DOI:
10.1039/D2TC00017B
(Paper)
J. Mater. Chem. C, 2022,
10, 6043-6049
Isomer engineering of dipyrido[3,2-a:3′,4′-c]phenazine-acceptor-based red thermally activated delayed fluorescent emitters†
Received
2nd January 2022
, Accepted 21st March 2022
First published on 21st March 2022
Abstract
To determine the effect of donor substitution pattern on emission properties, we designed and synthesized two isomeric red thermally activated delayed fluorescent (TADF) emitters, i.e., 11,12-bis(9,9-dimethylacridin-10(9H)-yl)dipyrido[3,2-a:2′,3′-c]phenazine (oDMAC–DPPZ) and 10,13-bis(9,9-dimethylacridin-10(9H)-yl)dipyrido[3,2-a:2′,3′-c]phenazine (pDMAC–DPPZ). Two 9,9-dimethyl-9,10-dihydroacridine (DMAC) donor units were attached at the ortho (11, 12) and para (10, 13) positions of the dipyrido[3,2-a:2′,3′-c]phenazine (DPPZ) acceptor to obtain oDMAC–DPPZ and pDMAC–DPPZ, respectively. Interestingly, the photoluminescence quantum yield, external quantum efficiency, and emission wavelength of the emitters varied depending on the substitution pattern of DMAC on DPPZ. The two emitters exhibited distinct photophysical and organic light-emitting diode (OLED) performances due to structural differences related to steric strain and dihedral angles. It is demonstrated that the ortho (11, 12) donor substitution on DPPZ is more effective in the design of high-performance red TADF OLEDs than the para (10, 13) substitution in terms of singlet–triplet energy gap and up-conversion rate constant. The ortho-substituted TADF emitter demonstrated a high EQE of 13.4% with color coordinates of (0.59, 0.40), whereas the para-substituted isomer displayed an EQE of 4.0% and color coordinates of (0.64, 0.35).
Introduction
Organic light-emitting diodes (OLEDs), proven to be superior to conventional liquid crystal displays, have been studied extensively for their solid-state lighting and display applications.1–6 Although state-of-the-art commercialized OLEDs are based on phosphorescent emitters, their demerits include the use of expensive noble metals, a low environmental sustainability, and short device lifetimes. Therefore, alternatives to phosphorescent OLEDs (Ph-OLEDs) are actively being sought. Organic-emitter-based thermally activated delayed fluorescent (TADF)-OLEDs are considered competent alternatives to Ph-OLEDs, as internal quantum efficiencies of up to 100% are achievable in both cases.7–13 Compared to precious-metal-based phosphorescent emitters, TADF emitters are inexpensive and allow for greater diversity in their design. Organic materials with a small singlet–triplet energy gap (ΔEST) are capable of converting non-radiative triplet excitons to radiative singlet excitons via a reverse intersystem crossing (RISC) process. A small ΔEST is achievable by minimizing the overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Another requirement for the high-performance TADF emitters is a high photoluminescence quantum yield (PLQY), attainable by maintaining a maximum overlap between the HOMO and LUMO in the molecular design. Therefore, two contradictory requirements, a small ΔEST and high PLQY, need to be balanced in a single organic molecule to realize a high external quantum efficiency (EQE).
Recently reported high-performance blue and green TADF emitters have been constructed either by introducing new donor/acceptor moieties or via novel molecular design approaches.14–20 As red is a primary color, red TADF OLEDs are equally important as the blue and green TADF OLEDs. However, the red TADF emitters deliver significantly lower EQEs than blue and green TADF emitters, primarily because of the fundamental limitation of the energy gap theory.21,22 Because of the small band gap of the red TADF emitters, internal conversion, that is, non-radiative decay, dominates over radiative decay in this spectral region. To resolve this issue, various molecular design strategies have been developed and novel electron-accepting units have been introduced to simultaneously achieve high PLQY and small ΔEST values and deliver efficient long-wavelength TADF emitters.23–27 To avoid non-radiative loss mechanisms resulting from the vibrational and rotational motions of phenyl-spacer-based flexible structures, molecular designs wherein the donor unit is directly attached to the planar and rigid acceptor unit are preferred for the construction of the red TADF emitters.28–33 Among the available electron-withdrawing acceptor units, dipyrido[3,2-a:2,3-c]phenazine (DPPZ) has been investigated for the design of highly efficient red TADF emitters because of its rigid and fully conjugated planar heteroaromatic structure.34–37 Recently, it was demonstrated that the PLQYs of the red TADF emitters can vary significantly according to the donor substitution pattern on planar and rigid acceptors, such as phenanthro[4,5-fgh]quinoxaline-10,11-benzene38,39 and dibenzo[a,c]phenazine.40 According to the energy gap law, the PLQY of TADF emitters decreases as their emission wavelength shifts toward the deep-red/near infrared region. Therefore, molecular designs that ensure high PLQYs in this region are crucial. To suppress non-radiative loss mechanisms and generate pure-color highly efficient TADF emitter-based OLEDs, the molecular designs wherein donor/acceptor rotation is restricted are preferred over those allowing for a free rotation.41–46 Recently, it was reported that the device performances of long wavelength TADF emitters can be largely varied depending upon the ortho/para donor substitution position.34,40,47 and the contribution of the locally excited triplet states in the emission process was studied to improve the RISC process of the ortho substituted TADF emitters over the para substituted derivatives.48,49
In this study, DPPZ, as an acceptor, was disubstituted with 9,9-dimethyl-9,10-dihydroacridine (DMAC), as a donor, in two distinct patterns to generate two red TADF emitters, namely 11,12-bis(9,9-dimethylacridin-10(9H)-yl)dipyrido[3,2-a:2′,3′-c]phenazine (oDMAC–DPPZ) and 10,13-bis(9,9-dimethylacridin-10(9H)-yl)dipyrido[3,2-a:2′,3′-c]phenazine (pDMAC–DPPZ), which are configurational isomers of each other. The photophysical performances of the two isomeric TADF emitters were then evaluated to determine the effects of the donor substitution pattern on the TADF properties. The EQEs and electroluminescence (EL) spectra of the two isomers differed significantly; the ortho-substituted TADF emitter, oDMAC–DPPZ, demonstrated a high EQE of 13.4% with color coordinates of (0.59, 0.40), whereas the para-substituted isomer, pDMAC–DPPZ, displayed an EQE of 4.0% and color coordinates of (0.64, 0.35). The effect of donor substitution pattern of the DPPZ acceptor derived TADF emitters was clearly revealed in this work.
