBET for the positive electrode used on the gel electrolyte. In the case of the negative electrodes, SBET is also slightly larger for the electrode used with the gel electrolyte (Table 2). The total volume adsorbed on the electrode used with liquid electrolyte is larger than the total volume adsorbed on the electrode used with the gel electrolyte. In other words, the external surface of the electrode used with the liquid electrolyte is more accessible than in the case of the gel electrolyte. This may be due to pore blocking with larger gel molecules, as washing cannot completely remove these species.
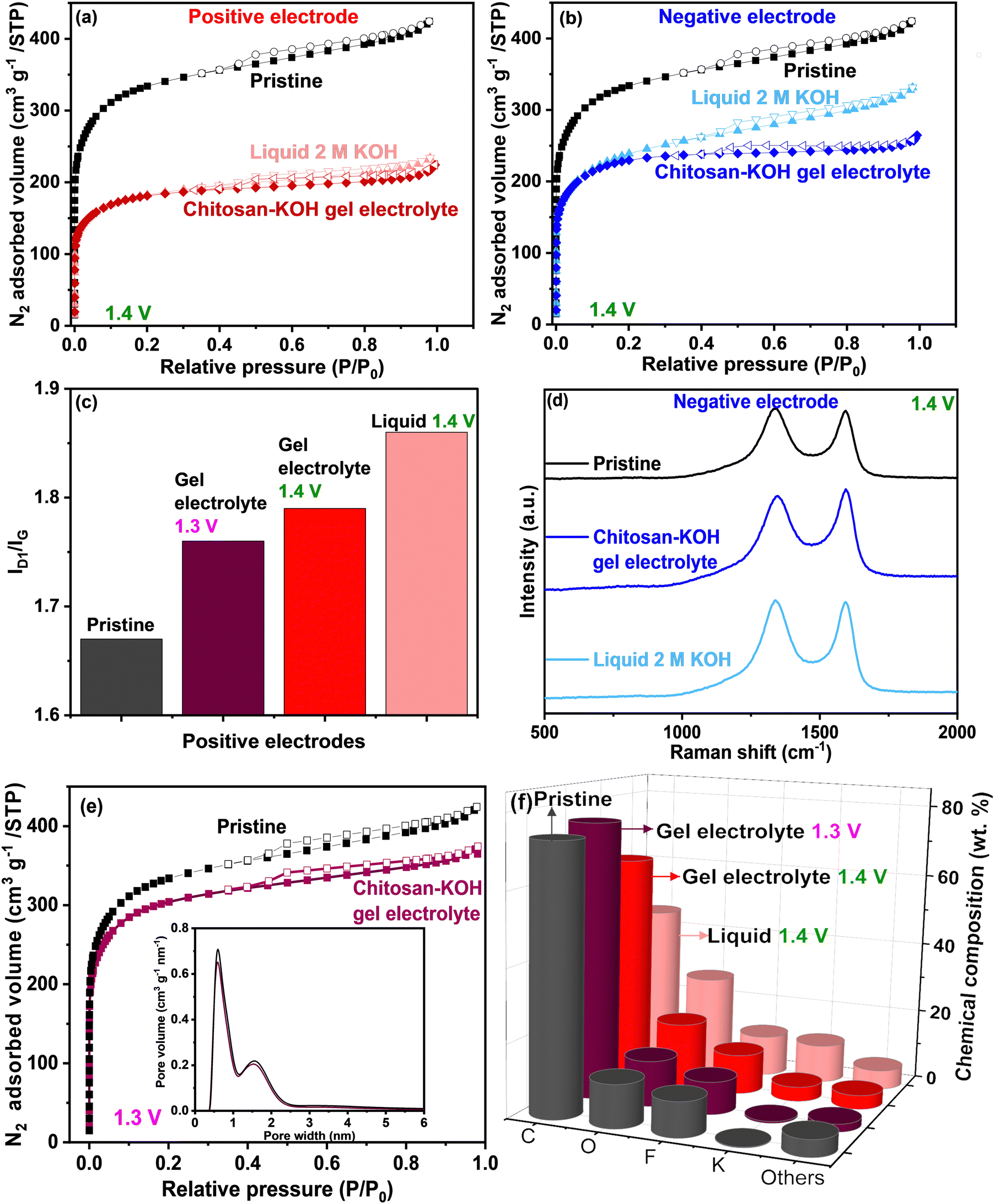 | ||
| Fig. 8 Post-mortem analysis for positive and negative electrodes after voltage window widening until 1.4 V for liquid 2 M KOH and chitosan–KOH gel electrolyte: (a) N2 adsorption/desorption for positive electrodes, (b) N2 adsorption/desorption for negative electrodes, (c) ID1/IG ratios obtained from Raman spectra for positive electrodes at 1.3 and 1.4 V, and (d) Raman spectra for negative electrodes at 1.4 V, (e) N2 adsorption/desorption for positive electrode after voltage window widening until 1.3 V for chitosan–KOH gel electrolyte, (f) chemical composition derived by EDX for pristine and positive electrodes after cycling in liquid KOH and chitosan–KOH gel electrolyte at 1.3 V and 1.4 V. | ||
| Electrode materials | S BET (m2 g−1) | I D1/IG | C (wt%) | O (wt%) |
|---|---|---|---|---|
| Pristine | 1249 | 1.67 | 73 | 12 |
| Positive – chitosan–KOH gel electrolyte 1.3 V | 1146 | 1.76 | 76 | 13 |
| Positive – chitosan–KOH gel electrolyte 1.4 V | 682 | 1.79 | 63 | 18 |
| Positive – liquid 2 M KOH 1.4 V | 672 | 1.86 | 45 | 27 |
| Negative – chitosan–KOH gel electrolyte 1.4 V | 867 | 1.62 | — | — |
| Negative – liquid 2 M KOH 1.4 V | 852 | 1.87 | — | — |
The window extension analysis performed leads to an important polarization between the cathodic and anodic cycling. This causes nascent hydrogen to be trapped more strongly on the electrode.70 Furthermore, the PSD is presented in Fig. S7, ESI,† and the same tendency is noticed. For the positive electrode (Fig. 8a, ESI†), the pore volume for the electrode used on the gel electrolyte is less than that for the electrode used with the liquid electrolyte. For the negative electrodes (Fig. 8b, ESI†), the pore size distribution is almost identical.
Raman spectroscopy was used to follow the structural modification. Qualitatively, the D1 and G bands are noticed at the same Raman shift, i.e., 1343 and 1585 cm−1, respectively, for all electrodes. Quantitatively, the ID1/IG ratio was calculated from the area under the curve, and the results are summarized in Table 2 and Fig. 8c for positive electrodes.
