DOI:
10.1039/D1TC06165H
(Paper)
J. Mater. Chem. C, 2022,
10, 7866-7874
Influence of charge transfer strength on emission bandwidth for multiple-resonance emitters via systematically tuning the acceptor–donor assembly†
Received
30th December 2021
, Accepted 7th March 2022
First published on 19th March 2022
Abstract
Multiple resonance (MR) thermally activated delayed fluorescence (MR-TADF) materials, categorized for the B/N and carbonyl/amine fragments, are now intensively and widely studied due to their color purity. However, the relationship between the MR molecular structure and full width at half maximum (FWHM) of the emission remains elusive. In order to probe the factor of determining the emission FWHM of the MR emitters, a series of relevant molecules were synthesized where the MR core acceptor moiety, phenylborane or acetophenone, is fixed, while the donor moiety is altered to adjust the charge transfer (CT) strength. These potential MR molecules provide sufficient photophysical data to shed light on the emission bandwidth influenced by CT strength. Meanwhile, OLEDs fabricated using all the studied compounds achieve maximum external quantum efficiencies (EQEmax) around 7.0–15.3% because of the TADF character induced by the host/guest interaction in the excited state. The results provide further understanding on the fundamentals of the MR emitters.
1. Introduction
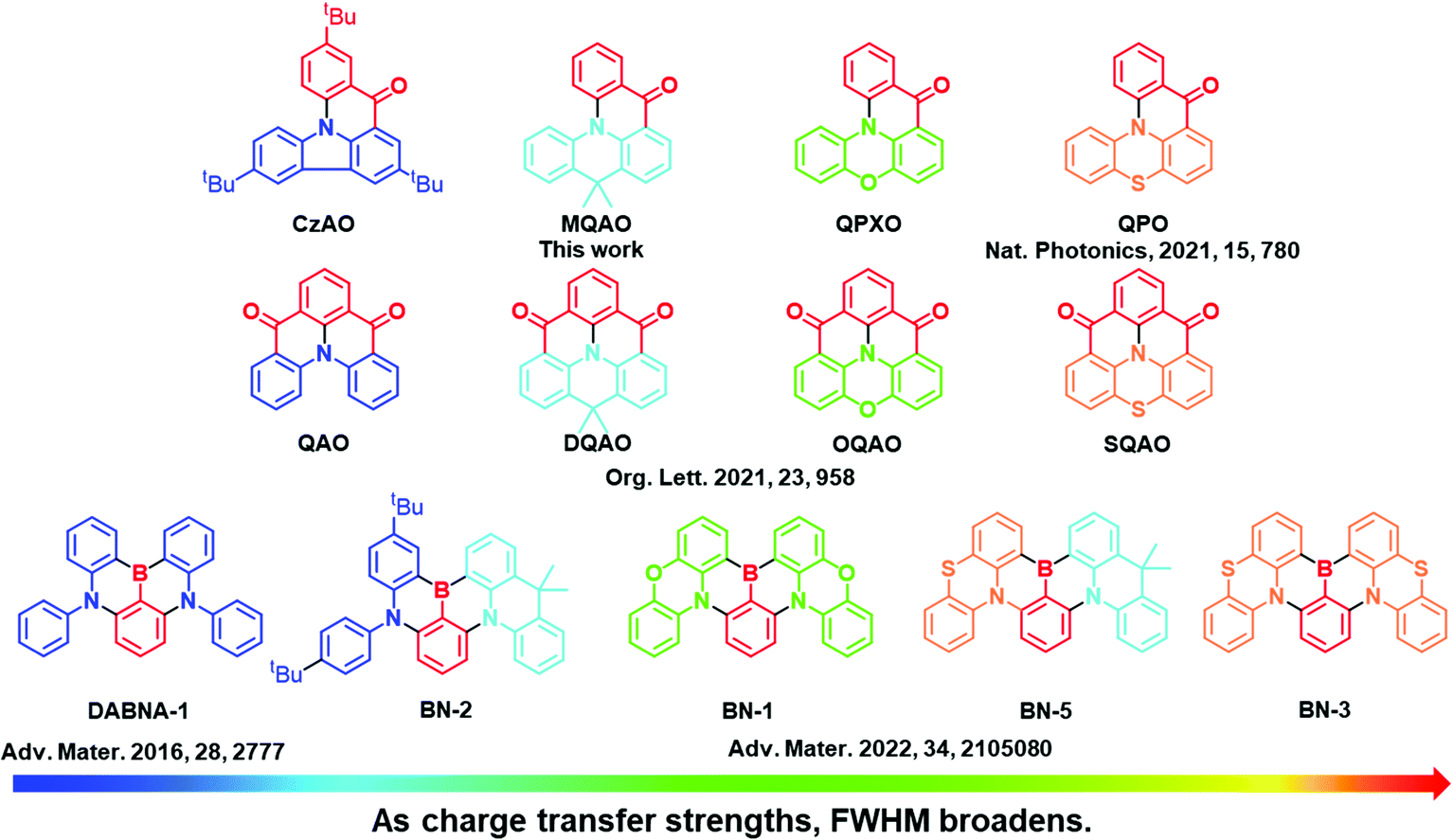
Highly efficient exciton harvesting and improved color fidelity of emitters are always top demands in the optoelectronic community. State-of-the-art OLEDs based on noble-metal phosphorescence and thermally activated delayed fluorescence (TADF) emitters have made great progress in exciton utilization efficiency (EUE).1–3 However, both types of emitters, to a certain extent, are subject to inferior color gamut. On the one hand, a key reason of a broad emission FWHM for transition metal complexes is the coordination geometry of noble-metal complexes, where the metal-to-ligand charge transfer (MLCT) contribution to the lowest lying optical transition is significant. On the other hand, to achieve a negligible S1–T1 energy gap (ΔEST) for typical TADF emitters requires large spatial separation between the donor and acceptor, compromising the broadening of the spectral FWHM. Until the discovery of emitters based on multiple resonance thermally activated delayed fluorescence (MR-TADF), their narrow FWHM was able to compete with LEDs based on, e.g., gallium nitrides (micro-LEDs) and CdS/ZnS or CdSe/ZnS quantum dots (QD-LEDs).4,5 Accordingly, MR-TADF emitters with panchromatic wavelength tuning from blue4–18 through green19–27 to red28–30 have sprung up and rushed. The key to high color purity of MR-TADF emitters, from the perspective of electronic structure and computational simulations,5,31 seems to lie in the strong ortho-positioned transition orbitals, i.e. the highest occupied molecular orbital (HOMO) filling the gaps in the lowest unoccupied molecular orbital (LUMO), while maintaining the rigid structure of polycyclic aromatic hydrocarbons. This unique property rationalizes the reduction of the FWHM, in a qualitative manner. Meanwhile, the locally excited (LE) π-conjugated transition is expected to give a larger ΔEST and hence impair the TADF properties.32–34 Such a trade-off leads to several puzzling phenomena for MR-TADF properties, where two main issues are raised. (1) In the photoluminescence study, some MR materials lack TADF in solution but reveal prominent TADF when doped in the host material. (2) The relationship between the MR