Results and discussion
Molecular simulation and synthesis
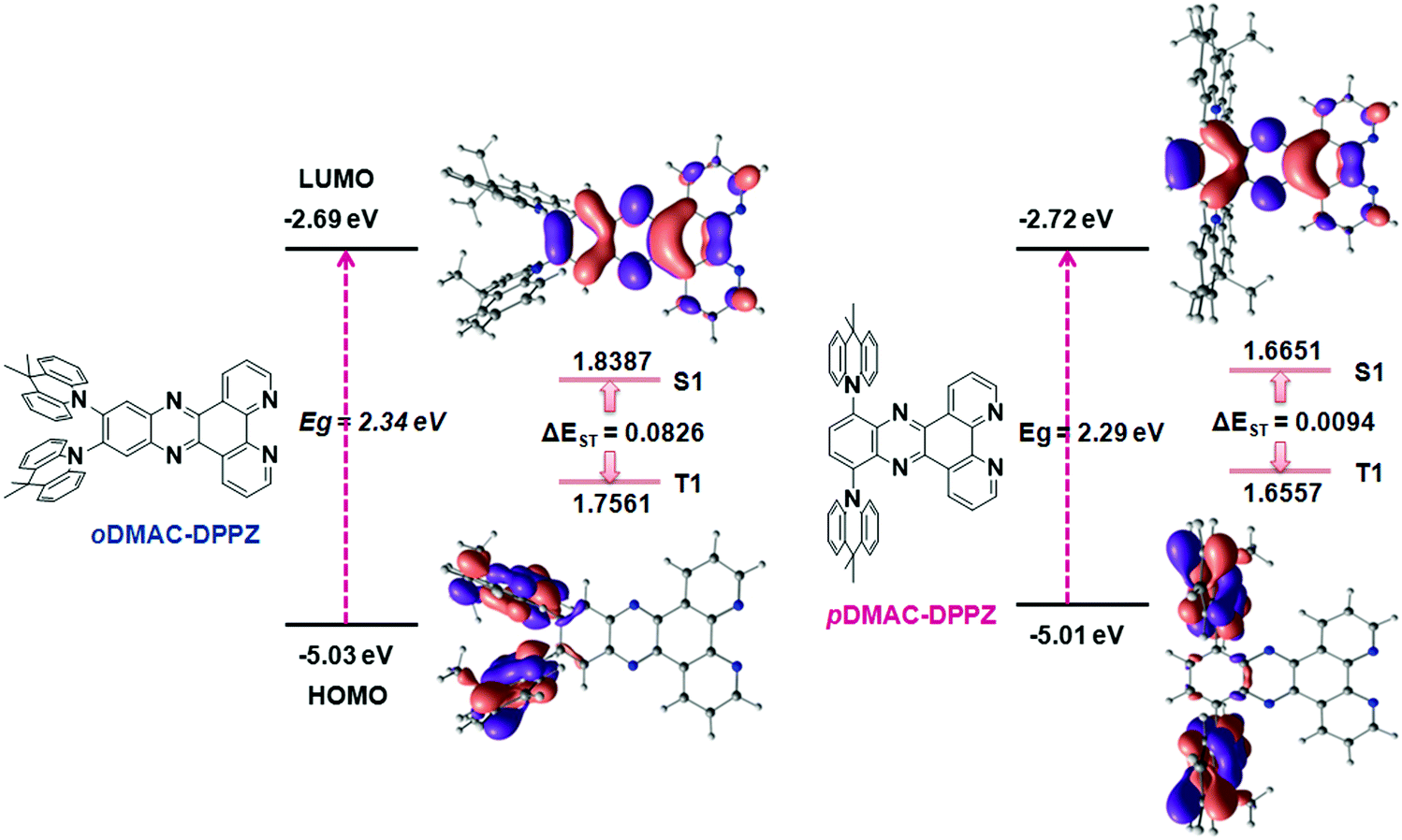
To estimate the effects of the DMAC/DPPZ substitution pattern, time-dependent density functional theory (TD-DFT) calculations were performed for oDMAC–DPPZ and pDMAC–DPPZ in the ground state using the B3LYP/6-31G (d,p) basis set in the gas phase. The frontier molecular orbital diagrams of the two emitters with their HOMO–LUMO energy gaps and singlet–triplet energies are shown in Fig. 1. As expected, the HOMOs and LUMOs of both compounds were largely located over the DMAC donor and DPPZ acceptor segments, respectively. The HOMO–LUMO gap values were calculated to be 2.34 and 2.29 eV for oDMAC–DPPZ and pDMAC–DPPZ, respectively, indicating a red-shifted emission for the latter. Based on their small ΔEST values (0.0826 eV and 0.0094 eV for oDMAC–DPPZ and pDMAC–DPPZ, respectively), the involvement of triplet excitons in the emission process can be predicted. Because the DMAC donor was directly attached to the DPPZ acceptor, large dihedral angles of 77.1° and 85.7° were obtained for oDMAC–DPPZ and pDMAC–DPPZ, respectively (Fig. S1 in ESI†). The small dihedral angle in oDMAC–DPPZ suggests a more prominent HOMO–LUMO overlap in this emitter, and consequently, a higher PLQY in the red region.