The positive electrodes have a larger ID1/IG than that of the pristine electrode, suggesting that there are more defects in the carbon after the electrochemical test, mainly due to –OH species. The negative electrodes have a slightly smaller ID1/IG than that of the pristine electrode, which is expected as they were less subjected to oxidation phenomena. If we compare the electrodes in each electrolyte, the ID1/IG values for the positive and negative electrodes used with the liquid electrolyte are higher than the values for the positive electrodes used with the gel electrolyte. It may be concluded that the carbon defects provoked by the voltage window extension in the case of liquid electrolyte are coarser than in the case of the gel electrolyte.
This has been further confirmed by the chemical composition of the pristine electrode and both positive electrodes determined by EDX (Fig. 8f). The two positive electrodes have a lower carbon content (63% for the gel and 45% for the liquid) than that of the pristine electrode (73%). Moreover, a higher oxygen amount is observed (18% for the gel and 27% for the liquid) vs. 12% for the pristine gel. This result reinforces the idea that drastic carbon oxidation occurs on the electrode during the voltage window extension. Clearly, in the case of liquid 2 M KOH, the electrode loses a large amount of carbon and gains oxygen. However, with the gel electrolyte, less carbon is consumed, which helps to have a longer device lifespan.
To further confirm that the 1.3 V voltage window does not induce the degradation of electrodes, post-mortem analyses similar to the previous ones were performed on the positive electrode (which is more subject to degradation).
It can be seen in Fig. 8e and Table 2, that the SSA of the positive electrode after the electrochemical test at 1.3 V, is close to the pristine electrode (1146 vs. 1249 m2 g−1, respectively) and significantly higher than the positive electrode cycled at 1.4 V (682 m2 g−1). The ID1/IG ratio of the positive electrode after the window widening to 1.3 V (Fig. 8c) is slightly smaller than the positive electrode after the test at 1.4 V (1.76 vs. 1.79, respectively), suggesting fewer defects are created when cycling at 1.3 V. Additionally, the EDX (Table 2 and Fig. 8f) reveals similar chemical compositions between the positive electrode cycled at 1.3 V and the pristine one: C (76 vs. 73%) and O (13 vs. 12%), attesting that oxidation phenomena are avoided. These results confirm that 1.3 V is an appropriate voltage window for the device, which allows to preserve the texture, the structure, and the surface chemistry of the electrodes (Table 2).