molecular structure and emission FWHM is of both fundamental and application importance, which unfortunately remains elusive. Based on the transient absorption studies, issue (1) has recently been deciphered, concluding the host/MR guest interaction via the charge transfer effect in the excited state.35 As for issue (2), parameters that influence the emission FWHM have been analyzed by the Huang–Rhys factor deduced from the Franck–Condon principle, whereas the generalization of MR molecules seems to be underexplored.36 Narrow emission bandwidth emitters are of importance in practical applications. Recently, the ternary architecture exploiting MR molecules as the terminal emitters, traditional host and TADF or noble-metal phosphorescence materials as the energy donor, is considered to be one of the prototypes for next generation OLEDs.10,37
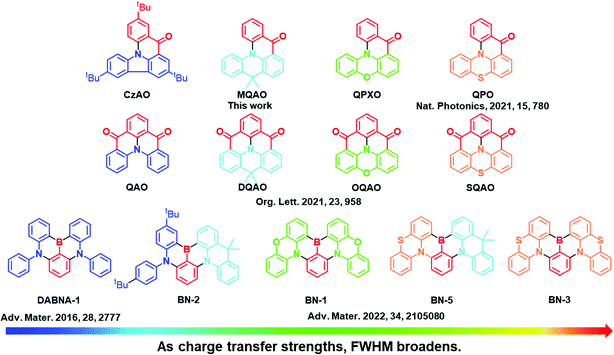
Herein, a series of potential MR emitters were synthesized and investigated in a systematic manner to probe issue (2), which are associated with the interplay between LE and charge transfer (CT) parameters of the MR materials (vide infra). In order to rationally probe the CT effect, the prototypical B/N and carbonyl/amine type MR core moieties, phenylborane and acetophenone, respectively, serve as the electron acceptor, while the electron donors are altered among diphenylamine (DPA), carbazole (Cz), 9,10-dihydro-9,9-dimethyl-acridine (DMAc), phenoxazine (PXO) and phenothiazine (PTZ). As a result, the B/N series including DADBA-1, BN-2, BN-1, BN-5 and BN-3, together with carbonyl/amine analogs containing CzAO, MQAO, QPXO and QPO (see Scheme 1), were prepared and investigated. Note that the carbonyl/amine analogs are all new, while most of the B/N analogs were synthesized in our recent report.38 The same acceptor for the analogs allows us to assess the trend of CT properties by tuning the electron donating strength. Notably, with increasing donor strength in the order of Cz < DMAc < PXO < PTZ,39,40 the stronger CT character contributes to a larger emission FWHM. The experimental results are in good agreement with the computational results. Accordingly, a mechanism based on the degree of charge transfer determining the FWHM is proposed.
 |
| | Scheme 1 MR molecules configured by regulating the strength of charge transfer. | |
In parallel, OLEDs based on the studied MR molecules have been fabricated. Remarkably, despite the lack of TADF in solution, all the studied emitters achieved a high EQEmax above 7%, indicating that reverse intersystem crossing (RISC) takes place in the host-doped film upon electronic excitation. Nevertheless, the rate of RISC is rather slow due to a large ΔEST, resulting in serious roll-off in the device performance. The details of the results and discussion are elaborated in the following sections.
2. Results and discussion
2.1. Synthesis and crystal structure analysis
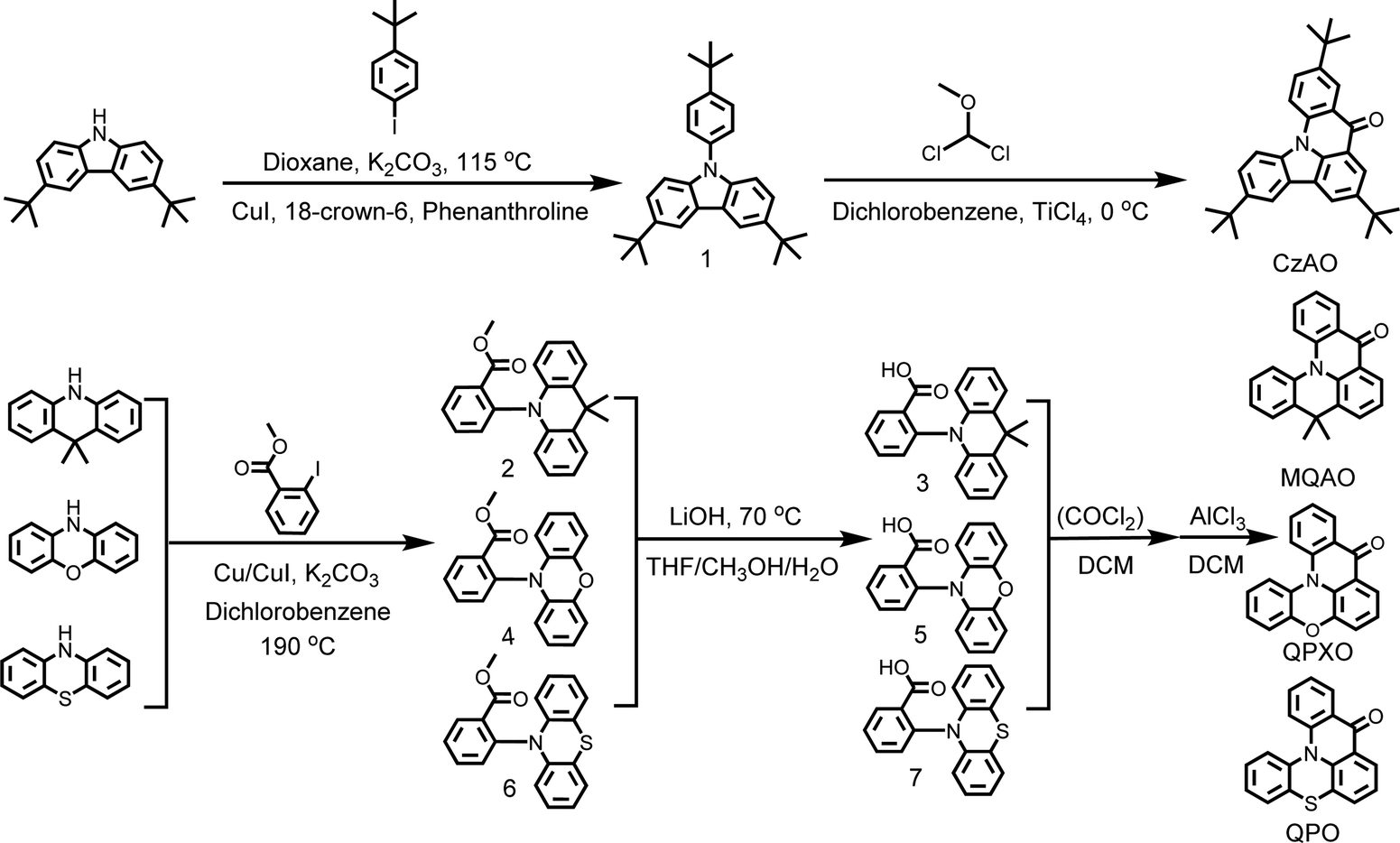
A series of new carbonyl/amine type MR emitters, MQAO, QPXO and QPO, were synthesized by Ullmann coupling reaction, followed by hydrolysis and Friedel–Crafts acylation in sequence.13 In contrast, CzAO was successfully synthesized by a novel Friedel–Crafts intramolecular cyclization in one step (Scheme 2).41 The advantages of the latter are that it is time-saving and high-yield. We believe that this method may be further expanded to the carbonyl/amine MR system. All the intermediates and final molecules were characterized by NMR, Fourier transform infrared (FT-IR) spectroscopy, mass spectrometry (MALDI-TOF-MS) and elemental analyses.