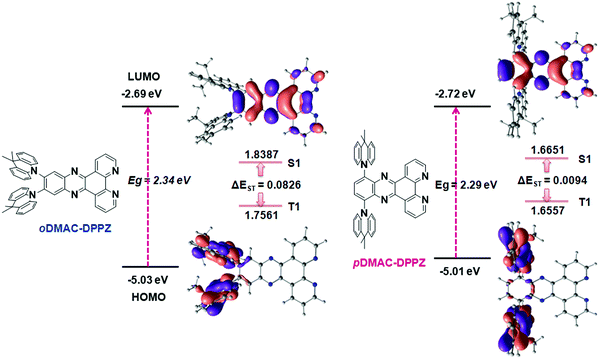 |
| | Fig. 1 Structures of oDMAC–DPPZ and pDMAC–DPPZ TADF emitters, and their ground state optimized geometries, HOMO–LUMO frontier molecular orbital diagrams, and HOMO–LUMO and singlet (S1)–triplet (T1) energies, calculated using DFT method. | |
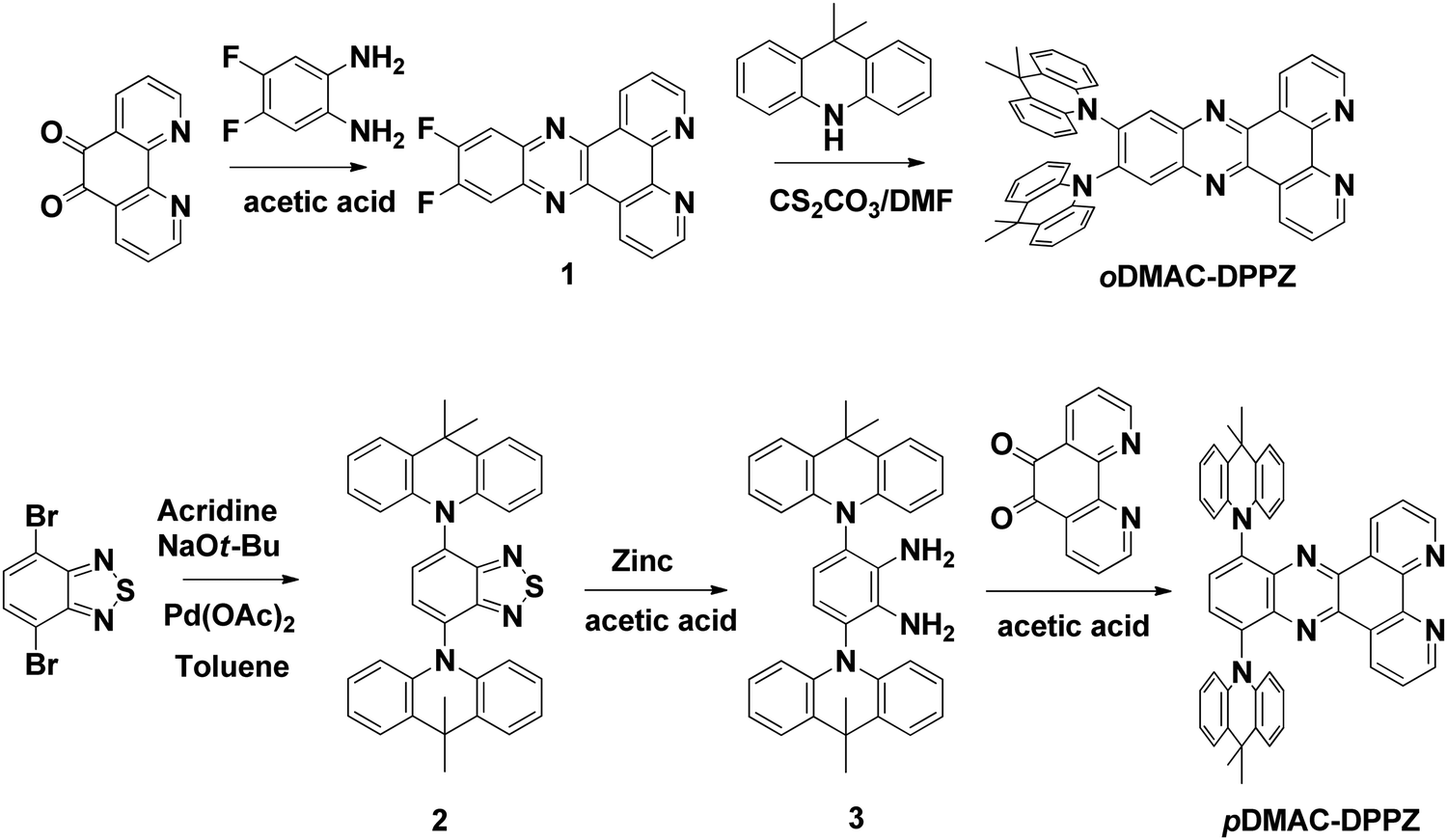
oDMAC–DPPZ and pDMAC–DPPZ are synthesized according to the reaction sequence presented in Scheme 1. Intermediate 1 was readily obtained in good yield by cyclizing 1,10-phenanthroline-5,6-dione and 4,5-difluorobenzene-1,2-diamine. Intermediate 1 was then heated with Cs2CO3 and DMAC in a sealed tube to obtain oDMAC–DPPZ. Intermediate 2 was synthesized according to a literature procedure,50 and subsequently reduced to intermediate 3. Finally, intermediate 3 was cyclized with 1,10-phenanthroline-5,6-dione to obtain pDMAC–DPPZ in good yield.
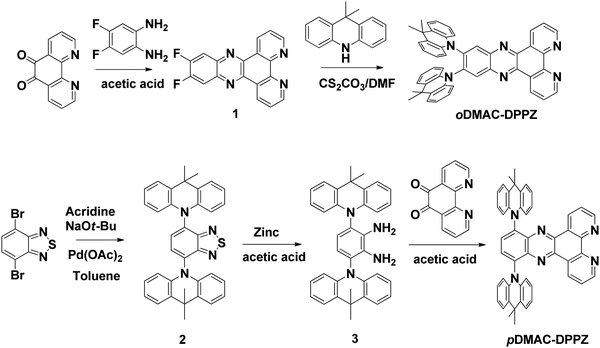 |
| | Scheme 1 Synthetic route to oDMAC–DPPZ and pDMAC–DPPZ TADF emitters. | |
Photophysical properties
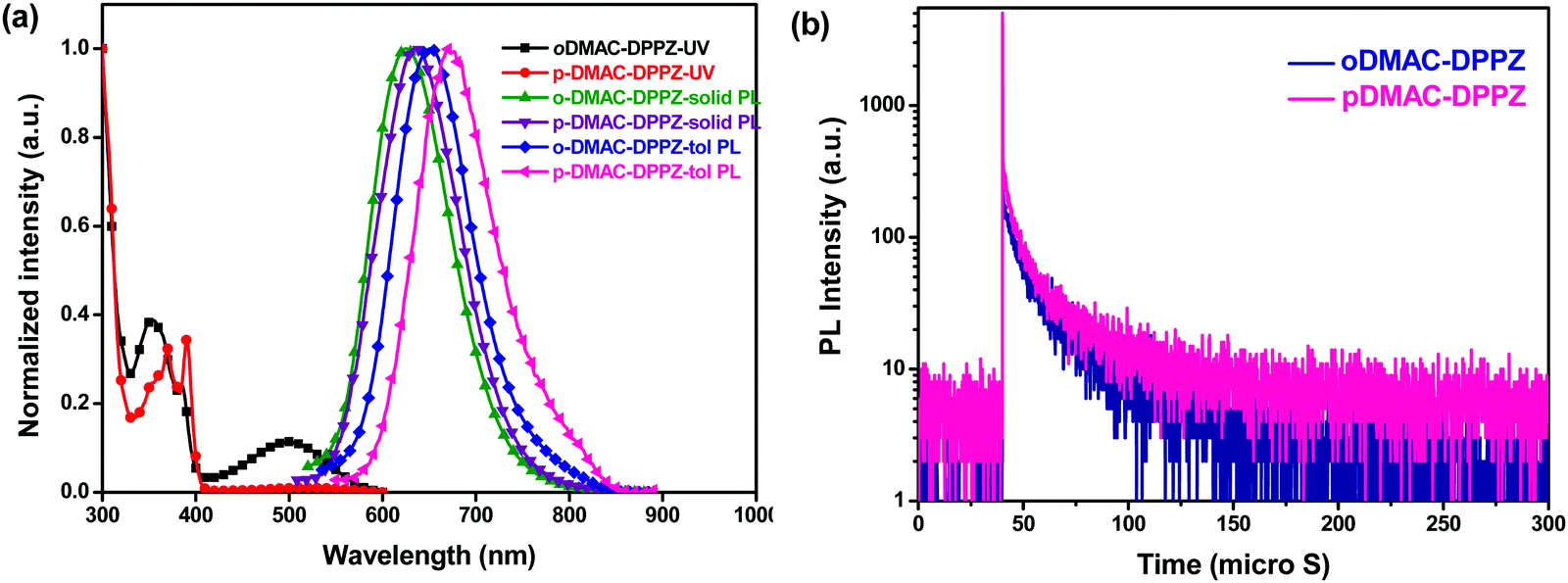
The photophysical properties of the two TADF emitters were investigated to determine the effect of the DMAC substitution position on their absorption and emission features, singlet–triplet energies, and the TADF phenomenon. Ultraviolet-visible (UV-vis) and photoluminescence (PL) spectra were recorded in toluene at room temperature (Fig. 2a and b). The high-energy peaks in their absorption spectra (330–400 nm) can be assigned to n–π* and π–π* transitions from the DMAC donor, as well as the DPPZ acceptor units, while the low-energy broad absorption peaks (420–600 nm) are attributed to the internal charge transfer (ICT) from the DMAC donor to the DPPZ acceptor unit. Notably, the intensity of the ICT absorption peak of oDMAC–DPPZ was higher than that of pDMAC–DPPZ, suggesting that the charge transfer (CT) properties of the two emitters were dependent on the substitution positions of the DMAC donor unit on the DPPZ acceptor. As predicted by DFT, the UV-vis absorption and PL spectra of pDMAC–DPPZ were slightly red-shifted compared to those of oDMAC–DPPZ. The singlet and triplet energies of both emitters were calculated from the onset of their fluorescence and phosphorescence, which were measured in toluene at 77 K (Fig. S2 in ESI†). The singlet–triplet energy gaps (ΔEST) calculated for oDMAC–DPPZ and pDMAC–DPPZ were 0.037 and 0.079 eV, respectively; these values were sufficiently small for an efficient RISC process to occur. The structureless and broad fluorescence and phosphorescence spectra of both emitters indicate ICT-dominated singlet and triplet excited states. The photophysical properties of the two emitters are summarized in Table 1.