To gain more insights into the performance of the capacitor device, the so-called Ragone plot and long-term cycling for 2 M KOH and chitosan–KOH electrolyte are presented in Fig. 9. When the calculations for the Ragone graph were conducted, the mass of the whole device was considered. At 0.8 V, a clear improvement in the energy density and power could be achieved with the chitosan/KOH gel electrolyte. At 1.3 V, the performance of the gel electrolyte was improved by three times, and an almost constant energy density vs. power confirmed the excellent capacitive behaviour of the supercapacitor. The energy density of the systems using gel electrolyte reached 5.1 W h kg−1 at 32.5 W kg−1. A comparison of the electrochemical performance of the different electrochemical capacitors reported in the literature using different gel electrolytes based on aqueous electrolytes is presented in Table S2, ESI.† As seen, the chitosan gel electrolyte has a good energy density compared to that of previously reported gel electrolytes.
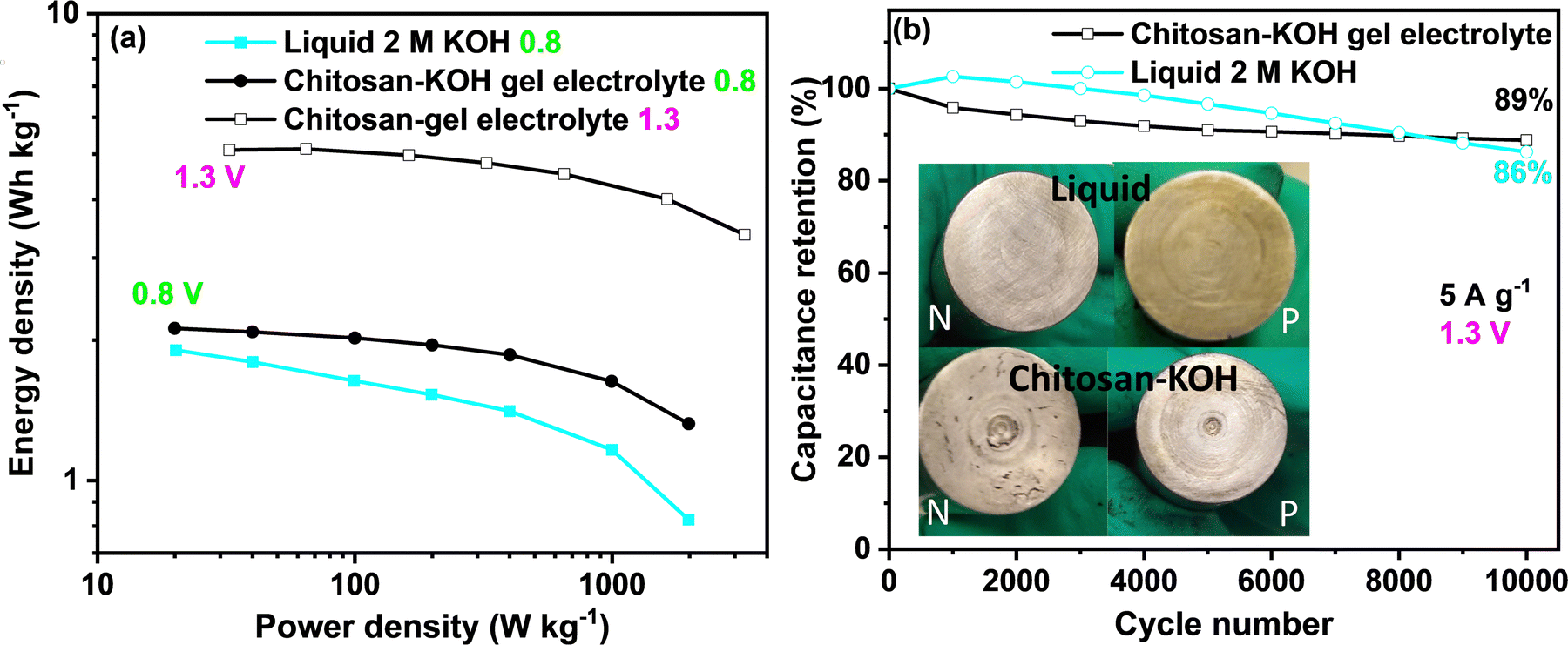 | ||
| Fig. 9 (a) Ragone plot of activated carbon R3-Extra at 0.8 V for the 2 M KOH liquid electrolyte and chitosan–KOH gel electrolyte and at 1.3 V. (b) Long cycling of liquid 2 M KOH electrolyte and chitosan–KOH gel electrolyte at 5 A g−1 and 1.3 V (inset: pictures of the current collector state after cycling for both electrochemical cells: N-negative and P-positive sides of the current collector). | ||
Long-term cycling was performed at a 5 A g−1 current load and 1.3 V for both liquid 2 M KOH and chitosan–KOH gel electrolyte (Fig. 9b). Two cells were launched and exhibited similar behaviour in both cases. With 2 M KOH, the device exhibits a slower increase then the decrease in retention, while the device with the chitosan–KOH gel electrolyte shows a steady stable low decrease over the cycles. By the end of 10000 cycles, the liquid electrolyte decreased sharply and retained 86% of its capacitance, whereas the chitosan–KOH gel electrolyte has an 89% retention.