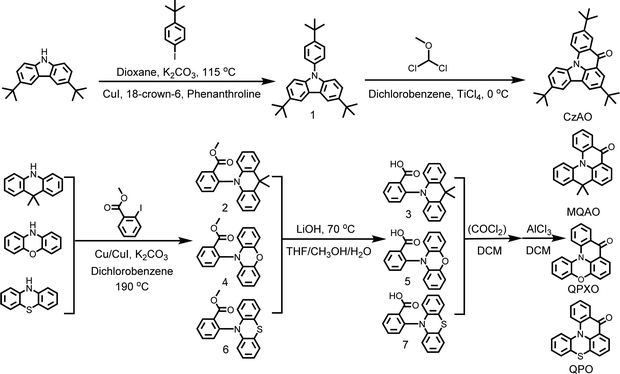 |
| | Scheme 2 Synthetic routes to CzAO, MQAO, QPXO and QPO. | |
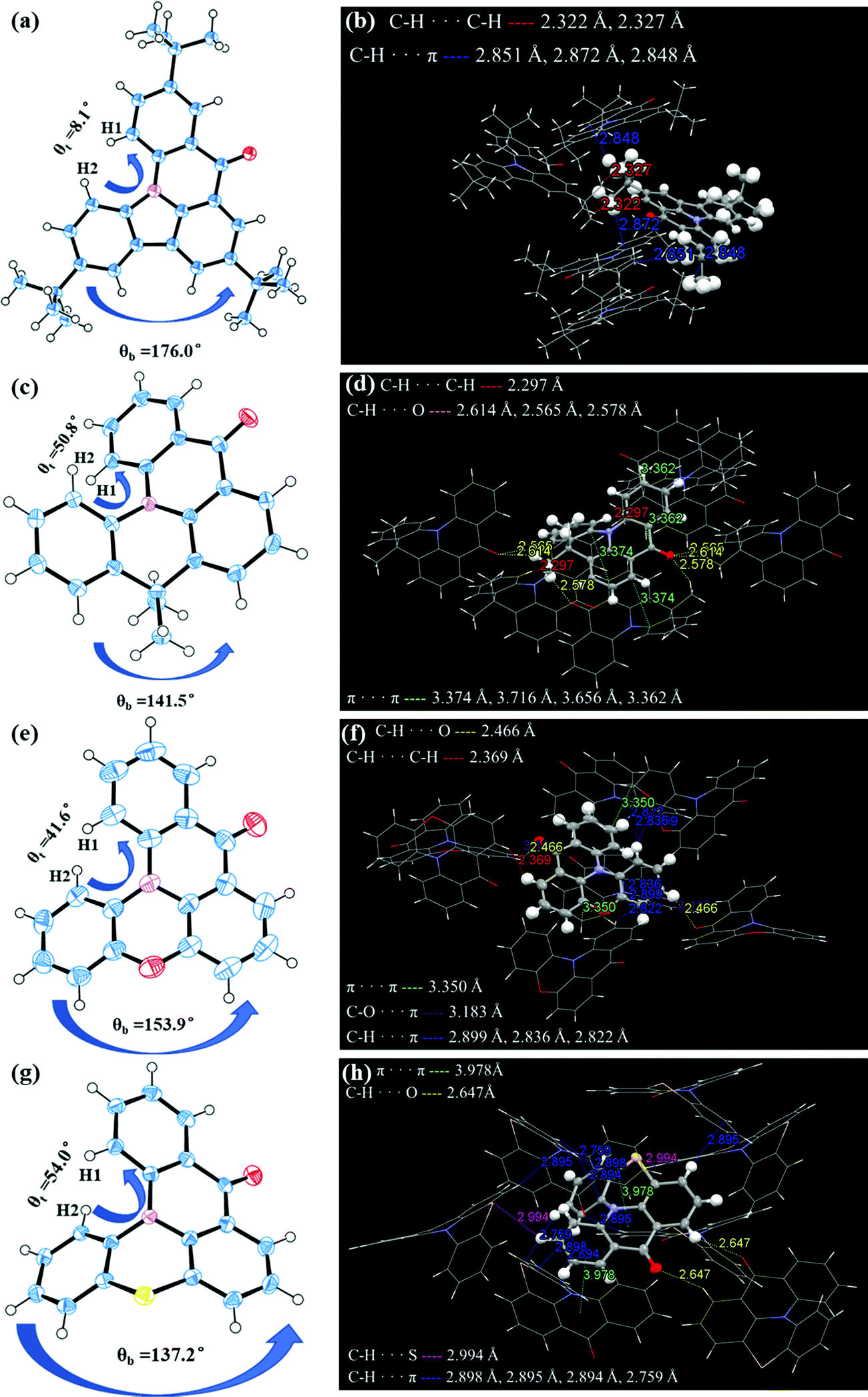
The single-crystal X-ray diffraction analysis further confirmed the structures of the four molecules and the related crystallographic data are listed in Table S1 (ESI†). Due to the steric hindrance introduced by hydrogen atoms (H1 and H2 in Fig. 1) and the boat conformation of PTZ, DMAc and PXO,42,43 all molecules exhibit different degrees of helical structure in the single crystal. With the transition from the plane conformation of Cz to the boat conformation of PTZ, their corresponding bending angles (denoted as θb) also decreased from 176.0° of CzAO to 153.9° of QPXO, to 141.5° of MQAO and to 137.2° of QPO. The results also imply that their torsion angle (denoted as θt) in the fjord regions increases from 8.1° of CzAO to 41.6° of QPXO, to 50.8° of MQAO and finally to 54.0° of QPO, which would increase the possibility of structural relaxation in excited states, affecting the emission FWHM (Fig. 1). It is worth noting that the introduction of hybrid atoms such as oxygen and sulfur to QPXO and QPO, respectively, results in more abundant short-range interaction forces (e.g., C–H⋯π, C–O⋯π, C–H⋯S and so on) compared to MQAO and CzAO.