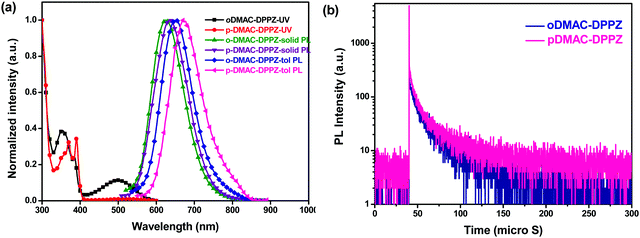 |
| | Fig. 2 (a) UV-vis absorption and PL spectra of oDMAC–DPPZ and pDMAC–DPPZ TADF emitters in toluene and those of 1% TADF-emitter-doped PS films at room temperature. (b) Transient PL decay curves of 1 wt% oDMAC–DPPZ- and pDMAC–DPPZ-doped thin PS films acquired at 300 K under a nitrogen atmosphere. | |
Table 1 Photophysical properties of oDMAC–DPPZ and pDMAC–DPPZ
| Emitter |
λ
abs
[nm] sol |
λ
PL
[nm] sol |
Φ
PL
[%] |
τ
p
[ns] |
τ
d
[μs] |
HOMOd [eV] |
LUMOd [eV] |
E
S/ETe [eV] |
ΔESTf [eV] |
|
UV-Vis absorption and photoluminescence (PL) λmax measured in toluene at 300 K.
Obtained by integrating sphere measurements.
PL lifetimes of prompt (τp) and delayed (τd) decay components measured in 1 wt% TADF-emitter-doped polystyrene films under a nitrogen atmosphere at 300 K.
Obtained from the oxidation and reduction onset potentials determined from the cyclic voltammograms.
Singlet (S1) and triplet (T1) energies.
ΔEST = ES − ET.
|
|
oDMAC–DPPZ
|
354, 499 |
652 |
63 |
27.02 |
1.76 |
−5.78 |
−3.59 |
2.26/2.23 |
0.037 |
|
pDMAC–DPPZ
|
390, 520 |
669 |
15 |
10.13 |
5.03 |
−5.73 |
−3.62 |
2.25/2.17 |
0.079 |
The TADF phenomenon was further investigated by measuring the photoluminescence quantum yield (ΦPL) and transient PL decay of polystyrene (PS) films doped with the emitters (1 wt%) under a nitrogen atmosphere at room temperature. Table 1 summarizes the ΦPL and PL lifetimes of the prompt (τp) and delayed (τd) decay components of the doped films. The ΦPL of pDMAC–DPPZ (15%) was lower than that of oDMAC–DPPZ (63%), presumably because the singlet energy of the former was lower, as per the energy-gap law, as well as the degree of HOMO–LUMO overlap, compared to that of the latter. Fig. 2b shows the transient PL decay curves of the two emitters, which exhibited typical TADF decay values with nanosecond-order, τp, and microsecond-order, τd, under degassed conditions at room temperature. The short-lifetime, τp, values are related to a direct emission from the singlet state, while the long-lifetime, τd, values are associated with TADF, involving ISC and RISC processes. The τp and τd lifetimes of the oDMAC–DPPZ-doped PS film were estimated to be 27.0 ns and 1.8 μs, respectively, while those of the pDMAC–DPPZ-doped PS film were 10.1 ns and 5.0 μs, respectively (Table 1). The short delayed decay lifetime (1.7 μs) of the oDMAC–DPPZ emitter can be correlated with its low ΔEST value (0.037 eV), suggesting that an efficient TADF process can be expected for this emitter.
