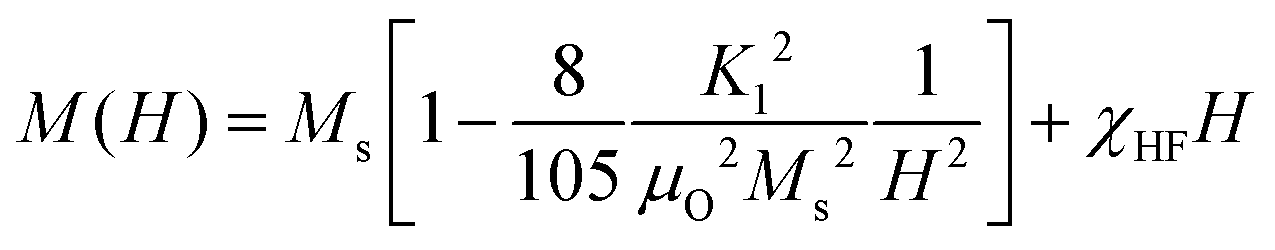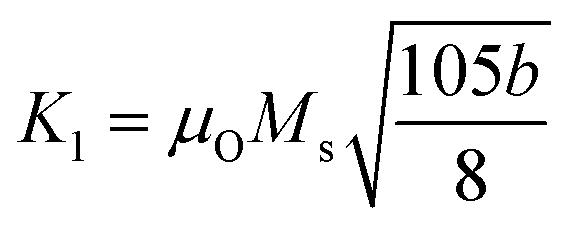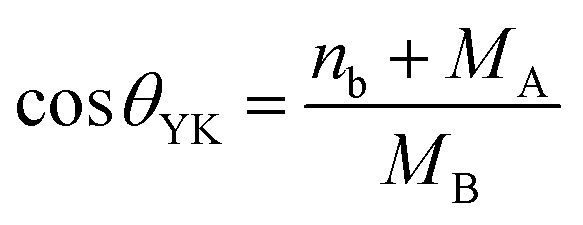DOI:
10.1039/D2TC02605H
(Paper)
J. Mater. Chem. C, 2022,
10, 12652-12679
Observation of intrinsic fluorescence in cobalt ferrite magnetic nanoparticles by Mn2+ substitution and tuning the spin dynamics by cation distribution†
Received
21st June 2022
, Accepted 3rd August 2022
First published on 5th August 2022
Abstract
In this work, we report the synthesis and detailed characterization of single-domain, optically active, manganese-substituted cobalt ferrite (CoFe2O4) magnetic nanoparticles without any surface functionalization as prospective fluorescent probes for bio-imaging. Generally, nanoferrites (NFs) do not show any intrinsic fluorescence and require surface modification to make them fluorescent by functionalization with fluorescent probes. Herein, we observed multi-band fluorescent emission in Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs synthesized via a one-pot hydrothermal method. The substitution of cobalt by manganese in CoFe2O4, which has an inner shell electronic transition between its d5 energy levels, and increase in the concentration of defect centers mainly contributed to the fluorescent characteristics of the as-synthesized NF samples. The two emission bands observed for the Co–Mn NFs are violet and blue bands. The violet band was observed due to the transfer of electrons from the shallow donor level to the valence band (i.e., near band edge (NBE) emission), while the emission in the blue region can be attributed to the band edge free and bound excitons. Also, the time-resolved photoluminescence studies indicated two decay times, which can be attributed to the blue and violet emission bands. Detailed structural modeling was performed using Rietveld refinement of the X-ray diffraction data and the cation distribution obtained from the modeling was corroborated by the optical properties and spin dynamics of the NFs. The cation distribution of the NF samples indicates that the blue band originates from the 3F → 3p transition in the octahedral sites between the Co2+/Mn2+ ions. Further, a strong ferromagnetic characteristic was observed for the NF samples and the optimized substitution of Mn2+ ions resulted in an improvement in the saturation magnetization from 68.51 to 80.30 emu g−1, which was corroborated by the Yafet–Kittel model. Further, imparting optical properties in magnetic materials opens a new horizon for the biomedical applications of these materials by capitalizing on their intrinsic fluorescence, which will not hamper their magnetic properties as in the case with external fluorescent probes.
1. Introduction
Currently, substantial growth in the biomedical and environmental applications of magnetic nanoparticles (MNPs) has been realized with the evolution of nanotechnology.1,2 Magnetic materials have a significant impact on modern-day materials science given that they have led to the discovery of many high-performance applications, resulting in an improvement in the quality of life.3 In this class, ferromagnetic materials (FM) are significantly important due to their superior properties such as low electromagnetic and eddy current losses, superparamagnetism (SPM), high magnetic dipolar strength, spin-glass behavior, and super exchange interactions (SEIs).4 These superior properties of FM materials have been employed to design and develop several applications in diverse fields. Ferrites (combination of metals and iron oxides) are generally FM in nature and their structure is based on the crystal structure of the oxygen (face-centered cubic “FCC”) in which the metal ions occupy the interstitial sites.5 The properties of these materials are mainly governed by their cation distribution, compositional variation, and synthetic methods.6 Thus, the properties of these ferrites can be effectively tuned by varying the shape and size of their particles, chemical composition, surface modification, doping, and site occupancies of their cations.7 Moreover, these materials have numerous current and prospective applications in the field of science and technology such as EMI shielding,8–10 high-density storage devices, magneto-optical devices, nanofluids, spintronic devices, transformer cores, and biomedical applications (magnetic resonance imaging, therapeutics, targeted drug delivery, and magnetic fluid hyperthermia11–16).
Moreover, the agglomeration of MNPs significantly affects the performance of these materials such as biomedical applications in the bloodstream.17 MNPs tend to form a cluster of MNPs, which can cause blockage of the smooth motion of the blood in the vein.18 The agglomeration of MNPs is mainly caused by their high surface-to-volume ratio and strong inter-particle dipolar interactions.8 Accordingly, in the last decade, special attention has been given to the development of surface-functionalized MNPs to avoid their agglomeration.19 However, surface functionalization affects the properties of MNPs, which can moderate the performance of the material.2,20,21 Thus, the development of strategies to prepare agglomeration-free MNPs remains at the forefront of contemporary research. Further, nano ferrites (NFs) are considered a special class of MNPs with a wide variety of applications. Especially, CoFe2O4 and MnFe2O4 present unique and remarkable properties such as spin canting, spin glass behavior, exchange bias, magnetic excitation, and magnetic field tunable optical characteristics.9 In the last decade, biomedical applications such as targeted drug delivery, bio-imaging, and hyperthermia treatment using these materials have been gaining significant attention.22 These materials show enhanced heating efficiency due to hysteresis loss and Néel and Brownian relaxations, which can be efficiently used to treat cancer cells.23 Generally, cancer cells are treated by heating them at elevated temperatures using chemotherapy and radiotherapy, where cancer cells are susceptible to these elevated temperatures, while healthy cells remain unharmed.18 Also, the other major applications of these materials include heavy metal ion removal, energy conversion, plastic degradation, magnetic resonance imaging (MRI), solar water splitting, photocatalysis, batteries, and memory devices.1,15,24–28 However, in all these applications, maintaining the nano features of the materials remains the biggest challenge and the current research in this field is focused on the large-scale production of MNPs with uniform and homogeneous properties.
In NFs, the distribution of cations at the tetrahedral (A) and octahedral (B) sites plays a key role in controlling their properties, which can be altered effectively by adopting a suitable synthetic method at the industrial scale to make it economically viable.29–31 The properties of NFs significantly depend on their synthetic method. Numerous methods such as the solvothermal, reverse micelle, co-precipitation, microwave synthesis, thermal decomposition, hydrolysis, and sol–gel methods have been explored in the past; however, the hydrothermal route is considered highly suitable for the large-scale synthesis of MNPs.9,32 Several research groups have investigated the preparation of CoFe2O4 using hydrothermal methods. However, the effect of Mn2+ substitution on the properties of Co–Mn mixed ferrites has rarely been investigated in detail.33–39 Also, none of the reports in the literature investigated the optical properties of Co–Mn mixed ferrites given that NFs are generally optically inactive materials.40–42 Consequently, imparting optical activity, which can be tuned by an external magnetic field, provides a unique opportunity to develop active control devices.13,16 Recently, Kumar et al.18 reported the preparation of optically active MnFe2O4 MNPs via a facile large-scale hydrothermal route and investigated their optical properties in detail. Also, they suggested that the optical activity of the MnFe2O4 MNPs is due to the cation distribution and increase in the concentration of defect centers, which are related to the synthesis method. Furthermore, they predicted that tuning the cation distribution can be an efficient way to control the optical properties of NFs. Balakrishnan et al. synthesized CoFe2O4 NFs with substituted manganese (Mn), nickel (Ni), zinc (Zn), and copper (Cu) ions and studied their cation distribution to provide a correlation between their structural and magnetic properties.43 However, they did not report any optical activity with the substitution of Mn, Ni, Zn, and Cu ions in CoFe2O4 MNPs. Also, Salunkhe et al. and Ansari et al. synthesized Mn-substituted CoFe2O4 MNPs via a hydrothermal method; however, they both focused on the structural and magnetic properties of the materials.39,42 There are also several reports on CoFe2O4 NFs with other transition metal ion substitutions; however, there are no reports on the optical and spin dynamic properties of Mn-substituted CoFe2O4 MNPs.39,44,45
Herein, we synthesized Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) MNPs and studied their optical properties in detail using photoluminescence (PL) spectroscopy and time-resolved PL. The motivation behind choosing Mn2+-substituted CoFe2O4 is that Mn has an inner shell electronic transition between its d5 energy levels, and thus it possesses good luminescent properties and has a large magnetic moment of 5 μB.46 The synthesis of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) MNPs was carried out via low-temperature hydrothermal synthesis and detailed structural modeling was conducted by Rietveld refinement of the X-ray diffraction (XRD) data. The effect of Mn substitution on the structure of CoFe2O4 is mainly responsible for the PL properties of the materials. Also, the spin dynamics of these materials play a key role in controlling the properties of the NFs, and thus we investigated the microwave spin resonance behavior of all the samples using ferromagnetic resonance (FMR). The correlation between the structure and spin dynamics of the NF samples was investigated by fitting their FMR spectra and obtaining different microwave spin resonance properties. The static magnetic properties of the NF samples were also investigated using a vibrating sample magnetometer (VSM) and the correlation between the cation distribution and magnetic properties was studied in detail. Further, we also studied the different properties such as structural, elastic, and morphological properties of the as-synthesized NF samples using different techniques including Fourier-transform infrared spectroscopy (FTIR), transmission electron microscopy (TEM), scanning electron microscopy (SEM) and energy dispersive x-ray (EDX), and UV-visible (UV-vis) spectroscopy.
2. Synthesis and characterization of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) nanoferrites
2.1 Synthesis of Mn-doped cobalt ferrites using the hydrothermal method
The synthesis of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF sample was carried out via a facile, low-cost, environmentally friendly, and scalable one-pot hydrothermal method. This method enables the fabrication of materials in bulk quantities at relatively moderate temperatures, which is the main bottleneck in the application of these nanomaterials.24,32 A schematic representation of the synthetic process adopted for the preparation of the NF samples is shown in Fig. 1. The synthesized NFs were capped with polymer (PEG-400), which acts as a primary surfactant to reduce the agglomeration of the NF particles. In a typical synthesis process, the precursor salts of iron (Fe3+), cobalt (Co2+), and manganese (Mn2+) were mixed in 50 mL solution (DI water 18.2 (MΩ cm) and ethanol), each with continuous stirring (500 rpm) until all the salt was completely dissolved in the solution. The amount of Co2+ and Mn2+ was varied for the different Co1−xMnxFe2O4 (x = 0, 0.2, 0.4, 0.6, and 0.8) samples in such a way that the molar ratio of the trivalent ion (Fe3+) to divalent ions (Co2+ + Mn2+) remained 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table S1, ESI†). All the salt solutions of each ion were mixed in a 500 mL beaker under constant stirring (400 rpm) and heating (60 °C).47,48 Further, 10 mL PEG-400 surfactant was added to the precursor salt solution, which prevented the agglomeration of the NF particles during their growth. Afterward, 10 mmol sodium hydroxide (NaOH) solution was added to the homogenized precursor solution dropwise until the pH reached a constant value of 12 and the resultant solution was kept stirring for 30 min. Subsequently, the obtained solution was transferred to a 200 mL Teflon-lined stainless-steel autoclave sealed at atmospheric pressure and kept inside a hot air oven for 12 h at 160 °C. Further, the autoclave was removed from the oven and allowed to cool to room temperature. Then, the obtained solution was filtered and washed several times using water and ethanol via magnetic decantation (separation of nanoparticles using NdFeB permanent magnet) to remove any trace of impurities, which significantly hampers the performance of magnetic systems. The black precipitate obtained after washing was centrifuged to remove the considerably smaller or bigger particles, with the resulting samples having a narrow size distribution. Finally, the centrifuged samples were dried under vacuum oven overnight to remove any trace of water present in the samples. The as-prepared samples were directly used for different characterizations. The reaction mechanism during the synthetic process is as follows:
:1 (Table S1, ESI†). All the salt solutions of each ion were mixed in a 500 mL beaker under constant stirring (400 rpm) and heating (60 °C).47,48 Further, 10 mL PEG-400 surfactant was added to the precursor salt solution, which prevented the agglomeration of the NF particles during their growth. Afterward, 10 mmol sodium hydroxide (NaOH) solution was added to the homogenized precursor solution dropwise until the pH reached a constant value of 12 and the resultant solution was kept stirring for 30 min. Subsequently, the obtained solution was transferred to a 200 mL Teflon-lined stainless-steel autoclave sealed at atmospheric pressure and kept inside a hot air oven for 12 h at 160 °C. Further, the autoclave was removed from the oven and allowed to cool to room temperature. Then, the obtained solution was filtered and washed several times using water and ethanol via magnetic decantation (separation of nanoparticles using NdFeB permanent magnet) to remove any trace of impurities, which significantly hampers the performance of magnetic systems. The black precipitate obtained after washing was centrifuged to remove the considerably smaller or bigger particles, with the resulting samples having a narrow size distribution. Finally, the centrifuged samples were dried under vacuum oven overnight to remove any trace of water present in the samples. The as-prepared samples were directly used for different characterizations. The reaction mechanism during the synthetic process is as follows:
| 2Fe3+ + xMn2+ + (1 − x)Co2+ + 8OH− → (1 − x)Co(OH)2xMn(OH)2·2Fe(OH)3 |
| xMn(OH)2·2Fe(OH)3(1 − x)Co(OH)2 → MnxCo1−xFe2O4·nH2O. |
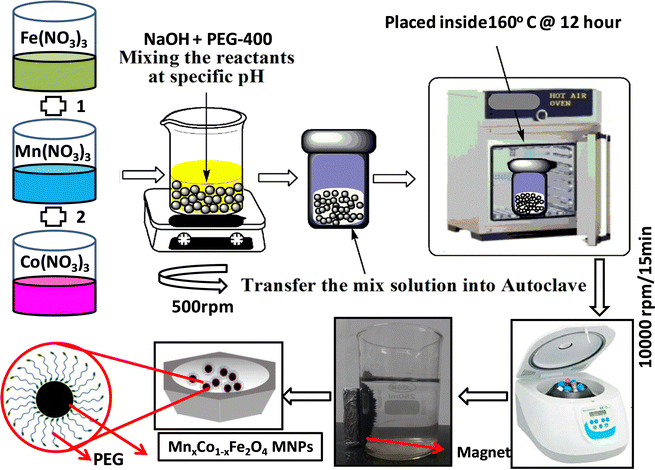 |
| | Fig. 1 Schematic diagram of hydrothermal synthesis of Co1−xMnxFe2O4 (x = 0, 0.2, 0.4, 0.6 and 0.8) nanoferrites. | |
2.2 Characterization techniques
The in-depth characterization of the structural, magnetic (static and dynamic), morphological, chemical, elastic, optical, and photoluminescence properties of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) was carried out via various sophisticated analytic measurement techniques including XRD, HRTEM, SEM & EDX, VSM, FMR, FTIR, UV-visible, PL, and TR-PL. The details of all the characterization tools used are as follows:
2.2.1 X-Ray diffraction (XRD).
The crystalline purity and phase identification of the as-synthesized Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) samples were investigated using a multi-purpose X-ray diffractometer (Rigaku Ultima-IV). The XRD patterns of all the sample compositions were obtained at 40 kV/40 mA voltage and current, respectively, using CuKα (λ = 1.54056 Å) radiation in the 2θ range of 20–80° (slow scan (1° min−1) and step size of 0.02°). Further, the detailed structural modeling was performed by Rietveld refinement using the FullProf software, and various theoretical parameters such as oxygen vacancy, bond length, edge length, packing efficiency, and bond angle were obtained for all the samples. Also, the 3-D crystal structure of the Co0.8Mn0.2Fe2O4 sample compositions was modeled using the VESTA software by different associated parameters such as bond angle, bond length, and cation-cation interaction among the A and B sites obtained from the structural modeling of the XRD patterns.
2.2.2 Transmission electron microscopy (TEM).
The morphology of the NFs and their size distribution were probed by TEM (model TECNAI F30). Also, all the samples were probed by selected area electron diffraction (SAED) and high-resolution transmission electron microscopy (HRTEM) to confirm the polycrystalline nature of the NFs. The sample for the measurement was prepared by dispersing the particles in ethanol under sonication for 3–4 min, followed by immediately dropping it on a 300 mesh carbon-coated copper grid and the grid was dried using an optical lamp.
2.2.3 Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS).
The surface morphology of the NF sample compositions and their element composition were obtained using a 10 keV SEM with EDS attachment (model EVO MA 10 variable pressure SEM). The SEM specimen for all the samples was prepared by dispersing the sample in ethanol under sonication and drop-casting it on a silicon wafer. The samples were dried in a vacuum oven at 60 °C followed by conducting coating. Further, EDS is a non-destructive technique used to determine the quantitative elemental composition of materials. The specimen for the EDS measurement was prepared using a pelletiser to make a 12 mm diameter and 2 mm thick pellet for all the samples.
2.2.4 Fourier transform infrared (FTIR) and UV-visible (UV-vis) spectroscopy.
The room-temperature FTIR spectra of all the NF compositions were recorded using an FTIR spectrometer (NICOLET 5700) in the wavenumber range of 4000–400 cm−1 in transmission mode to identify each functional group present in the sample. The specimen for the NF samples was prepared by mixing the dry powder sample with KBr matrix. The elastic parameters of all the sample compositions were obtained based on the vibrational frequency (ν) and force constant (K). The optical absorption spectra of all the NF compositions in the wavelength range of 200–900 nm were obtained using a UV-vis spectrophotometer (Lambda 950, PerkinElmer) with the step size of 0.1 nm to calculate the band gap energies and refractive indexes. The sample for the measurement was prepared by dispersing the powder sample in ethanol and sonication for 40 min.
2.2.5 Photoluminescence (PL) and time-resolved photoluminescence (TRPL).
The room-temperature PL and TRPL spectra of all the sample compositions were recorded using a fluorescence spectrometer (Edinburg Instrument (FLS-980.D2D2)). The PL spectra of all the samples were obtained at an excitation wavelength of 350 nm. Also, at room temperature, the TRPL measurements were performed for all the NF compositions to obtain their fluorescence decay time, which is in the ns range.
2.2.6 Vibrating sample magnetometer (VSM).
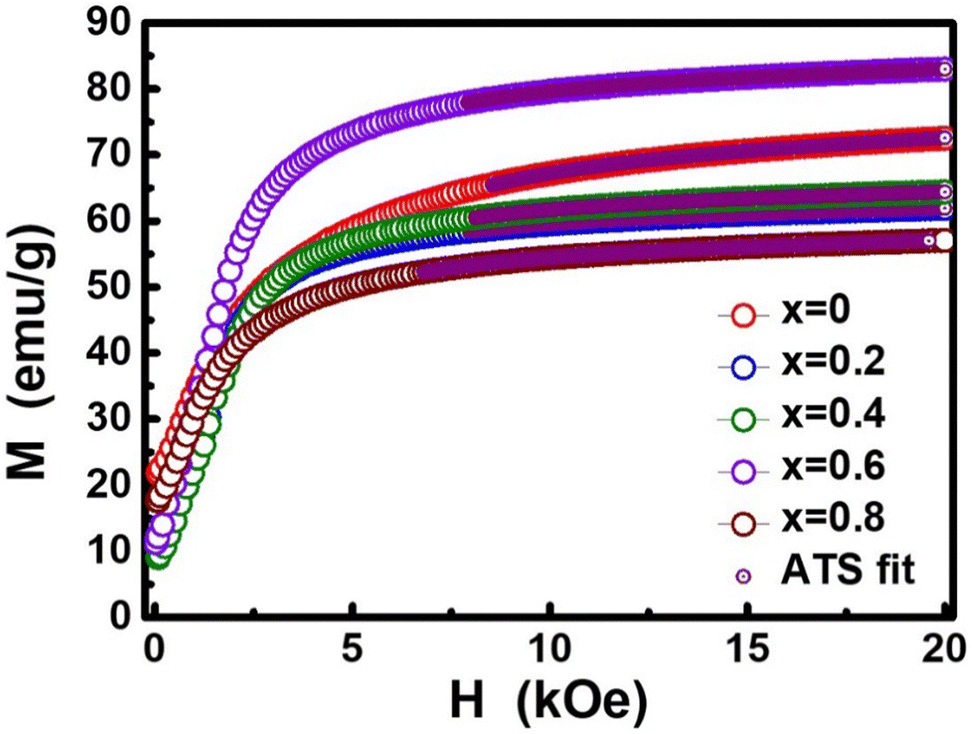
The dc magnetic properties of all the NF samples were obtained using a VSM (model Lakeshore 7410 VSM) in the magnetic field range of ±2T at room temperature (300 K). The magnetization curve obtained for the sample was further fitted to the theoretical predictions to calculate the anisotropy constant, initial permeability, and magnetic moment.
2.2.7 Ferromagnetic resonance (FMR).
The room-temperature spin dynamics study of the as-synthesized NF samples was conducted using an EPR spectrometer (Bruker, Biospin, Model A300) using the X-band microwave frequency (9.84 GHz) with a modulation frequency of 100 kHz in the magnetic field sweep range of 0–1.5 T. The FMR spectra obtained for all the samples were fitted to calculate their spin concentration and spin–spin relaxation time.
3. Results and discussion
3.1 X-Ray diffraction study
The phase confirmation and crystallinity of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs were probed by XRD measurements and the diffraction patterns of all the samples were obtained in the reflection mode. The XRD patterns of the pure and Mn2+-doped CFO samples are depicted in Fig. 2(a), which show multiple diffraction peaks, indicating the polycrystalline nature of the NFs. The sharp diffraction peaks for all the Co1−xMnxFe2O4 sample compositions indicate their good crystalline nature. The diffraction peaks of the pure CFO sample were observed at the Bragg angle (2θ) of 30.56°, 35.38°, 42.89°, 53.67°, 57.24°, and 62.93°, corresponding to the (220), (311), (400), (422), (511) and (440) lattice planes, respectively, which perfectly match the single cubic spinel structure (JCPDS card 22-1086).46,48,49 Also, all the other Mn-doped samples show diffraction peaks in close proximity, which suggests that the Mn ions occupy the lattice site and do not hamper the single cubic spinel structure of the CFO. Further, the incorporation of Mn ions resulted in the shifting of the peak systematically. Fig. 2(b) shows an enlarged view of the (311) plane of the XRD patterns of all the samples, which suggests that the diffraction peak shifted towards marginally lower values. This can be correlated with the exchange interactions and difference in radius between the Co and Mn ions, which induce strain in the material. Also, the lattice constant (LC- “a”) of the NFs was significantly affected by the distribution of cations from their respective sites. The LC was calculated from the interplanar spacing (d) and the respective (hkl) planes using eqn (1), as follows:| | 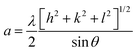 | (1) |
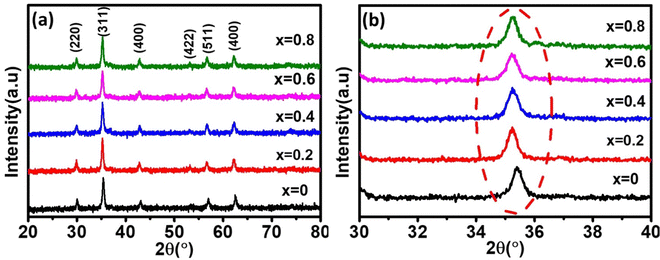 |
| | Fig. 2 (a) X-Ray diffraction spectra of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) nanoferrites. (b) Enlarged view of major diffraction peak of (311) plane XRD spectra of all the samples. All samples show good crystalline behavior and their major peaks shifted towards the lower 2θ values with an increase in Mn2+ ion concentration. | |
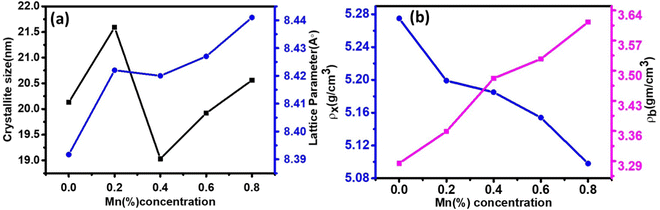
The LC for the Co1−xMnxFe2O4 NF sample for the x = 0 sample concentration is 8.391 Å, which increased to 8.44 Å for sample x = 0.8 with an increase in the Mn2+ concentration, as shown in Table 1. Similar behavior was also observed in the bulk samples, where the substitution of Mn2+ in place of Co2+ resulted in an increase in LC from 8.499 Å for MnFe2O4 (JCPDS card-10-0319) to 8.392 Å for CoFe2O4 (JCPDS-22-1086).50 The variation in LC with an increase in Mn2+ concentration is shown in Fig. 4(a). This increasing trend of LC with Mn2+ doping is due to the Mn2+ (0.83 Å) ions with a larger ionic radius replacing the Fe3+ (0.67) and Co2+ (0.78Å) ions with smaller ionic radii. It has been reported in the literature that the replacement of smaller radius host ions by larger radius ions results in an increase in the LC of the NFs. P. Monisha et al. reported similar behavior in the CFO, where Mn substitution resulted in an increase in the lattice parameters.49
Table 1 Crystallite size, strain, particle size, lattice constant and structural parameter (hopping length, specific surface area, and dislocation density) calculated from the X-ray diffraction studies of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) nanoferrites samples
| Sample name |
Crystallite size (nm) |
Strain (ε) |
Particle size TEM (nm) |
Lattice constant (Å) |
Lattice strain × 10−3 |
Volume (Å) |
L
A (Å) |
L
B (Å) |
ρ
x
(g cm−3) |
ρ
b (g cm−3) |
(P) % |
(S) m2 g−1 |
(δL) line per m2 |
| D–S method |
W–H method |
XRD |
Nelson Riley method |
|
X = 0 |
20.13 |
23.56 |
0.0045 |
23.47 |
8.391 |
8.386 |
9.9 |
590.80 |
3.633 |
2.966 |
5.275 |
3.285 |
37.726 |
56.505 |
0.0024 |
|
X = 0.2 |
21.59 |
25.82 |
0.0038 |
26.85 |
8.422 |
8.406 |
12.9 |
597.37 |
3.646 |
2.977 |
5.199 |
3.359 |
35.392 |
53.454 |
0.0021 |
|
X = 0.4 |
19.03 |
22.41 |
0.0027 |
22.41 |
8.424 |
8.412 |
12.7 |
596.94 |
3.645 |
2.976 |
5.185 |
3.482 |
32.845 |
60.808 |
0.0027 |
|
X = 0.6 |
19.92 |
24.92 |
0.0018 |
25.93 |
8.427 |
8.417 |
13.5 |
598.43 |
3.648 |
2.979 |
5.154 |
3.527 |
31.568 |
58.441 |
0.0025 |
|
X = 0.8 |
20.56 |
26.89 |
0.0012 |
27.48 |
8.441 |
8.473 |
15.2 |
601.42 |
3.655 |
2.984 |
5.098 |
3.613 |
29.137 |
57.244 |
0.0023 |
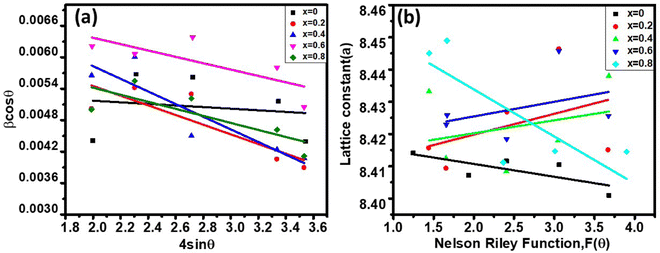
The theoretical and experimental LC was obtained using the Nelson-Riley function, as expressed in eqn (2):
| | 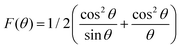 | (2) |
The LC obtained using the Nelson–Riley function (F(θ)) gives precise values and is in good agreement with that obtained using the XRD pattern directly using eqn (1).9 The LC values were obtained by assuming linear regression to determine the y-intercepts, by extrapolating the straight line to F(θ) = 0, as depicted in Fig. 3(b). The square blocks in Fig. 3(b) depict the experimental points corresponding to each θ value and the straight line represents the linear fit of the experimental points. The value of the LC obtained from the Nelson–Riley function is shown in Table 1 for all the sample compositions. The variation in the LC with varying Mn concentration is shown in Fig. 4(a), which is evident in the linear increase in the LC values with an increase in Mn concentration. The crystallite size (“d” – using the D–S method and “DWH” – using the W–H method) of all the sample compositions was obtained using the D–S equation (eqn (3)) and W–H method (eqn (5)). The D–S equation does not account for the instrumental and strain broadening, which is rectified in the W–H method giving a more precise value of the crystallite size of the samples. Also, the crystallite size in the D–S method is obtained by only considering the highest intensity peak (311), which sometimes results in an error. Thus, eqn (3) can be expressed as the sum of the full width at half maximum (FWHM) of the crystal and strain, as shown in eqn (4), which can be rewritten as eqn (5) (W–H method),51–53 as follows:
| | 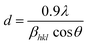 | (3) |
| | 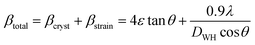 | (4) |
The above equation can also be expressed as:
| | 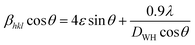 | (5) |
where
βhkl and
θ are the FWHM and Bragg angle of the (
hkl) plane, respectively,
λ is the wavelength of the X-ray source (
λ = 1.5406 Å), and
ε is the induced strain.