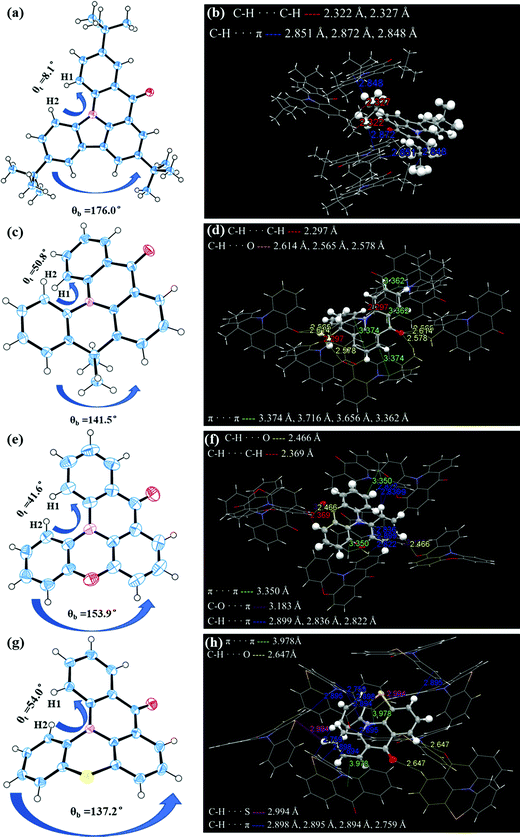 |
| | Fig. 1 Angle analysis in the X-ray structure and intermolecular interactions of single molecules with adjacent ones for (a and b) CzAO, (c and d) MQAO, (e and f) QPXO and (g and h) QPO. | |
2.2. Photophysical properties
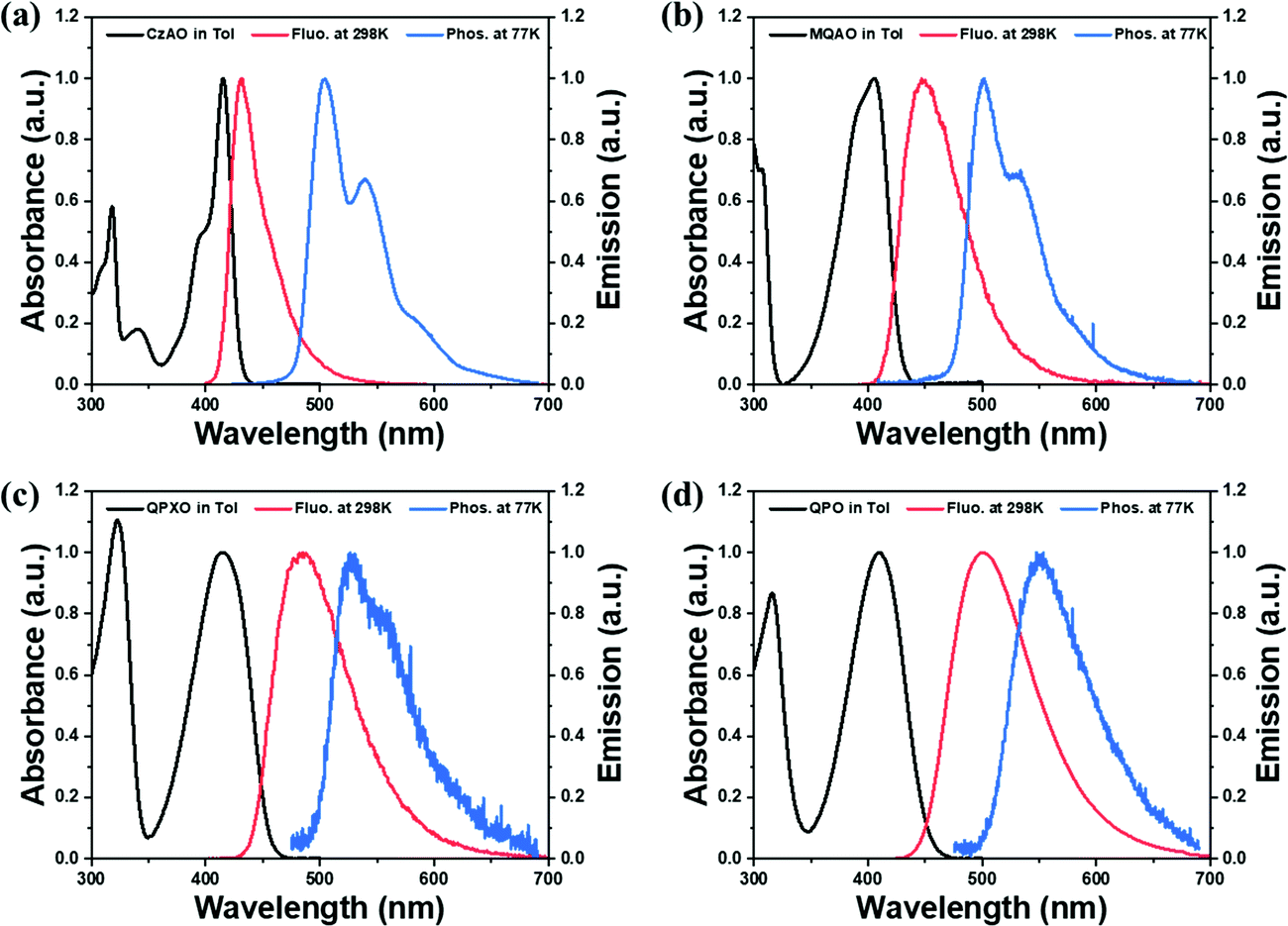
The ultraviolet-visible (UV-vis) absorption, photoluminescence (PL) at 298 K and phosphorescence at 77 K spectra of CzAO, MQAO, QPXO and QPO were measured in toluene (Fig. 2) and the results are summarized in Table 1. All emitters show a strong and sharp absorption peak around 400–420 nm, which can be assigned to the lowest lying electronic transition. Notably, the emission spectra of CzAO, MQAO, QPXO and QPO in toluene solution exhibited a bathochromic shift in the order of CzAO (431 nm) < MQAO (447 nm) < QPXO (485 nm) < QPO (501 nm), accompanied by the same increasing trend of spectral FWHM by 36 nm (0.23 eV) < 61 nm (0.36 eV) < 76 (0.39 eV) < 86 nm (0.41 eV). Furthermore, the Stokes shift also demonstrated the same trend, being in the order of 895 cm−1 (CzAO) < 2320 cm−1 (MQAO) < 3595 cm−1 (QPXO) < 4430 cm−1 (QPO) in toluene (Table S3, ESI†). The electron donor strengths of alternative donors, which are in the order of Cz < DMAc < PXO < PTZ,39 can reflect the CT strength for the targeted molecules. To further confirm the CT characteristics, the UV-vis absorption and PL spectra of these emitters were measured in different solvents at room temperature (Fig. S8–S11, ESI†). As a result, the onsets and peaks of absorption in the lowest lying electronic transition had an insignificant bathochromic shift with increasing solvent polarity. In sharp contrast, the PL spectra displayed a significant positive solvatochromic effect, where the difference in peak wavelength from n-hexane (the lowest polar solvent used in this study) to methanol was calculated to be 53, 76, 99 and 116 nm for CzAO, MQAO, QPXO and QPO, respectively. Importantly, following the increasing FWHM trend, the emission spectral profiles are gradually transformed from the MR-characterized fine structure to the CT-dominated structureless shape upon increasing the electron donating strength in the order of CzAO < MQAO < QPXO < QPO (Table S3, ESI†). Qualitatively, the CT strength seems to be crucial to the FWHM of the studied emitters. The same trend appears in the other series of QAO, DQAO, OQAO and SQAO (Scheme 1),44 though not as obvious, where the FWHM increases in the order of 33 nm (QAO), 33 nm (DQAO), 36 nm (OQAO) to 54 nm (SQAO) in toluene solution. Furthermore, the reported B/N type MR systems are also investigated to verify the CT–FWHM relationship (Fig. S6 and Table S2, ESI†). With the fixed acceptor of phenylborane and strengthened donors, the FWHM of DABNA-1, BN-2, BN-1, BN-5 and BN-3 (Scheme 1)5,38 becomes widened from 24, 34, 41, 44 to 46 nm for the B/N series in the toluene solution. Note that the emission FWHM calculated in the energy domain follows the same trend, being in the order of 0.140 (DABNA-1) < 0.178 (BN-2) < 0.197 (BN-1) < 0.215 (BN5) < 0.221 eV (BN3). In line with the broadening FWHM upon enhancing the CT strength, the ΔEST values, estimated from the fluorescence and phosphorescence spectra in toluene solution at 77 K, decrease in the order of 0.40, 0.31, 0.29 and 0.27 eV for CzAO, MQAO, QPXO and QPO, respectively (Fig. S4, ESI†).