This result is reinforced by the comparison between the state of the two device current collectors after cycling (Fig. 9b, inset). All current collectors on the positive and negative sides were intact, except for the positive collector used with the liquid electrolyte. It was previously demonstrated that an alkaline electrolyte leads to current collector oxidation after cycling69 and, compared to the negative ones, the positive current collector is more subjected to oxidation.40 Moreover, to confirm the stability of alkaline hydrogel for long cycling, the profiles of the first and last charge discharge cycles for the gel electrolyte are provided in Fig. S8, ESI,† and an almost overlapping can be seen. This indicates that the chitosan–KOH gel electrolyte is stable after cycling for 10000 cycles. In addition, post-cycling CV and EIS tests were conducted on both cells to confirm these observations (Fig. S9, ESI†). Concerning the CV, it may be seen that the two tested cells have quite similar behaviour with minor differences. The cell used with the chitosan–KOH gel electrolyte reach high current values more quickly. This originates from the faster ion transport within the electrode. For EIS, at high frequencies, the difference in ESR between the chitosan–KOH gel electrolyte and liquid 2 M KOH is maintained, as shown in Fig. 5d. However, for low frequencies in which the shape is controlled by the diffusion of ions, the line relative to the cell with liquid 2 M KOH is less vertical than that of the cell using chitosan–KOH gel electrolyte. This result comes purely from the increase in resistance created in the cell using liquid 2 M KOH and confirms the results mentioned above.71
The development of chitosan–KOH gel electrolyte using alkaline aqueous electrolyte is of great interest due to its potential applicability with pseudocapacitive materials. In particular, the extension of the voltage window and the improvement of the capacitance and energy density are desired. Thus, the developed gel was used with a synthesized nanocomposite C/Co3O4 reported recently in our works57 in an the extended voltage window up to 1.3 V. The material chosen was C/Co3O4 750–230 as it showed the optimal electrochemical performance in liquid KOH 0.8 V.57 In Fig. 10a, one can notice the difference in the rate capability for the presented devices. At 0.8 V, the performance of C/Co3O4 750–230 in 2 M KOH and chitosan–KOH gel electrolyte is similar, with almost the same value of rate capability. The capacitance of the device constructed with chitosan–KOH gel electrolyte is slightly above that of the device built with 2 M KOH. The possibility of extension of the voltage to 1.3 V definitely has a positive impact on the capacitance, which has doubled at 0.1 A g−1 from 54 A g−1 to 108 A g−1. This is maintained through the current density increase and a rate capability of 43.8% at 10 A g−1 is observed. These values are very promising for a capacitor, as with the chitosan–KOH gel electrolyte, a considerable increase was achieved for both the voltage window and the capacitance. As can be seen from the CV (Fig. S10, ESI†) at 1.3 V, the shape is rectangular for all sweep rates from 1 to 100 mV s−1, attesting to the capacitive behaviour. Moreover, the different voltammograms overlap almost completely, suggesting good electrochemical behaviour and fast charge transport in the device. This translates in an increase of energy density (Fig. 10b). In the case of C/Co3O4 used with the chitosan–KOH gel electrolyte, the energy increases 4 times, from 1.66 to 6.31 W h kg−1 by voltage window extension. This exceeds the Norit R3 extra carbon energy density at 1.3 V (5.1 W h kg−1).