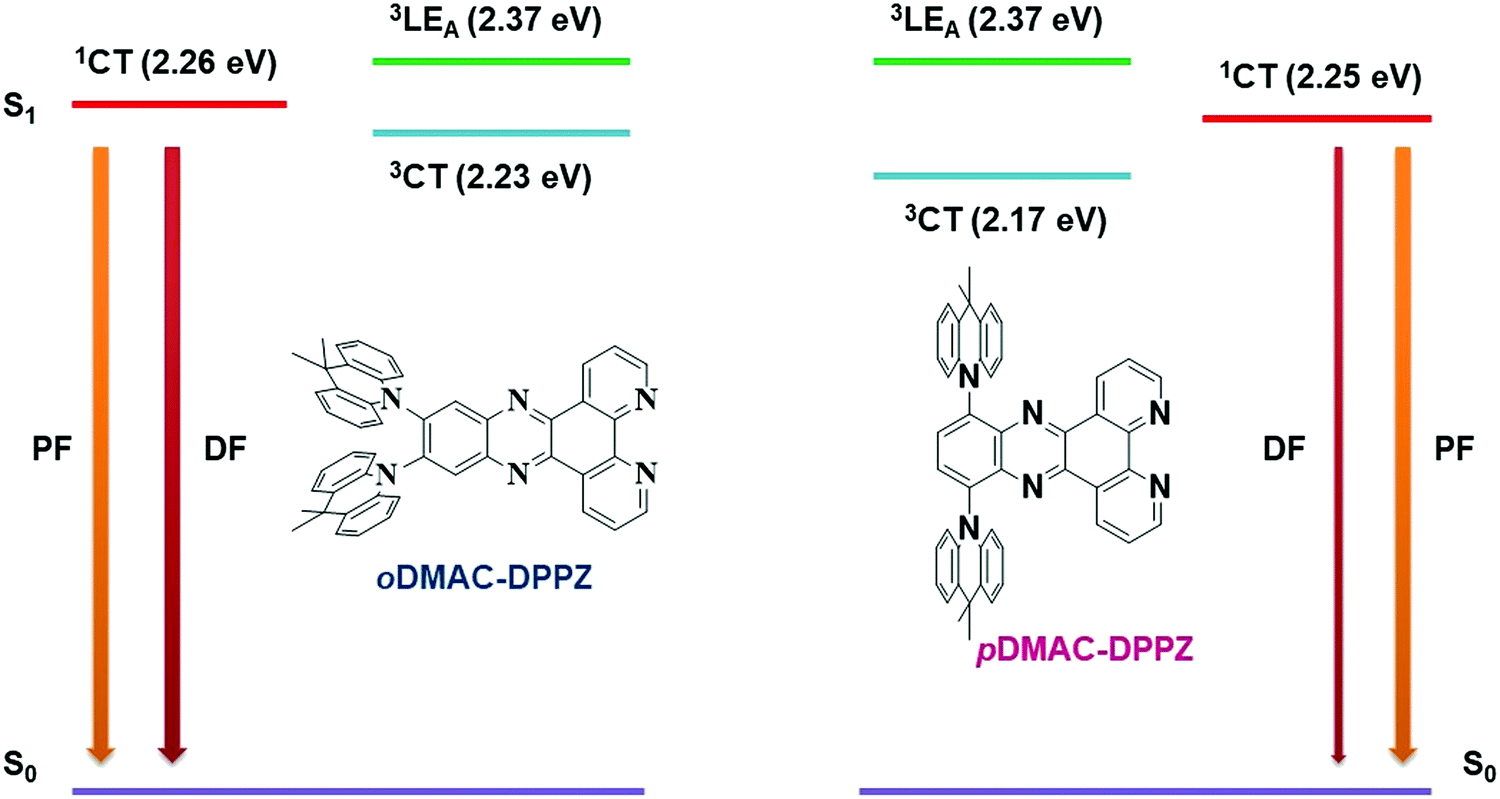
To find out the contribution of locally excited triplet state (3LEA) in the emission process, phosphorescence emission of DPPZ acceptor unit was measured in toluene solvent at low temperature (Fig. S10, ESI†). From the edge of the phosphorescence spectra, the triplet energy of the acceptor unit (3LEA) was calculated to be 2.37 eV. Emission mechanism of oDMAC–DPPZ- and pDMAC–DPPZ TADF emitters was drawn in Fig. 3. Though the prompt fluorescence (PF) of both the emitter is similar, their delayed fluorescence (DF) mechanism is different and mainly affected by the locally excited triplet state of the acceptor unit (3LEA) in the oDMAC–DPPZ TADF emitter. As compared to the pDMAC–DPPZ TADF emitter, the energy gap between charge transfer (3CT) and locally excited (3LEA) triplet states of oDMAC–DPPZ TADF emitter was decreased to enhance the RISC process between singlet and triplet excited charge transfer states (1CT and 3CT) through the contribution of 3LEA. The small energy gap for vibrational coupling between 3CT and 3LEA is responsible for the quick and efficient RISC of oDMAC–DPPZ.
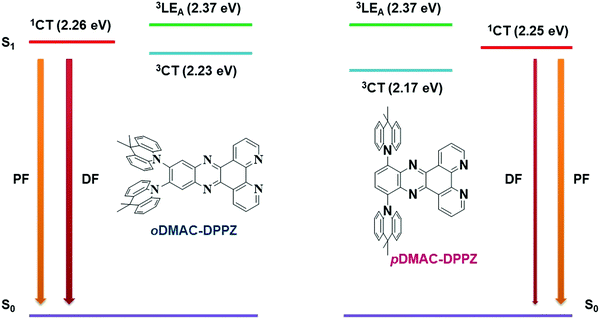 |
| | Fig. 3 Energy level diagram of oDMAC–DPPZ and pDMAC–DPPZ explaining the contribution of locally excited triplet state (3LEA) of DPPZ acceptor in the RISC process of the oDMAC–DPPZ TADF emitter. | |
The contribution of prompt (ΦF) and delayed component (ΦTADF) to the total ΦPL was calculated and the respective rate constants of prompt and delayed component (kp and kd) were calculated using the values of ΦF and ΦTADF. Similarly, the rate constant values of intersystem crossing and reverse intersystem crossing (kISC and kRISC) were calculated from the respective values of ΦF/kp and ΦTADF/kd, respectively. All the transient PL related parameters were calculated using the equations reported earlier26 and are summarized in Table 2. In the two TADF emitters, comparatively higher kd and kRISC values (5.68 × 105 and 4.01 × 106 s−1) were obtained for the oDMAC–DPPZ emitter because of its comparatively small ΔEST value (0.037 eV) and short delayed fluorescence lifetime (1.76 μs) relative to that of the pDMAC–DPPZ emitter (0.079 eV and 5.03 μs). The kRISC/kISC ratio of oDMAC–DPPZ TADF emitter was higher (0.14) than that of the pDMAC–DPPZ TADF emitter (0.02), confirming effective triplet exciton utilization in this emitter. The kRISC values (4.01 and 2.47 × 106 s−1) of both emitters were much higher than their respective knr values (2.29 and 1.71 × 105 s−1), suggesting effective utilization of triplet excitons by both emitters in the red region. However, the EQE of pDMAC–DPPZ was decreased because of its low PLQY value (15%).