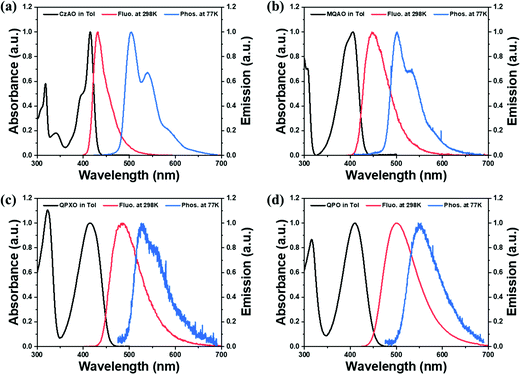 |
| | Fig. 2 Absorption (black line), fluorescence (red line) at 298 K and phosphorescence at 77 K (blue line) of (a) CzAO, (b) MQAO, (c) QPXO and (d) QPO in toluene solution and the frozen matrix (concentration: 10−5 M). | |
Table 1 Summary of the photophysical data and calculated HOMO/LUMO levels of four emitters
| Compound |
λ
abs
(nm) |
λ
em
(nm) |
ΔESTa (eV) |
FWHMa (nm eV−1) |
Stokes shifta (cm−1) |
PLQY (aerateda/degasseda/filmb) |
τ
p
(ns) |
τ
p
(ns) |
τ
d
(μs) |
HOMOc (eV) |
LUMOe (eV) |
E
g
(eV) |
|
Measured in degassed toluene solution (1 × 10−5 M).
Measured in the films of 3 wt% emitters doped in mCPCN.
Measured in neat films by ultraviolet photoelectron spectroscopy (UPS).
Estimated by the onset of the corresponding absorption spectra.
LUMO = −[Egap − HOMO].
|
| CzAO |
415 |
431 |
0.40 |
36/0.23 |
895 |
0.42/0.46/0.49 |
1.93 |
3.68 |
1340 |
−5.55 |
−2.66 |
2.89 |
| MQAO |
405 |
447 |
0.31 |
61/0.36 |
2320 |
0.72/0.85/0.61 |
6.62 |
9.90 |
1024 |
−5.45 |
−2.56 |
2.89 |
| QPXO |
413 |
485 |
0.29 |
76/0.39 |
3595 |
0.75/0.98/0.35 |
9.47 |
14.73 |
2394 |
−5.39 |
−2.66 |
2.73 |
| QPO |
410 |
501 |
0.27 |
86/0.41 |
4430 |
0.33/0.57/0.71 |
7.86 |
10.10 |
1436 |
−5.37 |
−2.61 |
2.76 |
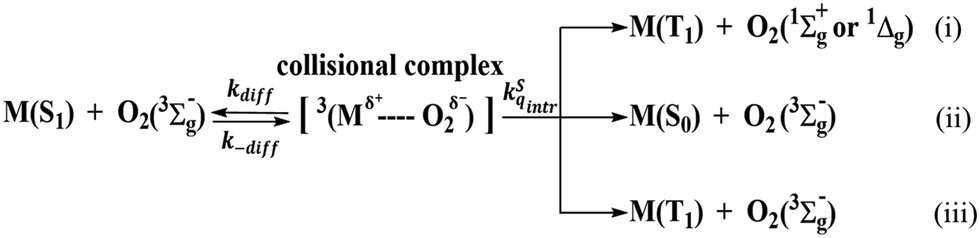
In the same series of derivatives where no heavy atom is involved, the trend of decreasing ΔEST is beneficial to both ISC and RISC rates.45 The former is due to the increase of singlet–triplet mixing according to the perturbation theory, while the latter is the combination of singlet–triplet mixing and the increase of Boltzmann population upon decreasing ΔEST. However, the calculated ΔEST values of ≥ 0.27 eV for the CzAO series is too large to proceed with RISC under regular ISC for organic compounds. This explains the lack of any TADF for CzAO, MQAO, QPXO and QPO in the fully degassed toluene solution (Fig. S1–S4, ESI†), similar to other CzAO analogues where no TADF is reported.11 Despite the lack of TADF in PL, however, we did observe the O2 concentration dependent fluorescence intensity, where the PLQY of CzAO, MQAO, QPXO and QPO jumped to 46%, 85%, 98% and 57% in degassed toluene solution, with an amplification by 4%, 13%, 23%, and 24%, respectively, compared with that in the aerated solution. Along with the steady state emission, the fluorescence decay time has been decreased in the aerated solution (Fig. S1–S4, ESI†). This observation is not uncommon and can be rationalized by the quenching of the S1 state by ground-state triplet oxygen depicted in Scheme 3,46 where pathway (i) is thermodynamically unfavorable due to the much smaller ΔEST of the studied compounds than the energy of singlet sigma or singlet delta oxygen. Both (ii) and (iii) are plausible quenching processes, which are termed as the O2 induced S1 radiationless transition and O2 induced intersystem crossing, respectively. The criterion for the O2 quenching S1 state lies in the relatively long S1 decay time which is typically >5 ns, as observed in MQAO, QPXO and QPO (see Fig. S1–S4, ESI†), so that the O2 collisional quenching can be efficient.
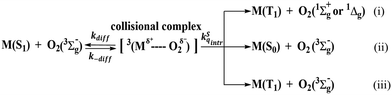 |
| | Scheme 3 The O2 quenching S1 state molecule M based on the spin conservation for the formation of the collisional complex. | |
The above elaborated fundamental concludes that steady-state fluorescence quenched by O2 is not sufficient to determine whether TADF occurs. It may simply indicate the quenching of the S1 state (Scheme 3) if the fluorescence lifetime is long enough as in the current case. In sharp contrast, upon photo-excitation, TADF is observed in their doped film of 9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole-3-carbonitrile (mCPCN) (Fig. S7, ESI†). The transient decay profile detected using an intensified charge-coupled device (ICCD) for doped films of 3% CzAO, MQAO, QPXO and QPO in mCPCN showed delayed fluorescence with a rather long decay component, τd, of 1340, 1024, 2394 and 1436 μs, respectively. Such a discrepancy between the solution and host/guest solid films in MR materials has been recently attributed to the excited-state interaction between the host material and emitters in the co-deposited solid mixture, facilitating both forward and reverse intersystem crossing efficiencies.35 Nevertheless, the delayed fluorescence of these compounds was long enough to cause serious efficiency roll-off in electroluminescence (vide infra). The PLQYs of CzAO, MQAO, QPXO and QPO films are 49%, 61%, 35% and 71%, respectively, where the abnormal decrease of QPXO may be attributed to the aggregation caused quenching (ACQ).