44,48 The strain in the material is considered to be uniform according to
eqn (5) in all the crystallographic directions, and thus is also known as the uniform deformation model. The average crystallite size and strain in the materials were calculated by plotting the liner fitted line of
βhklcos
θ vs. 4
sin
θ for the different Bragg angles obtained for each sample. The crystallite size was obtained from the
y-axis intercept
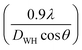
and the slope of the sample corresponds to the induced strain in the samples.
54,55 It was observed that the calculated average crystallite size of the CFO increased from 23.56 nm (
x = 0) to 26.89 nm (
x = 0.8) with an increase in the Mn
2+ ion concentration using the W–H method, as seen in
Table 1. The crystallite size values obtained using the D–S method show an almost similar trend, as shown in
Fig. 4(a) and
Table 1.
56 Further, the induced strain in the sample increased with an increase in the Mn concentration, which is obvious as the larger atoms (Mn
2+) substituted the smaller atoms (Co
2+), resulting in an increase in strain (from 9.9 × 10
−3 to 15.2 × 10
−3) in the samples. The positive slope of the W–H plot indicates the tensile strain in all the sample compositions given that the growth was restricted due to the larger ion substitution, which resulted in outward strain.
57 Koseoglu
et al. also reported a similar trend of increasing crystallite size upon an increase in Mn
2+ concentration. Further, the theoretical (
ρx) and experimental X-ray density (
ρb) of the samples were obtained using
eqn (6) and (7),
24,58 as follows:
| | 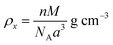 | (6) |
| | 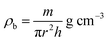 | (7) |
where
n is the number of atoms per unit cell (
n = 8),
M is the molecular weight,
NA is Avogadro's number, and
m is the mass,
r the radius, and
h the thickness of the specimen.
 |
| | Fig. 3 (a) Williamson Hall plots of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples for different Mn2+ ion concentration loadings. (b) Nelson Riley function F(θ) vs. lattice parameter plots of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. | |
 |
| | Fig. 4 (a) Variation in the crystallite size (d) and lattice parameters (a). (b) Variation in X-ray (ρx) and bulk densities (ρb) of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs with varying Mn2+ ion concentration. | |
The theoretical X-ray density of the samples decreased consistently with an increase in the Mn2+ ion concentration from 5.275 to 5.098 g cm−3. This can be well understood as ρx is inversely proportional to the LC of the materials. Alternatively, the experimental X-ray densities showed lower values than the theoretical values (bulk density) and increased from 3.285 to 3.613 g cm−3. The decrease in the density of the NFs from their bulk corresponds to the presence of pores, vacancies, and higher-order defects in the nanoregion.9,59 Further, the increase in Mn2+ ion concentration resulted in an increase in the density of the NFs due to the substitution of the larger-size Mn2+ ions in place of Co2+ ions. The variation in the theoretical and experimental X-ray densities (ρx and ρb, respectively) with an increase in Mn2+ ion concentration is shown in Fig. 4(b). Further, the percentage porosity (P) of all the sample compositions was obtained according to the empirical relationship in eqn (8), as follows:
| | 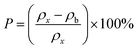 | (8) |
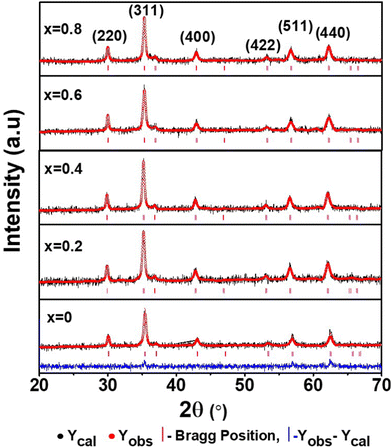
The porosity values obtained for all the sample compositions using eqn (8) are depicted in Table 1. The % porosity of the samples decreased with an increase in Mn2+ ion concentration from 37.726% to 29.137%. This resulted in an increase in the experimental X-ray density of the NF sample with an increase in Mn2+ ion concentration.20 Also, we obtained the hopping length (which is simply the distance between the magnetic ions from the A and B sites) corresponding to the tetrahedral (LA) and octahedral (LB) sites using eqn (9), as follows:
and
| | 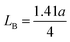 | (9) |
The values of the hopping length obtained for all the samples using
eqn (9) are summarized in
Table 1. The variation in CFO with Mn
2+ ion concentration indicates that the hopping lengths (
LA and
LB) increased with an increase in the Mn
2+ concentration. This can be associated with the fact that the hopping length corresponding to the A–B sites is longer compared to that of the A–A and B–B sites. Also, this can be well understood from
eqn (9),
33 which shows that the hopping lengths are directly proportional to the LC of the materials. Herein, we assume that our as-synthesized NFs have a spherical morphology. The specific surface area (
S) and dislocation density (
δL) of the NF samples were obtained using
eqn (10), as follows:
and
| | 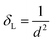 | (10) |
where
A′ = 6 for spherical nanoparticles.
9 The specific surface area is the area of the exposed surface of the nanoparticle per unit mass. The specific surface area and dislocation density values obtained for all the samples are shown in
Table 1. According to the obtained values of the specific surface area, we observed that the “
S” first increased, and then started to decrease with an increase in Mn
2+ ion concentration. According to
eqn (10), the specific surface area depends on both the theoretical density and crystallite size. The observed variation in the NF samples can be correlated with the presence of defect states in the samples.
48 Further, the dislocation density provides information about the presence of defects and purity of the crystal structure of the materials. It is defined as the number/length of dislocation sites per volume in the unit cell. The obtained values of the dislocation density using
eqn (10) indicate that.decreased with an increase in the Mn
2+ concentration. It is also evident from
eqn (10) that
δL varied inversely to the crystallite size and with an increase in Mn
2+ concentration, the crystallite size increased. Further, the magnitude of the values obtained for the Co
1−xMn
xFe
2O
4 samples is very low compared to the other NFs reported in the literature, making the prepared samples very useful for diverse applications in environmental remedial and biomedical applications.
23,35,44
3.2 Rietveld refinement of the diffraction data
The detailed structural investigation of the pure and Mn-substituted CFO NFs was carried out by Rietveld refinement using the powder XRD data and the full-proof refinement software. The refinement of the XRD patterns was performed by fixing the parameters such as “θ = 90°” assuming a cubic structure, while other parameters such as the LC, site occupancy, background, and scale factor were taken as free parameters. The parameter associated with the instrument was kept the same in all the refinement procedures and peak broadening was corrected using the reference standard of silicon powder (NIST standard reference material). The refinement was carried out by adopting a process similar to our previous work (please see the details in ref. 9, 18, 47 and 60). This process involves two steps and we adopted the least squares method to minimize the difference between the Bragg intensities of the experimental pattern and the proposed crystallographic structural model. The refinement was carried out by refining the global parameters such as background and scale factors followed by the refinement of LC, crystal orientation and asymmetries, metal ion occupancies, atomic coordinates, and isothermal parameters. The refinement of the XRD patterns was carried out by assuming the peak profile as pseudo-Voigt.46Fig. 5 shows the refined patterns of all the sample compositions, where the black line shows the experimental XRD curve and red circles show the refined pattern. The vertical lines show the Bragg position and the blue line at the bottom represents the difference between the refined patterns and experimental patterns. The Rietveld refinement procedure shows that the fitting of the extrinsic site inversion (α′) up to 10% maximum, which indicates the controlled synthesis of the Co1−xMnxFe2O4 NFs. It is evident from Fig. 5 that all the peaks in the experimental XRD patterns perfectly fit the standard data. Further, the R-factor is an important quantity that describes the extent of matching between the experimental and refined data (based on the crystallographic model) and calculated by taking the difference between them.35 The quality of the fitting of the refined data based on the crystallographic model was evaluated by determining various R-factors such as the profile (Rp and Rb), expected (Rexp), and weighted profile (Rwp). Further, we measured the goodness of fitting (χ2), which is the ratio of the weighted profile to the expected R-factor, further quantifying the quality of fitting.9,48,61 The value of χ2 is 1 for a perfect match and we obtained values in the range of 1.67 to 1.98, which are close and indicate very good matching with the structural model. The values of all the R-factors and χ2 obtained for all the samples are shown in Table 2, where it can be seen that an increase in Mn2+ concentration resulted in the distortion of the crystallographic model, increasing the χ2 value. However, after the critical value (x = 0.6), χ2 started to decrease given that the Mn2+ concentration now dominated the orientation and dictated the structural model. Further, different metal ions prefer different lattice sites in NFs, which influences the cation distribution significantly by the substitution of different metal ions in these materials. Also, the variation in the structural parameters was not systematic, which can be correlated with the difference in ionic radii of the metal ions such as Co2+ (0.74 Å), Mn2+ (0.80 Å), and Fe3+ (0.64 Å).36,44 This indicates that the optimized concentration of substitution holds the key to improving and governing the properties of these NFs.
 |
| | Fig. 5 La Bail Rietveld refinement XRD data of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs. The black circles show the experimental data and the red circles show the refined patterns. The vertical lines show the different XRD peaks obtained by the refinement of the pattern. | |
Table 2 Refinement (goodness of fit and R-factors) and structural parameter (lattice parameters and volume) of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs obtained by Rietveld refinement of the experimental XRD patterns
| Sample name |
χ
2
|
Bragg R-factor |
RF-Factor |
R
wp
|
R
exp
|
R
p
|
Cell volume (V) (Å)3 |
a (a = b = c) (Å) |
|
X = 0 |
1.67 |
2.89 |
2.27 |
44.6 |
34.6 |
98.5 |
593.41 |
8.403 |
|
X = 0.2 |
1.68 |
2.57 |
1.96 |
41.9 |
32.2 |
87.4 |
602.54 |
8.446 |
|
X = 0.4 |
1.80 |
1.82 |
1.16 |
34.9 |
26.0 |
51.4 |
601.03 |
8.439 |
|
X = 0.6 |
1.98 |
2.70 |
2.06 |
42.9 |
32.7 |
82.0 |
602.42 |
8.445 |
|
X = 0.8 |
1.72 |
2.99 |
2.17 |
43.6 |
33.2 |
91.7 |
601.67 |
8.442 |
3.3 Distribution of cations and crystal structure
The distribution of the cations in spinel NFs plays a crucial role in obtaining the desired properties in these materials. This becomes even more critical in the nano regime given that the slightest variation in metal ion concentration at the lattice sites results in a significant variation in the properties. Herein, we propose the cation distribution in the Mn2+ substituted CFO based on the preference of the metal ions at the octahedral and tetrahedral lattice sites. Generally, Mn2+ has a tendency to occupy the octahedral sites, whereas the Co2+ ions tend to occupy the tetrahedral sites and Fe3+ ions can occupy both the octahedral and tetrahedral sites. Considering the tendency of individual metal ions to occupy preferred lattice sites, the most appropriate cation distribution is shown in Table 3.62Fig. 7 shows the 3-D crystal structure of the Co0.8Mn0.2Fe2O4 NFs obtained using the VISTA software, where the orange color is the iron atom, the black color shows the cobalt atom, the red color shows the manganese atom, and the white color shows the oxygen atom. The cation distribution of the Co1−xMnxFe2O4 NF samples was obtained using the Bertaut method based on structural modeling. In this method, the cation distribution is obtained by calculating the refined XRD peak intensities. Further, the cation distribution proposed by the Bertaut method was verified by the lattice parameters obtained by fitting the crystallographic model. In the XRD patterns, some of the diffraction planes are sensitive to the cation distribution and their intensity ratios indicate the most appropriate metal ion distribution. In spinel NFs, the (220), (400), and (440) planes are considered the most suitable for the prediction of the cation distribution. The intensity ratio of these planes was calculated as I422/I400, I220/I422, and I220/I400 to verify the predicted cation distribution.33,42,63 The refinement of these lattice plane reflections was done by minimizing the R-factor and also calculated by distributing the intensities of the overlapping reflections, as given by eqn (11) and (12).| | 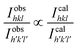 | (11) |
where| | 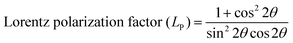 | (12) |
where IobshklIcalhkl, and Ihkl are the observed, calculated, and relative integrated intensities for the reflection plane (hkl), respectively, Fhkl is the structure factor, which is the sum of the individual ions present in each site, and P is the multiplicity factor. The estimated cation distribution of the A and B sites for the Co1−xMnxFe2O4 NFs is given in Table 3.
Table 3 Purposed cation distributions of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples
| Sample (MnxCo1−xFe2O4) |
Cation distribution at lattice sites (A and B sites) |
|
X = 0 |
(Co0.012+Fe0.993+)A(Co0.992+Fe1.013+)BO42− |
|
X = 0.2 |
(Co0.052+Fe0.953+)A(Co0.752+Fe1.053+Mn0.202+)BO42− |
|
X = 0.4 |
(Co0.032+Fe0.973+)A(Co0.572+Fe1.033+Mn0.402+)BO42− |
|
X = 0.6 |
(Co0.072+Fe0.933+)A(Co0.332+Fe1.073+Mn0.602+)BO42− |
|
X = 0.8 |
(Co0.042+Fe0.963+)A(Co0.292+Fe1.043+Mn0.802+)BO42− |
The average cation radius rA and rB of the A and B sites was obtained using the cation distribution shown in Table 3 and eqn (13) and (14), respectively, as follows:
| | | rA = (Cco2+)A(rco2+) + (CFe3+)A(rFe3+) + (CMn2+)A(rMn2+) | (13) |
| | 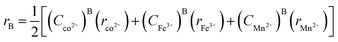 | (14) |
where
C and
r correspond to the ionic concentration and ionic radii of the cations distributed at the A and B sites, respectively. The obtained values of the average cation radius of both the A and B sites (
rA and
rB, respectively) are shown in
Table 4, which indicate a systematic increase in
rB with an increase in the Mn
2+ concentration. This can be well understood as the larger ionic radius Mn
2+ ions (0.80 Å) replaced the Co
2+ ions (0.74 Å) at the octahedral sites.
64 In contrast,
rA varied in a non-systematic manner with an increase in the Mn
2+ ion concentration as it has a strong tendency to occupy the B sites. This resulted in the movement of some of the Fe
3+ ions to the A sites, which is evident from the cation distribution shown in
Table 3. The higher concentration of Fe
3+ ions at A sites resulted in an increase in the average cation radius at the tetrahedral sites, as shown in
Table 4. Further, the theoretical lattice parameter (
ath) strongly depends on the ionic radius of the metal ions and obtained using
eqn (15).
9| | 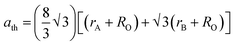 | (15) |
where
Ro = 1.32 Å (radius of O
2−). The value of
ath is obtained by substituting the mean ionic radius values obtained from
eqn (14)–(16) and summarized in
Table 4. The value of the theoretical LC increased consistently with an increase in Mn
2+ concentration, validating the experimental trend. The small variation in the theoretical values and experimental values originated from the consideration of an ideal unit cell in the theoretical analysis. The oxygen positional parameter (
u) is defined as the distance the oxygen ions need to be moved to incorporate the metal ions at the A-sites. The oxygen positional parameter was obtained using
eqn (16). Also, the inversion parameter (
δ), which is defined by the difference between the calculated oxygen parameter and ideal oxygen parameter (
uideal = 0.375),
18 was calculated using
eqn (17), as follows:
| | 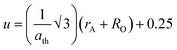 | (16) |
Table 4 Lattice parameters, ionic radii, bond lengths, oxygen and inversion parameters, tetrahedral and octahedral edge, shared and unshared lengths of as-synthesized NF samples obtained by analytical calculations
| Sample name |
a
th (Å) |
A–O (Å) |
B–O (Å) |
r
A1 (Å) |
r
B1 (Å) |
u
|
δ
|
R
|
R′ |
R′′ |
|
X = 0 |
8.5173 |
1.9900 |
2.0535 |
0.6711 |
0.7244 |
0.3849 |
0.0099 |
3.2425 |
2.7728 |
3.0149 |
|
X = 0.2 |
8.5248 |
1.9956 |
2.0536 |
0.6755 |
0.7246 |
0.3851 |
0.0101 |
3.2575 |
2.7704 |
3.0187 |
|
X = 0.4 |
8.5285 |
1.9941 |
2.0551 |
0.6733 |
0.7273 |
0.3850 |
0.0100 |
3.2589 |
2.7740 |
3.0201 |
|
X = 0.6 |
8.5346 |
1.9985 |
2.0551 |
0.6778 |
0.7271 |
0.3852 |
0.0102 |
3.2636 |
2.7712 |
3.0222 |
|
X = 0.8 |
8.6746 |
1.9937 |
2.1105 |
0.6744 |
0.7815 |
0.3827 |
0.0077 |
3.2658 |
2.8780 |
3.0693 |
The values of the oxygen positional parameter and inversion parameter for different Mn2+ concentrations in CFO are shown in Table 4. Both “u” and “δ ” show a non-systematic variation similar to the mean ionic radius corresponding to the A-sites. This can be well understood as the oxygen positional parameter is directly proportional to rA as ath, showing a consistent increasing trend. Further, the bond lengths (A–O and B–O) and ionic radii (rA1 and rB1) of the respective tetrahedral and octahedral sites were obtained using eqn (18)–(21).30,35
| | 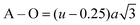 | (18) |
| | 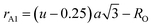 | (20) |
| | | rB1 = (0.625 − u)a − RO | (21) |
The values of the bond lengths and ionic radii obtained using the above-mentioned equations are presented in Table 4. The bond length value corresponding to the B-site is higher than that of the A-site. Similarly, the ionic radius of the B-site is also greater than that of the A-sites. The bond length corresponding to the B-sites showed a consistent increasing trend with an increase in Mn2+ concentration, which can be well understood as the ionic radius of the Mn2+ ions is greater than that of the substituted Co2+ ions. Alternatively, A–O and rA1 showed a similar trend, which first increased, and then decreased with a further increase in the Mn2+ concentration. The non-systematic variation in these quantities can be well described by their dependence on multiple factors, which counteracts these types of behaviours.31,65 The average bond length corresponding to the tetrahedral sites (RA) and octahedral sites (RB) was obtained using eqn (22) and (23), respectively.
| | | Tetrahedral bond length: RA = a√3(δ + 0.125) | (22) |
| | 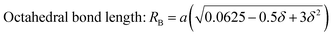 | (23) |
The values of RA and RB were obtained by the substitution of the inversion factor and experimental LC in the above-mentioned equations and shown in Table 5. Both the tetrahedral and octahedral bond lengths did not show any systematic variation with varying Mn2+ concentrations. The tetrahedral and octahedral bond lengths are more dependent on the inversion parameter compared to the lattice parameter, and thus follow a similar variation trend with an increase in Mn2+ concentration. The ionic packing coefficients corresponding to the tetrahedral (Pa) and octahedral (Pb) sites for the spinel NFs were calculated using eqn (24)–(27),49,66 as follows:
| | 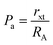 | (24) |
| | 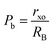 | (25) |
| | | rxt = a√3(u − 0.25) − RO | (26) |
| | | rxo = a(0.625 − u) − RO | (27) |
where
rxt and
rxo are the radius of the A and B interstitial sites, respectively. The calculated values of ionic packing coefficients are smaller than 1, suggesting the existence of ionic vacancies and larger overlapping between the cation and anion. The obtained packing coefficient values (P
a and P
b) using
eqn (25) and (26) are presented in
Table 5. Again, the variation in the value of P
a and P
b did not have any systematic change in behaviour with an increase in the Mn
2+ ion concentration. Further, the values of the ionic packing factor (
α) and total vacancy concentration existing in the sample (
β) were obtained for different sample concentrations using
eqn (28) and (29), respectively, and estimated by the fulfilment coefficient of the unit cell.
64| | 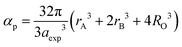 | (28) |
| | 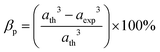 | (29) |
Table 5 The radii of the interstitial sites (A and B), average bond lengths, ionic packing fraction and total vacancy concentration of the as-synthesized NFs obtained by analytical calculations
| Sample name |
r
xt (Å) |
r
xo (Å) |
R
A
|
R
B
|
P
a
|
P
b
|
α
|
β
|
|
X = 0 |
0.6100 |
0.6650 |
1.9900 |
2.5185 |
0.3065 |
0.2640 |
0.6566 |
4.3827 |
|
X = 0.2 |
0.6148 |
0.6652 |
1.9948 |
2.5259 |
0.3082 |
0.2632 |
0.6496 |
3.5737 |
|
X = 0.4 |
0.6141 |
0.6648 |
1.9941 |
2.5227 |
0.3079 |
0.2643 |
0.6504 |
3.7690 |
|
X = 0.6 |
0.6185 |
0.6665 |
1.9985 |
2.5317 |
0.3094 |
0.2643 |
0.6491 |
3.7352 |
|
X = 0.8 |
0.6137 |
0.7218 |
1.9937 |
2.4802 |
0.3078 |
0.2910 |
0.6559 |
7.8636 |
The values of the ionic packing factors and total vacancy concentration are listed in Table 5. The value of α falls in the range of 0.64–0.66, which indicates the presence of two phases, i.e., normal and inverse spinel ferrite. Typically, a sharp change in the value of αp was observed at the boundary of the normal and inverse spinel ferrites. Further, the value of βp showed a two-fold increase with the addition of Mn2+ for a higher concentration Co1−xMnxFe2O4 (x = 0.8). Initially, with Mn2+ substitution, it started to decrease for the Co1−xMnxFe2O4 (x = 0.2) sample; however, it started to increase with the further addition of Mn2+ ions. The decrease in the βp value indicates a change in the mixed ferrite to normal ferrite and vice versa. According to Table 5, it is evident that the values of αp, Pa and Pb are lower than 1, which indicates the presence of vacancies at both the A and B sites. The values of the different edge lengths such as tetrahedral shared ®, shared octahedral (R′) and unshared octahedral (R′′) for all the NF samples were obtained using eqn (30)–(32), respectively,9,61 as follows:
| | 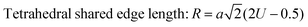 | (30) |
| | 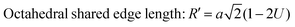 | (31) |
| | 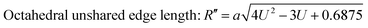 | (32) |
The values of the R obtained using eqn (30) are listed in Table 4, showing that the tetrahedral shared edge length consistently increased with an increase in the Mn2+ ion concentration in the CFO samples. This can be correlated with the increase in the mean radius of the tetrahedral site with Mn2+ substitution. Further, the cation distribution among the tetrahedral and octahedral sites is mainly responsible for the variation in R′ and R′′. A sharp increase in the value of the octahedral shared and unshared edge lengths was observed for the sample concentration x = 0.8 Mn2+ ions from the x = 0.6 sample concentration. This can be correlated with the transition of CFO towards MnFe2O4 due to the excessive substitution of Mn2+ ions. These lengths are a key indicator for predicting and controlling the magnetic properties of the NFs, as in the case of the Mn2+ ion-substituted CoFe2O4, which follows a mixed spinel structure, and the net magnet moment obtained by the exchange interaction between the tetrahedral and octahedral sites. The increase in these lengths resulted in a reduction in the exchange interaction; however, due to their ferromagnetic nature, the reduction in interaction has a different effect as the Mn2+ ions have a higher magnetic moment, which resulted in an overall increase in the net magnetic moment of the NF samples.
The two key parameters that control the magnitude of the magnetic exchange interactions in NFs are the bond angle and inter-ionic distance. An increase in bond angle directly results in an increase in exchange interaction, whereas the inter-ionic distances are inversely proportional. The interionic lengths, i.e., cation–anion and cation–cation distances, were obtained using eqn (33)–(41).42,49 Different magnetic interactions, i.e., A–B, A–A, and B–B, significantly depend on the bond lengths between the cation–cation and cation–anion. The values of the inter-ionic lengths obtained using eqn (33)–(41) were used for the calculation of the bond angles between the metal ions using eqn (42)–(46).
Cation-cation bond length
| | 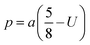 | (33) |
| | 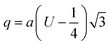 | (34) |
| | 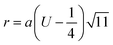 | (35) |
| | 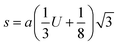 | (36) |
Cation–anion bond length
| | 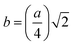 | (37) |
| | 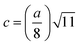 | (38) |
| | 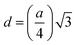 | (39) |
| | 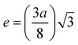 | (40) |
| | 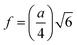 | (41) |
Interionic bond angle
| | 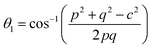 | (42) |
| | 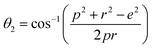 | (43) |
| | 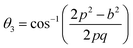 | (44) |
| | 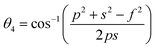 | (45) |
| | 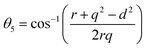 | (46) |
Further, the values of the bond length and bond angle obtained using the above-mentioned equations are listed in Tables 6 and 7, respectively. The bond angle indicates the strengthening and weakening of the exchange interaction. Fig. 6 shows a schematic of the different bond lengths and bond angles observed in the NF samples. It is evident from Table 7 that θ1, θ2, and θ5 increased, while θ3 and θ4 decreased with an increase in Mn2+ ion concentration. According to Fig. 6, θ1 and θ2 correspond to the A–B interaction, whereas θ5 corresponds to the A–A interaction. Thus, the increase in the values of θ1, θ2, and θ5 indicates the strengthening of the A–A and A–B interaction with an increase in Mn2+ ion concentration. Further, the values of θ3 and θ4 decreased with an increase in Mn2+ ion concentration, indicating the weakening of the B–B interaction. The increase in the A–B interaction can be correlated with the creation of an anti-parallel arrangement at the A and B sites by the substitution of the Co2+ ions with Mn2+ ions. Also, the Mn2+ ion substitution resulted in a reduction in the Fe3+ ions in the octahedral and tetrahedral sites. This resulted in the strengthening of the A–A interaction. Further, the overall reduction in the magnetic moment due to the substitution of the Co2+ (3μB) ions by Mn2+ (5μB) ions from the B to A sites resulted in a decrease in the B–B interaction.9,61 Further, the crystal structure of the Co0.8Mn0.2Fe2O4 sample is shown in Fig. 7.