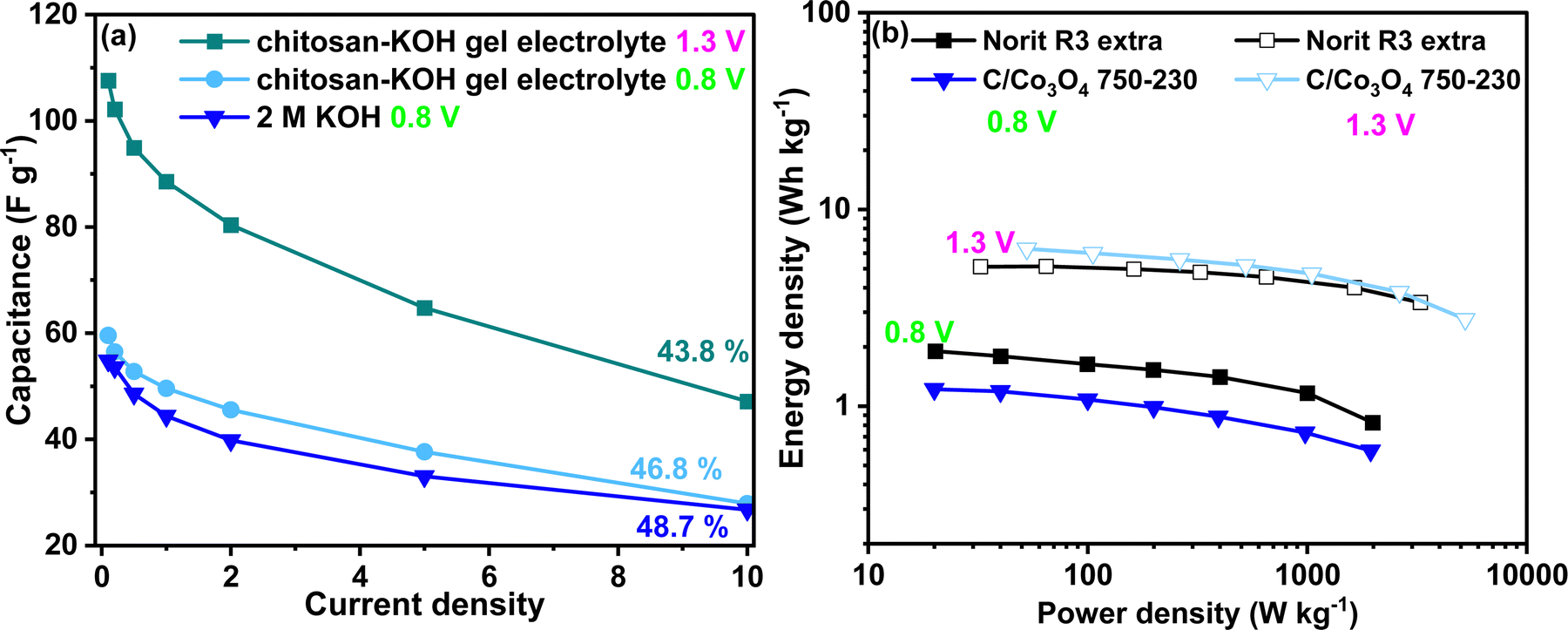 | ||
| Fig. 10 Electrochemical performance of C/Co3O4 750–230 nanocomposite using chitosan–KOH gel electrolyte: (a) rate capability for 2 M KOH at 0.8 V and for chitosan–KOH gel electrolyte at 0.8 and 1.3 V, (b) Ragone diagram of C/Co3O4 750–230 nanocomposite and Norit R3 extra using 2 M KOH at 0.8 V and chitosan–KOH gel electrolyte at 1.3 V. | ||
Thus, it can be clearly said that the chitosan–KOH gel electrolyte reduces the resistance created between the different element interfaces, inhibits the corrosion phenomena after long cycling, provides a higher capacitance retention compared to that of the liquid electrolyte and allows a higher energy density profile to be reached. All these results highlight that the chitosan–KOH gel is an inexpensive, ecological and sustainable alternative for liquid electrolytes. The use of the gel in symmetric capacitors gives an enhanced performance by widening the voltage window. This translates to an improved energy density and better retention after 10000 cycles.