2.3. Computational approach
We then performed the computational simulations to gain further insight into the material properties. Based on the natural transition orbital (NTO) analysis (Fig. S13–S16, ESI†), multiple Tn states were closer to the S1 state, which are more likely to strike the RISC process. Meanwhile, the n-orbital introduced by the carbonyl groups and hybrid atoms of oxygen/sulfur, according to the El-Sayed rule, largely promotes the spin–orbit coupling (SOC) between S1 and Tn (n = 1–5) states. All the studied molecules exhibit 〈S1|ĤSOC|T1〉 that are relatively lower than 〈S1|ĤSOC|Tn〉 (n ≥ 2), manifesting that the triplets may be harvested through Tn → S1. Thus, the carbonyl moiety might play a key role on the exciton dynamics with different spin multiplicities.
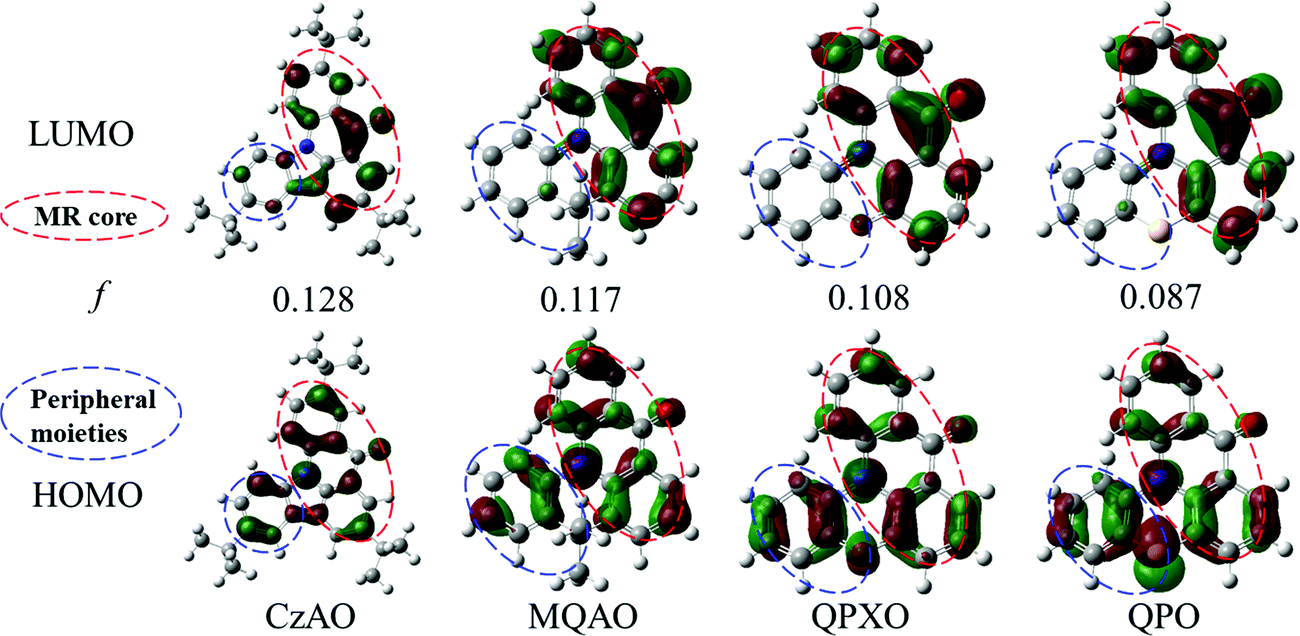
The HOMO and LUMO properties (Fig. 3) were then simulated by density functional theory (DFT). The structures of the four compounds are the combination of the MR core (red-dotted circle) and peripheral moieties (blue-dotted circle). The whole frontier molecular orbitals (FMOs) can be regarded as a hybridization of short-range CT in the MR core and intramolecular CT in peripheral moieties. The FMOs in the MR core are satisfied with the classic MR effect, i.e. atomic localization over the whole MR structure in an alternating pattern. While, at the peripheral moieties, as the electron donor strengthens, the deviation of atom contributions in peripheral moieties between the HOMO and LUMO gradually increases (Table S4, ESI†), which indicates that the CT effect is strengthened.47 Meanwhile, oscillator strengths (f) are estimated to be 0.128, 0.117, 0.108 and 0.087 for CzAO, MQAO, QPXO and QPO, respectively, which support that the intramolecular CT is strengthened with increasing donor ability. The CT analysis is in agreement with the photophysical (vide supra) and electronic (vide infra) experiments.
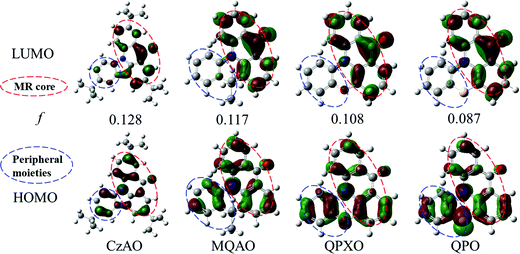 |
| | Fig. 3 The calculated electron density distributions (isovalue = 0.035) of FMOs and oscillator strengths (f) using the TD-B3LYP/6-31G(d) level in vacuum. The red-dotted circle means the MR core, and the blue-dotted circle means the peripheral moieties. | |
In theory, increasing various vibrational couplings may cause the broadening of the FWHM, which correlates with the molecular structural rigidity and symmetric factors such as planarity. We thus evaluate the structure of these four compounds from the viewpoints of planarity and rigidity. For the former, the calculation of the molecular planarity parameter (MPP)48 was conducted using the Multiwfn 3.8 program.49 The results are displayed in Fig. S17 (ESI†), where the atoms exactly on the fitted plane are shown in white, while those above and below the plane are shown in blue and red, respectively. Accordingly, the decrease of molecular planarity is in the order of CzAO < QPXO < MQAO < QPO, which is consistent with the increase of the corresponding torsion angle (vide supra). Furthermore, the rigidity of molecules can be quantified by the root-mean-square deviations (RMSDs) and reorganization energies (λ). Evidently, RMSDs are calculated to be 0.1078, 0.1234, 0.1139 and 0.1758 Å for CzAO, MQAO, QPXO and QPO, respectively (Fig. S18, ESI†). Additionally, the λ values are calculated to be 0.19, 0.82, 0.82 and 1.06 eV for CzAO, MQAO, QPXO and QPO, respectively (Fig. S19, ESI†). Similar to the reorganization energy, the Huang–Rhys factor50 is another measure to analyse the molecular vibrations upon electronic excitations (Fig. S20, ESI†). The reduction of high-frequency vibrations and the weakening of low-frequency vibrations are beneficial to the FWHM. Nevertheless, the results for these four compounds show that the contribution from the high-frequency vibration is not critical to the Huang–Rhys factor, while the contribution is from those low-frequency scissoring/twisting vibrations (<200 cm−1, Fig. S20, ESI†). As a result, the Huang–Rhys factor is calculated to be in the order of CzAO < QPXO < MQAO < QPO. In brief, both computational approaches in terms of molecular planarity (MPP) and rigidity (RMSDs, the Huang–Rhys factor and λ) are in mutual agreement, which all predict that the FWHM should follow the trend of CzAO < QPXO < MQAO < QPO. However, this prediction fails to actually agree with the right variation trend of the FWHM. This contradiction indicates that although the Huang factor has been widely reported to predict the FWHM,4,51 it only considers structural changes and ignores the CT effect. It can be inferred that, under a similar molecular structure, the CT effect is the dominant factor for the FWHM.