Table 2 Transient PL related photophysical parameters (ΦF, ΦTADF, kp, kd, kISC, kRISC, kr and knr) of oDMAC–DPPZ and pDMAC–DPPZ TADF emitters
| Emitter |
Φ
F [%] |
Φ
TADF [%] |
k
p [× 107 s−1] |
k
d [× 105 s−1] |
k
ISC [× 107 s−1] |
k
RISC [× 106 s−1] |
k
RISC
/k
ISC
|
k
r [× 106 s−1] |
k
nr [× 105 s−1] |
|
oDMAC–DPPZ
|
0.09 |
0.54 |
3.70 |
5.68 |
3.39 |
4.01 |
0.1384 |
3.12 |
2.29 |
|
pDMAC–DPPZ
|
0.01 |
0.14 |
9.87 |
1.99 |
9.76 |
2.47 |
0.0253 |
1.11 |
1.71 |
Thermal and electrochemical properties
The thermogravimetric analysis (TGA) of oDMAC–DPPZ and pDMAC–DPPZ was performed at a heating rate of 10 °C min−1 under a nitrogen atmosphere, to evaluate their thermal stability at elevated temperatures. Fig. S3 (ESI†) presents the TGA curves of the two TADF emitters. The decomposition temperature of both emitters was very high (>400 °C) at 5% weight loss, confirming their excellent thermal stability, and thus, their suitability for vacuum-evaporation-based device fabrication. Cyclic voltammetry measurements were performed to investigate the effect of the DMAC substitution pattern on the HOMO–LUMO energy levels of the two compounds using a conventional three-electrode system. As expected, lower oxidation/reduction potential values were obtained for pDMAC–DPPZ (0.92/−1.18 V) compared to those of oDMAC–DPPZ (0.97/−1.22 V) (Fig. S4, ESI†). The HOMO and LUMO energy levels of the TADF emitters were calculated from the corresponding oxidation and reduction onset potential values. The HOMO/LUMO energy levels of pDMAC–DPPZ and oDMAC–DPPZ were calculated to be −5.73/−3.62 eV and −5.78/−3.59 eV, respectively. A relatively small HOMO–LUMO energy gap was obtained for the pDMAC–DPPZ emitter (2.11 eV) than the oDMAC–DPPZ emitter (2.19 eV). Therefore, a slightly more red-shifted emission wavelength can be predicted for pDMAC–DPPZ, compared to that of oDMAC–DPPZ.
Electroluminescence performance
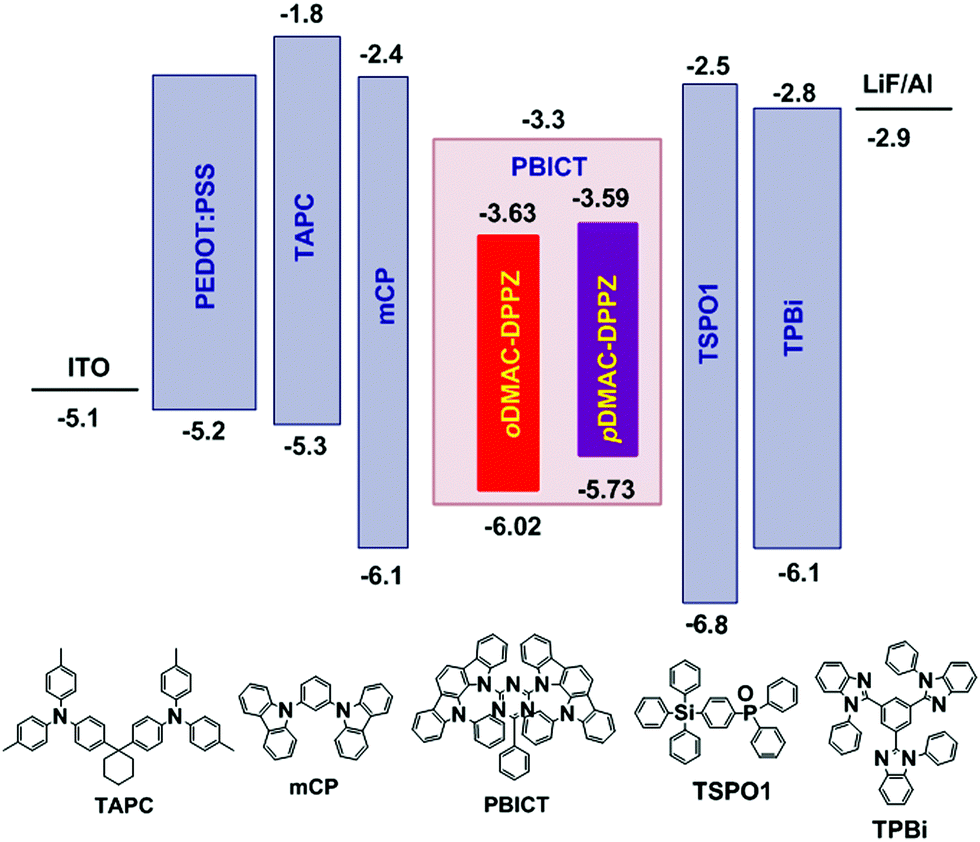
Finally, oDMAC–DPPZ- and pDMAC–DPPZ-doped OLEDs were fabricated, and their performances were evaluated. The OLED devices were fabricated using oDMAC–DPPZ or pDMAC–DPPZ as TADF emitters; poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), 4,4′-cyclohexylidenebis[N,N-bis(4-methylphenyl)aniline] (TAPC), and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) as the hole-injection layer, hole-transporting layer, and electron-transporting layer, respectively; N,N-dicarbazolyl-3,5-benzene (mCP) and diphenyl-4-triphenylsilyl phenyl phosphine oxide (TSPO1) as the high triplet energy and hole- and electron-transport type exciton blocking layers, respectively; and 2-phenyl-4,6-bis(12-phenylindolo[2,3-a]carbazole-11-yl)-1,3,5-triazine (PBICT) as a bipolar host material to ensure high EQEs. The devices were structured as follows: indium tin oxide (ITO, 50 nm)/PEDOT: PSS (60 nm)/TAPC (20 nm)/mCP (10 nm)/PBICT:TADF emitter (25 nm: 1, 3, and 5 wt%)/TSPO1 (5 nm)/TPBi (40 nm)/LiF (1.5 nm)/Al (200 nm). Fig. 4 shows the energy level diagrams and chemical structures of the materials used in the devices. The Current density–voltage–luminance (J–V–L), EQE–luminance and EL spectra of the oDMAC–DPPZ and pDMAC–DPPZ-doped OLEDs are shown in Fig. 5 and Fig. S5 (ESI†), and the values of key device parameters are summarized in Table 3.