4. Conclusions
Chitosan was used to develop a biodegradable gel electrolyte with KOH as the ion source. An optimization of the gelling process was performed to obtain a hydrogel with good mechanical and electrochemical properties. FTIR and NMR were used to follow the chemical and structural evolution during the ageing of the chitosan solution over 21 days. The cross-linking of the chitosan gel increased with time, directly impacting the gel mechanical properties and electrochemical performance, which improved after 2 days. The chitosan–KOH gel electrolyte that was tested in a symmetric carbon–carbon supercapacitor by different electrochemical techniques exhibited a better performance than that of liquid KOH. Moreover, the solution of the chitosan gel electrolyte was tested at different ageing days and was found to be functional and efficient from the 4th day until at least the 21st day of ageing. Through this ageing process, the gel electrolyte provides for C–C device at 0.8 V a higher capacitance of 95 F g−1 at 0.1 A g−1 and better capacitance retention, i.e., 59 F g−1 at 10 A g−1, than that of the liquid electrolyte. The stability of the gel electrolyte was demonstrated up to a cell voltage of 1.3 V. At this voltage, the capacitance reached 109 F g−1 at 0.1 A g−1 and 70% rate capability (76 F g−1 at 10 A g−1). Moreover, such a voltage extension window for alkaline gel electrolytes has rarely been reported, in contrast to neutral electrolytes, and allows a significant improvement (3 times) in the energy and power density compared to that of liquid electrolytes. Post-mortem analysis of the electrodes after window widening showed that at 1.4 V the positive carbon electrode is more subject to porosity loss, defect creation and oxidation and thus capacitance loss, however, at 1.3 V these electrode properties are well preserved. Moreover, the capacitor used with the gel electrolyte has a better retention than that of the liquid electrolyte (89% vs. 86%). Finally, the use of chitosan–KOH gel electrolyte with pseudocapacitive materials C/Co3O4 was proposed and showed enhanced capacitance, rate capability and energy density. Overall, the use of the chitosan–KOH gel electrolyte represents a safer and more sustainable approach for inhibiting corrosion issues in the cell. The use of this gel electrolyte concept can be beneficial in the development of organo-gels which might further allow the energy density increase.Author contributions
Sirine Zallouz: investigation, data curation, software, writing-original draft. Jean-Marc Le Meins: writing – review and editing, validation, conceptualization, supervision. Camélia Matei Ghimbeu: writing – review and editing, validation, conceptualization, supervision, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Université de Haute Alsace (UHA) for the financial support of the Ph.D. thesis of Sirine Zallouz. The French National Research Agency (STORE-EX Labex Project ANR-10-LABX-76-01) is acknowledged for financial support. We also thank Dr Cyril Vaulot (NMR analysis) and Mr Simon Gree (FTIR analysis) for technical help via IS2M technical platforms.References
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- E. Frackowiak, Q. Abbas and F. Béguin, J. Energy Chem., 2013, 22, 226–240 CrossRef.
- X. Cheng, J. Pan, Y. Zhao, M. Liao and H. Peng, Adv. Energy Mater., 2018, 8, 1702184 CrossRef.
- N. A. Choudhury, S. Sampath and A. K. Shukla, Energy Environ. Sci., 2009, 2, 55–67 RSC.
- S. Alipoori, S. Mazinani, S. H. Aboutalebi and F. Sharif, J. Energy Storage, 2020, 27, 101072 CrossRef.
- L. Cao, M. Yang, D. Wu, F. Lyu, Z. Sun, X. Zhong, H. Pan, H. Liu and Z. Lu, Chem. Commun., 2017, 53, 1615–1618 RSC.
- M. Liu, D. Zhou, Y.-B. He, Y. Fu, X. Qin, C. Miao, H. Du, B. Li, Q.-H. Yang, Z. Lin, T. S. Zhao and F. Kang, Nano Energy, 2016, 22, 278–289 CrossRef CAS.
- Z. Li, S. Gao, H. Mi, C. Lei, C. Ji, Z. Xie, C. Yu and J. Qiu, Carbon, 2019, 149, 273–280 CrossRef CAS.
- A. Jamaludin, Z. Ahmad, Z. A. Ahmad and A. A. Mohamad, Int. J. Hydrogen Energy, 2010, 35, 11229–11236 CrossRef CAS.
- S. Galliano, F. Bella, M. Bonomo, F. Giordano, M. Grätzel, G. Viscardi, A. Hagfeldt, C. Gerbaldi and C. Barolo, Sol. RRL, 2021, 5, 2000823 CrossRef CAS.
- J. C. de Haro, E. Tatsi, L. Fagiolari, M. Bonomo, C. Barolo, S. Turri, F. Bella and G. Griffini, ACS Sustainable Chem. Eng., 2021, 9, 8550–8560 CrossRef CAS PubMed.