Table 6 The calculated inter-ionic distance values, i.e., cation–anion (p, q, r, s) and cation–cation (b, c, d, e, f) distances of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples
| Sample name |
p (Å) |
q (Å) |
r (Å) |
s (Å) |
b (Å) |
c (Å) |
d (Å) |
e (Å) |
f (Å) |
|
X = 0 |
2.0450 |
1.9899 |
3.8104 |
3.7364 |
3.0112 |
3.5310 |
3.6879 |
5.5319 |
5.2155 |
|
X = 0.2 |
2.0450 |
1.9947 |
3.8197 |
3.7407 |
3.0139 |
3.5341 |
3.6912 |
5.5368 |
5.2201 |
|
X = 0.4 |
2.0468 |
1.9941 |
3.8185 |
3.7418 |
3.0152 |
3.5357 |
3.6927 |
5.5392 |
5.2223 |
|
X = 0.6 |
2.465 |
1.9985 |
3.8269 |
3.7455 |
3.0174 |
3.5382 |
3.6953 |
5.5430 |
5.2260 |
|
X = 0.8 |
2.1018 |
2.0237 |
3.8178 |
3.7944 |
3.0669 |
3.5962 |
3.7560 |
5.6341 |
5.3117 |
Table 7 The calculated bond angles between the cation-anion obtained by the cation distribution of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs
| Sample name |
θ
1 (°) |
θ
2 (°) |
θ
3 (°) |
θ
4 (°) |
θ
5 (°) |
|
X = 0 |
122.10 |
134.38 |
94.95 |
126.34 |
112.60 |
|
X = 0.2 |
122.25 |
136.48 |
94.48 |
126.30 |
112.84 |
|
X = 0.4 |
122.48 |
139.64 |
95.00 |
126.25 |
112.98 |
|
X = 0.6 |
122.86 |
139.76 |
95.10 |
126.18 |
113.42 |
|
X = 0.8 |
122.93 |
144.59 |
93.84 |
126.10 |
113.62 |
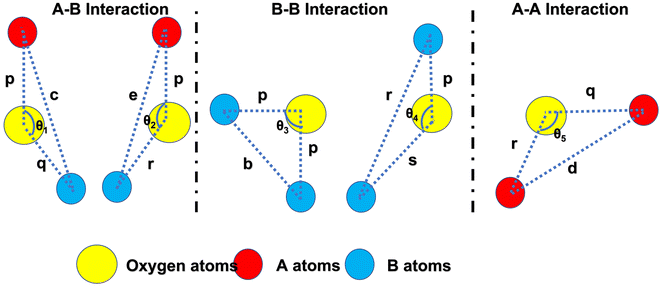 |
| | Fig. 6 Schematic representation of the different bond lengths and bond angles of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. | |
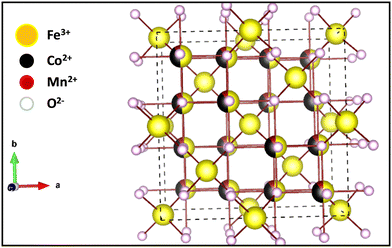 |
| | Fig. 7 3-D crystal structure of Co0.8Mn0.2Fe2O4 nano-ferrites. | |
3.4 Fourier-transform infrared spectroscopy (FTIR)
FTIR is a powerful tool to investigate the vibrational and absorption bands of the as-synthesized NFs samples. It provides vital information of the NFs regarding the formation of molecular bands, functional groups, and chemical residues. The FTIR spectra of the as-synthesized Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) samples obtained in the wavenumber range of 400–4000 cm−1 are depicted in Fig. 8(a). The chemical properties obtained by the absorption or emission IR spectra of the materials indicate that these NFs have a cubic spinel structure. The IR absorption band of the cubic spinel NFs is the signature of the chemical bands present in the ions of their lattice. Moreover, in the ferrites, there are two main absorption bands observed in the FTIR spectra, corresponding to the metal–oxygen band, which appeared in the range of 400–600 cm−1.67 According to Fig. 8(a), it is clear that two bands are observed for all the samples, which correspond to the tetrahedral-oxygen (“ν1” – Mtetr–O2−) and octahedral-oxygen (“ν2” – Moct–O2−) vibrations. In the cubic spinel structure, the absorption bands (ν1 and ν2) correspond to the intrinsic lattice vibrations of the A and B sites, respectively. The absorption band (ν2) at around 400–430 cm−1 is ascribed to the stretching vibration of Moct – O2− in the octahedral position and the stretching vibration of the absorption band (ν1) at around 500–600cm−1 is due to the stretching vibration of Moct – O2− at the tetrahedral sites in all the composition samples.68,69 The presence of these two stretching vibrations in the FTIR spectra gives evidence of the formation of the ferrite phase. The other bands observed at around 1500 and 3400 cm−1 are attributed to the –OH– stretching and bending, respectively, due to the presence of water molecules. These absorption bands originated from the mixing of KBr in the sample, which absorbed water molecules during the sample preparation in the open atmosphere. The absorption band at around 3000 cm−1 is due to the carboxyl group present in the materials, which confirms the presence of PEG-400 in all the samples. The calculated wavenumbers with the addition of Mn2+ ions in the cobalt ferrites at both A and B positions are shown in Table 8. Thus, it is evident from Table 8 that the value of ν1 decreased, whereas ν2 increased with an increase in Mn2+ ion concentration. The trend in the values of ν1 and ν2 can be well understood as the Mn2+ ions have a strong tendency to occupy the B sites of the spinel ferrites. Thus, the increase in Mn2+ ions at the B site resulted in the replacement of the Fe3+ ions in the A sites, as evident from the cation distribution shown in Table 3. Further, this change in the vibration frequency can also be correlated with the change in bond lengths between the oxygen molecules and metal ions. Also, the difference in the masses of Mn2+ and Co2+ ions resulted in a change in the vibration frequency along their band position.18 Moreover, the FTIR spectra also validate the transformation of the cubic inverse spinel to normal spinel structure.
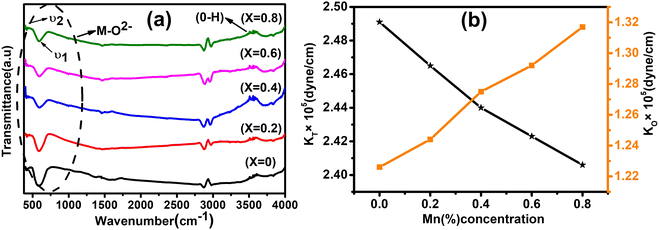 |
| | Fig. 8 (a) Room-temperature FTIR spectra of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) magnetic NFs in the 400–4000 cm−1 region. The two bands ν1 and ν2 corresponding to the metal ion–oxygen vibrations are the signature of the spinel ferrite phase. The stretching vibration at around 3000 cm−1 appearing for all the samples is due to PEG-400. (b) Variation in force constant (KO and KT) with varying Mn concentration in Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0, x = 0.2) NF samples. | |
Table 8 The absorption bands (ν1 and ν2), force constant (KT and KO), and Debye temperatures of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs
| Sample name |
ν
1 (cm−1) |
ν
2 (cm−1) |
Force constant (KT × 105) (dyne per cm) |
Force constant (KO × 105) (dyne per cm) |
Debye temperature (ΘD (K)) |
|
X = 0 |
583 |
409 |
2.491 |
1.226 |
713.24 |
|
X = 0.2 |
580 |
412 |
2.465 |
1.244 |
713.59 |
|
X = 0.4 |
577 |
417 |
2.440 |
1.275 |
714.68 |
|
X = 0.6 |
575 |
420 |
2.423 |
1.292 |
715.40 |
|
X = 0.8 |
573 |
424 |
2.406 |
1.317 |
716.84 |
3.5 Elastic study
The force constant (K) is the proportionality coefficient between the force exerted by the individual atoms on each other and the displacement from the mean position. This characterizes and provides knowledge about the chemical bonding and structural characteristic of molecules. The force constant (K) between metal–oxygen ions corresponding to the tetrahedral (Kt) and octahedral (Ko) sites was obtained using eqn (47), as follows:| | | Kt = 4π2c2ν12m and K0 = 4π2c2ν22m | (47) |
where c is the velocity of light (c = 3 × 108 cm s−1) and m is the reduced mass of metal–oxygen ions obtained by m = m1m2/m1 + m2, and its calculated value is 2.065 × 10−23 g.
The force constant values of all the Mn2+ concentration samples obtained using eqn (47) are depicted in Fig. 8(b) and listed in Table 8. The Ko value increased steadily from 1.226 × 105 dyne per cm to 1.317 × 105 dyne per cm, whereas Kt decreased from 2.491 × 105 dyne per cm to 2.406 × 105 dyne per cm with an increase in Mn2+ concentration. According to the calculated force constant values corresponding to the tetrahedral and octahedral sites, it can be concluded that Kt is higher than Kt for all the sample concentrations.66 The value of the elastic stiffness constant for the NF system was calculated using eqn (48) and (49), as follows:
| | 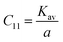 | (48) |
| | 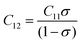 | (49) |
where
Kav is the average force constant
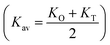
,
a is the experimental lattice parameter and
σ id Poisson's ratio, which depends on the porosity (
P) and calculated as
σ = 0.324(1 − 1.043·
P). The values of Poisson's ratio obtained for the different concentration samples are depicted in
Table 8 and indicate that it increased with an increase in Mn
2+ concentration. The Poisson's ratio varied from 0.19 to 0.22, which confirms the good elastic and isotropic characteristics of the samples.
30 Further, the values of the stiffness constant
C11 and
C12 of all the samples were obtained by putting the values of
Kav,
a and
σ in the above-mentioned equations and listed in
Table 9. The stiffness constant
C11 decreased, while
C12 increased systematically with an increase in Mn
2+ concentration. The stiffness constant
C11 mainly depends on two factors,
i.e., force constant and lattice parameter, whereas
C12 also depends on the porosity of the samples. Further, various elastic modulus constants such as Young's modulus (
E), modulus of rigidity (
G), and bulk modulus (
B) were calculated using the stiffness constant and Poisson's ratio and
eqn (50)–(52),
64 respectively, as follows:
| | 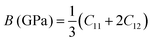 | (50) |
| | 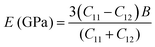 | (51) |
| | 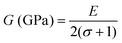 | (52) |
Table 9 The Poisson's ratio, stiffness constant, bulk, rigidity and young's moduli, wave velocity, longitudinal Vl (m s−1) and transverse Vt (m s−1) of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs
| Sample name |
σ
|
C
11 (GPa) |
C
12 (GPa) |
B (GPa) |
E (GPa) |
G (GPa) |
V
l (m s−1) |
V
t (m s−1) |
V
m (m s−1) |
|
X = 0 |
0.1965 |
221.6 |
51.4 |
108.1 |
202.1 |
84.4 |
2049.6 |
2152.9 |
1927.2 |
|
X = 0.2 |
0.2044 |
220.6 |
56.6 |
111.2 |
197.4 |
81.9 |
2059.4 |
2316.4 |
2032.0 |
|
X = 0.4 |
0.2130 |
220.4 |
59.7 |
113.4 |
195.3 |
80.5 |
2063.7 |
2115.3 |
1906.5 |
|
X = 0.6 |
0.2174 |
220.2 |
61.2 |
114.3 |
194.0 |
79.6 |
2068.3 |
2033.6 |
1852.1 |
|
X = 0.8 |
0.2255 |
219.6 |
63.9 |
115.8 |
190.7 |
77.8 |
2076.8 |
1897.7 |
1836.8 |
The values of all the elastic moduli (Young's modulus, modulus of rigidity and bulk modulus) for the varying Mn2+ concentration samples obtained using the above-mentioned equations are listed in Table 9. It is evident from this table that the bulk modulus increased, while the Young's modulus and modulus of rigidity decreased with an increase in Mn2+ concentration. The high values of the elastic moduli refer to the strong tendency of the material to recover from the deformation to reach an equilibrium state. The high values of Young's modulus and modulus of rigidity in the Mn-substituted cobalt ferrites are due to the strong interaction between the metal ions at their respective cation sites A and B. Further, the smaller crystallite size resulted in larger grain boundaries, which caused restrictive dislocation motion, improving the strength and toughness of the sample. Further, the wave velocities (longitudinal Vl and transverse Vt) were calculated using eqn (54) and (55), respectively, by substituting the value of GO, which show the modulus of rigidity with zero porosity (P = 0), as follows:
| | 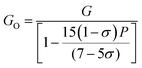 | (53) |
| | 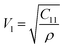 | (54) |
| | 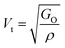 | (55) |
The mean elastic wave velocity (
Vm) was calculated using
eqn (56), as follows:
| | 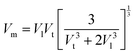 | (56) |
The values of all the wave velocities were calculated and presented in Table 9. It is evident from this table that the longitudinal wave velocity increased systematically, whereas the transverse and mean wave velocities firstly increased, and then started to decrease after a critical concentration of Mn2+ (x = 0.2). The highest value of the longitudinal wave velocity for the samples was observed at Mn2+ (x = 0.2) while the transverse and mean wave velocity peaked at Mn2+ (x = 0.2) as depicted in Table 9. The Debye temperature is a physical quantity that represents the temperature at which the maximum vibrations of ions take place in the crystal lattice. It signifies the thermal capacity of a material and calculated as the integral of the local heat capacity. It is also helpful to determine the thermodynamics properties such as entropy and specific heat of materials. The Debye temperature (θD) of all the samples with varying Mn concentrations was obtained using Anderson's formula, as shown in eqn (57).70,71
| | 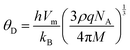 | (57) |
After rearranging the above equation, we get
| | 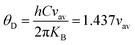 | (58) |
where
h is Planck's constant,
kB is Boltzmann's constant,
M is the molecular weight, and
q is the number of atoms per unit cell.
72 The calculated Debye temperature values for each sample composition are listed in
Table 8, which increased consistently with an increase in Mn
2+ concentration from 713.24 to 716.84 K. This is consistent with the specific heat theory, which suggests that the Debye temperature of the materials increased due to the decrease in the conduction of electrons (n-type) and increase in the conduction of holes (p-type). This type of increasing trend in the Debye temperature is due to the increase in the wavenumber of the bands in the FTIR spectra and the large interatomic bonding strength.
3.6 High-resolution transmission electron microscopy (HRTEM) study
The morphology, size distribution, and crystallinity of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0, x = 0.2) NF samples were investigated via bright-field imaging, HRTEM and SAED analysis. The TEM micrographs of all the as-synthesized samples are shown in Fig. 9(a–d), which indicate the spherical and uniform size distribution of the particles in all the samples. Also, all the NF samples did not show aggregation due to the PEG-400 coating, which prevented the formation of particle aggregates. The average size of the Co1−xMnxFe2O4 NF particles was calculated by taking an average of 100 particles of each sample of the different probing area and the images were analysed using the Image J software. The average particle size calculated for the samples varied in the range of 22 nm to 27 nm and follows a similar trend as the crystallite size obtained by the structural modeling, i.e., XRD, as depicted in Table 1. The small variation in the values of the TEM and crystallite size can be correlated with the agglomeration of particles due to the strong dipolar interactions, resulting in a higher particle size.73 Further, the particle size distribution was fitted with various distribution functions and it follows a Gaussian distribution. The particle size distribution histograms for the Co1−xMnxFe2O4x = 0.4 and x = 0.6 samples are shown in Fig. 9(e) and (f), respectively. Also, the crystallite statistical distribution of an average of 100 particles was estimated by using the TEM image, which was fitted using the log-normal distribution function given in the following relation:74| | 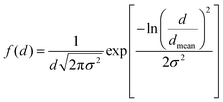 | (59) |
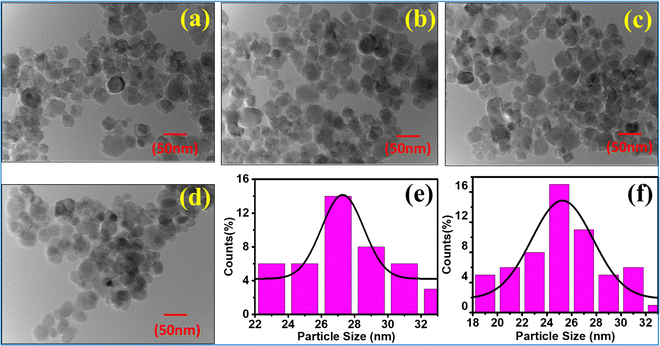 |
| | Fig. 9 TEM images of NF samples (a) CoFe2O4, (b) Co0.8Mn0.2Fe2O4, (c) Co0.6Mn0.4Fe2O4, (d) Co0.4Mn0.6Fe2O4 and (e) Co0.2Mn0.8Fe2O4. Particle size distribution (Gaussian distribution) of (e) Co0.4Mn0.6Fe2O4 and (f) Co0.6Mn0.4Fe2O4. | |
The mean value of dmean = 25 nm ± 1.5 nm, with a minimum size of 22 nm and maximum size of 27 nm, and standard deviation σ = 0.8 nm was obtained from the above relation. This is also consistent with the XRD data. Further, the crystalline nature of the samples was studied by SAED analysis for all the sample compositions. The SAED patterns of all the samples show multiple bright fringes, which indicate the polycrystalline nature of the samples, where each ring corresponds to a different crystalline plane.75–78Fig. 10(a) shows the SAED pattern of the sample composition Co0.8Mn0.2Fe2O4, which depicts the different planes of the spinel ferrites of (311), (220), (111), (422), and (511) obtained by the corresponding d-values of 0.25 nm, 0.29 nm, 0.35 nm, 0.17 nm, 0.16 nm, respectively.9,23,24 The d-values from the SAED plot were determined using the Image J software for accuracy. The SAED plots of all the samples indicate the high crystallinity of the as-synthesized samples. Further, the high-resolution TEM images of the samples were obtained to confirm the presence of various planes and defects in the samples. Fig. 10(b–d) show the HRTEM images of the Co0.2Mn0.8Fe2O4, Co0.6Mn0.4Fe2O4 and CoFe2O4 NF samples, respectively. Fig. 10(b) shows the d-values of 0.29 nm and 0.25 nm, which correspond to the (220) and (311) planes, respectively. Similarly, Fig. 10(c and d) show the d values of 0.25 nm, 0.21 nm, and 0.24 nm, which perfectly match the XRD planes of (311), (400), and (222), respectively.33,79,80 Also, a tilt boundary can be seen in Fig. 10(d), where the (311) planes were confirmed from the d-values at two places; however, they are oriented differently. According to the TEM analysis, we can conclude that the prepared samples are spherical, have a uniform size distribution, and are highly crystalline, which have an average size in the range of 22 nm to 27 nm.
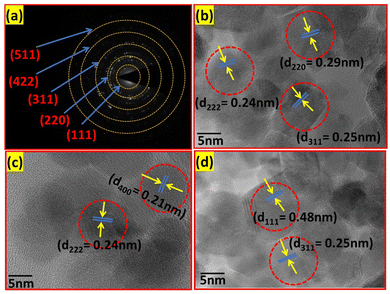 |
| | Fig. 10 (a) SAED pattern of the Co0.8Mn0.2Fe2O4 sample and HRTEM images of the (b) Co0.2Mn0.8Fe2O4, (c) Co0.6Mn0.4Fe2O4 and (d) CoFe2O4 samples. | |
3.7 UV-visible spectroscopy study
The room-temperature optical characteristics of all the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples were investigated by UV-Visible spectroscopy. The absorbance spectra of all the samples in the photon wavelength of 200–900 nm are depicted in Fig. 11(a) and the optical absorption band of each composition is listed in Table 10. According to the absorption peak values listed in Table 10, it is evident that the optical absorption band increased from 229 to 237 nm with an increase in Mn2+ concentration. The appearance of the absorption band in the UV range can be attributed to the photoexcitation of the electronic transition of the Fe3+(3d)–O2−(2p) electrons from the Fe3+ conduction band (CB) to the O2− valence band (VB). Interestingly, in the absorption spectra, a minor red shift in the absorption edge was observed with an increase in the Mn2+ concentration. The absorbance of materials depends on several factors such as their lattice parameters, particle size, band gap, crystal structure disorder, surface roughness, purity, and phase.81 The absorption spectra indicate that there is no absorption edge present in the visible range for all the samples. Also, the absorption spectra indicate that the absorption peak shifted to higher wavelengths with an increase in the doping concentration of Mn2+, indicating the change in optical energy band gap due to the effect of quantum confinement. This behavior is also due to the emergence of additional energy levels in both the CB and VB, which may be produced because of the decrease in the crystallite size and increase in intrinsic crystal defects. This type of behavior has also been reported in previous studies.40,82
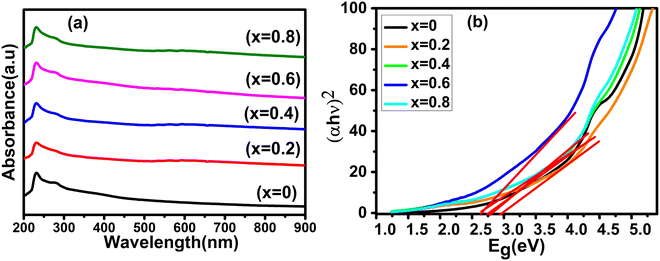 |
| | Fig. 11 (a) UV-visible absorbance spectra and (b) optical energy band gap (Tauc plots) spectra of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs. | |
Table 10 The absorption, optical band gap (direct), refractive index, and optical dielectric constant of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples
| Sample name |
Absorption λ (nm) |
Band gap (eV) direct |
Refractive index (n) |
Optical dielectric constant (εα) |
|
X = 0 |
229 |
2.71 |
2.40 |
5.76 |
|
X = 0.2 |
232 |
2.65 |
2.44 |
5.95 |
|
X = 0.4 |
234 |
2.68 |
2.42 |
5.85 |
|
X = 0.6 |
236 |
2.67 |
2.43 |
5.90 |
|
X = 0.8 |
237 |
2.66 |
2.42 |
5.85 |
The optical energy band gap for all the samples was obtained from their optical absorbance spectra assuming a direct energy band Eg following the Tauc plot relation,33,83i.e., αhν = A′(hν − Eg)1/2 by plotting αhν vs. hν, as depicted in Fig. 11(b), where A′ is a constant, α is the absorption coefficient and hν is the incident energy of photon. Furthermore, the absorption coefficient (α) of the MNPs was obtained using the relation 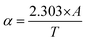 , where A is the absorbance and T is the thickness of the cuvette. The optical band gap, i.e., direct band gap of all the sample compositions, was obtained by putting (αhν)2 = 0 and extrapolation of the straight line segment. Thus, the obtained values of the optical energy band gap are listed in Table 10. The value of the band gap consistently decreased from 2.71 to 2.65 eV with an increase in Mn2+ concentration. The variation in the band gap values is due to the variation in crystallite size, lattice parameter, and cation distribution with a variation in the concentration of Mn2+ ions. Further, the variation in the band gap energies in NF samples may be due to the coordination of cations and the co-existence of low and high spin states. The decrease in the band gap energy with an increase in the Mn2+ concentration confirms the increase in the crystallite size and lattice strain in the sample. This is due to the quantum confinement effect.30,35 Also, some micro-strain was generated by the defects, which affected the crystal structure of the materials. In the case of the Mn2+substituted cobalt ferrite samples, the ionic radius of Mn2+ (0.80 Å) is larger than that of the Fe3+ (0.67 Å) ion, which caused a decrease in the optical band gap value. The decrease in the band gap with an increase in the particle size can be well understood by Brass’ equation84 given by eqn (60), as follows:
, where A is the absorbance and T is the thickness of the cuvette. The optical band gap, i.e., direct band gap of all the sample compositions, was obtained by putting (αhν)2 = 0 and extrapolation of the straight line segment. Thus, the obtained values of the optical energy band gap are listed in Table 10. The value of the band gap consistently decreased from 2.71 to 2.65 eV with an increase in Mn2+ concentration. The variation in the band gap values is due to the variation in crystallite size, lattice parameter, and cation distribution with a variation in the concentration of Mn2+ ions. Further, the variation in the band gap energies in NF samples may be due to the coordination of cations and the co-existence of low and high spin states. The decrease in the band gap energy with an increase in the Mn2+ concentration confirms the increase in the crystallite size and lattice strain in the sample. This is due to the quantum confinement effect.30,35 Also, some micro-strain was generated by the defects, which affected the crystal structure of the materials. In the case of the Mn2+substituted cobalt ferrite samples, the ionic radius of Mn2+ (0.80 Å) is larger than that of the Fe3+ (0.67 Å) ion, which caused a decrease in the optical band gap value. The decrease in the band gap with an increase in the particle size can be well understood by Brass’ equation84 given by eqn (60), as follows:
| | 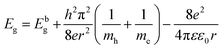 | (60) |
where
Eg and
Ebg are the energy and bulk energy gap,
r is the particle size,
mh and
me are the effective mass of electrons and holes,
e is charge of an electron,
ε is the relative permittivity, and
ε0 is the permittivity in free space, respectively. According to the above-mentioned relation, we can conclude that the optical energy band gap is affected by the lattice constant, shape and crystallite size. CFO materials are widely used in optoelectronic devices; however, detailed optical band gap studies on Mn
2+-substituted CFO are rarely reported in the literature. In this framework, Chen
et al. and Hema
et al. communicated the optical band gap values of 2.38 eV and 2.69 eV, respectively, for pure CFO.
35,85 Similarly, Monisha
et al. reported an optical band gap value of 3.84 eV for pure CFO, which increased with 1% Mn
2+ doping to 4.02 eV.
49 Also, some other reports published on pure CFO reported different optical band gap values, indicating their dependence on several factors such as particle size, crystallinity, surface functionality, doping concentration, and material composition. The refractive index (
n) is a property of materials that plays an important role in optical applications. We calculated the refractive index (
n) of all the sample compositions using
eqn (61) proposed by Kumar
et al.,
9 as follows:
| | 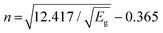 | (61) |
According to the above-mentioned relation, the calculated values of the refractive index of all the sample compositions are listed in Table 10. The refractive index of CoFe2O4 is 2.40, which initially increased for the x = 0.2 sample concentration, and then further decreased with an increase in the Mn2+ concentration in the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs. This type of behavior is also affected the optical band gap of the materials, which depends on the change in the electronic transition of Fe3+–O2− with Mn2+ ions on both the tetrahedral (A) and octahedral (B) sites. Furthermore, we calculated the high frequency optical dielectric constant (ε∝) with the refractive index (n) by using the following relation: ε∝ = n2 and the calculated values are listed in Table 10. Based on the UV-Vis data, we calculated the energy band gap, refractive index, and high-frequency dielectric constant of the Mn2+-doped CoFe2O4, which are listed in Table 10.
3.8 Photoluminescence spectroscopy (PL) study
PL spectroscopy is a vital tool to probe the optical characteristics of the as-synthesized NF samples, which gives key information about the carrier dynamics upon exposure to a light source. The light emission after recombination of excited-state charge carriers and dynamics of carriers produced during exposure to radiation from a light source was used to investigate the optical characteristics of the materials. Various optical characteristics such as electronic band gap (EBG), band structure, recombination phenomena, luminescent behavior, surface defects, defect states, and separation of charge carriers can be observed in material systems.46,61 Further, for the NF samples, PL spectroscopy gave information about their EBG relative to the energetic position of their sub-bandgap defect states and the effect of their size in the nanoregion based on the energies and kinetics of the photo-generated charge carriers. The room temperature PL spectra of all the as-synthesized Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples using a 350 nm excitation wavelength we recorded, as shown in Fig. 13 (left). Further, the right-hand side of Fig. 13 shows the Gaussian-fitted spectra, which are a combination of two different emissions, i.e., violet and blue band emissions in all the sample compositions. The cation distribution in the A and B sites of the NFs, the shallow and deep hole defects, and the presence of O2− vacancies are the main cause of the PL response of the NF samples. The PL spectra of the spinel ferrite samples were used to probe the site disordering, local site occupancy, and electronic defect states present in these materials. The PL processes in NFs are generally stimulated by the oxygen vacancies present at the interstitial sites.18,20 The O2− vacancies created at the octahedral and tetrahedral sites cause shallow and deep hole defects, which are mainly responsible for the PL properties. The band-edge emission of the electrons in the CB and holes in the VB is stimulated by the radioactive recombination. It is evident from Fig. 13 that all the NF samples have 2 primary bands, as follows: (i) a peak centered at 424 nm, which can be correlated with the violet band, i.e., near band edge (NBE) emission and (ii) a peak centred at 458 nm, which corresponds to the blue band. This band arises due to the 3F → 3p transition in the octahedral sites between the Co2+/Mn2+ ions. The emission in the violet region appeared due to the transfer of electrons from the shallow donor level to the VB and the emission in the blue region is attributed to the band edge free and bound excitons. Also, it is produced by another transition, i.e., electronic transitions occurring due to the near-conduction band-edge to the deep level acceptors and transitions from deep donor levels to the VB. The violet band emission appearing at around 424 nm for all the NF samples can be attributed to the NBE emission originating from the amalgamation of excited electrons from a localized level below the CB with the holes in VB. Generally, CFO strongly absorbs the impinged light in the UV range; however, it shifted to the violet band for the CFO MNPs. With the addition of Mn2+ ions in CoFe2O4, the intensity of its photoluminescence spectrum increased, which indicates that the new energy levels appeared between the VB and CB. Further, the occurrence of a direct transition between the CB and VB further resulted in an increase in the intensity of the PL emission.86,87 A schematic representation of the different bands and emissions describing the photoluminescence mechanism in the NFs is depicted in Fig. 12. Initially, the electrons present in the conduction band relax back to a shallower donor energy level due to the long lifetime and recombine with the holes present in the valence band. Further, the presence of surface and oxygen defects results in intermediate energy bands and recombination from these energy bands results in violet and blue emission. Further, the substitution of the Mn2+ ion in CoFe2O4 results in the creation of several dopants or defect centers on the surface of the MNPs, improving their emission efficiency. In the literature, there are mainly three types of emissions, i.e., violet, green and yellow band emissions, reported for CFO and other mixed NFs, as schematically shown in Fig. 12. Also, by adopting various surface modification techniques, the PL emission of the CFO MNPs can be tuned from 400 nm to 500 nm.88,89 Further, we also calculated the energy band gap from the PL spectra for all the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) sample compositions, which lies in the range of 2.90 to 2.93 eV and increased with the Mn2+ concentration. The obtained behaviour is the same as that of the optical energy band gap from the UV-visible data.