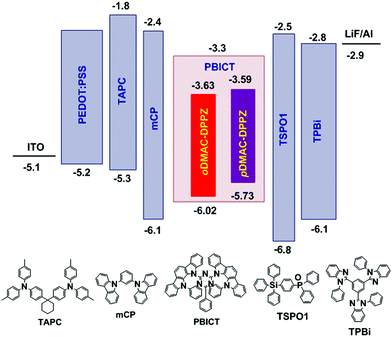 |
| | Fig. 4 Energy-level diagrams and chemical structures of the materials used in the OLEDs doped with oDMAC–DPPZ and pDMAC–DPPZ TADF emitters. | |
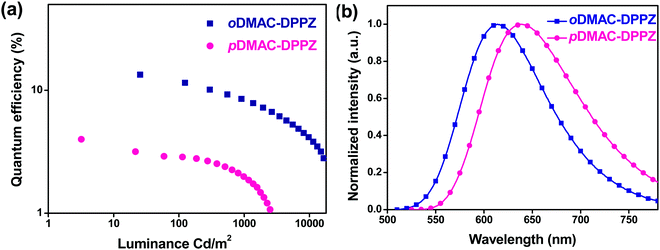 |
| | Fig. 5 (a) EQE–luminance characteristics, and (b) EL spectra of oDMAC–DPPZ- and pDMAC–DPPZ-doped OLEDs (1% doping concentration). | |
Table 3 Electroluminescence performances of the oDMAC–DPPZ- and pDMAC–DPPZ-emitter-based orange-red TADF-OLEDs at 1% doping concentration
| Emitter |
λ
EL [nm] |
CEmax [cd A−1] |
PEmax [lm W−1] |
EQEmax [%] |
CIE (x, y) |
|
oDMAC–DPPZ
|
614 |
21.1 |
18.9 |
13.4 |
(0.59, 0.40) |
|
pDMAC–DPPZ
|
638 |
4.2 |
3.8 |
4.0 |
(0.64, 0.35) |
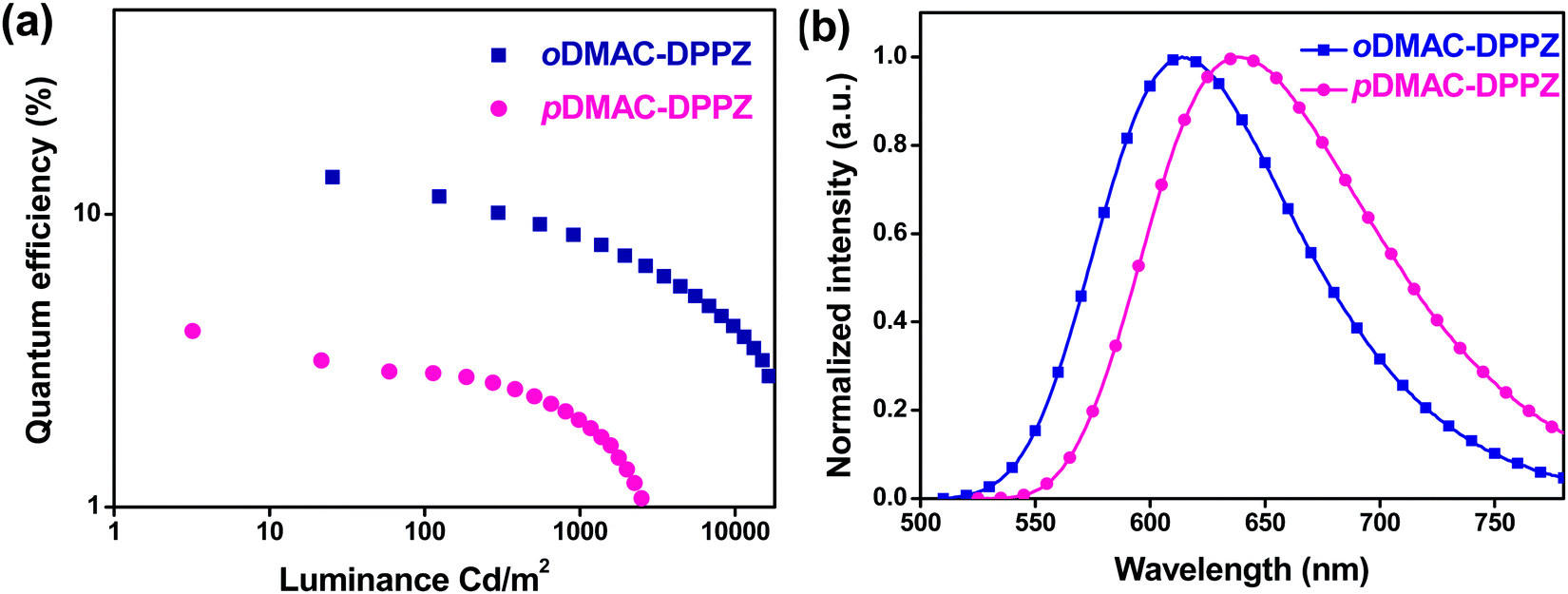
The EL spectra emitted by the OLED devices resembled those of TADF PL emission, confirming that the EL emission had originated exclusively from the emitter without any host material interference. oDMAC–DPPZ exhibited a red emission at 614 nm with the Commission Internationale de L’Eclairage (CIE) coordinates of (0.59, 0.40) and pDMAC–DPPZ exhibited a red-shifted emission at 638 nm with the CIE coordinates of (0.64, 0.35). Consistent with the high ΦPL value of oDMAC–DPPZ (63%), the OLED doped with this emitter exhibited a high current efficiency of 21.1 cd A−1, high power efficiency of 18.9 lm W−1, and high EQE of 13.4%, while for the pDMAC–DPPZ-doped OLED, these values were 4.2 cd A−1, 3.8 lm W−1, and 4.0%, respectively. Both the devices exhibit maximum EQE values at a low doping concentration of 1 wt%, decreasing with increasing doping concentrations owing to the concentration quenching effect, as shown in Fig. S6 and S7 (ESI†). The EL spectra of both the devices were further red shifted with increasing doping concentrations of 1 to 5 wt% (Fig. S8 and S9, ESI†). From the above results, it can be concluded that the ortho donor substitution at positions 11 and 12 of the DPPZ acceptor resulted in a better OLED performance than that of the para-substituted TADF emitter.
Conclusion
Two configurational isomers of long-wavelength TADF emitters were synthesized to study the effect of the donor substitution pattern on their OLED performances. The ortho-substituted TADF emitter, oDMAC–DPPZ, exhibited a higher PLQY (63%) than the para-substituted isomer, pDMAC–DPPZ (15%) due to efficient RISC process assisted by small ΔEST. The red TADF-OLED doped with oDMAC–DPPZ exhibited a considerably higher EQE (13.4%) than the pDMAC–DPPZ-doped OLED (4.0%). Therefore, it was demonstrated that donor substitution at the ortho (11, 12) position of the DPPZ acceptor is preferred to the para (10, 13) substitution in the design of high-performance long-wavelength TADF emitters.
Experimental
Synthesis
Detailed synthetic procedures for the TADF emitters are given in the Supporting Information.