- Y. Zuo, W. Zhang, M. Wei, P. Zhang, S. Zhao, P. Pei, L. Qiu, H. Wang, Z. Meng and K. Wang, Energy Storage Mater., 2022, 53, 136–147 CrossRef.
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7, 11782 CrossRef PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef.
- K. Sun, F. Ran, G. Zhao, Y. Zhu, Y. Zheng, M. Ma, X. Zheng, G. Ma and Z. Lei, RSC Adv., 2016, 6, 55225–55232 RSC.
- D. K. Kim, N. D. Kim, S.-K. Park, K. Seong, M. Hwang, N.-H. You and Y. Piao, J. Power Sources, 2018, 380, 55–63 CrossRef.
- X. Wu and M. Lian, J. Power Sources, 2017, 362, 184–191 CrossRef CAS.
- B. Karaman, E. Çevik and A. Bozkurt, Ionics, 2019, 25, 1773–1781 CrossRef CAS.
- E. Cevik and A. Bozkurt, J. Energy Chem., 2021, 55, 145–153 CrossRef CAS.
- G. Lee, S.-K. Kang, S. M. Won, P. Gutruf, Y. R. Jeong, J. Koo, S.-S. Lee, J. A. Rogers and J. S. Ha, Adv. Energy Mater., 2017, 7, 1700157 CrossRef.
- F. Tao, L. Qin, Z. Wang and Q. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15541–15548 CrossRef CAS.
- Y. N. Sudhakar, M. Selvakumar and D. K. Bhat, Mater. Renew. Sustain. Energy, 2015, 4, 10 CrossRef.
- M. Z. A. Yahya and A. K. Arof, Eur. Polym. J., 2003, 39, 897–902 CrossRef.
- M. H. Buraidah, L. P. Teo, S. R. Majid and A. K. Arof, Phys. B, 2009, 404, 1373–1379 CrossRef.
- A. Sivashanmugam, R. Arun Kumar, M. Vishnu Priya, S. V. Nair and R. Jayakumar, Eur. Polym. J., 2015, 72, 543–565 CrossRef.
- S. M. Nomanbhay and K. Palanisamy, Electron. J. Biotechnol., 2005, 8, 0 Search PubMed.
- S. Tripathi, G. K. Mehrotra and P. K. Dutta, Int. J. Biol. Macromol., 2009, 45, 372–376 CrossRef CAS PubMed.
- C. Matei Ghimbeu and V. A. Luchnikov, Microporous Mesoporous Mater., 2018, 263, 42–52 CrossRef CAS.
- M. Rayung, M. M. Aung, S. C. Azhar, L. C. Abdullah, M. S. Su’ait, A. Ahmad and S. N. A. M. Jamil, Materials, 2020, 13, 838 CrossRef CAS.
- Z. Osman and A. K. Arof, Electrochim. Acta, 2003, 48, 993–999 CrossRef CAS.
- N. A. Choudhury, J. Ma and Y. Sahai, J. Power Sources, 2012, 210, 358–365 CrossRef CAS.
- I. A. Fadzallah, S. R. Majid, M. A. Careem and A. K. Arof, J. Membr. Sci., 2014, 463, 65–72 CrossRef CAS.
- M. Yamagata, K. Soeda, S. Ikebe, S. Yamazaki and M. Ishikawa, Electrochim. Acta, 2013, 100, 275–280 CrossRef CAS.
- H. Yang, X. Ji, Y. Tan, Y. Liu and F. Ran, J. Power Sources, 2019, 441, 227174 CrossRef.
- Q. Zhang, L. Zhao, H. Yang, L. Kong and F. Ran, J. Membr. Sci., 2021, 629, 119083 CrossRef.
- M. S. Kumar and D. K. Bhat, J. Appl. Polym. Sci., 2009, 114, 2445–2454 CrossRef.
- L. Demarconnay, E. Raymundo-Piňero and F. Béguin, Electrochem. Commun., 2010, 12, 1275–1278 CrossRef.
- Q. Gao, L. Demarconnay, E. Raymundo-Piñero and F. Béguin, Energy Environ. Sci., 2012, 5, 9611 RSC.
- P. Ratajczak, K. Jurewicz, P. Skowron, Q. Abbas and F. Béguin, Electrochim. Acta, 2014, 130, 344–350 CrossRef CAS.