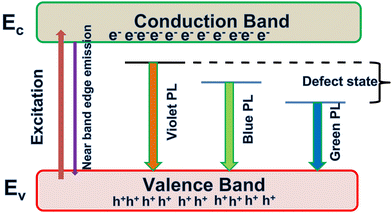 |
| | Fig. 12 Schematic representation of the different emission bands observed in the NF samples from their origin. | |
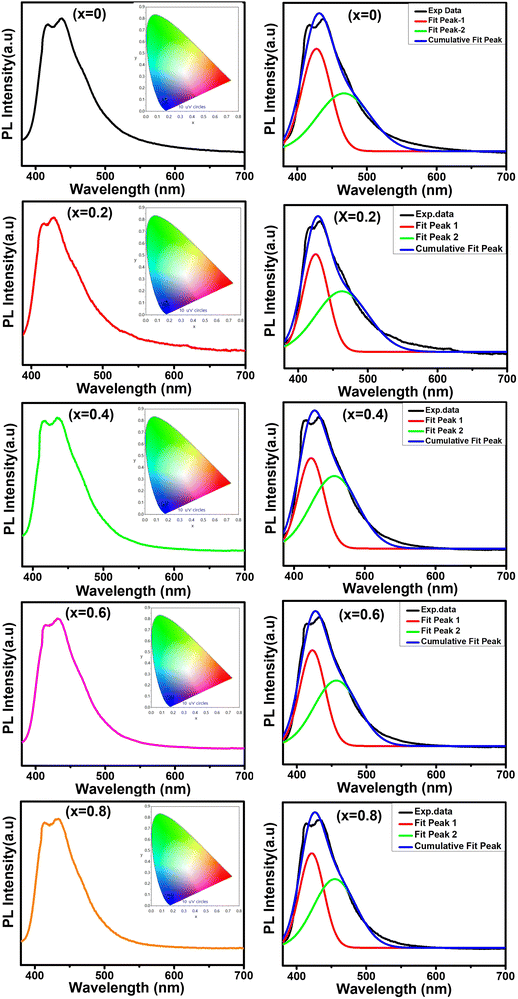 |
| | Fig. 13 Room temperature photoluminescence (PL) spectra (left) and fitted Gaussian fitting spectra (right) of the different Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. The inset of the left-hand figure shows the CIE plot of the respective sample. | |
3.9 Time-resolved photoluminescence spectroscopy (TRPL) investigation
The decay rate of the photons (charge carriers) inside the NFs and the dynamic rate of the luminescence materials were studied by TRPL. The decay lifetime of optical materials is an important parameter, which provides key information about the quality of material and device performance. Thus, we calculated the decay time of all the NF samples by fitting their TRPL spectra. Generally, for nanomaterials, the decay time mainly depends on the concentration of defects, which trap electrons/holes and increase their recombination process.90 It also depends on their morphology, size distribution, surface functionalization, and interactions with other materials. Given that the mixed ferrites material are a special class of materials in which a significant difference in properties was observed in the past, it is important to investigate the effect of different doping or substitution concentrations on their optical properties. The radioactive recombination efficiency associated with each transition is directly proportional to the decay time.18,91 The TRPL spectra (decay time vs. luminescence intensity) in the range of 0–80 ns of all the NF sample compositions are depicted in Fig. 14 (hollow symbols). The room temperature TRPL spectra of all the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples were obtained using a 266 nm laser as the excitation source. All the TRPL spectra of each sample composition were best fitted with the double exponential decay function and characterized by two decay times, i.e., τ1 and τ2, according to eqn (62),18 and the fitted graphs are shown in Fig. 14 in solid lines.| | 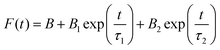 | (62) |
where F(T) is the luminescence intensity, B is the time constant, B1 and B2 are weighting constant parameters, t is the measured time and τ1 and τ2 are the decay time constants, which quantify the time taken by the excited state electron to return to its original state. These parameters were obtained by fitting the TRPL spectra using eqn (62) and listed in Table 11. The value of the goodness of fit (χTRPL2) falls in the range of 0.83–0.97, which indicates the good agreement between the experimental and fitted data. Two different decay times, i.e., τ1 and τ2, appeared for all the samples, which are attributed to the presence of different defect states in their CB. and VB. Besides, surface functionalities play a significant role in controlling the decay time. According to Table 11, it is evident that the pure cobalt ferrite has a slower rate (larger decay time), which increased (smaller decay time, i.e., fast decay) with the addition of Mn2+. The values of τ1 and τ2 are 9.36 ns and 4.56 ns, which decreased (faster decay) with the addition of Mn2+ ions to 1.04 and 4.56, respectively. The substitution of Mn2+ in the cobalt ferrites increased the defect concentration, which led to faster recombination, and thus a lower decay time. However, a further increase in Mn2+ concentration resulted in an increase in the decay time (τ1 = 1.25 and τ2 = 6.72) for the x = 0.4 sample concentration, which further decreased the decay time (τ1 = 1.03 and τ2 = 4.50) for the x = 0.6 sample concentration. This random variation in the decay time can be correlated with the variation in crystallite size given that they both follow the same trend. For the x = 0.4 sample, the crystallite size decreased compared to the x = 0.2 and 0.6 sample concentrations, which increased the surface defects, resulting in longer decay time. Further, for the x = 0.8 sample concentration, we observed a decay time comparable to that of the pure cobalt ferrite, which suggests that the (τ1 = 9.09 and τ2 = 3.82 ns) mixed or substituted ferrites have a shorter decay time, indicating a faster photoluminescence decay. Also, τ1 is greater than τ2 for the pure and x = 0.8 sample concentration, which was reverse for the other sample concentrations, proving that the optimized substitution of Mn2+ in the pure ferrites is the key for enhancing the optical characteristics of these materials. This result suggests that the charge carrier lifetime in the pure CFO and Co1−xMnxFe2O4 (x = 0.8) samples was longer in comparison with the other Mn2+-doped samples, which confirms the enhanced charge separation and transport performance of charge carriers in the pure CFO and Co1−xMnxFe2O4 (x = 0.8) samples. However, it is difficult to predict the exact cause of the different decay states, which are related to the recombination process, and there is no direct evidence to correlate the defect-induced emission energy with decay dynamics. Nevertheless, it can be correlated with the cubic spinel crystal structure of the Co1−xMnxFe2O4 NFs. All the material properties including physical, chemical, optical, and magnetic and efficiency of the radiative recombination are closely correlated with the decay time, especially the band-to-band and band-edge transitions.1 For the double-exponential decay, the average decay time (τavg) was determined using the following relation:| | 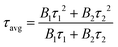 | (63) |
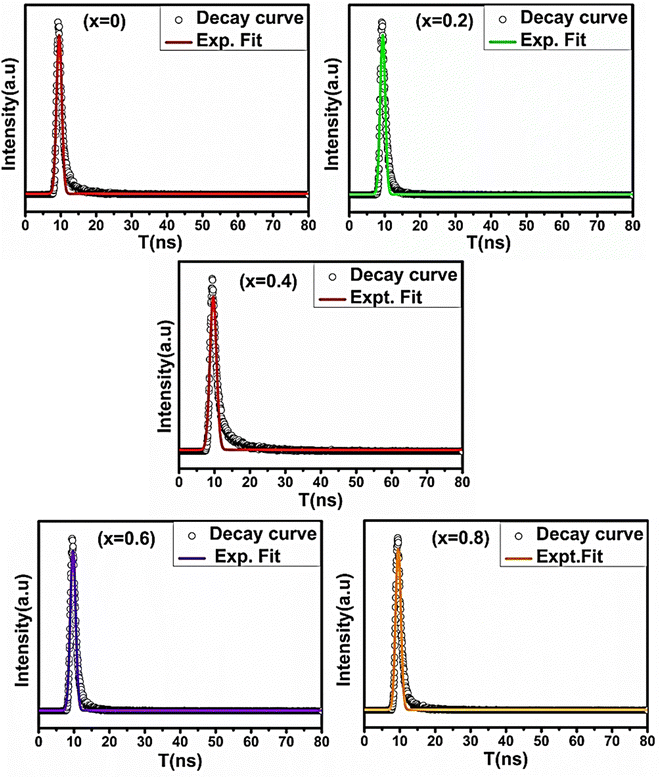 |
| | Fig. 14 Room temperature time-resolved photoluminescence spectra (hollow symbols) and double exponentially fitted spectrum (solid lines) of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. | |
Table 11 The decay lifetime (τ1 and τ2), average decay lifetime (τavg), goodness of fitting (χTRPL2) and other constant parameters of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs
| Sample name |
τ
1 (ns) |
τ
2 (ns) |
τ
avg (ns) |
B
|
B
1
|
B
2
|
χ
TRPL
2
|
|
X = 0 |
9.36 |
4.59 |
9.12 |
0.770 |
959.67 |
99.39 |
0.912 |
|
X = 0.2 |
1.04 |
4.56 |
1.32 |
0.277 |
1161.06 |
23.46 |
0.828 |
|
X = 0.4 |
1.25 |
6.72 |
3.67 |
0.724 |
958.58 |
142.45 |
0.913 |
|
X = 0.6 |
1.03 |
4.50 |
1.42 |
0.359 |
1155.80 |
33.86 |
0.925 |
|
X = 0.8 |
9.09 |
3.82 |
8.90 |
0.852 |
872.35 |
74.07 |
0.971 |
The obtained average decay (τavg) of all the sample concentrations using eqn (63)18 is listed in Table 11. The average decay time also followed a similar trend as of τ1, which is higher for the pure and high Mn concentration sample but lower for the other sample compositions. For the pure CFO, τavg = 9.12 ns, which decreased with an increase in Mn2+ concentration to x = 0.2. Also, it slightly increased with the further addition of Mn2+ for the x = 0.4 sample concentration to τavg = 3.67; however, immediate drops were observed after the further addition of Mn2+. At a high Mn2+ concentration, the average decay time showed a big jump, which is comparable to that of the pure CFO samples. This increase in decay lifetime at this particular concentration also indicates the reduction of non-radiative recombination centers. The reduction in the decay time of the Mn2+-doped CFO suggests an increase in defect density after the addition of Mn2+ ions, which can trap the energy of excitons and may have resulted in a decrease in the decay time. The observed decay time for the pure and Mn2+-substituted cobalt ferrite makes them ideal candidates for designing devices for fast optical sensing and switching and applications in the field of optoelectronic devices.42
Further, to understand the actual color emitted by the synthesized NF samples, it was vital to obtain their color coordinates marked on a Commission International d’Eclairage (CIE) diagram. The CIE plot of all the samples obtained by following our previous article18 is depicted in the inset of Fig. 14 together with the PL spectrum of each sample. The coordinates of the chromaticity plot were obtained as: 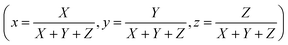 and its center was ascribed as the standard belonging to the white light emission (x = 0.34, y = 0.34). To determine the dominant emission colour for the Mn2+-doped CFO NFs, the emission spectra of the samples were converted to a CIE chromaticity diagram. The (x, y) coordinates of the chromaticity plot and the values of X, Y and luminescent intensity found for each sample composition are listed in Table 12.
and its center was ascribed as the standard belonging to the white light emission (x = 0.34, y = 0.34). To determine the dominant emission colour for the Mn2+-doped CFO NFs, the emission spectra of the samples were converted to a CIE chromaticity diagram. The (x, y) coordinates of the chromaticity plot and the values of X, Y and luminescent intensity found for each sample composition are listed in Table 12.
Table 12 The (x, y) coordinates of chromaticity plot, the X, Y values and luminescent intensity of each sample composition of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) (x = 0, 0.2, 0.4, 0.6 and 0.8) NFs
| Sample name |
X-coordinate |
Y-coordinate |
Intensity (Lum) |
|
X = 0 |
0.17250 |
0.10803 |
3.360 × 103 |
|
X = 0.2 |
0.17721 |
0.11502 |
3.437 × 103 |
|
X = 0.4 |
0.16023 |
0.08060 |
3.333 × 103 |
|
X = 0.6 |
0.16039 |
0.07497 |
1.298 × 104 |
|
X = 0.8 |
0.15816 |
0.06733 |
2.285 × 103 |
3.10 Static magnetic measurement (VSM)
The static magnetic measurements (M–H) of all the sample compositions of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs were carried out at 300 K using a VSM at the maximum field of ±2 T and depicted in Fig. 15. Various magnetic parameters such as saturation magnetization (Ms), coercivity (Hc), remanent magnetization (Mr), squareness ratio (R = Mr/Ms), magnetic moment (μB), Y–K angle, initial permeability, microwave frequency, magneto crystalline anisotropy (MCA) and anisotropy constant were obtained from the M–H loop of each sample and listed in Table 13. The S shape of the M–H loops indicates the strong ferromagnetic behavior of the prepared NFs. The Ms and Hc of all the prepared samples were depicted in Table 13, indicating the highest Ms of 80.30 emu g−1 for the x = 0.6 sample concentration. The Ms of all the NF samples varied in the range of 55.40 to 80.30 emu g−1 and did not follow any systematic trend given that the magnetic characteristics of the NFs strongly depend on several factors such as their synthetic route, crystallinity, defect state concentration, particle size, and porosity.92,93 The bulk Ms of MnFe2O4 and CoFe2O4 is 110 emu g−1 and 90 emu g−1, respectively; however, in the nano-regime, it is significantly lower due to the increase in defect state and surface spins, and non-stoichiometric cation distribution.46,64 The value of Ms obtained for Co0.4Mn0.6Fe2O4 is one of the highest reported to date for mixed ferrites of the same particle size.
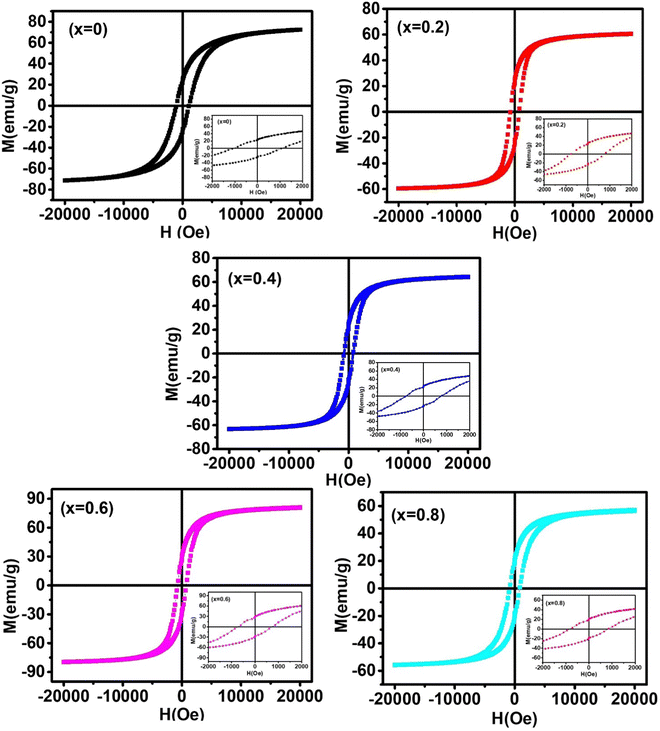 |
| | Fig. 15 Room temperature hysteresis (M–H) loops of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. The inset of the figure shows an enlarged view closer to the center. The lower values of coercivity indicate the single-domain particles. | |
Table 13 The room temperature magnetic parameters, i.e., saturation magnetization, coercivity, magnetic moments, remanent magnetization, squareness ratio, anisotropy constant, initial permeability, and Y–K angle of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF compositions
| Sample name |
M
s (emu g−1) |
H
c (Oe) |
(μB) exp |
(μB) the |
M
r (emu g−1) |
R = (Mr/MS) |
K
1 (erg cm−3) |
Initial permeability (μi) |
Y–K angle α (°) |
|
X = 0 |
68.51 |
1071.78 |
2.87 |
2.35 |
23.95 |
0.33 |
1.65 × 106 |
2.89 |
23.32 |
|
X = 0.2 |
59.88 |
787.80 |
2.50 |
2.98 |
23.80 |
0.39 |
0.96× 106 |
3.67 |
18.75 |
|
X = 0.4 |
62.23 |
803.01 |
2.59 |
2.72 |
23.60 |
0.36 |
1.07 × 106 |
3.32 |
29.24 |
|
X = 0.6 |
80.30 |
738.37 |
3.34 |
2.42 |
27.44 |
0.34 |
1.36 × 106 |
6.74 |
36.18 |
|
X = 0.8 |
54.40 |
784.36 |
2.25 |
2.04 |
20.14 |
0.35 |
0.89 × 106 |
3.65 |
43.48 |
According to Neel's sublattice theory of ferrimagnetism, the resultant magnetization (M(x)) of the NFs typically arises from the interactions of metal ions occupying the A and B lattice sites in NFs. Typically, there are 3 types of interactions possible in NFs, i.e., A–A, A–B, and B–B interactions. Among them, the A–B interaction is the predominant one and is mainly responsible for the ferro/ferri magnetism in NFs. In the case of ferrimagnets, the magnetic spins at the A and B sites are oppositely aligned, and thus the net magnetic moment of ferrimagnets is calculated by the difference in the magnetic moment of the B and A site lattice structure per formula unit, i.e., 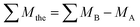 . The room temperature magnetic moments of Mn2+, Co2+, and Fe3+ are 5.0, 3.0, and 5.0 μB, respectively.42,46 The Fe3+ ions distribute equally on the A and B site lattice and the Co2+ ions only occupy the B-site lattice. The Mn2+ ions have a large ionic radius and magnetic moments compared to the Co2+ ions and they have a strong tendency to replace the Co2+ ions in the B-site lattice. This results in an increase in the saturation magnetization and a decrease in coercivity.44 For the pure cobalt ferrite sample, the saturation magnetization was 68.51 emu g−1, which is lower than that of its bulk counterpart due to the surface spin canting effects arising from the large surface area in the nanoregion.63
. The room temperature magnetic moments of Mn2+, Co2+, and Fe3+ are 5.0, 3.0, and 5.0 μB, respectively.42,46 The Fe3+ ions distribute equally on the A and B site lattice and the Co2+ ions only occupy the B-site lattice. The Mn2+ ions have a large ionic radius and magnetic moments compared to the Co2+ ions and they have a strong tendency to replace the Co2+ ions in the B-site lattice. This results in an increase in the saturation magnetization and a decrease in coercivity.44 For the pure cobalt ferrite sample, the saturation magnetization was 68.51 emu g−1, which is lower than that of its bulk counterpart due to the surface spin canting effects arising from the large surface area in the nanoregion.63
Further, the Mn2+ substitution in the pure CFO resulted in a decrease in Ms as Mn2+ ions occupy the A site, which resulted in a decrease in the AB interaction. This decrease in Ms with Mn2+ ions is ascribed to the higher magnetic moment of the Mn2+ (5 μB) ions, which replace the Co2+ (3 μB) ions. However, the further substitution of Mn2+ ions resulted in an increase in Ms and decrease in coercivity. The Ms and coercivity for the sample concentration of Co1−xMnxFe2O4 (x = 0.6) is 80.30 emu g−1 and 784.36 Oe, respectively. This is the highest Ms reported for nanomaterials in this particular size range. The highest Ms obtained for the sample concentration with the optimal Mn2+ substitution suggests the importance of the optimal concentration for achieving the desired properties. This increase in the Ms of the Mn2+-substituted CFO samples can be correlated with the reduced MCA due to the substitution of Co2+ ions by Mn2+ and the lower surface spin disorder. The amount of surface spin disorder in the Mn2+-substituted CFO increased with a decrease in crystallite size.39 Further, the coercivity of the NF samples obtained from the M–H plot indicates that the coercivity decreased from 1071.78 to 738.37 Oe with an increase in the Mn2+ ion concentration from x = 0 to x = 0.6. However, it further increased with an increase in Mn2+ concentration. As is known, the magnetic moment of Co2+ (3d7) in the CFO sample has lower ferromagnetic order compared to the Mn2+ (3d5) in manganese ferrites. Thus, there are some intrinsic parameters such as Bohr magneton, number of cations, MCA, magnetic domain, and SEI that are responsible for the variations in the coercivity.34,45
The remanent magnetization of the sample decreased with an increase in Mn2+ concentration; however, for the Co1−xMnxFe2O4 (x = 0.6) sample, it slightly increased. Further, we measured the squareness ratio (R) by taking the ratio of remanent magnetization and coercivity. The squareness ratio indicates the degree of squareness of the hysteresis loop. According to the Stoner–Wohlfarth (S–W) model, for single-domain nanoparticles with a random orientation, the value of “R” should be lower than 0.5. This parameter is extremely useful from a material point of view, while selecting an appropriate ferromagnetic material system. For applications such as nanomagnetic fluids and biomedical applications, the value of R should be as low as possible, while for memory devices, a high value of R is desirable.9,33 The squareness ratio primarily depends on factors such as particle size, morphology, magnetic interactions, crystallinity, and anisotropy of the particles. Thus, for most materials, the value of R is different from 0.5 (which is the perfect squareness).29 The obtained value of R for the pure and Mn2+-doped CFO falls in the range of 0.33–0.39, which indicates the randomly oriented single-domain nature of all the samples. Given that the anisotropy symmetry of the prepared NFs is cubic, the M–H loop of the sample is governed by the shape anisotropy of the nanorods. The magnetic moments of the NF samples were obtained in terms of the Bohr magneton per formula unit using 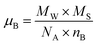 ,60 where MW is the molecular weight of each sample composition, NA is Avogadro's number, and nB is the Bohr magneton. To calculate the magnetic moments, we have to consider the strong interaction between A–B and their distribution of cations.24 The obtained magnetic moment value for all the sample concentrations is listed in Table 13 together with the theoretical values. With the substitution of Mn2+ ions in CFO, the μB started to decrease, which is also evident from the Ms values. This can be well understood as the Mn2+ incorporation in CFO results in a decrease in the A–B interaction. However, the further substitution of Mn2+ in place of Co2+ results in an increase in the A–B interaction and a significant increase in magnetic moment is observed. The magnetic moment values obtained by experimental and theoretical calculation differ slightly given that the NFs do not follow the ideal cation distribution assumed for the theoretical calculations.65,94 Further, the initial permeability (μi) of the pure and Mn2+-doped CFO were obtained as follows:95
,60 where MW is the molecular weight of each sample composition, NA is Avogadro's number, and nB is the Bohr magneton. To calculate the magnetic moments, we have to consider the strong interaction between A–B and their distribution of cations.24 The obtained magnetic moment value for all the sample concentrations is listed in Table 13 together with the theoretical values. With the substitution of Mn2+ ions in CFO, the μB started to decrease, which is also evident from the Ms values. This can be well understood as the Mn2+ incorporation in CFO results in a decrease in the A–B interaction. However, the further substitution of Mn2+ in place of Co2+ results in an increase in the A–B interaction and a significant increase in magnetic moment is observed. The magnetic moment values obtained by experimental and theoretical calculation differ slightly given that the NFs do not follow the ideal cation distribution assumed for the theoretical calculations.65,94 Further, the initial permeability (μi) of the pure and Mn2+-doped CFO were obtained as follows:95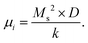 The initial permeability also follows the same trend as the saturation magnetization with Mn2+ substitution and varies in the range of 2.89 to 6.74.
The initial permeability also follows the same trend as the saturation magnetization with Mn2+ substitution and varies in the range of 2.89 to 6.74.
The saturation magnetization (Ms) and magneto-crystalline anisotropy (MCA) constant (K1) were calculated from the M–H loop by fitting the high-field magnetization data (Fig. 16) by the law of approach to saturation (LAS):18
| | 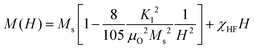 | (64) |
where
| | 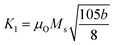 | (65) |
where
M is the magnetization,
H is the externally applied magnetic field, and
χHF – is the high field susceptibility. The constant values of 105/8 in
eqn (64) and (65) are related to the calculations for randomly oriented polycrystalline cubic materials.
23 The calculated values of
Ms and
K1 by ATS fitting are listed in
Table 13. The MCA arises because of the spin–orbital (L–S) coupling of electrons and the strength of spin–orbit coupling is determined by the value of MCA. The value of
K1 primarily depends on the distribution of the magnetic moments of the ions on the surface of MNPs. In the case of cobalt ferrites, they generally have a high MCA due to the presence of Co
2+ ions at the B-site in the spinel ferrite structures, which is evident from
Table 13. For the pure CFO samples, the MCA constant is 1.65 × 10
6 erg cm
−3, which decreased with the Mn
2+ ion concentration and the values lie in the range of 0.96–1.36 × 10
6 erg cm
−3 for the different sample concentrations.
96,97 This decrease can well be understood as in the case of the pure CFO, where the Co
2+ ions generally occupy both the A and B lattice sites and the presence of Co
2+ ions at the B-sites contributes to a higher MCA. With the addition of Mn
2+ ions, they have a strong tendency to occupy the B-sites, and thus substitute the Fe
3+ ions from the A-site to B-site, resulting in a decrease in the MCA and also increase in the
Ms in the Mn
2+-substituted CFO samples. A similar trend in the decrease in MCA for the Mn
2+-substituted CFO samples has also been reported in the literature.
39,45 This decrease in MCA also denotes the presence of individual interactions between the grains of the MNPs. Further, with the reduction in the size of the MNPs, their surface-to-volume ratio increases, which results in a decrease in the surface spin disorder. This decrease in surface spin disorder due to the size reduction of MNPs results in a decrease in the MCA constant.
48
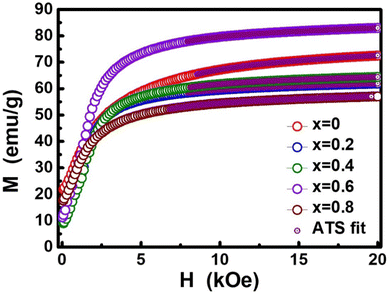 |
| | Fig. 16 ATS fitting (approach to saturation) of the M–H loop of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs. | |
In NFs, the spin canting effect is predominant, and thus the net magnetic moment cannot be explained using the N’eel Brown model because of the non-zero values of the Yafet–Kittel Y–K angles (θYK) for the NF samples. A schematic of the N’eel Brown model, which assumes a 2D collinear arrangement of metal ions, is shown in Fig. 17. However, due to the non-zero θYK angle, the model fails to predict the correct estimate of the magnetic characteristics of the samples. The spin canting effect in the samples results in a non-collinear arrangement of the metal ions, which are at an angle of θYK from each other. This results in an increase in the AB interaction, whereas a decrease in the AA and BB interaction. To study the spin canting effect on the magnetic moment, the Yafet–Kittel (Y–K) model has been proposed.61 The Y–K non-collinear theory suggests that the θYK of the NF samples can be calculated using eqn (66), as follows:
| | 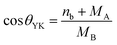 | (66) |
where
nb is the experimentally calculated magnetic moments. The calculated
θYK angles for all the NF composition samples are listed in
Table 13. According to
Table 13, it can be observed the pure CFO has a
θYK of 23.32°, where with Mn
2+ substitution up to
x = 0.2, the
θYK angle decreased to 18.75°. This also explains the higher value of M
s for the pure CFO sample compared to the Co
1−xMn
xFe
2O
4 (
x = 0.2) sample. However, a further increase in Mn
2+ concentration led to an increase in the
θYK angle to 43.48° for the Co
1−xMn
xFe
2O
4 (
x = 0.8) sample. The theoretically calculated bond angle based on the purposed distribution of cations also indicates the enhanced SEI among the magnetic ions with an increase in the concentration of Mn
2+ ions.
46,74 The 3-D non-collinear (Y–K angle) model and collinear 2-D N’eel model of the spin arraignments are shown in
Fig. 17. Further, to investigate the utility of the prepared NFs for high-frequency microwave device applications, we calculated the operating microwave frequency (
ωm) using the following relation:
ωm = 8π
γ2Ms, where
γ = 2.8 MHz Oe
−1 is the gyro-magnetic factor of the NFs. The calculated value of the operating microwave frequency lies in the range of 12.01 to 17.73 GHz, suggesting that the NF samples are highly suitable for high-frequency microwave applications.