Device fabrication and measurements
The respective layers of the device were deposited via vacuum thermal evaporation under a high pressure of 3.0 × 10−7 Torr. Following the thermal evaporation of each material, the device was placed under a glass lid in a nitrogen-filled glove box to exclude moisture and oxygen. The electrical characterization of the devices was performed using a Keithley 2400 source meter, and optical characterization was conducted using a CS 2000 spectroradiometer.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Ministry of Trade, Industry, and Energy (20006464).
References
- K. Goushi and C. Adachi, Appl. Phys. Lett., 2012, 101, 23306 CrossRef.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- S. S. Kothavale and J. Y. Lee, Adv. Opt. Mater., 2020, 8, 2000922 CrossRef CAS.
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2021, 2106954 Search PubMed.
- Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li and L. Duan, Adv. Mater., 2021, 33, 2103293 CrossRef CAS PubMed.
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B.-M. Seo, J.-Y. Kim, C. W. Han and J. H. Kwon, Adv. Mater., 2021, 33, 2007724 CrossRef CAS PubMed.
- C.-C. Peng, S.-Y. Yang, H.-C. Li, G.-H. Xie, L.-S. Cui, S.-N. Zou, C. Poriel, Z.-Q. Jiang and L.-S. Liao, Adv. Mater., 2020, 32, 2003885 CrossRef CAS PubMed.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32, 2003911 CrossRef CAS PubMed.
- S. Kothavale, K. H. Lee and J. Y. Lee, Chem. – Eur. J., 2020, 26, 845–852 CrossRef CAS PubMed.
- S. Kothavale, K. H. Lee and J. Y. Lee, Chem. – Asian J., 2020, 15, 122–128 CrossRef CAS PubMed.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- D. R. Lee, B. S. Kim, C. W. Lee, Y. Im, K. S. Yook, S.-H. Hwang and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 9625–9629 CrossRef CAS PubMed.
- C. Li, C. Duan, C. Han and H. Xu, Adv. Mater., 2018, 30, 1804228 CrossRef PubMed.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- D. R. Lee, M. Kim, S. K. Jeon, S.-H. Hwang, C. W. Lee and J. Y. Lee, Adv. Mater., 2015, 27, 5861–5867 CrossRef CAS PubMed.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed.
- I. S. Park, S. Y. Lee, C. Adachi and T. Yasuda, Adv. Funct. Mater., 2016, 26, 1813–1821 CrossRef CAS.
- S. Kothavale, K. H. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 17583–17591 CrossRef CAS PubMed.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2021, 9, 528–536 RSC.
- S. Kothavale, W. J. Chung and J. Y. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 18730–18738 CrossRef CAS PubMed.
- S. Wang, Y. Miao, X. Yan, K. Ye and Y. Wang, J. Mater. Chem. C, 2018, 6, 6698–6704 RSC.
- C. Zhou, C. Cao, D. Yang, X. Cao, H. Liu, D. Ma, C.-S. Lee and C. Yang, Mater. Chem. Front., 2021, 5, 3209–3215 RSC.
- U. Balijapalli, Y.-T. Lee, B. S. B. Karunathilaka, G. Tumen-Ulzii, M. Auffray, Y. Tsuchiya, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 19364–19373 CrossRef CAS PubMed.
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed.
- S. Kothavale, J. Lim and J. Yeob Lee, Chem. Eng. J., 2022, 431, 134216 CrossRef CAS.
- B. Zhao, H. Wang, C. Han, P. Ma, Z. Li, P. Chang and H. Xu, Angew. Chem., Int. Ed., 2020, 59, 19042–19047 CrossRef CAS PubMed.
- H. Wang, B. Zhao, P. Ma, Z. Li, X. Wang, C. Zhao, X. Fan, L. Tao, C. Duan, J. Zhang, C. Han, G. Chen and H. Xu, J. Mater. Chem. C, 2019, 7, 7525–7530 RSC.
- J.-X. Chen, K. Wang, C.-J. Zheng, M. Zhang, Y.-Z. Shi, S.-L. Tao, H. Lin, W. Liu, W.-W. Tao, X.-M. Ou and X.-H. Zhang, Adv. Sci., 2018, 5, 1800436 CrossRef PubMed.
- J.-X. Chen, W.-W. Tao, W.-C. Chen, Y.-F. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F.-X. Geng, C. Adachi, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14660–14665 CrossRef CAS PubMed.
- A. Shang, T. Lu, H. Liu, C. Du, F. Liu, D. Jiang, J. Min, H. Zhang and P. Lu, J. Mater. Chem. C, 2021, 9, 7392–7399 RSC.
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li and Y. Wang, Adv. Funct. Mater., 2020, 30, 2002681 CrossRef CAS.
- C. Zhou, W.-C. Chen, H. Liu, X. Cao, N. Li, Y. Zhang, C.-S. Lee and C. Yang, J. Mater. Chem. C, 2020, 8, 9639–9645 RSC.
- Y. J. Cho, S. K. Jeon, S.-S. Lee, E. Yu and J. Y. Lee, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- G. Meng, X. Chen, X. Wang, N. Wang, T. Peng and S. Wang, Adv. Opt. Mater., 2019, 7, 1900130 CrossRef.
- D.-Y. Chen, W. Liu, C.-J. Zheng, K. Wang, F. Li, S. Tao, X.-M. Ou and X.-H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 16791–16798 CrossRef CAS PubMed.
- Z.-P. Chen, D.-Q. Wang, M. Zhang, K. Wang, Y.-Z. Shi, J.-X. Chen, W.-W. Tao, C.-J. Zheng, S.-L. Tao and X.-H. Zhang, Adv. Opt. Mater., 2018, 6, 1800935 CrossRef.
- J. Jayakumar, T.-L. Wu, M.-J. Huang, P.-Y. Huang, T.-Y. Chou, H.-W. Lin and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 21042–21048 CrossRef CAS PubMed.
- T.-L. Wu, S.-H. Lo, Y.-C. Chang, M.-J. Huang and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 10768–10776 CrossRef CAS PubMed.
- W. Xie, M. Li, X. Peng, W. Qiu, Y. Gan, Z. Chen, Y. He, W. Li, K. Liu, L. Wang, Q. Gu and S.-J. Su, Chem. Eng. J., 2021, 425, 131510 CrossRef CAS.
- J.-X. Chen, Y.-F. Xiao, K. Wang, D. Sun, X.-C. Fan, X. Zhang, M. Zhang, Y.-Z. Shi, J. Yu, F.-X. Geng, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS PubMed.
- H. Wang, L. Zhou, Y.-Z. Shi, X.-C. Fan, J.-X. Chen, K. Wang, J. Yu and X.-H. Zhang, Chem. Eng. J., 2022, 433, 134423 CrossRef CAS.
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma and C. Yang, J. Mater. Chem. C, 2017, 5, 1363–1368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2tc00017b |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *
*