2.4. Thermostability and electronic property analyses
The thermostability of the four emitters was measured by thermogravimetric analysis and differential scanning calorimetry under a nitrogen atmosphere (Fig. S22, ESI†). As a result, CzAO, MQAO, QPXO and QPO exhibited good thermal stability with a high decomposition temperature (Td, corresponding to 5% weight loss) of 310, 259, 284 and 290 °C, while the melting temperatures (Tm) of these emitters were 261, 194, 204 and 230 °C, respectively. The glass transition temperature (Tg) appeared in CzAO and MQAO with dangling alkyl chains, but was absent in QPXO and QPO. The good thermostability guaranteed the fabrication and applications of OLEDs. Ultraviolet photoelectron spectroscopy (UPS) was used to investigate the electronic properties. As shown in Fig. S22 (ESI†), the tangents of the curves showed the position of the Fermi and the cut-off edges.52,53 Based on the EFermi and the Ecut-off, the HOMO levels were calculated to be −5.55, −5.45, −5.39 and −5.37 eV for CzAO, MQAO, QPXO and QPO (Table S9, ESI†), respectively, which are consistent with the photophysical data and theoretical calculations (vide supra). LUMO levels were achieved from the band gap obtained from the corresponding UV-vis absorption spectrum. All the energy levels of the four emitters were located between the redox potentials of mCPCN, indicating that mCPCN is a suitable host in the OLED application.
2.5. Electroluminescence properties
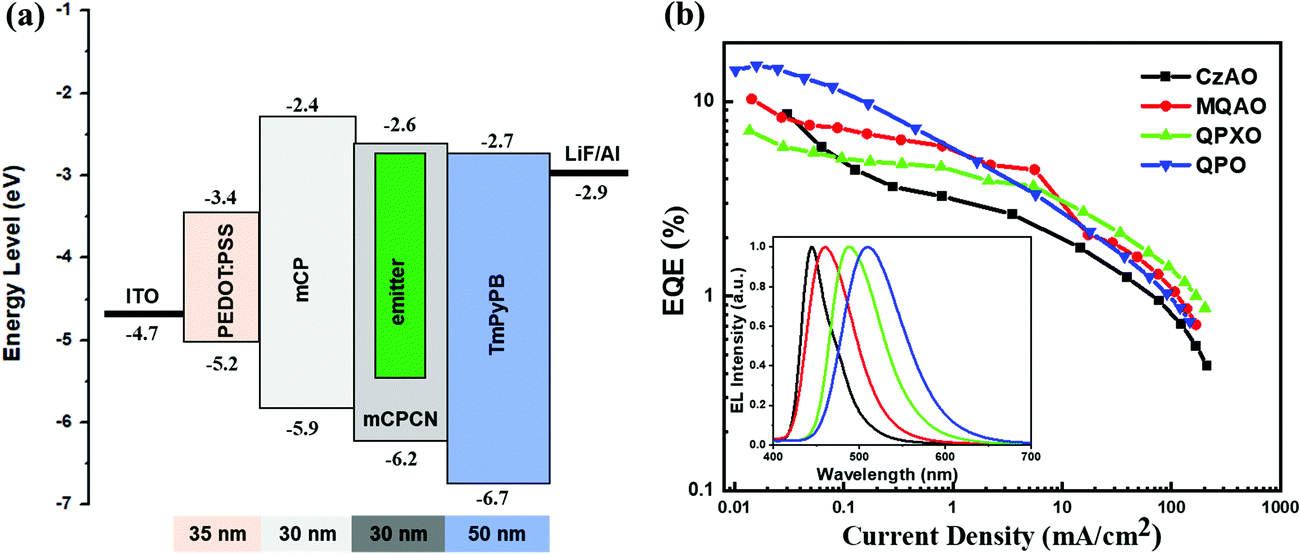
Since these molecules exhibit delayed fluorescence when doped in mCPCN, their electroluminescence properties were evaluated via the fabrication of OLEDs with the following configuration: ITO/PEDOT:PSS (35 nm)/mCP (30 nm)/mCPCN:3 wt% enantiomers (30 nm)/TmPyPB (50 nm)/LiF (0.5 nm)/Al (120 nm), where PEDOT:PSS, mCP and TmPyPB acted as the hole-injection, hole-transport and electron-transport layers, respectively (Fig. S23, ESI†). As shown in Fig. 4, the EL emission peak shows the same trend as the PL peak, being shifted from 444 nm of CzAO to 460 nm of MQAO, 486 nm of QPXO, and 510 nm of QPO, which correspond to Commission Internationale de l'éclairage (CIE) coordinates of (0.148, 0.065), (0.142, 0.139), (0.166, 0.406) and (0.237, 0.529), respectively. The EQEmax values are measured to be 8.62%, 10.28%, 7.05% and 15.29% for CzAO, MQAO, QPXO and QPO, respectively (Table 2), which are consistent with their PLQYs in solid films. All the devices had a severe efficiency roll-off with increasing driving voltage, which may be caused by the long delayed lifetime of exciton (vide supra) induced triplet–triplet annihilation as well as unwanted chemical reactions and hence defect accumulation.
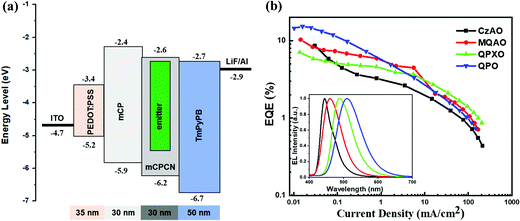 |
| | Fig. 4 (a) The energy level diagram of the devices and (b) EQE versus current density with electroluminescence spectra shown in the inset. | |
Table 2 EL performance of CzAO, MQAO, QPXO and QPO
| Enantiomer |
V
on (V) |
L
max (cd m−2) |
CEmax (cd A−1) |
PEmax (lm W−1) |
EQEmax (%) |
Peak (nm) |
FWHM (nm eV−1) |
CIE (x, y) |
| CzAO |
4.0 |
636 |
5.46 |
4.28 |
8.62 |
444 |
43/0.26 |
(0.148, 0.065) |
| MQAO |
3.6 |
1368 |
11.02 |
9.60 |
10.28 |
460 |
62/0.34 |
(0.142, 0.139) |
| QPXO |
3.6 |
4044 |
16.20 |
14.11 |
7.05 |
486 |
67/0.34 |
(0.166, 0.406) |
| QPO |
5.2 |
3217 |
45.56 |
25.96 |
15.29 |
510 |
82/0.38 |
(0.237, 0.529) |
3. Conclusion
In summary, we have successfully synthesized a series of carbonyl/amine compounds by fixing the acceptor of carbonyl and changing the donor. These emitters exhibited a unique electron density distribution, which is a hybridization of short-range CT in the MR core and intramolecular CT in peripheral moieties. OLEDs based on these emitters exhibited narrowband emissions with EQEs of 7.0–15.3%. Through detailed analyses of photophysical data and theoretical calculations, a new point of view is proposed that, under a similar molecular structure, the CT effect is the dominant factor for the FWHM, instead of structural changes. The CT strength increase will inevitably be accompanied by broadening of the FWHM but a decrease of ΔEST. The former is detrimental to color purity, while the latter is beneficial to RISC and TADF. The results thus provide insight into MR emitters with a narrow FWHM and a small ΔEST by regulating the molecular parameters.