42,50
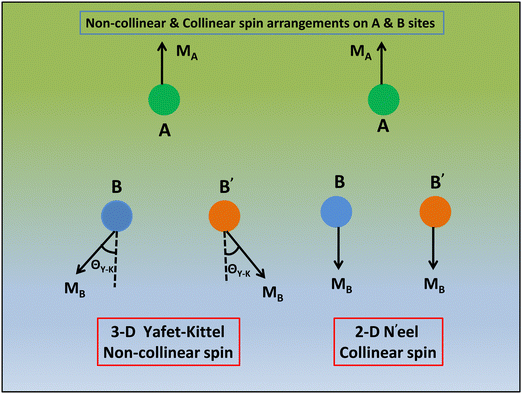 |
| | Fig. 17 Schematic representation of the 3-D non-collinear (Y–K angle) model, collinear 2-D N’eel model of the spin arraignments and Y–K angle (degree) of Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NFs. | |
3.11 Microwave spin resonance investigation using FMR
Ferromagnetic resonance (FMR) spectroscopy is a unique and highly sensitive technique to determine the properties of material systems such as paramagnetic species, oxygen defects, local electronic structure, spin dynamics, and oxidation state. The room temperature FMR spectra of the Co1−xMnxFe2O4 (0 ≤ x ≤ 0.8) NFs were recorded using the X-band (9.8 GHz) microwave frequency, as shown in Fig. 18. The interaction of the high-frequency electromagnetic radiation with the electronic spin results in a resonance, which provides key details that can be used to study the spin dynamics of these material system.24,48 The resonance peak position and its width signify the thermal motion and spins of the lattice. The FMR spectra of all the Co1−xMnxFe2O4 (0 ≤ x ≤ 0.8) NF samples were fitted using a Lorentzian distribution function and different spin dynamic parameters including resonance field (Hr), peak-to-peak line width (ΔHpp), Lande g-factor, spin glass relaxation (Ns), and spin–spin relaxation time (T2) were obtained using eqn (67)–(70)23 and presented in Table 14, as follows:| | 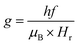 | (67) |
| | 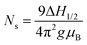 | (68) |
| | 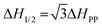 | (69) |
| | 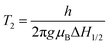 | (70) |
where h is Plank's constant (6.626 × 10−34 J s), f is the electromagnetic radiation frequency, μB is the Bohr magneton (9.27 × 10−24 J T−1), ΔH1/2 is the half of the line width corresponding to the absorption peak. A broad FMR spectrum was observed for all the sample compositions, confirming their strong ferromagnetic behavior. For NFs, when the size of the magnetic nanoparticles is less than the critical diameter, then only N’eel's rotation takes place; otherwise, a bulk rotation is observed, which is generally evident for bulk single-domain particles. Given that the prepared NF samples are in the single-domain range with sizes varying in the range of 20–30 nm, bulk rotation was observed in these materials systems.9,67 For the pure CFO NF, a resonance field of 2398.7 G with a peak-to-peak line width of 3061.9 G was observed, which is consistent with the previously reported values.48 A distorted FMR spectrum was observed for the CFO NF due to the large MCA (magnetic crystalline anisotropy) of the samples. Further, the broadness of the FMR spectra for the ferromagnetic samples is due to the fact that the magnetic moments of larger particles will be prolonged in the easy direction of magnetization, having a larger relaxation time, and thus broadening the ΔHpp. The two major factors that influence the FMR spectrum are magnetic dipole–dipole interactions and SEI among the magnetic and oxygen ions.18 If the width of the FMR spectrum increases, it indicates the dominance of dipolar interactions, whereas a sharp FMR spectrum suggests that the SEI is dominant. With the substitution of Mn2+ ions in the CFO NFs, for the Co1−xMnxFe2O4 (x = 0.2) sample, the sharpness of its FMR spectrum increased, as evident from Fig. 18; however, the resonance field shifted towards the higher magnetic field of 3497.6 G with ΔHpp of 925.9 G. This can be attributed to the increase in particle size, which resulted in an interaction between the individual domains separated by a domain wall.20,98 A further increase in the Mn2+ concentration led to an increase in the broadness of the FMR spectrum; however, the resonance field decreased for the Co1−xMnxFe2O4 (x = 0.4 and 0.6) samples and increased for the Co1−xMnxFe2O4 (x = 0.8) sample concentration. The change in the resonance field and peak-to-peak width of the Mn2+-substituted NFs can also be correlated with the cation distribution and change in the crystallite size. The substation of the Mn2+ ions at the B-sites results in a reduction in the MCA, which also contributed to the reduction in the resonance field value. The increase in the broadness of the peaks can further be correlated with the strong dipole–dipole interaction between the Fe3+–O2−Co2+ ions.73
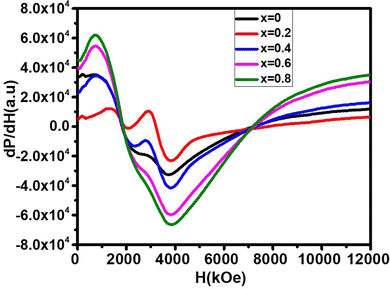 |
| | Fig. 18 Room temperature FMR spectra of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples. The broad FMR spectrum for all the samples indicates a high MCA, which was reduced with an increase in the Mn2+ concentration in the cobalt ferrite. | |
Table 14 Room temperature spin resonance parameters of the Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF samples
| Sample name |
H
r (G) |
ΔHPP (G) |
ΔH1/2 (G) |
Lande g-factor |
T
2 × (10−12) s |
N
s × 1022 (spin per g) |
|
X = 0 |
2398.7 |
3061.9 |
5303.3 |
2.40 |
8.92 |
5.43
|
|
X = 0.2 |
3497.6 |
925.9 |
1603.7 |
2.01 |
3.53 |
1.96 |
|
X = 0.4 |
3006.9 |
3034.4 |
5255.7 |
2.34 |
9.25 |
5.25 |
|
X = 0.6 |
2707.8 |
3061.8 |
5303.1 |
2.60 |
8.25 |
5.02 |
|
X = 0.8 |
2854.6 |
3080.2 |
5335.0 |
2.82 |
7.56 |
4.65 |
The g-factor of all the NF sample compositions was calculated using eqn (67) and depicted in Table 14. According to eqn (67), it is evident that the g-factor varies inversely to the resonance field and varies from 2.01 (for x = 0.2) to 2.82 (for x = 0.8) in the samples, while for the pure CFO, the g-factor is 2.40. This is due to the strengthening of the magnetic moment and stronger dipole–dipole interaction between the magnetic ions through the O2− ions, leading to an increase in the g-value with an increase in Mn2+ concentration.23 Further, the anisotropic arrangement of magnetic moments also led to an increase in the g-factor value. The spin concentration of the NF samples with different concentrations was obtained using eqn (68)24 and listed in Table 14. The spin concentration of the pure CFO sample is 5.43 × 1022 spin per g, which sharply decreased for the Co1−xMnxFe2O4 (x = 0.2) sample to 1.96 × 1022 spin per g. However, a further increase in the Mn2+ concentration (Co1−xMnxFe2O4 (x = 0.4)) sharply increased the spin concentration, which again started to decrease for the Co1−xMnxFe2O4 (x = 0.6 and 0.8) samples. Further, the spin–spin relaxation time quantifies the rate of absorption or dissipation of the microwave energy by the NF system and is a function of the applied magnetic field. The spin–spin relaxation time is correlated with the peak-to-peak line width of the resonance signal. The relaxation time was calculated using eqn (69) and (70) and its values are in the range of 3.53 to 9.25 Ps, which are listed in Table 14. It was observed that initially, the relaxation time decreased from 8.92 Ps for the pure cobalt ferrites to 3.53 Ps for the Co1−xMnxFe2O4 (x = 0.2) sample. However, a further increase in Mn2+ concentration resulted in an increase in the relaxation time to 9.25 Ps for the concentration of x = 0.4 and further decreased to 7.56 Ps at a higher Mn2+ concentration (x = 0.8). The spin-spin relation time of the Mn2+-substituted CFO samples is in Ps, which makes them ideal candidates for several applications such as magnetic switches, magneto-optical devices, magnetic chips, and sensors.9,61,63
4. Conclusion
Herein, we reported the synthesis of optically active Co–Mn ferrites, which usually do not show intrinsic fluorescence. NFs are widely used in many biomedical applications; however, they are generally surfaced functionalized using external fluorescence probes to impart fluorescence characteristics. In this work, we observed intrinsic fluorescence in the Co–Mn NFs due to the cation distribution and increase in the defect concentration of the hydrothermally synthesized Co1−xMnxFe2O4 (0.8 ≤ x ≤ 0) NF single-domain particles. To understand the correlation between the optical properties and structure of the Co–MN NFs, detailed structural modeling was carried out by Rietveld refinement of the XRD patterns of each sample. Moreover, the cation distribution indicates that the Mn2+-substituted CoFe2O4 samples are partially inverse spinel ferrites, where the Mn2+ ions primarily occupy the B-sites. The substitution of the Mn2+ ions initially resulted in the weakening of the A–B interaction, resulting in a decrease in the saturation magnetization initially; however, further substation of the Mn2+ ions significantly improved the saturation magnetization from 59.88 to 80.30 emu g−1. Further, the reduction in MCA and improved magnetic properties with optimized Mn2+ ion substitution indicate that the cation distribution plays a key role in controlling the properties of the NFs. The increase in the defect concentration also serves as the major reason for the observation of fluorescence characteristics, which can be related to the synthetic method. The total vacancy concentration increased significantly from 4.38 to 7.86 with the Mn2+ ion substitution. Further, the TRPL investigation showed two decay times, which can be correlated with the two emission spectra observed for all the NF samples. The intensity of the two emission bands, i.e., blue and violet, observed for all the samples increased significantly with the substitution of Mn2+ ions. The decay time of all the samples is in the range of 2–9 ns, which makes them a suitable candidate for optoelectronic devices. Further, the operating frequency range of the Mn2+ ion-substituted CoFe2O4 is in the range of 12–17 GHz, which is ideal for high-frequency microwave devices. The TEM images of all the samples depict the spherical shape of all the samples and their particle size in the range of 15–35nm, which confirm the single-domain nature of the samples. A single broad FMR spectrum was observed for the samples, which indicates ferromagnetic behavior, and with the incorporation of Mn2+ ions, the reduction in MCA resulted in a more symmetrical FMR spectrum. Further, we theoretically calculated the different bond angles and lengths, which indicate the strengthening of the A–A and A–B interaction; however, the BB interaction decreased. All the samples showed strong ferromagnetic characteristics and the optimized substitution of Mn2+ ions resulted in an improvement in the saturation magnetization from 68.51 to 80.30 emu g−1. Further, the net magnetic moment in the prepared samples was explained using the Yafet-Kittel model given that the N’eel Brown model assumes a 2D collinear arrangement of the metal ions. The spin canting effect is predominant in the prepared samples and results in a non-collinear arrangement of metal ions and N’eel Brown model fails to predict the magnetic behavior appropriately. The detailed investigation of Co–Mn ferrites opens a new horizon for the application of these materials in biomedical applications and magneto-optical devices using their magnetically controlled optical properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to the Director, CSIR-NPL for his encouragement to carry out this research work. Prashant Kumar is grateful to University Grant Commission (UGC), India for Senior Research Fellowship (SRF), Ref No. (19/06/2016(i) EU-V) to carry out this research. H. Khanduri acknowledges the Department of Science and Technology, New Delhi, India, for DST INSPIRE Faculty award [DST/INSPIRE/04/2017/002826]. Also, the authors are thankful to the CSIR, New Delhi, India for a research grant under the Emeritus scientist scheme to carry out this research work. S. Pathak and S. K. Kim acknowledges National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant No. NRF-2021R1A2C2013543) and Institute of Engineering Research at Seoul National University for additional fund support.
References
- R. Verma, S. Pathak, A. K. Srivastava, S. Prawer and S. Tomljenovic-Hanic, J. Alloys Compd., 2021, 876, 160175 CrossRef CAS.
- S. Gbadamasi, M. Mohiuddin, V. Krishnamurthi, R. Verma, M. W. Khan, S. Pathak, K. Kalantar-Zadeh and N. Mahmood, Chem. Soc. Rev., 2021, 50, 4684–4729 RSC.
- A. Mishra, S. Pathak, P. Kumar, A. Singh, K. Jain, R. Chaturvedi, D. Singh, G. A. Basheed and R. P. Pant, IEEE Trans. Magn., 2019, 55, 1–7 Search PubMed.
- T. Dippong, E. A. Levei, F. Goga, I. Petean, A. Avram and O. Cadar, J. Sol-Gel Sci. Technol., 2019, 92, 736–744 CrossRef CAS.
- M. Stefanescu, M. Stoia, T. Dippong, O. Stefanescu and P. Barvinschi, Acta Chim. Slov., 2009, 59, 379–385 Search PubMed.
- M. Stefanescu, M. Stoia, C. Caizer, T. Dippong and P. Barvinschi, J. Therm. Anal. Calorim., 2009, 97, 245 CrossRef CAS.
- S. Pathak, R. Verma, P. Kumar, A. Singh, S. Singhal, P. Sharma, K. Jain, R. P. Pant and X. Wang, Nanomaterials, 2021, 11, 3009 CrossRef CAS PubMed.
- S. Pathak, K. Jain, P. Kumar, X. Wang and R. P. Pant, Appl. Energy, 2019, 239, 1524–1535 CrossRef.
- P. Kumar, S. Pathak, A. Singh, H. Khanduri, K. Jain, J. Tawale, L. Wang, G. Basheed and R. Pant, J. Alloys Compd., 2021, 887, 161418 CrossRef CAS.
- S. P. Dalawai, S. Kumar, M. A. S. Aly, M. Khan, H. Zaved, R. Xing, P. N. Vasambekar and S. Liu, J. Mater. Sci.: Mater. Electron., 2019, 30, 7752–7779 CrossRef CAS.
- P. Das, M. Colombo and D. Prosperi, Colloids Surf., B, 2019, 174, 42–55 CrossRef CAS PubMed.
- M. Pardavi-Horvath, J. Magn. Magn. Mater., 2000, 215, 171–183 CrossRef.
- S. Pathak, R. Zhang, B. Gayen, V. Kumar, H. Zhang, R. Pant and X. Wang, Sustainable Energy Technol. Assess., 2022, 52, 102024 CrossRef.
- S. Pathak, R. Zhang, K. Bun, H. Zhang, B. Gayen and X. Wang, Sustainable Energy Technol. Assess., 2021, 48, 101641 CrossRef.
- P. Sharma, V. Alekhya, S. Pathak, K. Jain, P. Tomar, G. Basheed, K. Maurya and R. Pant, J. Magn. Magn. Mater., 2021, 534, 168024 CrossRef CAS.
- S. Pathak, K. Jain, V. Kumar and R. P. Pant, IEEE Sens. J., 2017, 17, 2670–2675 CAS.
- J. Li, A. Khalid, R. Verma, A. Abraham, F. Qazi, X. Dong, G. Liang and S. Tomljenovic-Hanic, Nanomaterials, 2021, 11, 695 CrossRef CAS PubMed.
- P. Kumar, S. Pathak, K. Jain, A. Singh, G. Basheed and R. Pant, J. Alloys Compd., 2022, 904, 163992 CrossRef CAS.
- J.-H. Lee, Y. Kim and S.-K. Kim, Sci. Rep., 2022, 12, 5232 CrossRef CAS PubMed.
- P. Monisha, P. Priyadharshini, S. Gomathi and K. Pushpanathan, J. Alloys Compd., 2021, 856, 157447 CrossRef CAS.
- K. Jain, S. Pathak and R. Pant, RSC Adv., 2016, 6, 70943–70946 RSC.
- J.-H. Lee, B. Kim, Y. Kim and S.-K. Kim, Sci. Rep., 2021, 11, 4969 CrossRef CAS PubMed.
- A. Singh, S. Pathak, P. Kumar, P. Sharma, A. Rathi, G. Basheed, K. Maurya and R. P. Pant, J. Magn. Magn. Mater., 2020, 493, 165737 CrossRef CAS.
- P. Kumar, H. Khanduri, S. Pathak, A. Singh, G. Basheed and R. Pant, Dalton Trans., 2020, 49, 8672–8683 RSC.
- V. Jeseentharani, M. George, B. Jeyaraj, A. Dayalan and K. Nagaraja, J. Exp. Nanosci., 2013, 8, 358–370 CrossRef CAS.
- H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 2476–2487 CrossRef CAS PubMed.
- J. Philip and J. M. Laskar, J. Nanofluids, 2012, 1, 3–20 CrossRef CAS.
- I. Sharifi, H. Shokrollahi and S. Amiri, J. Magn. Magn. Mater., 2012, 324, 903–915 CrossRef CAS.
- F. Liu, S. Laurent, A. Roch, L. Vander Elst and R. N. Muller, J. Nanomater., 2013, 462540 Search PubMed.
- R. Kershi, Mater. Chem. Phys., 2020, 248, 122941 CrossRef CAS.
- J. Msomi, H. Abdallah, T. Moyo and A. Lančok, J. Magn. Magn. Mater., 2011, 323, 471–474 CrossRef CAS.
- S. Pathak, R. Verma, S. Singhal, R. Chaturvedi, P. Kumar, P. Sharma, R. P. Pant and X. Wang, Sci. Rep., 2021, 11, 3799 CrossRef CAS PubMed.
- P. Kumar, S. Pathak, A. Singh, H. Khanduri, X. Wang, G. Basheed and R. P. Pant, Mater. Chem. Phys., 2021, 265, 124476 CrossRef CAS.
- A. Manikandan, M. Durka and S. A. Antony, J. Supercond. Novel Magnet., 2014, 27, 2841–2857 CrossRef CAS.
- E. Hema, A. Manikandan, P. Karthika, S. A. Antony and B. Venkatraman, J. Supercond. Novel Magnet., 2015, 28, 2539–2552 CrossRef CAS.
- A. L. Gurgel, A. E. Martinelli, O. L. de Aquino Conceição, M. M. Xavier Jr, M. A. M. Torres and D. M. de Araújo Melo, J. Alloys Compd., 2019, 799, 36–42 CrossRef CAS.
- Y. Gao, Y. Zhao, Q. Jiao and H. Li, J. Alloys Compd., 2013, 555, 95–100 CrossRef CAS.
- B. Issa, I. M. Obaidat, B. A. Albiss and Y. Haik, Int. J. Mol. Sci., 2013, 14, 21266–21305 CrossRef CAS PubMed.
- A. Salunkhe, V. Khot, M. R. Phadatare, N. Thorat, R. Joshi, H. Yadav and S. Pawar, J. Magn. Magn. Mater., 2014, 352, 91–98 CrossRef CAS.
- P. Monisha, P. Priyadharshini, S. S. Gomathi and K. Pushpanathan, J. Phys. Chem. Solids, 2021, 148, 109654 CrossRef CAS.
- N. F. Andrade Neto, L. E. Nascimento, M. Correa, F. Bohn, M. R. D. Bomio and F. V. Motta, Mater. Chem. Phys., 2020, 242, 122489 CrossRef CAS.
- S. M. Ansari, K. C. Ghosh, R. S. Devan, D. Sen, P. U. Sastry, Y. D. Kolekar and C. Ramana, ACS Omega, 2020, 5, 19315–19330 CrossRef CAS PubMed.
- P. Balakrishnan and P. Veluchamy, J. Appl. Phys., 2016, 8, 19–29 Search PubMed.
- N. Adeela, K. Maaz, U. Khan, S. Karim, A. Nisar, M. Ahmad, G. Ali, X. Han, J. Duan and J. Liu, J. Alloys Compd., 2015, 639, 533–540 CrossRef CAS.
- Y. Köseoğlu, F. Alan, M. Tan, R. Yilgin and M. Öztürk, Ceram. Int., 2012, 38, 3625–3634 CrossRef.
- M. Atif, R. S. Turtelli, R. Grössinger, M. Siddique and M. Nadeem, Ceram. Int., 2014, 40, 471–478 CrossRef CAS.
- P. Kumar, S. Pathak, A. Singh, Kuldeep, H. Khanduri, X. Wang, G. A. Basheed and R. P. Pant, Mater. Chem. Phys., 2021, 124476, DOI:10.1016/j.matchemphys.2021.124476.
- P. Kumar, S. Pathak, A. Singh, H. Khanduri, G. A. Basheed, L. Wang and R. P. Pant, Nanoscale Adv., 2020, 2, 1939–1948 RSC.
- P. Monisha, P. Priyadharshini, S. Gomathi and K. Pushpanathan, J. Phys. Chem. Solids, 2021, 148, 109654 CrossRef CAS.
- S. Yadav, S. Shinde, P. Bhatt, S. Meena and K. Rajpure, J. Alloys Compd., 2015, 646, 550–556 CrossRef CAS.
- S. Pathak, K. Jain and R. Pant, Colloids Surf., A, 2018, 539, 273–279 CrossRef.
- Noorjahan, G. A. Basheed, K. Jain, S. Pathakd and R. P. Pant, J. Nanosci. Nanotechnol., 2018, 18, 2746–2751 CrossRef CAS PubMed.
- R. Verma, S. Pathak, K. K. Dey, S. Sikarwar, B. C. Yadav and A. K. Srivastava, Nanoscale Adv., 2022, 4, 2902–2912 RSC.
- T. Dippong, E.-A. Levei, C. L. Lengauer, A. Daniel, D. Toloman and O. Cadar, Mater. Charact., 2020, 163, 110268 CrossRef CAS.
- T. Dippong, E. A. Levei, O. Cadar, F. Goga, G. Borodi and L. Barbu-Tudoran, J. Therm. Anal. Calorim., 2017, 128, 39–52 CrossRef CAS.
- M. El-Okr, M. Salem, M. Salim, R. El-Okr, M. Ashoush and H. Talaat, J. Magn. Magn. Mater., 2011, 323, 920–926 CrossRef CAS.
- T. Dippong, E. A. Levei, O. Cadar, A. Mesaros and G. Borodi, J. Anal. Appl. Pyrolysis, 2017, 125, 169–177 CrossRef CAS.
- S. Talukdar, D. Mandal and K. Mandal, Chem. Phys. Lett., 2017, 672, 57–62 CrossRef CAS.
- N. M. Basith, J. J. Vijaya, L. J. Kennedy and M. Bououdina, Mater. Sci. Semicond. Process., 2014, 17, 110–118 CrossRef CAS.
- A. Singh, S. Pathak, P. Kumar, P. Sharma, A. Rathi, G. A. Basheed, K. K. Maurya and R. P. Pant, J. Magn. Magn. Mater., 2020, 493, 165737 CrossRef CAS.
- R. Sharma, P. Thakur, P. Sharma and V. Sharma, J. Alloys Compd., 2017, 704, 7–17 CrossRef CAS.
- R. Safi, A. Ghasemi, R. Shoja-Razavi, E. Ghasemi and T. Sodaee, Ceram. Int., 2016, 42, 6375–6382 CrossRef CAS.
- S. Chakrabarty, A. Dutta and M. Pal, J. Alloys Compd., 2015, 625, 216–223 CrossRef CAS.
- T. R. Tatarchuk, N. D. Paliychuk, M. Bououdina, B. Al-Najar, M. Pacia, W. Macyk and A. Shyichuk, J. Alloys Compd., 2018, 731, 1256–1266 CrossRef CAS.
- R. Jabbar, S. H. Sabeeh and A. M. Hameed, J. Magn. Magn. Mater., 2020, 494, 165726 CrossRef CAS.
- K. Mohammed, A. Al-Rawas, A. Gismelseed, A. Sellai, H. Widatallah, A. Yousif, M. Elzain and M. Shongwe, Phys. B: Condens. Matter, 2012, 407, 795–804 CrossRef CAS.
- M. Deepty, C. Srinivas, E. R. Kumar, N. K. Mohan, C. Prajapat, T. C. Rao, S. S. Meena, A. K. Verma and D. Sastry, Ceram. Int., 2019, 45, 8037–8044 CrossRef CAS.
- L. Kumar, P. Kumar and M. Kar, J. Alloys Compd., 2013, 551, 72–81 CrossRef CAS.
- R. Waldron, Phys. Rev., 1955, 99, 1727 CrossRef CAS.
- E. V. Gopalan, P. Joy, I. Al-Omari, D. S. Kumar, Y. Yoshida and M. Anantharaman, J. Alloys Compd., 2009, 485, 711–717 CrossRef CAS.
- S. Joshi, V. B. Kamble, M. Kumar, A. M. Umarji and G. Srivastava, J. Alloys Compd., 2016, 654, 460–466 CrossRef CAS.
- T. Tatarchuk, M. Bououdina, W. Macyk, O. Shyichuk, N. Paliychuk, I. Yaremiy, B. Al-Najar and M. Pacia, Nanoscale Res. Lett., 2017, 12, 1–11 CrossRef CAS PubMed.
- S. Pathak, R. Verma, P. Kumar, A. Singh, S. Singhal, P. Sharma, K. Jain, R. P. Pant and X. Wang, Nanomaterials, 2021, 11, 3009 CrossRef CAS PubMed.
- A. Bajorek, C. Berger, M. Dulski, P. Łopadczak, M. Zubko, K. Prusik, M. Wojtyniak, A. Chrobak, F. Grasset and N. Randrianantoandro, J. Phys. Chem. Solids, 2019, 129, 1–21 CrossRef CAS.
- R. Verma, J. Gangwar and A. K. Srivastava, RSC Adv., 2017, 7, 44199–44224 RSC.
- R. Verma, S. Singh, M. K. Dalai, M. Saravanan, V. V. Agrawal and A. K. Srivastava, Mater. Des., 2017, 133, 10–18 CrossRef CAS.
- N. Marwaha, B. K. Gupta, R. Verma and A. K. Srivastava, J. Mater. Sci., 2017, 52, 10480–10484 CrossRef CAS.
- R. Verma, A. Awasthi, P. Singh, R. Srivastava, H. Sheng, J. Wen, D. J. Miller and A. K. Srivastava, J. Colloid Interface Sci., 2016, 475, 82–95 CrossRef CAS PubMed.
- T. Dippong, D. Toloman, E.-A. Levei, O. Cadar and A. Mesaros, Thermochim. Acta, 2018, 666, 103–115 CrossRef CAS.
- T. Dippong, E.-A. Levei, I. G. Deac, F. Goga and O. Cadar, J. Anal. Appl. Pyrolysis, 2019, 144, 104713 CrossRef CAS.
- M. M. N. Ansari and S. Khan, Phys. B: Condens. Matter, 2017, 520, 21–27 CrossRef CAS.
- Suman, S. Chahal, A. Kumar and P. Kumar, Crystals, 2020, 10, 273 CrossRef CAS.
- A. Manikandan, L. J. Kennedy, M. Bououdina and J. J. Vijaya, J. Magn. Magn. Mater., 2014, 349, 249–258 CrossRef CAS.
- S. Agrawal, A. Parveen and A. Azam, J. Magn. Magn. Mater., 2016, 414, 144–152 CrossRef CAS.
- J. Chen, Y. Wang and Y. Deng, J. Alloys Compd., 2013, 552, 65–69 CrossRef CAS.
- P. Monisha, P. Priyadharshini, S. Gomathi, M. Mahendran and K. Pushpanathan, Appl. Phys. A, 2019, 125, 1–16 CrossRef CAS.
- A. Ravindra, M. Chandrika, C. Rajesh, P. Kollu, S. Ju and S. Ramarao, Eur. Phys. J. Plus, 2019, 134, 1–10 CrossRef.
- J. Revathi, M. J. Abel, C. L. Pearline, T. Sumithra and P. F. H. Inbaraj, Inorg. Chem. Commun., 2020, 121, 108186 CrossRef CAS.
- J. Yang, E.-K. Lim, H. J. Lee, J. Park, S. C. Lee, K. Lee, H.-G. Yoon, J.-S. Suh, Y.-M. Huh and S. Haam, Biomaterials, 2008, 29, 2548–2555 CrossRef CAS PubMed.
- N. Jain, N. Marwaha, R. Verma, B. K. Gupta and A. K. Srivastava, RSC Adv., 2016, 6, 4960–4968 RSC.
- M. Pal, R. Rakshit and K. Mandal, ACS Appl. Mater. Interfaces, 2014, 6, 4903–4910 CrossRef CAS PubMed.
- T. Dippong, E. A. Levei and O. Cadar, Materials, 2021, 14, 1139 CrossRef CAS PubMed.
- T. Dippong, O. Cadar, E. A. Levei, C. Leostean and L. Barbu Tudoran, Ceram. Int., 2017, 43, 9145–9152 CrossRef CAS.
- S. Kakati, M. Rendale and S. Mathad, Int. J. Self-Propag. High-Temp. Synth., 2021, 30, 189–219 CrossRef CAS.
- R. S. Yadav, I. Kuřitka, J. Vilcakova, J. Havlica, L. Kalina, P. Urbánek, M. Machovsky, D. Skoda, M. Masař and M. Holek, Ultrason. Sonochem., 2018, 40, 773–783 CrossRef CAS PubMed.
- A. J. Rondinone, A. C. Samia and Z. J. Zhang, Appl. Phys. Lett., 2000, 76, 3624–3626 CrossRef CAS.
- J. Slonczewski, Phys. Rev., 1958, 110, 1341 CrossRef CAS.