- J. H. Chae and G. Z. Chen, Electrochim. Acta, 2012, 86, 248–254 CrossRef CAS.
- P. Bujewska, B. Gorska and K. Fic, Synth. Met., 2019, 253, 62–72 CrossRef CAS.
- C. M. Ghimbeu, R. Gadiou, J. Dentzer, L. Vidal and C. Vix-Guterl, Adsorption, 2011, 17, 227–233 CrossRef.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef.
- A. J. Eugene, S.-S. Xia and M. I. Guzman, J. Phys. Chem. A, 2016, 120, 3817–3826 CrossRef PubMed.
- H. Xu and S. Matysiak, Chem. Commun., 2017, 53, 7373–7376 RSC.
- Z. Wang, L. Zheng, C. Li, D. Zhang, Y. Xiao, G. Guan and W. Zhu, Carbohydr. Polym., 2015, 117, 973–979 CrossRef CAS.
- D. Ailincai, L. Marin, S. Morariu, M. Mares, A.-C. Bostanaru, M. Pinteala, B. C. Simionescu and M. Barboiu, Carbohydr. Polym., 2016, 152, 306–316 CrossRef CAS PubMed.
- Z. Shariatinia and A. M. Jalali, Int. J. Biol. Macromol., 2018, 115, 194–220 CrossRef CAS.
- M. O. Ahmed, A. Shrpip and M. Mansoor, Processes, 2020, 8, 246 CrossRef CAS.
- R. L. Redington and C.-K. J. Liang, J. Mol. Spectrosc., 1984, 104, 25–39 CrossRef CAS.
- L. Racine, I. Texier and R. Auzély-Velty, Polym. Int., 2017, 66, 981–998 CrossRef CAS.
- M. A. Hassan, A. M. Omer, E. Abbas, W. M. A. Baset and T. M. Tamer, Sci. Rep., 2018, 8, 11416 CrossRef.
- I. Ledeţi, A. Alexa, V. Bercean, G. Vlase, T. Vlase, L.-M. Şuta and A. Fuliaş, Int. J. Mol. Sci., 2015, 16, 1711–1727 CrossRef.
- Y. C. Wei, S. M. Hudson, J. M. Mayer and D. L. Kaplan, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2187–2193 CrossRef CAS.
- S. Zallouz, B. Réty, L. Vidal, J.-M. Le Meins and C. Matei Ghimbeu, ACS Appl. Nano Mater., 2021, 4, 5022–5037 CrossRef CAS.
- J. Menzel, E. Frąckowiak and K. Fic, Electrochim. Acta, 2020, 332, 135435 CrossRef CAS.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 CrossRef.
- N. Batisse and E. Raymundo-Piñero, J. Power Sources, 2017, 348, 168–174 CrossRef.
- K. Fic, A. Platek, J. Piwek and E. Frackowiak, Mater. Today, 2018, 21, 437–454 CrossRef.
- Y. Guo, J. Bae, F. Zhao and G. Yu, Trends Chem., 2019, 1, 335–348 CrossRef.
- A. Platek-Mielczarek, C. Nita, C. Matei Ghimbeu, E. Frackowiak and K. Fic, ACS Appl. Mater. Interfaces, 2021, 13, 2584–2599 CrossRef CAS.
- J. Menzel, E. Frackowiak and K. Fic, J. Power Sources, 2019, 414, 183–191 CrossRef CAS.
- J. Menzel, K. Fic and E. Frackowiak, Prog. Nat. Sci.: Mater. Int., 2015, 25, 642–649 CrossRef CAS.
- M. Yu, Y. Lu, H. Zheng and X. Lu, Chem. – Eur. J., 2018, 24, 3639–3649 CrossRef CAS.
- J. Piwek, A. Platek, K. Fic and E. Frackowiak, Electrochim. Acta, 2016, 215, 179–186 CrossRef CAS.
- J. Piwek, A. Platek, E. Frackowiak and K. Fic, J. Power Sources, 2019, 438, 227029 CrossRef CAS.
- K. Fic, M. Meller, J. Menzel and E. Frackowiak, Electrochim. Acta, 2016, 206, 496–503 CrossRef CAS.
- K. Fic, E. Frackowiak and F. Béguin, J. Mater. Chem., 2012, 22, 24213–24223 RSC.
- B.-A. Mei, O. Munteshari, J. Lau, B. Dunn and L. Pilon, J. Phys. Chem. C, 2018, 122, 194–206 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00250g |
| This journal is © The Royal Society of Chemistry 2022 |