Author contributions
Jing-Wei Huang, Yu-Chieh Hsu and Xiugang Wu contributed equally to this work. Xiugang Wu, Weiguo Zhu and Pi-Tai Chou conceived the project. Jing-Wei Huang, Yu-Chieh Hsu and Xiugang Wu conducted the experiments, analyzed the data, and wrote the paper. Sai Wang and Wen-Yi Hung fabricated the devices. Xiang-Qin Gan, Wei-Qiong Zheng, Hu Zhang and Yin-Zhi Gong assisted in the characterization of the structure and provided helpful discussions. All the authors commented on the paper.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (52073035 and U1663229), the Natural Science Foundation of Jiangsu Province (BK20211335), the Research Innovation Program for Postgraduates of Jiangsu Province (KYCX21_2792, KYCX21_2805 and SJCX21_1247), the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Provincial Talents Project of High-Level Innovation and Entrepreneurship, the Science and Technology Project of Changzhou University (ZMF20020429), the Analysis and Testing Center, NERC Biomass of Changzhou University, and the Ministry of Science and Technology (MOST), Taiwan (110-2639-M-002-001-ASP).
References
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed.
- X. Liang, Z. P. Yan, H. B. Han, Z. G. Wu, Y. X. Zheng, H. Meng, J. L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed.
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 CrossRef CAS PubMed.
- S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 3082–3089 RSC.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311–9314 CrossRef CAS PubMed.
- J. A. Knoller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier, A. Kohler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- Y. Yuan, X. Tang, X. Y. Du, Y. Hu, Y. J. Yu, Z. Q. Jiang, L. S. Liao and S. T. Lee, Adv. Opt. Mater., 2019, 7, 1801536 CrossRef.
- X. Li, Y. Z. Shi, K. Wang, M. Zhang, C. J. Zheng, D. M. Sun, G. L. Dai, X. C. Fan, D. Q. Wang, W. Liu, Y. Q. Li, J. Yu, X. M. Ou, C. Adachi and X. H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 13472–13480 CrossRef CAS PubMed.
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2019, 8, 1901627 CrossRef.
- F. Chen, L. Zhao, X. Wang, Q. Yang, W. Li, H. Tian, S. Shao, L. Wang, X. Jing and F. Wang, Sci. China: Chem., 2021, 64, 547–551 CrossRef CAS.
- S. Y. Yang, S. N. Zou, F. C. Kong, X. J. Liao, Y. K. Qu, Z. Q. Feng, Y. X. Zheng, Z. Q. Jiang and L. S. Liao, Chem. Commun., 2021, 57, 11041–11044 RSC.
- H. Min, I. S. Park and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 7643–7648 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912 CrossRef CAS PubMed.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, 2004072 CrossRef CAS PubMed.
- Y. Wang, Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai and J. Wei, Angew. Chem., Int. Ed., 2020, 59, 17442–17446 CrossRef PubMed.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen and L. Duan, Angew. Chem., Int. Ed., 2020, 59, 17499–17503 CrossRef CAS PubMed.
- D. Sun, S. M. Suresh, D. Hall, M. Zhang, C. Si, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, X. Zhang and E. Zysman-Colman, Mater. Chem. Front., 2020, 4, 2018–2022 RSC.
- J. Zhang, L. Xu and W.-Y. Wong, Coord. Chem. Rev., 2018, 355, 180–198 CrossRef CAS.
- J. Zhang, L. Wang, A. Zhong, G. Huang, F. Wu, D. Li, M. Teng, J. Wang and D. Han, Dyes Pigm., 2019, 162, 590–598 CrossRef CAS.
- J. Zhang, H. Ye, Y. Jin and D. Han, Top. Curr. Chem., 2021, 380, 6 CrossRef PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 CrossRef CAS PubMed.
- X. C. Fan, K. Wang, Y. Z. Shi, J. X. Chen, F. Huang, H. Wang, Y. N. Hu, Y. Tsuchiya, X. M. Ou, J. Yu, C. Adachi and X. H. Zhang, Adv. Opt. Mater., 2022, 10, 2101789 CrossRef CAS.
- A. Pershin, D. Hall, V. Lemaur, J. C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- H. L. Lee, W. J. Chung and J. Y. Lee, Small, 2020, 16, 1907569 CrossRef CAS PubMed.
- V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W. J. Son, S. O. Jeon and J. Y. Lee, Adv. Sci., 2021, 8, 2101137 CrossRef CAS PubMed.
- J. Wei, C. Zhang, D. Zhang, Y. Zhang, Z. Liu, Z. Li, G. Yu and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 12269–12273 CrossRef CAS PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
- X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo and Y. Ma, Adv. Opt. Mater., 2020, 9, 2001845 CrossRef.
- D. Zhang and L. Duan, Nat. Photonics, 2021, 15, 173–174 CrossRef CAS.
- X. Wu, J. W. Huang, B. K. Su, S. Wang, L. Yuan, W. Q. Zheng, H. Zhang, Y. X. Zheng, W. Zhu and P. T. Chou, Adv. Mater., 2022, 34, 2105080 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- R. Ansari, W. Shao, S. J. Yoon, J. Kim and J. Kieffer, ACS Appl. Mater. Interfaces, 2021, 13, 28529–28537 CrossRef CAS PubMed.
- T. Yamato, M. Komine and K. Matsuo, J. Chem. Res., Synop., 1997, 82–83, 10.1039/a605730f.
- K. Wang, C. J. Zheng, W. Liu, K. Liang, Y. Z. Shi, S. L. Tao, C. S. Lee, X. M. Ou and X. H. Zhang, Adv. Mater., 2017, 29, 1701476 CrossRef PubMed.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- S. N. Zou, C. C. Peng, S. Y. Yang, Y. K. Qu, Y. J. Yu, X. Chen, Z. Q. Jiang and L. S. Liao, Org. Lett., 2021, 23, 958–962 CrossRef CAS PubMed.
- P. T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed.
- Y.-H. Cheng, A. Belyaev, M.-L. Ho, I. O. Koshevoy and P.-T. Chou, Phys. Chem. Chem. Phys., 2020, 22, 27144–27156 RSC.
- Y. Tsuchiya, Y. Ishikawa, S. H. Lee, X. K. Chen, J. L. Brédas, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2021, 9, 2002174 CrossRef CAS.
- T. Lu, J. Mol. Model., 2021, 27, 263 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. P. Yan, T. T. Liu, R. Wu, X. Liang, Z. Q. Li, L. Zhou, Y. X. Zheng and J. L. Zuo, Adv. Funct. Mater., 2021, 31, 2103875 CrossRef CAS.
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, CCS Chem., 2021, 3, 2077–2091 CrossRef.
- A. Kahn, Mater. Horiz., 2016, 3, 7–10 RSC.
- M. R. Habib, H. Li, Y. Kong, T. Liang, S. M. Obaidulla, S. Xie, S. Wang, X. Ma, H. Su and M. Xu, Nanoscale, 2018, 10, 16107–16115 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2119761–2119764. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc06165h |
| ‡ J.-W. H., Y.-C. H. and X. W. made equal contributions. |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *c,
Pi-Tai
Chou
*c,
Pi-Tai
Chou