- Y. Sánchez, S. Briceño, J. Larionova, J. Long, Y. Guari and P. Silva, J. Mater. Res., 2021, 36, 3329–3338 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |

 *d,
Arjun
Singh
be,
Komal
Jain
*d,
Arjun
Singh
be,
Komal
Jain
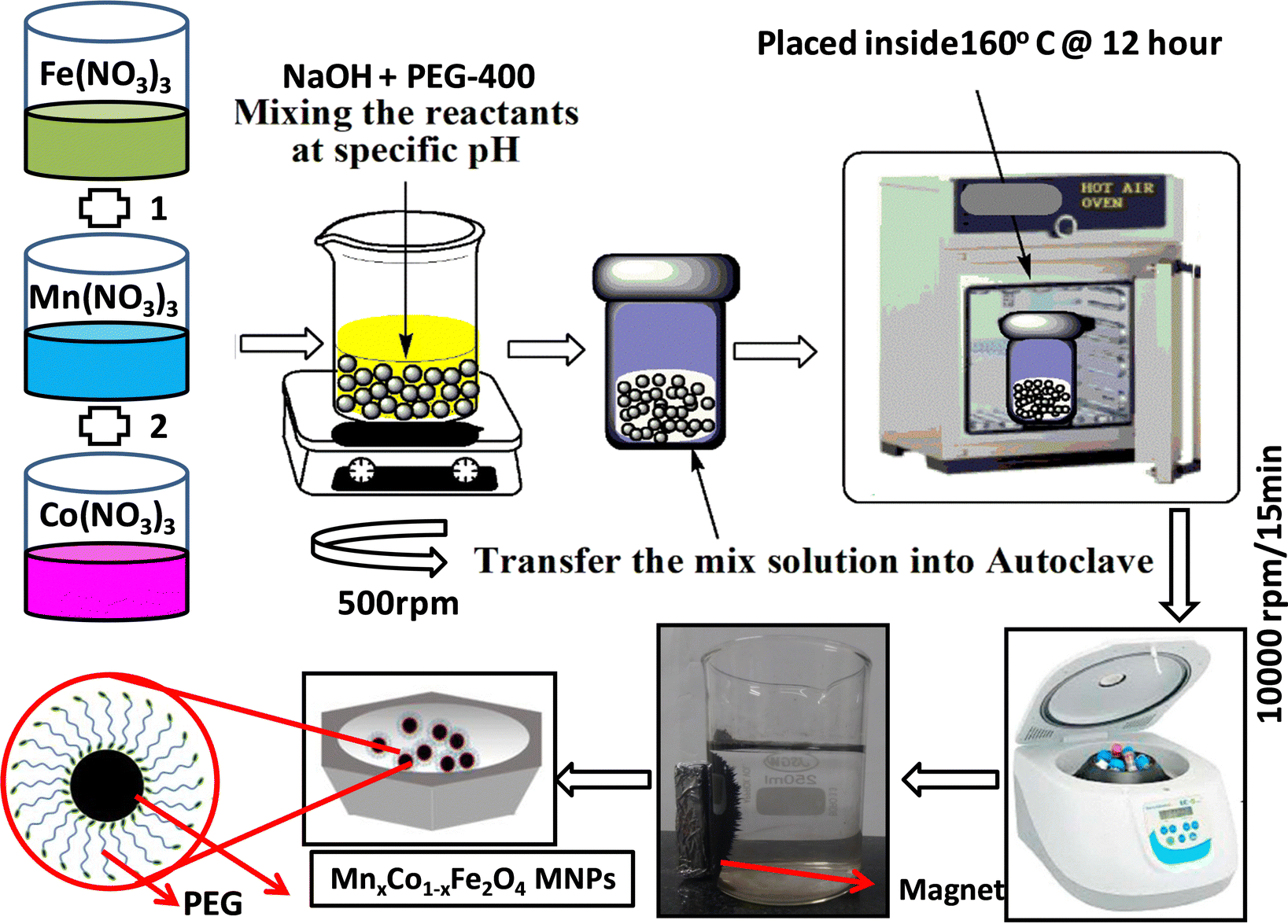
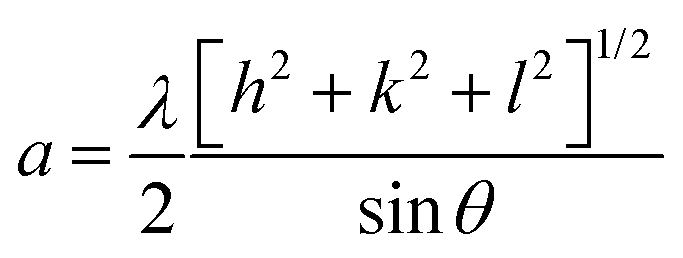
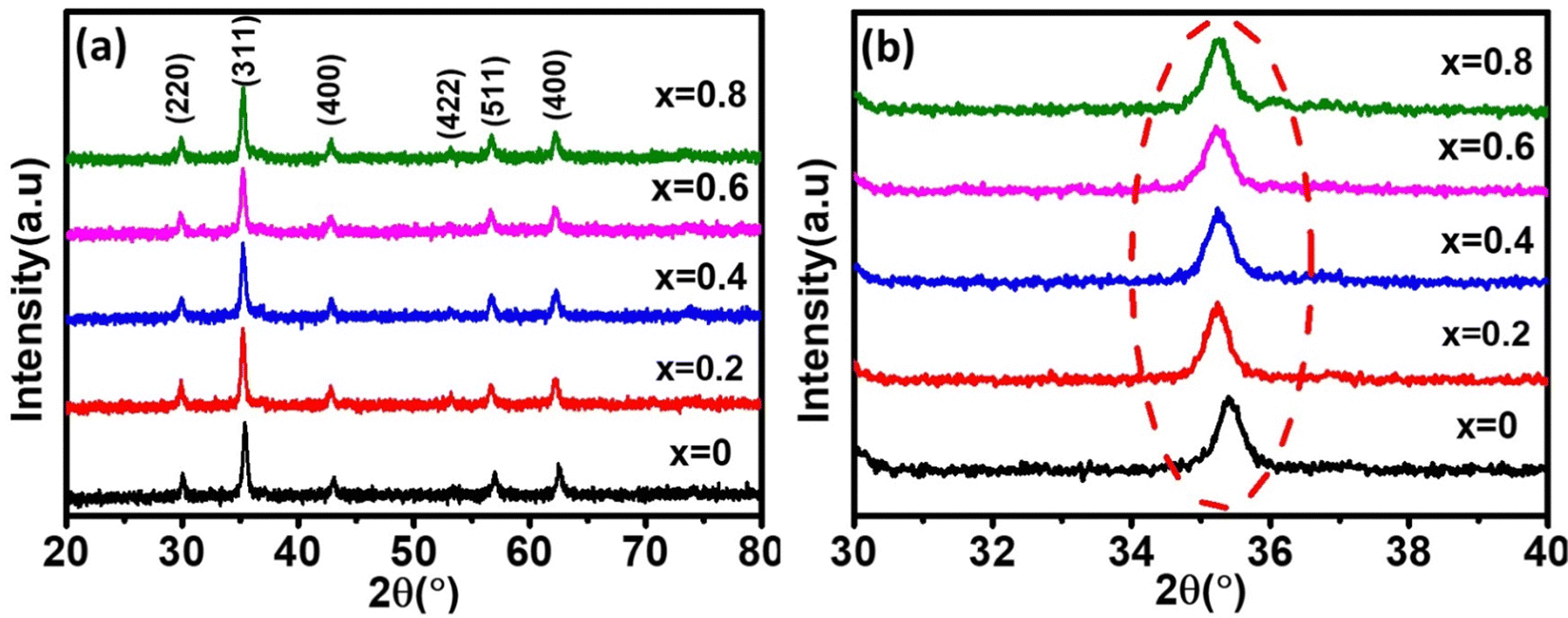
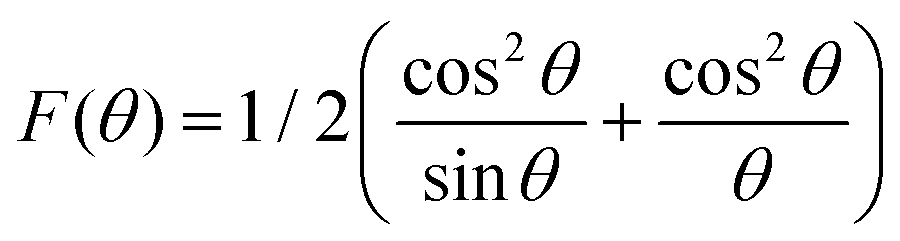
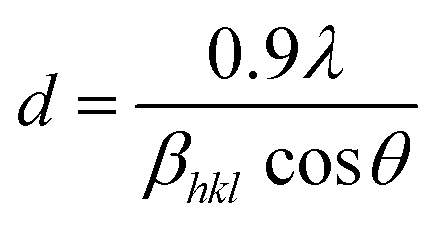
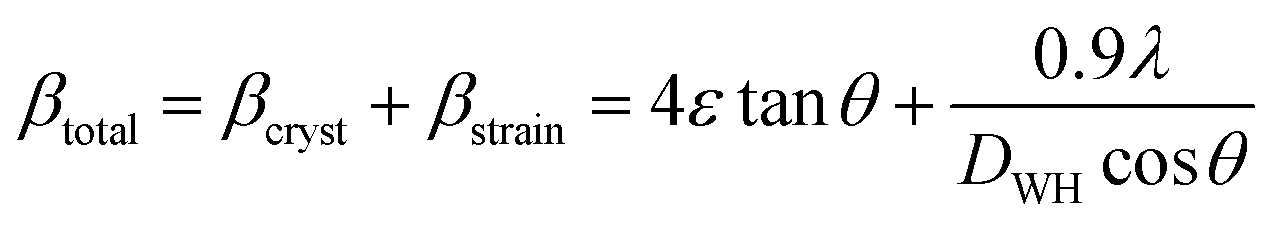
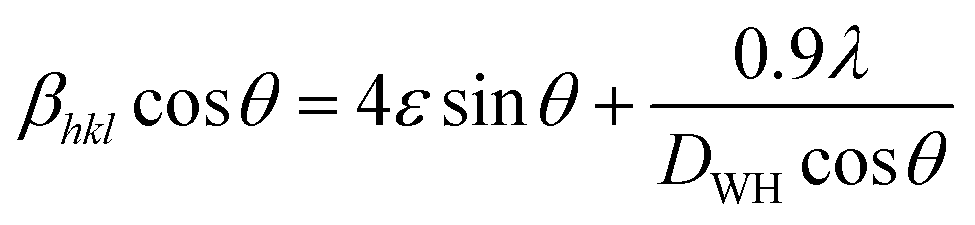
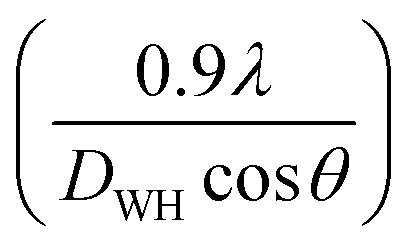 and the slope of the sample corresponds to the induced strain in the samples.54,55 It was observed that the calculated average crystallite size of the CFO increased from 23.56 nm (x = 0) to 26.89 nm (x = 0.8) with an increase in the Mn2+ ion concentration using the W–H method, as seen in Table 1. The crystallite size values obtained using the D–S method show an almost similar trend, as shown in Fig. 4(a) and Table 1.56 Further, the induced strain in the sample increased with an increase in the Mn concentration, which is obvious as the larger atoms (Mn2+) substituted the smaller atoms (Co2+), resulting in an increase in strain (from 9.9 × 10−3 to 15.2 × 10−3) in the samples. The positive slope of the W–H plot indicates the tensile strain in all the sample compositions given that the growth was restricted due to the larger ion substitution, which resulted in outward strain.57 Koseoglu et al. also reported a similar trend of increasing crystallite size upon an increase in Mn2+ concentration. Further, the theoretical (ρx) and experimental X-ray density (ρb) of the samples were obtained using eqn (6) and (7),24,58 as follows:
and the slope of the sample corresponds to the induced strain in the samples.54,55 It was observed that the calculated average crystallite size of the CFO increased from 23.56 nm (x = 0) to 26.89 nm (x = 0.8) with an increase in the Mn2+ ion concentration using the W–H method, as seen in Table 1. The crystallite size values obtained using the D–S method show an almost similar trend, as shown in Fig. 4(a) and Table 1.56 Further, the induced strain in the sample increased with an increase in the Mn concentration, which is obvious as the larger atoms (Mn2+) substituted the smaller atoms (Co2+), resulting in an increase in strain (from 9.9 × 10−3 to 15.2 × 10−3) in the samples. The positive slope of the W–H plot indicates the tensile strain in all the sample compositions given that the growth was restricted due to the larger ion substitution, which resulted in outward strain.57 Koseoglu et al. also reported a similar trend of increasing crystallite size upon an increase in Mn2+ concentration. Further, the theoretical (ρx) and experimental X-ray density (ρb) of the samples were obtained using eqn (6) and (7),24,58 as follows: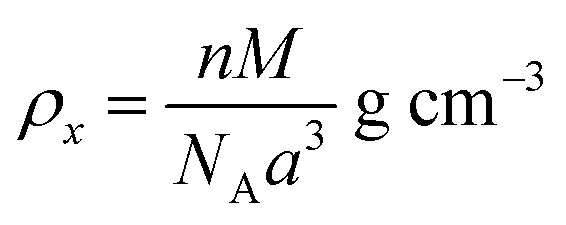
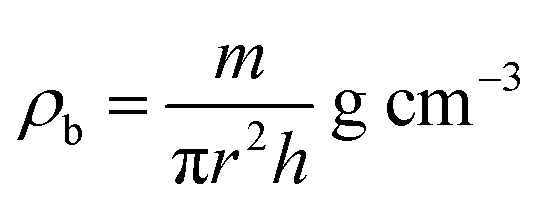
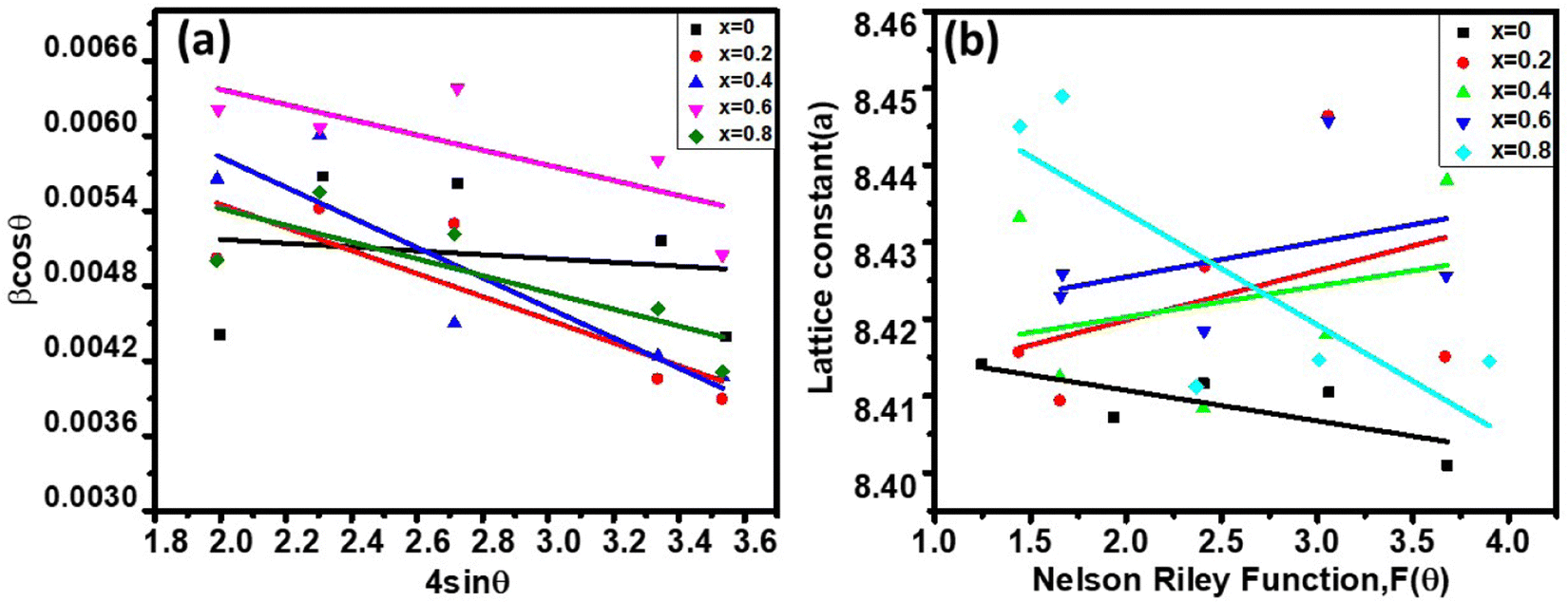
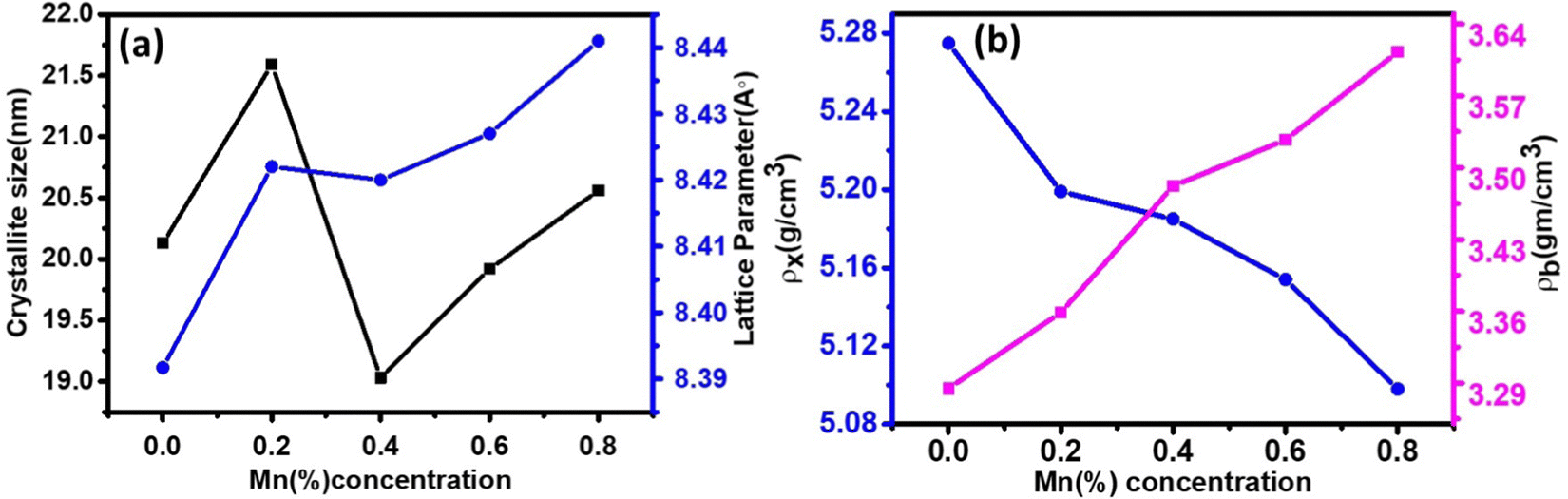
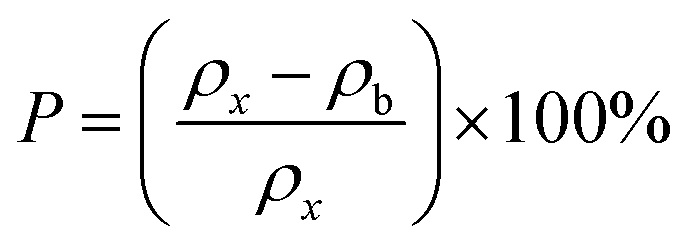
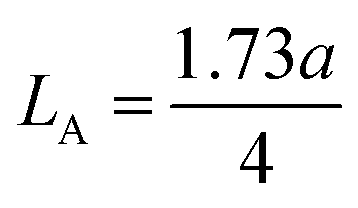
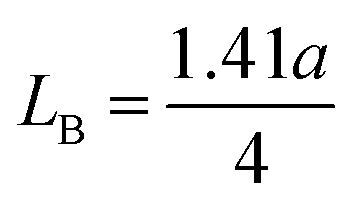
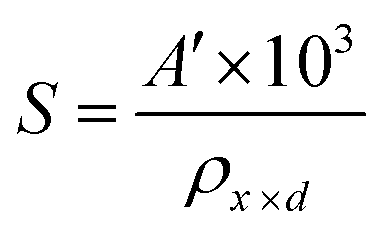
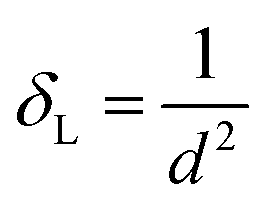
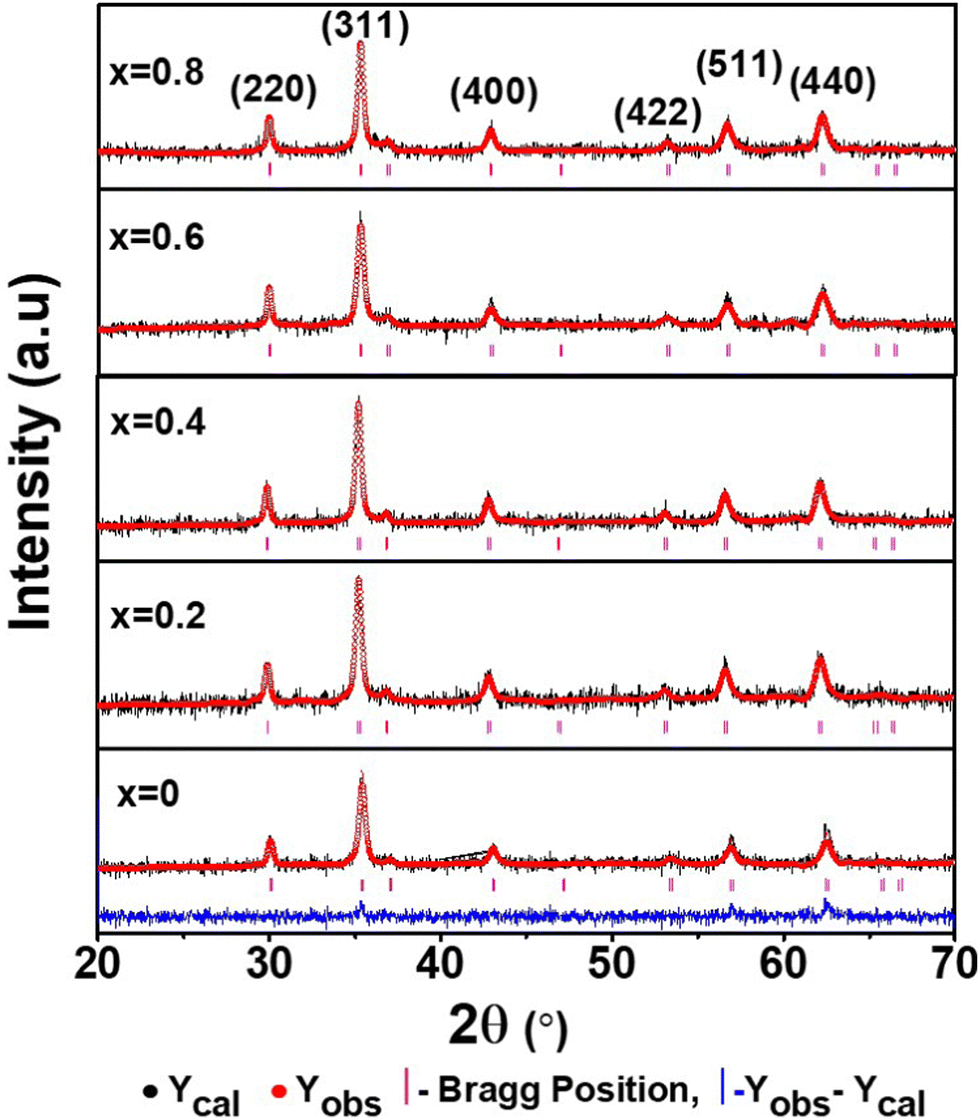
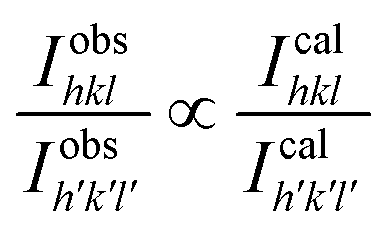
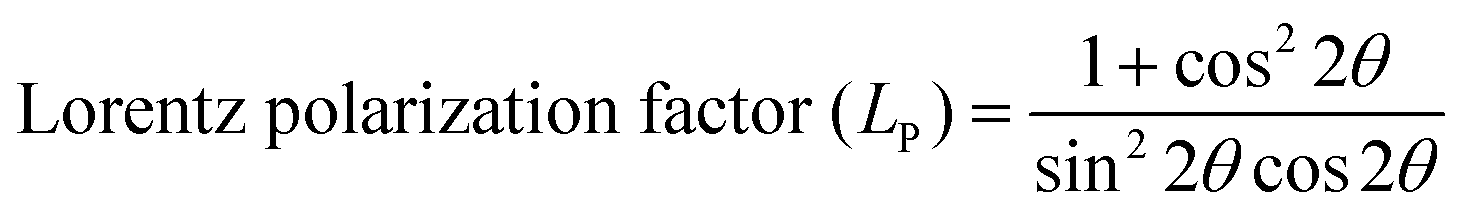
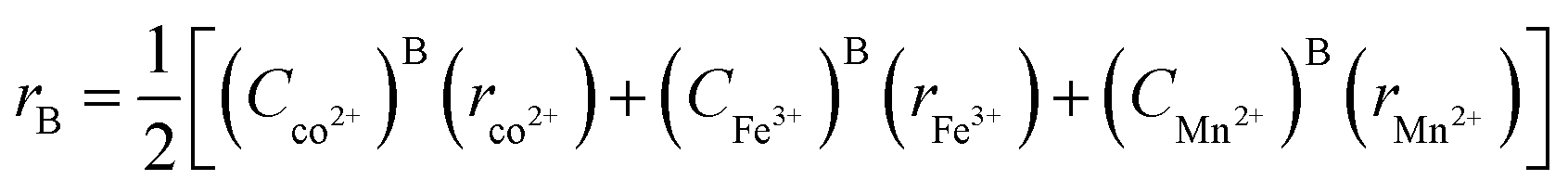
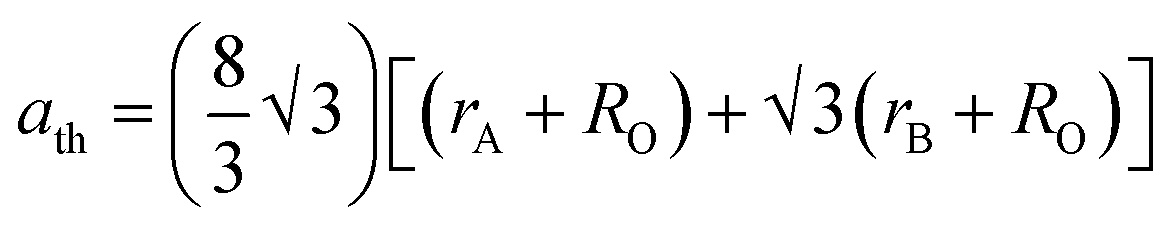
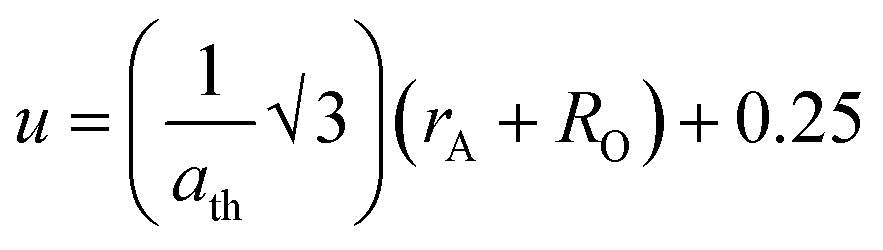
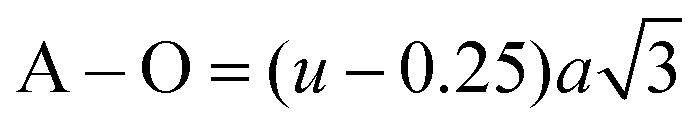
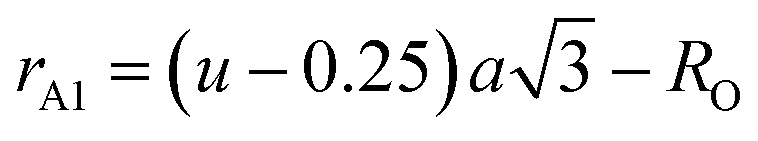
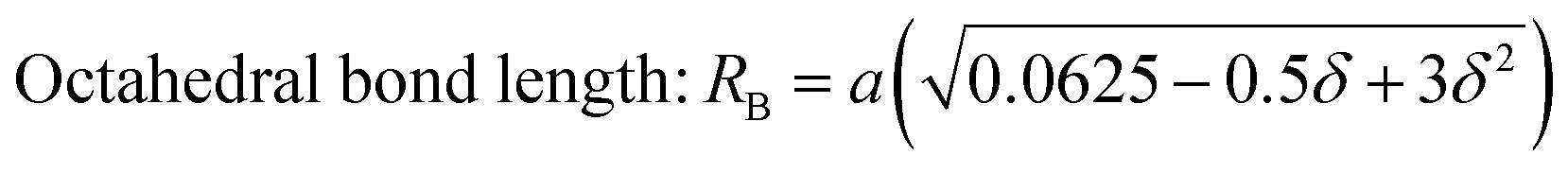
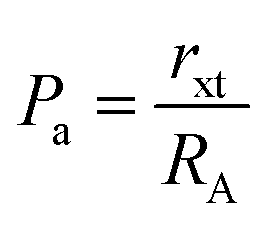
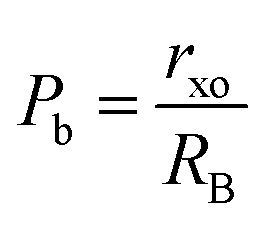
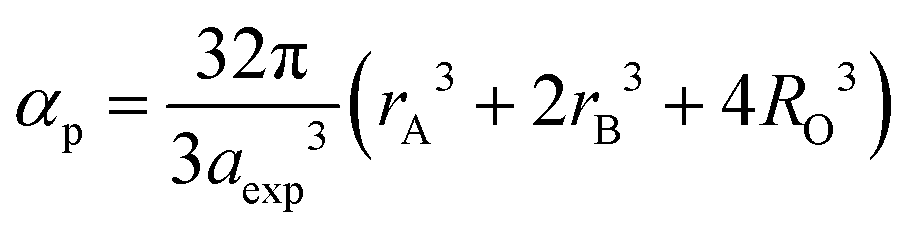
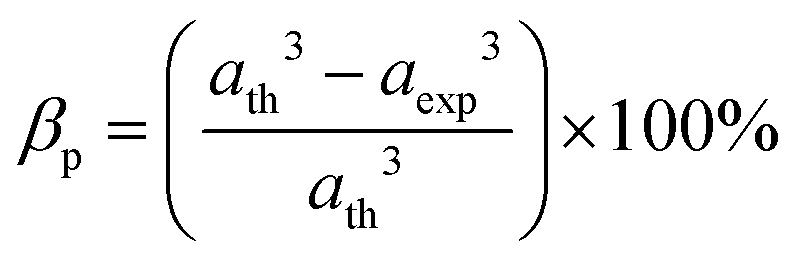
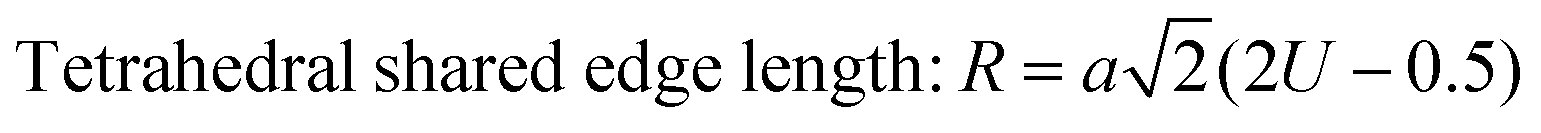
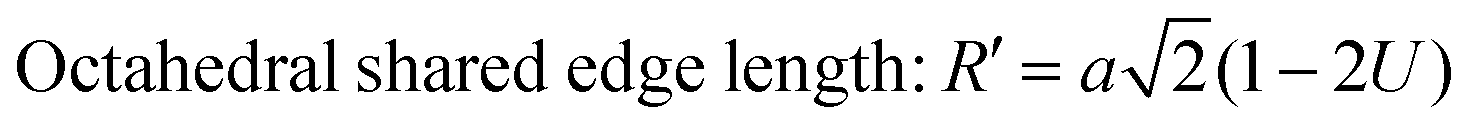
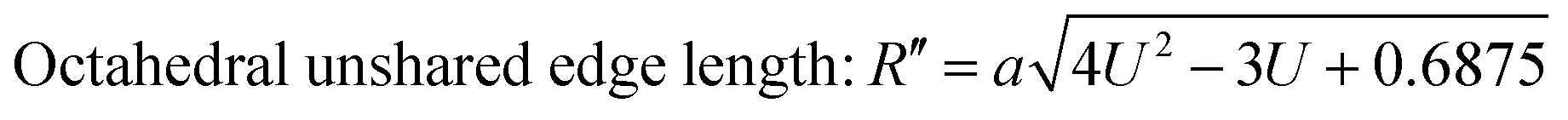
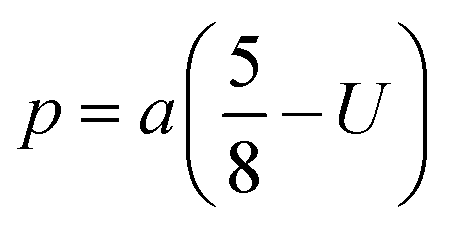
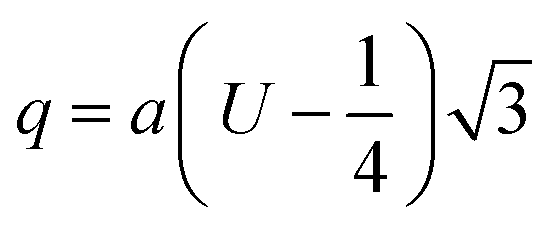
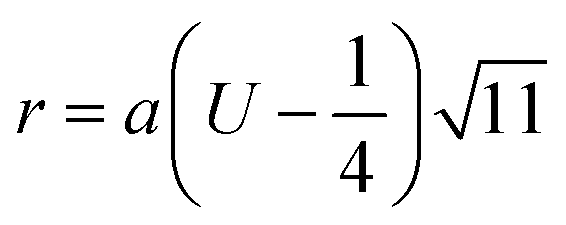
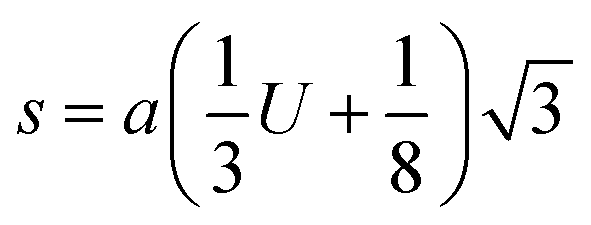
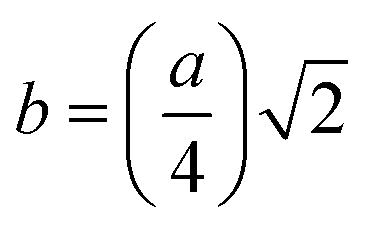
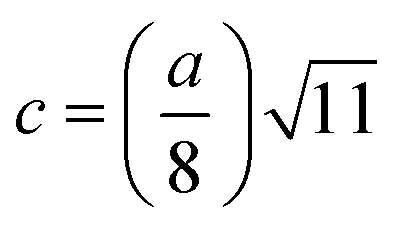
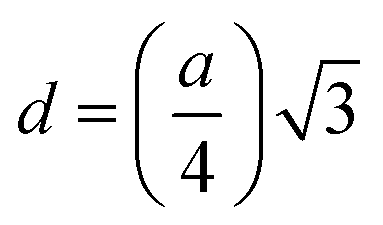
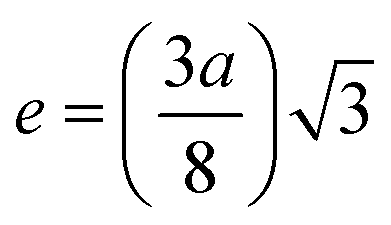
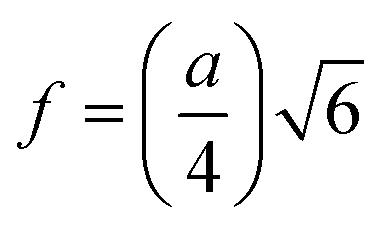
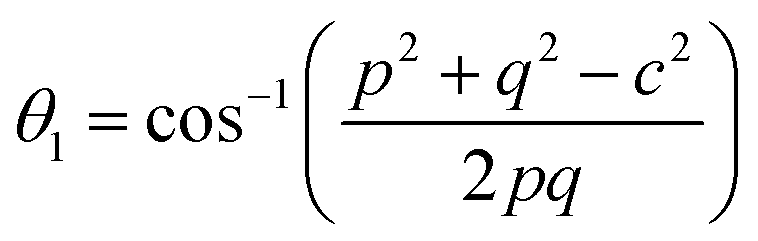
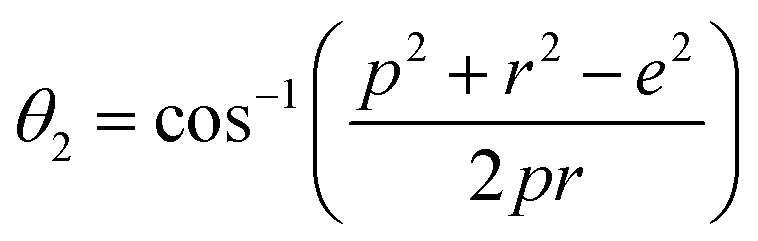
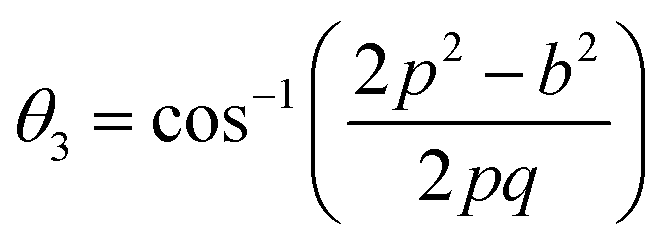
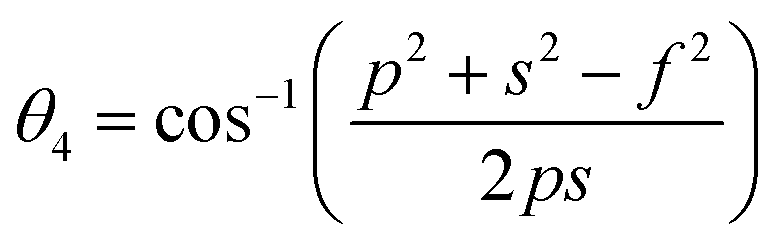
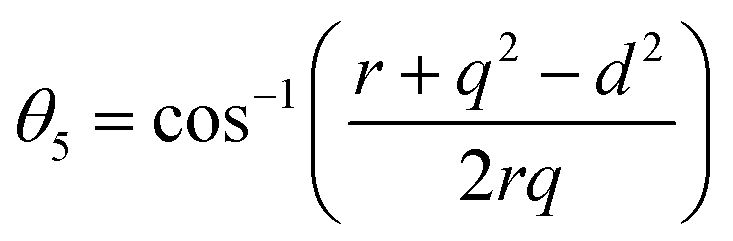
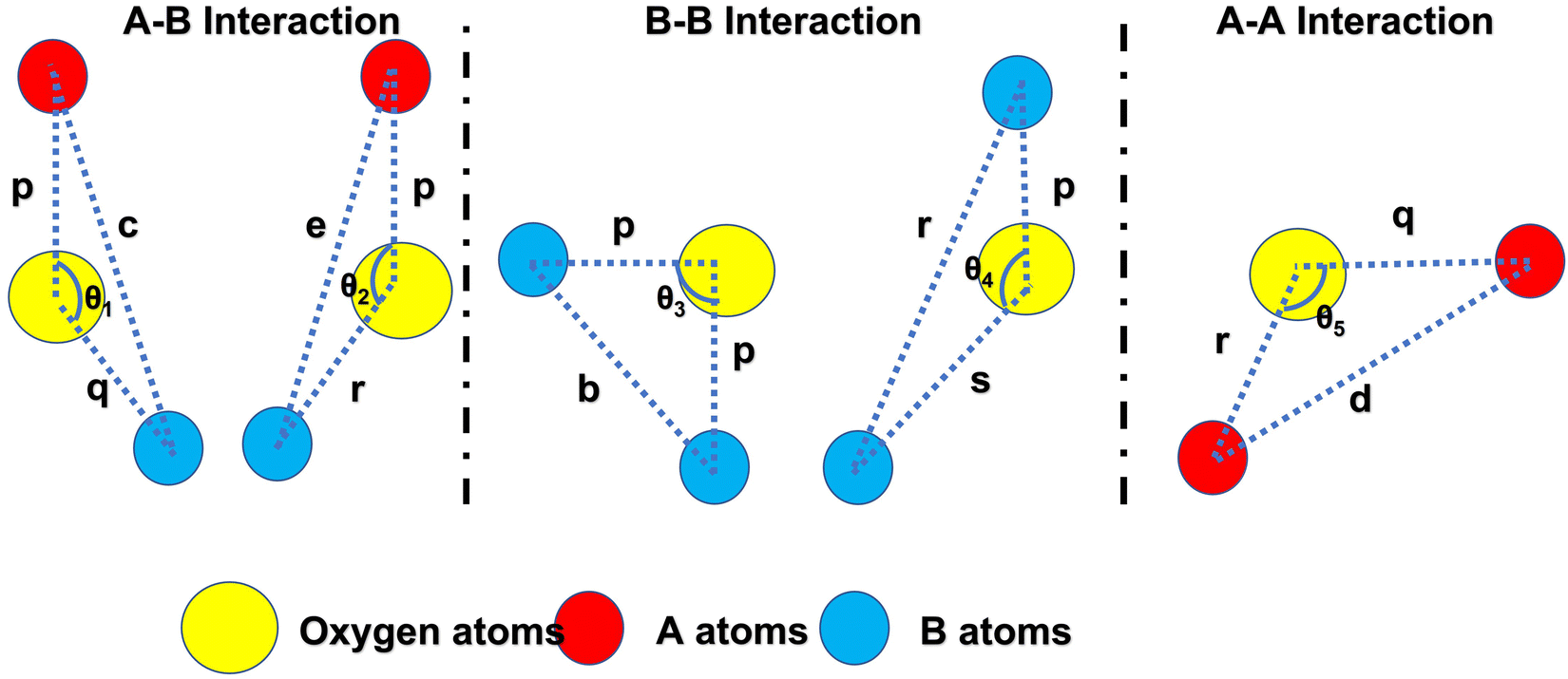
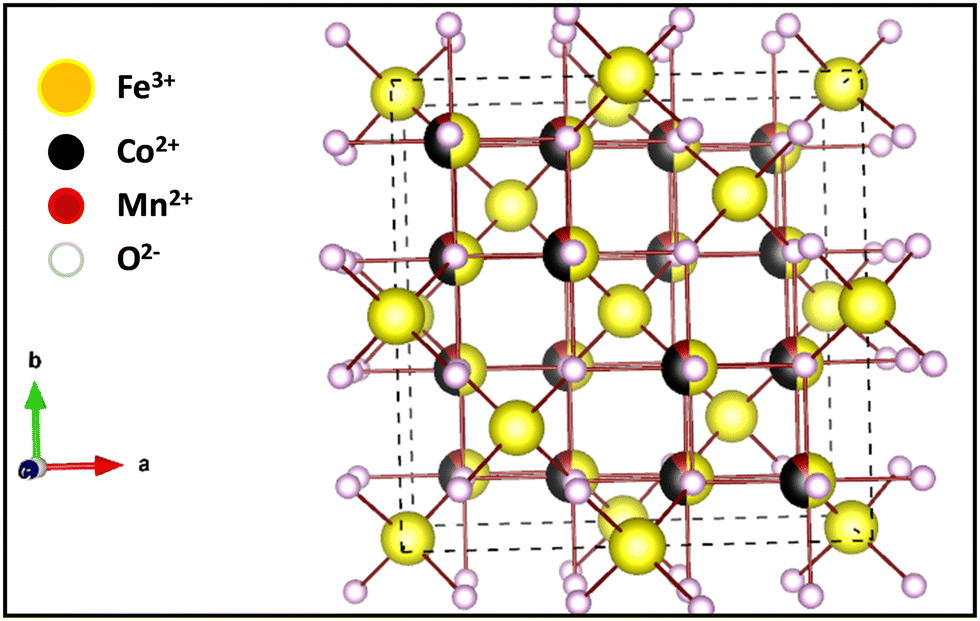
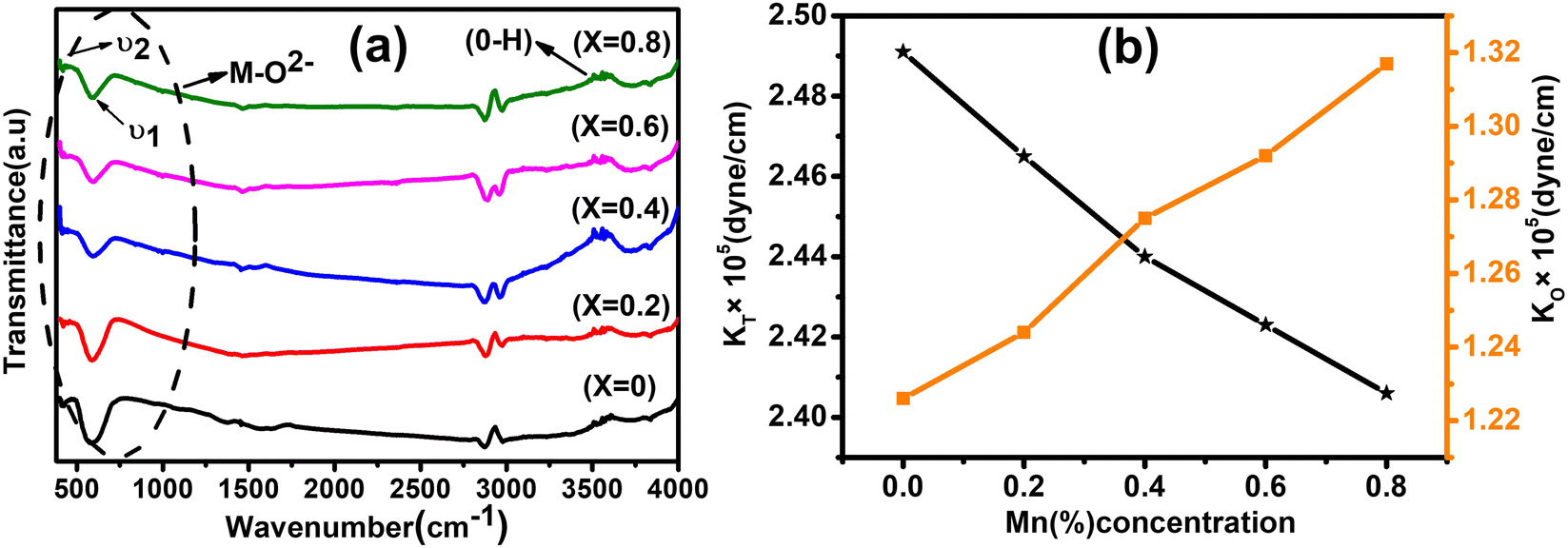
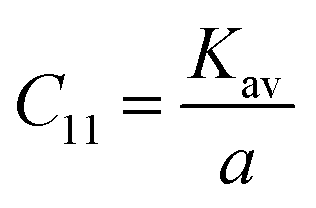
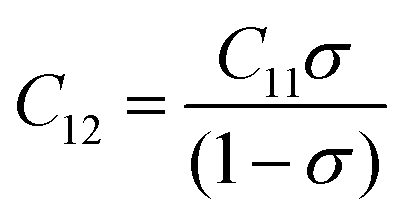
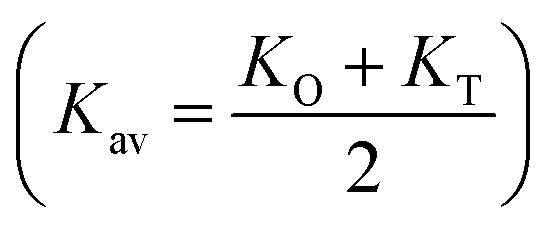 , a is the experimental lattice parameter and σ id Poisson's ratio, which depends on the porosity (P) and calculated as σ = 0.324(1 − 1.043·P). The values of Poisson's ratio obtained for the different concentration samples are depicted in Table 8 and indicate that it increased with an increase in Mn2+ concentration. The Poisson's ratio varied from 0.19 to 0.22, which confirms the good elastic and isotropic characteristics of the samples.30 Further, the values of the stiffness constant C11 and C12 of all the samples were obtained by putting the values of Kav, a and σ in the above-mentioned equations and listed in Table 9. The stiffness constant C11 decreased, while C12 increased systematically with an increase in Mn2+ concentration. The stiffness constant C11 mainly depends on two factors, i.e., force constant and lattice parameter, whereas C12 also depends on the porosity of the samples. Further, various elastic modulus constants such as Young's modulus (E), modulus of rigidity (G), and bulk modulus (B) were calculated using the stiffness constant and Poisson's ratio and eqn (50)–(52),64 respectively, as follows:
, a is the experimental lattice parameter and σ id Poisson's ratio, which depends on the porosity (P) and calculated as σ = 0.324(1 − 1.043·P). The values of Poisson's ratio obtained for the different concentration samples are depicted in Table 8 and indicate that it increased with an increase in Mn2+ concentration. The Poisson's ratio varied from 0.19 to 0.22, which confirms the good elastic and isotropic characteristics of the samples.30 Further, the values of the stiffness constant C11 and C12 of all the samples were obtained by putting the values of Kav, a and σ in the above-mentioned equations and listed in Table 9. The stiffness constant C11 decreased, while C12 increased systematically with an increase in Mn2+ concentration. The stiffness constant C11 mainly depends on two factors, i.e., force constant and lattice parameter, whereas C12 also depends on the porosity of the samples. Further, various elastic modulus constants such as Young's modulus (E), modulus of rigidity (G), and bulk modulus (B) were calculated using the stiffness constant and Poisson's ratio and eqn (50)–(52),64 respectively, as follows: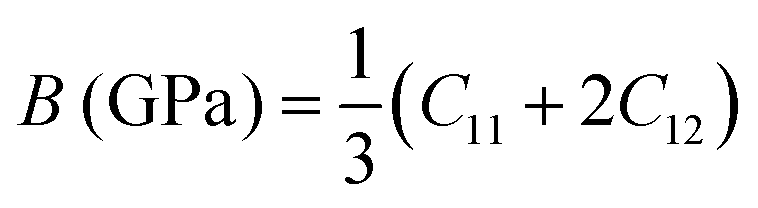
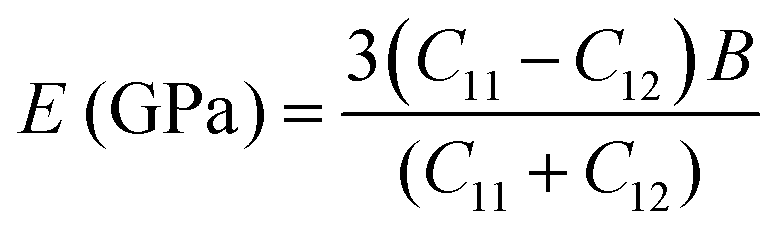
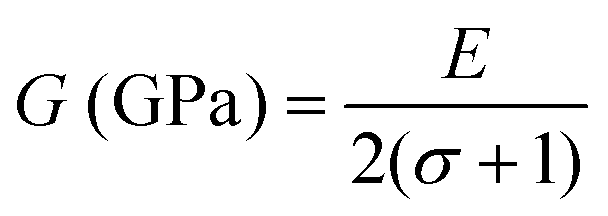
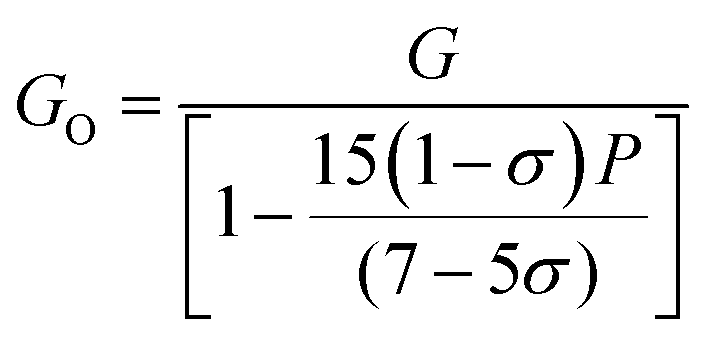
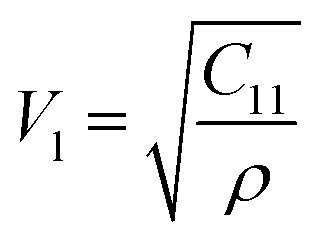
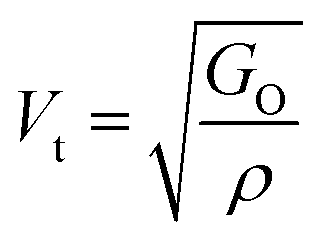
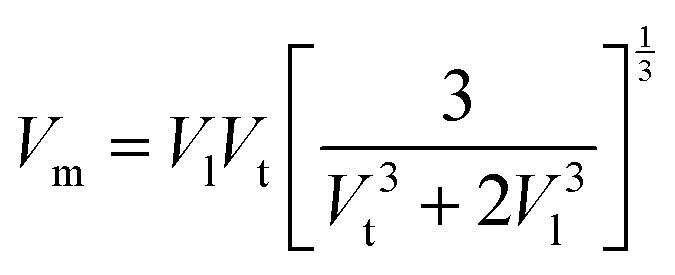
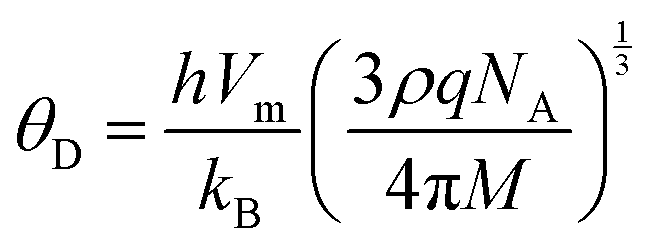
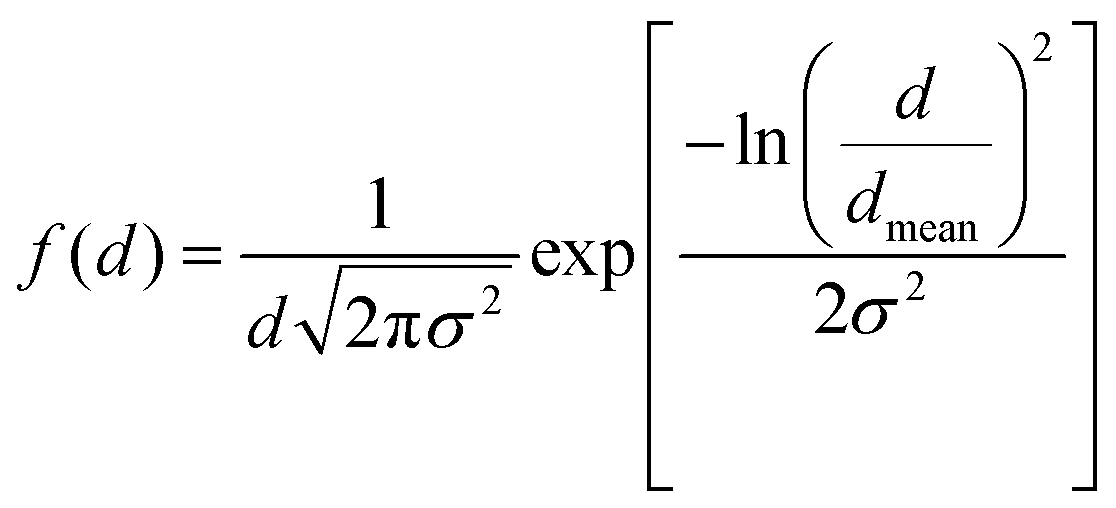
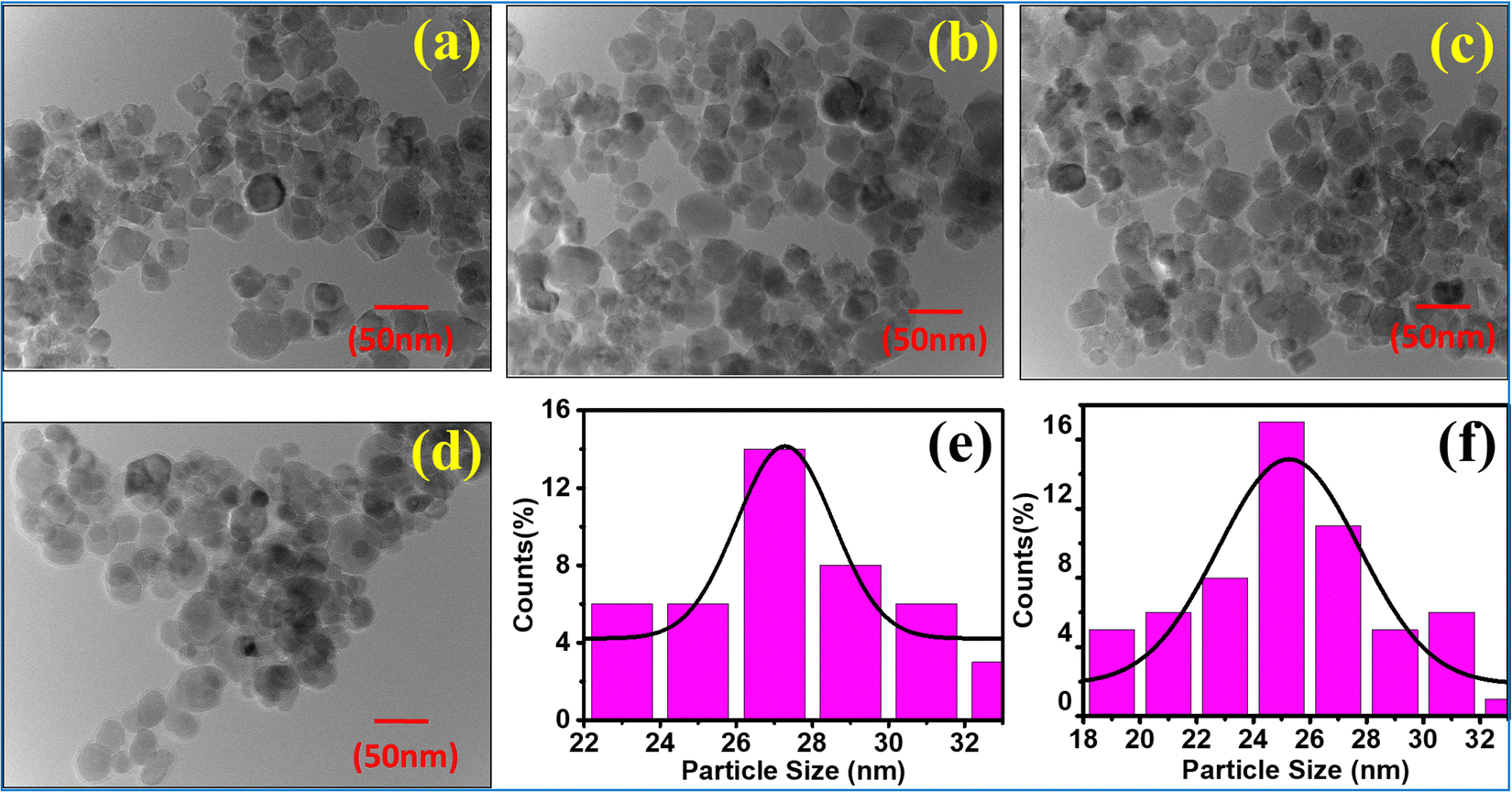
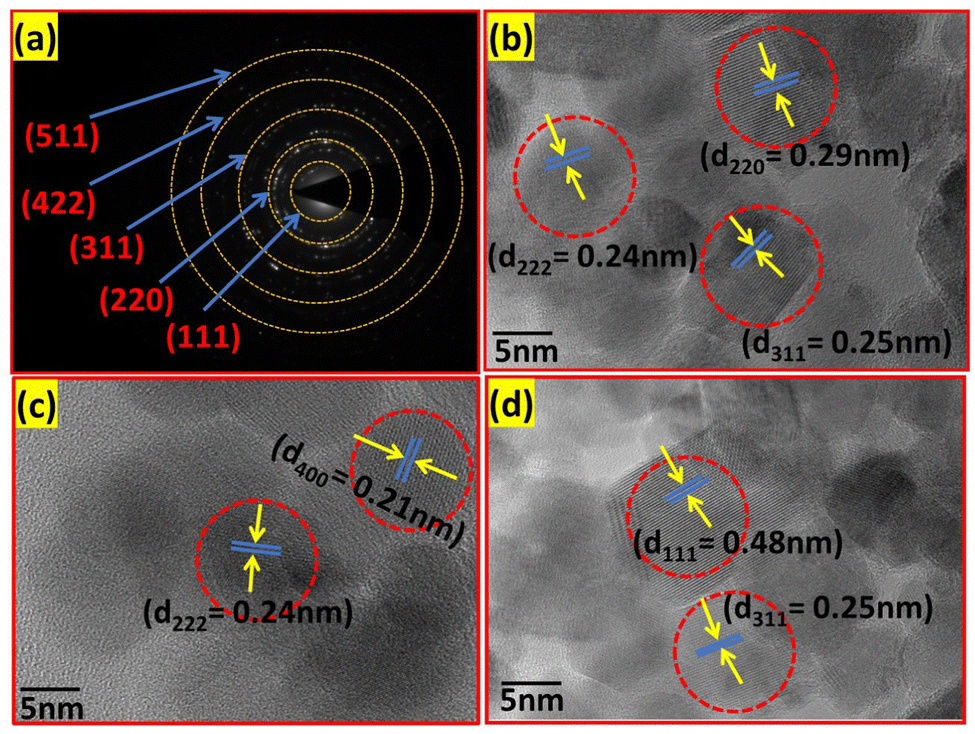
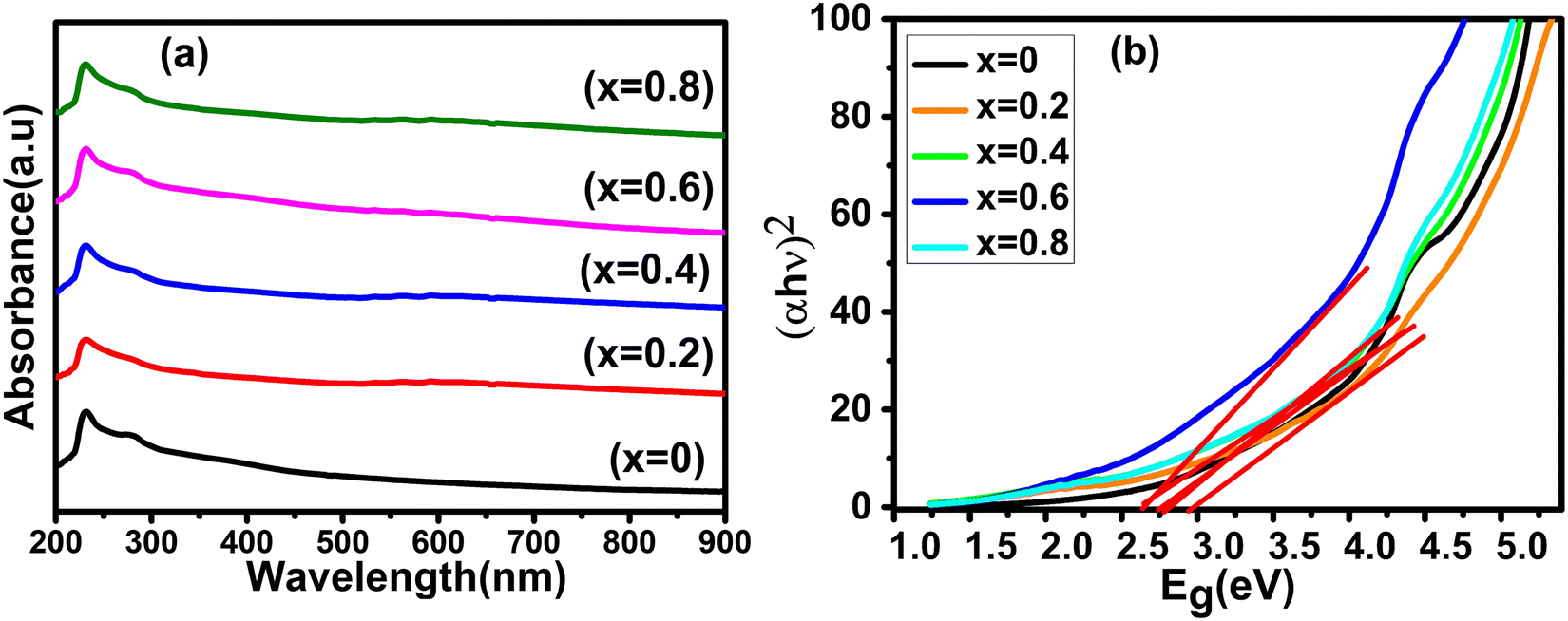
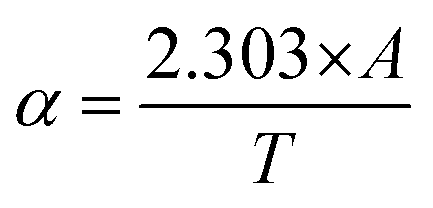 , where A is the absorbance and T is the thickness of the cuvette. The optical band gap, i.e., direct band gap of all the sample compositions, was obtained by putting (αhν)2 = 0 and extrapolation of the straight line segment. Thus, the obtained values of the optical energy band gap are listed in Table 10. The value of the band gap consistently decreased from 2.71 to 2.65 eV with an increase in Mn2+ concentration. The variation in the band gap values is due to the variation in crystallite size, lattice parameter, and cation distribution with a variation in the concentration of Mn2+ ions. Further, the variation in the band gap energies in NF samples may be due to the coordination of cations and the co-existence of low and high spin states. The decrease in the band gap energy with an increase in the Mn2+ concentration confirms the increase in the crystallite size and lattice strain in the sample. This is due to the quantum confinement effect.30,35 Also, some micro-strain was generated by the defects, which affected the crystal structure of the materials. In the case of the Mn2+substituted cobalt ferrite samples, the ionic radius of Mn2+ (0.80 Å) is larger than that of the Fe3+ (0.67 Å) ion, which caused a decrease in the optical band gap value. The decrease in the band gap with an increase in the particle size can be well understood by Brass’ equation84 given by eqn (60), as follows:
, where A is the absorbance and T is the thickness of the cuvette. The optical band gap, i.e., direct band gap of all the sample compositions, was obtained by putting (αhν)2 = 0 and extrapolation of the straight line segment. Thus, the obtained values of the optical energy band gap are listed in Table 10. The value of the band gap consistently decreased from 2.71 to 2.65 eV with an increase in Mn2+ concentration. The variation in the band gap values is due to the variation in crystallite size, lattice parameter, and cation distribution with a variation in the concentration of Mn2+ ions. Further, the variation in the band gap energies in NF samples may be due to the coordination of cations and the co-existence of low and high spin states. The decrease in the band gap energy with an increase in the Mn2+ concentration confirms the increase in the crystallite size and lattice strain in the sample. This is due to the quantum confinement effect.30,35 Also, some micro-strain was generated by the defects, which affected the crystal structure of the materials. In the case of the Mn2+substituted cobalt ferrite samples, the ionic radius of Mn2+ (0.80 Å) is larger than that of the Fe3+ (0.67 Å) ion, which caused a decrease in the optical band gap value. The decrease in the band gap with an increase in the particle size can be well understood by Brass’ equation84 given by eqn (60), as follows: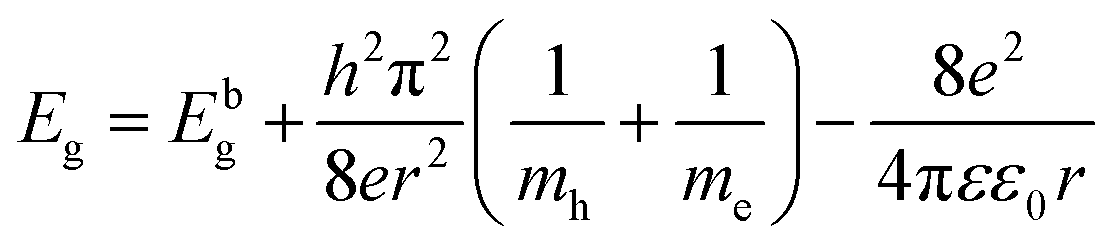
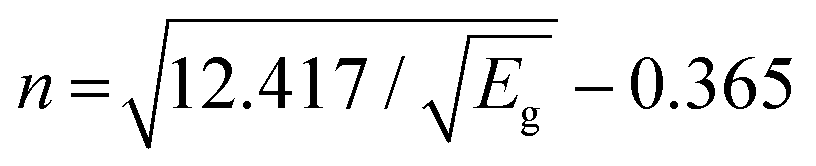
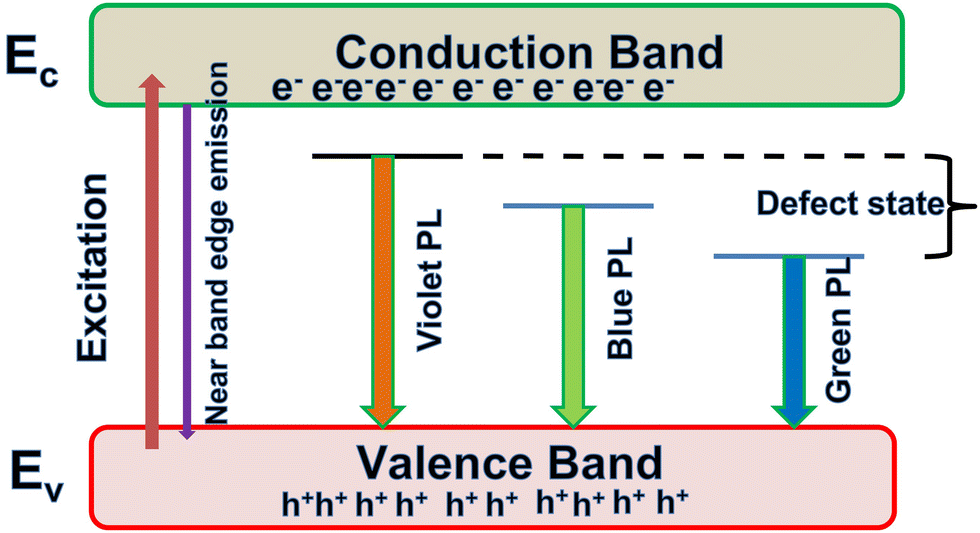
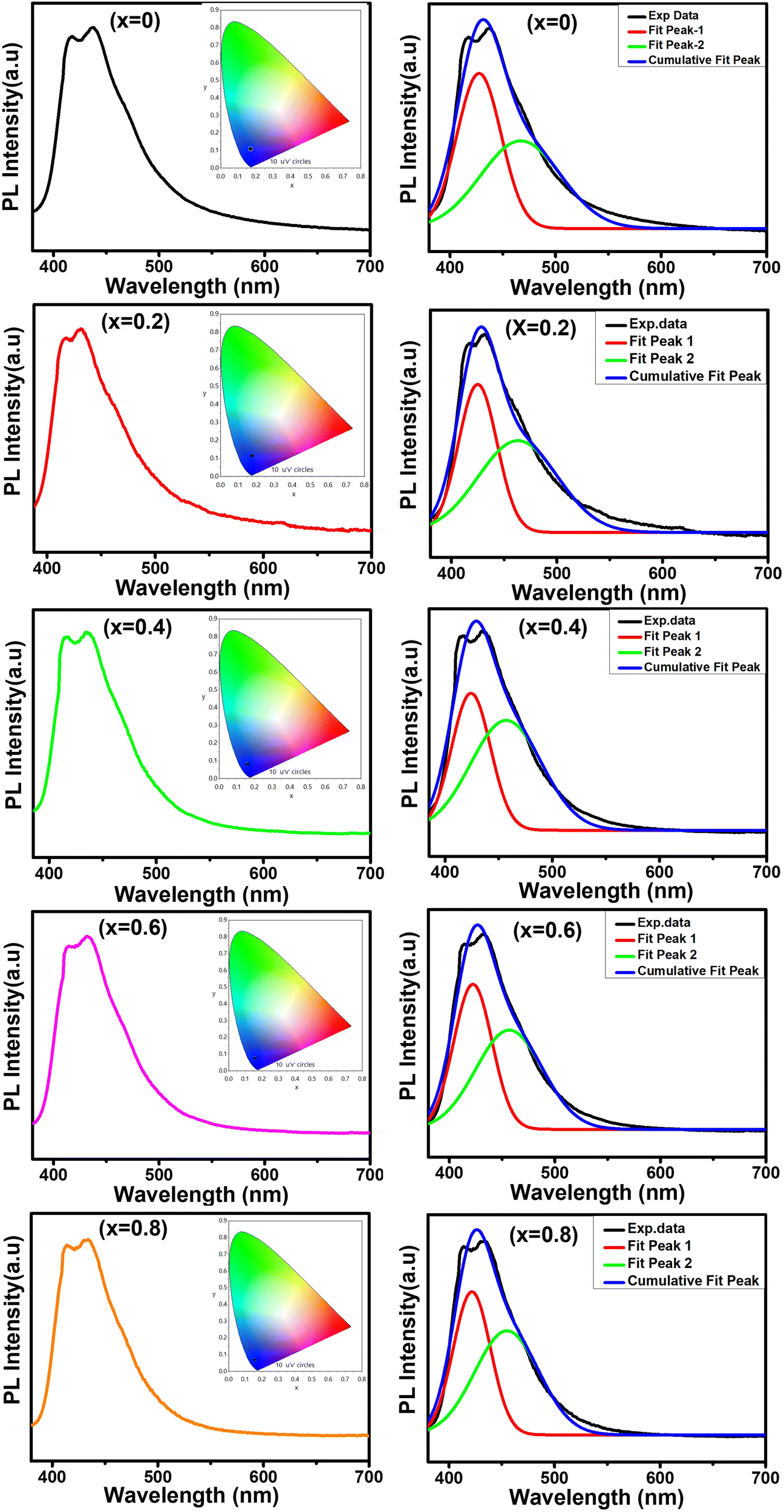
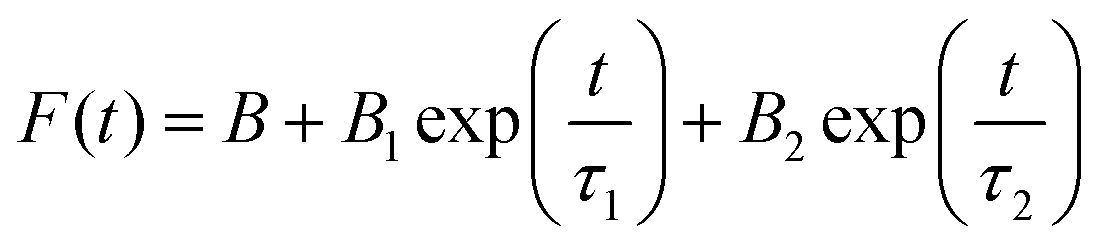
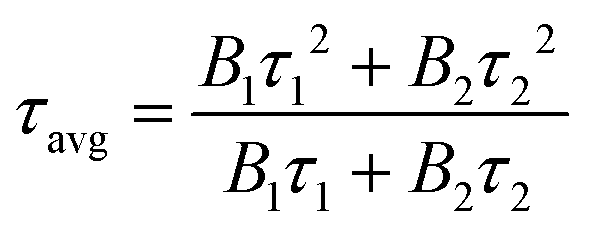
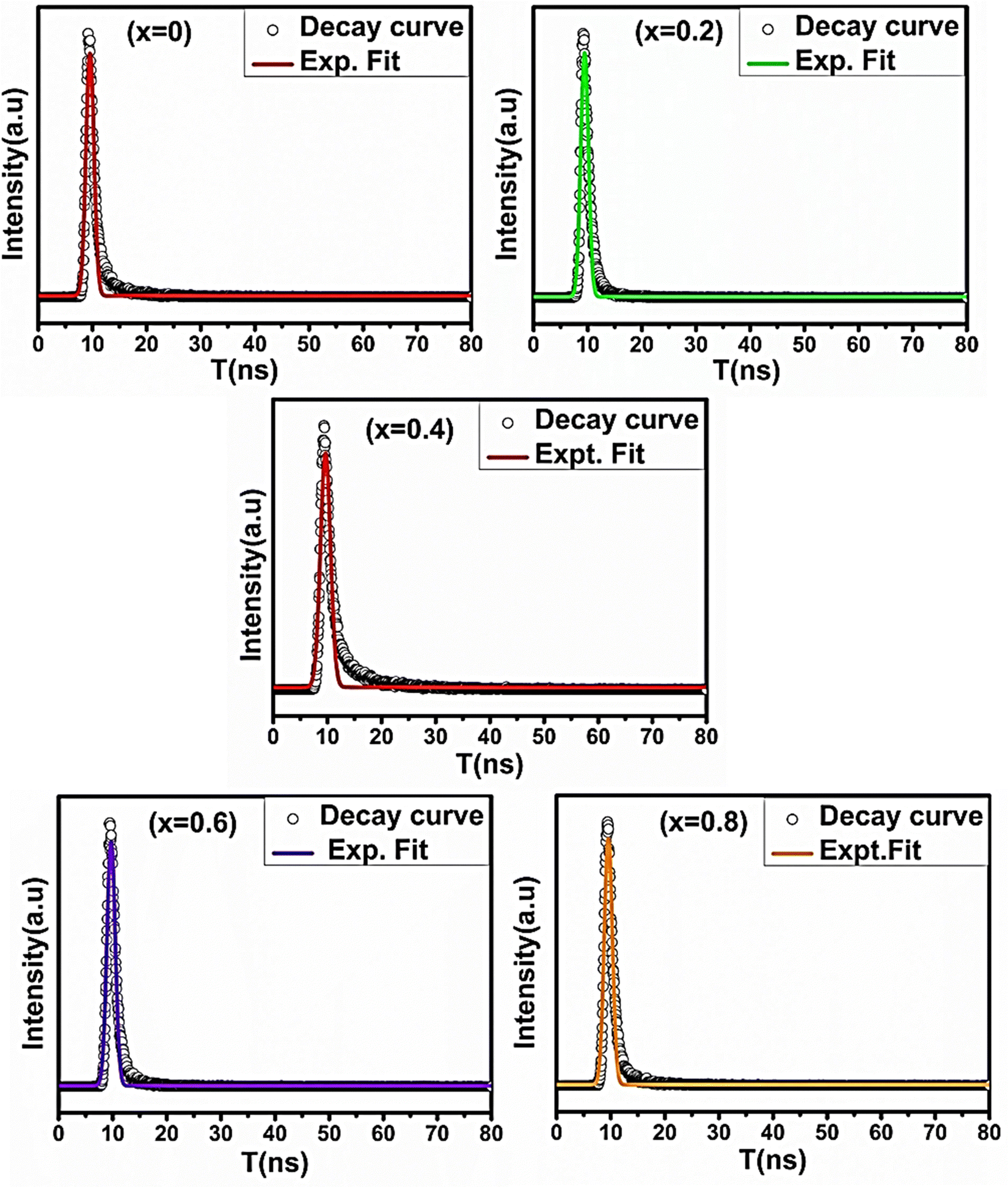
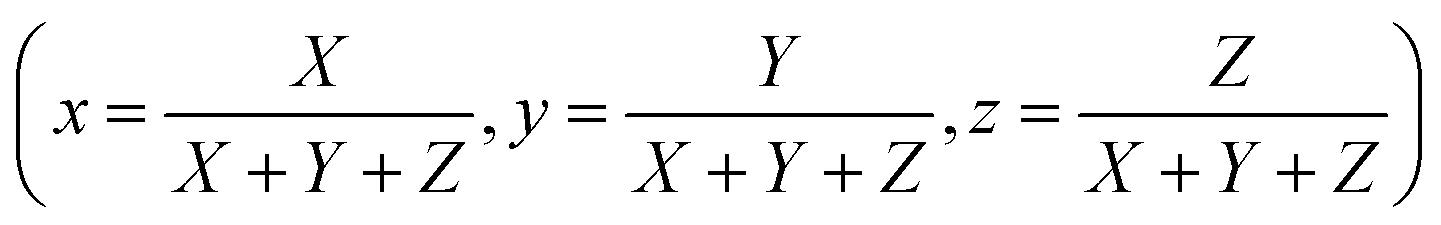 and its center was ascribed as the standard belonging to the white light emission (x = 0.34, y = 0.34). To determine the dominant emission colour for the Mn2+-doped CFO NFs, the emission spectra of the samples were converted to a CIE chromaticity diagram. The (x, y) coordinates of the chromaticity plot and the values of X, Y and luminescent intensity found for each sample composition are listed in Table 12.
and its center was ascribed as the standard belonging to the white light emission (x = 0.34, y = 0.34). To determine the dominant emission colour for the Mn2+-doped CFO NFs, the emission spectra of the samples were converted to a CIE chromaticity diagram. The (x, y) coordinates of the chromaticity plot and the values of X, Y and luminescent intensity found for each sample composition are listed in Table 12.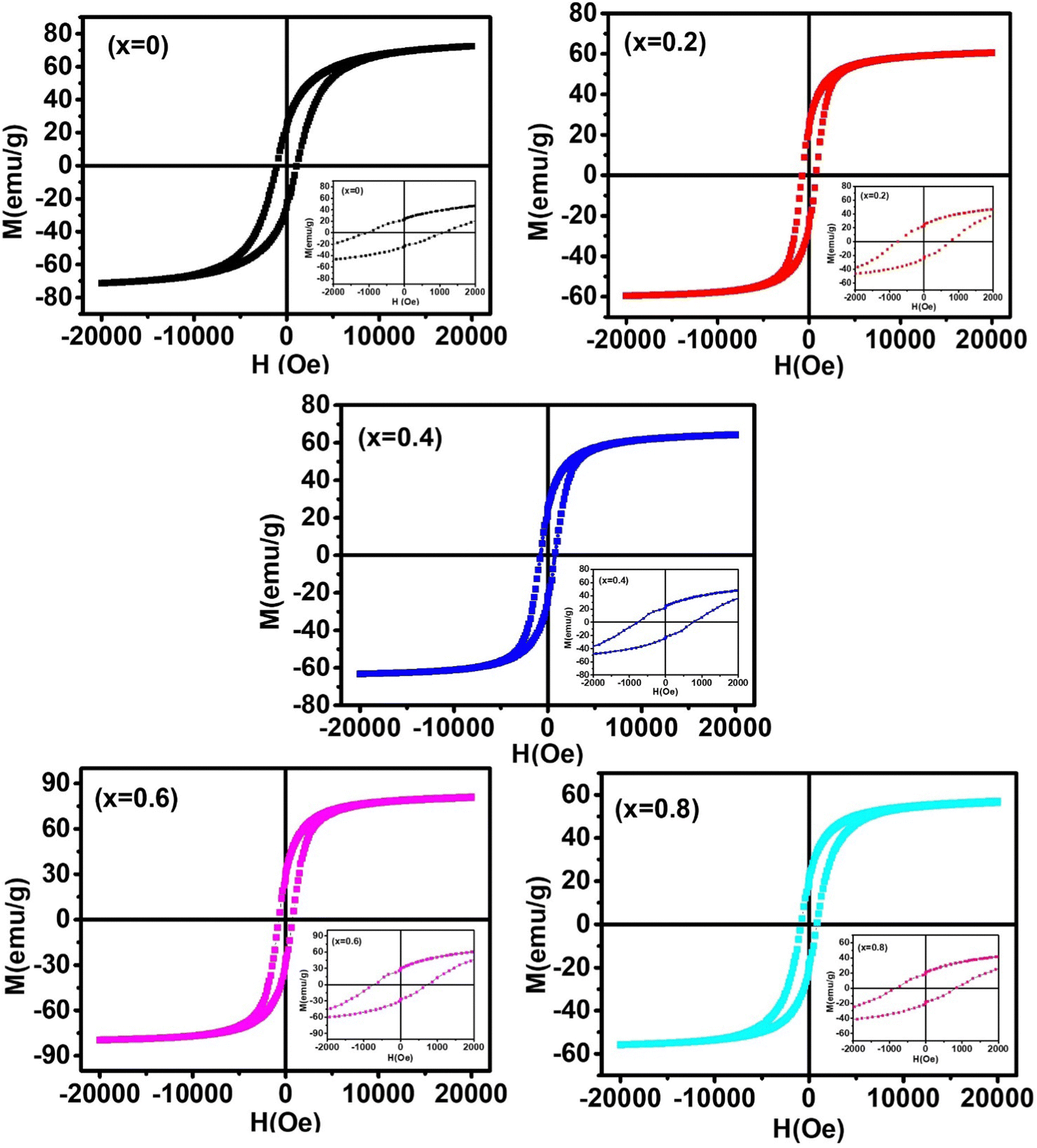
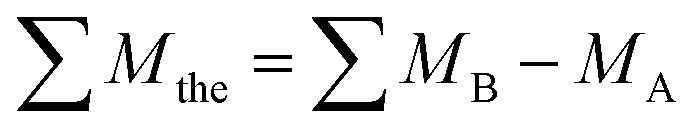 . The room temperature magnetic moments of Mn2+, Co2+, and Fe3+ are 5.0, 3.0, and 5.0 μB, respectively.42,46 The Fe3+ ions distribute equally on the A and B site lattice and the Co2+ ions only occupy the B-site lattice. The Mn2+ ions have a large ionic radius and magnetic moments compared to the Co2+ ions and they have a strong tendency to replace the Co2+ ions in the B-site lattice. This results in an increase in the saturation magnetization and a decrease in coercivity.44 For the pure cobalt ferrite sample, the saturation magnetization was 68.51 emu g−1, which is lower than that of its bulk counterpart due to the surface spin canting effects arising from the large surface area in the nanoregion.63
. The room temperature magnetic moments of Mn2+, Co2+, and Fe3+ are 5.0, 3.0, and 5.0 μB, respectively.42,46 The Fe3+ ions distribute equally on the A and B site lattice and the Co2+ ions only occupy the B-site lattice. The Mn2+ ions have a large ionic radius and magnetic moments compared to the Co2+ ions and they have a strong tendency to replace the Co2+ ions in the B-site lattice. This results in an increase in the saturation magnetization and a decrease in coercivity.44 For the pure cobalt ferrite sample, the saturation magnetization was 68.51 emu g−1, which is lower than that of its bulk counterpart due to the surface spin canting effects arising from the large surface area in the nanoregion.63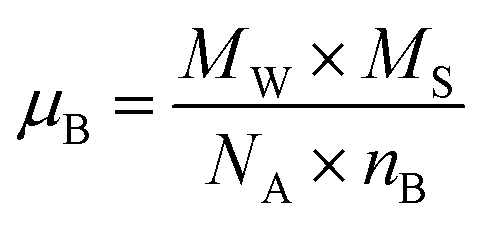 ,60 where MW is the molecular weight of each sample composition, NA is Avogadro's number, and nB is the Bohr magneton. To calculate the magnetic moments, we have to consider the strong interaction between A–B and their distribution of cations.24 The obtained magnetic moment value for all the sample concentrations is listed in Table 13 together with the theoretical values. With the substitution of Mn2+ ions in CFO, the μB started to decrease, which is also evident from the Ms values. This can be well understood as the Mn2+ incorporation in CFO results in a decrease in the A–B interaction. However, the further substitution of Mn2+ in place of Co2+ results in an increase in the A–B interaction and a significant increase in magnetic moment is observed. The magnetic moment values obtained by experimental and theoretical calculation differ slightly given that the NFs do not follow the ideal cation distribution assumed for the theoretical calculations.65,94 Further, the initial permeability (μi) of the pure and Mn2+-doped CFO were obtained as follows:95
,60 where MW is the molecular weight of each sample composition, NA is Avogadro's number, and nB is the Bohr magneton. To calculate the magnetic moments, we have to consider the strong interaction between A–B and their distribution of cations.24 The obtained magnetic moment value for all the sample concentrations is listed in Table 13 together with the theoretical values. With the substitution of Mn2+ ions in CFO, the μB started to decrease, which is also evident from the Ms values. This can be well understood as the Mn2+ incorporation in CFO results in a decrease in the A–B interaction. However, the further substitution of Mn2+ in place of Co2+ results in an increase in the A–B interaction and a significant increase in magnetic moment is observed. The magnetic moment values obtained by experimental and theoretical calculation differ slightly given that the NFs do not follow the ideal cation distribution assumed for the theoretical calculations.65,94 Further, the initial permeability (μi) of the pure and Mn2+-doped CFO were obtained as follows:95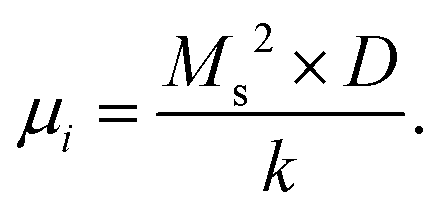 The initial permeability also follows the same trend as the saturation magnetization with Mn2+ substitution and varies in the range of 2.89 to 6.74.
The initial permeability also follows the same trend as the saturation magnetization with Mn2+ substitution and varies in the range of 2.89 to 6.74.